

Abstract
Inflammation can be resolved by pro-homeostatic lipids called specialized proresolving mediators (SPMs) upon activation of their receptors. Dysfunctional inflammatory resolution is now considered as a driver of chronic neuroinflammation and Alzheimer’s disease (AD) pathogenesis. We have previously shown that SPM levels were reduced and also that SPM-binding receptors were increased in patients with AD compared to age-matched controls. Individuals with Down syndrome (DS) exhibit accelerated acquisition of AD neuropathology, dementia, and neuroinflammation at an earlier age than the general population. Beneficial effects of inducing resolution in DS have not been investigated previously. The effects of the SPM resolvin E1 (RvE1) in a DS mouse model (Ts65Dn) were investigated with regard to inflammation, neurodegeneration, and memory deficits. A moderate dose of RvE1 for 4 weeks in middle-aged Ts65Dn mice elicited a significant reduction in memory loss, along with reduced levels of serum pro-inflammatory cytokines, and reduced microglial activation in the hippocampus of Ts65Dn mice but had no effects in age-matched normosomic mice. There were no observable adverse side effects in Ts65Dn or in normosomic mice. These findings suggest that SPMs may represent a novel drug target for individuals with DS and others at risk of developing AD.
Keywords: Alzheimer’s disease, cognition, Down syndrome, memory, neuroinflammation, specialized proresolving mediators, Ts65Dn
1 |. INTRODUCTION
Neuroinflammation accompanies the neurodegeneration of Alzheimer’s disease (AD) (Eikelenboom & Stam, 1982) and is believed to promote secondary molecular pathogenesis, by an intimate relationship with both β-amyloid and phosphorylated-Tau pathology (Kinney et al., 2018). For individuals with Down syndrome (DS), inflammation occurs early in life (Wierzba-Bobrowicz, Lewandowska, Schmidt-Sidor, & Gwiazda, 1999) with increased glial activation (Wilcock, 2012; Wilcock et al., 2015) and elevated pro-inflammatory cytokines in serum from both adults (Convertini et al., 2016; Iulita et al., 2016) and children (Zhang et al., 2017). AD prevalence in the population with DS is thought to be highly influenced by the location of the amyloid precursor protein gene (APP) on the triplicated chromosome, and individuals with DS exhibit high penetration for AD early in life, with the incidence of AD nearing 70–80% in those with DS over the age of 50 (Head, Lott, Wilcock, & Lemere, 2016).
Inflammation is normally terminated by resolution upon completed neutralization of a pathogenic insult (Buckley, Gilroy, & Serhan, 2014) by a dynamic process induced by specialized proresolving mediators (SPMs), derived from long-chain polyunsaturated fatty acids (PUFAs) through several enzymatic steps, promoting a return to homeostasis (i.-e., healing) (Serhan, Chiang, & Van Dyke, 2008). PUFAs were shown to be enriched in the brain (Dyall, 2015), where the SPM-producing enzymes have also been observed to be abundantly expressed (Zhang et al., 2006) but how SPM biosynthesis is coordinated in neural tissues is unknown. SPMs bind to several receptor classes shown to be expressed in the brain, including peroxisome proliferator-activated receptor-gamma (PPAR-γ), a nuclear receptor expressed in neurons and glia that modulates lipid homeostasis (Heneka, Reyes-Irisarri, Hull, & Kummer, 2011), and several membrane-bound G-protein coupled receptors (GPCRs). Resolvin E1 (RvE1) is a potent agonist for PPAR-γ, as well as for the GPCR chemerin-like receptor-1 (ChemR23) and for the leukotriene B4 receptor (BLT1), both of which were observed to be ubiquitously expressed in human neurons and microglia (Emre et al., 2020; Wang et al., 2015). ChemR23 and BLT1 signal through the mammalian target of rapamycin (mTOR) and extracellular signal-regulated kinase (Erk1/2) pathways which were shown to be abnormally activated in individuals with DS (Iyer et al., 2014; Perluigi et al., 2014) and in mouse models for DS (Ahmed et al., 2013; Tramutola et al., 2016). The authors and others have previously demonstrated deficits in the SPM cascade in patients with AD or in animal models of AD (Lukiw et al., 2005; Wang et al., 2015; Zhao et al., 2011; Zhu et al., 2016), but it is not known whether SPMs can provide in vivo therapeutic benefits for AD or DS-related AD (DS-AD) patients. Recent studies have revealed that RvE1 imparts a significant decrease in inflammatory cytokines peripherally (Flesher, Herbert, & Kumar, 2014) which has prompted several clinical trials for RvE1 for peripheral inflammatory conditions (PubChem, 2018; CID=10473088), but the effects of RvE1 on AD pathology or memory loss have not yet been investigated in the context of DS.
Ts65Dn mice represent the most studied mouse model for DS-related AD, exhibiting age-related memory loss coupled with degeneration of several neuronal populations, including the locus coeruleus (LC), basal forebrain cholinergic neurons, and specific hippocampal neuronal populations (Hamlett et al., 2016). Chronic inflammation is also exacerbated in the Ts65Dn mouse model of DS, which recapitulates APP triplication along with behavioral phenotypes and AD-like neuropathologies associated with T21 in humans (Lockrow, Fortress, & Granholm, 2012). Our previous data have shown that the microglial marker CD45 was progressively elevated in an age-dependent manner in the hippocampus and the frontal cortex of Ts65Dn mice (Lockrow et al., 2012), and that this increase in microglial activation was accompanied by elevated protein and mRNA levels of pro-inflammatory cytokines with age in the same brain regions (Hunter, Bachman, & Granholm, 2004; Hunter, Quintero, Gilstrap, Bhat, & Granholm, 2004). In the same study, we also found that the tetracycline derivative minocycline could prevent microglial activation, neuronal cell loss and age-related memory loss in Ts65Dn mice, strongly suggesting that neuroinflammation plays an important role for the development of AD pathology in DS, at least in this particular mouse model. Thus, Ts65Dn mice represent a plausible model to test new interventions that may augment or decrease inflammatory conditions. In this study, we investigated the effects of chronic administration of the proresolving factor RvE1 in the Ts65Dn mouse model of DS, hypothesizing that RvE1 could restore memory performance and halt the chronic inflammatory phenotype observed in these mice via reduced activation of microglial cells.
2 |. METHODS
2.1 |. Animal cohorts, behavioral tasks, and experimental design
Male Ts65Dn mice (B6EiC3Sn.BLiA-Ts(1716)65Dn/DnJ, stock No: 005252), n = 16, with partial trisomy for a segment of murine Chromosomes 16 and 17 (Reeves et al., 1995) and normosmic control mice (NS, B6EiC3SnF1/J, n = 18) were obtained from Jackson Laboratory (Bar Harbor, ME). All mice were single-housed and were maintained on a 12 hr light/dark cycle. All experimental procedures were approved by the Institutional Animal Care and Use Committee of MUSC in accordance with the National Institutes of Health guide for the care and use of laboratory animals (NIH Publications No. 8023, revised 1978). At 8 months of age, the mice received a subcutaneous mini-osmotic pump (Alzet, Cupertino, CA) delivering one of two sterile solutions: vehicle (Veh, 5% EtOH, 95% saline) or RvE1, (10 ng/g body weight/day) dissolved in Veh. The pump was calibrated to deliver approximately 2.3 μL per day for a maximum of 35 days to deliver a daily dose of 300 ng to a 30-g mouse per day. After implantation, the minor incision was closed using sterile monofilament nylon suture and topical antibiotic ointment was applied. Stitches were removed 10 days following the implantation. Following the completion of the behavioral tasks (see below), the mice were euthanized using an over-dose of isoflurane and the brains rapidly removed. From the left hemisphere, the hippocampus, frontal cortex, and parietal cortex were dissected and prepared for biochemistry using our previously published protocol (Fortress et al., 2015). The right hemisphere was fixed in 4% paraformaldehyde and sectioned at 40 μm thickness for subsequent experiments.
After 25 days of treatment, a behavioral battery was administered to all groups. The behavioral battery included spontaneous activity, novel object recognition task (NORT), and water radial arm maze (WRAM) (Lockrow, Boger, Bimonte-Nelson, & Granholm, 2011; Lockrow, Boger, Gerhardt, et al., 2011). Spontaneous activity was measured using the Digiscan Animal Activity Monitor system and horizontal and vertical activity were collected as described previously (Lockrow et al., 2012). NORT, a working memory task, was based on familiar and novel object recognition. For each study, two dependent measures were calculated: the percent time with the novel object and the discrimination index (DI) (Lockrow, Boger, Bimonte-Nelson, & Granholm, 2011; Lockrow, Boger, Gerhardt, et al., 2011), which represents the net time spent with the novel object relative to the total time spent with both objects. We have previously shown that Ts65Dn mice exhibit progressive deficits in this task regardless of the intertrial interval (Lockrow et al., 2009; Lockrow, Boger, Bimonte-Nelson, & Granholm, 2011; Lockrow, Boger, Gerhardt, et al., 2011). The WRAM utilized here was a 3-day spatial reference memory task in which one platform in an eight-arm maze was kept in the same position throughout testing using a win-stay paradigm, and the platform was placed approximately 1 cm below the water surface (Lockrow, Boger, Bimonte-Nelson, & Granholm, 2011; Lockrow, Boger, Gerhardt, et al., 2011). On each day, 12 trials were run as two blocks of six trials, with mice from all groups run in cohorts of six per block, permitting a short intertrial rest period (5 min). On day 3 of the WRAM task, the hidden platform was switched to a new arm while extramaze cues remained unchanged to examine perseverance and cognitive flexibility.
2.2 |. Immunoblotting
Samples were prepared for immunoblotting using our standard protocol (Fortress et al., 2015). A common loading control (made from a fraction of each sample) enabled signal normalization between multiple gels. Briefly, the membranes were incubated with the primary antibodies for 1 hr at room temperature and then washed with trisbuffered saline (TBS) pH 7.6, 3 × 5 min. The antibodies and dilutions are outlined in Table 1. The membrane was incubated with secondary antibodies conjugated with either IRDye 680RD or 800CW (Li-COR Biosciences, Lincoln, NE) for 30 min and then washed with TBS, 3 × 5 min. All probe signals were measured with a Li-COR Odyssey scanner and quantified with Image Studio Lite (Li-COR Biosciences).
TABLE 1.
Antibody details
| Antibody | Manufacturer | Method | Dilution |
|---|---|---|---|
| Rb anti-leukotriene B4 receptor 1 (BLT1) | Cayman Chemical, Ann Arbor, MI | Immunoblot | 1:1,000 |
| Ms anti-chemokine-like receptor 1 (ChemR23) | Santa Cruz Biotechnology, Dallas, TX | Immunoblot | 1:200 |
| Rb anti-Peroxisome proliferator-activated receptor gamma (PPAR-γ) | R&D Systems, Minneapolis, MN | Immunoblot | 1:1,000 |
| Rb anti-N-formyl peptide receptor 2 (LxA4R) | Santa Cruz Biotechnology, Dallas TX | Immunoblot | 1:500 |
| Rb anti-phospho (Thr202/Tyr204) extracellular-signal-regulated kinase (Erk1/2) | Cell Signaling, Danvers, MA | Immunoblot | 1:1,000 |
| Ms anti-total extracellular-signal-regulated kinase (Erk1/2) | Cell Signaling, Danvers, MA | Immunoblot | 1:1,000 |
| Rb anti-15-lipoxygenase-2 (15LOX2) | Cayman Chemical, Ann Arbor, MI | Immunoblot | 1:200 |
| Rb anti-calbindin-D28 | Bio-Rad, Hercules, CA | Immunoblot | 1:200 |
| Rb anti-ionized calcium binding adaptor molecule 1 (Iba1) | MilliporeSigma, Burlington, MA | Immunohistochemistry | 1:200 |
| Rb anti-protein tyrosine phosphatase, receptor type, C (CD45) | Wako-Chem, Richmond, VA | Immunohistochemistry | 1:200 |
2.3 |. Immunohistochemistry
Coronal brain sections were incubated with antibodies against ionized calcium-binding adaptor molecule 1 (Iba1) or protein tyrosine phosphatase, receptor type, C (CD45) to visualize microglia (see Table 1). After washing with TBS 3 × 10 min, sections were incubated with Alexa488-conjugated secondary antibodies for Iba1 or biotin-conjugated secondary antibody amplified by ABC solution (Vector, Burlingame, CA) and diaminobenzidine (DAB, Millipore Sigma, St. Louis, MO) for CD45. All images were captured using a Nikon Eclipse 80i microscope (Nikon Instruments, Melville, NY) and densitometry was performed using Fiji image analysis software (ImageJ, v1.52 c). Quantification of Iba1-positive microglial cell body size was achieved with MorphoLibJ (v1.3.1) integrated library and FIJI plugin (Legland, Arganda-Carreras, & Andrey, 2016).
2.4 |. Multiplex enzyme-linked immunosorbent assay
A 31-plex enzyme-linked immunosorbent assay was utilized for the detection of mouse serum cytokines and chemokines (Eve Technologies, Calgary, Canada). Serum samples were diluted 1:2 with TBS, and cytokines were incubated with a panel of beads that each had unique fluorophore signatures. After washing, a multiplex panel of bio-tinylated detection antibodies was added to the bead-captured cytokine targets resulting in antibody-target complexes, which were visualized by incubation with a streptavidin-conjugated phycoerythrin probe. Both bead and probe signals were quantified using a dual-laser flow cytometry system (Bio-Plex 200; Bio-Rad, Hercules, CA).
2.5 |. Statistical analyses
We tested all data sets for outliers using Grubb’s test and confirmed normality with the Shapiro–Wilk test. Behavior data and protein measurements were analyzed by two-way ANOVAs to test for differences between the four groups (karyotype × treatment), followed by Tukey’s test for post hoc analysis. Data from the 3-day WRAM task were tested by repeated-measures mixed ANOVAs with time as within-subject factors, and karyotype and treatment as between-subject factors. All data were correlated using Pearson correlation matrix and graphically presented as mean ± SEM. All statistics were performed with GraphPad Prism 7.03 (GraphPad Software, La Jolla, CA) or IBM SPSS v25 (IBM, Armonk, NY).
3 |. RESULTS
3.1 |. RvE1 ameliorated deficits in memory and hyperactivity in Ts65Dn mice
Veh(icle)-treated NS groups and both RvE1-treated groups showed significant learning in all tasks, while Veh-treated Ts65Dn mice exhibited decreased performance across all days and trials. For the WRAM task, a repeated-measures mixed ANOVA on performance over 3 days showed significant effects of karyotype, treatment and time, with no significant time-karyotype or time–treatment interactions (Figure 1a). A one-way ANOVA was used to separately test trial blocks (one for each day), revealing that Veh-treated Ts65Dn mice made significantly more errors compared to Veh-treated NS mice on all 3 days of testing (p < .001) (Figure 1b). RvE1-treated Ts65Dn mice showed a significant decrease in the number of errors on the last 2 days of the WRAM test when compared to Veh-treated Ts65Dn mice, strongly suggesting that the RvE1 treatment resulted in improved WRAM performance and/or prevention of age-related impairment on this spatial reference memory task (Day 2: p = .006 and Day 3: p = .001).
FIGURE 1.

RvE1-mediated effects on memory. (a) Two-way ANOVA results by karyotype and RvE1 treatment on behavior in the WRAM and NORT tests. (b) Over 3 days, Ts65Dn mice displayed significantly more errors than the NS mice throughout the WRAM task, and RvE1 treatment significantly enhanced Ts65Dn performance to that observed in all NS mice. (c) In a cognitive flexibility task, RvE1 treatment significantly enhanced Ts65Dn performance to that observed in all NS mice. (d) In the locomotion test, Veh-treated Ts65Dn mice were hyperactive (distance in meters traveled over 60 min) relative to Veh-treated NS mice and RvE1 treatment normalized this behavioral phenotype with no effect on NS mice. (e,f) In the NORT task, Veh-treated Ts65Dn mice displayed severe deficits that were normalized by RvE1 treatment at 90 min (e) and 24 hr (f). The RvE1 treatment had no effects on NS mice at any interval. Tukey’s post hoc p values are shown for group comparisons and error bars represent mean ± SEM. NS, normosomic; NORT, novel object recognition task; RvE1, resolvin E1; TS, Ts65Dn; Veh, vehicle; WRAM, water radial maze task
In the reversal task, during which the platform was switched to another location, NS mice rapidly learned the new platform location, but the Veh-treated Ts65Dn mice continued to show high error rates throughout the testing period, contrary to the RvE1-treated Ts65Dn mice, suggesting perseverance to previous task conditions (Figure 1c). A repeated-measures mixed ANOVA confirmed significant effects of karyotype, treatment and time as well as a significant time-karyotype interaction (F1,24 = 5.58, p = .027) and a time–treatment interaction (F1,24 = 11.52, p < .001, see Figure 1a). A one-way ANOVA showed that all NS mice (regardless of treatment) and RvE1-treated Ts65Dn mice had a significant decrease in errors as trials progressed (p < .001 for both). At completion, Veh-treated Ts65Dn mice made significantly more errors than Veh-treated NS mice (p < .001) and RvE1-treated Ts65Dn mice made significantly fewer errors than the Veh-treated Ts65Dn mice (p < .001), suggesting increased cognitive flexibility in the reversal component of the WRAM in the Ts65Dn mice treated with RvE1.
In an open-field task, spontaneous locomotion was examined for 60 min while animals were traveling at will on a platform recording vertical and horizontal movement. Veh-treated Ts65Dn mice exhibited elevations in spontaneous locomotion (distance traveled) relative to Veh-treated NS mice (Figure 1d) as previously reported (Fortress et al., 2015; Lockrow, Boger, Bimonte-Nelson, & Granholm, 2011; Lockrow, Boger, Gerhardt, et al., 2011), and RvE1 treatment decreased hyperactivity seen in Ts65Dn mice almost to the activity levels observed in NS mice. Two-way ANOVA confirmed significant main effects of karyotype and RvE1 treatment with a significant interaction between these factors. Tukey’s post hoc analysis further confirmed hyperactivity in the Veh Ts65Dn group relative to Veh-treated NS mice (p < .001) and also that RvE1 treatment corrected the hyperactive phenotype in Ts65Dn mice (p = .035) without affecting NS mice, suggesting that the RvE1 treatment corrected hyperactivity in Ts65Dn mice.
We observed deficits in short-term working memory in the NORT at both 90 min and 24 hr intertrial intervals in Ts65Dn mice, which were alleviated by the RvE1 administration. A two-way ANOVA confirmed a significant effect of karyotype as well as a significant main effect of treatment for both testing intervals (Figure 1a). Tukey’s post hoc analysis also confirmed that Veh-treated Ts65Dn mice had significantly reduced performance at 90 min compared to NS mice (p < .001) with no observable behavioral effects on NS mice (Figure 1e). RvE1 treatment significantly increased the performance of Ts65Dn mice in this task (p < .001) with a DI comparable to that observed in both NS groups. At 24 hr (Figure 1f), the mean DI was significantly less in Veh-treated Ts65Dn mice compared to Veh-treated NS mice (p < .001). Again, RvE1 treatment significantly increased performance in Ts65Dn mice (p < .001). Collectively, RvE1 treatment enhanced Ts65Dn working memory at two different intertrial intervals with no effects observed in this behavioral task on age-matched NS control mice.
3.2 |. RvE1-mediated receptor responses
We investigated levels of receptors that bind RvE1 including ChemR23, BLT1, and PPAR-γ, along with Erk1/2, a primary signaling effector for all known RvE1 receptors in frontal cortex and hippocampus (Figures 2 and 3). As seen in the sample blots (Figure 2a,b), Veh-treated NS and Ts65Dn mice had equivalent ChemR23 and BLT1 receptor levels, with a trend towards increased ChemR23 levels observed in the frontal cortex of Ts65Dn mice (Figure 2d), while chronic RvE1 treatment led to significant reductions in levels of both receptors in the RvE1 treated groups. A two-way ANOVA showed that the main significant effect was treatment in both the frontal cortex and hippocampus (Figure 2c). Tukey’s post hoc analysis confirmed that RvE1 administration significantly decreased ChemR23 levels (Figure 2d) in both NS and Ts65Dn mice for both frontal cortex (NS, p = .018; Ts65Dn, p = .003), and hippocampus (NS, p = .036; Ts65Dn, p = .033). Similarly, Tukey’s post hoc analysis confirmed that RvE1 administration significantly decreased BLT1 levels (Figure 2e) in all mice for both frontal cortex (NS, p = .006; Ts65Dn, p = .024), and hippocampus (NS, p = .014; with a trend for Ts65Dn, p = .051). Lipoxin A4 receptor (LXA4R), a resolution receptor with high sequence homology to ChemR23 (Chiang et al., 2006) was not affected by RvE1 administration in NS nor in Ts65Dn mice in any region analyzed (data not shown), suggesting receptor-specific effects in the brain following RvE1 treatment.
FIGURE 2.

Effects of RvE1 on levels of RvE1-binding receptors. Immunoblot signals in the frontal cortex (a) and hippocampus (b) revealed that RvE1 treatment significantly reduced ChemR23, BLT1, and PPAR-γ. (c) Two-way ANOVA effects by karyotype and resolvin (Rv) E1 treatment on levels of ChemR23 and BLT1 were attributed mainly to RvE1 treatment. (d,e) Densitometric analysis showed significant compensatory responses to RvE1-treatment in both NS and Ts65Dn mice for ChemR23 (d) and BLT1 (e) in the frontal cortex and hippocampus. The results of immunoblot signals were normalized to levels in Veh-treated NS mice. Tukey’s post hoc p values are shown for group comparisons and error bars represent the average ± SEM. BLT1, leukotriene B4 receptor 1; ChemR23, chemokine-like receptor 1; NS, normosomic; RvE1, resolvin E1; TS, Ts65Dn; Veh, vehicle
FIGURE 3.
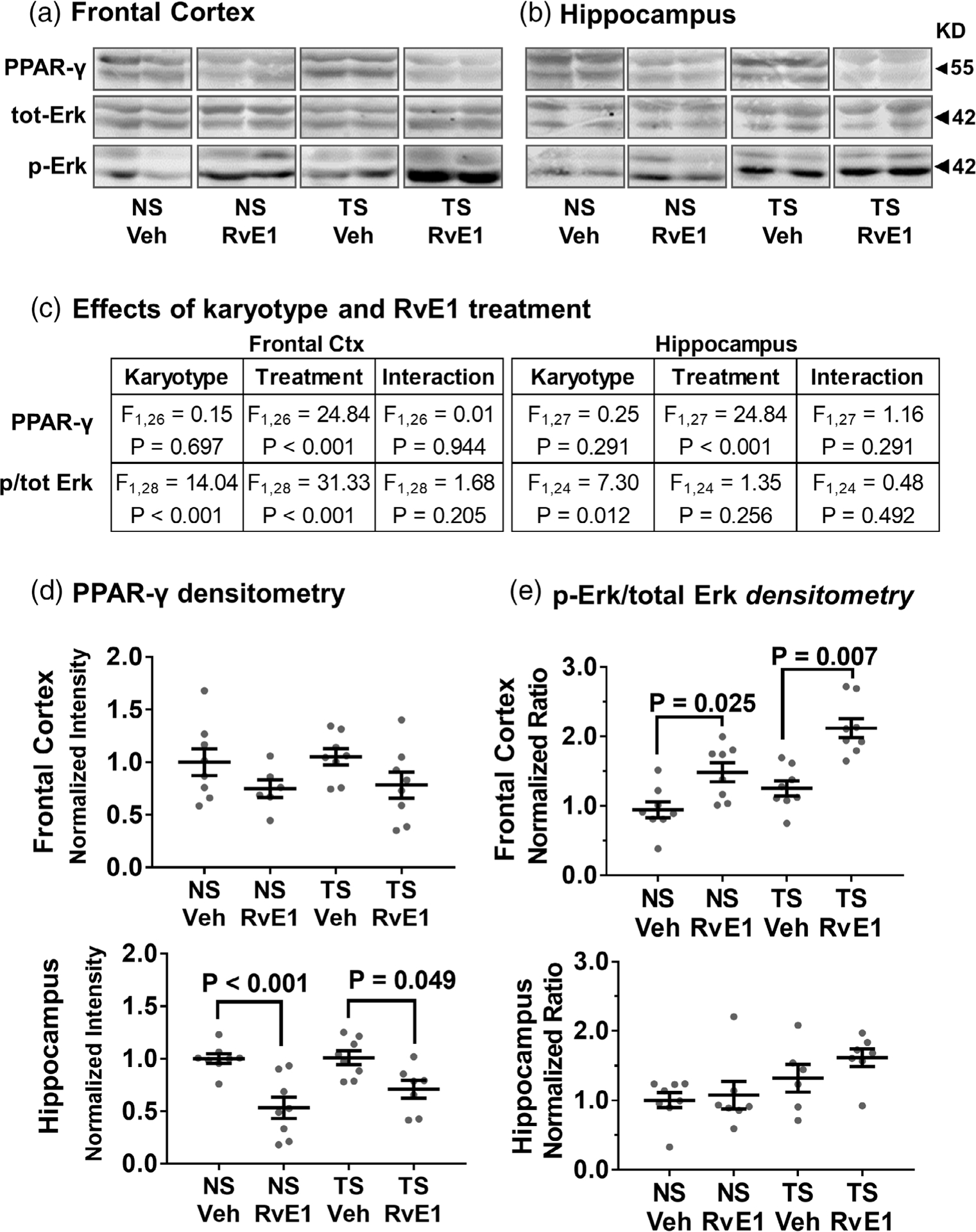
Effects of RvE1 on PPAR-γ levels and Erk1/2 phosphorylation. RvE1 treatment significantly reduced PPAR-γ levels and increased phosphorylated Erk1/2 immunoblot signals without significantly affecting total Erk1/2 levels in the frontal cortex (a) and hippocampus (b). (c) Two-way ANOVA effects by karyotype and RvE1 treatment on PPAR-y, ERK1/2 and phosphorylated (p) ERK1/2 were attributed mainly to RvE1 treatment for PPAR-γ and to treatment and karyotype for relative Erk signaling. (d,e) Densitometric analysis showed significant responses to RvE1-treatment in both NS and Ts65Dn mice for PPAR-γ (d) and relative Erk (e) signaling in frontal cortex and hippocampus. The lower band (Erk2) displayed a greater response compared to the upper band (Erk1). Tukey’s post hoc p values are shown for group comparisons. Relative Erk signaling reflects the ratio p-Erk over total Erk and error bars represent mean ± SEM. Erk1/2 = extracellular-signal-regulated kinase isoform 1 and 2; NS, normosomic; RvE1, resolvin E1; PPAR-γ, peroxisome proliferator-activated receptor gamma; TS, Ts65Dn; Veh, vehicle
There was a treatment effect for PPAR-γ upon RvE1 in all mice as seen from the two-way ANOVA (Figure 3a,b) and analysis by Tukey’s post hoc test confirmed that RvE1 treatment significantly decreased PPAR-γ levels in both NS and Ts65Dn mice in the hippocampus, whereas the reduction did not reach statistical significance for the frontal cortex (Figure 3c,d). Ts65Dn mice initially had higher levels of phosphorylated (p)-Erk relative to NS mice, and p-Erk levels were further increased in all mice that received RvE1 treatment, with the Erk2 isoform (lower band, semunostaining. As seen ie Figure 3a,b) showing the greatest response. RvE1 treatment significantly increased the p-Erk/total-Erk ratio in both NS and Ts65Dn mice (Figure 3c,e) in frontal cortex (NS: p = .025; Ts65Dn: p = .007), but not with statistical significance in the hippocampus. Thus, RvE1 treatment elicited decreases in all three associated receptors, and an increase in the pErk/tErk ratio, indicating selective effects on beneficial pathways in the brain following systemic administration.
3.3 |. RvE1 decreased microglial activation in Ts65Dn mice
Microglial activation has been associated with an increase in cell body size and a decrease in process territory, reflecting the morphological transition from a ramified “resting stage” to an activated phagocyte-like phenotype. We morphologically characterized Iba1-positive microglia in the hippocampal region by immunostaining. As seen in Figure 4a–d, microglial cells with an activated morphology (larger soma) were more abundant in Veh-treated Ts65Dn mice compared to NS mice. RvE1 treatment reduced the abundance of large soma microglia in Ts65Dn mice. We performed a segmentation analysis of cell body area (Figure 4e) and determined the frequency area distribution for each cohort. Approximately 13% of all microglial cell somas were larger than observed in either NS cohort. Only 2% of the microglia in RvE1-treated Ts65Dn mice had this activated phenotype.
FIGURE 4.

Microglial morphology changes following RvE1 treatment. Morphological assessment of Iba1 immunostaining of microglia revealed small somas for both NS cohorts (a,b). However, for Veh-treated Ts65Dn mice (c), microglia with enlarged soma was more abundant, indicating increased activation (arrowheads). (d) The enlarged soma phenotype was rarely observed in RvE1-treated Ts65Dn mice. Morphometric quantitation of cell body area (e) using segmentation analysis established the typical distribution of healthy hippocampal microglia and revealed that 13% of microglia exhibited much larger cell bodies in Veh-treated Ts65Dn, while only 2% of microglia with larger cell bodies were observed in RvE1-treated Ts65Dn mice. Iba1, ionized calcium-binding adaptor molecule 1; NS, normosomic; RvE1, resolvin E1; TS, Ts65Dn; Veh, vehicle
CD45 staining of microglia Ts65Dn mice showed groups of cells with an activated morphology, including short, stubby processes and increased cell body size, as compared to NS controls. This was especially evident in the CA1 and dentate gyrus of the hippocampus (Figure 5a–d). RvE1 treatment significantly reduced the hippocampal CD45 staining density in Ts65Dn mice toward staining density levels observed in NS mice. A two-way ANOVA of staining density confirmed that the main effects were due to both karyotype and treatment with significant interaction between the two factors, and post hoc analysis confirmed that Veh-treated Ts65Dn mice had significantly higher levels of CD45 staining density compared to Veh-treated NS mice (p < .001), and that the CD45 staining density was significantly reduced with RvE1 treatment in Ts65Dn mice (p < .001) (Figure 5e).
FIGURE 5.
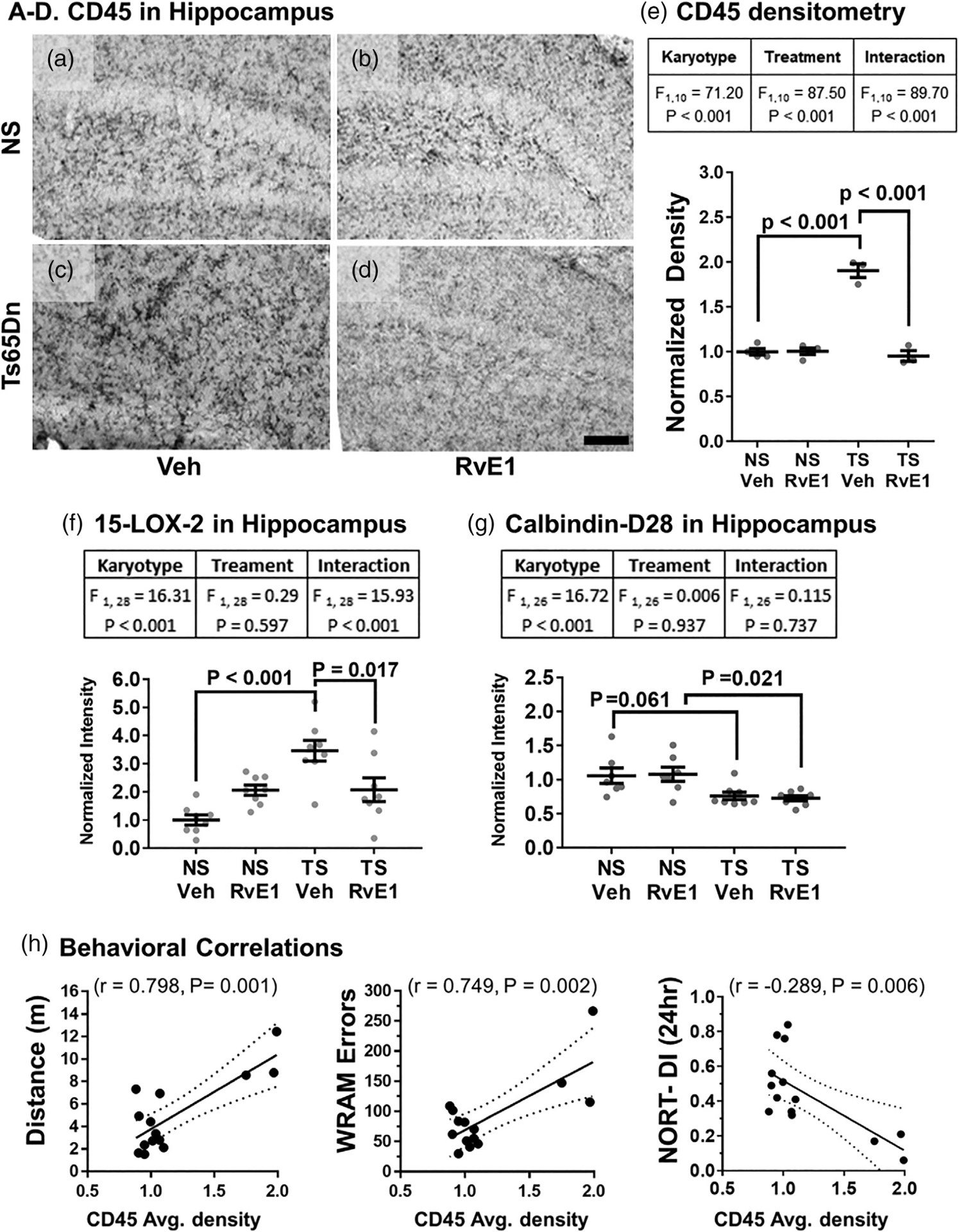
Microglial inflammatory response to RvE1 treatment. (a) Typical CD45-staining seen in Veh-treated NS mice showed multiple resting microglia with small cell somas and long, thin processes, which were not affected by RvE1 treatment (b). (c) Veh-treated Ts65Dn mice displayed elevated CD45-staining of microglial cells displaying an activated morphology compared to the NS mice (a). (d) RvE1 treatment reversed the microglial CD45 phenotype of Ts65Dn mice to staining patterns observed in NS cohorts. Scale bar equals 500 μm. (e) Two-way ANOVA effects by karyotype and RvE1 treatment for CD45 densitometry confirmed that RvE1 treatment significantly reduced CD45 staining density in Ts65Dn mice. (f) RvE1 treatment also reversed elevated 15-LOX-2 levels in the hippocampus of Ts65Dn mice. Two-way ANOVA effects by karyotype and RvE1 treatment of 15-LOX-2 densitometry confirmed that RvE1 treatment significantly reduced 15-LOX-2 levels in Ts65Dn mice. (g) Calbindin-D28 was significantly reduced in Ts65Dn mice and not affected by RvE1 treatment. Two-way ANOVA effects by karyotype only on averaged calbindin-D28 densitometry confirmed that RvE1 treatment had no effects on calbindin-D28 levels in Ts65Dn mice. Tukey’s post hoc p values are shown for group comparisons and error bars represent mean ± SEM. (h) CD45 density correlated significantly with spontaneous locomotion (positive correlation), total WRAM errors (positive correlation), and with NORT performance (negative correlation). Dotted lines represent 95% confidence interval. CD45, protein tyrosine phosphatase, receptor type, C; 15-LOX-2, 15-Lipoxygenase-2; NS, normosomic; NORT-DI, novel object recognition discrimination index; RvE1, resolvin E1; TS, Ts65Dn; Veh, vehicle; WRAM, water radial arm maze task
We quantified 15-LOX-2 by immunoblot, a lipoxygenase converting fatty acids to 15-hydroxyderivatives, that was found to be upregulated in astrocytes and microglia in postmortem hippocampi from subjects with AD (Wang et al., 2015). Ts65Dn mice had over two-fold higher 15-LOX-2 staining density compared to NS mice and RvE1 administration reduced 15-LOX-2 to levels observed in NS mice. A two-way ANOVA of 15-LOX-2 staining density (Figure 5f) confirmed that the main effects were due to both karyotype and treatment with significant interaction between the two factors. Subsequent post hoc analysis confirmed that levels of 15-LOX-2 were significantly higher in Veh-treated Ts65Dn mice compared to Veh-treated NS mice (p < .001), and that the 15-LOX-2 staining density was significantly reduced by RvE1 treatment in Ts65Dn mice (p = .017). Altogether, administration of RvE1 decreased microglial activation in Ts65Dn mice, as evidenced by Iba1 and CD45 staining, and lowered 15-LOX-2 levels suggesting an overall reduction in inflammation within the hippocampal formation.
Previously, the authors and others have shown that Ts65Dn mice have significantly reduced levels of calbindin-D28, a major calcium-binding/buffering protein that prevents neuronal death (Lockrow, Boger, Bimonte-Nelson, & Granholm, 2011; Lockrow, Boger, Gerhardt, et al., 2011). We quantified calbindin-D28 levels by immunoblot and found that RvE1 administration had no effect on this protein in Ts65Dn mice. A two-way ANOVA of staining density (Figure 5g) confirmed that the main effects were due only to karyotype with no treatment effects suggesting that calbindin-D28 may not respond to RvE1 treatment.
Analysis of possible correlation between behavior and CD45 staining by Pearson correlation test revealed a positive correlation with regard to spontaneous locomotion and total WRAM errors and a negative correlation with NORT performance, both at 90 min (data not shown) and at 24 hr (Figure 5h), indicating that RvE1-mediated changes in microglial activation in Ts65Dn mice may contribute to increased hippocampal-dependent memory performance.
3.4 |. RvE1 normalized serum cytokine levels in Ts65Dn mice
Elevated peripheral inflammation has not been described in Ts65Dn mice previously but was seen in humans with DS already in the fetal stage, and may contribute to the elevated microglial activation observed in the hippocampus. Since RvE1 administration was subcutaneous and thereby could exert its effects systemically, we sought to quantify peripheral effects that can influence brain functions. Using a multiplex cytokine array, we assessed whether RvE1 administration affected peripheral cytokine levels, including interleukin (IL) −1α, IL-1β, IL-6, and tumor necrosis factor-α (TNF-α) (Figure 6). For all four cytokines, a two-way ANOVA confirmed that the main effects were due to both karyotype and treatment with significant interaction between the two factors (Figure 6e). As can be seen in Figure 6a–d, Ts65Dn mice exhibited significant elevations of several pro-inflammatory cytokines in serum, and RvE1 treatment restored the serum cytokine levels in the Ts65Dn group to levels comparable to NS mice (Figure 6b–d). Veh-treated Ts65Dn mice had significantly higher levels of the four cytokines when compared to Veh-treated NS mice (see F and p values in Figure 6e), and the RvE1 treatment significantly reduced the cytokine levels in Ts65Dn mice, demonstrating that RvE1 treatment was effective in reducing peripheral inflammation in Ts65Dn mice to normal levels. Interestingly, most serum cytokine levels showed a highly significant negative correlation with NORT DI both at 90 min and at 24 hr (Figure 6e), suggesting a causative function of peripheral inflammation in the working memory deficits observed.
FIGURE 6.

Serum cytokine levels in response to RvE1. Analysis of serum levels of interleukin (IL)-1α (a), IL-1β (b), IL-6 (c), and tumor necrosis factor (TNF)-α (d) showed that Veh-treated Ts65Dn mice had significantly higher levels relative to the NS cohorts. All cytokines were significantly reduced in Ts65Dn mice upon RvE1-treatment, whereas no treatment effect was observed in NS mice. (e) Two-way ANOVA analyses showed that the main effects were shared between karyotype and treatment. Tukey’s post hoc analysis confirmed that Veh-treated Ts65Dn mice had significantly higher levels of the four cytokines when compared to Veh-treated NS mice (IL-1α: p = .037, IL-1β: p = .017, IL-6: p < .001 and TNF-α: p < .001), and the RvE1 treatment significantly reduced the cytokine levels in Ts65Dn mice (IL-1α: p = .025, IL-1β: p = .013, IL-6: p = .011, and TNF-α: p < .001). All of the cytokines significantly correlated with NORT performance at both testing intervals. Tukey’s post hoc p values are shown for group comparisons and error bars represent mean ± SEM. NS, normosomic; NORT-DI, novel object recognition discrimination index; RvE1, resolvin E1; TNF-α, tumor necrosis factor-alpha; TS, Ts65Dn; Veh, vehicle
4 |. DISCUSSION
We investigated the effects of chronic administration of the SPM RvE1 in the Ts65Dn mouse model of DS. The results presented here have demonstrated that the administration of RvE1 improved memory performance in Ts65Dn mice, both in terms of spatial reference and working memory. Ts65Dn mice also displayed a significant decrease in hyperactivity in response to RvE1 treatment, which may lead to an improvement in attention to memory tasks. In addition to the strong behavioral effects observed with this SPM, RvE1 treatment also reduced hippocampal microglial activation phenotypes observed in Ts65Dn mice toward resting-state phenotypes frequently observed in NS mice. Our observations corroborate other studies from the authors and others showing that SPMs reduce microglial activation in vivo (Kantarci et al., 2018; Lee et al., 2018) and in vitro models (Rey et al., 2016; Zhu et al., 2016). Also, within the hippocampus, we observed elevated levels of 15-LOX-2 in Ts65Dn mice and RvE1 treatment significantly reduced the levels of 15-LOX-2 in Ts65Dn without altering levels in NS mice. RvE1 treatment had no effects on the reduced levels of calbindin-D28, a Ts65Dn phenotype thought to be involved in neuronal injury. Despite this lack of effect, RvE1 treatment led to significant multimodal effects on microglia and behavior. Earlier administration of RvE1 may impact the calbindin-D28 phenotype in Ts65Dn mice.
Along with these significant effects on microglial cells, RvE1 also alleviated elevations in serum pro-inflammatory cytokine levels in Ts65Dn mice and reduced the protein levels of RvE1-binding receptors in both frontal cortex and hippocampus, strongly indicating that RvE1 crossed the blood–brain barrier and exerted targeted effects in discrete brain regions. ChemR23 and BLT1 receptors have been shown to be abnormally elevated in postmortem brain specimens from individuals with AD (Emre et al., 2020; Wang et al., 2015), but a significant elevation in ChemR23 or BLT1 receptors was not observed in the Ts65Dn mice. However, treatment with RvE1 in the current study produced a significant decrease in the levels of both ChemR23 and BLT1 in all brain regions examined in both NS and Ts65Dn mice, with no effects on the levels of LXA4R, binding LXA4 and RvD1 but not RvE1 (Rey et al., 2016). RvE1 also has high affinity to PPAR-γ (Muralikumar, Vetrivel, Narayanasamy, & U, 2017), a nuclear receptor already targeted in AD clinical trials (Tyagi, Gupta, Saini, Kaushal, & Sharma, 2011) due to modulatory effects on nuclear factor (NF)-κB driven cytokine expression (Heneka et al., 2011; Wu, Tsai, Cheung, Hsu, & Lin, 2016). We observed a significant reduction of PPAR-γ levels in the RvE1-treated groups, similar to effects observed after the administration of synthetic PPAR-γ agonists (Hauser et al., 2000). This specific response was notable since PPAR-γ mRNA levels have been observed to be upregulated in postmortem brain specimens isolated from individuals with AD (de la Monte & Wands, 2006).
Dysfunction of Erk signaling has been observed in the AD brain and was postulated as a signaling effector of neurodegeneration (Kirouac, Rajic, Cribbs, & Padmanabhan, 2017). Here, we found significant increases in Erk2 signaling upon RvE1 administration, which recapitulates other findings of resolution signaling for both RvE1 (Keyes et al., 2010) and LXA4 (Prieto et al., 2010). Interestingly, Erk2 has previously been shown to be a compensatory response that regulates downstream PPAR-γ transcriptional activity in vitro (Adams, Reginato, Shao, Lazar, & Chatterjee, 1997). Since Erk signaling enhances levels of brain-derived neurotrophic factor (Zhang et al., 2015) or modulates APP and tau phosphorylation (Kirouac et al., 2017), further studies will be necessary to better understand the relationship between RvE1 administration and the specific the Erk2 activation mechanism. The results reported here will be expanded to more in-depth studies of the Erk signaling cascade in DS, both in neurons and glia, since all three RvE1 receptors were observed to be expressed in these cells (Emre et al., 2020; Wang et al., 2015; Zhu et al., 2016).
Ts65Dn mice exhibit elevated activation of microglia and increased levels of pro-inflammatory cytokines (Hamlett et al., 2018; Lockrow, Boger, Bimonte-Nelson, & Granholm, 2011; Lockrow, Boger, Gerhardt, et al., 2011; Rueda, Florez, & Martinez-Cue, 2012), which mimics observations made from analysis of blood in persons with DS (Arita et al., 2005; Dunn & Swiergiel, 1998; Roberson, Kuddo, Horowitz, Caballero, & Spong, 2012; Wilson, Finch, & Cohen, 2002). In the current study, serum cytokine levels were elevated in Ts65Dn mice and we further demonstrated that RvE1 administration restored normal levels of several cytokines involved in innate immunity (IL-1ɑ, IL-1β, IL-6, and TNF-ɑ). These results further indicate the potency of RvE1 in reducing inflammation in different tissues and corroborate previous findings in other mouse models of AD (Baker et al., 2018; Kantarci et al., 2018). Furthermore, modulating inflammation by chronically upregulating proresolving mechanisms should be considered as a novel alternative to anti-inflammatory approaches, such as chronic minocycline treatment (Hunter, Bachman, & Granholm, 2004; Hunter, Quintero, et al., 2004).
In summary, we here provide critical behavioral and new molecular evidence pointing to RvE1 as a potential candidate to reduce neuroinflammation and memory loss in persons with DS-AD. The administration of RvE1 had no adverse side effects in the mice and was able to reach the brain in physiologically meaningful quantities, and thus provides a much-needed step toward a preclinical assessment of RvE1 as an AD therapy, especially since inflammation has been shown to be an important contributor to neuropathology in AD, as well as in DS-related AD (Iulita et al., 2016; Y. Zhang et al., 2017, Hamlett et al., 2018).
ACKNOWLEDGMENTS
The authors would like to acknowledge the technical expertise provided by Ms Laura Columbo and Ms Ceren Emre. The work was supported by a grant from the LeJeune Foundation, a grant from the Alzheimer’s Association (DSADIIP-13–284845), an R21 grant from the National Institutes on Aging (R21AG048631) to A.C.G. and M.S., a grant from the Swedish Brain Foundation (Hjärnfonden) to M.S. and A.C.G., and grants from the Swedish Research Council (22743, 22744) to M.S.
Funding information
Alzheimer’s Association, Grant/Award Number: DSADIIP-13–284845; Fondation Jérôme Lejeune; National Institute on Aging, Grant/Award Number: R21AG048631; Swedish Brain Foundation; Swedish Research Council, Grant/Award Numbers: 22743, 22744
Footnotes
CONFLICT OF INTEREST
The authors declare no potential conflict of interest.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Adams M, Reginato MJ, Shao D, Lazar MA, & Chatterjee VK (1997). Transcriptional activation by peroxisome proliferator-activated receptor gamma is inhibited by phosphorylation at a consensus mitogen-activated protein kinase site. The Journal of Biological Chemistry, 272(8), 5128–5132. 10.1074/jbc.272.8.5128 [DOI] [PubMed] [Google Scholar]
- Ahmed MM, Dhanasekaran AR, Tong S, Wiseman FK, Fisher EM, Tybulewicz VL, & Gardiner KJ (2013). Protein profiles in Tc1 mice implicate novel pathway perturbations in the Down syndrome brain. Human Molecular Genetics, 22(9), 1709–1724. 10.1093/hmg/ddt017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arita M, Yoshida M, Hong S, Tjonahen E, Glickman JN, Petasis NA, … Serhan CN (2005). Resolvin E1, an endogenous lipid mediator derived from omega-3 eicosapentaenoic acid, protects against 2,4,6-trinitrobenzene sulfonic acid-induced colitis. Proceedings of the National Academy of Sciences of the United States of America, 102(21), 7671–7676. 10.1073/pnas.0409271102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker LA, Martin NRW, Kimber MC, Pritchard GJ, Lindley MR, & Lewis MP (2018). Resolvin E1 (Rv E1 ) attenuates LPS induced inflammation and subsequent atrophy in C2C12 myotubes. Journal of Cellular Biochemistry, 119(7), 6094–6103. 10.1002/jcb.26807 [DOI] [PubMed] [Google Scholar]
- Buckley CD, Gilroy DW, & Serhan CN (2014). Proresolving lipid mediators and mechanisms in the resolution of acute inflammation. Immunity, 40(3), 315–327. 10.1016/j.immuni.2014.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang N, Serhan CN, Dahlen SE, Drazen JM, Hay DW, Rovati GE, … Brink C (2006). The lipoxin receptor ALX: Potent ligand-specific and stereoselective actions in vivo. Pharmacological Reviews, 58(3), 463–487. 10.1124/pr.58.3.4 [DOI] [PubMed] [Google Scholar]
- Convertini P, Menga A, Andria G, Scala I, Santarsiero A, Castiglione Morelli MA, … Infantino V (2016). The contribution of the citrate pathway to oxidative stress in Down syndrome. Immunology, 149(4), 423–431. 10.1111/imm.12659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Monte SM, & Wands JR (2006). Molecular indices of oxidative stress and mitochondrial dysfunction occur early and often progress with severity of Alzheimer’s disease. Journal of Alzheimer’s Disease, 9 (2), 167–181. 10.3233/JAD-2006-9209 [DOI] [PubMed] [Google Scholar]
- Dunn AJ, & Swiergiel AH (1998). The role of cytokines in infection-related behavior. Annals of the New York Academy of Sciences, 840, 577–585. 10.1111/j.1749-6632.1998.tb09596.x [DOI] [PubMed] [Google Scholar]
- Dyall SC (2015). Long-chain omega-3 fatty acids and the brain: A review of the independent and shared effects of EPA, DPA and DHA. Frontiers in Aging Neuroscience, 7, 52 10.3389/fnagi.2015.00052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eikelenboom P, & Stam FC (1982). Immunoglobulins and complement factors in senile plaques. An immunoperoxidase study. Acta Neuropathologica, 57(2–3), 239–242. 10.1007/BF00685397 [DOI] [PubMed] [Google Scholar]
- Emre C, Hjorth E, Bharani K, Carroll S, Granholm A, & Schultzberg M (2020). Receptors for proresolving mediators are increased in Alzheimer’s disease brain. Brain Pathology, 30 10.1111/bpa.12812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flesher RP, Herbert C, & Kumar RK (2014). Resolvin E1 promotes resolution of inflammation in a mouse model of an acute exacerbation of allergic asthma. Clinical Science (London, England), 126(11), 805–814. 10.1042/CS20130623 [DOI] [PubMed] [Google Scholar]
- Fortress AM, Hamlett ED, Vazey EM, Aston-Jones G, Cass WA, Boger HA, & Granholm AC (2015). Designer receptors enhance memory in a mouse model of Down syndrome. The Journal of Neuroscience, 35 (4), 1343–1353. 10.1523/JNEUROSCI.2658-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamlett ED, Boger HA, Ledreux A, Kelley CM, Mufson EJ, Falangola MF, … Granholm AC (2016). Cognitive impairment, neuroimaging, and Alzheimer neuropathology in mouse models of Down syndrome. Current Alzheimer Research, 13(1), 35–52. 10.2174/1567205012666150921095505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamlett ED, Ledreux A, Potter H, Chial HJ, Patterson D, Espinosa JM, … Granholm AC (2018). Exosomal biomarkers in Down syndrome and Alzheimer’s disease. Free Radical Biology & Medicine, 114, 110–121. 10.1016/j.freeradbiomed.2017.08.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser S, Adelmant G, Sarraf P, Wright HM, Mueller E, & Spiegelman BM (2000). Degradation of the peroxisome proliferator-activated receptor gamma is linked to ligand-dependent activation. The Journal of Biological Chemistry, 275(24), 18527–18533. 10.1074/jbc.M001297200 [DOI] [PubMed] [Google Scholar]
- Head E, Lott IT, Wilcock DM, & Lemere CA (2016). Aging in Down syndrome and the development of Alzheimer’s disease neuropathology. Current Alzheimer Research, 13(1), 18–29. 10.2174/1567205012666151020114607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, Reyes-Irisarri E, Hull M, & Kummer MP (2011). Impact and therapeutic potential of PPARs in Alzheimer’s disease. Current Neuropharmacology, 9(4), 643–650. 10.2174/157015911798376325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter CL, Bachman D, & Granholm AC (2004). Minocycline prevents cholinergic loss in a mouse model of Down’s syndrome. Annals of Neurology, 56(5), 675–688. 10.1002/ana.20250 [DOI] [PubMed] [Google Scholar]
- Hunter CL, Quintero EM, Gilstrap L, Bhat NR, & Granholm AC (2004). Minocycline protects basal forebrain cholinergic neurons from mu p75-saporin immunotoxic lesioning. The European Journal of Neuroscience, 19(12), 3305–3316. 10.1111/j.0953-816X.2004.03439.x [DOI] [PubMed] [Google Scholar]
- Iulita MF, Ower A, Barone C, Pentz R, Gubert P, Romano C, … Cuello AC (2016). An inflammatory and trophic disconnect biomarker profile revealed in Down syndrome plasma: Relation to cognitive decline and longitudinal evaluation. Alzheimer’s & Dementia, 12 (11), 1132–1148. 10.1016/j.jalz.2016.05.001 [DOI] [PubMed] [Google Scholar]
- Iyer AM, van Scheppingen J, Milenkovic I, Anink JJ, Adle-Biassette H, Kovacs GG, & Aronica E (2014). mTOR hyper-activation in Down syndrome hippocampus appears early during development. Journal of Neuropathology and Experimental Neurology, 73(7), 671–683. 10.1097/NEN.0000000000000083 [DOI] [PubMed] [Google Scholar]
- Kantarci A, Aytan N, Palaska I, Stephens D, Crabtree L, Benincasa C, … Dedeoglu A (2018). Combined administration of resolvin E1 and lipoxin A4 resolves inflammation in a murine model of Alzheimer’s disease. Experimental Neurology, 300, 111–120. 10.1016/j.expneurol.2017.11.005 [DOI] [PubMed] [Google Scholar]
- Keyes KT, Y Y, Lin Y, Zhang C, Perez-Polo JR, Gjorstrup P, & Birnbaum Y (2010). Resolvin E1 protects the rat heart against reperfusion injury. American Journal of Physiology - Heart and Circulatory Physiology, 299(1), H153–H164D. 10.1152/ajpheart.01057.2009 [DOI] [PubMed] [Google Scholar]
- Kinney JW, Bemiller SM, Murtishaw AS, Leisgang AM, Salazar AM, & Lamb BT (2018). Inflammation as a central mechanism in Alzheimer’s disease. Alzheimer’s & Dementia, 4, 575–590. 10.1016/j.trci.2018.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirouac L, Rajic AJ, Cribbs DH, & Padmanabhan J (2017). Activation of Ras-ERK signaling and GSK-3 by amyloid precursor protein and amyloid beta facilitates neurodegeneration in Alzheimer’s disease. eNeuro, 4(2), ENEURO.0149–16.2017. 10.1523/ENEURO.0149-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Han SH, Park MH, Baek B, Song IS, Choi MK, … Jin HK (2018). Neuronal SphK1 acetylates COX2 and contributes to pathogenesis in a model of Alzheimer’s disease. Nature Communications, 9(1), 1479 10.1038/s41467-018-03674-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legland D, Arganda-Carreras I, & Andrey P (2016). MorphoLibJ: Integrated library and plugins for mathematical morphology with ImageJ. Bioinformatics, 32(22), 3532–3534. [DOI] [PubMed] [Google Scholar]
- Lockrow J, Boger H, Bimonte-Nelson H, & Granholm AC (2011). Effects of long-term memantine on memory and neuropathology in Ts65Dn mice, a model for Down syndrome. Behavioural Brain Research, 221(2), 610–622. 10.1016/j.bbr.2010.03.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockrow J, Boger H, Gerhardt G, Aston-Jones G, Bachman D, & Granholm AC (2011). A noradrenergic lesion exacerbates neurodegeneration in a Down syndrome mouse model. Journal of Alzheimer’s Disease, 23(3), 471–489. 10.3233/JAD-2010-101218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockrow J, Fortress A, & Granholm AC (2012). Age-related neurodegeneration and memory loss in Down syndrome. Current Gerontology and Geriatrics Research, 2012, 463909 10.1155/2012/463909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockrow J, Prakasam A, Huang P, Bimonte-Nelson H, Sambamurti K, & Granholm AC (2009). Cholinergic degeneration and memory loss delayed by vitamin E in a Down syndrome mouse model. Experimental Neurology, 216(2), 278–289. 10.1016/j.expneurol.2008.11.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukiw WJ, Cui JG, Marcheselli VL, Bodker M, Botkjaer A, Gotlinger K, … Bazan NG (2005). A role for docosa hexaenoic acid-derived neuroprotectin D1 in neural cell survival and Alzheimer disease. Journal of Clinical Investigation, 115(10), 2774–2783. 10.1172/JCI25420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muralikumar S, Vetrivel U, Narayanasamy A, & U ND (2017). Probing the intermolecular interactions of PPARgamma-LBD with polyunsaturated fatty acids and their anti-inflammatory metabolites to infer most potential binding moieties. Lipids in Health and Disease, 16(1), 17 10.1186/s12944-016-0404-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perluigi M, Pupo G, Tramutola A, Cini C, Coccia R, Barone E, … Di Domenico F (2014). Neuropathological role of PI3K/Akt/mTOR axis in Down syndrome brain. Biochimica et Biophysica Acta, 1842(7), 1144–1153. 10.1016/j.bbadis.2014.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prieto P, Cuenca J, Traves PG, Fernandez-Velasco M, Martin-Sanz P, & Bosca L (2010). Lipoxin A4 impairment of apoptotic signaling in macrophages: Implication of the PI3K/Akt and the ERK/Nrf-2 defense pathways. Cell Death and Differentiation, 17(7), 1179–1188. 10.1038/cdd.2009.220 [DOI] [PubMed] [Google Scholar]
- PubChem. National Center for Biotechnology Information. Compound Database. CID=10473088. https://pubchem.ncbi.nlm.nih.gov/compound/10473088, 2018. [Google Scholar]
- Reeves RH, Irving NG, Moran TH, Wohn A, Kitt C, Sisodia SS, … Davisson MT (1995). A mouse model for Down syndrome exhibits learning and behaviour deficits. Nature Genetics, 11(2), 177–184. 10.1038/ng1095-177 [DOI] [PubMed] [Google Scholar]
- Rey C, Nadjar A, Buaud B, Vaysse C, Aubert A, Pallet V, … Joffre C (2016). Resolvin D1 and E1 promote resolution of inflammation in microglial cells in vitro. Brain, Behavior, and Immunity, 55, 249–259. 10.1016/j.bbi.2015.12.013 [DOI] [PubMed] [Google Scholar]
- Roberson R, Kuddo T, Horowitz K, Caballero M, & Spong CY (2012). Cytokine and chemokine alterations in Down syndrome. American Journal of Perinatology, 29(9), 705–708. 10.1055/s-0032-1314892 [DOI] [PubMed] [Google Scholar]
- Rueda N, Florez J, & Martinez-Cue C (2012). Mouse models of Down syndrome as a tool to unravel the causes of mental disabilities. Neural Plasticity, 2012, 584071 10.1155/2012/584071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan CN, Chiang N, & Van Dyke TE (2008). Resolving inflammation: Dual anti-inflammatory and proresolution lipid mediators. Nature Reviews. Immunology, 8(5), 349–361. 10.1038/nri2294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tramutola A, Lanzillotta C, Arena A, Barone E, Perluigi M, & Di Domenico F (2016). Increased mammalian target of rapamycin signaling contributes to the accumulation of protein oxidative damage in a mouse model of Down syndrome. Neurodegenerative Diseases, 16 (1–2), 62–68. 10.1159/000441419 [DOI] [PubMed] [Google Scholar]
- Tyagi S, Gupta P, Saini AS, Kaushal C, & Sharma S (2011). The peroxisome proliferator-activated receptor: A family of nuclear receptors role in various diseases. Journal of Advanced Pharmaceutical Technology & Research, 2(4), 236–240. 10.4103/2231-4040.90879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhu M, Hjorth E, Cortes-Toro V, Eyjolfsdottir H, Graff C, … Schultzberg M (2015). Resolution of inflammation is altered in Alzheimer’s disease. Alzheimer’s & Dementia, 11(1), 40–50 e41–42. 10.1016/j.jalz.2013.12.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierzba-Bobrowicz T, Lewandowska E, Schmidt-Sidor B, & Gwiazda E (1999). The comparison of microglia maturation in CNS of normal human fetuses and fetuses with Down’s syndrome. Folia Neuropathologica, 37(4), 227–234. [PubMed] [Google Scholar]
- Wilcock DM (2012). Neuroinflammation in the aging Down syndrome brain; lessons from Alzheimer’s disease. Current Gerontology and Geriatrics Research, 2012, 170276 10.1155/2012/170276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock DM, Hurban J, Helman AM, Sudduth TL, McCarty KL, Beckett TL, … Head E (2015). Down syndrome individuals with Alzheimer’s disease have a distinct neuroinflammatory phenotype compared to sporadic Alzheimer’s disease. Neurobiology of Aging, 36(9), 2468–2474. 10.1016/j.neurobiolaging.2015.05.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson CJ, Finch CE, & Cohen HJ (2002). Cytokines and cognition—The case for a head-to-toe inflammatory paradigm. Journal of the American Geriatrics Society, 50(12), 2041–2056. 10.1046/j.1532-5415.2002.50619.x [DOI] [PubMed] [Google Scholar]
- Wu JS, Tsai HD, Cheung WM, Hsu CY, & Lin TN (2016). PPAR-gamma ameliorates neuronal apoptosis and ischemic brain injury via suppressing NF-kappaB-driven p22phox transcription. Molecular Neurobiology, 53(6), 3626–3645. 10.1007/s12035-015-9294-z [DOI] [PubMed] [Google Scholar]
- Zhao Y, Calon F, Julien C, Winkler JW, Petasis NA, Lukiw WJ, & Bazan NG (2011). Docosa hexaenoic acid-derived neuroprotectin D1 induces neuronal survival viasecretase- and PPARγ-mediated mechanisms in Alzheimer’s disease models. PLoS One, 6(1), e15816 10.1371/journal.pone.0015816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Fang Y, Xu Y, Lian Y, Xie N, Wu T, … Wang Z (2015). Curcumin improves amyloid beta-peptide (1–42) induced spatial memory deficits through BDNF-ERK signaling pathway. PLoS One, 10(6), e0131525 10.1371/journal.pone.0131525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, W Z, Hu H, Wang ML, Sheng WW, Yao HT, … Wei EQ (2006). Expression patterns of 5-lipoxygenase in human brain with traumatic injury and astrocytoma. Neuropathology, 26(2), 99–106. 10.1111/j.1440-1789.2006.00658.x [DOI] [PubMed] [Google Scholar]
- Zhang Y, Che M, Yuan J, Yu Y, Cao C, Qin XY, & Cheng Y (2017). Aberrations in circulating inflammatory cytokine levels in patients with Down syndrome: A meta-analysis. Oncotarget, 8(48), 84489–84496. 10.18632/oncotarget.21060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu M, Wang X, Hjorth E, Colas RA, Schroeder L, Granholm AC, … Schultzberg M (2016). Proresolving lipid mediators improve neuronal survival and increase Aβ phagocytosis. Molecular Neurobiology, 53, 2733–2749. 10.1007/s12035-015-9544-0 [DOI] [PMC free article] [PubMed] [Google Scholar]