

The stability of transposase-encoding vectors during cloning and propagation is crucial for the reliable application of transposons. Here, we increased the stability of the mariner delivery vehicle in E. coli. Moreover, the TnFLX transposon system will improve the application of forward genetic methods with an increased number of antibiotic resistance markers and the ability to generate unbiased green fluorescent protein (GFP) fusions to report on protein translation and subcellular localization.
KEYWORDS: Bacillus subtilis, mutagenesis, transposons
ABSTRACT
Random transposon mutagenesis is a powerful and unbiased genetic approach to answer fundamental biological questions. Here, we introduce an improved mariner-based transposon system with enhanced stability during propagation and versatile applications in mutagenesis. We used a low-copy-number plasmid as a transposon delivery vehicle, which affords a lower frequency of unintended recombination during vector construction and propagation in Escherichia coli. We generated a variety of transposons allowing for gene disruption or artificial overexpression, each in combination with one of four different antibiotic resistance markers. In addition, we provide transposons that will report gene/protein expression due to transcriptional or translational coupling. We believe that the TnFLX system will help enhance the flexibility of future transposon modification and application in Bacillus and other organisms.
IMPORTANCE The stability of transposase-encoding vectors during cloning and propagation is crucial for the reliable application of transposons. Here, we increased the stability of the mariner delivery vehicle in E. coli. Moreover, the TnFLX transposon system will improve the application of forward genetic methods with an increased number of antibiotic resistance markers and the ability to generate unbiased green fluorescent protein (GFP) fusions to report on protein translation and subcellular localization.
INTRODUCTION
Transposon mutagenesis is a powerful method in molecular genetics, as it combines random insertional mutagenesis with the creation of linked antibiotic markers and rapid insertion site identification. Typically, an antibiotic resistance cassette is cloned between two inverted terminal repeat (ITR) elements that define the transposon. Next, the transposase enzyme is provided either as purified protein in vitro or expressed from a gene in vivo to recognize and mobilize the transposon to another genetic location (1–3). Transposon systems have different transposition frequencies, insertional biases, and mechanisms for providing the transposase. Moreover, the functionality of transposons can be extended by cloning additional genetic material between the ITR elements. For instance, transposon derivatives have been generated that insert not only the antibiotic resistance cassette but other genes and promoters for the formation of randomly inserted reporter fusions and artificial expression systems (4–8).
Bacillus subtilis is a model genetic organism, and a number of transposon systems have been developed to aid in genetic analyses. The earliest transposon system developed for B. subtilis was Tn917, which integrated in a nucleotide sequence-independent manner but exhibited a strong bias toward insertions near the chromosome’s terminus (9–11). The next system used transposon Tn10, which both improved insertion diversity over the chromosome and provided an easier method for insertion site identification but was still prone to insertions at particular locations or “hot spots” (12–16). Later, the first generation of systems was developed using the transposon mariner, which inserted at two-base-pair “TA” sequences in the chromosome, thus increasing the potential number of insertion sites in low-G+C content organisms (2). The sequencing of transposon pools (TnSeq) revealed that mariner-based transposition is highly random and has reduced “hot spotting” issues exhibited by the Tn10- and Tn917-based systems (17–19). Our group constructed second-generation derivatives of the mariner system in which we added additional antibiotic cassettes and specialized transposons for generating transcriptional reporter insertions of the β-galactosidase gene lacZ and artificial expression insertions with an outward-facing isopropyl-β-d-thiogalactopyranoside (IPTG)-inducible promoter (20).
It became clear that there was a problem with the second-generation system after we started sharing Escherichia coli strains that carried the transposon delivery plasmids. Other labs had difficulty recovering functional plasmids from E. coli, and isolation of plasmid DNA from different colonies of the same frozen E. coli stock gave different plasmid digestion patterns, none of which were correct. We retracted the plasmids from the Bacillus Genetic Stock Center (BGSC) (The Ohio State University) and provided alternative solutions for obtaining the transposon system we had created (21). Here, we report the construction of a third generation of mariner-based transposons that affords increased stability in E. coli through the use of a low-copy-number delivery plasmid. We also redesigned all of the second-generation plasmids using the new platform, added an additional antibiotic resistance cassette conferring resistance to tetracycline, and created a new transposon derivative for the formation of C-terminal translational fusions to green fluorescent protein (GFP).
RESULTS AND DISCUSSION
Redesign of the transposon delivery system.
himar-based systems for random transposon mutagenesis were already described for B. subtilis (2, 20). Unfortunately, we detected a high frequency of genetic reorganization in the delivery plasmid during cloning and propagation in an E. coli DH5α host while working with the second-generation TnKRM series of himar-based transposon system. Endonuclease digestion analyses of plasmids derived from separate clonal isolates of a frozen E. coli stock revealed a heterogeneous pattern of fragments, none of which resembled the digestion pattern predicted for the intact plasmid (Fig. 1A). We suspected that the plasmid rearrangements were due to recombination events during transposition of the plasmid-borne transposon mediated by the plasmid-encoded transposase. Transposase expression might be high in E. coli, as the himar gene was under control of the conserved and constitutive σ70/σA promoter sequence of E. coli/B. subtilis. Moreover, the replication origin of the transposon delivery plasmid was derived from a high-copy-number pUC plasmid (22), further amplifying the potential for himar expression. We infer that expression of the himar transposase likely contributed to high-frequency transposition, plasmid rearrangements, and loss of plasmid sequence integrity, including the loss of the transposon itself.
FIG 1.

Plasmid stability of the TnKRM and TnFLX series. Restriction digests of individually isolated plasmids from colonies isolated from a clonal E. coli culture grown overnight at 37°C in LB amp were resolved on a 1% agarose gel. (A) pTnKRM kan (pKB178) was digested with NheI and EcoRI. Fragment sizes of approximately 3,900, 2,850, and 1,450 bp are expected. (B) pTnFLX kan (pFK131) was digested with EcoRV. Fragments sizes of approximately 7,650, 1,050, 750, and 200 bp are expected. A 1-kb DNA ladder (New England Biolabs) was provided for size determination, with marker sizes indicated in kbp. Arrowheads indicate expected fragment sizes.
In an effort to increase plasmid fidelity, we chose to redesign the transposon delivery system, based on a low-copy-number plasmid and a modified promoter for the transposase-encoding gene. We chose the plasmid pGB2 (23) for the vector backbone (a kind gift of T. Bernhardt, Harvard University). Replication of pGB2 is regulated by an origin of replication (oriEc) derived from pSC101, which maintains approximately 5 copies per cell (24, 25). A spectinomycin resistance cassette (specREc) particular for conferring resistance in E. coli allows for selection of plasmid incorporation and maintenance when cloning. An erythromycin resistance cassette (ermR) was also integrated to allow for the selection of plasmid uptake in the B. subtilis host. Next, we integrated the himar expression construct from the plasmid pMarB (2), such that transposase expression was under the control of a promoter recognized by the B. subtilis-specific stress response sigma factor σB, further reducing the likelihood of expression in E. coli. To complete our transposon delivery system (pFK7) (Fig. 2), we integrated a temperature-sensitive origin of replication specific for B. subtilis (oriBsTs) to allow replication in the host at permissive temperatures (<30°C) (26–29). A single intergenic SmaI restriction site was included for blunt-end linearization, allowing for the integration of various transposable elements into this system by isothermal assembly (ITA) (30).
FIG 2.
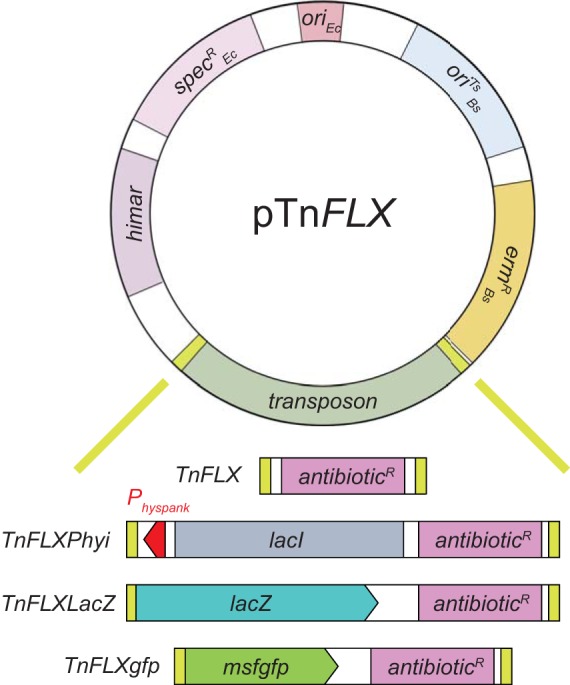
Diagram of the delivery plasmid and TnFLX transposons. A cartoon representation of the TnFLX series of plasmids is shown. oriEc, E. coli origin of replication; specREc, spectinomycin resistance cassette for selection in E. coli; himar, transposase gene expressed from a B. subtilis σB-directed promoter, ermRBs, erythromycin resistance cassette for selection in B. subtilis; oriTSBs, temperature-sensitive origin of replication in B. subtilis; transposon, transposon between two inverted terminal repeats (ITR) (yellow). Note that for the empty delivery vehicle pFK7, a unique SmaI site would sit between the ermRBs gene and the himar transposase gene and is destroyed upon insertion of the transposon. TnFLX, standard transposon with modular antibiotic resistance; TnFLXPhyi, transposon with outward-facing IPTG-inducible Physpank promoter (red); TnFLXLacZ, transposon with inward-facing promoterless lacZ gene (blue); TnFLXgfp, transposon with inward facing msfgfp gene that lacks a Shine-Dalgarno ribosome binding site.
Construction of TnFLX.
The first-generation mariner-based transposon system included a mariner transposon containing a single antibiotic resistance cassette conferring kanamycin resistance (TnYLB-1) (2). The second generation of modified mariner transposons, TnKRM, provided a transposon that had the kanamycin resistance cassette as well as two other transposons separately carrying resistance to spectinomycin and chloramphenicol (20). Here, we constructed transposons that separately carry genes that confer resistance to kanamycin (kanR), spectinomycin (specRBs), and chloramphenicol (catR) and a fourth cassette conferring resistance to tetracycline (tetR) (Fig. 2). In addition, all antibiotic resistance cassettes were flanked by a common nucleotide sequence, which will allow for transposon insertion site identification with the same pair of oligonucleotides (with the exception of the TnFLXPhyi series, which requires a distinct set of oligonucleotides) (Table 1).
TABLE 1.
Primers for inverse PCR and transposon insertion site identification
| No. | Name | Sequence |
|---|---|---|
| 6212 | TnFLX F | CAAAAGCTGGGTACCGGG |
| 6420 | TnFLX R | GGCTCTTTAAGAATTCTATCAGGCACTTCCGGACTCAATGGTGAGGGGAGACCGGGGAC |
| 7195 | TnFLXPhyi F | GGCTCTTTAAGAATTCTATCAGCTTTATCTACAAGGTGTGGC |
| 7196 | TnFLXPhyi R | GCAAAATGAATTGTGAGTGC |
| 6224 | TnFLX seq (universal) | GGCTCTTTAAGAATTCTATCAG |
One objective for our redesigned system was to increase the stability of the delivery system during propagation in E. coli. To assess plasmid stability, we transformed the plasmid pTnFLX kan into E. coli, and one transformant was restruck on lysogeny broth (LB) agar supplemented with spectinomycin. Ten independent cultures inoculated with 10 separate colonies were grown overnight under spectinomycin selection to maintain the plasmid. Plasmids were purified from each culture in parallel, restriction digested, and separated on an agarose gel. All 10 independently propagated plasmids had identical digestion products (Fig. 1B). While only 10 clonal colonies were tested, the data suggest that the frequency of plasmid rearrangement in E. coli is low. We note, however, that propagation of any plasmid bearing a potentially active transposon and transposase provides an inherent and unavoidable risk of rearrangement, and we recommend minimal propagation whenever possible. While the experiment for Fig. 1B was performed after overnight growth in an attempt to observe rare rearrangements, our standard operating procedure was to purify plasmids after short-term (8-h) growth of E. coli. While we cannot guarantee that TnFLX rearrangement has been eliminated at the subpopulation level, we nonetheless conclude that the overall plasmid backbone of the TnFLX transposon series is more stable than the backbone previously used to deliver the TnKRM series (Fig. 1A).
To compare transposition efficiencies of these third-generation transposon plasmids, each plasmid was transformed into a naturally competent derivative of B. subtilis NCIB3610, DK1042 (31, 32). Cells were propagated in LB medium at room temperature overnight, diluted and regrown in LB medium at the nonpermissive temperature of 42°C for 6 h in the presence of the corresponding transposon-specific antibiotic, dilution plated on LB agar plates with antibiotic, and incubated overnight at 42°C. The transposition frequency appeared to be consistent with that of other himar-based systems such as TnYLB-1 (2) and TnKRM (20). Whereas wild-type colonies have a dull, rough architecture, mutant colonies that exhibited a shiny, smooth colony morphology indicative of a biofilm defect were isolated (33). Inverse PCR (IPCR) was used to identify the location of each transposon insertion, and each strain was found to be disrupted in a gene previously reported to be involved in biofilm formation (Table 2). One mutant with a hyperrough phenotype indicative of biofilm extracellular polysaccharide overproduction was isolated, and one mutant with a mucoid colony morphology indicative of poly-γ-glutamate overproduction was isolated. Again, each mutant was disrupted in a gene previously reported to confer the associated phenotype (Table 2). We conclude that the TnFLX series of transposons insert in the chromosome and create loss-of-function mutations.
TABLE 2.
TnFLX mutants
| Strain | Phenotype | Insertion | Taga | Annotation | Reference(s)b |
|---|---|---|---|---|---|
| DK5707 | Smooth | epsC::TnFLX cat | TAAATGCCC | EPSc glycosyltransferase | 67 |
| DK5708 | Smooth | recF::TnFLX cat | TATAAGTTC | Recombination | 68 |
| DK5784 | Smooth | epsE::TnFLX cat | TATGTAATC | EPS glycosyltransferase | 69 |
| DK5706 | Smooth | ymdB::TnFLX kan | TAGTAGGAA | Phosphodiesterase | 70, 71 |
| DK5916 | Smooth | epsN::TnFLX kan | TATATGGGC | EPS aminotransferase | 72 |
| DK5918 | Smooth | epsG::TnFLX kan | TATACAATC | EPS, unknown | 73, 74 |
| DK5740 | Smooth | epsI::TnFLX spec | TATACCGTT | EPS glycosyltransferase | 75 |
| DK5705 | Rough | abrB::TnFLX spec | TATGTTGAT | Biofilm repressor | 76, 77 |
| DK5793 | Smooth | epsN::TnFLX spec | TATGACATA | EPS aminotransferase | 72 |
| DK5880 | Smooth | epsC::TnFLX tet | TAACACCTG | EPS glycosyltransferase | 67 |
| DK5881 | Smooth | epsD::TnFLX tet | TAAAAGAAA | EPS glycosyltransferase | 73, 74 |
| DK5883 | Mucoid | fliI::TnFLX tet | TATATGCCG | Flagellar secretion | 78, 79 |
The tag is the first 9 bases immediately adjacent to the transposon inverted terminal repeat and is included to allow precise location of the insertion.
Reference(s) supporting the colony phenotype obtained.
EPS, exopolysaccharide.
In addition to gene disruption, the artificial expression of a gene in a nonphysiological context can reveal valuable information about the function of a gene product. To generate unbiased insertions that provide artificial control over gene expression, the second-generation transposon TnHyJump was created with an antibiotic resistance cassette and a strong Physpank promoter (20) cloned facing outward from one arm of the transposon. Thus, TnHyJump insertions would create constitutive high-level transcription on one side of the integration site. Optionally, the lacI gene encoding the Physpank repressor protein LacI could be separately integrated at an ectopic location in the chromosome to provide IPTG-inducible control. Here, we created the TnFLXPhyi series of transposons in which an antibiotic resistance cassette was also combined with the outward-facing Physpank promoter (a kind gift from D. Rudner, Harvard University). Unlike TnHyJump, however, TnFLXPhyi also carried the lacI gene between the ITR elements such that IPTG-inducible control was inherent with, and linked to, each insertion (Fig. 2).
The TnFLXPhyi system was tested by performing a transposon mutagenesis in the domesticated laboratory B. subtilis strain PY79 (34) in a “blue/white” screen, and cells were spread on plates containing the transposon-specific antibiotic, the inducer IPTG, and the chromogenic substrate 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal) and incubated overnight at a nonpermissive temperature. It had previously been shown that mutagenesis with the second-generation TnHyJump system in a similar screen would produce blue colonies when the transposon inserted upstream of, and colinear with, cryptic β-galactosidase genes (20). After mutagenesis with TnFLXPhyi, blue colonies were observed at a low frequency of approximately one per thousand (Fig. 3A). We isolated 12 blue colonies (3 for each transposon) (Table 3) and identified the transposon insertion site by IPCR (Table 1). In 9 out of 12 mutants, the insertions were found to be within the large yes operon and oriented such that the Physpank promoter could drive artificial expression of the yesZ gene, encoding a cryptic β-galactosidase (Table 3) (20, 35). For one strain, the transposon was integrated such that the Physpank promoter was inserted in the gan operon and oriented such that the Physpank promoter could drive artificial expression of the ganA gene, encoding another β-galactosidase, GanA (formerly LacA) (36). Finally, two transposons were found to be inserted within the ganR gene, and unlike with the other 10 insertions, the blue color was IPTG independent (Fig. 3B). Consistent with IPTG independence, disruption of ganR, encoding GanR (formerly LacR), has been shown to result in constitutive GanA derepression (36). Thus, each TnFLXPhyi transposon insertion was capable of generating insertional disruptions and/or artificial expression of downstream adjacent genes.
FIG 3.

Blue/white transposon mutagenesis screens for TnFLXPhyi and TnFLXLacZ. (A) Plate from a transposon mutagenesis screen conducted with TnFLXPhyi on medium containing X-Gal. The caret indicates a blue colony. (B) TnFLXPhyi mutant strains DK7532 (top) and DK7530 (bottom) were plated on X-Gal-containing medium that either contained or lacked 1 mM IPTG. (C) Plate from a transposon mutagenesis screen conducted with TnFLXLacZ on medium containing X-Gal. The carets indicate blue colonies.
TABLE 3.
TnFLXPhyi mutants
| Strain | Phenotypea | IPTGb | Insertion | Tagc | Annotation |
|---|---|---|---|---|---|
| DK7507 | Blue | Yes | yesX::TnFLXPhyi cat | TATCATCGG | Induces yesZ |
| DK7508 | Blue | Yes | yesS::TnFLXPhyi cat | TATGGCCGC | Induces yesZ |
| DK7509 | Blue | Yes | yesS::TnFLXPhyi cat | TATGGAGAG | Induces yesZ |
| DK7510 | Blue | Yes | ganQ::TnFLXPhyi spec | TAACCTTTA | Induces ganA |
| DK7525 | Blue | Yes | yesW::TnFLXPhyi spec | TAGTTGCCT | Induces yesZ |
| DK7527 | Blue | Yes | yesX::TnFLXPhyi spec | TATGGTGTA | Induces yesZ |
| DK7505 | Blue | No | ganR::TnFLXPhyi tet | TATGTGGGC | Derepresses ganA |
| DK7692 | Blue | Yes | yesQ::TnFLXPhyi tet | TAATGCTCG | Induces yesZ |
| DK7506 | Blue | Yes | yesR::TnFLXPhyi tet | TACAAAATA | Induces yesZ |
| DK7528 | Blue | Yes | yesQ::TnFLXPhyi kan | TATATTATG | Induces yesZ |
| DK7530 | Blue | No | ganR::TnFLXPhyi kan | TAAGTCATC | Derepresses ganA |
| DK7532 | Blue | Yes | yesT::TnFLXPhyi kan | TACCTTGTT | induces yesZ |
Blue/white phenotype on plates containing X-Gal.
Whether IPTG is required in the medium to obtain a blue colony phenotype on X-Gal.
The tag is the first 9 bases immediately adjacent to the transposon inverted terminal repeat and is included to allow precise location of the insertion.
The second-generation mariner system introduced another functional application to transposon mutagenesis in which the lacZ gene, encoding the β-galactosidase LacZ, was cloned with an antibiotic resistance cassette within the boundaries of the transposon ITR elements, creating TnLacJump (20). Thus, insertions of the transposon into, and cooriented with, an actively transcribed region of the chromosome would create a transcriptional fusion to lacZ in addition to possible gene disruption. To recreate TnLacJump functionality, the lacZ gene was cloned into the TnFLX system along with an antibiotic resistance cassette, generating TnFLXLacZ (Fig. 2). As proof of concept, TnFLXLacZ was introduced to the domesticated strain B. subtilis PY79, and after mutagenesis, cells were spread on plates containing the transposon-specific antibiotic with the chromogenic substrate X-Gal and incubated overnight at the nonpermissive temperature. Blue colonies were common (Fig. 3C); 12 blue colonies (3 for each transposon) were isolated, and the insertion site was identified by IPCR. In each case, the transposon had inserted in or near a different open reading frame and was cooriented with the direction of transcription, consistent with the generation of an inserted transcriptional reporter (Table 4). We conclude that the TnFLXLacZ system produces high-frequency, random lacZ reporter insertions.
TABLE 4.
TnFLXLacZ mutants
| Strain | Phenotypea | Insertion | Tagb | Annotationc |
|---|---|---|---|---|
| DK7773 | Blue | yvaF::TnFLXLacZ spec | TAAAGATCA | Unknown |
| DK7774 | Blue | TnFLXLacZ spec < ald | TACAGGAGG | Insert upstream of ald |
| DK7775 | Blue | azlB::TnFLXLacZ spec | TATATAGCTT | Transcriptional regulator |
| DK7776 | Blue | TnFLXLacZ tet < yvyD | CATCAACCT | Insert upstream of yvyD |
| DK7777 | Blue | gltT::TnFLXLacZ tet | TAACTGGTG | Glutamate/aspartate transporter |
| DK7778 | Blue | mcsB::TnFLXLacZ tet | TAGAAATGA | Protein arginine kinase |
| DK7779 | Blue | yneN::TnFLXLacZ kan | TATACGGGA | Unknown |
| DK7780 | Blue | pksS::TnFLXLacZ kan | TAAAAAATG | Polyketide synthesis |
| DK7781 | Blue | yvlB::TnFLXLacZ kan | TAAAAAAGT | Unknown |
| DK7782 | Blue | trmB::TnFLXLacZ cat | TATATTGGTA | tRNA methyltransferase |
| DK7783 | Blue | mfd::TnFLXLacZ cat | TATTTGACG | Transcription-coupled DNA repair |
| DK7784 | Blue | ctaD::TnFLXLacZ cat | TATGCACGG | Cytochrome c oxidase subunit |
Blue/white phenotype on plates containing X-Gal.
The tag is the first 9 bases immediately adjacent to the transposon inverted terminal repeat and is included to allow precise location of the insertion.
Unknown, protein of unknown function. The insertions upstream of ald and yvyD are cooriented with the indicated gene.
Development of TnFLXgfp.
Fluorescent translational fusion proteins are a powerful tool to investigate temporal and spatial protein localization, but fluorescent fusions are available for only a subset of the B. subtilis proteome. Here, we introduce a new family of transposons, in which a GFP gene was cloned into the TnFLX system with an antibiotic resistance cassette (Fig. 2). We chose to base the TnFLXgfp series on the monomeric version of the superfolder green fluorescent protein (msfGFP) for reasons of high photostability, quantum yield, and enhanced folding capability (37, 38). A coding sequence of the msfgfp gene, which was optimized for B. subtilis codon usage, was combined with an antibiotic cassette and flanked by himar-specific ITRs. The ITR and msfgfp gene were positioned in a way that no base triplet upstream of the fluorophore-encoding region would lead to premature translational termination. Moreover, the msfgfp gene sequence lacked a Shine-Dalgarno ribosome binding site, ensuring that GFP would be expressed only when inserted in frame within an actively transcribed and translated open reading frame as a C-terminal GFP fusion protein at the native site in the chromosome.
For an initial test, each TnFLXgfp transposon was transformed into B. subtilis DK1042, and transposon mutageneses were performed by selecting for the corresponding antibiotic resistance at the nonpermissive temperature. Approximately 100,000 colonies were harvested per pool, and aliquots of each pool were observed by fluorescence microscopy. A small number of cells in the initial pools (approximately one in a thousand) displayed a detectable msfGFP-specific signal, indicating that integration into an actively translated open reading frame was rare (Fig. 4A). To enrich for cells expressing detectable levels of msfGFP, each transposon pool was subjected to fluorescence-activated cell sorting (FACS). Cells displaying fluorescence levels higher than that of the wild-type strain were separated, recovered by incubation on LB agar plates, and pooled to generate the respective msfGFP-positive mutant library (Fig. 4A). A total of 12 mutants (3 from each transposon) were isolated, the localization pattern was assessed, and the transposon insertion site was determined by IPCR (Table 5).
FIG 4.
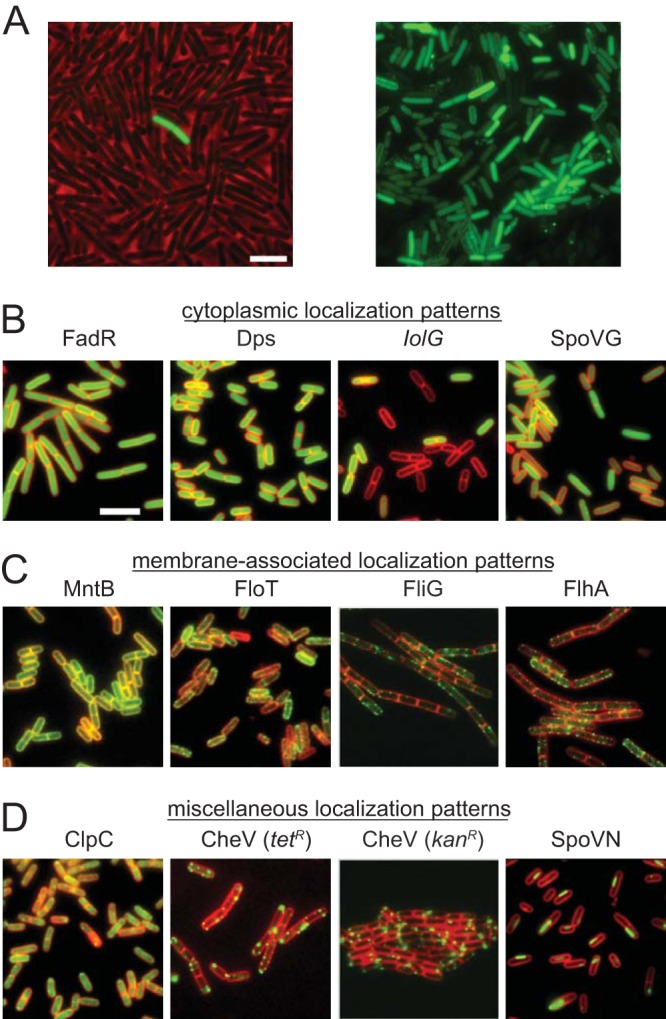
TnFLXgfp generates translational GFP fusions. (A) Left, fluorescence microscopy of an aliquot of a TnFLXgfp transposon mutant pool. GFP is false colored green. The phase-contrast background is false colored red. Right, fluorescence microscopy of a TnFLXgfp transposon mutant pool that had been enriched for GFP fluorescent cells by FACS for fluorescence. GFP is false colored green. (B to D) Analysis of clonal isolates from the enriched GFP library. GFP localization patterns were classified as cytoplasmic (FadR-GFP, DK7547; Dps-GFP, DK7755; IolG-GFP, DK7657; SpoVG-GFP, DK7560) (B), membrane associated (MntB-GFP, DK7554; FloT-GFP, DK7558; FliG-GFP, DK7662; FlhA-GFP, DK7549) (C) or miscellaneous (ClpC-GFP, DK7658; CheV-GFP, DK7548 and DK7552; SpoVN-GFP, DK7559) (D). GFP is false colored green. The membrane was stained with FM4-64 and is false colored red. The bar represents 5 μm, and all panels are at the same magnification.
TABLE 5.
Fluorescent TnFLXgfp mutants
| Strain | Localization | Insertion | Taga | Annotation |
|---|---|---|---|---|
| DK7554 | Membrane | mntB::TnFLXgfp cat | TAAGGCTGA | Manganese transporter |
| DK7657 | Diffuse | iolG::TnFLXgfp cat | TATCAATCT | Inositol dehydrogenase |
| DK7658 | Diffuse/punctate | clpC::TnFLXgfp cat | TAGAAGATG | Protease subunit |
| DK7755 | Diffuse | dps::TnFLXgfp kan | TAGGTTAAG | Iron storage protein |
| DK7552 | Punctate | cheV::TnFLXgfp kan | TAGAGGACT | Chemotaxis protein |
| DK7662 | Membrane | fliG::TnFLXgfp kan | TATCTTCTG | Flagellar rotor protein |
| DK7547 | Diffuse | fadR::TnFLXgfp tet | TATCCAAAA | Transcriptional repressor |
| DK7548 | Punctate | cheV::TnFLXgfp tet | TATCAAAAC | Chemotaxis protein |
| DK7549 | Membrane | flhA::TnFLXgfp tet | TAGATCCGA | Flagellar secretion protein |
| DK7560 | Diffuse | spoVG::TnFLXgfp spec | TAGATTCGA | RNA-binding protein |
| DK7559 | Filament | spoVN::TnFLXgfp spec | TAGGCTATG | Alanine dehydrogenase |
| DK7558 | Membrane | floT::TnFLXgfp spec | TAATTGCAC | Membrane scaffold protein |
The tag is the first 9 bases immediately adjacent to the transposon inverted terminal repeat and is included to allow precise location of the insertion.
A variety of subcellular localization patterns were observed in the mutants containing fluorescent GFP fusions. One pattern of expression was the diffuse localization of green fluorescence in the cytoplasm. Diffuse cytoplasmic localization was observed in strains in which the transposon had inserted into the fadR gene, encoding the transcriptional repressor FadR (39, 40), and the dps gene, encoding the iron storage protein Dps (41, 42) (Fig. 4B). Either these proteins are normally diffusely distributed in the cytoplasm or insertion of the transposon and premature truncation of the coding sequence by the GFP fusion altered localization. At most, we can infer that the expression of these genes occurs during vegetative growth and is uniform in the population. In contrast, two TnFLXgfp fusions in the iolG gene, encoding the inositol 2-dehydrogenase IolG (43), and the spoVG gene, encoding the RNA binding regulatory protein SpoVG (44, 45), gave rise to diffuse localization in the cytoplasm but only highly expressed in a subset of cells in the population. Again, while lack of a subcellular localization pattern cannot be interpreted with confidence based on loss-of-function insertion, the expression of the iolG and spoVG genes appears to be heterogeneous in the population. We conclude that TnFLXgfp is useful for generating translational reporters that can also reveal subpopulation-level gene expression in some cases.
Four TnFLXgfp insertion mutant strains showed fluorescence that was enriched at the cell periphery (Fig. 4C). Insertion of the transposon into the mntB gene, encoding the membrane-associated manganese transport protein MntB (46), exhibited fluorescence that was uniformly distributed throughout the periphery consistent with the membrane (47). Insertion of the transposon into three other loci, including the gene floT, encoding the flotillin homolog FloT (48), the fliG gene, encoding the flagellar rotor protein FliG, and the flhA gene, encoding a component of flagellar type III export apparatus FlhA, exhibited a nonuniform or punctate localization at the membrane. We note that the localization pattern is consistent with previous observations in that FloT was reported to have a punctate localization (48). Moreover, FliG (49) and FlhA (50) are integral parts of the flagellar basal body that also localizes as membrane-associated puncta (51). We conclude that TnFLXgfp can generate translational fusions within an open reading frame and can reproduce subcellular localization patterns that have been reported previously for the corresponding gene.
The last four TnFLXgfp mutants showed alternate localization patterns (Fig. 4D). For example, one mutant strain with an insertion into the coding region of the protease subunit ClpC (52) showed both diffuse cytoplasmic localization and concentrations of more intense puncta in the cytoplasm. Both the diffuse and punctate localization patterns of ClpC are consistent with subcellular localization reports for this protein (53, 54). Two different insertions were found in the coding region of the chemotaxis protein CheV (55), and each insertion gave rise to intense cytoplasmic foci of variable size that sometimes appeared to be membrane-associated. Again, reports on B. subtilis and Helicobacter pylori support a punctate localization for CheV (56, 57). Finally, a strain harboring an insertion in the coding sequence of the alanine dehydrogenase SpoVN (58) produced filament-like structures that seemed to be localized near the cell periphery. Filamentous localization is an unusual pattern in bacteria, and while the localization of SpoVN in particular has not been directly investigated in B. subtilis, we note that other metabolic enzymes, e.g., the CTP synthase in the alphaproteobacterium Caulobacter crescentus, have been reported to form filamentous structures (59, 60).
Conclusion.
Here, we generated a transposon delivery vehicle with increased stability in E. coli and one that is amenable to easy modification by molecular biological approaches. Moreover, we reproduced the previous-generation series of modified mariner-based transposons and developed a new reporter tool in the form a TnFLXgfp for making nondirected fluorescent translational fusions in vivo. All plasmids will be donated to the Bacillus Genetic Stock Center (BGSC) for community acquisition. We note that while the plasmids of the third-generation mariner transposon system are substantially more stable than the previous ones, propagation of transposons is inherently destabilizing due to spontaneous transposition, and thus we recommend minimal propagation and careful handling as a precaution.
MATERIALS AND METHODS
Strain construction.
For the construction of the transposon delivery vectors, all plasmids were amplified in E. coli DH5α (Table 6 shows all strains used in this study). Plasmids were isolated from late-log-phase cultures using the QIAprep Spin miniprep kit (Qiagen), yielding on average 3 μg of DNA per 1.5 ml of bacterial culture. To allow for transformation of B. subtilis, E. coli TG1 cells were transformed with the plasmids to generate dimers and higher-order oligomers. If not stated differently, all B. subtilis strains derived from the naturally competent modified undomesticated strain DK1042. Cells were incubated in lysogeny broth medium (5% [wt/vol] yeast extract, 10% [wt/vol] tryptone, 0.09 M NaCl) or on plates containing this medium fortified with agar (1.5% [wt/vol]). Antibiotics were supplemented when appropriate as follows: 100 μg/ml spectinomycin, 5 μg/ml kanamycin, 10 μg/ml tetracycline, 5 μg/ml chloramphenicol, and MLS (macrolide-lincosamide-streptogramin B, 1 μg/ml erythromycin, 25 μg/ml lincomycin). For Physpank promoter-dependent gene expression, 1 mM IPTG was added to the medium. For colorimetric assays, using β-galactosidase activity as a readout, 0.5 mM X-Gal was supplemented.
TABLE 6.
Strains
| Species and strain | Genotype (reference) |
|---|---|
| B. subtilis | |
| DK1042 | comIQ12L (32) |
| DK5705 | DK1042 abrB::TnFLX spec |
| DK5706 | DK1042 ymdB::TnFLX kan |
| DK5707 | DK1042 epsC::TnFLX cat |
| DK5708 | DK1042 recF::TnFLX cat |
| DK5740 | DK1042 epsI::TnFLX spec |
| DK5784 | DK1042 epsE::TnFLX cat |
| DK5793 | DK1042 epsN::TnFLX spec |
| DK5880 | DK1042 epsC::TnFLX tet |
| DK5881 | DK1042 epsD::TnFLX tet |
| DK5883 | DK1042 fliI::TnFLX tet |
| DK5916 | DK1042 epsN::TnFLX kan |
| DK5918 | DK1042 epsG::TnFLX kan |
| DK7505 | PY79 ganR::TnFLXPhyi tet |
| DK7506 | PY79 yesR::TnFLXPhyi tet |
| DK7507 | PY79 yesX::TnFLXPhyi cat |
| DK7508 | PY79 yesS::TnFLXPhyi cat |
| DK7509 | PY79 yesS::TnFLXPhyi cat |
| DK7510 | PY79 ganQ::TnFLXPhyi spec |
| DK7525 | PY79 yesW::TnFLXPhyi spec |
| DK7527 | PY79 yesX::TnFLXPhyi spec |
| DK7528 | PY79 yesQ::TnFLXPhyi kan |
| DK7530 | PY79 ganR::TnFLXPhyi kan |
| DK7532 | PY79 yesT::TnFLXPhyi kan |
| DK7547 | DK1042 fadR::TnFLXgfp tet |
| DK7548 | DK1042 cheV::TnFLXgfp tet |
| DK7549 | DK1042 flhA::TnFLXgfp tet |
| DK7550 | DK1042 dps::TnFLXgfp kan |
| DK7552 | DK1042 cheV::TnFLXgfp kan |
| DK7554 | DK1042 mntB::TnFLXgfp cat |
| DK7558 | DK1042 floT::TnFLXgfp spec |
| DK7559 | DK1042 spoVN::TnFLXgfp spec |
| DK7560 | DK1042 spoVG::TnFLXgfp spec |
| DK7657 | DK1042 iolG::TnFLXgfp cat |
| DK7658 | DK1042 clpC::TnFLXgfp cat |
| DK7662 | DK1042 fliG::TnFLXgfp kan |
| DK7692 | PY79 yesQ::TnFLXPhyi tet |
| DK7773 | PY79 yvaF::TnFLXLacZ spec |
| DK7774 | PY79 dhA::TnFLXLacZ spec |
| DK7775 | PY79 azlB::TnFLXLacZ spec |
| DK7776 | PY79 yvyD::TnFLXLacZ tet |
| DK7777 | PY79 gltT::TnFLXLacZ tet |
| DK7778 | PY79 mcsB::TnFLXLacZ tet |
| DK7779 | PY79 yneN::TnFLXLacZ kan |
| DK7780 | PY79 pksS::TnFLXLacZ kan |
| DK7781 | PY79 yvlB::TnFLXLacZ kan |
| DK7782 | PY79 trmB::TnFLXLacZ cat |
| DK7783 | PY79 mfd::TnFLXLacZ cat |
| DK7784 | PY79 catD::TnFLXLacZ cat |
| E. coli | |
| DE238 | pDG1515 tet amp (61) |
| DE260 | pAH54 spec amp (62) |
| DE727 | pAC225 cat amp (63) |
| DE236 | pDG780 kan amp (61) |
| DE3712 | pGB2 amp (23) |
| DE3092 | pDP111 amyE::Physpank kan amp |
| DE1961 | pMarB TnYLB-1 kan oriTSBs erm amp (2) |
| DE1888 | pKB178 pTnKRM kan oriTSBs erm amp (20) |
| DE3713 | pFK175 oriTSBs erm specEc |
| DE3714 | pFK7 oriTSBs erm specEc |
| DE3715 | pFK129 pTnFLX spec oriTSBs erm specEc |
| DE3716 | pFK130 pTnFLX tet oriTSBs erm specEc |
| DE3717 | pFK131 pTnFLX kan oriTSBs erm specEc |
| DE3718 | pFK132 pTnFLX cat oriTSBs erm specEc |
| DE3719 | pFK145 pTnFLXLacZ oriTSBs erm specEc |
| DE3720 | pFK146 TnFLXLacZ spec oriTSBs erm specEc |
| DE3721 | pFK147 TnFLXLacZ tet oriTSBs erm specEc |
| DE3722 | pFK148 TnFLXLacZ kan oriTSBs erm specEc |
| DE3723 | pFK149 TnFLXLacZ cat oriTSBs erm specEc |
| DE3724 | pFK161 TnFLXgfp oriTSBs erm specEc |
| DE3725 | pFK162 TnFLXgfp spec oriTSBs erm specEc |
| DE3726 | pFK163 TnFLXgfp tet oriTSBs erm specEc |
| DE3727 | pFK164 TnFLXgfp kan oriTSBs erm specEc |
| DE3728 | pFK165 TnFLXgfp cat oriTSBs erm specEc |
| DE3729 | pFK199 Physpank oriTSBs erm specEc |
| DE3730 | pFK200 TnFLXPhyi oriTSBs erm specEc |
| DE3731 | pFK205 TnFLXPhyi spec oriTSBs erm specEc |
| DE3732 | pFK206 TnFLXPhyi tet oriTSBs erm specEc |
| DE3733 | pFK207 TnFLXPhyi kan oriTSBs erm specEc |
| DE3734 | pFK208 TnFLXPhyi cat oriTSBs erm specEc |
Plasmid construction.
The low-copy-number plasmid pGB2 was linearized by SmaI treatment, and an erythromycin resistance cassette (ermR) and a temperature-sensitive origin of replication (oriBsTs) amplified from pMarB (2) using oligonucleotides 5211 and 4991 were introduced by isothermal assembly (ITA) (Table 7 shows the oligodeoxyribonucleotides used in this study), giving rise to pFK175. In a next step, pFK175 was linearized with SmaI, and the mariner-himar1 transposase-coding region controlled by a σB-dependent promoter, amplified from pMarB with oligonucleotides 5212 and 5214, was integrated by ITA, creating pFK7. This plasmid was used as a universal acceptor of all transposons described in this study. The different transposons were constructed by PCR and integrated, as one or two DNA fragments, respectively, into the SmaI-linearized plasmid pFK7 by ITA.
TABLE 7.
Primers
| No. | Sequence |
|---|---|
| 4991 | GTCTTACTGTCGGAATTCCCCGATAGAAAGCGTGAGAAAC |
| 5211 | CTGCAGGTCGACGGATCCCCGGGCACACTAGACTTATTTACTTCG |
| 5212 | GAAGTAAATAAGTCTAGTGTGCCCGGGGATCCTACACTTGCTGC |
| 5314 | CTGCAGGTCGACGGATCCCCTTTATTATTCAACATAGTTCC |
| 5759 | GTAAATAAGTCTAGTGTGCCCACAGGTTGGCTGATAAGTCC |
| 5760 | AGCAGCAAGTGTAGGATCCCCACAGGTTGGCTGATAAGTCC |
| 6140 | ACAGGTTGGCTGATAAGTCCCCGGTCTTTGTTATCCGCTCACAATTACAC |
| 6142 | ACAGGTTGGCTGATAAGTCCCCGGTCTCCCCTCACTAAAGGGAACAAAAGCTGG |
| 6143 | ACAGGTTGGCTGATAAGTCCCCGGTCTCCCACGACTCACTATAGGGCGAATTG |
| 6925 | GTAAATAAGTCTAGTGTGCCCACAGGTTGGCTGATAAGTCC |
| 6960 | GTGTGCCCACAGGTTGGATGATAAGTCCCCGGTCTGATACACTAATGCTTTTATATAGG |
| 6962 | GGATCCCCACAGGTTGGCTGATAAGTCCCCGGTCTCCCGGGATTTCCTTACGCGAAATAC |
| 6977 | CCACAGGTTGGCTGATAAGTCCCCGGTCTCCCGGGTTATTTATATAACTCGTCCATCC |
| 6978 | GGGATGGACGAGTTATATAAATAACCCACGACTCACTATAGGGCGAATTG |
| 7183 | GCCTCAAGCTAGAGAGTCGCCCACGACTCACTATAGGGCGAA |
| 7184 | GCAGCAAGTGTAGGATCCCCGGGCGACTCTCTAGCTTGAGGC |
| 7185 | CGTTTCCACCGAATTAGCCCCCTCACTAAAGGGAACAAAAGC |
| 7186 | CCTCAAGCTAGAGAGTCGCCCGGGGCTAATTCGGTGGAAACGAG |
| 7187 | ACAGGTTGGCTGATAAGTCCCCGGTCTCCTTACGCGAAATACGGGC |
For all transposons the antibiotic resistance cassettes were amplified from pDG1515 (61) (tetracycline resistance cassette [tetR]), pAH54 (62) (spectinomycin resistance cassette [specR]), pAC225 (63) (chloramphenicol resistance cassette [catR]), and pDG780 (61) (kanamycin resistance cassette [kanR]). For the TnFLX series, the antibiotic resistance cassettes were amplified and extended with the inverted terminal repeats (ITRs) using oligonucleotides 6142 and 6143. To allow for ITA, sequences overlapping with the SmaI-linearized pFK7 plasmid were introduced by PCR with oligonucleotides 5759 and 5760.
The TnFLXLacZ transposon series was assembled in a sequential manner. To allow transcriptional coupling, first, the lacZ gene as well as a ribosomal binding site was amplified from pEP22 (64) and extended by ITR regions using oligonucleotides 6960 and 6962, also integrating a SmaI recognition site. Homologies to the linearized pFK7 plasmid were introduced by an amplification step with oligonucleotides 5759 and 5760. The resulting fragment was introduced into the SmaI-linearized acceptor plasmid (pFK7) by ITA, giving rise to pFK145. In a second step, antibiotic resistance cassettes were equipped with homologies to the acceptor plasmid by amplification with 6961 and 6142 and integrated into SmaI-linearized pFK145 by ITA.
Transposons of the TnFLXPhyi series were assembled in three steps. First, the promoter-encoding sequence was amplified from the plasmid pDP111 and extended by an ITR region by PCR with oligonucleotides 6140 and 7184. The resulting fragment was equipped with homologies to SmaI-linearized pFK7 by a second PCR using the primers 6925 and 7184 and integrated into the acceptor plasmid via ITA, creating pFK199. In a second step, the repressor-encoding gene, lacI, was amplified from pDP111 and equipped with an ITR sequence using the primers 7186 and 7187. Regions overlapping with SmaI-linearized pFK199 were introduced by PCR amplification with oligonucleotides 7186 and 5760. Insertion of the resulting product into pFK199 was performed via ITA, giving rise to pFK200. The antibiotic resistance cassettes were introduced in a third step into SmaI-linearized pFK200. Therefore, the coding regions were amplified and flanked by pFK200 homologies by PCR with oligonucleotides 7183 and 7185 and integrated into pFK200 by ITA.
To generate a transposon allowing for the translational fusion of proteins with monomeric superfolder green fluorescent protein (msfGFP), the coding sequence for msfGFP was analyzed, modified for optimal tRNA utilization according to the IDT codon optimization tool (Integrated DNA Technologies [IDT], Coralville, IA), and synthesized by IDT. The obtained fragment, which already contained a single proximal ITR sequence, was appended with a second, distal, ITR sequence by amplification with oligonucleotides 6925 and 6977. To allow for integration into the acceptor plasmid, flanking homologies to pFK7 were introduced by PCR with oligonucleotides 5759 and 5760 following an ITA reaction with SmaI-linearized pFK7, creating pFK161. Antibiotic resistance cassettes were introduced into pFK161 by amplification of the coding region with oligonucleotides 6978 and 5760 and a subsequent ITR reaction with SmaI-linearized pFK161.
All plasmid sequences are included in the supplemental material.
Transposon mutagenesis.
The transposon-harboring plasmids were introduced into the B. subtilis acceptor strain via transformation (65), selecting for MLS resistance, and incubated at 30°C for 48 h. Subsequently, cells were incubated for 16 h at room temperature in LB medium containing MLS. The cell suspension was diluted 1:100 in LB medium containing the transposon-specific antibiotic. The culture was grown for 6 h at 42°C to cure the cells of the plasmid. To obtain individual colonies, the cell suspension was serially diluted, spread on LB plates containing the appropriate antibiotic, and incubated overnight at 42°C. In order to separate the transposon insertion from other mutations that might have occurred during transposition, an SPP1 phage lysate was prepared from cells of candidate colonies. Wild-type cells were transduced with the lysates and the culture transferred to LB plates containing the transposon-specific antibiotic.
Transposon insertion site identification by IPCR.
Cells were grown in LB medium at 37°C for 6 h. DNA was isolated, and 6 μg was digested with 2 U/μg DNA of the restriction endonuclease Sau3AI (NEB). Ligation with 400 U of T4 DNA ligase (NEB) was performed at room temperature for 2 h with 1.5 μg of the digested DNA. Inverse PCR (IPCR) was performed with 0.5 μl of ligated DNA using oligonucleotides 6212 and 6420 (or 7195 and 7196 in the case of the TnFLXPhyi series) in a standard PCR program with a 4-min extension time using a Mastercycler thermocycler (Eppendorf). Phusion DNA polymerase (NEB) was used for PCR amplification. The amplified fragment was analyzed by Sanger sequencing (IDT) using the universal sequencing oligonucleotide 6224.
Fluorescence microscopy.
Microscopy was performed with a Nikon eclipse 80i microscope equipped with a Plan Apo 100× Ph 3 objective (numerical aperture [NA], 1.4). Cells were mounted on 1% (wt/vol) agarose pads containing S750 minimal medium (66) on object slides. Images were acquired with a Cool Snap HQ2 camera (Photometrix) and were processed with MetaMorph 7.7.9 software (Universal Imaging Corp.). Membranes were stained with FM4-64 (final concentration, 1 nM; Molecular Probes).
Flow cytometry.
Cells were grown in LB medium at 37°C to an optical density at 600 nm (OD600) of 0.7, sedimented, and resuspended in phosphate-buffered saline (PBS) at a concentration of 5 × 106 cells/ml. Analysis and cell sorting were performed, using a FACSAriaII flow cytometer (BD Biosciences) equipped with an 85-μm nozzle. Cells that were separated by FACS were collected in a reaction tube containing LB medium that was supplemented with the appropriate antibiotic and transferred onto fortified medium for further incubation.
Supplementary Material
ACKNOWLEDGMENTS
We thank Marta Perego for bringing to our attention the plasmid instability issue when plasmids carrying the TnKRM series were propagated in E. coli. We are grateful for the plasmids and the constructs received from the Bernhardt lab (Harvard University) and the Rudner lab (Harvard University). We thank Christiane Hassel (IUB Flow Cytometry Core Facility) for her support concerning bacterial cell sorting.
This work was supported by NIH grant R35 GM131783 to D.B.K.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Lampe DJ, Churchill ME, Robertson HM. 1996. A purified mariner transposase is sufficient to mediate transposition in vitro. EMBO J 15:5470–5479. doi: 10.1002/j.1460-2075.1996.tb00930.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Le Breton Y, Mohapatra NP, Haldenwang WG. 2006. In vivo random mutagenesis of Bacillus subtilis by use of TnYLB-1, a mariner-based transposon. Appl Environ Microbiol 72:327–333. doi: 10.1128/AEM.72.1.327-333.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wilson AC, Perego M, Hoch JA. 2007. New transposon delivery plasmids for insertional mutagenesis in Bacillus anthracis. J Microbiol Methods 71:332–335. doi: 10.1016/j.mimet.2007.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Perkins JB, Youngman PJ. 1986. Construction and properties of Tn917-lac, a transposon derivative that mediates transcriptional gene fusions in Bacillus subtilis. Proc Natl Acad Sci U S A 83:140–144. doi: 10.1073/pnas.83.1.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Castilho BA, Olfson P, Casadaban MJ. 1984. Plasmid insertion mutagenesis and lac gene fusion with mini-Mu bacteriophage transposons. J Bacteriol 158:488–495. doi: 10.1128/JB.158.2.488-495.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Calamia J, Manoil C. 1990. lac permease of Escherichia coli: topology and sequence elements promoting membrane insertion. Proc Natl Acad Sci U S A 87:4937–4941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gregory JA, Becker EC, Jung J, Tuwatananurak I, Pogliano K. 2010. Transposon assisted gene insertion technology (TAGIT): a tool for generating fluorescent fusion proteins. PLoS One 5:e8731. doi: 10.1371/journal.pone.0008731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zagorec M, Steinmetz M. 1991. Construction of a derivative of Tn917 containing an outward-directed promoter and its use in Bacillus subtilis. J Gen Microbiol 137:107–112. doi: 10.1099/00221287-137-1-107. [DOI] [PubMed] [Google Scholar]
- 9.Youngman PJ, Perkins JB, Losick R. 1983. Genetic transposition and insertional mutagenesis in Bacillus subtilis with Streptococcus faecalis transposon Tn917. Proc Natl Acad Sci U S A 80:2305–2309. doi: 10.1073/pnas.80.8.2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garsin DA, Urbach J, Huguet-Tapia JC, Peters JE, Ausubel FM. 2004. Tn917-mediated-gene-disruption library offers insight into Tn917 insertion patterns. J Bacteriol 186:7280–7289. doi: 10.1128/JB.186.21.7280-7289.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shi Q, Huguet-Tapia JC, Peters JE. 2009. Tn917 targets the region where DNA replication terminates in Bacillus subtilis, highlighting a difference in chromosome processing in the Firmicutes. J Bacteriol 191:7623–7627. doi: 10.1128/JB.01023-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Halling SM, Kleckner N. 1982. A symmetrical six-base-pair target site sequence determines Tn10 insertion specificity. Cell 28:155–163. doi: 10.1016/0092-8674(82)90385-3. [DOI] [PubMed] [Google Scholar]
- 13.Petit MA, Bruand C, Janniere L, Ehrlich SD. 1990. Tn10-derived transposons active in Bacillus subtilis. J Bacteriol 172:6736–6740. doi: 10.1128/jb.172.12.6736-6740.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bender J, Kleckner N. 1992. Tn10 insertion specificity is strongly dependent upon sequences immediately adjacent to the target-site consensus sequence. Proc Natl Acad Sci U S A 89:7996–8000. doi: 10.1073/pnas.89.17.7996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Steinmetz M, Richter R. 1994. Easy cloning of mini-Tn10 insertions from the Bacillus subtilis chromosome. J Bacteriol 176:1761–1763. doi: 10.1128/jb.176.6.1761-1763.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pribil PA, Haniford DB. 2003. Target DNA bending is an important specificity determinant in target site selection in Tn10 transposition. J Mol Biol 330:247–259. doi: 10.1016/s0022-2836(03)00588-6. [DOI] [PubMed] [Google Scholar]
- 17.Van Opijnen T, Camilli A. 2013. Transposon insertion sequencing: a new tool for systems-level analysis of microorganisms. Nat Rev Microbiol 11:435–442. doi: 10.1038/nrmicro3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meeske AJ, Rodrigues CDA, Brady J, Lim HC, Bernhardt TG, Rudner DZ. 2016. High-throughput genetic screens identify a large and diverse collection of new sporulation genes in Bacillus subtilis. PLoS Biol 14:e1002341. doi: 10.1371/journal.pbio.1002341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnson CM, Grossman AD. 2014. Identification of host genes that affect acquisition of an integrative and conjugative element in Bacillus subtilis. Mol Microbiol 93:1284–1301. doi: 10.1111/mmi.12736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pozsgai ER, Blair KM, Kearns DB. 2012. Modified mariner transposons for random inducible-expression insertions and transcriptional reporter fusion insertions in Bacillus subtilis. Appl Environ Microbiol 78:778–785. doi: 10.1128/AEM.07098-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pozsgai ER, Blair KM, Kearns DB. 2020. Correction for Pozsgai et al., “Modified mariner transposons for random inducible-expression insertions and transcriptional reporter fusion insertions in Bacillus subtilis.” Appl Environ Microbiol 86:e00355-20. doi: 10.1128/AEM.00355-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yanisch-Perron C, Vieira J, Messing J. 1985. M13 phage cloning vectors and host strains: nucleotide sequences of the M13mpl8 and pUC19 vectors. Gene 33:103–119. doi: 10.1016/0378-1119(85)90120-9. [DOI] [PubMed] [Google Scholar]
- 23.Churchward G, Belin D, Nagamine Y. 1984. A pSC101-derived plasmid which shows no sequence homology to other commonly used cloning vectors. Gene 31:165–171. doi: 10.1016/0378-1119(84)90207-5. [DOI] [PubMed] [Google Scholar]
- 24.Hasunuma K, Sekiguchi M. 1977. Replication of plasmid pSC101 in Escherichia coli K12: requirement for dnaA function. Mol Gen Genet 154:225–230. doi: 10.1007/bf00571277. [DOI] [PubMed] [Google Scholar]
- 25.Cabello F, Timmis K, Cohen SN. 1976. Replication control in a composite plasmid constructed by in vitro linkage of two distinct replicons. Nature 259:285–290. doi: 10.1038/259285a0. [DOI] [PubMed] [Google Scholar]
- 26.Horinouchi S, Weisblum B. 1982. Nucleotide sequence and functional map of pE194, a plasmid that specifies inducible resistance to macrolide, lincosamide, and streptogramin type B antibiotics. J Bacteriol 150:804–814. doi: 10.1128/JB.150.2.804-814.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gryczan TJ, Hahn S, Contente DD. 1982. Replication and incompatibility properties of plasmid pE194 in Bacillus subtilis. J Bacteriol 152:722–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Villafane R, Bechhofer DH, Narayanan CS, Dubnau D. 1987. Replication control genes of plasmid pE194. J Bacteriol 169:4822–4829. doi: 10.1128/jb.169.10.4822-4829.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maguin E, Duwat P, Hege T, Ehrlich D, Gruss A. 1992. New thermosensitive plasmid for gram-positive bacteria. J Bacteriol 174:5633–5638. doi: 10.1128/jb.174.17.5633-5638.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA, Smith HO. 2009. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 31.Nye TM, Schroeder JW, Kearns DB, Simmons LA. 2017. Complete genome sequence of undomesticated Bacillus subtilis strain NCIB 3610. Genome Announc 5:12–13. doi: 10.1128/genomeA.00364-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Konkol MA, Blair KM, Kearns DB. 2013. Plasmid-encoded comi inhibits competence in the ancestral 3610 strain of Bacillus subtilis. J Bacteriol 195:4085–4093. doi: 10.1128/JB.00696-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vlamakis H, Chai Y, Beauregard P, Losick R, Kolter R. 2013. Sticking together: building a biofilm the Bacillus subtilis way. Nat Rev Microbiol 11:157–168. doi: 10.1038/nrmicro2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Youngman P, Perkins JB, Losick R. 1984. Construction of a cloning site near one end of Tn917 into which foreign DNA may be inserted without affecting transposition in Bacillus subtilis or expression of the transposon-borne erm gene. Plasmid 12:1–9. doi: 10.1016/0147-619x(84)90061-1. [DOI] [PubMed] [Google Scholar]
- 35.Carneiro L, Yu L, Dupree P, Ward RJ. 2018. Characterization of a β-galactosidase from Bacillus subtilis with transgalactosylation activity. Int J Biol Macromol 120:279–287. doi: 10.1016/j.ijbiomac.2018.07.116. [DOI] [PubMed] [Google Scholar]
- 36.Daniel RA, Haiech J, Denizot F, Errington J. 1997. Isolation and characterization of the lacA gene encoding β-galactosidase in Bacillus subtilis and a regulator gene, lacR. J Bacteriol 179:5636–5638. doi: 10.1128/jb.179.17.5636-5638.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aronson DE, Costantini LM, Snapp EL. 2011. Superfolder GFP is fluorescent in oxidizing environments when targeted via the Sec translocon. Traffic 12:543–548. doi: 10.1111/j.1600-0854.2011.01168.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pédelacq JD, Cabantous S, Tran T, Terwilliger TC, Waldo GS. 2006. Engineering and characterization of a superfolder green fluorescent protein. Nat Biotechnol 24:79–88. doi: 10.1038/nbt1172. [DOI] [PubMed] [Google Scholar]
- 39.Matsuoka H, Hirooka K, Fujita Y. 2007. Organization and function of the YsiA regulon of Bacillus subtilis involved in fatty acid degradation. J Biol Chem 282:5180–5194. doi: 10.1074/jbc.M606831200. [DOI] [PubMed] [Google Scholar]
- 40.Tojo S, Satomura T, Matsuoka H, Hirooka K, Fujita Y. 2011. Catabolite repression of the Bacillus subtilis FadR regulon, which is involved in fatty acid catabolism. J Bacteriol 193:2388–2395. doi: 10.1128/JB.00016-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Antelmann H, Engelmann S, Schmid R, Sorokin A, Lapidus A, Hecker M. 1997. Expression of a stress- and starvation-induced dps/pexB-homologous gene is controlled by the alternative sigma factor σB in Bacillus subtilis. J Bacteriol 179:7251–7256. doi: 10.1128/jb.179.23.7251-7256.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Höper D, Völker U, Hecker M. 2005. Comprehensive characterization of the contribution of individual SigB-dependent general stress genes to stress resistance of Bacillus subtilis. J Bacteriol 187:2810–2826. doi: 10.1128/JB.187.8.2810-2826.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ramaley R, Fujita Y, Freese E. 1979. Purification and properties of Bacillus subtilis inositol dehydrogenase. J Biol Chem 254:7684–7690. [PubMed] [Google Scholar]
- 44.Segall J, Losick R. 1977. Cloned Bacillus subtilis DNA containing a gene that is activated early during sporulation. Cell 11:751–761. doi: 10.1016/0092-8674(77)90289-6. [DOI] [PubMed] [Google Scholar]
- 45.Matsuno K, Sonenshein AL. 1999. Role of SpoVG in asymmetric septation in Bacillus subtilis. J Bacteriol 181:3392–3401. doi: 10.1128/JB.181.11.3392-3401.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Que Q, Helmann JD. 2000. Manganese homestasis in Bacillus subtilis is regulated by MntR, a bifunctional regulator related to the diphtheria toxin repressor family of proteins. Mol Microbiol 35:1454–1468. doi: 10.1046/j.1365-2958.2000.01811.x. [DOI] [PubMed] [Google Scholar]
- 47.Quentin Y, Fichant G, Denizot F. 1999. Inventory, assembly and analysis of Bacillus subtilis ABC transport systems. J Mol Biol 287:467–484. doi: 10.1006/jmbi.1999.2624. [DOI] [PubMed] [Google Scholar]
- 48.Dempwolff F, Schmidt FK, Hervás AB, Stroh A, Rösch TC, Riese CN, Dersch S, Heimerl T, Lucena D, Hülsbusch N, Stuermer CAO, Takeshita N, Fischer R, Eckhardt B, Graumann PL. 2016. Super resolution fluorescence microscopy and tracking of bacterial flotillin (Reggie) paralogs provide evidence for defined-sized protein microdomains within the bacterial membrane but absence of clusters containing detergent-resistant proteins. PLoS Genet 12:e1006116–29. doi: 10.1371/journal.pgen.1006116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Morimoto YV, Nakamura S, Kami-Ike N, Namba K, Minamino T. 2010. Charged residues in the cytoplasmic loop of MotA are required for stator assembly into the bacterial flagellar motor. Mol Microbiol 78:1117–1129. doi: 10.1111/j.1365-2958.2010.07391.x. [DOI] [PubMed] [Google Scholar]
- 50.Li H, Sourjik V. 2011. Assembly and stability of flagellar motor in Escherichia coli. Mol Microbiol 80:886–899. doi: 10.1111/j.1365-2958.2011.07557.x. [DOI] [PubMed] [Google Scholar]
- 51.Guttenplan SB, Shaw S, Kearns DB. 2013. The cell biology of peritrichous flagella in Bacillus subtilis. Mol Microbiol 87:211–229. doi: 10.1111/mmi.12103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Frees D, Savijoki K, Varmanen P, Ingmer H. 2007. Clp ATPases and ClpP proteolytic complexes regulate vital biological processes in low GC, Gram-positive bacteria. Mol Microbiol 63:1285–1295. doi: 10.1111/j.1365-2958.2007.05598.x. [DOI] [PubMed] [Google Scholar]
- 53.Kirstein J, Strahl H, Molière N, Hamoen LW, Turgay K. 2008. Localization of general and regulatory proteolysis in Bacillus subtilis cells. Mol Microbiol 70:682–694. doi: 10.1111/j.1365-2958.2008.06438.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Simmons LA, Grossman AD, Walker GC. 2008. Clp and Lon proteases occupy distinct subcellular positions in Bacillus subtilis. J Bacteriol 190:6758–6768. doi: 10.1128/JB.00590-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fredrick KL, Helmann JD. 1994. Dual chemotaxis signaling pathways in Bacillus subtilis: a σD-dependent gene encodes a novel protein with both CheW and CheY homologous domains. J Bacteriol 176:2727–2735. doi: 10.1128/jb.176.9.2727-2735.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wu K, Walukiewicz HE, Glekas GD, Ordal GW, Rao CV. 2011. Attractant binding induces distinct structural changes to the polar and lateral signaling clusters in Bacillus subtilis chemotaxis. J Biol Chem 286:2587–2595. doi: 10.1074/jbc.M110.188664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Johnson KS, Ottemann KM. 2018. Colonization, localization, and inflammation: the roles of H. pylori chemotaxis in vivo. Curr Opin Microbiol 41:51–57. doi: 10.1016/j.mib.2017.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Siranosian KJ, Ireton K, Grossman AD. 1993. Alanine dehydrogenase (ald) is required for normal sporulation in Bacillus subtilis. J Bacteriol 175:6789–6796. doi: 10.1128/jb.175.21.6789-6796.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Aughey GN, Liu JL. 2015. Metabolic regulation via enzyme filamentation. Crit Rev Biochem Mol Biol 51:282–293. doi: 10.3109/10409238.2016.1172555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Werner JN, Chen EY, Guberman JM, Zippilli AR, Irgon JJ, Gitai Z. 2009. Quantitative genome-scale analysis of protein localization in an asymmetric bacterium. Proc Natl Acad Sci U S A 106:7858–7863. doi: 10.1073/pnas.0901781106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Guérout-Fleury AM, Shazand K, Frandsen N, Stragier P. 1995. Antibiotic-resistance cassettes for Bacillus subtilis. Gene 167:335–336. doi: 10.1016/0378-1119(95)00652-4. [DOI] [PubMed] [Google Scholar]
- 62.Wach A. 1996. PCR-synthesis of marker cassettes with long flanking homology regions for gene disruptions in S. cerevisiae. Yeast 12:259–265. doi: 10.1002/(SICI)1097-0061(19960315)12:3<259::AID-YEA901>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 63.Kain J, He GG, Losick R. 2008. Polar localization and compartmentalization of ClpP proteases during growth and sporulation in Bacillus subtilis. J Bacteriol 190:6749–6757. doi: 10.1128/JB.00589-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Park E, Guzder SN, Koken MHM, Jaspers-Dekker I, Weeda G, Hoeijmakers JHJ, Prakash S, Prakash L. 1992. RAD25 (SSL2), the yeast homolog of the human xeroderma pigmentosum group B DNA repair gene, is essential for viability. Proc Natl Acad Sci U S A 89:11416–11420. doi: 10.1073/pnas.89.23.11416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Koo BM, Kritikos G, Farelli JD, Todor H, Tong K, Kimsey H, Wapinski I, Galardini M, Cabal A, Peters JM, Hachmann AB, Rudner DZ, Allen KN, Typas A, Gross CA. 2017. Construction and analysis of two genome-scale deletion libraries for Bacillus subtilis. Cell Syst 4:291–305. doi: 10.1016/j.cels.2016.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jaacks KJ, Healy J, Losick R, Grossman AD. 1989. Identification and characterization of genes controlled by the sporulation-regulatory gene spo0H in Bacillus subtilis. J Bacteriol 171:4121–4129. doi: 10.1128/jb.171.8.4121-4129.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.McLoon AL, Guttenplan SB, Kearns DB, Kolter R, Losick R. 2011. Tracing the domestication of a biofilm-forming bacterium. J Bacteriol 193:2027–2034. doi: 10.1128/JB.01542-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Winkelman JT, Blair KM, Kearns DB. 2009. RemA (YlzA) and RemB (YaaB) regulate extracellular matrix operon expression and biofilm formation in Bacillus subtilis. J Bacteriol 191:3981–3991. doi: 10.1128/JB.00278-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Guttenplan SB, Blair KM, Kearns DB. 2010. The EpsE flagellar clutch is bifunctional and synergizes with EPS biosynthesis to promote Bacillus subtilis biofilm formation. PLoS Genet 6:e1001243–12. doi: 10.1371/journal.pgen.1001243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Diethmaier C, Pietack N, Gunka K, Wrede C, Lehnik-Habrink M, Herzberg C, Hübner S, Stülke J. 2011. A novel factor controlling bistability in Bacillus subtilis: the Ymdb protein affects flagellin expression and biofilm formation. J Bacteriol 193:5997–6007. doi: 10.1128/JB.05360-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Diethmaier C, Newman JA, Kovács ÁT, Kaever V, Herzberg C, Rodrigues C, Boonstra M, Kuipers OP, Lewis RJ, Stülke J. 2014. The YmdB phosphodiesterase is a global regulator of late adaptive responses in Bacillus subtilis. J Bacteriol 196:265–275. doi: 10.1128/JB.00826-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kaundinya CR, Savithri HS, Krishnamurthy Rao K, Balaji PV. 2018. EpsN from Bacillus subtilis 168 has UDP-2,6-dideoxy 2-acetamido 4-keto glucose aminotransferase activity in vitro. Glycobiology 28:802–812. doi: 10.1093/glycob/cwy063. [DOI] [PubMed] [Google Scholar]
- 73.Branda SS, González-Pastor JE, Ben-Yehuda S, Losick R, Kolter R. 2001. Fruiting body formation by Bacillus subtilis. Proc Natl Acad Sci U S A 98:11621–11626. doi: 10.1073/pnas.191384198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kearns DB, Chu F, Branda SS, Kolter R, Losick R. 2005. A master regulator for biofilm formation by Bacillus subtilis. Mol Microbiol 55:739–749. doi: 10.1111/j.1365-2958.2004.04440.x. [DOI] [PubMed] [Google Scholar]
- 75.Roux D, Cywes-Bentley C, Zhang YF, Pons S, Konkol M, Kearns DB, Little DJ, Howell PL, Skurnik D, Pier GB. 2015. Identification of poly-N-acetylglucosamine as a major polysaccharide component of the Bacillus subtilis biofilm matrix. J Biol Chem 290:19261–19272. doi: 10.1074/jbc.M115.648709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hamon MA, Lazazzera BA. 2001. The sporulation transcription factor Spo0A is required for biofilm development in Bacillus subtilis. Mol Microbiol 42:1199–1209. doi: 10.1046/j.1365-2958.2001.02709.x. [DOI] [PubMed] [Google Scholar]
- 77.Hamon MA, Stanley NR, Britton RA, Grossman AD, Lazazzera BA. 2004. Identification of AbrB-regulated genes involved in biofilm formation by Bacillus subtilis. Mol Microbiol 52:847–860. doi: 10.1111/j.1365-2958.2004.04023.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chan JM, Guttenplan SB, Kearns DB. 2014. Defects in the flagellar motor increase synthesis of poly-γ-glutamate in Bacillus subtilis. J Bacteriol 196:740–753. doi: 10.1128/JB.01217-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Calvo RA, Kearns DB. 2015. FlgM Is secreted by the flagellar export apparatus in Bacillus subtilis. J Bacteriol 197:81–91. doi: 10.1128/JB.02324-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.