

Abstract
Recent studies show gut microbiota modulate antitumor immune responses; one proposed mechanism is cross-reactivity between antigens expressed in commensal bacteria and neoepitopes. We found that T cells targeting an epitope called SVYRYYGL (SVY), expressed in the commensal bacterium Bifidobacterium breve (B. breve), cross-react with a model neoantigen, SIYRYYGL (SIY). Mice lacking B. breve had decreased SVY-reactive T cells compared with B. breve–colonized mice, and the T cell response was transferable by SVY immunization or by cohousing mice without Bifidobacterium with ones colonized with Bifidobacterium. Tumors expressing the model SIY neoantigen also grew faster in mice lacking B. breve compared with Bifidobacterium-colonized animals. B. breve colonization also shaped the SVY-reactive TCR repertoire. Finally, SVY-specific T cells recognized SIY-expressing melanomas in vivo and led to decreased tumor growth and extended survival. Our work demonstrates that commensal bacteria can stimulate antitumor immune responses via cross-reactivity and how bacterial antigens affect the T cell landscape.
Keywords: Immunology, Oncology
Keywords: Antigen, Cancer immunotherapy, T cells

Antigen homology between commensal bacteria and tumor can lead to T cell cross-reactivity and a beneficial antitumor T cell response.
Introduction
The basis for differential patient responses to cancer immunotherapy involves many biological processes, including genetic variability among different patients, differences in tumor mutational load, and differential microbiome composition. Higher tumor mutational load is correlated with beneficial responses to checkpoint blockade outcomes in melanoma and non-small cell lung carcinoma (1). Mechanistically mutations in the tumor can result in novel peptides, called neoantigens, which can be targeted by T cells. These neoantigen-specific responses are critical for tumor clearance after checkpoint blockade immunotherapy (CPI) (2, 3). In addition to the quantity of potential neoantigens, several studies have also considered their quality to help identify which patients will be responsive to CPI (4, 5).
Another important parameter for patient outcome is the gut microbiota. Responders to checkpoint blockade have a different gut bacterial composition than nonresponders (6, 7); however, the potential mechanisms underlying antitumor effects are unclear. Evidence has emerged that the gut microbiota modulates the efficacy of various tumor therapies, possibly by engaging the innate and/or adaptive immune systems (8–10). Retrospective analysis of pancreatic cancer long-term survivors revealed that some high-quality neoepitope responses bear sequence homology to pathogenic or commensal bacteria (4). However, the effect of homologous epitopes between the tumor and microbiota on T cell populations is unknown. Therefore, identifying antigens expressed by microbiota and their potential cross-reactivity with T cells recognizing tumor-specific neoantigens will provide insight into tumor-reactive T cell populations that can be activated by cancer immunotherapy. In addition to identifying potential homologous antigens in microbiota, mechanistic studies must address how cross-reactive T cell populations react to tumor cells and the effect of bacterial colonization on the cross-reactive T cell populations. These studies will provide insight into how antigen mimicry between the commensal bacteria and tumor antigens can lead to beneficial antitumor responses.
Here, we identify an epitope, SVYRYYGL (SVY), found in the Bifidobacterium breve (B. breve) genome, with homology to the tumor model neoepitope antigen SIYRYYGL (SIY) expressed in B16.SIY tumors. Molecular dynamics simulations show that SVY peptide loaded on the murine H2-Kb MHC binds to a model TCR specific for the KbSIY complex, the 2C transgenic TCR, but with an altered configuration. Heterogeneous CD8+ T cell populations, expanded from wild-type C57BL/6 animals specific for either SIY or SVY, cross-reacted with both the bacterial and tumor antigens. Indeed, Bifidobacteria colonization, as seen by differences in C57BL/6 mice obtained from different suppliers, The Jackson Laboratory versus Taconic Biosciences, boosted expansion of SVY-specific T cell populations in vitro, and differences in commensal bacteria influenced the SVY population in vivo. In addition, immunization of Taconic mice with the SVY antigen led to activation of SVY-specific T cells that cross-reacted and killed SIY-pulsed target cells in vivo. Furthermore, C57BL/6 mice, obtained from the different commercial sources, had differences in their antitumor response to B16.SIY tumors, which grew more slowly in Jackson than Taconic mice. C57BL/6 mice also had an altered TCR repertoire depending on their source. Finally, in an adoptive cellular therapy (ACT) treatment model, SVY-expanded T cells targeted SIY-expressing tumor cells in vivo, inhibited tumor growth, and extended survival. These studies, for the first time to our knowledge, mechanistically identify that commensal bacteria can augment antitumor immunity by amplifying T cells that cross-react with model tumor neoantigens.
Results
B. breve contains a peptide epitope (SVY) that is homologous to the SIY epitope.
To identify commensal bacteria epitopes that may influence the B16.SIY antitumor response, we conducted a search of microbial genomes with the BLAST (http://www.ncbi.nlm.nih.gov/blast/), using the SIY peptide sequence as the query. Genetic alignment (Figure 1A) identified a single peptide sequence, SVYRYYGL, that varies by only 1 amino acid from the murine H2-Kb MHC–restricted model neoepitope SIY, valine (Val) versus isoleucine (Ile) at position 2. The SVY epitope sequence, derived from the B. breve EBP, has a 126-fold stronger predicted binding constant for H2-Kb compared with the next highest predicted epitope sequence from the EBP (Table 1). Processing of B. breve in vitro was studied by incubating heat-killed B. breve with splenocytes, and the resultant antigen-specific T cell expansions were analyzed (Figure 1B). By day 11, approximately 3.5% of CD8+ T cells were cross-reactive with the H2-Kb SIY complex (KbSIY) (Figure 1B and Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/jci.insight.135597DS1). In contrast, a control commensal bacterium, Lactobacillus rhamnosus, did not stimulate a KbSIY cross-reactive response. Thus, B. breve expresses an antigen that can be processed and presented and can stimulate KbSIY cross-reactive CD8+ T cells.
Figure 1. Commensal bacteria B. breve harbors the CD8+ T cell epitope SVY.

(A) Genetic map of B. breve, highlighting the homology of a peptide derived from B. breve exopolysaccharide biosynthesis protein (EBP) to the model epitope KbSIY. (B) Jackson mice splenocytes and mesenteric lymph node cells were cultured with or without heat-killed bacteria and tested for SIY-specific T cell expansion by staining with SIY peptide–loaded Kb-Ig dimer on day 11. Live CD8+ lymphocytes were analyzed by flow cytometry for KbSIY binding, with frequency determined by subtracting unloaded Kb-Ig staining frequency. P value = 0.011 by 1-way ANOVA and Dunnett’s post hoc test for multiple comparisons. N = 7. Data represent mean ± SEM. (C) MHC stabilization assay: RMA-S cells were incubated overnight with peptide as indicated. Cell surface expression of H2-Kb was determined by flow cytometry. Reported values are relative to the H2-Kb mean fluorescence intensity (MFI) observed with 10 μM OVA peptide. mCMV, a non–Kb-restricted peptide, was used as a negative control. Data trended toward no difference between SIY and SVY groups. Data trended with no difference between SIY and SVY groups. N = 2. Data represent mean. (D) CD8+ T cells were isolated from the spleens of 2C TCR (SIY-reactive) transgenic mice and stained with 1 μg of cognate KbSIY-Ig, cross-reactive KbSVY-Ig, or irrelevant KbOVA-Ig. Representative data shown from 1 of 3 separate experiments. (E) Competitive off-rate binding assay of 2C CD8+ T cells with KbSIY or KbSVY peptide MHC dimer over time by the addition of 1B2 TCR-binding antibody. Cells were gated on CD8+. Cells were stained with KbOVA as a negative control or experimental pMHC to gate on antigen-specific cells over time. This competitive binding assay was performed twice, with similar koff rates determined each time.
Table 1. IEDB predictions for Bifidobacteria EBP.

The biophysical interaction of the antigens was analyzed by comparing the ability of SIY and SVY to stabilize the Kb MHC complex on RMA-S cells. Both SVY and SIY peptides stabilized the H2-Kb MHC molecule to a similar extent, with half-maximal stabilization seen at approximately 100 nM (Figure 1C), indicating that SIY and SVY bind H2-Kb MHC with equal affinity. Using T cells from the 2C transgenic mouse, which are specific for the KbSIY peptide MHC (pMHC) complex, we found that this model TCR was cross-reactive with the KbSVY complex (Figure 1D), and 2C lymphocytes proliferated equally well as KbSIY and KbSVY (Supplemental Figure 2). Quantitative assessment of TCR affinity, using a competitive off-rate assay, showed that KbSVY has an approximately 4-fold lower affinity, koff 12.59e-4/s, compared with 3.12e-4/s (Figure 1E), and functionally lower trends for cytokine production, TNF-α, and IL-2, and CD107a expression were seen (Supplemental Figure 3, A–C). Thus, in a model 2C TCR system, there is cross-reactivity due to the single amino acid change, which still results in TCR binding, proliferation, and cytokine production, albeit at lower levels.
Modeling the interaction between KbSVY and KbSIY with the 2C TCR.
We investigated the change in Kb-peptide-TCR binding for the Val-to-Ile mutation at position 2 in the epitope sequence using molecular dynamics (MD) simulation. Figure 2A shows the constructed 3-way binding complex HLA-epitope-TCR, using the published individual x-ray structures of the Kb-epitope and TCR (10, 11) (see more details in Methods). Although the 2 epitope sequences differ only in the second position, the root mean square deviation (RMSD) per residue calculated between the highest populated binding pose of each epitope shows that Tyr3 (4.6 Å), Arg4 (3.6 Å), and Leu8 (4.3 Å) deviate more than the mutated residue (Ile2/Val2) (2.9 Å) (see Supplemental Table 1). The overlapping binding poses of KbSIY and KbSVY show that the side chains of the 3 residues with larger RMSDs deviate between the 2 epitopes (Supplemental Figure 4). Although the binding poses of the 2 epitopes differ from each other, the binding poses from the same epitope are generally consistent among the top 5 populated clusters, which account for over 98% of the MD trajectories (Supplemental Figure 5, A and B). A contact analysis found that the contacts at the interface between the peptide epitope, Kb, and TCR in the 3-way complex vary between epitopes because of conformational changes (Supplemental Figure 6, A and C). The frequencies of either Kb or TCR atoms, making close contact (less than 6 Å) with the epitopes in the MD trajectories, are presented on the surfaces of Kb and TCR (Supplemental Figure 6, A–D). The difference in SIY and SVY contact frequency (Figure 2B) shows the contact frequency on both the Kb and TCR surfaces. The contact frequency difference is greater than 80% at several regions on the TCR-epitope interface, specifically where TCR atoms contact Tyr3, Arg4, and Leu8 of the epitope. These data are consistent with the RMSD analysis, shown in Supplemental Table 1, indicating that the structures of the 2 epitopes deviate the most at these residues (Figure 2C). Meanwhile, the contact profile at the Kb-epitope interface is more similar between epitopes, without the large differences observed at the TCR-epitope interface, indicating the altered peptide configuration is mainly on the TCR side.
Figure 2. Modeling highlights differences in SIY and SVY binding to MHC and TCR.

(A) Structure of the H2-Kb/epitope/2C TCR complex. The α and β chains of 2C TCR and H2-Kb are represented by ribbon models in yellow, blue, red, and orange, respectively, and are transparent at the epitope interface. The epitope in the box at the center with a dotted line is represented by a stick model. (B) The difference in contact frequency between the SIY and the SVY epitopes on the 2C TCR-epitope (left) and the Kb-epitope (right) interfaces. The SIY and the SVY epitopes are colored in red and blue, respectively. The surfaces of TCR and Kb proteins are colored from blue to red, as the difference of the contact frequency is changed from –100% (SVY) to 100% (SIY). The red color indicates that protein atoms contact the SIY epitope more often than the SVY epitope, while the blue color indicates the opposite. (C) A zoomed-in image of the binding poses of the SIY (red) and the SVY (blue) epitopes, which are interfaced with H2-Kb and 2C TCR on the top and the bottom sides of the figure. The structure of each epitope is taken from the frame at the center of the most populated cluster in the MD trajectory. Ile2 of the SIY epitope and Val2 of the SVY epitope are colored in yellow and green, respectively. The α and β chains of 2C TCR and the α chain of H2-Kb are represented by ribbon models in yellow, blue, and red, respectively. (D) The relative free energies for the binding of the epitope to Kb (ΔΔGKb+epi), and the binding of TCR to the Kb-epitope complex (ΔΔGKb/epi+TCR), between the SIY and the SVY epitopes. Each free energy value was calculated by the FEP method.
We next computed, using the free energy perturbation (FEP) method, the binding affinity for each of 2 intermediate steps in the formation of the ternary complex, where each epitope is loaded onto the Kb protein, and then the Kb-epitope binary complex binds to the TCR. We found a change in binding affinity (SVY → SIY) of 0.09 ± 0.36 and –1.2 ± 0.2 kcal/mol for the epitope loaded into Kb, and the subsequent binding to TCR, respectively (Figure 2D). The FEP calculation results indicate that the binding affinity of the SVY binding to H2-Kb MHC is similar to that of the SIY binding; however, the binding affinity of the KbSVY complex to TCR is somewhat weaker than KbSIY (about 2 kBT, the Boltzmann factor times temperature, or equivalent to ~7.6-fold difference in the binding equilibrium constant at room temperature). These results are in excellent agreement with the biophysical and functional data shown in Figure 1, C and E, indicating that epitopes indeed have similar binding affinity for Kb, but KbSVY has somewhat weaker interactions with the 2C TCR than KbSIY because of the altered peptide configuration. Thus, the processing and presentation of EBP from B. breve leads to an SIY cross-reactive response, and in fact the SVY antigen can load nicely onto the MHC molecule. However, the amino acid substitution alters TCR recognition, creating slightly lower biophysical binding of 2C TCR to pMHC, which may affect T cell population cross-reactivity.
KbSIY- and KbSVY-specific T cell populations are cross-reactive.
To study cross-reactivity in endogenous, heterogeneous T cell populations, we analyzed SVY- and SIY-specific T cells from naive mice. CD8+ T cells from mice were enriched and stimulated with KbSIY- or KbSVY-based artificial antigen-presenting cells (aAPCs). The resultant day 7 cultures represent a heterogeneous mix of CD8+ T cells specific for each individual antigen, as previously described (12) (see Figure 3A). Expansion with KbSIY aAPCs led to approximately 50.4% KbSIY-specific T cells as seen by cognate KbSIY dimer staining (Figure 3B, top row, left). These were cross-reactive with KbSVY dimer, which had a similar 49.8% staining and an average expansion frequency of 36.4% (Figure 3B, top row, middle; and Supplemental Figure 7A). MFIs of the antigen-reactive populations were 685 for the cognate KbSIY dimer and 549 (i.e., ~20% lower) for the cross-reactive response to KbSVY pMHC (Figure 3B, top row). Background noncognate staining was less than 1%. Similarly, expansion of T cells with KbSVY aAPCs led to a cognate KbSVY-bound T cell population of 58.1% and a cross-reactive KbSIY-bound population of 54.4% of CD8+ T cells with similar MFIs and an average expansion frequency of 31.7% (Figure 3B, lower row, and Supplemental Figure 7A).
Figure 3. KbSVY- and KbSIY-specific T cells are cross-reactive.

(A) Schematic for isolation, expansion, and staining of antigen-specific T cells from mouse splenocytes. Nanoparticles tethered to both peptide-Kb complexes and agonistic anti-CD28 antibodies were used as aAPCS to enrich and expand antigen-specific T cells. Expanded T cells were then stained for SIY or SVY epitope reactivity. (B) Representative flow cytometry results of KbSIY and KbSVY staining of antigen-specific CD8+ T cells enriched from Jackson mouse splenocytes and expanded for 7 days. Cells were gated on live CD8+ cells. Representative data shown from 1 of 5 separate experiments. (C) Jackson mouse splenocytes were isolated and expanded as in A. KbSIY stimulations are on top and KbSVY stimulations are below. On day 7, CD8+ T cells were harvested and stained with a titration of pMHC-Ig as indicated and analyzed by flow cytometry. Results are reported as percentage of peptide-MHC positivity relative to that observed at 10 μg pMHC. KbSIY stimulations have a difference in cognate (red) and cross-reactive (black) binding with a P value of 0.016 by 2-way ANOVA with Bonferroni’s post hoc test for multiple comparisons. KbSVY stimulation shows no difference. P = 0.016, and N = 3 for each antigen expansion. Data represent mean ± SEM. (D) KbSVY-reactive T cells were isolated from Jackson mouse splenocytes and expanded for 7 days as in A. T cells were then stimulated with B16.SIY or parental B16.F10 tumor cells and measured for cytokine response by intracellular cytokine staining. P value = 0.0038 by 2-way ANOVA for B16.SIY and B16.F10 response. **P ≤ 0.01. N = 3. Data represent mean ± SEM.
Antigen-specific T cells were stained with varying concentrations of Kb-Ig molecules to determine the IC50 of the expanded T cells for each peptide-MHC complex and overall T cell avidity (Supplemental Figure 7B and Figure 3C). Similar to the findings from the 2C transgenic T cells, KbSIY-expanded T cells had a higher IC50 (0.1 μg) for KbSVY compared with KbSIY(0.01 μg) (Figure 3C, top). Differences in overall TCR affinity were also seen functionally as KbSIY-expanded T cells exhibited a lower trend of cytokine production for the cross-reacting KbSVY antigen restimulation (15% IL-2) than for the cognate SIY-pulsed RMA-S cells (30% IL-2) (Supplemental Figure 7C), but this was not significantly different.
Interestingly, T cells expanded with KbSVY-based aAPCs had equal affinity (0.1 μg) for both KbSVY and KbSIY pMHC-Ig complexes (Figure 3C, lower), and KbSVY-expanded T cells produced similar amounts of cytokines in response to both SIY and SVY stimulations (Supplemental Figure 7D). Cross-reactivity was also tested by analyzing the ability of SVY-expanded cells to recognize naturally processed KbSIY antigen on B16.SIY murine melanoma cells. Recognition of the naturally processed tumor antigen SIY was seen by cytokine release (Figure 3D). Therefore, heterogeneous KbSVY-specific T cells had equal affinity for both SIY and SVY and recognized SIY-expressing tumor cells. These data highlight the differences in T cell populations expanded by SIY or SVY antigens, which result in subtle differences in the cross-reactive T cell responses. Together, this demonstrates the overall cross-reactivity of polyclonal, heterogeneous T cell populations that recognize 2 distinct but closely related antigens.
Bifidobacterium colonization enhances SVY-specific T cell expansion.
Previously commensal Bifidobacterium was shown to promote antitumor immunity, and the bacterium associated with antitumor response was most similar to B. breve, B. longum, and B. adolescentis (99% identity) (13). Based on the sequence homology to the cross-reactive antigen between B16.SIY and B. breve (Figure 1A), we focused our studies on the SVY antigen and examined the presence or absence of Bifidobacterium in mice obtained from Jackson or mice obtained from Taconic, lacking all Bifidobacterium species (Supplemental Figure 8A) (13). KbSVY induced more antigen-specific T cells with a higher average MFI from Jackson than Taconic mice (Figure 4A). On average we were able to expand only approximately 8% SVY-specific T cells from Taconic mice versus 22% from Jackson mice and overall obtained about 4-fold fewer SVY-specific T cells from Taconic mice (Figure 4, B and C). A lower MFI, 109 versus 262, was also seen in cells stimulated from Taconic mice. Similarly, T cells from germ-free mice showed reduced frequency and total number of SVY-stimulated cells compared with Jackson mice (Supplemental Figure 8B).
Figure 4. SVY antigen in B.breve bacteria stimulates an immunogenic T cell response.

(A) CD8+ T cells were isolated and expanded from Jackson and Taconic splenocytes with KbSVY aAPCS. On day 7, cells were harvested and stained for antigen specificity. Data represent 1 of 5 independent experiments. (B) CD8+ T cells were harvested and expanded as in A. Data show percentage of KbSVY+ CD8+ T cells. P value = 0.0025, measured by a 1-tailed, paired t test. N = 5. (C) Total cell numbers for CD8+ T cell enrichment and expansion with KbSVY aAPCs. P value = 0.0214, measured by 1-tailed, paired t test. N = 5. (D) Jackson and Taconic mouse CD8+ T cells stimulated as in A. On day 7, cells were harvested and stained with a titration of KbSVY. Results are reported as percentage of peptide-MHC positivity relative to that observed at 10 μg pMHC. Jackson (blue) and Taconic (red) peptide stains were compared at a given concentration. P value = 0.0071, measured by 2-way ANOVA and Bonferroni’s post hoc test for multiple comparisons. N = 3. (E and F) Taconic mice were immunized by intraperitoneal injection of αCD40 on day –8 and subcutaneous injection of KbSVY on day –7. (E) On day 0, control or immunized mice were injected intravenously with target and nontarget autologous splenocytes mixed at a 1:1 ratio. Spleens were harvested from mice on day 1, and the ratio of target to nontarget cells was analyzed. P < 0.0001 by 2-tailed, unpaired t test. N = 3 mice/group. (F) Taconic mice were sacrificed on day 1. Spleens and draining lymph nodes were harvested and stained for KbSVY specificity. P = 0.019 by 2-tailed, unpaired t test. N = 6 immunized, 3 not immunized. (G) Jackson and Taconic mice were cohoused for 7 days or separately housed with littermates. CD8+ T cells from splenocytes stimulated as in A and harvested and stained for antigen specificity on day 7. Antigen specificity measured by subtracting noncognate antigen reactivity. P value = 0.0014 by 1-tailed t test. N = 5 Taconic only; N = 8 cohoused animals. (H) Jackson and Taconic mice were either cohoused for 7 days or housed with their respective littermates. CD8+ T cells were isolated and enriched and expanded with KbOVA aAPCS and harvested and stained for antigen specificity on day 7. Antigen specificity was measured by subtracting the noncognate antigen specificity. No statistical difference seen by 1-tailed, unpaired t test. N = 4. All cells gated on live CD8+ cells. All data represent mean ± SEM.
Overall KbSVY-specific effector T cells from Taconic mice had a lower TCR avidity compared with KbSVY-specific T cell populations from Jackson mice (Figure 4D). However, KbSVY-specific T cells from both populations produced cytokines upon restimulation with SVY peptide–loaded RMA-S cells (Supplemental Figure 8C). The impact of Bifidobacterium colonization was also seen in analysis of SVY-specific precursor frequency. Jackson mice trended to have more KbSVY-specific splenocytes compared with Taconic mice (Supplemental Figure 9, A and B). Thus, B. breve exposure increases the expansion capacity of KbSVY-reactive cells, leading to higher affinity responses and more robust KbSVY T cell expansion.
SVY antigen is immunogenic and response is transferable via gut colonization.
If SVY is indeed a cross-reactive antigen, one should be able to immunize mice and test for cross-reactive responses in vivo. Immunization of Bifidobacterium-negative Taconic mice with SVY-loaded Kb complexes, as previously described (14), led to approximately 60% killing of SVY target cells in vivo and a cross-reactive killing of approximately 50% of SIY-pulsed targets in vivo (Figure 4E). This was associated with an increased number of KbSVY effector T cells, from 0.4% to 0.6% (Figure 4F and Supplemental Figure 9C). Thus, immunization with SVY elicited an in vivo response that was cross-reactive with SIY.
To investigate whether the differences seen between Jackson and Taconic mice were due to the microbiome, we studied the effect of microbiome transfer on the differences in SVY reactivity. Taconic mice cohoused with Jackson mice for a week had a significant increase in KbSVY expansion compared with Taconic mice that were not cohoused (Figure 4G). This effect was SVY antigen specific because control KbOVA responses were not influenced by cohousing before T cell expansion (Figure 4H). This antigen-specific T cell response based on microbiome colonization shows CD8+ T cells primed in vivo lead to increased expansion capabilities of SVY-specific T cells.
Jackson mice have delayed growth of B16.SIY melanoma tumors.
An implication that differences in the commensal bacteria between Taconic and Jackson mice drive a cross-reactive antigen-specific targeting of B16.SIY tumors is that there should be differences in tumor growth between the 2 strains of mice. Indeed B16.SIY tumors grew more slowly in Jackson mice than in Taconic mice (Figure 5A) while B16.F10 tumors grew identically in the 2 strains (Supplemental Figure 10). Of note, initially it appeared that tumors grew slightly more rapidly in Jackson than Taconic mice (Figure 5A), but by day 10 tumors in Jackson mice were consistently smaller than in Taconic mice. By day 24, Taconic mice had an average tumor size of approximately 146 mm2, whereas tumors of Jackson mice were, on average, only 54 mm2, and individual spider plots display the slower tumor progression (Supplemental Figure 11, A and B). By day 32, all Taconic mice succumbed to disease, while 71% of the Jackson mice survived (Figure 5B).
Figure 5. Jackson mice have an increased antitumor response and antigen-selective pressure on the tumor.

(A) Jackson and Taconic mice were injected with 2 × 106 B16.SIY cells subcutaneously on day 0. Tumor growth curves show Jackson mice have significantly delayed tumor growth as compared with Taconic mice. P value = 0.0455. Significance was measured by 2-way ANOVA with Bonferroni’s post hoc test for multiple comparisons. N = 7/group. (B) Additionally, Jackson mice had significantly increased survival compared with the Taconic mice. P = 0.0065. Significance was measured by the log-rank test (Mantel-Cox test). N = 7/group. (C) Jackson and Taconic mice were injected with 2 × 106 B16.SIY cells subcutaneously on day 0. Tumors were harvested at an approximate size of 200–250 mm2 and analyzed by flow cytometry for GFP expression, as SIY and GFP are expressed as a fusion protein in the B16.SIY cell line (15). Jackson have a significantly lower GFP expression (P value = 0.0030) by unpaired, 2-tailed t test (n = 5 TAC, and n = 6 JAX). (D) Jackson and Taconic mice were injected with 2 × 106 B16.SIY cells subcutaneously on day 0. Tumor-infiltrating lymphocytes were harvested on day 24 and analyzed by flow cytometry for CD8+ T cell specificity, gated on live CD8+ T cells. N = 2/group.
We hypothesized that if differences between growth of B16.SIY tumors in Jackson versus Taconic mice were driven by antigen-specific recognition of the SIY neoantigen, we should see evidence for pressure on SIY antigen in tumors from Jackson mice. Tumors were collected from individual mice and analyzed for GFP expression, which is encoded by the same vector as SIY in the B16.SIY tumors. This approach has been used for a surrogate marker of anti-SIY antigen-specific T cell responses (15). B16.SIY tumors in Jackson mice had a significant loss of GFP expression, from 50% in Taconic mice tumors to 10% in Jackson mice tumors (Figure 5C and Supplemental Figure 11C).
Tumor-infiltrating T cells in B16.SIY tumors show cross-reactivity for KbSIY and KbSVY in Jackson mice tumors.
Jackson mice tumor-infiltrating T lymphocytes (TILs) were stained for KbSIY specificity or cross-reactivity for KbSVY and showed substantial staining over background KbOVA, which is not expected to be present in the B16.SIY tumor (Figure 5D, Supplemental Figure 11D). Notably, when TCR β chain sequencing was performed on TILs derived from B16.SIY tumors, we found that 22% of TIL clones were identical to those detected independently in splenocyte populations sorted and expanded by KbSVY. Overlapping TIL clones consisted of 9 of the 198 KbSVY-reactive sequences identified by in vitro enrichment and expansion of healthy mouse splenocytes (P = 3.42 × 10–30 for enrichment of SVY-reactive clones in the TIL population by the hypergeometric test). Taken together, Jackson mice have a beneficial antitumor response that is antigen driven and have higher selective pressure against the tumor antigen, and KbSVY cross-reactive T cells can be detected in the tumor, all highlighting the beneficial cross-reactive T cell population that is present in the Jackson mice.
Bifidobacterium alters the composition of the SVY-responsive TCR repertoire.
The impact of B. breve colonization on T cell specificity was also studied by analyzing TCR repertoires expanded by KbSIY or KbSVY from B. breve–colonized (Jackson) mice and those lacking B. breve (Taconic). KbSIY- and KbSVY-specific T cells from Jackson and Taconic mice were stimulated and sorted based on KbSIY or KbSVY staining (see Figure 6A schematic). The 8 T cell populations were analyzed by TCR β chain sequencing to determine how B. breve shapes KbSVY TCR repertoires.
Figure 6. B. breve colonization shapes the KbSVY TCR repertoire.

(A) Schematic setup of TCR repertoire analysis. CD8+ T cells from Jackson and Taconic mice were stimulated with KbSIY or KbSIY aAPCs. On day 7, cells from each stimulation were stained and sorted by antigen reactivity (KbSIY or KbSVY) and processed for TCR β chain deep sequencing. (B) Exact TCR clone overlap between all repertoires. Numbers indicate the number of unique overlapping clones between 2 TCR repertoires, and the color scale indicates the percentage contribution of the overlapping sequences to the total combined repertoire for each pair. (C) TCR clone overlap between all repertoires based on TCR homology using ImmunoMap algorithm. Numbers indicate the number of homologous clusters shared between 2 TCR repertoires, and the color scale indicates the percentage contribution of shared clusters to the total combined repertoire for each pair. (D) Overlap between Jackson and Taconic mice of T cell repertoires stimulated by SIY or SVY based on the frequency of homologous TCR clones and overlap between SIY and SVY stimulations for Jackson or Taconic mice groups. Data included the cognate and cross-reactive sorted populations, as indicated by the black boxes in C. P value = 0.0016 by 2-tailed t test. N = 4. Data represent mean ± SEM. (E) PCA of homology-based TCR clusters. TCR repertoires were analyzed by CDR3 sequence homology and separated into dominant motifs. (F) TCR β chain deep sequencing was performed on Jackson mouse splenocytes incubated with or without heat-killed Bifidobacterium. TCR clones with a Hamming distance of 1 were defined as homologous to clones from aAPC-expanded KbSIY- or KbSVY-specific T cells. Productive frequency of these homologous clones from Bifidobacterium-stimulated and unstimulated cells is shown. P value = 0.0001 by 2-tailed t test.
All 4 expanded TCR repertoires showed significant overlap when stained with cognate or cross-reactive Kb-peptide dimer (Figure 6B). Independent of B. breve colonization, we found many overlapping clones when comparing the cognate and cross-reactive sorted populations. As seen in the heatmap analysis (Supplemental Figure 12, A and B), the overlapping pairs were in distinct groups with little overlap with other repertoires and led to a selection of 4 distinct TCR populations of SIY and SVY cross-reactive clones (Figure 6B). Within Jackson mice T cell repertoires expanded by KbSIY, the KbSIY-sorted and KbSVY-sorted populations shared 47 identical clones comprising 79.9% of the response (Figure 6B). These populations also shared the same V and J allele expression based on heatmap analysis (Supplemental Figure 13, A and B). Within Jackson mice T cell repertoires expanded by KbSVY, the KbSVY-sorted and KbSIY-sorted populations shared 93 unique clones comprising 95.6% of the response (Figure 6B). In Taconic mice, T cell populations expanded with KbSIY and sorted on KbSIY or KbSVY had 45 overlapping clones that made up 85.9% of the response, while the T cells expanded with KbSVY and sorted on KbSVY or KbSIY had 49 shared clones comprising 77.1% of the response. Thus, identical T cell clones from cognate and cross-reactive staining of expanded cells showed that T cells are cross-reactive for both antigens, regardless of the commensal bacteria colonization.
The relationship between TCR repertoires was also examined using ImmunoMap, a TCR homology program we recently developed (16). ImmunoMap characterizes the overall homology patterns between different T cell repertoires based on homologous complementarity-determining region 3 (CDR3) sequences. In this fashion multiple repertoires can be overlaid to compare homology among various TCR repertoires (Supplemental Table 3). The effect of B. breve on TCR repertoire composition can be seen when comparing the overlap of SIY or SVY stimulation in Jackson and Taconic mice (Figure 6, C and D). B. breve–negative mice (Taconic) cells stimulated with KbSVY reactivity have only 51.1% homology to the KbSVY-stimulated repertoire from B. breve–positive (Jackson) mice (Figure 6C, note highlighted boxes; Figure 6D; and Supplemental Figure 12B). In contrast, the KbSIY TCR repertoire had a significantly overlapping response between Jackson and Taconic stimulations, with 71% overlap between both repertoires, which highlights that B. breve colonization has a greater effect on the composition of the KbSVY-stimulated TCR repertoire than on that of the KbSIY-stimulated repertoire (Figure 6D and Supplemental Figure 12).
Principal component analysis (PCA) was used to compare the TCR clones based on homology of dominant motifs identified from repertoires (16). Although individual repertoires all shared similar diversity metrics (Supplemental Table 3), KbSIY- or KbSVY-specific TCR motifs from B. breve–colonized (Jackson) or noncolonized (Taconic) mice revealed 4 distinct pairings, each driven by the supplier and the epitope used for expansion; however, the separation within and between pairs was qualitatively greater for Taconic mice than for Jackson mice (Figure 6E). Taken together, TCR repertoire sequencing indicates that the majority of SIY- or SVY-stimulated T cell clones are cross-reactive with both epitopes and that B. breve colonization is associated with a significantly greater shift in the clonal composition of the SVY-stimulated repertoire than in that of the SIY-stimulated repertoire.
The effect of Bifidobacteria stimulation on the TCR repertoire was also analyzed on T cells stimulated in vitro with heat-killed Bifidobacteria. After bacterial stimulation, TCR repertoire analysis of the expanded T cells identified 255 unique clones that overlapped with Jackson KbSIY- or KbSVY-stimulated and sorted repertoires by a Hamming distance of 1. The TCR clones similar to SIY- or SVY-peptide–reactive TCR repertoires were at a significantly higher productive frequency compared with clones present in non–Bifidobacteria-stimulated controls (Figure 6F). Thus, similar to our in vivo findings, in vitro stimulation with Bifidobacteria expanded a KbSIY-reactive TCR repertoire that could have antitumor activity.
KbSVY-reactive T cells inhibit growth of established SIY-expressing tumors in vivo.
One critical question is whether KbSVY-reactive T cells cross-react with the SIY epitope in vivo and mediate an effective antitumor response. This was studied using KbSVY effector T cells from B. breve–positive mice (Jackson) for ACT. Effector T cell populations were generated in vitro and adoptively transferred into B16.SIY tumor–bearing mice. KbSVY-expanded T cells inhibited B16.SIY tumor growth and extended survival in tumor-bearing mice. By day 22, untreated mice had an average tumor size of approximately 89.87 mm2, whereas SVY ACT–treated mice were, on average, only 22.37 mm2 (Figure 7A and Supplemental Figure 14A). When we analyzed survival data, 60% of KbSVY ACT–treated mice survived past 41 days while no control mice survived that long (Figure 7A).
Figure 7. Commensal bacteria epitope cross-reactivity mediates an antitumor response.

(A) Jackson mice (n = 5 SVY ACT; n = 4 no treatment) were injected with 2 × 106 B16.SIY cells subcutaneously on day 0. CD8+ T cells were harvested from spleens of independent Jackson mice and stimulated with KbSVY/anti-CD28 nanoparticles. On day 8, 1.3 × 105 of the resultant KbSVY-specific CD8+ T cells were injected intravenously into the tumor-bearing mice. Tumor growth curves show SVY ACT treatment significantly delayed tumor growth as compared with no treatment. P value = 0.0136, measured by 2-way ANOVA with Bonferroni’s post hoc test for multiple comparisons. Additionally, KbSVY ACT significantly increased survival compared with the no treatment group. Significance was measured by the log-rank test. P value = 0.0015. (B) Jackson mice were injected with 2 × 106 B16.SIY cells subcutaneously on day 0 and treated on day 8 with 1.3 × 105 KbSVY-specific CD8+ T cells via intravenous injection. Tumors were harvested on day 24 and analyzed by flow cytometry for GFP expression. Orange and blue indicate SVY T cell–treated mice and red indicates untreated mice. (C). Jackson mice (n = 3 SIY ACT, and n = 3 no treatment) were injected with 2 × 106 B16.SIY cells subcutaneously on day 0. CD8+ T cells were harvested from spleens of independent Jackson mice and stimulated with KbSIY/anti-CD28 nanoparticles. On day 8, 1.3 × 105 of the resultant SIY-reactive T cells were injected intravenously into tumor-bearing mice. Tumor growth curves show SIY ACT treatment significantly delayed tumor growth as compared with no treatment. Significance was measured by 2-way ANOVA with Bonferroni’s post hoc test for multiple comparisons (P < 0.001). ACT also significantly extended survival. Significance was measured by the log-rank test. P value = 0.024. N = 3/group.
ACT-treated mice, which eventually developed resistant tumors, were analyzed for antigen loss. Tumors were isolated at approximately 135 mm2 and measured for GFP intensity as a marker of SIY expression (17). The resistant tumors had a decrease in GFP expression compared with tumors isolated from an untreated mouse (Figure 7B), demonstrating that KbSVY-specific cells placed selective pressure on B16.SIY tumors, resulting in loss of antigen expression. As expected, KbSIY-expanded T cells also delayed tumor progression in tumor-bearing mice and extended survival (Figure 7C), showing that the repertoire present in KbSIY-specific T cells also has antitumor activity. Thus polyclonal, cross-reactive T cell populations expanded with KbSVY decreased tumor growth and increased survival, demonstrating the potential benefits of commensal bacteria–responsive T cell cross-reactivity on antitumor immunity.
Discussion
Understanding the role of antigen mimicry leading to T cell–mediated antitumor responses is essential in understanding how microbiota can lead to antitumor effects. B. breve contains a peptide with homology to the SIY epitope, which influences anti–B16.SIY melanoma-specific responses. Use of B. breve as a source of homologous antigen does not limit the possibility of other sources of antigen, although throughout the paper the B. breve antigen sequence shows robust cross-reactivity and ability to induce the antitumor T cell response both in vitro and in vivo. Looking closely, the isoleucine substitution to valine at the second position did not affect peptide-MHC binding; however, the substitution appears to shift the orientation of more distant amino acids in the binding pocket, leading to altered TCR recognition. Although differences in binding with the 2C transgenic TCR illustrated that an individual TCR can display somewhat lower affinity for KbSVY than KbSIY, endogenous murine polyclonal SIY and SVY T cell responses exhibit striking cross-reactivity. On closer examination, TCR β sequencing reveals the same clones can bind to both the SIY and SVY antigens after a single stimulation. The endogenous KbSIY response is cross-reactive and recognizes both antigens, SIY and SVY, albeit with a lower affinity and less robust functional response to the SVY antigen. In contrast, KbSVY-reactive T cells recognize both peptide antigens with similar affinity as well as functional ability, as assessed by their ability to release cytokines in response to tumor cells. This difference in cross-reactivity relative to the initial T cell stimulation suggests that C57BL/6 mice possess a heterogeneous SIY/SVY T cell population that can be stimulated by B. breve and that the polyclonal population contains a range of affinities for SIY and SVY based on ability to bind to the slightly altered amino acid orientation in the MHC pocket.
One of the most interesting findings is that the gut microbiota primes an SVY-reactive T cell response. Although the mechanism of T cell activation and selection by components of the gut microbiota is not understood, analysis of mice with and without Bifidobacterium indicates that in vivo priming occurs and boosts the ability to expand SVY-reactive T cells. The SVY antigen is immunogenic and can induce effector T cells that are cross-reactive in vivo with the SIY antigen. In addition, the antigen-specific T cell response is transferable, as demonstrated by cohousing experiments. Comparing the TCR repertoires between colonized and noncolonized animals revealed SVY-reactive T cells that express TCRs with little or no overlap between the B. breve–naive (Taconic) and B. breve–experienced (Jackson) mouse repertoires. An altered SVY-expandable TCR repertoire developed in the colonized mice, while the SIY-expandable populations showed markedly greater homology between Taconic and Jackson mice. The SVY-stimulated TCR repertoires from B. breve–colonized (Jackson) mice contained homologous TCR clones that are shared with the Jackson KbSIY stimulation. These findings suggest that differences in B16.SIY and B16.F10 tumor growth between Taconic and Jackson mice may be driven in part by a cross-reactive, SVY-primed subset of the TCR repertoire. In addition, stimulating T cells directly with B. breve expands T cell clones that are similar to the Jackson KbSIY or KbSVY T cell populations. These data highlight how commensal bacterial antigens may affect the host immune repertoire landscape through antigen mimicry. An important characteristic of the SVY-reactive T cells was their ability to kill SIY-expressing tumors in vivo. Treatment of B16.SIY tumor–bearing animals with adoptively transferred KbSVY-reactive T cells showed cross-reactive antitumor activity of an endogenous, polyclonal commensal bacteria antigen–stimulated T cell population.
Importantly, our study illustrates that antigen mimicry from gut microbes influences T cell immunity and helps generate a cross-reactive antitumor response. Current work identifying beneficial bacteria from responders to immunotherapy as well the benefits of fecal transplant in patients has yet to isolate a clear mechanism of action; antigen mimicry may play a role in patient responses. Here we confirmed that antigen homology between Bifidobacterium and the murine melanoma SIY “neoantigen” stimulates cross-reactive T cells. Furthermore, the presence of B. breve in the microbiome boosts the KbSVY T cell population, and KbSVY response can target and slow tumor progression. CD8+ T cell populations driven by commensal bacteria stimulation have shown functional responses (7, 18–21), and we now reveal how changing the microbiome can alter cross-reactive T cell responses, which highlights the importance of the microbiome for CD8+ T cell responses. Although the overall diversity of the microbiome has been linked to positive clinical responses in patients, identifying homologous antigens found in commensal bacteria relevant to specific patients’ tumors will require more extensive studies with potentially a more robust, larger array of antigens to stimulate the T cell response (7, 22, 23). With this mechanism in mind, we believe that a rigorous panspecies genomic analysis of commensal organisms would aid a more precise understanding of the effects of microbiota on antitumor immunotherapy. Tumor neoantigen homology to pathogen or commensal bacteria may provide an effective way to isolate unique T cell populations that can be stimulated by both gut microbes and tumor cells and to generate robust antitumor T cell responses.
Methods
Mice.
C57BL/6 mice were purchased from The Jackson Laboratory (Bar Harbor, Maine, USA) and Taconic Biosciences (Germantown, New York, USA). 2C TCR transgenic mice were a gift from the Gajewski laboratory (University of Chicago, Chicago, Illinois, USA) and bred as heterozygotes on the C57BL/6 background. Male and female mice, 8–12 weeks old, were used and maintained according to Johns Hopkins University’s (JHU) Institutional Review Board.
Cell lines.
RMA-S cells were a gift from the Edidin laboratory (JHU, Baltimore, Maryland, USA). B16.F10 cells were a gift from the Mackensen laboratory (Alfred Wegener Institute, Bremerhaven, Germany). B16.SIY cells were a gift from the Drake laboratory (JHU, Baltimore, Maryland, USA).
Peptide stabilization assay.
RMA-S cells were held at 25°C overnight and pulsed with peptide (GenScript) for 2 hours, then incubated at 37°C for 2 hours. Cells were stained with anti-Kb (M1/42, BioLegend) and run on flow cytometry.
2C functional analysis.
2C splenocytes were stimulated with peptide for 5 days, harvested and restimulated with RMA-S cells, and peptide pulsed overnight. T cells and RMA-S cells were incubated (1:1) with GolgiStop (554724, BD Biosciences), GolgiPlug (555029, BD Biosciences), and CD107a (FITC, 1D4B, BD Pharmingen). After 6 hours, cells were stained with viability stain (L23101, Thermo Fisher Scientific) and anti-CD8+ (PerCP, 53-6.7, BioLegend). Cells were fixed and permeabilized (Fixation/Permeabilization Solution Kit 554714, BD Biosciences) and stained for IFN-γ (PE, XMG1.2, BD Pharmingen), TNF-α (PE-Cy7, MP6-XT22, BioLegend), and IL-2 (APC, JES6-5H4, BD Pharmingen).
Competitive binding assay.
2C T cells were stimulated with SIY peptide for 5 days and stained with fluorescent peptide-MHC multimer/(in-house) for 1 hour. Afterward, saturating amounts of an unconjugated competitor anti-mouse 2C TCR (1B2, Bio X Cell) was added and measured by flow cytometry over time.
Bifidobacteria quantification.
Fecal DNA samples were isolated from Jackson and Taconic mice (R2002, Zymo Research). Bifidobacteria colonization relative to total eubacteria was quantified by multiplexed quantitative real-time PCR analysis of microbial rRNA genes. The following primers and TaqMan probe were used for detection of Bifidobacteria rRNA: 5′-CGGGTGAGTAATGCGTGACC-3′, 5′-TGATAGGACGCGACCCCA-3′, and 5′-6FAM-CTCCTGGAAACGGGTG-QSY-3′ (13). The following primers and TaqMan probe were used for detection of total eubacteria rRNA: 5′-TCCTACGGGAGGCAGCAGT-3′, 5′-GGACTACCAGGGTATCTAATCCTGTT-3′, and 5′-JUN-CGTATTACCGCGGCTGCTGGCAC-QSY-3′ (24).
Bifidobacteria stimulation of murine T cells.
Splenocytes and mesenteric lymph node cells were isolated from Jackson mice. Cells were suspended in medium (RPMI containing 10% heat-inactivated FBS, 50 μM β-mercaptoethanol, 1 unit/mL penicillin, 1 μg/mL streptomycin, and 20 IU/mL recombinant murine IL-2 from R&D Systems, Bio-Techne) at 2 × 106 viable cells/mL, and 4 × 106 cells were seeded per well in a 24-well plate. Bifidobacterium and Lactobacillus probiotic capsules were obtained from SeekingHealth. One capsule was resuspended in 10 mL of PBS and heated at 57°C for 1 hour. Splenocytes were stimulated with 160 μL of the heat-killed resuspended bacteria (approximately 50–100 bacterial cfu per splenocyte) or left unstimulated. Cultures were maintained at 37°C at 5% CO2 for 11 days. Fresh medium and IL-2 were added every other day. Cultures were then stained with anti-CD8a (APC-Cy7, 53-6.7, BioLegend), DAPI, and either PE-Kb-SIY dimer or a control PE-Kb dimer (in-house). Cells were analyzed on an LSRII flow cytometer (BD). Antigen-specific frequency was calculated by subtracting the noncognate background staining. DNA was extracted from 4 × 105 remaining cells using the DNeasy Blood and Tissue Kit (69504, QIAGEN). Extracted DNA was submitted to Adaptive Biotechnologies for survey-level TCR β chain sequencing.
Preparation of MHC-Ig dimers and nanoparticles.
Soluble MHC-Ig dimers Kb-Ig (in-house) were prepared in-house and loaded with peptides (GenScript) as described (25). Anti-CD28 antibody was purchased from BioLegend (37.51). In-house particles were manufactured by directly conjugating peptide-Kb-Ig and anti-CD28 to amine-coated, labeled, 80- to 100-nm super paramagnetic iron-oxide particles purchased from Micromod and functionalized according to the manufacturer’s recommendations. Briefly, amines on particles reacted with Sulfo SMCC (Proteochem) and then were magnetically washed. Soluble peptide-MHC-Ig and anti-CD28 were modified with 2-iminothiolane HCl purchased from MilliporeSigma. Excess 2-iminothiolane was washed away from protein solution by a Vivaspin 20, 50 kDa MWCO concentrator (GE Healthcare). Peptide-MHC-Ig and anti-CD28 were mixed to a 1:1 ratio and added to washed particles and mixed overnight at 25°C. Particles were then washed and stored at 4°C. Nano-aAPCs were stored at a concentration of 5 × 1012 particles/mL.
T cell enrichment and expansion stimulations and cross-reactivity.
Spleen and lymph nodes were harvested and isolated with CD8 kits (130-104-075, Miltenyi Biotec) as per the manufacturer’s instructions. CD8+ T cells were incubated with nanoparticles at 10 μL of particles per 107 CD8+ T cells at 4°C for 1 hour, and cells were washed over MS Miltenyi column. The positive fraction was eluted and plated at 2.5 × 105 cells/mL and cultured for 7 days in 96-well round-bottom plates in complete RPMI 1640 medium supplemented with 10% human autologous serum and 3% T cell growth factor, a cytokine cocktail derived from stimulated PBMCs as described in the literature (12), in a humidified 5% CO2, 37°C, incubator. Specificity of cytotoxic T lymphocytes was monitored on day 7, by FACS analysis following viability stain (L34970, Invitrogen, Thermo Fisher Scientific), dimeric MHC-Ig staining (PE, in-house), and anti-CD8a (APC, 53-6.7, BioLegend). The number of antigen-specific cells was calculated by multiplying the number of total live cells by the fractions of CD8+ and antigen-specific cells; the fraction of antigen-specific cells was calculated after subtracting the noncognate MHC staining from cognate MHC staining.
T cell cytokine response.
After 7 days of stimulation, T cells were incubated with GolgiStop (554724, BD Biosciences) and GolgiPlug (555029, BD Biosciences) and anti-CD107a (FITC, 1D4B, BD Pharmingen) in RPMI 1640 medium supplemented with 10% FBS. RMA-S pulsed overnight at 25°C with peptide at 1 mg/mL were washed and added to cells at a 1:2 ratio (RMA-S/T cells). Cells were then incubated at 37°C for 6 hours. Cells were washed and stained with anti-CD8a, (PerCP, 53-6.7, BioLegend) and viability stain (L34970, Thermo Fisher Scientific) for 15 minutes at 4°C. Cells were then washed with PBS, and 100 μL of Cytofix/Cytoperm Fixation and Permeabilization Solution (51-2090KZ, BD Biosciences) was added to the cells and incubated overnight at 4°C. Cells were washed with Perm/Wash Buffer (51-2090KZ, BD Biosciences). Cells were stained with anti–IFN-γ (PE, XMG1.2, BD Pharmingen), anti–IL-2 (APC, JES6-5H4, BD Pharmingen), and anti–TNF-α (PE-Cy7, MP6-XT22, BioLegend) for 1 hour at 4°C. Cells were washed and read on an LSRII flow cytometer (BD). Background cytokine staining was accounted for by subtracting cytokine-positive cells in nonstimulated conditions from the restimulated cells.
MD simulation setup.
Each system consisted of a protein complex in one of the following 3 states: (a) the epitope only, (b) the epitope bound to H2-Kb MHC (Kb/epitope binary complex), and (c) 2C TCR bound to the H2-Kb/epitope complex (Kb/epitope/2C TCR ternary complex). The epitope sequence of each system was chosen to be either SIYRYYGL (SIY) or SVYRYYGL (SVY). The initial coordinates of H2-Kb and 2C TCR were taken from the x-ray crystal structure of each (Protein Data Bank [PDB] 3P9L, ref. 10; H2-Kb, PDB 2OI9, ref. 11: 2C TCR). The initial configuration of the epitope was built based on the OVA257-264 epitope (SIINFEKL), which is complexed with H2-Kb in the crystal structure. The OVA epitope was mutated to either SIY or SVY epitopes using VMD mutator plugin (26). The H2-Kb/epitope/2C TCR complex was constructed in the following steps: the coordinates of 2C TCR in the crystal structure were adjusted by aligning the α-carbons of H2-Kb and MHC protein between the 2 crystal structures; then the structures of H2-Kb, epitope, and 2C TCR were combined into one system. The protein complex was placed in the center of a rectangular box with a periodic boundary condition. The rest of the empty space of the box was filled with water molecules using the GROMACS tool solvate (27). The sizes of the simulation box for the protein complex in states 1, 2, and 3 were 60 Å × 60 Å × 60 Å, 83 Å × 90 Å × 114 Å, and 86 Å × 90 Å × 145 Å, respectively. The length of each side of the box was set to be sufficiently large that proteins at the center of the box were separated at least 30 Å away from their periodic images. The protonation states of His residues of the proteins were determined based on an optimal hydrogen bonding conformation, determined by the GROMACS tool pdb2gmx. His93 of the α chain of H2-Kb MHC, and His29 of the β chain of 2C TCR, were singly protonated at the Nδ1 atom. His84 of the β chain of H2-Kb was doubly protonated at the Nδ1 and the Nε2 atoms. All other His residues were singly protonated at the Nε2 atom. Standard protonation states were chosen for all other residues. Na+ and Cl– ions were added in the solvent using the GROMACS tool genion, to neutralize the protein systems, as well as mimic the physiological ionic concentration, 150 mM. The CHARMM36 force field (28) was employed for the protein and ions and TIP3P model (29) for waters. The MD simulations were performed with GROMACS package version 5.14 (27). Long-ranged electrostatic interactions were treated with the Particle Mesh Ewald method (30). The cutoff distance for both the Lennard-Jones and the real space Coulomb interaction was set to be 12 Å. All covalent bonds involving the hydrogen atoms of the protein and water molecules were constrained by the LINC algorithm (31). Each system was minimized at 0 K for 1000 steps, then equilibrated in NPT (constant pressure and temperature) ensemble with a temperature of 300 K and a pressure of 1 atm for 1 ns. The time step was set to be 2.0 fs. After the total volume of the system was stabilized in NPT ensemble, the system was equilibrated again in NVT (constant volume and temperature) ensemble at 300 K for 100 ns. Then, the NVT simulation was extended for another 500 ns for the production run. The temperature and the pressure were controlled by Nose-Hoover (32) and Berendsen (33) schemes. The system coordinates were written every 100 ps during the production run; then all MD frames were clustered by the RMSD of the protein complex using gromos method (34). The RMSD cutoff value of each cluster was set to be 2.5 Å.
Free energy perturbation.
The FEP calculation was performed to calculate the relative free energy changes of the systems in 3 states, while Val2 of the SIY epitope was mutated to Ile2. The hybrid structure and topology of each system were generated by PMX software (35). The alchemical transformation from Val2 to Ile2 was governed by the hybrid Hamiltonian H(λ), as a function of the coupling parameter λ (10), where H(λ) = (1 – λ) × HV + λ × HI + H0.
HV and H1 are the Hamiltonians for the atoms of Val2 and Ile2, respectively, which undergo the alchemical transformation during the simulation, and H0 is the Hamiltonian for the rest of the system, with a fixed topology. Thirty-four λ-windows were generated between λ = 0 and λ = 1, where the λ-interval between neighboring windows was fixed to 0.04 between λ = 0.02 and λ = 0.98 and gradually decreased from 10–2 to 10–5, as λ approaches both ends, λ = 0 and λ = 1. To circumvent numerical instability caused by insertion or deletion of atoms in the simulation, the soft-core potential (36) was applied to both the van der Waals and the electrostatic interactions for the atoms in the alchemical domain. The λ value for the electrostatic interaction was fixed to 0 at the windows between λ = 0 and λ = 0.1, then rescaled linearly to [0, 1] at the windows between λ = 0.1 and λ = 1, while the λ value for the van der Waals and the bonded interactions was set to be equal to that of the window. At each λ-window, the system was equilibrated for 100 ps, followed by the production run for 500 ps. The simulation was carried out sequentially from λ = 0 to λ = 1 so that the initial coordinates and velocities of each window were taken from the last MD frame of the previous window. The setup for the MD simulation part was the same used in the NPT simulation in the earlier section. All other FEP setups were the same used in our previous studies (37–40) on other MHC-epitope-TCR systems.
The free energy change (ΔG) was calculated using the thermodynamic integration method (26, 41, 42), in the equation as follows (11):
(Equation 1),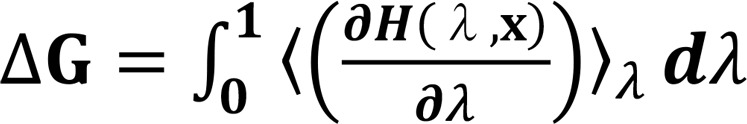
where ‹(əH(λ, x)/əλ)›λ is the ensemble average of the derivative of the hybrid potential with respect to λ. əH(λ, x)/əλ was written every 0.1 ps. ΔG was calculated for each of 3 systems with different protein complexes as listed above (ΔGepi, ΔGKb/epi, and ΔGKb/epi/TCR). Statistical error of ΔG was estimated using the block average method by dividing the trajectory of each window into 4 consecutive blocks. To better sample the conformational change of the protein complex, ΔGKb/epi and ΔGKb/epi/TCR were averaged over 5 independent runs, starting from different initial configurations. Each initial configuration was taken from the frame at the center of each of the top 5 most populated clusters from the MD simulations, as shown in Supplemental Figure 3. ΔG of each individual run was weighted by the population of each cluster. Due to its small system size, ΔGepi was calculated from a single run. The calculation details are shown in Supplemental Table 2. The relative free energy changes for the 2 reaction steps during the formation of the protein complex, ΔΔGKb+epi and ΔΔGKb/epi+TCR, defined in the main text, were calculated as follows (26, 27):
(Equation 2)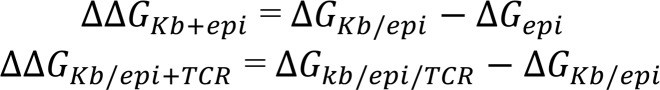
Immunization assay.
Recipient mice were injected intraperitoneally with 10 μg/mouse of anti-CD40 mAb (clone 3/23; BioLegend) and a day later immunized subcutaneously with 250 μg as indicated of either OVA−Kb-Ig or SVY−Kb-Ig dimer (14).
In vivo killing assay.
Target cells were collected from splenocytes of naive mice, cleaned from erythrocytes by osmotic lysis, washed, and split into 2 populations. The control population was incubated at 37°C for 20 minutes and labeled with a high concentration of CFSE (Invitrogen, Thermo Fisher Scientific) (2.5 μM) (unlabeled−CFSEhi cells). The target population was pulsed with 1 μM SVY, SIY, or OVA peptide and was labeled with a low concentration of CFSE (0.25 μM) (target−CFSElo cells). The 2 populations were mixed together at a 1:1 ratio and intravenously injected into mice (10 × 106/population/mouse) 7 days after immunization with KbSVY-Ig dimer, KbOVA-Ig dimer, or no immunization control mice. After 18 hours the mice were sacrificed and spleens harvested. The cell suspensions were analyzed by FACS for the presence of 2 differentially CFSE-labeled target populations. The recovery and percentage killing of the various CFSE-labeled, peptide-pulsed target cells were calculated as follows: % of in vivo killing = 100 − ([(% specific peptide–pulsed cells in immunized/% unspecific peptide–pulsed cells in immunized)/(% specific peptide–pulsed cells in naive/% unspecific peptide–pulsed cells in naive)] × 100).
Cohousing experiment.
Taconic and Jackson mice, 8 weeks old, were cohoused in fresh cages for 7 days. On day 7, age-matched Taconic mice, cohoused Taconic mice, and cohoused Jackson mice were sacrificed and spleens isolated for CD8+ T cell enrichment and expansion for KbSVY and KbOVA T cell stimulations.
TCR repertoire analysis.
A custom R script was developed to analyze CDR3 data obtained from Adaptive Biotech based on both exact and homology overlaps between groups. Only sequences with a productive frequency more than 0.01% were included in the analysis. For exact overlap analysis, CDR3 reads were normalized within samples and then compared across all samples. First, the number of unique CDR3s and the overall contribution from shared sequences were compared. Next, PCA was performed and samples were plotted in terms of their top 2 principal components to examine sample clustering behavior in the lower dimensional space. Additionally, a heatmap representation was drawn using row-wise Z scores. Finally, the Jensen Shannon Divergence was computed in terms of the Shannon’s entropy of each CDR3 distribution as (43):
(Equation 3)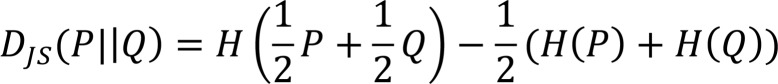 ,
,
where the Shannon’s entropy in terms of the frequency of all N unique CDR3s, p(i) is (11):
(Equation 4)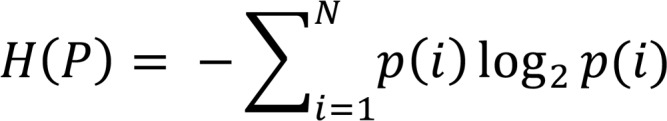
For homology overlap analysis, the ImmunoMap algorithm was used (16). Briefly, pairwise distance scores between all unique CDR3s were calculated using a PAM10 substitution matrix, a gap opening penalty of 30, and a gap extension penalty of 0, and the ImmunoMap distance metric was calculated. Then, average hierarchical clustering was performed, and a homology threshold of 0.35 was used. Finally, dominant motifs were defined as homology clusters with a frequency greater than 3%. Homology overlaps were represented in a similar manner as above.
Tumor growth and ACT.
Jackson and Taconic mice, 8 weeks old, were injected with B16.SIY (2 × 106 cells) or B16.F10 (1 × 105 cells) in the flank and measured for tumor growth with calipers over time and monitored for survival. Jackson mice were injected subcutaneously with B16.SIY tumor cell line (2 × 106 cells). At 7 days after tumor injection, mice were partially ablated with 5 cGy to create room for adoptively transferred cells. On day 8, day 7–stimulated KbSIY- or KbSVY-specific CD8+ T cells from Jackson mice were transferred into tumor-bearing animals by retro-orbital injection with 1.3 × 105 antigen-specific cells/mouse. On day 8 and day 9, mice received IL-2 (30,000 units by intraperitoneal injection). Mice were measured 3 times/wk for tumor growth and survival, and tumor volume was reported in square millimeters.
TIL isolation.
TILs were obtained from tumors by manual digestion and washing, followed by a density gradient centrifugation (Mouse, Lympholyte Cell Separation Media, CEDARLANE). Cells were counted and used for flow cytometry analysis. All cell lines underwent testing for mycoplasma contamination.
Data and materials availability.
All data are available in the main text or the supplemental materials.
Statistics.
Information on statistical tests is present in all figure legends. When making multiple comparisons, 1- and 2-way ANOVA were used. Bonferroni’s posttests were performed when comparing all groups, and Dunnett’s posttests were performed when the hypotheses being tested involved comparison against a single group. When comparing 2 groups, 1-tailed and 2-tailed t tests were used, as indicated in figure legends. All data sets were assumed to fit a normal distribution. All graphs show mean and error bars represent SEM. All n values are present within figure legends, and representative data include numbers of repeated experiments. Mice with outlier tumor size before the beginning of treatment were removed from the studies, with outlier size determined by no detected tumor or tumor larger than mean plus standard deviation from mean tumor size of all animals used in the experiment. Randomization was performed by cage in all animal studies. All statistical analysis was performed using GraphPad Prism software. P value less than 0.05 was considered significant.
Study approval.
The present studies in mice have been reviewed and approved by the Johns Hopkins University Institutional Review Board.
Author contributions
JJH and TAC conceived the project, and CAB, AI, JJH, TAC, JPS all designed and discussed the project. CAB, AI, JJH, JWH, WC, JGB, RS, FK, and TP all helped with the investigation. CAB, AI, and JJH acquired and analyzed data and helped write the manuscript. JWH, WC, RS, FK, and TP helped acquire data. JGB helped with analyzing data and writing the manuscript. AI conducted formal analysis and visualization of the TCR repertoire analysis. SL, DRB, and RZ conducted formal analysis for the TCR-modeling simulations. CAB led writing and AI, JJH, SL, RZ, TAC, and JPS edited the manuscript. RZ, TAC, and JPS supervised the project.
Supplementary Material
Acknowledgments
We would like to thank Jeffrey Weber and Binquan Luan for their various help with the modeling part of the work. We thank all members of the Chan lab for helpful discussions. We thank the Marie-Josée and Henry R. Kravis Center for Molecular Oncology for excellent suggestions. We also thank Memorial Sloan Kettering Cancer Center core laboratories, including Integrated Genomics Operations, Molecular Cytology, Cytogenetics, and Flow Cytometry cores. We thank the Sears lab for access to germ-free mice for experiments. We thank Eliot Sachsenmeier for his help. We thank the Johns Hopkins Bloomberg School of Public Health Bloomberg Flow Cytometry and Immunology Core and Dr. Hao Zhang. RZ acknowledges the support of the IBM Blue Gene Science Program (W1258591, W1464125, W1464164). This study was funded in part through the NIH/National Cancer Institute Cancer Center Support Grant P30 CA008748. We acknowledge funding sources, including Pershing Square Sohn Cancer Research grant (to TAC), the PaineWebber Chair (to TAC), Stand Up 2 Cancer (to TAC), NIH R01 CA205426 (to TAC), NIH R35 CA232097 (to TAC), the STARR Cancer Consortium (to TAC), and the Precision Immunotherapy Kidney Cancer Fund (to TAC, RJM). JWH thanks the NIH Cancer Nanotechnology Training Center at the Johns Hopkins Institute for NanoBioTechnology, the National Science Foundation Graduate Research Fellowship (DGE-1232825), and the ARCS Foundation for fellowship support. AI is supported by the National Science Foundation Graduate Research Fellowship. This work was funded by support from the NIH R01-CA108835 to JPS, R21-EB023411 to JPS, R33-CA229042 to JPS, and P41-EB028239 to JPS; a grant from AstraZeneca (to JPS); and a gift from the Troper Wojcicki Foundation (to JPS).
Version 1. 04/23/2020
Electronic publication
Footnotes
RZ’s present address is: Institute of Quantitative Biology, Zhejiang University, Hangzhou, China, and Department of Chemistry, Columbia University, New York, New York, USA.
Conflict of interest: TAC is a cofounder of Gritstone Oncology and holds equity. TAC holds equity in An2H. TAC acknowledges grant funding from Bristol Myers Squibb, AstraZeneca, Illumina, Pfizer, An2H, and Eisai. TAC has served as an advisor for Bristol Myers Squibb, Illumina, Eisai, and An2H. Under a licensing agreement between NexImmune and the Johns Hopkins University, JPS is entitled to shares of royalty received by the university on sales of artificial antigen-presenting cell products described in this article. He also owns NexImmune stock, which is subject to certain restrictions under university policy. JPS is a member of the company’s Scientific Advisory Board. The terms of this arrangement are being managed by the Johns Hopkins University in accordance with its conflict-of-interest policies. JPS acknowledges grant funding from AstraZeneca.
Copyright: © 2020, American Society for Clinical Investigation.
Reference information: JCI Insight. 2020;5(8):e135597.https://doi.org/10.1172/jci.insight.135597.
Contributor Information
Ariel Isser, Email: aisser1@jhu.edu.
Jonathan J. Havel, Email: jonathan.havel@gmail.com.
Sangyun Lee, Email: sangyun0708@gmail.com.
David R. Bell, Email: D.R.Bell@ibm.com.
John W. Hickey, Email: jhickey8@jhmi.edu.
Worarat Chaisawangwong, Email: wchaisa1@jhu.edu.
Joan Glick Bieler, Email: jbieler@jhmi.edu.
Fengshen Kuo, Email: kuof@mskcc.org.
Ruhong Zhou, Email: rz24@columbia.edu.
Timothy A. Chan, Email: chant@mskcc.org.
References
- 1.Rizvi NA, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348(6230):124–128. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gubin MM, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. 2014;515(7528):577–581. doi: 10.1038/nature13988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gros A, et al. Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nat Med. 2016;22(4):433–438. doi: 10.1038/nm.4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Łuksza M, et al. A neoantigen fitness model predicts tumour response to checkpoint blockade immunotherapy. Nature. 2017;551(7681):517–520. doi: 10.1038/nature24473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Balachandran VP, et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature. 2017;551(7681):512–516. doi: 10.1038/nature24462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Routy B, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science. 2018;359(6371):91–97. doi: 10.1126/science.aan3706. [DOI] [PubMed] [Google Scholar]
- 7.Gopalakrishnan V, et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science. 2018;359(6371):97–103. doi: 10.1126/science.aan4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Iida N, et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science. 2013;342(6161):967–970. doi: 10.1126/science.1240527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Viaud S, et al. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science. 2013;342(6161):971–976. doi: 10.1126/science.1240537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Denton AE, et al. Affinity thresholds for naive CD8+ CTL activation by peptides and engineered influenza A viruses. J Immunol. 2011;187(11):5733–5744. doi: 10.4049/jimmunol.1003937. [DOI] [PubMed] [Google Scholar]
- 11.Colf LA, et al. How a single T cell receptor recognizes both self and foreign MHC. Cell. 2007;129(1):135–146. doi: 10.1016/j.cell.2007.01.048. [DOI] [PubMed] [Google Scholar]
- 12.Perica K, et al. Enrichment and expansion with nanoscale artificial antigen presenting cells for adoptive immunotherapy. ACS Nano. 2015;9(7):6861–6871. doi: 10.1021/acsnano.5b02829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sivan A, et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science. 2015;350(6264):1084–1089. doi: 10.1126/science.aac4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schütz C, Zoso A, Peng S, Bennett JD, Schneck JP, Oelke M. MHC-Ig induces memory T cell formation in vivo and inhibits tumour growth. Immun Inflamm Dis. 2014;2(3):181–192. doi: 10.1002/iid3.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blank C, et al. PD-L1/B7H-1 inhibits the effector phase of tumor rejection by T cell receptor (TCR) transgenic CD8+ T cells. Cancer Res. 2004;64(3):1140–1145. doi: 10.1158/0008-5472.CAN-03-3259. [DOI] [PubMed] [Google Scholar]
- 16.Sidhom JW, Bessell CA, Havel JJ, Kosmides A, Chan TA, Schneck JP. ImmunoMap: a bioinformatics tool for T-cell repertoire analysis. Cancer Immunol Res. 2018;6(2):151–162. doi: 10.1158/2326-6066.CIR-17-0114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blank C, Brown I, Kacha AK, Markiewicz MA, Gajewski TF. ICAM-1 contributes to but is not essential for tumor antigen cross-priming and CD8+ T cell-mediated tumor rejection in vivo. J Immunol. 2005;174(6):3416–3420. doi: 10.4049/jimmunol.174.6.3416. [DOI] [PubMed] [Google Scholar]
- 18.Matson V, et al. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science. 2018;359(6371):104–108. doi: 10.1126/science.aao3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gil-Cruz C, et al. Microbiota-derived peptide mimics drive lethal inflammatory cardiomyopathy. Science. 2019;366(6467):881–886. doi: 10.1126/science.aav3487. [DOI] [PubMed] [Google Scholar]
- 20.Ruff WE, et al. Pathogenic autoreactive T and B cells cross-react with mimotopes expressed by a common human gut commensal to trigger autoimmunity. Cell Host Microbe. 2019;26(1):100–113.e8. doi: 10.1016/j.chom.2019.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tanoue T, et al. A defined commensal consortium elicits CD8 T cells and anti-cancer immunity. Nature. 2019;565(7741):600–605. doi: 10.1038/s41586-019-0878-z. [DOI] [PubMed] [Google Scholar]
- 22.Routy B, Gopalakrishnan V, Daillère R, Zitvogel L, Wargo JA, Kroemer G. The gut microbiota influences anticancer immunosurveillance and general health. Nat Rev Clin Oncol. 2018;15(6):382–396. doi: 10.1038/s41571-018-0006-2. [DOI] [PubMed] [Google Scholar]
- 23.Zitvogel L, Ma Y, Raoult D, Kroemer G, Gajewski TF. The microbiome in cancer immunotherapy: diagnostic tools and therapeutic strategies. Science. 2018;359(6382):1366–1370. doi: 10.1126/science.aar6918. [DOI] [PubMed] [Google Scholar]
- 24.Haarman M, Knol J. Quantitative real-time PCR assays to identify and quantify fecal Bifidobacterium species in infants receiving a prebiotic infant formula. Appl Environ Microbiol. 2005;71(5):2318–2324. doi: 10.1128/AEM.71.5.2318-2324.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Frenkel D, Smit B. Understanding Molecular Simulation: From Algorithms to Applications. Cambridge, Massachusetts, USA: Elsevier; 2001. [Google Scholar]
- 26.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14(1):33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 27.Abraham MJ, et al. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1:19–25. [Google Scholar]
- 28.Best RB, et al. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ(1) and χ(2) dihedral angles. J Chem Theory Comput. 2012;8(9):3257–3273. doi: 10.1021/ct300400x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79(2):926–935. doi: 10.1063/1.445869. [DOI] [Google Scholar]
- 30.Darden T, York D, Pedersen L. Particle mesh Ewald: An N∙ log (N) method for Ewald sums in large systems. J Chem Phys. 1993;98(12):10089–10092. doi: 10.1063/1.464397. [DOI] [Google Scholar]
- 31.Hess B, Bekker H, Berendsen HJC, Fraaije JGEM. LINCS: a linear constraint solver for molecular simulations. J Comput Chem. 1997;18(12):1463–1472. doi: 10.1002/(SICI)1096-987X(199709)18:12<1463::AID-JCC4>3.0.CO;2-H. [DOI] [Google Scholar]
- 32.Nosé S. A unified formulation of the constant temperature molecular dynamics methods. J Chem Phys. 1984;81(1):511–519. doi: 10.1063/1.447334. [DOI] [Google Scholar]
- 33.Berendsen HJC, Postma JPM van, van Gunsteren WF, DiNola A, Haak JR. Molecular dynamics with coupling to an external bath. J Chem Phys. 1984;81(8):3684–3690. doi: 10.1063/1.448118. [DOI] [Google Scholar]
- 34.González-Alemán R, Hernández-Castillo D, Caballero J, Montero-Cabrera LA. Quality threshold clustering of molecular dynamics: a word of caution. J Chem Inf Model. 2020;60(2):467–472. doi: 10.1021/acs.jcim.9b00558. [DOI] [PubMed] [Google Scholar]
- 35.Gapsys V, Michielssens S, Seeliger D, de Groot BL. pmx: automated protein structure and topology generation for alchemical perturbations. J Comput Chem. 2015;36(5):348–354. doi: 10.1002/jcc.23804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Beutler TC, Mark AE, van Schaik RC, Gerber PR, van Gunsteren WF. Avoiding singularities and numerical instabilities in free energy calculations based on molecular simulations. Chem Phys Lett. 1994;222(6):529–539. doi: 10.1016/0009-2614(94)00397-1. [DOI] [Google Scholar]
- 37.Xia Z, et al. The complex and specific pMHC interactions with diverse HIV-1 TCR clonotypes reveal a structural basis for alterations in CTL function. Sci Rep. 2014;4:4087. doi: 10.1038/srep04087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hölzemer A, et al. Selection of an HLA-C*03:04-restricted HIV-1 p24 gag sequence variant is associated with viral escape from KIR2DL3+ natural killer cells: data from an observational cohort in South Africa. PLoS Med. 2015;12(11):e1001900; discussion e1001900. doi: 10.1371/journal.pmed.1001900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chowell D, et al. Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science. 2018;359(6375):582–587. doi: 10.1126/science.aao4572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Joglekar AV, et al. T cell receptors for the HIV KK10 epitope from patients with differential immunologic control are functionally indistinguishable. Proc Natl Acad Sci U S A. 2018;115(8):1877–1882. doi: 10.1073/pnas.1718659115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kirkwood JG. Statistical mechanics of fluid mixtures. J Chem Phys. 1935;3(5):300–313. doi: 10.1063/1.1749657. [DOI] [Google Scholar]
- 42.Straatsma TP, McCammon JA. Multiconfiguration thermodynamic integration. J Chem Phys. 1991;95(2):1175–1188. doi: 10.1063/1.461148. [DOI] [Google Scholar]
- 43.Russ BE, Denton AE, Hatton L, Croom H, Olson MR, Turner SJ. Defining the molecular blueprint that drives CD8(+) T cell differentiation in response to infection. Front Immunol. 2012;3:371. doi: 10.3389/fimmu.2012.00371. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.