

Abstract
This work provides a proposal for proper determination of matrix effects and extraction efficiencies as an integral part of full validation of liquid chromatography coupled to tandem mass spectrometry-based multiclass methods for complex feedstuff. Analytical performance data have been determined for 100 selected analytes in three compound feed matrices and twelve single feed ingredients using seven individual samples per matrix type. Apparent recoveries ranged from 60–140% for 52–89% of all compounds in single feed materials and 51–72% in complex compound feed. Regarding extraction efficiencies, 84–97% of all analytes ranged within 70–120% in all tested feed materials, implying that signal suppression due to matrix effects is the main source for the deviation from 100% of the expected target deriving from external calibration. However, the comparison between compound feed and single feed materials shows great variances regarding the apparent recoveries and matrix effects. Therefore, model compound feed formulas for cattle, pig, and chicken were prepared in-house in order to circumvent the issue of the lack of a true blank sample material and to simulate compositional uncertainties. The results of this work highlight that compound feed modeling enables a more realistic estimation of the method performance and therefore should be implemented in future validation guidelines.
Keywords: residues, mycotoxins, veterinary drugs, pesticides, compound feed, recovery, model matrices
1. Introduction
A number of influencing factors such as storage and climate conditions, cultivation practices, and processing contribute to the presence of a large variety of undesired substances in the food and feed chain. Besides anthropogenic inputs, by purpose-related use such as pesticides and veterinary drugs, food safety is additionally challenged by the occurrence of natural contaminants such as secondary fungal metabolites or plant toxins.1 Liquid chromatography coupled to tandem mass spectrometry (LC–MS/MS) gained more and more attention within the last decades and has become the instrumental technique of choice for a precise and reliable determination of trace compounds in complex food and feed material.2 However, the high sample complexity and substance-related physicochemical diversity hamper quantitative extraction of target molecules from the sample material. Although in routine pesticide analysis modified QuEChERS (quick, easy, cheap, effective, rugged, and safe) extraction procedures are most commonly applied, recent sample preparation protocols in multiclass analysis follow an even more straightforward and economic extraction approach which is applicable for multiple analytes from various substance classes.3,4 These generic extraction protocols are based on a simple dilution of the sample extract after a fast solid–liquid extraction, which represents an optimal compromise in terms of work and resource consumption, extraction efficiency, and analytical quality. To ensure advanced laboratory quality assurance measures in a routine-orientated environment, a precise characterization of analytical performance parameters in target matrices is inevitable. However, the maintenance of such extended quality assurance is significantly hampered by increasing sample heterogeneity. Particularly, in the field of animal feed analysis, the sample matrix is often characterized by a highly complex nature and exact specifications of feed rations are therefore not given. Based on the European Commission regulation 767/2009, animal feed is differentiated as feed materials and compound feed. Feed materials are defined as products of vegetable or animal origin, whose principal purpose is to meet animals’ nutritional needs, in their natural state, fresh or preserved, and products derived from industrial processing.5 These products are intended for use in oral animal feeding either directly as such, or after processing, or in preparation of compound feed. This category includes cereal grains (e.g., barley, maize, triticale, and wheat), oil seeds and oil fruits (rape seed, soy, sesame, and sunflower), legume seeds (horse beans, lentils, peas, and vetches), tubers and roots (sugar beet, beet pulp, carrots, and potato), other seeds and fruits (acorn, buckwheat, red clover, and fruit pulp), forages and roughage (beet leaves, alfalfa, silages, and straw), other plants (algae, barks, leaves, and mint), milk products (butter, casein, milk fat, and whey), as well as land animal products, fish products, minerals, and products obtained by fermentation using microorganisms.6 In contrast, compound feed is defined as a mixture of at least two feed materials whether or not containing feed additives, for oral animal feeding in the form of complete or complementary feed. By reason of its composition, complete feed on the one hand is sufficient for a daily ration, whereby complementary feed on the other hand is only sufficient if used in combination with other feed sources.5 Considering animal species-specific properties and growth status, the individual feed rations are prepared in order to meet the animal-related physiological requirements, leading to high compositional differences in feed formulas.7 Because standardization of the global feed production is not feasible and the compound feed market is growing steadily (+58% compound feed production in EU28 between 1989 and 2018), extensive validation processes are necessary in order to meet the high demands on feed and food safety. So far, trace analysis in compound feed has been rather neglected, but the growing production figures show that there is a rising need for action in the field of method validation and guideline regulations.
Current validation guidelines of the German accreditation body (DAkkS) are exclusively focusing on the validation of single feed material, leading to a potential discrepancy between analytical performance measures derived from validation data and data based on real world samples.8
In this work, method performance data for 80 fungal metabolites, 11 pesticides, and 9 pharmaceutical active agents in three different compound feed and twelve different single feed matrices were determined. Based on these data, the applicability of the current animal feed validation guidelines to multiclass methods is discussed. This study presents the first comprehensive overview and comparison of analytical performance data in complex compound feed and its single feed ingredients and differs significantly from studies which exclusively evaluated data on individual feed components.
2. Materials and Methods
2.1. Chemicals and Reagents
LC gradient-grade methanol and acetonitrile and MS-grade ammonium acetate and glacial acetic acid (p.a.) were purchased from Sigma-Aldrich (Vienna, Austria). A Purelab Ultra system (ELGA Lab Water, Celle, Germany) was used for further purification of reverse osmosis water.
Standards of fungal and bacterial metabolites, pesticides, and pharmaceuticals were either purchased from Romer Labs Inc. (Tulln, Austria), Sigma-Aldrich (Vienna, Austria), Iris Biotech GmbH (Marktredwitz, Germany), Axxora Europe (Lausanne, Switzerland), NEOCHEMA GmbH (Bodenheim, Germany), Restek GmbH (Bad Homburg, Germany), BioAustralis (Smithfield, Australia), AnalytiCon Discovery (Potsdam, Germany), Adipogen AG (Liestal, Switzerland), and LGC Promochem GmbH (Wesel, Germany) or were obtained as gifts from various research groups. Each analyte was dissolved in acetonitrile (primarily), acetonitrile/water 1:1 (v/v), methanol, methanol/water 1:1 (v/v), or water.
By mixing the stock solutions of the corresponding analyte, 74 combined working solutions were prepared for fungal toxins, 9 working solutions for pesticides, and 8 for pharmaceutical active agents and were stored at −20 °C. For spiking purposes, a liquid multi-analyte standard was freshly prepared by combining the intermediate working mixtures deriving from liquid stock solutions.
2.2. Instrumental Conditions
A detailed description of the analytical procedure for this study was published elsewhere.9 Briefly, the detection was carried out with a QTrap 5500 MS/MS system (SCIEX, Foster City, CA, USA) equipped with a TurboV source and an electrospray ionization (ESI) probe coupled to a 1290 series UHPLC system (Agilent Technologies, Waldbronn, Germany). Chromatographic separation was performed at 25 °C on a Gemini C18-column, 150 × 4.6 mm i.d. and a particle size of 5 μm (Phenomenex, Torrance, CA, US). The column was equipped with a C18 security guard cartridge, 4 × 3 mm i.d. (Phenomenex, Torrance, CA, US).
The autosampler program included an injection volume of 5 μL, and elution was carried out in the binary gradient mode following a flow rate of 1 mL/min. Mobile phase A was composed of methanol/water/acetic acid 10:89:1 (v/v/v) and mobile phase B was composed of methanol/water/acetic acid 97:2:1 (v/v/v). Both mobile phases contained 5 mM ammonium acetate. Gradient conditions started with 100% A after an initial time of 2 min. After 3 min, the proportion of B was increased linearly to 50%. Within 9 min, mobile phase B was increased to 100% followed by a hold time of 4 and 3.5 min column re-equilibration at 100% A.
The analytical measurement was carried out in two successive chromatographic runs in the positive and negative polarity mode following a scheduled multiple reaction monitoring (sMRM) algorithm with a run time of 21 min each. For increased confidence in compound identification, two sMRM transitions per analyte were acquired according to the SANTE/11813/2017 validation guideline.10
2.3. Data Evaluation
2.3.1. Calibration and Quantitation
External neat solvent calibration was performed by diluting suitable volumes of multi-analyte standard working solutions. The final calibrant solution contained 300 μL of multitoxin working solution, 120 μL of pesticide solution, 120 μL of veterinary drug solution, 20 μL of a certified liquid standard of fumonisin B1 and B2, and 20 μL of a certified liquid standard of fumonisin B3. Because the concentration of fumonisins does not remain stable in the almost pure acetonitrile multi-analyte solution, they were added at this late stage.
Serial dilution was performed with acetonitrile/water/formic acid (49.5/49.5/1, v/v/v) to obtain calibration levels of 1:3, 1:10, 1:30, 1:100, 1:300, and 1:1000. Linear 1/x weighted calibration curves were obtained for the solvent standards in order to check the linearity of the response. MultiQuant 3.0.3 (SCIEX, Foster City, CA, USA) software was used to construct the calibration curve and perform peak integration. Final data evaluation was performed in Microsoft Excel 2013. Graphical content was prepared using the open access visualization tool Flourish (Kiln Enterprises Ltd, London, UK).
2.3.2. Performance Parameters
Performance characteristics of the method were evaluated by the apparent recovery (RA), the matrix effects, expressed by signal suppression/enhancement (SSE), and the recovery of the extraction step (RE). The parameters were calculated from the peak areas of the samples spiked before and after the extraction and the neat solvent standards.
2.4. Set of Analytes
The described analytical approach was originally designed for the determination of 39 mycotoxins in cereals in the year 2006.11 Since then, the method has been extended and improved continuously to a wide range of additional secondary metabolites of fungi and bacteria, plant toxins, pesticides, and veterinary drugs.9,12,13 In order to ensure an adequate workload and time management, a set of 100 analytes, including 80 secondary fungal metabolites (including all regulated mycotoxins), 11 pesticides, and 9 pharmaceutical active agents, was chosen. To guarantee a high representativeness, the selected analytes were evenly distributed over the whole chromatogram covering differences in physicochemical characterization such as acidity, hydrophobicity, functional groups, and ESI polarity. An overview of the selected set of representative analytes is depicted in Table 1.
Table 1. Overview of the Investigated Analytes, Categorized by the Substance Class, Including Spiking Concentrations in μg/kga.
| analyte | substance class | polarity | concentration [μg/kg] |
|---|---|---|---|
| lotaustralin | FM | neg | 604 |
| altertoxin-I | FM | neg | 388 |
| agistatin E | FM | neg | 287 |
| gibberellic acid | FM | neg | 259 |
| 3-nitropropionsäure | FM | neg | 223 |
| pseurotin A | FM | neg | 165 |
| alpha-zearalenol | FM | neg | 95 |
| macrosporin | FM | neg | 87 |
| cladosporin | FM | neg | 79 |
| moniliformin | FM | neg | 78 |
| alternariolmethylether | FM | neg | 53 |
| fusarenon-X | FM | neg | 51 |
| 3-acetyldeoxynivalenol | FM | neg | 51 |
| deoxynivalenol | FM | neg | 51 |
| nivalenol | FM | neg | 51 |
| zearalenone | FM | neg | 51 |
| averantin | FM | neg | 49 |
| norsolorinic acid | FM | neg | 48 |
| malformin C | FM | neg | 43 |
| curvularin | FM | neg | 34 |
| ternatin | FM | neg | 34 |
| altersetin | FM | neg | 30 |
| amidepsin B | FM | neg | 30 |
| andrastin A | FM | neg | 30 |
| averufin | FM | neg | 30 |
| dihydrocitrinone | FM | neg | 30 |
| meleagrin | FM | neg | 30 |
| phomalone | FM | neg | 30 |
| thielavin B | FM | neg | 30 |
| equisetin | FM | neg | 28 |
| fumiquinazolin A | FM | neg | 27 |
| ilicicolin A | FM | neg | 27 |
| cercosporamide | FM | neg | 27 |
| alternariol | FM | neg | 27 |
| emodin | FM | neg | 27 |
| pinselin | FM | neg | 24 |
| versicolorin A | FM | neg | 24 |
| cylindrocarpon A4 | FM | neg | 12 |
| atpenin | FM | neg | 10 |
| asperphenamate | FM | neg | 3 |
| bentazon | P | neg | 50 |
| dinoseb | P | neg | 50 |
| fluazinam | P | neg | 50 |
| novaluron | P | neg | 50 |
| sulfoxaflor | P | neg | 50 |
| carprofen | VD | neg | 50 |
| florfenicol | VD | neg | 50 |
| flumethasone | VD | neg | 50 |
| mefenamic acid | VD | neg | 50 |
| chloramphenicol | VD | neg | 34 |
| fumonisin B1 | FM | pos | 404 |
| fumonisin B2 | FM | pos | 400 |
| 15-acetyldeoxynivalenol | FM | pos | 286 |
| chetomin | FM | pos | 286 |
| neosolaniol | FM | pos | 191 |
| secalonic acid D | FM | pos | 145 |
| gliotoxin | FM | pos | 129 |
| fumigaclavine C | FM | pos | 121 |
| mycophenolic acid | FM | pos | 75 |
| 15-hydroxyculmorin | FM | pos | 73 |
| cytochalasin B | FM | pos | 72 |
| cytochalasin J | FM | pos | 72 |
| roquefortine C | FM | pos | 72 |
| griseofulvin | FM | pos | 65 |
| sulochrine | FM | pos | 65 |
| aflatoxin M1 | FM | pos | 52 |
| diacetoxyscirpenol | FM | pos | 51 |
| HT-2 toxin | FM | pos | 51 |
| T-2 toxin | FM | pos | 51 |
| monoacetoxyscirpenol | FM | pos | 42 |
| penitrem A | FM | pos | 40 |
| 3-methylsterigmatocystin | FM | pos | 39 |
| cyclopenin | FM | pos | 39 |
| ochratoxin A | FM | pos | 38 |
| brevianamid F | FM | pos | 36 |
| questiomycin A | FM | pos | 32 |
| sterigmatocystin | FM | pos | 27 |
| destruxin A | FM | pos | 24 |
| ochratoxin B | FM | pos | 20 |
| anisomycin | FM | pos | 18 |
| aflatoxin B1 | FM | pos | 17 |
| aflatoxin B2 | FM | pos | 17 |
| aflatoxin G1 | FM | pos | 17 |
| aflatoxin G2 | FM | pos | 17 |
| fungerin | FM | pos | 12 |
| quinolactacin A | FM | pos | 12 |
| herquline A | FM | pos | 8 |
| ergine | FM | pos | 3 |
| ergocristine | FM | pos | 3 |
| enniatin A1 | FM | pos | 0.55 |
| aspon | P | pos | 50 |
| cyromazine | P | pos | 50 |
| dithiopyr | P | pos | 50 |
| ethirimol | P | pos | 50 |
| permethrin | P | pos | 50 |
| prometon | P | pos | 50 |
| rofecoxib | VD | pos | 50 |
| sulfamethoxazole | VD | pos | 50 |
| tiamulin | VD | pos | 50 |
| tilmicosin | VD | pos | 50 |
Fungal metabolite (FM), pesticide (P), and veterinary drug (VD).
2.5. Spiking and Extraction Procedure
The extraction procedure is used for the routine analysis of contaminated food and feedstuff, basically the animal feed material. Therefore, 5 g of the sample is extracted with 20 mL of the extraction solvent (acetonitrile/water/acetic acid 79:20:1, v/v/v) and shaken using a rotary shaker (GFL 3017, Burgwedel, Germany) for 90 min under horizontal conditions. To improve the extraction for fumonisins, the pH value of the extraction solvent was lowered to pH 4 using formic acid instead of acetic acid, following the original dilution ratio. The improved extraction under strong acidic conditions is apparently structure-related because fumonisins contain several carboxyl groups.14
For spiking purposes, an appropriate amount of multi-analyte working solutions (50 μL of multi-toxin solution, 25 μL of pesticide solution, 25 μL of veterinary drug solution, and 20 μL of fumonisin solution) was added to 0.25 g of homogenized samples. The miniaturization of the spiking protocol was performed for the economical use of standards. In order to isolate matrix effects, the obtained spike concentrations were matched to calibrant standard dilution levels of the higher working range, such as 1:10 and 1:30. For mycotoxins addressed by regulatory limits, the spiking concentrations were far below the guidance values and in the range of the regulatory limit for aflatoxins in feed.15,16 The difference between the lowest and highest concentration levels (0.55 μg/kg for enniatin A1 and 604 μg/kg for lotaustralin) investigated in this study amounted to a factor of 100.
To avoid analyte degradation and to ensure solvent evaporation, the spiked samples were stored in darkness and at room temperature overnight. This step ensures proper equilibration between matrix and analytes. On the next day, the samples were extracted using 1 mL of the extraction solvent and were shaken for 90 min using a rotary shaker. Finally, the samples were centrifuged at 3500 rpm for 5 min. After transferring the supernatant (300 μL) into high-performance liquid chromatography (HPLC) vials, the same volume of a complementary dilution solvent (acetonitrile/water/formic acid 20:79:1, v/v/v) was added and mixed properly. Finally, 5 μL of the diluted raw extract was injected into the LC–MS/MS system without further cleanup.
For post extraction spiking, 5 g of the sample was extracted with 20 mL of the extraction solvent. The supernatant (400 μL) was fortified with an appropriate amount of spiking solution (20 μL of multi-toxin solution, 10 μL of pesticide solution, 10 μL of veterinary drug solution, and 8 μL of fumonisin solution), diluted with 352 μL of the dilution solvent and injected as described above.
2.6. Samples
Three matrices of real and model compound feed (with distinct differences in their composition) and twelve matrices of single feed material including alfalfa, barley, maize, horse beans (broad beans), distiller’s dried grains with solubles (DDGS), rapeseed, silage, soy, sunflower cake, triticale, wheat, and wheat bran were chosen for this study. Cattle feed was taken as a matrix with high amounts of forage crops. Matrices with high grain content were represented by pig and chicken feed. Between four and seven different lots of each matrix type were collected, in order to maximize the intrasubject variation and challenge the reproducibility of the extraction protocol. The heterogeneous set of individual raw samples was provided by the companies BIOMIN GmbH (Getzersdorf, Austria), LVA GmbH (Klosterneuburg, Austria), Garant-Tiernahrung GmbH (Pöchlarn, Austria), Romer Labs Diagnostic GmbH (Tulln, Austria), and Bipea (Paris, France). The model compound feed formulas were prepared following the information provided by our company partners BIOMIN GmbH and Garant-Tiernahrung GmbH and are illustrated in Table 2 (compositional information might vary from country to country and has to be collected by national feed producers in order to apply this approach in other laboratories). In total, 42 compound feed samples (21 real and 21 model) and 73 single feed matrix replicates were evaluated. The detailed model matrix composition is illustrated in the work sheet “samples” in the Supporting Information (Table S1). Homogenization of the samples was carried out using an Osterizer blender (Sunbeam Oster Household Products, Fort Lauderdale, FL, USA).
Table 2. Compositional Information of the Real-World Samples and in-House-Prepared Prehomogenized Model Matrices, Expressed as the Maximum Share in Percenta.
| maximum share (%) |
||||||
|---|---|---|---|---|---|---|
| cattle |
pig |
chicken |
||||
| real | model | real | model | real | model | |
| additives | 7 (4) | |||||
| barley | 18 (6) | 24 (7) | 29 (4) | 30 (5) | ||
| broad beans | 22 (5) | 22 (7) | ||||
| corn meal | 4 (1) | |||||
| DDGS | 35 (3) | 10 (4) | 10 (5) | 10 (5) | ||
| maize | 20 (3) | 20 (7) | 44 (4) | 44 (7) | 62 (7) | 74 (7) |
| peas | 7 (3) | |||||
| rapeseed | 0.8 (1) | 3.5 (2) | 5 (7) | |||
| rice bran | 15 (1) | |||||
| rye | 25 (4) | |||||
| silage | 26 (7) | 26 (7) | ||||
| soy | 27 (4) | 35 (7) | 30 (7) | 30 (7) | ||
| sunflower cake | 10 (5) | 10 (6) | ||||
| triticale | 18 (4) | 21 (6) | 15 (1) | 10 (3) | ||
| unknown | 37 (6) | 100 (4) | 17 (6) | |||
| wheat | 29.7 (4) | 30 (5) | 20 (1) | |||
| wheat bran | 18 (6) | 20 (7) | ||||
Numbers in brackets represent the absolute prevalence of compound feed samples containing the respective individual single feed ingredient.
3. Results and Discussion
3.1. Validation of Multiresidue Methods in Feed
Multimethods covering dozens or even hundreds of analytes are characterized using a high number of compounds, which differ in polarity, structural formulas, and physicochemical properties. With single-residue methods, compounds may be extracted almost quantitatively, and optionally, after clean up determined with the help of one and/or several specific detectors. In contrast, a high level of compromise is needed for the development of multiple methods, especially accounting for complex sample materials, in which the applicability of analyte-specific extraction and purification steps is extremely limited. Because of its compositional variability, feed represents one of the most complex sample materials and therefore requires powerful and reliable analytics. In routine laboratories, multiple methods are frequently covering more than 300 individual compounds which are subject to matrix validation procedures. Based on the validation guide from the German accreditation body for multiresidue methods in feed, the matrix validation can be conducted in groups for the specific feed type (Table 3). To obtain accreditation for feed matrices in general, the analysis of the active substances in each group must be validated by selecting at least one matrix from the corresponding feed group.8 In order to include a multimethod in the scope of accreditation, the laboratory must be able to determine at least 75% of the target analytes with a satisfactory performance per group, following SANTE criteria for pesticide analysis in terms of reproducibility and repeatability.8,17
Table 3. Overview of Animal Feed Groups for Validation Purposes of Multimethods8.
| no. | feed group | characteristics | matrix example |
|---|---|---|---|
| F1 | cabbage vegetable | high water content | kale |
| forage plant | weeds, alfalfa, clover, rape | ||
| leaves from root and tuber vegetables | sugar beet leaves | ||
| silage | maize, clover, weeds | ||
| F2 | fruit pulp | high acidic and high water content | citrus fruits |
| F3 | extraction cake | high sugar and low water content | rape extraction cake |
| F4a | oils and oilseeds | high fat and very low water content | sunflower, rapeseeds |
| F4b | oil fruits | high fat and moderate water content | soybeans, olives |
| F5 | cereals | low water and low fat content, high starch, and/or protein content | wheat, rye, barley, maize, rice, oat grains |
| hay | weeds | ||
| legumes | horse bean, lenses | ||
| straw | wheat, rye, barley, oat | ||
| F6 | special matrices | ||
| F7 | meat, fish and shellfishes | animal-based compound feed | feed from fish farms |
| F8 | milk and milk products | ||
| F9 | eggs | ||
| F10 | fat from animal-based compound feed | fat-based compound feed |
Related to the method performance, significant variations may occur because of the high number of analyte/matrix combinations.9 Variations within the analytical performance data have to be collected in the validation process and, if necessary, reduced by adequate adaptations of the extraction step and/or chromatographic conditions.
3.2. Influencing Criteria on the Method Performance
3.2.1. Valid Analytical Methods
For routine laboratories working in the food and feed sector, the use of confirmatory methods which comply with the requirements of international standardizing organizations, such as Codex Alimentarius Commission, the European Committee for Standardization (CEN), AOAC International (AOACI), or the European Union, is essential. Therefore, valid analytical methods require the determination of accuracy, covering trueness, and precision.18 The accuracy is defined as the closeness of the measurement result to the true or accepted reference value and thus combines both, precision and trueness.10 In this study, a comprehensive spike-and-recovery experiment was carried out in order to assess the accuracy of the method.
3.2.1.1. Apparent Recovery
The apparent recovery is a parameter combining the recovery of the analyte from the matrix by the sample extraction procedure and matrix effects and has also been termed as “process efficiency”.19 Generally, the apparent “recovery” should be in the range of 70–120%.10,20 In routine analysis, recovery rates between 60 and 140% are still acceptable.8 If the recovery rate is outside this range, it must be shown that the method variability allows a reliable quantitative statement.8,9 In particular, low apparent recoveries show adverse effects on the accuracy, especially affecting the limit of quantification.21
The distribution of apparent recoveries for 100 analytes in 6 grain-based feed matrices (A) and 6 matrices including legumes, oilseeds, and forage crops (B) is displayed in Figure 1. Absolute apparent recoveries for each matrix commodity are expressed as average values of the individual lots measured under repeatability conditions. The variety of matrices allows a comprehensive collection of different matrix characteristics such as low water and low fat content, represented by group A commodities such as wheat, barley, maize, or triticale. In contrast, group B is characterized by matrices with high water content such as alfalfa and silages, high fat and very low water content such as sunflower cake and rapeseeds, high fat and moderate water content such as soybeans, and high starch and/or protein content such as horse beans. The spike concentration corresponds to a 1:10 and 1:30 dilution range of the final working solution of the analytical reference standards. RA values are expressed as the mean apparent recovery derived from 4 to 7 different lots of each feed type and were calculated according to equation RA described in 2.3.2. Regarding the RA results, 72% of analytes in maize, 89% in barley, 82% in wheat bran, 52% in DDGS, 88% in triticale, 84% in wheat, 66% in rapeseed, 52% in alfalfa, 52% in silage, 61% in sunflower, 56% in soy, and 84% in horse beans were in the range of 60–140% as described by DAkkS.8 For the analytes outside the acceptance criteria, a combination of low extraction efficiency and high signal suppressions or enhancements was observed.
Figure 1.

Distribution of apparent recoveries through the set of 100 analytes in grains and byproducts (A) and legumes, oilseeds, and forage crops (B).
3.2.1.2. Extraction Efficiency
Currently, there is no official guidance document available which is focusing on the validation of analytical methods for the determination of multiple analytes in compound feed in general.9 This nonavailability opens some gaps in the interpretation of results, which counts, in particular, for the definition of the term recovery. An exact definition is missing and therefore two possible interpretations exist. First, the previously described apparent recovery and the recovery of the analyte from the matrix using the sample extraction procedure.19 Based on the DAkkS guideline, the recovery has to be determined using a single or multi-analyte standard prepared in the respective matrix, which implies the second definition mentioned above.8
The distribution of extraction efficiencies (according to equation RE described in 2.3.2) for 100 analytes in 12 tested feed materials is depicted in Figure 2. Absolute extraction recoveries for each matrix commodity are expressed as average values of the individual lots measured under repeatability conditions. Regarding the RE results, 94% of analytes in maize, 91% in barley, 89% in wheat bran, 90% in DDGS, 94% in triticale, 96% in wheat, 86% in rapeseed, 83% in alfalfa, 91% in silage, 90%, in sunflower, 83% in soy, and 89% in horse beans were in the range of 60–100%. Only 2–4% of analytes in group A (grains and byproducts) and 3–12% of analytes in group B (legumes, oilseeds, and forage crops) show lower extraction recovery than 60%. Low extraction efficiencies were especially observed for altersetin, andrastin A, chetomin, and cyromazine. These compounds share a number of specific alkaline functional groups which might decrease the solubility in the acidified apolar extraction mixture. Performing the extraction process at low pH is necessary for the majority of secondary fungal metabolites as approximately 40% of them contain an acidic moiety.22 Nevertheless, excellent extraction recoveries were observed for the majority of compounds, leading to the conclusion that matrix effects are the main causes for not achieving the required method performance criteria of isolated analytes.
Figure 2.

Distribution of extraction efficiencies through the set of 100 analytes in grains and byproducts (A) and legumes, oilseeds, and forage crops (B).
3.2.1.3. Matrix Effects
In HPLC–ESI–MS/MS, matrix effects are combined consequences between the influence of the matrix entering the electrospray ion source and the chemical nature of the target compound.23,24 The heterogenous environment of feed matrices results in a competition between analyte ions and nonvolatile matrix components. This competition leads to an effective decrease (ion suppression) or increase (enhancement) in the ionization process, expressed as the absolute matrix effect and shows high analyte/matrix-dependent differences.19
An overview of absolute matrix effects in 12 single feed matrices is depicted in Figure 3.25 Moderate absolute matrix effects were particularly observed in grain-based feed materials with median values of 104, 102, 99, 97, and 96% in wheat, triticale, barley, bran, and maize, respectively. In contrast, higher signal suppressions were observed in crops and oilseeds. With 85, 85, 81, 75, and 61% in soy, rapeseed, sunflower, silage, and alfalfa, respectively, matrix effects were considerably more-pronounced in this category. Contrasting effects within their specific feed group were observed for DDGS and horse bean with median values of 72.5 and 100%, respectively. Although the majority of compounds were primarily affected by signal suppressions, some compounds were influenced by an enhancement of the signal (>20%) in almost all matrices. In general, the ion enhancement can be caused by matrix components which act as a dopant, increasing the ionization efficiency of analytes with high ionization energy.26 Furthermore, especially polar analytes in the positive ionization mode are more susceptible to undergo ion suppression.27 The observed signal enhancements in this experiment were evident for rather apolar analytes in the negative ionization mode such as dihydrocitrinone (Rt: 10.0 min), amidepsin B (Rt: 11.1 min), cercosporamide (Rt: 11.5 min), carprofen (Rt: 12.3 min), dinoseb (Rt: 12.6 min), ternatin (Rt: 12.7 min), atpenin (Rt: 13.1 min), novaluron (Rt: 13.2 min), mefenamic acid (Rt: 13.4 min), fluazinam (Rt: 13.7 min), equisetin (Rt: 14.7 min), altersetin (Rt: 15.1 min), and norsolorinic acid (Rt: 16.6 min). Additionally, with moniliformin (Rt: 3.3 min) and gibberellic acid (Rt: 7.1 min), two polar representatives in the negative ionization mode showed similar signal enhancement patterns, which could be caused either by concomitant matrix components or target analytes in the same ion mode.28 The work sheet “single feed material” in the Supporting Information (Table S1) gives a detailed overview about matrix effects, extraction recoveries, and apparent recoveries of the individual single feed matrices.
Figure 3.

Scatter plot illustrating matrix effects in percent (x-axis) expressed as SSE for 12 single feed matrices (y-axis). Each target analyte is depicted by a colored dot. The outlier-corrected box plot includes an interquartile range of 1.5, representing John Tukey’s standard value.25
The obtained results for RA and SSE reflect the high variation in the exact composition of different lots/brands of animal feed which counts for both, single feed material and consequently also for complex feed. Because there is no uniform recipe in the production of complex compound feed, validation protocols of routine-based confirmation methods and scientific focus is mainly set on single feed matrices, for example, grains or silages, as described in several studies.17,29−31 However, because of its variability in composition, complex feedstuff should also be considered in validation approaches for this matrix type. As the exemplary comparison of pseurotin A between real complex cattle feed samples and their main single ingredients in Figure 4 shows, great variances in RA and SSE can be observed. The relative standard deviation derived from 7 different cattle feed lots either for RA (RSD: 32%) or SSE (RSD: 31%) indicates that validation data obtained from individual feed material cannot guarantee a correct and reliable estimation of complex animal feedstuff.
Figure 4.
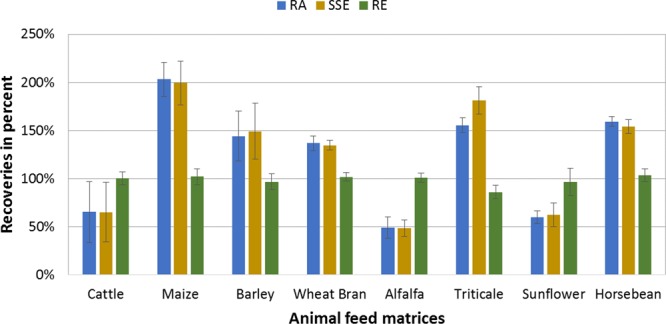
Comparison between real cattle feed and its main individual ingredients for pseurotin A. Apparent recoveries (RA, blue bar), matrix effects (SSE, yellow bar), and extraction efficiencies (RE, green bar) in percent include the error indicator expressed as the relative standard deviation.
This is aggravated by the fact that a comprehensive validation of an analytical approach for animal feed is associated with a very high workload. A complete validation of an average multimethod in each of the listed feed groups in Table 3 would lead to an evaluation of about 60,000 signals (300 compounds × 200 chromatograms, deriving from 10 matrices × 5 lots × 2 concentration levels × 2 (RA, SSE)), blank and calibration data excluded.
Therefore, reconsideration of the current analytical approach must be taken into account, including the economization of resources (standards, measurement time, workload, etc.) and the complexity of compound feed material.
3.3. Preparation of Model Matrices
In order to account for information gaps about the composition of complex feed, model matrices were prepared in-house for three different compound feed types (cattle, chicken, and pig) with seven different lots each. Information regarding the compositional nature of real compound feedstuff was provided by the companies listed in 2.5. In order to minimize the workload and because of the nonavailability of specific feed ingredients, only the main compound feed elements were used for modeling purposes. Furthermore, the proportions of unknown feed ingredients were complemented by increasing the share of the selected known elements.
Beside knowledge of the exact compositional formula, in-house matrix modeling has the advantage to use blank single feed material for the preparation of the individual lots because it is almost impossible to obtain complex feedstuff that is entirely free from charge of natural contaminants.
With seven individual ingredients, cattle feed was the most heterogeneous matrix representative. In contrast, chicken feed mainly consists of maize and soy, leading to the hypothesis that cattle feed is more prone to intrasubject variations than chicken, or pig feed, whose main components are maize, soy, and wheat. In general, no differences were expected between real and model samples in terms of RA, SSE, and RE. Furthermore, accurate intrasubject variations can be simulated by preparing nonidentical individual lots, which better reflect the real conditions in a routine-orientated laboratory, instead of using a single replicate prepared multiple times.
3.3.1. Intrasubject Variation
Multimethod validation procedures are commonly performed based on a single lot of a matrix because there are no particular regulations existing for this matter. However, not considering the intrasubject variation could lead to an additional component of uncertainty during the method validation process. Neglecting the intrasubject variation leads to an underestimation of the measurement uncertainty,32 especially relevant for complex matrices such as compound feed, because of their heterogeneous composition. In official guidance documents, a statement of intrasubject variation or specific performance criteria for this parameter is either limited or completely missing. Only the validation guide of the US Food and Drug Administration for chemical methods requires a minimum number of three different sources per matrix type for the analysis of contaminants.33 In the official validation guidelines of the European Union (EU), the phenomenon of a matrix mismatch is mentioned as a potential source of uncertainty; specific requirements, however, are not formulated.34,35 To avoid an underestimation of the measurement uncertainty and to obtain an accurate estimation of the method performance, the aspect of intramatrix variations was implemented in this study by replicate analysis of seven different matrix lots.
3.3.1.1. Absolute Matrix Effects
Strong matrix effects (>20% SSE) were observed for all complex feed matrices. The distribution of SSE in real and model feed samples is visualized in Figure 5. A detailed overview of the numerical SSE values for real and model matrices is displayed in the work sheets “real compound feed” and “model compound feed” within Table S1. Smaller matrix effects were observed in pig and chicken feed. Concerning pig feed, 42% of analytes in real samples and 43% of analytes in model samples were suppressed/enhanced by <20%. In chicken feed, for both types of samples, 39% of analytes for model and real samples were in the SSE range between 80 and 120% and therefore not affected by matrix effects according to SANTE/11813/2017.10 In contrast, higher matrix effects were observed in cattle feed. In this matrix, only 28% of analytes in real samples and 31% of analytes in model samples were not affected by SSE, indicating that the analysis of cattle feed suffers the most from matrix effects. In general, matrix-related signal suppression was observed more frequently than signal enhancement. A higher number of analytes were suppressed in pig (47% real and 44% model) and in chicken feed (48% real and 49% model) than enhanced in pig (11% real and 13% model) and chicken feed (13% real and 12% model). Furthermore, even more analytes were suppressed in cattle feed 63% (real) and 61% (model), compared to 9% (real) and 8% (model) of analytes showing an enhancement of the signal in this matrix. As already observed within the matrix categories of single feed material, signal enhancement is strongly correlated with compounds analyzed in the negative mode such as altersetin, equisetin, dihydrocitrinone, and fluazinam in all compound feed formulas. All average values for SSE, RA, and RE for the positive and negative mode, respectively, are shown in the Supporting Information (Table S1).
Figure 5.

Box-plot comparison of matrix effects in complex compound feed. The x-axis represents the matrix effects expressed as SSE in percent, and the y-axis shows the different sets of real and model compound feed samples.
However, model and real sample materials are well-comparable in terms of absolute matrix effects. Median values for SSE in chicken feed are at 82% in real samples and 81% in model samples. In pig feed, 82 and 83% median values were observed for real and model matrices, respectively, and 70% in each case for cattle feed. Furthermore, T-test statistics (Table S1, work sheet t-test and F-test statistics) revealed no significant difference between model and real samples for all species. Null hypothesis is not rejected because t-stats for cattle feed (0.616), pig feed (0.898), and chicken feed (1.611) are lower than the critical value 1.66. Additionally, PT≤t values for cattle feed (0.270), pig feed (0.186), and chicken feed (0.055) are not falling below α (0.05).
A visualized correlation analysis between matrix effects derived from the sample sets of real and model matrices is displayed in Figure 6. With a Pearson correlation coefficient of 0.987 in cattle, 0.990 in pig, and 0.992 in chicken feed, all categories showed a high positive correlation, which indicates a strong connection between modeled and real matrices.
Figure 6.

Basic scatter plot for correlation analysis between absolute matrix effects from real compound feed samples (x-axis) and model compound feed samples (y-axis). Analytes are represented by a colored dot. Cattle feed is pictured by green, pig feed is pictured by red, and chicken feed is pictured by yellow dots.
3.3.1.2. Relative Matrix Effects
A matrix mismatch is typically the result of the heterogeneous nature of the tested sample material. Analyte-specific variabilities in SSE in samples from different sources, but from the same type, can be considered as a measure of relative matrix effects.36,37 In general, an acceptable deviation from a nominal value expressed as a percentage (RSDSSE) should be ≤15% to be considered as not affected by intramatrix variations.38 We observed the highest relative matrix effects in cattle feed, followed by pig and chicken feed. Concerning real samples, in cattle feed, 50% of analytes were affected by high intramatrix variations, compared to 34 and 15% in pig and chicken feed. In contrast, model feed matrices were less prone to relative matrix effects. Only 7% of analytes in cattle and in each case, 4% of analytes in pig and chicken feed did not comply with the RSDSSE criterion of ≤15%. A summary of relative matrix effects for compound and single feed matrices is shown in the respective work sheet of Table S1.
The high intramatrix variability of the SSE in real samples, particularly in cattle feed, can be a result of the nature of the samples or by the feed ration, which may pose an interference. Because the model feed matrices were basically prepared using blank single feed ingredients, the relative matrix effects were significantly reduced. F-test statistics (Table S1, work sheet t-test and F-test statistics) gives a detailed explanation of the statistical characteristics for relative matrix effects in model and real compound feed samples. F values are higher for cattle feed (4.120), pig feed (2.428), and chicken feed (1.532), compared to the critical F value 1.394. Additionally, all PF≤f values are lower for cattle feed (6.78 × 10–12), pig feed (7.37 × 10–6), and chicken feed (1.74 × 10–2), compared to α (0.05), indicating that the null hypothesis is rejected.
Thus, shown by the statistical T-test (Table S1/work sheet t-test and F-test statistics), modeling different feed lots reveals a suitable technique to obtain an accurate estimation of the method performance and ensure high compliance with validation acceptance criteria. In contrast, as shown by the statistical F-test (Table S1/work sheet t-test and F-test statistics), results obtained under repeatability conditions (n = 7) from one identical replicate indicate an overestimation of the method performance, graphically exemplified for cattle feed in Figure 7.
Figure 7.

Scatter plot comparison of matrix effects (x-axis) for extracts from a single cattle feed replicate, as well as from a different model, and real cattle feed samples (y-axis) under repeatability conditions.
Consistently, strong relative matrix effects in real and model feed samples were observed for alternariol (20% real and 16% model), alternariolmonomethylether (16% real and 15% model), brevianamid F (19% real and 17% model), cytochalasin J (15% real and 18% model), ergine (26% real and 15% model), fumigaclavine C (17% real and 16% model), and ilicicolin A (20% real and 19% model), while these compounds were much less-affected under repeatability conditions based on an identical matrix replicate. In general, concerning RSDSSE, we observed high differences between the different cattle feed sample sets. Median RSDSSE values of 3.7, 5.7, and 15% for a single sample replicate, model samples, and real samples, respectively, imply an increasing overestimation of the method performance through the application of replicates derived from a single sample material.
3.3.1.3. Compatibility of the Extraction Protocol
Apparent recoveries and extraction efficiencies for all three modeled compound feed formulas are depicted in Figure 8. Predominant extraction efficiencies between 60 and 100% prove the applicability of the extraction protocol with complex feed material, while, in particular, signal suppression leads to low numerical values of apparent recoveries for some analyte/matrix combinations. Lower extraction efficiencies (≤60%) were observed for cyromazin (57%), andrastin A (49%), and ilicicolin A (60%) in cattle feed. In pig feed, gliotoxin (54%), chetomin (45%), and andrastin A (54%) and in chicken feed, only chetomin (45%) and andrastin A (56%) showed RE values lower than 60%. Low extraction efficiencies for andrastin A, chetomin, and cyromazin were also observed in the single feed materials, while low extraction yield for ilicicolin A in cattle feed and for gliotoxin in pig feed is associated with a poor extraction efficiency of ilicicolin A in sunflower and gliotoxin in soy, as components of the respective compound feed formula.
Figure 8.

Basic scatter plot visualizing apparent recoveries (x-axis) and extraction efficiencies (y-axis) for selected analytes in complex model matrices. Each target analyte is represented by a colored dot. Retention times are reflected by different colors from purple (polar compounds) to green (apolar compounds).
Concerning RA-values, 47% of analytes in cattle feed and 66 and 59% in pig and chicken feed were in the RE criteria range of 60–140%. This implies that deviations from 100% of the external calibration are, in particular, a result of adverse matrix contributions. For a significant reduction of these effects, validation guidelines recommend a preparation of calibration standards with the corresponding matrix extract. However, because of the high sample complexity, a correction between different matrix lots is not applicable, graphically illustrated in Figure 7. In addition, the natural sample background contamination complicates the applicability of this approach.8
The extraction variability under repeatability conditions for the model compound feeds is shown in Figure 9. With regard to the acceptance criteria of RSD ≤15%, extraction efficiency complies similarly to relative matrix effects. The fraction of analytes not complying to this criterion was 6, 4, and 10% for cattle, pig, and chicken feed, respectively. However, the majority of analytes show excellent extraction behavior under repeatability conditions, indicating the high efficacy of the extraction protocol for complex feed material.
Figure 9.

Scatter plot comparison of repeatability conditions of the extraction protocol (x-axis) for model compound feed matrices (y-axis).
3.4. Validation Proposal for Complex Feed Material
Based on considerable analyte/matrix-dependent differences between performance criteria for compound feed formulas and their single feed ingredients, the requirements of future validation guidelines for feed should be extended.
Validation guidelines such as the DAkkS document (71SD4012) are exclusively focusing on the validation of single feed ingredients or are completely neglecting these matrices.8 Therefore, we propose an extension of validation guidelines with the most important compound feed formulas, depicted in Figure 10. Based on the European animal feed production data provided by FEFAC, more than 90% of the total compound feed production (253.6 million tons in 2018) is accounting for chicken, pig, and cattle.39 Taking the market share as a reference, these three compound feed types should be included within the validation scope of laboratories conducting routine analysis for animal feed material. Because the natural background contamination of compound feed possesses a particular problem in order to validate these matrices, we further propose to perform validation processes using in-house model matrices, based on true blank single feed ingredients. We have shown that there is no significant difference between real and model matrices with respect to absolute effects such as extraction efficiency and matrix effects.
Figure 10.

Validation proposal scheme for complex feed material.
In order to simulate the heterogenic nature of compound feed, we suggest preparing at least 5 lots with different compositional patterns. Feed formula variations for animals at different growth stages should be taken into account. As elaborated in chapter 3.3.1.2, simulating the intramatrix variation leads to a more realistic estimation of the method precision.
To conclude, this work presents the first comprehensive evaluation of analytical parameters for complex compound feed based on in-house-prepared model matrices in LC–MS/MS analysis. We have shown that substantial differences between RA, SSE, and RE values occur, when comparing single feed material with complex compound feed formulas. A straightforward and economical procedure for the validation of compound feed was applied which ensures an accurate estimation of real-life conditions in routine-based laboratories. The method performance was estimated based on spiking experiments for a representative set of analytes in seven different lots (compound feed) of each matrix type. Performance criteria in current animal feed validation guidelines exclusively focus on single feed material without consideration of intramatrix variation, which facilitates the compliance of the corresponding criteria regarding trueness and precision. Discrepancies in RSDRA and RSDSSE for compound feed and its single feed ingredients indicate a noncompliance of validation data based on individual feed material with complex feedstuff. However, recoveries outside the range of 70–120% can be accepted if they are consistent (RSD ≤20%) and a recovery correction is applied.10 Model matrices for three different animal species (cattle, pig, and chicken) were prepared in-house based on the compositional information provided by animal feed producers. Analytical parameters for extraction efficiency, matrix effects, and apparent recovery were compared between modeled feed material and equivalent real samples. High absolute and relative matrix effects were the major negative contributor to the overall analytical outcome. Excellent comparability for absolute matrix effects between model and real samples was observed, while model matrices were less-prone to influences of sample inhomogeneity. It was further demonstrated that neglecting the intrasubject variation by following a validation protocol based on one single matrix replicate leads to an overestimation of the method performance and subsequently underestimates the measurement uncertainty. The major outcomes are summarized as follows
in-house model matrices allow a high comparability of real-life conditions,
background information about the individual ratios of ingredients in different lots of compound feed is required in order to prepare the model matrix for validation (may differ from country to country),
ensure an accurate but not overestimated method performance,
simulate intrasubject variations,
economize workload and resources, and
retain no uncertainties regarding the composition of the complex material.
In summary, the work describes a fit-for-purpose validation proposal for LC–MS/MS multiclass methods in complex feed matrices.
Acknowledgments
This work was created within a research project of the Austrian Competence Centre for Feed and Food Quality, Safety and Innovation (FFoQSI). The COMET-K1 competence centre FFoQSI is funded by the Austrian ministries BMVIT and BMDW and the Austrian provinces Niederoesterreich, Upper Austria, and Vienna within the scope of COMET—Competence Centers for Excellent Technologies. The program COMET is handled by the Austrian Research Promotion Agency FFG.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jafc.9b07706.
Title page; full list of investigated analytes including following information: quantifier/qualifier, substance class, polarity, retention, and acquisition parameter (q1, q3, DP, CE, and CXP); compositional information about tested real and model compound feed samples, graphical illustration of model feed samples, and list of investigated single feed material with origin information; overview on data of RA, SSE, and RE and the respective RSD values for tested real compound feed samples; overview on data of RA, SSE, and RE and the respective RSD values for tested model compound feed samples; overview on data of RA, SSE, and RE and the respective RSD values for tested single feed material; T-test and F-test statistics for the comparison of model and real compound feed samples; relative standard deviations for the extraction protocol for single feed material and model compound feed; and overview on data of RA, SSE, and RE and the respective RSD values for the tested single technical replicate of real cattle feed (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Danezis G. P.; Anagnostopoulos C. J.; Liapis K.; Koupparis M. A. Multi-Residue Analysis of Pesticides, Plant Hormones, Veterinary Drugs and Mycotoxins Using HILIC Chromatography – MS/MS in Various Food Matrices. Anal. Chim. Acta 2016, 942, 121–138. 10.1016/j.aca.2016.09.011. [DOI] [PubMed] [Google Scholar]
- Hird S. J.; Lau B. P.-Y.; Schuhmacher R.; Krska R. Liquid Chromatography-Mass Spectrometry for the Determination of Chemical Contaminants in Food. TrAC, Trends Anal. Chem. 2014, 59, 59–72. 10.1016/j.trac.2014.04.005. [DOI] [Google Scholar]
- Alder L.; Greulich K.; Kempe G.; Vieth B. Residue Analysis of 500 High Priority Pesticides: Better by GC-MS or LC-MS/MS?. Mass Spectrom. Rev. 2006, 25, 838–865. 10.1002/mas.20091. [DOI] [PubMed] [Google Scholar]
- Mol H. G. J.; Plaza-Bolaños P.; Zomer P.; De Rijk T. C.; Stolker A. A. M.; Mulder P. P. J. Toward a Generic Extraction Method for Simultaneous Determination of Pesticides, Mycotoxins, Plant Toxins, and Veterinary Drugs in Feed and Food Matrixes. Anal. Chem. 2008, 80, 9450–9459. 10.1021/ac801557f. [DOI] [PubMed] [Google Scholar]
- REGULATION (EC) No 767/2009 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 13 July 2009 on the Placing on the Market and Use of Feed, Amending European Parliament and Council Regulation (EC) No 1831/2003 and Repealing Council Directive 79/373/EEC, Commi. Off. J. Eur. Union 2002, 1–42. [Google Scholar]
- Commission Regulation (EU) No 68/2013 of 16 January 2013 on the Catalogue of Feed Materials. Off. J. Eur. Union 2013, 29, 1–64. [Google Scholar]
- FEFAC . FEFAC Code of Good Labelling Practices for Compound Feed for Food Producing Animals, 2018, https://www.fefac.eu/files/86306.pdf (accessed date: 2019-09-05).
- Deutsche Akkreditierungsstelle . Anforderungen an Laboratorien im Gesundheitlichen Verbraucherschutz–Wirkstoff–Multimethoden zur Pestizidanalytik in Lebens- und Futtermitteln, 2015, https://www.din-dakks-portal.de/de/wdc-beuth:din21:269802721/directPdf-2637323/dakksDownload (accessed date: 2019-10-01).
- Malachová A.; Sulyok M.; Beltrán E.; Berthiller F.; Krska R. Optimization and Validation of a Quantitative Liquid Chromatography-Tandem Mass Spectrometric Method Covering 295 Bacterial and Fungal Metabolites Including All Regulated Mycotoxins in Four Model Food Matrices. J. Chromatogr. A 2014, 1362, 145–156. 10.1016/j.chroma.2014.08.037. [DOI] [PubMed] [Google Scholar]
- European Commission . SANTE/11813/2017—Guidance Document on Analytical Quality Control and Method Validation Procedures for Pesticide Residues and Analysis in Food and Feed. Off. J. Eur. Union 2017, 461−42. [Google Scholar]
- Sulyok M.; Berthiller F.; Krska R.; Schuhmacher R. Development and Validation of a Liquid Chromatography/Tandem Mass Spectrometric Method for the Determination of 39 Mycotoxins in Wheat and Maize. Rapid Commun. Mass Spectrom. 2006, 20, 2649–2659. 10.1002/rcm.2640. [DOI] [PubMed] [Google Scholar]
- Varga E.; Glauner T.; Köppen R.; Mayer K.; Sulyok M.; Schuhmacher R.; Krska R.; Berthiller F. Stable Isotope Dilution Assay for the Accurate Determination of Mycotoxins in Maize by UHPLC-MS/MS. Anal. Bioanal. Chem. 2012, 402, 2675–2686. 10.1007/s00216-012-5757-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulyok M.; Krska R.; Schuhmacher R. A Liquid Chromatography/Tandem Mass Spectrometric Multi-Mycotoxin Method for the Quantification of 87 Analytes and Its Application to Semi-Quantitative Screening of Moldy Food Samples. Anal. Bioanal. Chem. 2007, 389, 1505–1523. 10.1007/s00216-007-1542-2. [DOI] [PubMed] [Google Scholar]
- Meister U. Effect of Extraction and Extract Purification on the Measurable Fumonisin Content of Maize and Maize Products. Tests on the Efficiency of Acid Extraction and Use of Immunoaffinity Columns. Mycotoxin Res. 1999, 15, 13–23. 10.1007/BF02945210. [DOI] [PubMed] [Google Scholar]
- COMMISSION RECOMMENDATION of 17 August 2006 on the Presence of Deoxynivalenol, Zearalenone, Ochratoxin A, T-2 and HT-2 and Fumonisins Inproducts Intended for Animal Feeding. Off. J. Eur. Union 2006, 229, 7–9. [Google Scholar]
- Directive of The European Parliament and of the Council of 7 May 2002 on Undesirable Substances in Animal Feed 2002/32. Off. J. Eur. Communities 2002, 140, 10–21. [Google Scholar]
- Alonso V. A.; Pereyra C. M.; Keller L. A. M.; Dalcero A. M.; Rosa C. A. R.; Chiacchiera S. M.; Cavaglieri L. R. Fungi and Mycotoxins in Silage: An Overview. J. Appl. Microbiol. 2013, 115, 637–643. 10.1111/jam.12178. [DOI] [PubMed] [Google Scholar]
- Wood R. How to Validate Analytical Methods. TrAC, Trends Anal. Chem. 1999, 18, 624–632. 10.1016/S0165-9936(99)00150-8. [DOI] [Google Scholar]
- Taylor P. J. Matrix Effects: The Achilles Heel of Quantitative High-Performance Liquid Chromatography-Electrospray-Tandem Mass Spectrometry. Clin. Biochem. 2005, 38, 328–334. 10.1016/j.clinbiochem.2004.11.007. [DOI] [PubMed] [Google Scholar]
- CEN/TR 16059Food Analysis—Performance Criteria for Single Laboratory Validated Methods of Analysis for the Determination of Mycotoxins, 2010.
- Buhrman D. L.; Price P. I.; Rudewicz P. J. Quantitation of SR 27417 in Human Plasma Using Electrospray Liquid Chromatography-Tandem Mass Spectrometry: A Study of Ion Suppression. J. Am. Soc. Mass Spectrom. 1996, 7, 1099–1105. 10.1016/S1044-0305(96)00072-4. [DOI] [PubMed] [Google Scholar]
- Nielsen K. F.; Larsen T. O. The Importance of Mass Spectrometric Dereplication in Fungal Secondary Metabolite Analysis. Front. Microbiol. 2015, 6, 1–15. 10.3389/fmicb.2015.00071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King R.; Bonfiglio R.; Fernandez-Metzler C.; Miller-Stein C.; Olah T. Mechanistic Investigation of Ionization Suppression in Electrospray Ionization. J. Am. Soc. Mass Spectrom. 2000, 11, 942–950. 10.1016/S1044-0305(00)00163-X. [DOI] [PubMed] [Google Scholar]
- Bonfiglio R.; King R. C.; Olah T. V.; Merkle K. The Effects of Sample Preparation Methods on the Variability of the Electrospray Ionization Response for Model Drug Compounds. Rapid Commun. Mass Spectrom. 1999, 13, 1175–1185. 10.1002/(SICI)1097-0231(19990630)13:12<1175::AID-RCM639>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Tukey J. W.Exploratory Data Analysis, 1st ed.; Pearson, 1977. [Google Scholar]
- Zhou W.; Yang S.; Wang P. G. Matrix Effects and Application of Matrix Effect Factor. Bioanalysis 2017, 9, 1839–1844. 10.4155/bio-2017-0214. [DOI] [PubMed] [Google Scholar]
- Gosetti F.; Mazzucco E.; Zampieri D.; Gennaro M. C. Signal Suppression/Enhancement in High-Performance Liquid Chromatography Tandem Mass Spectrometry. J. Chromatogr. A 2010, 1217, 3929–3937. 10.1016/j.chroma.2009.11.060. [DOI] [PubMed] [Google Scholar]
- Leverence R.; Avery M. J.; Kavetskaia O.; Bi H.; Hop C. E. C. A.; Gusev A. I. Signal Suppression/Enhancement in HPLC-ESI-MS/MS from Concomitant Medications. Biomed. Chromatogr. 2007, 21, 1143–1150. 10.1002/bmc.863. [DOI] [PubMed] [Google Scholar]
- Kononenko G.; Burkin A.; Gavrilova O.; Gagkaeva T. Fungal Species and Multiple Mycotoxin Contamination of Cultivated Grasses and Legumes Crops. Agric. Food Sci. 2015, 24, 323–330. 10.23986/afsci.52313. [DOI] [Google Scholar]
- Zachariasova M.; Dzuman Z.; Veprikova Z.; Hajkova K.; Jiru M.; Vaclavikova M.; Zachariasova A.; Pospichalova M.; Florian M.; Hajslova J. Occurrence of Multiple Mycotoxins in European Feedingstuffs, Assessment of Dietary Intake by Farm Animals. Anim. Feed Sci. Technol. 2014, 193, 124–140. 10.1016/j.anifeedsci.2014.02.007. [DOI] [Google Scholar]
- Kokkonen M. K.; Jestoi M. N. A Multi-Compound LC-MS/MS Method for the Screening of Mycotoxins in Grains. Food Anal. Methods 2009, 2, 128–140. 10.1007/s12161-008-9051-2. [DOI] [Google Scholar]
- Stadler D.; Sulyok M.; Schuhmacher R.; Berthiller F.; Krska R. The Contribution of Lot-to-Lot Variation to the Measurement Uncertainty of an LC-MS-Based Multi-Mycotoxin Assay. Anal. Bioanal. Chem. 2018, 410, 4409–4418. 10.1007/s00216-018-1096-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- US Food & Drug Administration . Guidelines for the Validation of Chemical Methods for the FDA Foods Program, 2nd ed.; US Food & Drug Administration Office of Foods and Veterinary Medicine, 2015. [Google Scholar]
- European Commission . Report on the Relationship between Analytical Results, Measurement Uncertainty, Recovery Factors and the Provisions of Eu Food and Feed Legislation, with Particular Reference to Community Legislation Concerning, 2002.
- Commission Recommendation 401/2006 of 23 February 2006 Laying Down the Methods of Sampling and Analysis for the Official Control of the Levels of Mycotoxins in Foodstuffs. Off. J. Eur. Union 2006, 70, 12–34. [Google Scholar]
- Matuszewski B. K.; Constanzer M. L.; Chavez-Eng C. M. Matrix Effect in Quantitative LC/MS/MS Analyses of Biological Fluids: A Method for Determination of Finasteride in Human Plasma at Picogram Per Milliliter Concentrations. Anal. Chem. 1998, 70, 882–889. 10.1021/ac971078+. [DOI] [PubMed] [Google Scholar]
- Matuszewski B. K.; Constanzer M. L.; Chavez-Eng C. M. Strategies for the Assessment of Matrix Effect in Quantitative Bioanalytical Methods Based on HPLC-MS/MS. Anal. Chem. 2003, 75, 3019–3030. 10.1021/ac020361s. [DOI] [PubMed] [Google Scholar]
- Viswanathan C. T.; Bansal S.; Booth B.; DeStefano A. J.; Rose M. J.; Sailstad J.; Shah V. P.; Skelly J. P.; Swann P. G.; Weiner R. Quantitative Bioanalytical Methods Validation and Implementation: Best Practices for Chromatographic and Ligand Binding Assays. Pharm. Res. 2007, 24, 1962–1973. 10.1007/s11095-007-9291-7. [DOI] [PubMed] [Google Scholar]
- FEFAC . Resource Efficiency Champions of the Food Chain for 60 Years. https://www.fefac.eu/files/88220.pdf (accessed date: 2019-09-05).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.