

Abstract
Gas-phase ion/ion chemistry was coupled to ion mobility/mass spectrometry analysis to correlate the structure of gaseous ubiquitin to its solution structures with selective covalent structural probes. Collision cross section (CCS) distributions were measured to ensure the ubiquitin ions were not unfolded when they were introduced to the gas phase. Aqueous solutions stabilizing the native state of ubiquitin yielded folded ubiquitin structures with CCS values consistent with previously published literature. Denaturing solutions favored several families of unfolded conformations for most of the charge states evaluated. Gas-phase covalent labeling via ion/ion reactions was followed by collision induced dissociation of the intact, labeled protein to determine which residues were labeled. Ubiquitin 5+ and 6+ electrosprayed from aqueous conditions were covalently modified preferentially at the lysine 29 and arginine 54 positions, indicating that elements of three-dimensional structure were maintained in the gas phase. On the other hand, most ubiquitin ions produced in denaturing conditions were labeled at various other lysine residues, likely due to the availability of additional sites following methanol and low pHinduced unfolding. These data support the conservation of ubiquitin structural elements in the gas phase. The research presented here provides the basis for residue-specific characterization of biomolecules in the gas phase.
Keywords: Ion Mobility, native mass spectrometry, ion/ion reactions, covalent labeling
Graphical Abstract

Caption: A gas-phase ion/ion reaction covalent modification and ion mobility/mass spectrometry workflow for determining three-dimensional structural information.
Introduction
Characterization of protein structures is critical for understanding their function.1 The development of “soft” ionization mass spectrometry in proteomics led to assays capable of preserving non-covalent bonds as proteins transition from solution to the gas phase.2–3 Therefore, a branch of biological mass spectrometry referred to as ‘native mass spectrometry’ (native MS) has rapidly expanded, driven by the implicit hypothesis that specific interactions formed by biomolecules in solution can be maintained under carefully controlled conditions for MS analysis in the gas phase.4 Applications of native MS, ion mobility/mass spectrometry (IM/MS), and tandem MS (MS/MS) involve probing proteins to obtain information such as higher order subunit architecture, stoichiometry, shape, and sequence information.5–7
Ion/ion reaction chemistries have been exploited for analytical applications since the beginning of the adoption of electrospray ionization (ESI), using mass spectrometers as the gas-phase analog to the chemist’s wet bench.8–11 The increasing use and versatility of ion/ion reactions within the past half-decade has resulted from the development and commercial availability of novel instrumentation equipped to perform such experiments.12 Covalent labeling analyzed by mass spectrometry (CLMS) is an example of a reaction that has been transferred from solution13–15 to the gas phase.16–19 Covalent modification by gas-phase ion/ion reactions relies on long-lived complex formation between oppositely charged protein and reagent. In addition to containing an electrostatically ‘sticky’ group (e.g., sulfonate or phosphate), reagents for covalent modification require a reactive site that will undergo chemical reactions with the analyte ion. Several examples of nucleophilic addition, utilizing electrophilic reagents such as reactive esters, have been successfully applied.20 Solution CLMS provides insight about protein conformations,21 dynamics, and amino acid residue reactivity and microenvironment.22 CLMS, conducted in a tandem mass spectrometer through ion/ion reactions, has the advantages of independent control/optimization of reactant species, well-defined reaction conditions, reagent purification through mass-to-charge isolation, and tandem MS capabilities in conjunction with ion/ion reactions.12 Hence, ion/ion covalent labeling coupled to IM-MS/MS can, in principle, provide for the three-dimensional characterization of gaseous protein ions.23–24
Though most CLMS approaches have relied on ‘bottom-up’ proteomics, utilizing enzymatic digestion to enable the identification of modification sites, the ‘top-down’ approach in proteomics was developed in order to obtain primary structural information directly from the gas-phase dissociation of intact protein ions without the need for extensive separations or digestion prior to MS/MS analysis.25 During a typical ‘top-down’ experiment, protein identification is made by analyzing the sequence fragments of intact proteins from tandem MS, which allows for the examination of the entire amino acid sequence, thereby characterizing intact proteins and identifying the number and type of post-translational and other modifications in various so-called proteoforms.26
Solvent-free, gaseous proteins can maintain their solution structures with careful control of experimental parameters.27–29 Pioneering studies from the laboratories of David Clemmer and Michael Bowers revealed that ubiquitin solution structures can be preserved as kinetically trapped intermediates in the gas phase after evaporative cooling associated with the electrospray process. Their data suggested minor structural changes occur during desolvation of low charge states ions (z ≅ 7) for native-like conformations, and unfolded gas-phase structure happens for higher charge states (z ≅ 13) caused by rapid unfolding (<10 ms).30 Additional studies evaluated the abundance of different conformations of ubiquitin in the gas phase as a function of methanol content in solution, where the native state was favored in aqueous solutions and more elongated states of ubiquitin were dominant in solutions of 20:80 water:methanol content.31 The importance in revealing the behavior and overall structure of native proteins in the gas phase is a consequence of the increasing number of MS-related techniques applied in the field of structural biology.32 Hence, it is essential to evaluate protein structures in vacuo after their transition from solution into the gas phase with tools of higher structural specificity than ion mobility alone.
In this study, we focus on the three-dimensional characterization of gaseous protein ions with CLMS performed completely inside the mass spectrometer. The structures of gaseous ubiquitin generated from both aqueous and denaturing conditions were evaluated using ion/ion chemistry, top-down tandem mass spectrometry, and ion mobility-derived collision cross section measurements. Covalent labeling reactions between ubiquitin and sulfo-benzoyl-1-hydroxy-7-azabenzotriazole ester (HOAt) were performed in the trap cell of a quadrupole IM-MS. The reaction results in the formation of amide bonds with primary amines and guanidine in the gas-phase. The protein ions are covalently modified by multiple additions of the reagent, separated by ion mobility, and fragmented with mass analysis of the fragmentation products. Mass shifts in the sequence fragments due to the covalent addition of the sulfo-benzoyl moiety allow for the identification of covalently labeled sites. The results demonstrate the power of combining collision cross section and covalent labeling approach to detect changes induced by solution conditions, with measurements conducted entirely in the gas phase.
Experimental
Materials.
Methanol, N,N-dimethyl formamide (DMF), and formic acid were purchased from Fisher Scientific (Fairmont, NJ). Ubiquitin from bovine erythrocytes, myoglobin from horse heart, cytochrome c from equine heart, and ammonium acetate were purchased from Sigma-Aldrich (St. Louis, MO). 1-Hydroxy-7-azabenzonitrazole (HOAt) was purchased from TCI America (Portland, OR). 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC) was purchased from Thermo Scientific (Rockford, IL). 3-Sulfobenzoic acid monosodium salt was purchased from Alfa Aesar (Ward Hill, MA).
Sample Preparation.
For the experiments performed in denaturing conditions, ubiquitin was dissolved in a 50/50/0.1 vol/vol solution of water/methanol/formic acid at 1 μM. For analysis using aqueous conditions, ubiquitin was dissolved in an aqueous 10 mM ammonium acetate solution at 1 μM. The reagent used for the ion/ion reactions, sulfobenzoyl-HOAt, was synthesized following a previously published procedure.33 The calibrant mix used for CCS calculations consisted of 1 μM ubiquitin, cytochrome C, and myoglobin in 50:50:0.1 (v/v) solution of water/methanol/formic acid.
Traveling Wave Ion Mobility Spectrometry – CCS Calibration.
Calibration of drift time measurements to known collision cross section values is necessary for traveling wave-type IM instruments that use time-varying electric fields within the drift region. Traveling-wave drift times were calibrated by measuring TWIMS profiles of a calibrant mix for each set of experiments following a previously published protocol.34–36 A calibration curve (Fig. S3) was obtained by plotting natural logarithm of the nitrogen CCS to charge ratios versus the calibrant ion drift times.36 The data was fit with a power function of the form given by Equation 1 where CCSN2 is the calibrant nitrogen CCS value, z is the charge state of the ion, and td is the drift time.
| Equation 1 |
Nitrogen TWIMS CCS values were determined from measured drift times according to Equation 2.
| Equation 2 |
The CCS values were reported as the average obtained from triplicate measurements in Table S1. The instrument settings used in CCS measurements and ion/ion reactions are summarized in Table S2. All the CCS calibration calculations and results were reported as recommended by recently introduced criteria.37
Mass Spectrometry and Ion/Ion Reactions.
Experiments were performed on a Synapt G2-Si High Definition Mass Spectrometer (Waters Corporation, Wilmslow, U.K.) furnished with electron transfer dissociation (ETD) and a NanoLockspray source. The instrumental arrangement for the ion/ion reactions performed has been previously described.38 Briefly, the source contains two nanoelectrospray (nESI) probes positioned normal to each other and the sampling cone. The nESI baffle was removed. Sequential anion (sulfobenzoyl-HOAt) and cation (ubiquitin) ionization was enabled by a WRENS (Waters Research Enabled Software) script coupled with ETD mode to synchronize ion injection with the polarity of the instrument optics and ETD refill times (1s each) for reagent and cation fills, respectively. Infusion flow rates were 500 nl/min or lower.
The control sequence consists of injecting ions through the stepwave region with m/z isolation in the quadrupole. Anions are trapped in the trap cell in the first step, followed by introduction of a specific analyte (cationic) charge state (again, m/z isolated by the quadrupole) into the trap. Next, reaction products are pulsed out of the trap, separated by their mobilities, and then traverse the transfer cell where the transfer collision energy is increased allowing for collision induced dissociation after the reaction products exit the mobility cell. Thus, ion/ion reactions products and their sequence fragments share identical drift times since fragments were not generated until after IM separation. Ions were mass analyzed by the time-of-flight mass spectrometer in Resolution Mode (nominal resolving power of 20,000 FWHM). Tandem mass spectra were internally calibrated against the monoisotopic mass of the y182+ fragment ion from ubiquitin (m/z 1049.0997).
Data Analysis.
Mobility-selected mass spectra were extracted with the instrument control software MassLynx V4. Extracted mass spectra were converted into .mgf (Mascot Generic Format) files and imported into Mash Explorer,39 where spectra were deconvoluted by the eThrash algorithm40 with a S/N threshold of 3, peak background ratio of 1, peptide minimum background ratio of 1, and minimum isotopic fit % of 80. The covalently modified and unmodified CID fragments obtained for all experiments were investigated against the ubiquitin primary sequence by applying custom PTMs equal to the mass of the covalent modification formed by the ion/ion reactions (i.e., 182.98 Da) at the N and C termini. Covalently modified peaks were annotated with a mass error tolerance of 20 ppm.41 The annotations were then manually confirmed.
Results and Discussion
Protein Mass Spectra.
The ions produced by nESI ionization of ubiquitin from both aqueous and denaturing conditions exhibit characteristic distributions (Fig. S1) when analyzed with the “softest” conditions that allowed enough ion transmission to collect mass and mobility spectra (Table S2). A profile of high m/z signals with lower charge states (i.e., 6 ≥ z ≥ 4) peaks for ubiquitin was observed for the sample sprayed from aqueous conditions. A distribution of higher charge state peaks (13 ≥ z ≥ 5) with considerably higher relative intensities was obtained using denaturing conditions. The charge state distributions suggest that ubiquitin ions electrosprayed under aqueous conditions have a compact solution structure, as supported by the literature.32, 42 The compact native state of ubiquitin has a limited number of amino acid residues accessible for protonation. On the other hand, the higher charge states exhibited for denaturing conditions are evidence of the disruption of the tertiary structure of ubiquitin.43–45 The observed transition in charge state distributions indicates that methanol induces structural transitions for ubiquitin.
Gas-Phase Ubiquitin Conformations in the Trap Cell from Native and Denaturing Conditions.
To compare ubiquitin conformations generated from different solution conditions, calibrated collision cross sections were measured for each of the charge states that was investigated by covalent labeling with both denaturing and aqueous conditions (Table S1). The experimental conditions applied for CCS calibration and ion/ion reactions were identical (with exception of the gas flows into the helium and mobility cells) and are summarized in Table S2. Ubiquitin conformers originating from aqueous and denaturing conditions were assessed by converting the peaks in the ion mobility arrival time distributions (ATDs) to CCS values, allowing for the characterization of ubiquitin populations that undergo ion/ion reaction chemistry. Thus, we are chiefly concerned with the ion populations present in the trap cell prior to the ion mobility separation, as these are the populations directly probed by the ion/ion reactions. Therefore, we minimized the trap and mobility voltages to prevent unintended activation. The %CV values for the calibrated CCS values measured on three different days were less than 2.5%. Figures 1A and 1B show the ATDs for ubiquitin 5+ and 6+ in aqueous and denaturing conditions. In solution, aqueous conditions of ubiquitin favor the N-state (native state) while the partially unfolded so-called A-state is dominant in solutions containing 40% methanol or more.46–48 Ions generated from aqueous conditions presented a narrow structural region with similar cross section values (TWCCSN2 – 1193 Å2 and 1233 Å2, for ubiquitin 5+ and 6+, respectively) corresponding to compact conformations.49 For aqueous ubiquitin 6+ a minor peak is present at ~1371 Å2, which is likely composed of partially folded states. Previous reports of the 6+ charge state generated from solutions of ubiquitin in aqueous ammonium acetate with ATDs measured by both drift tube and TWIMS instruments also display this feature.34, 50 The presence of these states is best explained by the increase in Coulombic repulsion from the additional proton bound to the 6+ charge state versus the 5+, as the 5+ charge state lacks this more extended feature.31 Similarly, the distribution for ubiquitin 5+ in denaturing conditions (Fig. 1) displays a distribution of compact ions (~1228 Å2) that extends into the region corresponding to partially folded ions (~1333 Å2). Ubiquitin 6+ in denaturing conditions gives a broad distribution (from ~1300 Å2 to 1900 Å2) that can be related to multiple stable, elongated forms. Although this distribution is broad, there are 2 features with maxima at ~1398 Å2 and ~1676 Å2, corresponding to a partially unfolded intermediate state and partially unfolded structure arising from the A state, respectively. Figure S2 presents the CCS distributions for all charge states of electrosprayed ubiquitin ions from aqueous and denaturing solutions. The distributions for ubiquitin 7+ and 8+ prepared in denaturing conditions are dominated by relatively sharper features at ~1834 Å2 and ~1906 Å2, respectively. Sharper features in protein ATDs indicate that the ion conformer population is collapsed into relatively few stable structures that exist over a narrow region of the available cross section space and appear as a result of protein unfolding.50
Figure 1.
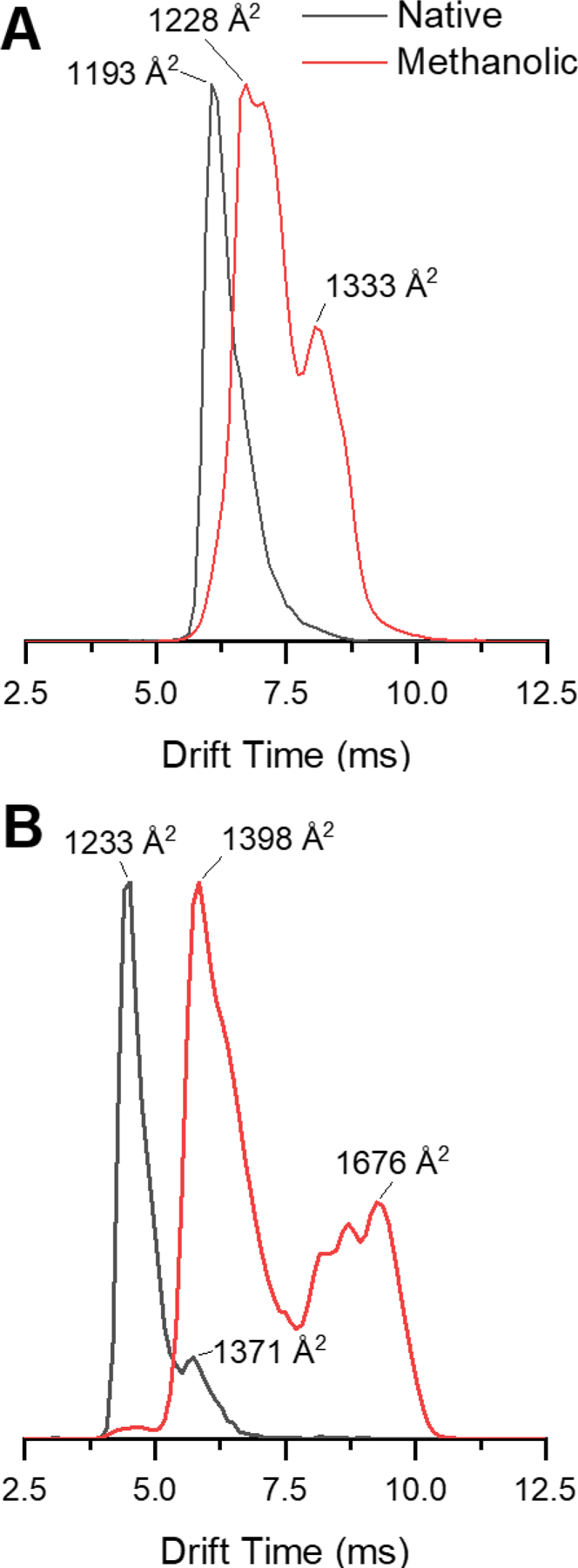
Intensity normalized arrival time distributions (ATDs) of ubiquitin 5+ (A) and 6+ (B) charge states sprayed from native (black trace) and denaturing (red trace) conditions.
Characterization of Gaseous Ubiquitin Structures with Ion/Ion Reactions.
Covalent modification of ubiquitin via ion/ion reaction in the gas phase.
Covalent bond formation occurs via ion/ion reactions by a three-step process: 1) Formation of a stable, long-lived electrostatically bound complex; 2) Activation of the complex; and 3) Dissociation of the leaving group from the complex. The first step is completed by trapping both reagent anions and protein cations in the trap cell. A minimal amplitude trap traveling wave (< 0.2 V) is used to promote better mixing and, in effect, increases the effective reaction time.51 The product is observed by a shift in m/z equal to a reduction in charge by the number of reagents electrostatically attached and an increase in mass equal to the molecular mass of the reagent. Next, the complex is activated. The pressures and voltages from the source and into the trap cell were kept identical to the conditions used for our CCS measurements to prevent gas-phase unfolding prior to the ion/ion reaction. Thus, the protein ions that were labeled structurally correlate with the observed arrival time distributions and CCS values. The transition state for a covalent reaction between a model amine and sulfobenzoyl-HOAt has been calculated to be 17.4 kcal/mol higher in energy than the electrostatic product.33 The sulfonate is expected to be electrostatically attached to a protonated arginine, lysine, or histidine residue. The proton transfer barrier for transfer from guanidinium to sulfonate was calculated to be 61 kcal/mol and for transfer from ammonium to sulfonate was calculated to be 28 kcal/mol higher in energy than the complex. Since collisional activation on a mass spectrometry timescale is kinetically controlled, enough collisional energy must be applied to form the covalent reaction transition state but not high enough to result in proton transfer without covalent bond formation or fragmentation of the protein.
Though the application of this energy may lead to coulombically-driven unfolding of the protein, the strong electrostatic “anchor” holds the reagent in place. The through-bond distance from the reactive carbonyl carbon to the sulfonate oxygens in the reagent is approximately 6.4 Å. Thus, the reactive side chain must be close by the charged anchoring residue (i.e., on the surface of the protein) and a reactive nucleophile. Therefore, though collision-induced unfolding or intramolecular proton transfer may occur during the activation of the complex, these processes are not expected to affect the ability of the ion/ion reaction to report on surface accessible regions of the protein that are nearby external, protonated side chains. The fact that the reagent to protonated side chain noncovalent bond is not fragmented under these conditions illustrates that the applied activation to form the covalent product is mild. The applied collisional energy will drive off the weakly-bound leaving group after the covalent product is formed. The covalent reaction is observed by a decrease in m/z equal to neutral loss of the leaving group.
Ion/ion reactions were used to probe the gas phase microenvironment and relative reactivity of lysine and arginine side chains in ubiquitin cations formed from the aqueous and denaturing solutions. Previously, histidine was found to only react with low energy activation applied over long time periods.33 These conditions cannot be accessed with the instrument used in this study as CID is performed in transmission mode (beam-type CID). Therefore, we do not expect to observe histidine modification. Ion/ion reactions were performed under similar ion optics voltage conditions as the CCS measurements from the source up to and including the trap cell (vide supra). The choice of the sulfobenzoyl-HOAt reagent (versus, e.g., sulfobenzoyl-N-hydroxysuccinimide) was based on its relatively low activation energy for covalent reactions in the gas phase, its simple and one-pot synthesis, and the ability of sulfo-benzoyl-HOAt to react with amino acids side chains such as arginine and lysine.33
Figure 2A displays the ion/ion reaction of ubiquitin 6+ electrosprayed from aqueous conditions and sulfobenzoyl-HOAt−. The amide bond formation between ubiquitin and 3-sulfobenzoate is characterized by the neutral loss of HOAt (Molecular mass = 135.1235 g/mol) from the ion/ion reaction product. The peak [M+6H+♦]5+ represents the electrostatic product formed between ubiquitin 6+ and the reagent, [M+5H+*]5+ is covalently modified ubiquitin, and the [M + 5H]5+ peak is the proton transfer product corresponding to the loss of the electrostatically attached reagent. In order to favor covalent product formation (as opposed to proton transfer) several parameters were optimized aiming to apply energy below the threshold for proton transfer product formation but above the transition state energy for covalent bond formation.52 With the helium cell and IM pressures used to measure CCS, the only observed product upon collisional activation was loss of the reagent from the ion/ion product complex. This is due to intentional rapid thermalization of ions by many low-energy collisions as they enter the mobility cell, preventing unintended activation of ions.53 However, rapid thermalization results in the need to use much higher voltages to achieve ion activation, with the consequence of not being able to access the neutral loss of HOAt channel, as the loss of the entire reagent is kinetically favorable. Previous work has shown that the transition state for loss of an electrostatically bound reagent is very loose compared the transition state for covalent reaction,54 restraining the appearance of the covalent reaction to activation energies below the threshold for loss of the entire reagent. Therefore, the gas flows into the helium and IM cells were set to 20 mL/min each (0.59 and 0.66 mbar pressures for each of the cells. respectively). This way, the injection energy into the mobility cell was able to be reduced (center of mass energy of 3.6 kcal/mol for 5+, Table S4) and fewer energizing collisions occur. The result is efficient formation of the -HOAt without a dominant channel for loss of the entire reagent. The tune parameters used during ion/ion reactions are presented in Table S2. The trap pressure was kept the same. In this way, the ratio of the covalently modified product to the proton transfer (reagent loss) peak was maximized to yield the mass spectrum in Figure 2A.38 The ATD in Figure 2B was obtained under these conditions and represents the ion mobility separation of different numbers of sequential ion/ion reactions between ubiquitin 6+ and sulfobenzoyl-HOAt−. The peak at 65 ms is related to the precursor ubiquitin 6+, the peak at ~78 ms corresponds to the attachment of one sulfobenzoyl-HOAt, and the peaks at ~96 and 120 ms correspond to attachment of two and three sulfobenzoyl-HOAt, respectively. Figure 2C displays the mass spectrum at extracted from drift time 72 – 83 ms resulting from CID of the ion/ion reaction covalent modified product. Figure 3 shows the mass spectra related to the peaks in the ATD which correspond to the ion/ion reactions products obtained for ubiquitin 7+ in denaturing conditions, with up to three covalent additions of sulfobenzoyl-HOAt reagents. Fragments from CID of the labeled protein ions were only investigated for addition of a single label to help prevent labelinduced structural changes from affecting our analysis.15
Figure 2.
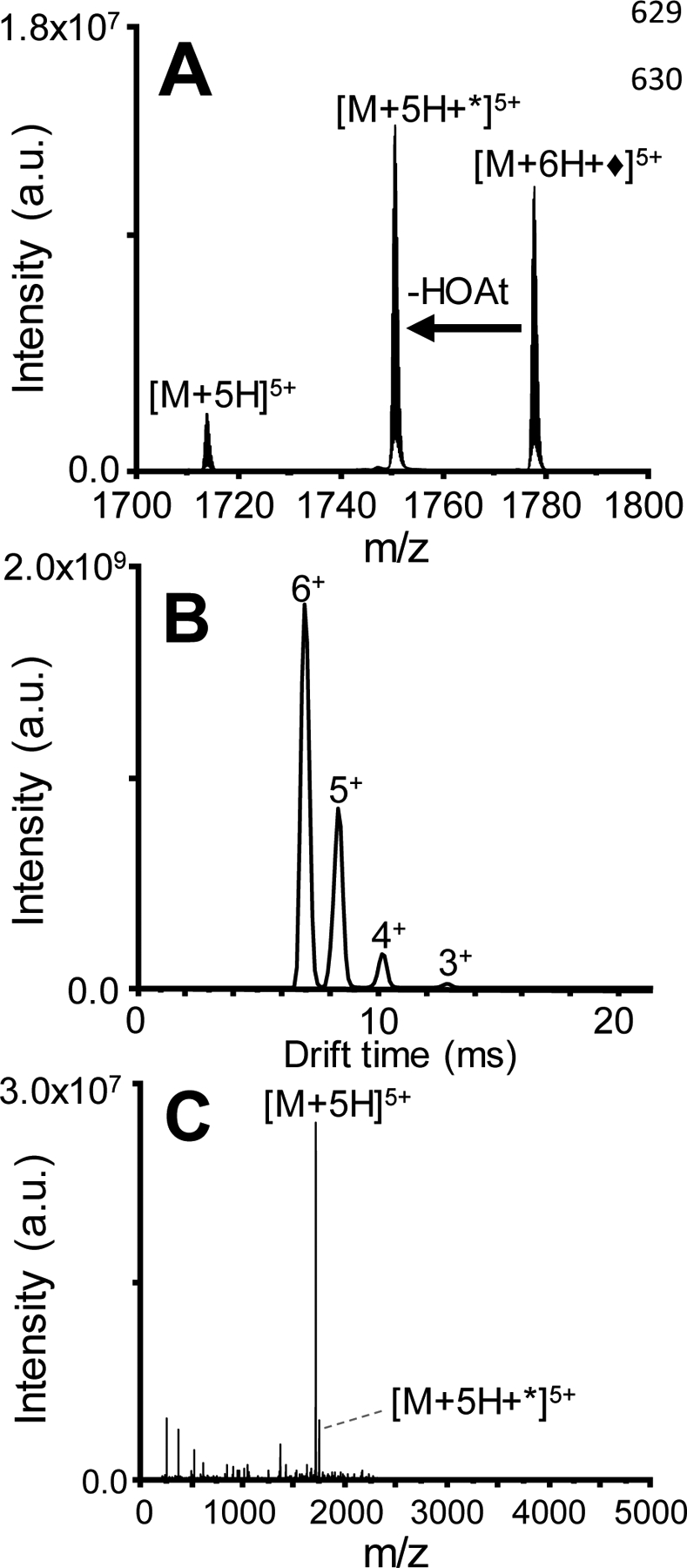
Covalent modification of [ubiquitin+6H]6+ ionized from native conditions with [sulfo-HOAt]−. (A) Product ion spectrum of the ion/ion reaction between [ubiquitin + 6H] 6+ and [sulfo-HOAt – H]− prior to activation. ♦ refers to electrostatic attachment of the reagent and * refers to covalent modification. (B) ATD of the full scan (mass range of 100 to 500 m/z) corresponding to ion/ion reactions between [ubiquitin + 6H]6+ and [sulfo-HOAt – H]-revealing the mobility separation of covalently modified products generated with different extents of modification. (C) Mass spectrum resulting from CID of the ion/ion reaction product (corresponding to 72 – 83 ms in the ATD).
Figure 3.
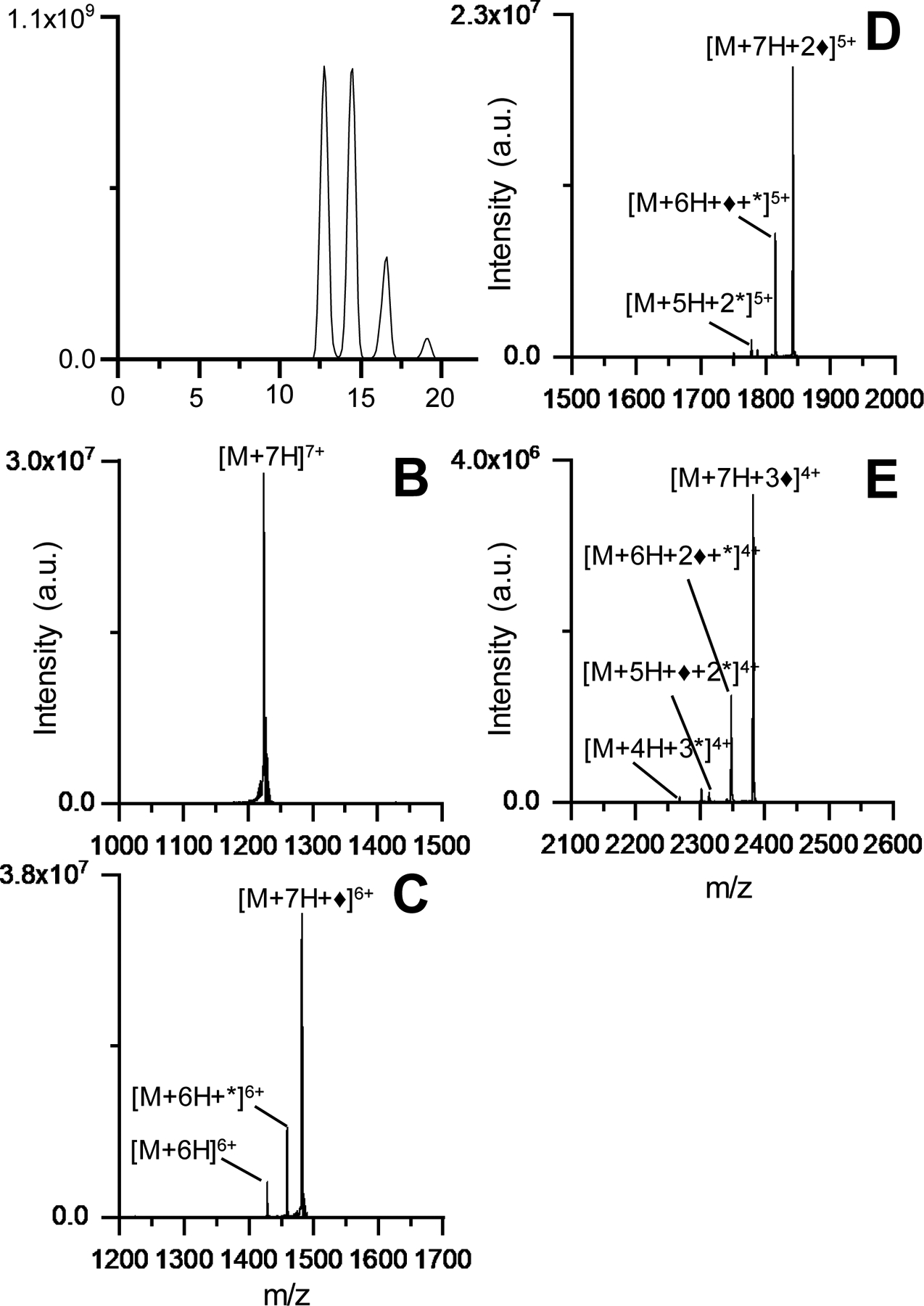
(A) Post-ion/ion reaction IM spectrum and mass spectra from zero (B), one (C), two (D), and three (E) anion attachments. ♦ refers to electrostatic attachment of the reagent and * refers to covalent modification.
The charge states 5+ and 6+ ionized from aqueous conditions and 5+, 6+, 7+, and 8+ all displayed a neutral loss of m/z 136 (the mass of the leaving group, HOAT) following ion/ion reactions with sulfobenzoyl-HOAt. However, 7+ and 8+ from aqueous conditions and 9+ from denaturing conditions did not show neutral loss of HOAt. The only products were the electrostatic addition of sulfobenzoyl-HOAt and loss of the entire reagent. This observation is attributed to the lack of unprotonated lysine or arginine residues available on the exterior of the protein with 7+ and 8+ ionized from aqueous conditions and 9+ ionized from denaturing conditions. The difference in reactivity between the 7+ and 8+ charge states ionized from aqueous solution and 7+ and 8+ from denaturing solution indicate that their protonation sites and gas-phase structures are likely different. The injection energy was controlled to prevent fragmentation of the protein backbone. No fragments other than the loss of HOAt or the entire reagent were observed without adding collisional energy in the transfer cell.
Comparison and characterization of the ubiquitin ion structures obtained from aqueous and denaturing solutions.
CID was performed upon injection into the transfer cell to form covalent modification sequence fragments originating from different charge states of ubiquitin in both aqueous (ubiquitin 5+ and 6+) and denaturing (ubiquitin 5+ to 8+) conditions. Table S3 summarizes the collision energy voltages applied to the transfer cell for each CID experiment. The covalent product ions generated b (N-terminal) and y (C-terminal) fragment ions that matched drift times of their precursors. Figure 2C shows the fragment mass spectrum resulting from CID of the covalent product [M+5H+*]5+ that was used to determine the sites of covalent modification. The fragment ion annotations from the solution condition and charge state-dependent ion/ion gas-phase covalent modification of ubiquitin are shown in Tables 1 and 2.
Table 1.
Sequence Ladder for Aqueous Ubiquitin in different charge states displaying the covalently modified fragmentation sites and the modified residues.

|
Table 2.
Sequence Ladder for Denatured Ubiquitin in different charge states displaying the covalently modified fragmentation sites and the modified residues.

|
For ubiquitin 5+ and 6+ electrosprayed from aqueous conditions the modified fragment ions generated suggested covalent modifications to lysine 29 (modified b29) and arginine 54 (modified y24) which is in agreement with previously published work.38 The residues available for covalent modification must be accessible to the reagent – which excludes side chains buried in the interior of the protein – and reactive towards the reagent, precluding protonated and non-nucleophilic sites. Modification sites were annotated based on the smallest terminal (b- or y-ion) fragment that has a m/z shift corresponding to covalent addition. The process of assigning labeled sites is as follows: b- and y-ions that matched the m/z of sequence fragments plus the mass of the covalent label were annotated as covalently labeled fragments and manually validated. Next, the mass spectra were manually compared against spectra resulting from CID of unmodified ubiquitin at the same charge. Fragments that were originally annotated as covalently labeled that matched the m/z and isotopic distribution of fragments resulting from CID of unmodified ubiquitin were thrown out and considered false positives. Side chains were assigned as covalently labeled only if there was no evidence for covalent labeling of amino acid residues N-terminal (for b-ions) or C-terminal (for y-ions) to the assigned site (i.e., no labeled sequence fragments that include these residues). For example, Table 1 shows that the smallest labeled b-ion was modified b29, but unmodified fragments are observed for b27 and b28, ions that include the N-terminus, K6, K11, and K27, but not K29. Therefore, there is no evidence for labeling of any of these amino acids, but the observation of b-ions matching the mass of the addition of the covalent label that include K29 suggests that K29 is the labeled side chain. These results correlate to the crystal structure of ubiquitin (PDB 1UBQ)55 where the suggested modified residues are exposed and accessible to the reagent (Figure 4). Recently, results from 193 nm ultraviolet photodissociation (UVPD) were used to determine the protonation sites for different native charge states of ubiquitin in the gas phase.56 The possible protonation sites for the 5+ and 6+ charge state were determined to be Q2, P19, K33, R42, K48, K63, and R74. For both charge states, K29 and R54 are not protonated, rendering them reactive to sulfobenzoyl-HOAt. The solvent-accessible surface area (SASA) was calculated from the crystal structure with a probe size of 1.4 Å (i.e., the van der Waals radius of water) with the GETAREA program.57 Side chains with a SASA ratio above 30% were considered solvent accessible.58 Including the accessible arginine and lysine side chains from the SASA calculation and excluding the UVPD-determined protonated side chains limits the remaining available sites for labeling by sulfobenzoyl-HOAt to K6, K11, K29, R54, and R72, although K11 (and K27) participates in a salt bridge and thus may not be labeled if these salt bridges are not disrupted under our labeling conditions.59 The observed labeling of K29 and R54 (Fig. 4) suggests that ubiquitin structures electrosprayed from aqueous conditions retain elements of solution structure, as predicted by molecular dynamics60 and the structure relaxation approximation60–61. K27 is not labeled, although it is only two residues away from K29, and is also not protonated. This may be evidence that elements of solution structure can be maintained, as K27 and K29 are in an alpha helix. Although the side chain of K27 faces the interior of the protein, the alpha helix positions K29 to be oriented outwards.55 Another interpretation of these results could suggest that the label is electrostatically bound to a side chain that is greater than 6.4 Å from the primary amine of the K27 side chain. Nonetheless, the labeling of K29 and K27 is not random (it occurs repeatably for both 5+ and 6+ charge states electrosprayed from aqueous solution) and does correlate with the region of the protein including K29 being accessible. The combination of CCS data, mass spectra, identified covalently modified residues, and modeling for native ubiquitin 5+ and 6+ suggests that ubiquitin structures remain compact in the gas phase when electrosprayed from aqueous conditions.60
Figure 4.
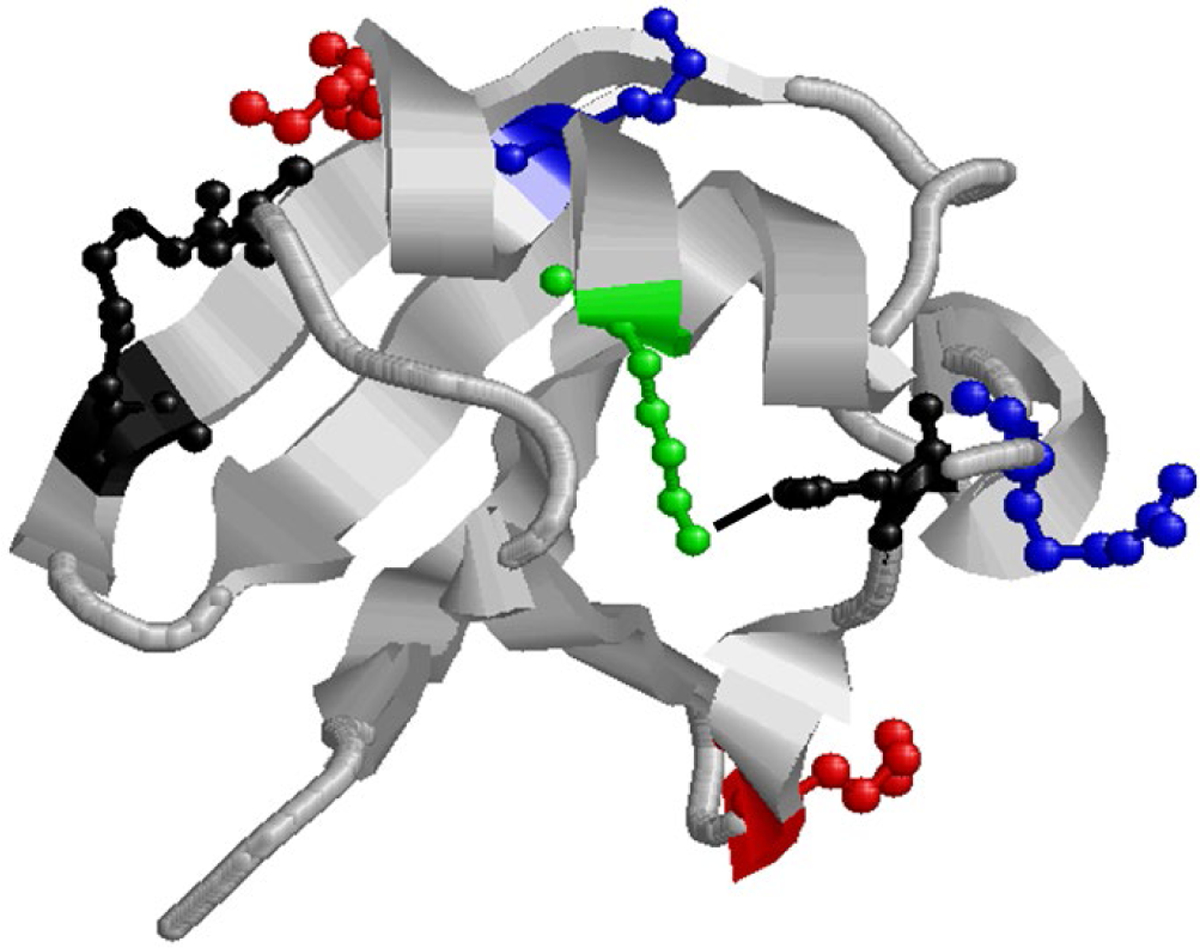
X-ray structure of ubiquitin (1ubq). The blue residues (K29, R54) are labeled under native conditions and the red (K33, K48) and green residue (K27) are labeled only under denaturing conditions. The red residues are protonated under native conditions and the green residue is buried and participates in a salt bridge with D52 (black). K11 is black as it participates in a salt bridge but in not labeled under any conditions. The black line between K27 and D52 represents the salt bridge.
Ubiquitin has been shown to undergo an alcohol-induced transition to a partially folded state (A state). For the A state, NMR experiments performed in a 40:60 water:methanol solution suggested that it retains a majority of its native secondary structural elements in the N-terminal half, whereas the structure of the C-terminal half unfolds to a highly helical more elongated state.31, 62–64 For the 5+ ion sprayed from a denaturing solution, our ion/ion reaction results show that K29 and R54 are labeled (Table 2), the same results as determined for the 5+ ions from aqueous conditions, consistent with CCS distribution being very similar between the 5+ sprayed from denaturing conditions and the 5+ and 6+ sprayed from native conditions. The ion/ion covalent labeling also illustrates that the peak around 1400 Å in the aqueous 6+ and denaturing 5+ likely reflects compact structures, since the labeled sites are identical for native 5+/6+ and denaturing 5+. This is consistent with molecular dynamics data that show reversible unfolding and folding for ubiquitin 6+ ions generated from native conditions for 1 μs in the gas phase.60 Additionally, the 6+ and 7+ charge state fragments include modified y24, also indicating that R54 was labeled. The labeling of R54 under various conditions indicates that for charge states 5+−7+, R54 is unprotonated, accessible, and sufficiently reactive under all these conditions.
However, the 6+, 7+, and 8+ charge states of ubiquitin sprayed from denaturing solution were all labeled at different lysine residues, with no evidence for labeling at the K29 residue. As previously illustrated, these ions all produced ATDs showing more extended conformations. This suggests that K29 is no longer the most reactive accessible lysine side chain for these charge states. The 6+ fragmentation data shows that K48 is likely labeled (modified b52), the 7+ fragmentation data shows labeling likely occurs on K33 (modified b36), and the 8+ data may provide evidence for the labeling of K27, though the lack of labeled b-ions for the 8+ charge state gives some ambiguity to this assignment. The reduced number of labeled sequence fragments for the 8+ ions is likely a consequence of most of the reactive residues in ubiquitin being protonated, diminishing the overall reactivity and the number of available sites for labeling. The labeling of 6+ at K48 and 7+ at K33 is likely due to changes in preferred protonation sites following the unfolding of the protein, as are K33 and K48 can both be protonated when sprayed from aqueous conditions. NMR measurements have demonstrated that a characteristic of the A-state is that the solution salt bridge between K27 and D52, which stabilizes the fold of the protein and buries K27 in the interior of the protein, is disrupted.63–64 Therefore, our results for 6+ and 7+ ionized from denaturing conditions correlate with at least partially disrupted solution states. Covalent labeling by ion/ion reactions is expected to be a powerful tool for protein structural analysis.
Conclusions
Ubiquitin ions electrosprayed from aqueous and denaturing solutions have been analyzed by IM-MS/MS and covalent structural probes delivered by ion/ion reactions inside of the mass spectrometer. Ubiquitin conformational populations were evaluated prior to performing ion/ion reactions by IM-MS, ensuring that energy imparted on the ions between the source and trap cell did not lead to collision induced unfolding. Examination of the conformation types as function of the solution conditions and charge states allowed for solution structures to be correlated to gas-phase measurements, suggesting the preservation of solution-like structures in the gas phase. Ions generated from aqueous solution had CCS values corresponding to compact conformations while ubiquitin 6+ also exhibited a minor peak at ~1371 Å2, which has been attributed to partially folded states due to the increase in Coulombic repulsion over the 5+ charge state. On the other hand, arrival time distributions for ubiquitin in denaturing conditions presented much higher CCS values which have been previously correlated to multiple elongated stable conformations.44–45, 65
The covalent modification data revealed distinct characteristics for ions originating from either aqueous or denaturing conditions. For aqueous conditions, the modified fragment ions suggested covalent modifications to lysine 29 (modified b32) and arginine 54 (modified y24) It is possible that elements of secondary structure as well as tertiary structure are conserved explained by the covalent modification of K29 instead of the buried and salt-bridged K27.51–52 These results correlate to the crystal structure of ubiquitin (PDB 1UBQ)55, molecular dynamics results57, and UVPD data,48 where the modified residues are exposed and accessible to the reagent. Ion/ion reaction results for ubiquitin 5+ sprayed from denaturing solutions also reveal the labeling of K29 and R54, agreeing with the CCS data, and suggesting that aqueous 6+ and denaturing 5+ are structurally very similar. Therefore, the denaturing 5+ ion is produced from the remaining compact ubiquitin population in denaturing solutions. The 6+, 7+, and 8+ charge states of ubiquitin sprayed from denaturing solutions were labeled at various lysines, accessible most likely due to the changes in possible protonation sites as a result disruption of the salt bridge between K27 and D52 after methanol-induced unfolding.55–56 Overall, the analysis of protein structures by covalent modification in the gas phase analyzed by IM-MS/MS suggests that the gas phase is a suitable environment for probing protein structure if care is taken to ensure gentle ion introduction.
Supplementary Material
Acknowledgement
Portions of this work were funded by the National Institutes of Health (NIH) NIGMS Grant R21 GM134408 (I.K.W.). The authors would like to acknowledge Lindsay Morrison and Jeffery Brown of Waters Corporation for helpful discussions.
References
- 1.Ishima R; Torchia DA, Protein dynamics from NMR. Nat Struct Biol 2000, 7 (9), 740–3. [DOI] [PubMed] [Google Scholar]
- 2.Loo JA; Loo RRO; Udseth HR; Edmonds CG; Smith RD, Solvent-Induced Conformational-Changes of Polypeptides Probed by Electrospray-Ionization Mass-Spectrometry. Rapid Commun Mass Sp 1991, 5 (3), 101–105. [DOI] [PubMed] [Google Scholar]
- 3.Chowdhury SK; Katta V; Chait BT, An electrospray-ionization mass spectrometer with new features. Rapid Commun Mass Spectrom 1990, 4 (3), 81–7. [DOI] [PubMed] [Google Scholar]
- 4.Ishii K; Zhou M; Uchiyama S, Native mass spectrometry for understanding dynamic protein complex. Biochim Biophys Acta Gen Subj 2018, 1862 (2), 275–286. [DOI] [PubMed] [Google Scholar]
- 5.Lanucara F; Holman SW; Gray CJ; Eyers CE, The power of ion mobility-mass spectrometry for structural characterization and the study of conformational dynamics. Nat Chem 2014, 6 (4), 281–294. [DOI] [PubMed] [Google Scholar]
- 6.Resing KA; Ahn NG, Proteomics strategies for protein identification. FEBS Letters 2005, 579 (4), 885–889. [DOI] [PubMed] [Google Scholar]
- 7.Konijnenberg A; Butterer A; Sobott F, Native ion mobility-mass spectrometry and related methods in structural biology. Biochim. Biophys. Acta 2013, 1834 (6), 1239–1256. [DOI] [PubMed] [Google Scholar]
- 8.Loo RRO; Udseth HR; Smith RD, Evidence of charge inversion in the reaction of singly charged anions with multiply charged macroions. The Journal of Physical Chemistry 1991, 95 (17), 6412–6415. [Google Scholar]
- 9.Ogorzalek Loo RR; Udseth HR; Smith RD, A new approach for the study of gas-phase ion-ion reactions using electrospray ionization. J Am Soc Mass Spectrom 1992, 3 (7), 695–705. [DOI] [PubMed] [Google Scholar]
- 10.Herron WJ; Goeringer DE; McLuckey SA, Product Ion Charge State Determination via Ion/Ion Proton Transfer Reactions. Anal Chem 1996, 68 (2), 257–262. [DOI] [PubMed] [Google Scholar]
- 11.Stephenson JL; McLuckey SA, Simplification of Product Ion Spectra Derived from Multiply Charged Parent Ions via Ion/Ion Chemistry. Anal Chem 1998, 70 (17), 3533–3544. [DOI] [PubMed] [Google Scholar]
- 12.Foreman DJ; McLuckey SA, Recent Developments in Gas-Phase Ion/Ion Reactions for Analytical Mass Spectrometry. Anal Chem 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oetjen J; Rexroth S; Reinhold-Hurek B, Mass spectrometric characterization of the covalent modification of the nitrogenase Fe-protein in Azoarcus sp. BH72. The FEBS Journal 2009, 276 (13), 3618–3627. [DOI] [PubMed] [Google Scholar]
- 14.Guan J-Q; Chance MR, Structural proteomics of macromolecular assemblies using oxidative footprinting and mass spectrometry. Trends in Biochemical Sciences 2005, 30 (10), 583–592. [DOI] [PubMed] [Google Scholar]
- 15.Mendoza VL; Vachet RW, Probing protein structure by amino acid-specific covalent labeling and mass spectrometry. Mass Spectrom. Rev 2009, 28 (5), 785–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peng Z; McLuckey SA, C-terminal peptide extension via gas-phase ion/ion reactions. International Journal of Mass Spectrometry 2015, 391, 17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McGee WM; McLuckey SA, Efficient and directed peptide bond formation in the gas phase via ion/ion reactions. Proceedings of the National Academy of Sciences 2014, 111 (4), 1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Prentice BM; McGee WM; Stutzman JR; McLuckey SA, Strategies for the gas phase modification of cationized arginine via ion/ion reactions. International Journal of Mass Spectrometry 2013, 354–355, 211–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mentinova M; McLuckey SA, Covalent Modification of Gaseous Peptide Ions with N-Hydroxysuccinimide Ester Reagent Ions. J Am Chem Soc 2010, 132 (51), 18248–18257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Prentice BM; McLuckey SA, Gas-phase ion/ion reactions of peptides and proteins: acid/base, redox, and covalent chemistries. Chem. Commun 2013, 49 (10), 947–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Loo JA; Loo RRO; Goodlett DR; Smith RD; Fuciarelli AF; Springer DL; Thrall BD; Edmonds CG, Elucidation of Covalent Modifications and Noncovalent Associations in Proteins by Electrospray Ionization Mass Spectrometry In Techniques in Protein Chemistry IV, Angeletti RH, Ed. Academic Press: 1993; pp 23–31. [Google Scholar]
- 22.Limpikirati P; Pan X; Vachet RW, Covalent Labeling with Diethylpyrocarbonate: Sensitive to the Residue Microenvironment, Providing Improved Analysis of Protein Higher Order Structure by Mass Spectrometry. Anal. Chem 2019, 91 (13), 8516–8523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Webb IK; Mentinova M; McGee WM; McLuckey SA, Gas-phase intramolecular protein crosslinking via ion/ion reactions: ubiquitin and a homobifunctional sulfo-NHS ester. J. Am. Soc. Mass. Spectrom 2013, 24 (5), 733–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pitts-McCoy AM; Harrilal CP; McLuckey SA, Gas-Phase Ion/Ion Chemistry as a Probe for the Presence of Carboxylate Groups in Polypeptide Cations. J. Am. Soc. Mass. Spectrom 2019, 30 (2), 329–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reid GE; McLuckey SA, ‘Top down’ protein characterization via tandem mass spectrometry. J Mass Spectrom 2002, 37 (7), 663–75. [DOI] [PubMed] [Google Scholar]
- 26.Smith LM; Kelleher NL; Linial M; Goodlett D; Langridge-Smith P; Ah Goo Y; Safford G; Bonilla L; Kruppa G; Zubarev R; Rontree J; Chamot-Rooke J; Garavelli J; Heck A; Loo J; Penque D; Hornshaw M; Hendrickson C; Pasa-Tolic L; Borchers C; Chan D; Young N; Agar J; Masselon C; Gross M; McLafferty F; Tsybin Y; Ge Y; Sanders I; Langridge J; Whitelegge J; Marshall A; The Consortium for Top Down, P., Proteoform: a single term describing protein complexity. Nat. Methods 2013, 10 (3), 186–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ruotolo BT; Giles K; Campuzano I; Sandercock AM; Bateman RH; Robinson CV, Evidence for Macromolecular Protein Rings in the Absence of Bulk Water. Science 2005, 310 (5754), 1658. [DOI] [PubMed] [Google Scholar]
- 28.Skinner OS; McLafferty FW; Breuker K J. J. o. t. A. S. f. M. S., How ubiquitin unfolds after transfer into the gas phase. 2012, 23 (6), 1011–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Loo JA; He JX; Cody WL J. J. o. t. A. C. S., Higher order structure in the gas phase reflects solution structure. 1998, 120 (18), 4542–4543. [Google Scholar]
- 30.Wyttenbach T; Bowers MT, Structural Stability from Solution to the Gas Phase: Native Solution Structure of Ubiquitin Survives Analysis in a Solvent-Free Ion Mobility–Mass Spectrometry Environment. The Journal of Physical Chemistry B 2011, 115 (42), 12266–12275. [DOI] [PubMed] [Google Scholar]
- 31.Shi HL; Clemmer DE, Evidence for Two New Solution States of Ubiquitin by IMS-MS Analysis. J Phys Chem B 2014, 118 (13), 3498–3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ruotolo BT; Robinson CV, Aspects of native proteins are retained in vacuum. Curr. Opin. Chem. Biol 2006, 10 (5), 402–408. [DOI] [PubMed] [Google Scholar]
- 33.Bu J; Peng Z; Zhao F; McLuckey SA, Enhanced Reactivity in Nucleophilic Acyl Substitution Ion/Ion Reactions Using Triazole-Ester Reagents. J. Am. Soc. Mass. Spectrom 2017, 28 (7), 1254–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun Y; Vahidi S; Sowole MA; Konermann L J. J. o. T. A. S. f. M. S., Protein Structural Studies by Traveling Wave Ion Mobility Spectrometry: A Critical Look at Electrospray Sources and Calibration Issues. 2016, 27 (1), 31–40. [DOI] [PubMed] [Google Scholar]
- 35.Ruotolo BT; Benesch JL; Sandercock AM; Hyung S-J; Robinson CV J. N. p., Ion mobility–mass spectrometry analysis of large protein complexes. 2008, 3 (7), 1139. [DOI] [PubMed] [Google Scholar]
- 36.Bush MF; Hall Z; Giles K; Hoyes J; Robinson CV; Ruotolo BT, Collision Cross Sections of Proteins and Their Complexes: A Calibration Framework and Database for Gas-Phase Structural Biology. Anal. Chem 2010, 82 (22), 9557–9565. [DOI] [PubMed] [Google Scholar]
- 37.Gabelica V; Shvartsburg AA; Afonso C; Barran P; Benesch JL; Bleiholder C; Bowers MT; Bilbao A; Bush MF; Campbell JL J. M. s. r., Recommendations for reporting ion mobility mass spectrometry measurements. 2019. [DOI] [PMC free article] [PubMed]
- 38.Webb IK; Morrison LJ; Brown J J. I. J. o. M. S., Dueling electrospray implemented on a traveling-wave ion mobility/time-of-flight mass spectrometer: Towards a gas-phase workbench for structural biology. 2019, 444, 116177. [Google Scholar]
- 39.Cai W; Guner H; Gregorich ZR; Chen AJ; Ayaz-Guner S; Peng Y; Valeja SG; Liu X; Ge Y, MASH Suite Pro: A Comprehensive Software Tool for Top-Down Proteomics. Mol. Cell. Proteomics 2016, 15 (2), 703–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Horn DM; Zubarev RA; McLafferty FW, Automated reduction and interpretation of. J. Am. Soc. Mass. Spectrom 2000, 11 (4), 320–332. [DOI] [PubMed] [Google Scholar]
- 41.Donnelly DP; Rawlins CM; DeHart CJ; Fornelli L; Schachner LF; Lin Z; Lippens JL; Aluri KC; Sarin R; Chen B; Lantz C; Jung W; Johnson KR; Koller A; Wolff JJ; Campuzano IDG; Auclair JR; Ivanov AR; Whitelegge JP; Paša-Tolić L; Chamot-Rooke J; Danis PO; Smith LM; Tsybin YO; Loo JA; Ge Y; Kelleher NL; Agar JN, Best practices and benchmarks for intact protein analysis for top-down mass spectrometry. Nat. Methods 2019, 16 (7), 587–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wyttenbach T; Bowers MT, Structural stability from solution to the gas phase: native solution structure of ubiquitin survives analysis in a solvent-free ion mobility-mass spectrometry environment. J. Phys. Chem. B 2011, 115 (42), 12266–75. [DOI] [PubMed] [Google Scholar]
- 43.Konermann L; Douglas DJ, Unfolding of proteins monitored by electrospray ionization mass spectrometry: a comparison of positive and negative ion modes. J. Am. Soc. Mass. Spectrom 1998, 9 (12), 1248–1254. [DOI] [PubMed] [Google Scholar]
- 44.Shi H; Pierson NA; Valentine SJ; Clemmer DE, Conformation Types of Ubiquitin [M+8H]8+ Ions from Water:Methanol Solutions: Evidence for the N and A States in Aqueous Solution. The Journal of Physical Chemistry B 2012, 116 (10), 3344–3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shi H; Clemmer DE, Evidence for two new solution states of ubiquitin by IMS-MS analysis. The journal of physical chemistry. B 2014, 118 (13), 3498–3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brutscher B; Brüschweiler R; Ernst RR J. B., Backbone dynamics and structural characterization of the partially folded A state of ubiquitin by 1H, 13C, and 15N nuclear magnetic resonance spectroscopy. 1997, 36 (42), 13043–13053. [DOI] [PubMed] [Google Scholar]
- 47.Lenkinski RE; Chen DM; Glickson JD; Goldstein G J. B. e. B. A.-P. S., Nuclear magnetic resonance studies of the denaturation of ubiquitin. 1977, 494 (1), 126–130. [DOI] [PubMed] [Google Scholar]
- 48.Wilkinson KD; Mayer A N. J. A. o. b.; biophysics, Alcohol-induced conformational changes of ubiquitin. 1986, 250 (2), 390–399. [DOI] [PubMed] [Google Scholar]
- 49.May JC; Jurneczko E; Stow SM; Kratochvil I; Kalkhof S; McLean JA, Conformational Landscapes of Ubiquitin, Cytochrome c, and Myoglobin: Uniform Field Ion Mobility Measurements in Helium and Nitrogen Drift Gas. Int J Mass Spectrom 2018, 427, 79–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.May JC; Jurneczko E; Stow SM; Kratochvil I; Kalkhof S; McLean JA, Conformational landscapes of ubiquitin, cytochrome c, and myoglobin: Uniform field ion mobility measurements in helium and nitrogen drift gas. Int. J. Mass spectrom 2018, 427, 79–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lermyte F; Verschueren T; Brown JM; Williams JP; Valkenborg D; Sobott F, Characterization of top-down ETD in a travelling-wave ion guide. Methods 2015, 89, 22–29. [DOI] [PubMed] [Google Scholar]
- 52.Bu J; Fisher CM; Gilbert JD; Prentice BM; McLuckey SA J. J. o. T. A. S. f. M. S., Selective covalent chemistry via gas-phase ion/ion reactions: an exploration of the energy surfaces associated with N-hydroxysuccinimide ester reagents and primary amines and guanidine groups. 2016, 27 (6), 1089–1098. [DOI] [PubMed] [Google Scholar]
- 53.Giles K; Williams JP; Campuzano I, Enhancements in travelling wave ion mobility resolution. Rapid Commun. Mass Spectrom 2011, 25 (11), 1559–1566. [DOI] [PubMed] [Google Scholar]
- 54.Bu J; Fisher CM; Gilbert JD; Prentice BM; McLuckey SA, Selective Covalent Chemistry via Gas-Phase Ion/ion Reactions: An Exploration of the Energy Surfaces Associated with N-Hydroxysuccinimide Ester Reagents and Primary Amines and Guanidine Groups. J. Am. Soc. Mass. Spectrom 2016, 27 (6), 1089–1098. [DOI] [PubMed] [Google Scholar]
- 55.Vijaykumar S; Bugg CE; Cook WJ, Structure of Ubiquitin Refined at 1.8 a Resolution. J Mol Biol 1987, 194 (3), 531–544. [DOI] [PubMed] [Google Scholar]
- 56.Morrison LJ; Brodbelt JS, Charge site assignment in native proteins by ultraviolet photodissociation (UVPD) mass spectrometry. Analyst 2016, 141 (1), 166–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fraczkiewicz R; Braun W, Exact and efficient analytical calculation of the accessible surface areas and their gradients for macromolecules. J. Comput. Chem 1998, 19 (3), 319–333. [Google Scholar]
- 58.Mendoza VL; Vachet RW, Protein Surface Mapping Using Diethylpyrocarbonate with Mass Spectrometric Detection. Anal. Chem 2008, 80 (8), 2895–2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Harding MM; Williams DH; Woolfson DN, Characterization of a Partially Denatured State of a Protein by 2-Dimensional Nmr - Reduction of the Hydrophobic Interactions in Ubiquitin. Biochemistry 1991, 30 (12), 3120–3128. [DOI] [PubMed] [Google Scholar]
- 60.Bakhtiari M; Konermann L, Protein Ions Generated by Native Electrospray Ionization: Comparison of Gas Phase, Solution, and Crystal Structures. The Journal of Physical Chemistry B 2019, 123 (8), 1784–1796. [DOI] [PubMed] [Google Scholar]
- 61.Bleiholder C; Liu FC, Structure Relaxation Approximation (SRA) for Elucidation of Protein Structures from Ion Mobility Measurements. The Journal of Physical Chemistry B 2019, 123 (13), 2756–2769. [DOI] [PubMed] [Google Scholar]
- 62.Cox MJ; Haas AL; Wilkinson KD, Role of ubiquitin conformations in the specificity of protein degradation: iodinated derivatives with altered conformations and activities. Arch Biochem Biophys 1986, 250 (2), 400–9. [DOI] [PubMed] [Google Scholar]
- 63.Pan YQ; Briggs MS, Hydrogen-Exchange in Native and Alcohol Forms of Ubiquitin. Biochemistry-Us 1992, 31 (46), 11405–11412. [DOI] [PubMed] [Google Scholar]
- 64.Stockman BJ; Euvrard A; Scahill TA, Heteronuclear three-dimensional NMR spectroscopy of a partially denatured protein: the A-state of human ubiquitin. J Biomol NMR 1993, 3 (3), 285–96. [DOI] [PubMed] [Google Scholar]
- 65.Shi H; Atlasevich N; Merenbloom SI; Clemmer DE, Solution Dependence of the Collisional Activation of Ubiquitin [M + 7H]7+ Ions. J. Am. Soc. Mass. Spectrom 2014, 25 (12), 2000–2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.