

Abstract
The heat shock protein 70 (Hsp70) family of molecular chaperones are highly expressed in tumors. Inhibitors containing a pyridinium-modified benzothiazole, such as JG-98, bind to a conserved, allosteric site in Hsp70, showing promising anti-proliferative activity in cancer cells. When bound to Hsp70, the charged pyridinium makes favorable contacts; however, this moiety also increases the inhibitor’s fluorescence, giving rise to undesirable interference in biochemical and cell-based assays. Here, we explore whether the pyridinium can be replaced with a neutral pyridine. We report that pyridine-modified benzothiazoles, such as compound 17h (JG2–38), have reduced fluorescence, yet retain promising anti-proliferative activity (EC50 values ~0.1 to 0.07 μM) in breast and prostate cancer cell lines. These chemical probes are expected to be useful in exploring the roles of Hsp70s in tumorigenesis and cell survival.
Keywords: allosteric inhibitor, molecular chaperone, proteostasis, prostate cancer, breast cancer, chemical probe, anti-cancer agents, fluorescence
Members of the Hsp70 family of molecular chaperones, including heat shock protein 72 (Hsp72, HSPA1A) and heat shock cognate 70 (Hsc70, HSPA8), are over-expressed in many cancers.1–3 In tumor cells, these Hsp70s are part of a multi-protein complex that is not present in non-transformed cells,4 and which seems to stabilize oncoproteins and provide resistance to chemotherapy.1, 5, 6 Consistent with this role, knockdown of both Hsp72 and Hsc70 leads to preferential cell death in cancer cells, but not normal epithelia.7 Interestingly, knockdown of only one family member (e.g. Hsp72 or Hsc70) is typically not sufficient to initiate apoptosis, suggesting that pan-inhibitors (hereafter referred to as “Hsp70 inhibitors”) may hold particular promise. Fortunately, members of the Hsp70 family are highly conserved, with up to 95% sequence identity.8, 9
Multiple Hsp70 inhibitors with different mechanisms and binding sites have been explored. For example, VER-15500810, 11 and its analogs12–14 bind to Hsp70’s ATP-binding cleft, while YK5 and its analogs15–17 bind a nearby allosteric site. Despite their different binding sites, these compounds all seem to limit Hsp70’s function by interrupting its nucleotide cycling. Another strategy is to target the protein-protein interactions between Hsp70 and the other factors that are present in cancer-associated, multi-protein complexes. For example, the dihydropyridine, MAL3–10118, 19 and its analogs20–22, disrupt binding of Hsp70 to J-domain proteins (JDPs).
Whadwa and colleagues identified the benzothiazole, MKT-077 (Figure 1), as another promising inhibitor of Hsp70s. This compound has anti-proliferative activity in multiple cancer cells, with minimal toxicity in non-transformed cells.23 MKT-077 progressed to a Phase I clinical trial for solid tumors,24 however, modest efficacy (~5 μM) and metabolic instability limited its further exploration. Subsequent studies revealed that MKT-077 binds a distinct allosteric site in Hsp70,25 which is highly conserved amongst the family members.26 Binding at this allosteric site blocks binding to nucleotide-exchange factors (NEFs), although the pocket does not overlap with the NEF-interaction surface.27, 28 Leveraging this structural knowledge, a series of medicinal chemistry campaigns were designed to improve MKT-077, resulting in analogs such as JG-98 and JG-231 (Figure 1), with improved anti-proliferative activity and longer lifetimes in rodents.29–31
Figure 1.
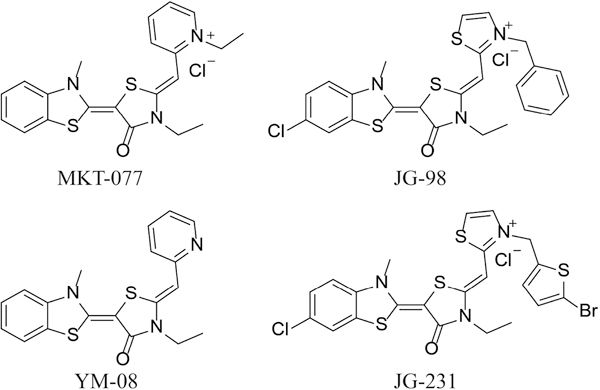
Previously reported benzothiazole rhodacyanine probes.
While JG-98 and its analogs have been useful chemical probes, these compounds are fluorescent (λexcitation 470 nm / λemission 560 nm). This photochemical property likely originates from the conjugated π electron system that encompasses the rhodacyanine and charged pyridinium. This fluorescence property is not favorable, because it interferes with many assays; for example, these compounds cannot be used in biochemical assays that utilize fluorescence, such as fluorescence polarization (FP). Here, we set out to design neutral, non-fluorescent analogs. Specifically, based on findings with the analog YM-08 (Figure 1),32 we reasoned that replacing the rhodacyanine and/or the pyridinium might sufficiently reduce fluorescence and enable use of these chemical probes in a wider range of applications.
Towards this goal, we first replaced the central rhodacyanine with either a benzene (compound 4a-b) or thiazole (compound 8a-b). Based on structural information obtained with JG-98, the central rhodacyanine is relatively solvent exposed, yet it sets the position of the other ring systems (i.e. the benzothiazole) to optimally interact with two deep pockets.26 Thus, compounds 4a-b and 8a-b were created to understand whether the rhodacyanine might be replaced without interfering with these contacts. Briefly, the synthesis of compounds 4a-b started from cyclization of 2-aminothiophenol and 3-bromobenzaldehyde,33 followed by Buchwald-Hartwig amination with anilines (Scheme 1). In contrast, compounds 8a-b were synthesized by reacting 2-aminothiophenol with lactic acid, leading to the alcohol which was then oxidized to ketone 6 with manganese dioxide. Intermediate 6 was brominated with copper(II) bromide and then reacted with substituted thiourea to obtain the final products 8a-b. The purified compounds (>95% HPLC) were then tested in anti-proliferative assays using cancer cells from breast (MCF7) and prostate (22Rv1 and PC3). However, we found that 4a-b and 8a-b tended to have worse activity than JG-98 (Table 1), with IC50 values between ~0.7 and 13 μM. Based on this result, we decided to retain the rhodacyanine in subsequent analogs and turn our attention to the pyridinium.
Scheme 1.
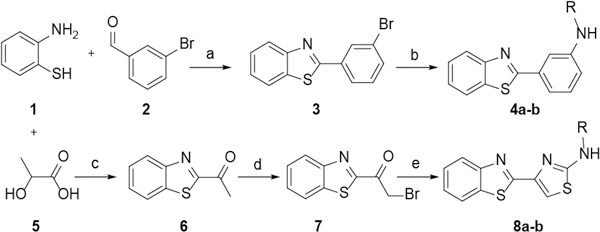
Reagents and conditions: (a) PTSA, H2O, 70 °C, 12 h, 60%; (b) Pd(OAc)2 (10 mol%), (±)BINAP (10 mol%), Cs2CO3 (2 mol), toluene, 25–34%; (c) (i) 4 N HCl aq., reflux, 24 h, 94%; (ii) 10 eq. MnO2, CHC13, 72%; (d) Cu(II)Br, CHC13 + EtOAc, reflux, 12 h, 78%; (e) Substituted thiourea, EtOH, reflux, 2 h, 52–60%.
Table 1:
Antiproliferative Activities of compounds 4a-b and 8a-b
| Compd | R | MCF-7 IC50/μM | 22RV1 IC50/μM | PC3 IC50/μM |
|---|---|---|---|---|
| JG-98 | - | 0.71 ± 0.22 | - | - |
| 4a | 13 ± 0.90 | 13 ± 2.0 | 4.8 ± 1.3 | |
| 4b | 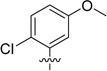 |
0.71 ± 0.20 | 2.1 ± 0.15 | 2.2 ± 0.43 |
| 8a | 7.6 ± 0.67 | 6.9 ± 1.4 | 5.5 ± 0.74 | |
| 8b | 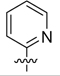 |
3.6 ± 0.60 | 3.2 ± 0.28 | 6.7 ± 0.76 |
Specifically, we assembled analogs (compounds 17a-o) in which the pyridinium was replaced with a neutral pyridine, using a previously reported route (Scheme 2).26, 29 Briefly, substituted anilines 9 were treated with potassium ethyl xanthate, followed by methylation with iodomethane under basic conditions. The resulting benzothiazoles 10 were first reacted with methyl p-toluenesulfonate and then coupled with 3-ethylrhodanine to obtain intermediate 12. Intermediate 12 was further activated by methyl p-toluenesulfonate, followed by condensation with 1-((1,3-dioxoisoindolin-2-yl)methyl)-2-methylpyridin-1-ium bromide (14) to yield compound 15. Deprotection of 15 with aqueous ammonium hydroxide yielded 16, which was coupled with anilines to obtain the final products 17a-o. These products were purified by flash chromatography (>95% HPLC) and characterized by LC-MS/MS and 1H-NMR (see Supporting Information).
Scheme 2.
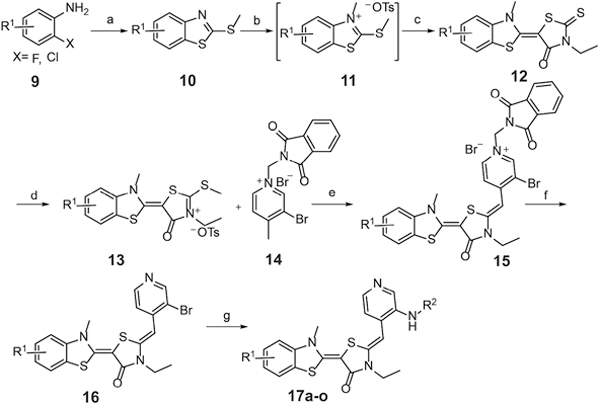
Reagents and conditions: (a) (i) Potassium ethylxanthate, DMF, 140 °C, 4 h; (ii) MeI, NEt3, EtOH, 80 °C, 1 h; 40–80%; (b) p-Ts0Me, anisole, 125 °C, 4 h, used directly for next step; (c) 3-Ethyl rhodanine, Net3, MeCN, 25 °C, 4 h, 30–75%; (d) p-Ts0Me, DMF, 135 °C, 3 h, 50–85%; (e) Net3, MeCN, 70 °C, 3 h, 70–83%; (f) DCM/MeOH (1:3), aq. NH3, r.t., l h, 79–87%; (g) anilines, Pd2(dba)3/BINAP/t-BuONa (0.05:0.15:2), toluene, 100 °C, overnight, 38–55%.
A key goal of this synthetic effort was to reduce the compound’s intrinsic fluorescence. To specifically test this idea, compounds 17a-o (50 μM) were dissolved in aqueous buffer (100 mM Tris buffer with 10% DMSO, pH 7.4), and the emission spectra of these solutions measured ((λexcitation 470 nm / λemission 520 to 750 nm). We found that all of the neutral analogs were significantly less fluorescent than JG-98 (Figure 2).
Figure 2.
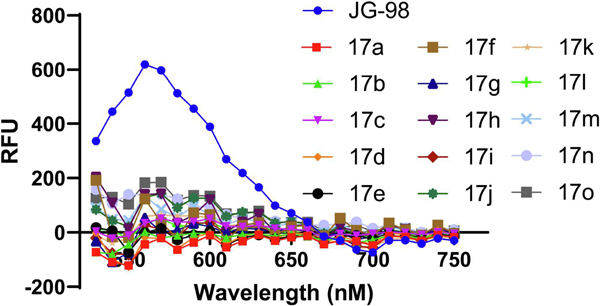
Compounds 17a-o are less fluorescent than JG-98. The emission spectrum of compounds 17a-o when excited at 470 nM.
With these compounds in-hand, we measured their anti-proliferative activity against the breast (MCF7) and prostate cancer (22Rv1 and PC3) cell lines. Our previous report shows that ortho- and meta-modification of the benzene ring in JG-98 can increase potency,29 so we initially tested compounds with dimethyl (17a) or difluoro (17b) substituents at the o- and m’- positions. Indeed, both had modest activity against all three cancer cell lines, with IC50 values in the range of ~1 to 2 μM. Removal of the methyl group at the meta position (17c) further improved activity, especially against 22Rv1 cells (IC50 ~0.5 μM). Accordingly, we tested ortho-substituted analogs with either electron withdrawing or donating groups at the ortho-position (17c-e), revealing that the o-fluorine compound 17e was the most potent of these analogs, with IC50 values ~0.3 μM.
Using these ortho-substituents at the benzene ring, we turned our attention to modifications of the benzothiazole. In JG-98, small alkyl and halogen substitutions have been reported to increase activity.26 Similarly, we found that a methyl group at the 4-position (17f-h) resulted in approximately 2-fold improvements. For example, compound 17h (JG2–38) had IC50 values of 0.1 μM against MCF7 cells, 0.15 μM in 22Rv1 cells and 0.07 μM for PC3 cells. These values represent a ~7 to 10-fold improvement on JG-98. Replacement of methyl with methoxy (17i-k) resulted in similar trends, with the o-fluoro analog 17k again having the best activity (IC50 values ~ 0.1 to 0.19 μM). In JG-98 analogs, the benzothiazole substitution is tolerated on either the 4- or 5- position.26 Accordingly, we tested the effects of replacing the 4-methoxy of 17i-k with 5-fluoro groups (17l-n). However, this modification generally decreased activity (IC50 values 2.3 to 0.15 μM). Similarly, larger substitutions, such as 5-ethyl (17o), did not improve activity (IC50 values ~2.1 to 0.18 μM).
Based on these results, we selected compound 17h (JG2–38) for further exploration. First, we studied its possible binding pose, using molecular docking in Schrodinger, as previously described.29 In these studies, we used the structure of a truncated form of human Hsc70 (PDB code 3HSC). In the best-predicted models (Figure 3), compound 17h (JG2–38) was anchored in a deep pocket formed by residues R72, K71, T13, F150, P147 and T204. In this orientation, the pyridine ring of 17h (JG2–38) was largely solvent-exposed, but it positioned the benzyl group into an adjacent hydrophobic pocket. This binding pose might explain the preference for only small, ortho-substitutions, because this group is nestled against Y149 and the β-sheet composed of residues 222–226.
Figure 3.
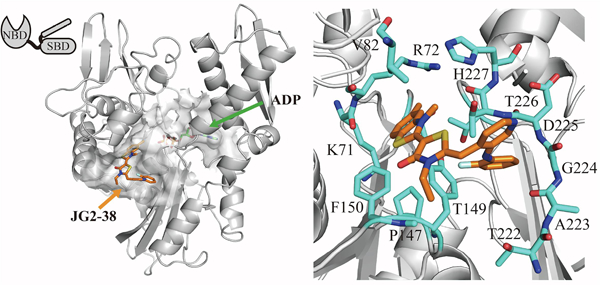
Compound 17h (JG2–38) binds to an allosteric site in the nucleotide binding domain of Hsp70. Compound 17h (JG2–38) and the residues within 5 angs are shown as stick representation. Carbon atoms of the protein and 17h (JG2–38) are shown in gray and orange, respectively. Oxygen, nitrogen and sulfur are shown in red, blue and yellow, respectively. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Finally, it is known that treatment of cancer cells with Hsp70 inhibitors leads to degradation of pro-survival proteins, likely because chaperone function is needed to maintain their folding. To confirm whether 17h (JG2–38) might share this activity on Hsp70 biomarkers, we treated 22Rv1 cells for 24 h and then performed western blots for oncogenic kinases, Akt1, c-Raf-1 and CDK4, and the translation factor HuR (Figure 4). The levels of each protein were reduced by ~50%, consistent with Hsp70 inhibition. Moreover, consistent with previous findings on other Hsp70 inhibitors,29 we found that treatment with 17h (JG2–38) did not cause a stress response, as measured by the constant levels of Hsp72 and Hsp90 (Figure 4). This result is important because compensatory stress responses have been hypothesized to reduce sensitivity to chaperone inhibitors.
Figure 4.
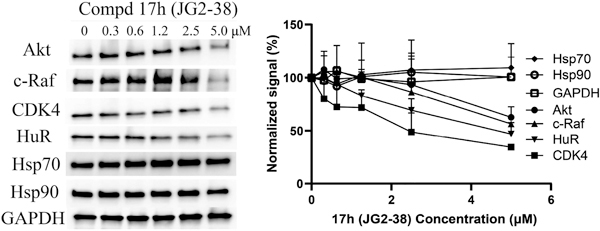
Compound 17h (JG2–38) led to degradation of oncoproteins without eliciting a heat shock response. 22-RV1 cells were treated with 17h (JG2–38) for 24 h at the indicated concentration, lysed and western blots performed. Results are representative of at least two independent experiments. Quantifications shown are averages of at least two independent experiments and the error bars represent S.E.
In summary, we identified 17h (JG2–38) as a less fluorescent Hsp70 inhibitor, which retains anti-proliferative activity (IC50 ~0.1 μM) and the expected effects on Hsp70 “client” proteins, such as oncogenic kinases. Further work will be needed to optimize the metabolic stability and solubility of this analog for use in animal models, but its improved photochemical properties are expected to be beneficial in a range of applications.
Supplementary Material
Table 2:
Structure and Activity Relationship of Allosteric Hsp70 inhibitors
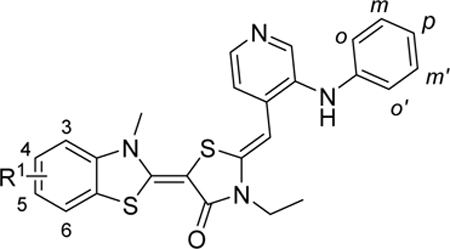 | |||||
|---|---|---|---|---|---|
| Compd | R1 | R2 | MCF-7 IC50/μM | 22RV1 IC50/μM | PC3 IC50/μM |
| 17a | H | o,m’-Me | 1.7 ± 0.05 | 1.8 ± 0.43 | 2.0 ± 0.60 |
| 17b | H | o, m’-F | 1.5 ± 0.49 | 1.1 ± 0.55 | 1.2 ± 0.23 |
| 17c | H | o-Me | 0.99 ± 0.01 | 0.47 ± 0.20 | 1.2 ± 0.11 |
| 17d | H | o-Cl | 0.75 ± 0.11 | 0.46 ± 0.25 | 0.78 ± 0.07 |
| 17e | H | o-F | 0.33 ± 0.06 | 0.30 ± 0.04 | 0.36 ± 0.02 |
| 17f | 4-Me | o-Me | 0.40 ± 0.10 | 0.48 ± 0.15 | 0.33 ± 0.02 |
| 17g | 4-Me | o-Cl | 0.65 ± 0.01 | 0.35 ± 0.05 | 0.50 ± 0.10 |
| 17h (JG2–38) | 4-Me | o-F | 0.10 ± 0.01 | 0.15 ± 0.02 | 0.07 ± 0.01 |
| 17i | 4-OCH3 | o-Me | 0.42 ± 0.03 | 0.41 ± 0.08 | 0.37 ± 0.03 |
| 17j | 4-OCH3 | o-Cl | 0.71 ± 0.30 | 0.50 ± 0.08 | 0.76 ± 0.01 |
| 17k | 4-OCH3 | o-F | 0.13 ± 0.01 | 0.19 ± 0.02 | 0.10 ± 0.01 |
| 17l | 5-F | o-Me | 1.3 ± 0.35 | 2.3 ± 0.94 | 2.3 ± 0.31 |
| 17m | 5-F | o-Cl | 1.3 ± 0.15 | 0.49 ± 0.03 | 1.2 ± 0.31 |
| 17n | 5-F | o-F | 0.23 ± 0.04 | 0.15 ± 0.07 | 0.26 ± 0.06 |
| 17o | 5-ethyl | o-F | 1.2 ± 0.02 | 1.2 ± 0.10 | 2.2 ± 0.01 |
Acknowledgements.
This work was generously supported by grants from the NIH (NS059690) and the U.S. Department of Defense (PC180716).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Evans CG, Chang L, Gestwicki JE. Heat Shock Protein 70 (Hsp70) as an Emerging Drug Target. J Med Chem 2010;53(12): 4585–4602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Srinivasan SR, Cesa LC, Li X, et al. Heat Shock Protein 70 (Hsp70) Suppresses RIP1-Dependent Apoptotic and Necroptotic Cascades. Molecular cancer research : MCR 2018;16(1): 58–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Joshi S, Wang T, Araujo TLS, Sharma S, Brodsky JL, Chiosis G. Adapting to stress - chaperome networks in cancer. Nature reviews Cancer. 2018;18(9): 562–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang T, Rodina A, Dunphy MP, et al. Chaperome heterogeneity and its implications for cancer study and treatment. Journal of Biological Chemistry. 2019;294(6): 2162–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Murphy ME. The HSP70 family and cancer. Carcinogenesis 2013;34(6): 1181–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sherman MY, Gabai VL. Hsp70 in cancer: back to the future. Oncogene 2015;34(32): 4153–4161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Powers MV, Clarke PA, Workman P. Dual targeting of HSC70 and HSP72 inhibits HSP90 function and induces tumor-specific apoptosis. Cancer cell. 2008;14(3): 250–262. [DOI] [PubMed] [Google Scholar]
- 8.Hunt C, Morimoto RI. Conserved features of eukaryotic hsp70 genes revealed by comparison with the nucleotide sequence of human hsp70. Proceedings of the National Academy of Sciences of the United States of America. 1985;82(19): 6455–6459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Daugaard M, Rohde M, Jaattela M. The heat shock protein 70 family: Highly homologous proteins with overlapping and distinct functions. FEBS letters. 2007;581(19): 3702–3710. [DOI] [PubMed] [Google Scholar]
- 10.Williamson DS, Borgognoni J, Clay A, et al. Novel Adenosine-Derived Inhibitors of 70 kDa Heat Shock Protein, Discovered Through Structure-Based Design. J Med Chem 2009;52(6): 1510–1513. [DOI] [PubMed] [Google Scholar]
- 11.Macias AT, Williamson DS, Allen N, et al. Adenosine-derived inhibitors of 78 kDa glucose regulated protein (Grp78) ATPase: insights into isoform selectivity. J Med Chem 2011;54(12): 4034–4041. [DOI] [PubMed] [Google Scholar]
- 12.Pettinger J, Le Bihan YV, Widya M, van Montfort RLM, Jones K, Cheeseman MD. An Irreversible Inhibitor of HSP72 that Unexpectedly Targets Lysine-56. Angew Chem Int Edit. 2017;56(13): 3536–3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moseng MA, Nix JC, Page RC. 2- and N6-functionalized adenosine-5’-diphosphate analogs for the inhibition of mortalin. FEBS letters. 2019;593(15): 2030–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pettinger J, Carter M, Jones K, Cheeseman MD. Kinetic Optimization of Lysine-Targeting Covalent Inhibitors of HSP72. J Med Chem 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rodina A, Patel PD, Kang YL, et al. Identification of an Allosteric Pocket on Human Hsp70 Reveals a Mode of Inhibition of This Therapeutically Important Protein. Chem Biol 2013;20(12): 1469–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Taldone T, Kang YL, Patel HJ, et al. Heat Shock Protein 70 Inhibitors. 2. 2,5 ‘-Thiodipyrimidines, 5-(Phenylthio)pyrimidines, 2-(Pyridin-3-ylthio)pyrimidines, and 3-(Phenylthio)pyridines as Reversible Binders to an Allosteric Site on Heat Shock Protein 70. J Med Chem 2014;57(4): 1208–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kang YL, Taldone T, Patel HJ, et al. Heat Shock Protein 70 Inhibitors. 1. 2,5 ‘-Thiodipyrimidine and 5-(Phenylthio)pyrimidine Acrylamides as Irreversible Binders to an Allosteric Site on Heat Shock Protein 70. J Med Chem 2014;57(4): 1188–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fewell SW, Smith CM, Lyon MA, et al. Small molecule modulators of endogenous and co-chaperone-stimulated Hsp70 ATPase activity. J Biol Chem 2004;279(49): 51131–51140. [DOI] [PubMed] [Google Scholar]
- 19.Wisen S, Bertelsen EB, Thompson AD, et al. Binding of a Small Molecule at a Protein-Protein Interface Regulates the Chaperone Activity of Hsp70-Hsp40. Acs Chem Biol 2010;5(6): 611–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Manos-Turvey A, Al-Ashtal HA, Needham PG, et al. Dihydropyrimidinones and -thiones with improved activity against human polyomavirus family members. Bioorganic & medicinal chemistry letters. 2016;26(20): 5087–5091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chiang AN, Liang M, Dominguez-Meijide A, et al. Synthesis and evaluation of esterified Hsp70 agonists in cellular models of protein aggregation and folding. Bioorganic & medicinal chemistry. 2019;27(1): 79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wright CM, Chovatiya RJ, Jameson NE, et al. Pyrimidinone-peptoid hybrid molecules with distinct effects on molecular chaperone function and cell proliferation. Bioorganic & medicinal chemistry. 2008;16(6): 3291–3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wadhwa R, Sugihara T, Yoshida A, et al. Selective toxicity of MKT-077 to cancer cells is mediated by its binding to the hsp70 family protein mot-2 and reactivation of p53 function. Cancer research. 2000;60(24): 6818–6821. [PubMed] [Google Scholar]
- 24.Propper DJ, Braybrooke JP, Taylor DJ, et al. Phase I trial of the selective mitochondrial toxin MKT077 in chemo-resistant solid tumours. Annals of oncology : official journal of the European Society for Medical Oncology. 1999;10(8): 923–927. [DOI] [PubMed] [Google Scholar]
- 25.Rousaki A, Miyata Y, Jinwal UK, Dickey CA, Gestwicki JE, Zuiderweg ER. Allosteric drugs: the interaction of antitumor compound MKT-077 with human Hsp70 chaperones. Journal of molecular biology. 2011;411(3): 614–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li XK, Srinivasan SR, Connarn J, et al. Analogues of the Allosteric Heat Shock Protein 70 (Hsp70) Inhibitor, MKT-077, As Anti-Cancer Agents. Acs Med Chem Lett 2013;4(11): 1042–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Young ZT, Rauch JN, Assimon VA, et al. Stabilizing the Hsp70-Tau Complex Promotes Turnover in Models of Tauopathy. Cell chemical biology. 2016;23(8): 992–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rinaldi S, Assimon VA, Young ZT, et al. A Local Allosteric Network in Heat Shock Protein 70 (Hsp70) Links Inhibitor Binding to Enzyme Activity and Distal Protein-Protein Interactions. ACS chemical biology. 2018;13(11): 3142–3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shao H, Li XK, Moses MA, et al. Exploration of Benzothiazole Rhodacyanines as Allosteric Inhibitors of Protein-Protein Interactions with Heat Shock Protein 70 (Hsp70). J Med Chem 2018;61(14): 6163–6177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moses MA, Kim YS, Rivera-Marquez GM, et al. Targeting the Hsp40/Hsp70 Chaperone Axis as a Novel Strategy to Treat Castration-Resistant Prostate Cancer. Cancer research. 2018;78(14): 4022–4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eftekharzadeh B, Banduseela VC, Chiesa G, et al. Hsp70 and Hsp40 inhibit an inter-domain interaction necessary for transcriptional activity in the androgen receptor. Nature communications. 2019;10(1): 3562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miyata Y, Li X, Lee HF, et al. Synthesis and initial evaluation of YM-08, a blood-brain barrier permeable derivative of the heat shock protein 70 (Hsp70) inhibitor MKT-077, which reduces tau levels. ACS chemical neuroscience. 2013;4(6): 930–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Azizi N, Amiri AK, Baghi R, Bolourtchian M, Hashemi MM. PTSA catalyzed simple and green synthesis of benzothiazole derivatives in water. Monatshefte für Chemie - Chemical Monthly. 2009;140(12): 1471. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.