

Summary
The gut microbiota is a complex network of diverse organisms that exhibits plasticity and is capable of impacting host immunity. The malleability of the microbiota presents microbial alteration as an avenue for tuning the immune system to different set points, an approach that has the potential to enable a wide range of therapeutic applications, and ultimately enable disease prevention. Despite the tremendous potential in harnessing the microbiome-immune axis to benefit human health, key barriers must be addressed to hasten translation. Studies using mouse models have identified numerous specific interactions between the gut microbiota and both local and systemic immunity, in several cases using gnotobiotics and other highly controlled approaches to establish causal relationships. Recent advances in our ability to perform expansive profiling of both the microbiota and the immune system now enable exploring human-gut microbiota connections more thoroughly than ever before. An important next step in realizing the power of microbiome reprogramming is to elucidate a human-relevant “map” of microbial-immune wiring, while focusing on animal studies to probe the subset of interactions likely to be relevant to human biology. Such efforts have the potential for revealing new paradigms in immune function as it relates to the microbiome, and for harnessing this connection to improve human health. Here we provide an overview of this field’s current status and discuss two approaches for establishing priorities for detailed investigation: (i) longitudinal intervention studies in humans probing the dynamics of both the microbiota and the immune system, and (ii) the study of traditional populations to assess lost features of human microbial identity whose absence may be contributing to the rise of immunological disorders. These human centered approaches offer a judicious path forward to capitalize on the potent power of the microbiota as a driver in immune health.
Introduction
The gut microbiota is comprised of a diverse set of species with individual-specific makeup and metabolic output, and has emerged as a potent regulator of the host’s metabolism and immune system(Kundu et al., 2017; Lynch and Pedersen, 2016). Our gut microbiota has cospeciated with us over millions of years, likely shaping our immune system and becoming intertwined with our physiology (Davenport et al., 2017; Falush et al., 2003; Ley et al., 2008; Moeller et al., 2016; Moeller et al., 2014). This complex community both contributes and responds to conditions in the gut in settings of health and pathology; providing signals critical to intestinal and metabolic homeostasis(Rakoff-Nahoum et al., 2004; Schroeder and Bäckhed, 2016). These effects are not confined to local responses but rather are integrated with systemic immunity(Belkaid and Hand, 2014; Plunkett and Nagler, 2017; Trompette et al., 2014). Recent work demonstrated that microbiota composition was predictive of the efficacy of cancer immunotherapy(Helmink et al., 2019). These far-reaching impacts of the microbiota suggest that a focus on microbiota-immune interactions may provide insight into principals of immune system function while facilitating precision therapeutics for systemic disease.
The malleability of the gut microbiota presents an exciting opportunity for mechanistic exploration and therapeutic potential. Recent evidence, points to environmental factors as important determinants beyond host genetics in driving community composition(Carmody et al., 2015; David et al., 2014; Rothschild et al., 2018; Turnbaugh et al., 2009). Genetically similar populations have shown distinct microbiotas over a gradient of lifestyle differences such as farming practices and water sources(Fragiadakis et al., 2018; Gomez et al., 2016; Jha et al., 2018; Morton et al., 2015). Diet in particular has emerged as a key driver of microbiota composition and function(Zmora et al., 2019). Short-term changes in diet produce rapid changes in the microbiota, and long-term dietary patterns are associated with distinct microbial phenotypes(David et al., 2014; De Filippo et al., 2010; Smits et al., 2017; Wu et al., 2011). The dynamic nature of the microbiota also leaves it vulnerable to perturbing forces, such as antibiotics or industrialized diets, which result in persistent or compounding deterioration of the gut community over generations in mouse models(Dethlefsen and Relman, 2011; Korpela et al., 2016; Schulfer et al., 2018; Sonnenburg et al., 2016). Rapid gut microbiota change in humans occurs upon immigration to an industrialized setting, and the loss of functions and species becomes more severe over time and subsequent generations(Vangay et al., 2018). As a result of this, the microbiota that shaped our human genome over millennia, has been remodeled by forces of modernization. One hypothesis is that the “industrialized microbiota” is a major contributor to the rising rates of inflammatory bowel disease (IBD) and asthma as well as chronic inflammatory conditions such as diabetes, obesity, and cardiovascular disease (Bach, 2002; Blaser and Falkow, 2009; Sonnenburg and Sonnenburg, 2014). Therefore harnessing, and potentially repairing lost, but beneficial aspects of the microbiota will be a key path forward toward slowing trends in chronic disease.
Foundational work has been done in mouse models to elucidate interactions between the microbiota and the immune system. However, translating these findings to humans has been challenging; differences in gut-immune interaction between host species separated by 100 million years of evolution is compounded by the expense and time required to test if candidate interactions are relevant to human health and pathology. A movement for “immunology taught by humans” advocates for reversing this approach of using animal models to reveal mechanism: start with immune monitoring in humans as a means for establishing hypothesis informed by human-relevant aspects and variation of immune dynamics that can then be subjected to detailed investigation in mouse models (Davis and Brodin, 2018). This general approach to translational immunology can extend to reveal microbiota-immune interactions relevant to humans. The strength of a human-centric approach for microbiota profiling was shown in recent work using the microbiota to determine personalized glycemic responses to diet (Bashiardes et al., 2018; Zeevi et al., 2015). New technologies both for profiling the immune system and the microbiota enable omics profiling of both systems from human samples to make this a feasible reality for studies moving forward.
In this review, we start with a description of the landscape of the gut microbiota, presenting concepts from ecology that are used as an informative framework for its complex community dynamics. We next give an overview of some of the foundational principles of microbiota-immune interactions revealed through mouse models, including considerations for incorporating the microbiota as a parameter in studies of murine immunity. We then present two avenues for a human centered approach to microbiota-immune interaction, including longitudinal dietary intervention studies and the study of traditional verses industrialized populations, using ethical and culturally-sensitive human based studies as an important tool for understanding critical microbe-host interactions.
The Complexity of the Microbial Landscape
Significant effort in the field has focused on understanding the complexity of the gut microbiota—the types of organisms present, their localization, their genetics, their metabolic output, and the dynamics of this community in response to perturbation (Figure 1, Table 1)(Dethlefsen and Relman, 2011; Gilbert et al., 2018). Parallels can be drawn to the study and framework for the immune system: a series of specialized, interacting cell types with distinct functionality, localization, and output, with coordinated reactions to various stimuli. A set of tools and technologies have emerged that can be used as complementary methods for understanding these various levels of complexity that will be discussed below(Debelius et al., 2016; Knight et al., 2018).
Figure 1: The complexity of the microbial landscape.
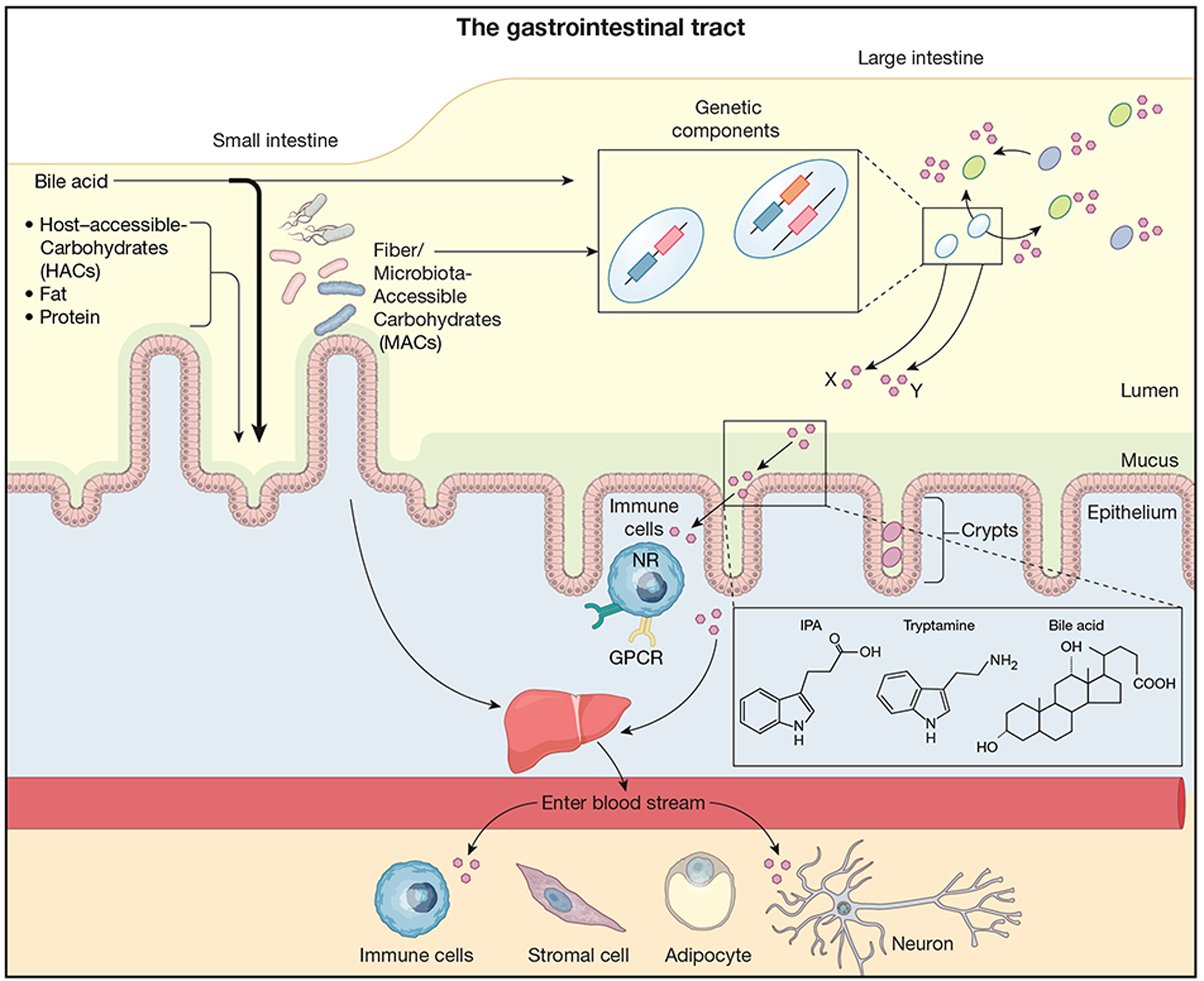
An array of bacterial taxa (different colors represent different species) colonize the length of the gastrointestinal tract, the majority of which reside in the large intestine. Bacteria also may preferentially colonize the intestinal lumen, the mucus, or intestinal crypts in the epithelium. Bacteria of the same species may contain different genetic components as well as differential expression of genes (zoomed in box) allowing it to differentially process substrates and produce distinct metabolites, denoted by X and Y below box. Bacteria produce a variety of metabolites (pink hexagons) that modulate other bacteria as well as traffic across the intestinal barrier to influence the host. These diverse metabolites, including short-chain fatty acids (SCFA), indolepropionic acid (IPA)(Dodd et al., 2017) tryptamine(Bhattarai et al., 2018) and secondary bile acids (example chemical structures shown) modulate host cells both locally and systemically to influence host physiology as well as immune activation. Host receptors for sensing metabolites are broadly expressed: SCFA (formate, acetate, propionate, butyrate) via GPCR41, GPR43 and GPR109A, IPA via pregnane X receptor (PXR)(Venkatesh et al., 2014), tryptamine via GPCR serotonin receptor-4 (5-HT4R)(Bhattarai et al., 2018), secondary bile acids via the GPCR TGR5, the farnesoid X receptor (FXR), the liver X receptor (LXR), and the vitamin D receptor (VDR)(Fiorucci and Distrutti, 2015; Ridlon et al., 2006), microbial derived histamine via the GPCR histamine receptors (H1–H4)(Palm et al., 2014), commendamide via GPCR132(Cohen et al., 2015), tryptophan metabolites via aryl hydrocarbon receptor (AhR)(Zelante et al., 2013). Additional metabolites likely activate a large and diverse number of other GPCRs in interactions that remain to be identified(Chen et al., 2019).
Table 1: Key studies providing insight into human gut microbiota stability, malleability, and composition using high-throughput sequencing.
An abbreviated listing of impactful studies regarding intestinal microbiome composition in various settings and disease states, its stability over time in the absence of intervention, and its response to various interventions (antibiotics, weight loss, FMT etc.)
| Study | Findings | Cohort size (*longitudinal aspect to study) | Related Studies |
|---|---|---|---|
| (Eckburg et al., 2005) | Stool microbiota composition is individualized and shares similarity with mucosal microbiota; mucosal microbiota shows regional variation; 16S rRNA sequencing | 3 | (Hold et al., 2002) |
| (Ley et al., 2006) | Gut microbiota is different between lean and obese individuals; obese microbiota normalizes with weight loss; 16S rRNA sequencing | 12* (4 samplings over 12 months) | (Turnbaugh et al., 2009) |
| (Palmer et al., 2007) | Gut microbiota development is dynamic and individual during the first year of life; 16S rRNA microarrays | 14 | |
| (Frank et al., 2007) | IBD associated mucosal microbiota is distinct from healthy controls and is depleted in commensal anaerobes while enriched in facultative anaerobes (Proteobacteria); 16S rRNA sequencing | 129 IBD and 61 controls | (Halfvarson et al., 2017; Schirmer et al., 2018) |
| (Dethlefsen et al., 2008) | Longitudinal dynamics of gut microbiota depletion and recovery during and antibiotic treatment; 16S rRNA sequencing | 3* (5 samples over 8 months) | (Dethlefsen and Relman, 2011) |
| (De Filippo et al., 2010) | Gut microbiota from children in non-industrialized setting is distinct from industrialized children (more diverse, increased SCFA production); 16S rRNA sequencing and mass spectrometry | 30 (15 Burkina Faso, 15 Italy) | |
| (Qin et al., 2010) | MetaHIT (Metagenomics of the Human Intestinal Tract) project established a gene catalogue of >3 million genes present in the human intestinal microbiota; 16S rRNA sequencing and metagenomics | 124 (European; lean, overweight, Obese, IBD) | |
| (Yatsunenko et al., 2012) | Traditional populations are distinct from industrialized, while similar across wide geographic differences (Africa vs South America); infant microbiomes are less complex and diversify with age in all populations; 16S rRNA sequencing and metagenomics | 326 (148 traditional, Malawian or Amerindian and 178 USA) | |
| (H.M.P. Consortium, 2012) | Gut microbiota of healthy subjects from USA. 242 of 300 from HMP cohort; 16S rRNA sequencing as well as metagenomics | 242 (131* at additional timepoint) | |
| (Faith et al., 2013) | Adult gut microbiota stable over time; strain tracking via culture and sequencing suggest individuals harbor same species over decades. Weight loss perturbs microbiome to greater degree than variation over time; 16S rRNA sequencing and bacterial culture | 37* (Up to 5 year sampling length), 4* (sampled over 34 week weight loss intervention) | (Caporaso et al., 2011) |
| (Clemente et al., 2015) | Gut microbiota of uncontacted Amerindians has unprecedented high diversity; despite no exposure to synthetic antibiotics, antibiotic resistance genes present; metagenomics reveal high functional diversity as well; 16S rRNA sequencing and metagenomics | 34 | |
| (Staley et al., 2016) | Gut microbiota analysis of patients after FMT for C diff infection, demonstrate chimeric microbiota enriched for secondary bile acid metabolism; 16S rRNA sequencing | 24* (pre-FMT, 2 and 8 weeks after FMT) | (Buffie et al., 2015; Smillie et al., 2018) |
| Moeller et al., 2016) | Strains within the human gut microbiota have been associated with humans for at least 15 million years; DNA gyrase subunit B (gyrB) gene sequencing | 16 humans, 47 chimpanzees, 24 bonobos, 24 gorillas | |
| (Smits et al., 2017) | An analysis of aggregated data demonstrating difference between industrialized and non-industrialized samples across all studies; Hadza hunter-gatherers exhibit seasonal cycling in gut microbiome; 16S rRNA sequencing and metagenomics | 2064, 19–54* samples at 4 timepoints over 24 months | (Pasolli et al., 2019) |
| (Jha et al., 2018) | A lifestyle gradient corresponds to degree of microbiota shift from traditional to industrialized; 16S rRNA sequencing | 54 | |
| (Vangay et al., 2018) | Gut microbiota diversity in non-industrialized humans is lost upon migration to industrialized setting; 16S rRNA sequencing and metagenomics | 550, 19* samples prior to and over 9 months after immigration to USA |
An important component of all microbiome studies is to first assess which organisms are present and define aspects of the community structure. Accordingly, the majority of studies profiling the gut microbiota to date have focused on “census-taking”: an identification of which organisms are present and at what abundance. Metrics derived from this often focus on composition at different taxonomic levels from phylum-level differences down to the genus and species levels, as well as some recent studies focusing on strain-level variation(De Filippis et al., 2019; Morowitz et al., 2011; Smillie et al., 2018; Yassour et al., 2018). The field has adapted concepts from ecology to describe and help understand microbial communities(Costello et al., 2012; Dethlefsen et al., 2007; Foster et al., 2017; Miller et al., 2018; Relman, 2012; Sprockett et al., 2018). There has been a particular focus on diversity, including alpha diversity, the diversity within a community; beta diversity, the level of similarity between communities from different hosts; and gamma diversity, the collective diversity among a collection of hosts (Box 1: Terms)(Magurran, 1988; Reese and Dunn, 2018; Whittaker et al., 2001). Different metrics of alpha diversity (hereafter, “diversity”) integrates aspects of the community including bacterial presence-absence, abundance, and phylogenetic relatedness. A strong and common association between diversity of the intestinal microbiota and healthy states exist, while, conversely, industrialization and inflamed states often have decreased diversity(Sonnenburg and Sonnenburg, 2014). Diet is a well-established driver of microbiota composition and function, and specific diets appear to be related to maintaining microbiota diversity, particularly diets high in fiber(De Filippo et al., 2010; Sonnenburg et al., 2016). Immunologic mechanisms underlying diversity are emerging and host immunity is likely integral to maintaining a diverse microbiota(Campbell et al., 2018; Donaldson et al., 2018; Foster et al., 2017; Levy et al., 2017; Sonnenburg et al., 2016). The contributions of microbiota diversity to host physiology are less clear, however, one possibility is that a diverse microbiota with expanded metabolic repertoire for complex diet-derived carbohydrate (i.e., dietary fiber, or microbiota accessible carbohydrates, MACs) degradation is a marker of a diet high in plant-based fiber, a nutrient severely deficient in the industrialized diet. This correlation may extend to causation, with evidence that microbial metabolite production (short-chain fatty acids or SCFAs and others) will reduce inappropriate inflammation and promote metabolic health.
Box 1: Terms.
A listing of important terms in gut microbiome research with definitions
| Term: Definition |
|---|
| Alpha diversity: The diversity within a community |
| Beta diversity: The level of similarity between communities from different hosts |
| Gamma diversity: The collective diversity among a collection of hosts |
| Operational Taxonomic Unit (OTU): Groups to which sequencing reads (i.e., 16S rRNA) are assigned based on their similarity; A representative unit of microbial sequence variation |
| Amplicon Sequence Variant (ASV): Inferred exact sequence (i.e., 16S rRNA) that can be used to classify members when assessing a community. A newer method that preserves sequence information and can offer higher resolution as well as consistency across studies |
| Carbohydrate-Active Enzyme (CAZyme): Enzymes and proteins involved in recognition, metabolism, and synthesis of complex carbohydrates (http://www.cazy.org/) |
| Short Chain Fatty Acid (SCFA): Volatile fatty acids with less than six carbon atoms that are the result of microbial fermentation of complex carbohydrates (i.e. formate, acetate, propionate, butyrate) |
| Microbial-Accessible Carbohydrate (MAC): A carbohydrate, often structurally complex, unable to be degraded/absorbed during typical mammalian digestion, can be degraded by intestinal bacteria as an energy source with resultant SCFA production (i.e., inulin) |
| Host-Accessible Carbohydrate (HAC) : A structurally simple carbohydrate degraded by mammalian enzymes and absorbed in the small intestine (i.e., glucose, sucrose) |
| Fluorescent In-Situ Hybridization (FISH): An imaging based technique using molecular probes, that can be bacteria-specific, to examine bacterial localization in a given tissue |
| Germ-free/Gnotobiotic: An animal devoid of known microbes (germ-free) and with a known/defined community (gnotobiotic), most commonly mice, that serve as critical tools to elucidate mechanisms of host-microbe interactions |
| Volatile and/or Associated Negatively with Industrialized Societies of Humans (VANISH) taxa: Taxa of bacteria that are found in non-industrialized settings, but lost or reduced in abundance in industrialized humans |
While the majority of focus has been on bacteria within the gut microbiota, there is a growing interest in the eukaryotic (protozoa, parasitic and fungal) and viral components, though notably eukaryotes are largely diminished in samples from industrialized countries(Laforest-Lapointe and Arrieta, 2018; Nash et al., 2017). However, how the relationship between helminths, protozoa, and fungus with bacterial components modulate mucosal and systemic immunity deserves substantial investigation. Intestinal fungal microbiota composition is associated with the development of atopy and childhood asthma(Fujimura et al., 2016). Intestinal Candida albicans drives systemic Th17 responses and can influence inflammation and anti-fungal responses at distal sites(Bacher et al., 2019; Shao et al., 2019).
For organismal profiling, a common tool used for census taking is to analyze genes that code the ribosomal subunit sequences (16S rRNA for bacteria and 18S rRNA for eukaryotic), which due to the combination of hairpin and loop structural units, provide excellent sites from primer annealing and slowly evolving regions, respectively, enable identification of bacteria by PCR-enabled sequencing— generally providing phylogenetic resolution to the genus level(Fox et al., 1977; Hugenholtz and Pace, 1996; Woese, 1987). Thus, via PCR amplification and sequencing of the 16S or 18S rRNA gene alone, using conserved priming sites (i.e., “universal” primers), community structure can be determined. These 16S rRNA gene sequences can be grouped using a variety of computational methods, the most prevalent being assignment into bins of operational taxonomic units (OTUs) or preserving sequence information and using error-correction models to identify amplicon sequence variants (ASVs)(Callahan et al., 2017). Analogous experimental methods for profiling eukaryotes use other genes similarly, including the 18S rRNA gene for eukaryotes, or the internal transcribed spacer (ITS) gene for fungi(Laforest-Lapointe and Arrieta, 2018).
Shot-gun metagenomics is a method in which all DNA in a sample is extracted and sequenced, distinct from the region- and kingdom-specific targeted sequencing of 16S rRNA analysis. Metagenomic sequencing can be used to determine both the abundance of microbial taxa, including viral and eukaryotic components, as well as functional capacity of a community, inferred by gene content(Sharpton, 2014). Metagenomic approaches are a more reliable method to discern strain-level differences, allowing for genetic content of specific bacteria to be collected and tracked over time(Smillie et al., 2018). As sequencing costs decline and technology continues to improve, metagenomic based methods to analyze microbiota will become more widely used. Meta-transciptomics, similar to metagenomics but with sequencing focused on the pool of microbial mRNA, will also be propelled by sequencing trends.
Culture-based methods for bacterial identification are becoming an increasingly important compliment to genetic based techniques(Bilen et al., 2018; Lagier et al., 2018; Lagier et al., 2016). As techniques improve for culturing fastidious organisms, previously “unculturable” microbes are now being isolated in a high throughput manner, an endeavor known as “culturomics”(Lagier et al., 2016). Culturomic approaches to studying the microbiota enable discrimination between viable and dead organisms as well as the detection of low abundance organisms, which can be overlooked using sequencing-based approaches(Lagier et al., 2018). Culture based approaches can be used to discriminate the true presence of viable organisms detected by sequencing-based approaches versus contamination, with the caveat that false-negatives can also result from organisms that are viable but difficult to isolate in culture(Bushman, 2019). Perhaps most important of all, culturomics leads to the creation of strain libraries of immense value for subsequent functional/mechanistic studies as well as potential therapeutic uses(Forster et al., 2019). With hundreds of newly identified species isolated from the human GI tract via recent culturomic efforts, a proportional scaling of investigation into the function of these microbes in needed.
In addition to defining which microbiota are present, there are diverse locations and niches within the GI tract to occupy and identifying microbiota location is integral to understand its physiology and impact on the host. Gut microbes are differentially distributed along the length of the gastrointestinal tract. While the majority of microbes reside in the colon, certain organisms preferentially colonize the small intestine and certain sections of the upper GI tract, different sections favoring organisms with particular metabolic constraints. In addition, microbes colonize at different points radially, and this localization appears to be non-random, with a strain’s location dictated by its functional attributes. Most microbial cells are present in the lumen, rich in diet-derived nutrients but not in host immune effectors. Some species are known to localize to the mucus layer, and others can be closely associated with host tissue, in intestinal crypts and transverse folds(Donaldson et al., 2016; Tropini et al., 2017). Crypt colonization have become an important area of focus as a microhabitat that could serve as a protective reservoir of bacterial cells to enable exclusion of competitors from the gut ecosystem and to enable recolonization after perturbations in the gut(Donaldson et al., 2018; Pédron et al., 2012; Shepherd et al., 2018). Notably, many studies profiling the microbiota use stool samples which, while providing a useful survey of the microbes present in the gut, obscures this spatial information and has been noted to have certain biases(Zmora et al., 2018). Imaging techniques have been most straightforward means for understanding the spatial distribution of microbes(Earle et al., 2015; Tropini et al., 2017). Fluorescent In-Situ Hybridization (FISH) is a technique used to examine bacterial location within tissue samples and serves a critical tool in evaluating bacterial localization(Dejea et al., 2018; Swidsinski et al., 2005; Wagner and Haider, 2012). Mucosal biopsies and washings can also provide important information regarding mucosal associate communities, and new approaches that couple omics technologies with co-localized elements in a microbiome provide a way to couple high-resolution functional analysis with spatial information(Sheth et al.; Zmora et al., 2018). Crypt resident and mucosal associated microbiota may, despite decreased representation in homogenized stool samples, have a disproportionate impact on the host; including spatial information into studies of intestinal microbiota can reveal important aspects of host-microbe interactions(Campbell et al., 2018; Dejea et al., 2018; Donaldson et al., 2018).
There is tremendous value in extending the profiling of gut microbiota beyond composition to an analysis of community functional capacity and metabolic output(Skelly et al., 2019; Vernocchi et al., 2016; Zierer et al., 2018). Shot-gun metagenomic sequencing provides information about the transcriptional and metabolic capacity of the microbiota. Analysis of encoded functional attributes, such as genes involved in carbohydrate metabolism (e.g., Carbohydrate-Active Enzymes (CAZymes)) or biosynthetic gene clusters, can provide insight into nutritional input and metabolic output of a complex microbial ecosystem(El Kaoutari et al., 2013). The detection of functional capacity to produce metabolites that have been shown to modulate immune function, such as the SCFAs, enables sequencing data of a microbiota to inform potential interactions with the host. Efforts are underway in metabolomics for improved high-throughput profiling and structural identification of microbial-derived metabolites using mass spectrometry(Dorrestein et al., 2014; Skelly et al., 2019). These techniques will offer key functional insight, as these metabolites are a major avenue by which the microbiota communicates with the host immune system. Detailed study of single strains including molecular genetic approaches to assign gene-to-function relationships, and the use of gnotobiotic (Greek for “known life”, includes germ-free mice) animal models complement the application of diverse approaches to understand the functionality of intact communities(Dodd et al., 2017).
The Microbiota and the Immune System: Lessons from animal models of human disease
Animal models have served to demonstrate several foundational concepts about host-microbe interactions in the intestine. Germ-free animals, devoid of known living organisms, and gnotobiotic animals, with defined colonization states, are critical tools for understanding host-microbe interactions that have been studied for over 80 years(Gordon and Pesti, 1971; Smith et al., 2007). These animal models have demonstrated that the gut microbiota influences a wide range of biological processes in the host including nutrient absorption and metabolism, and serves as a critical factor driving mucosal and systemic immune system development in mice(Cebra, 1999; Crabbé et al., 1968). Some disease processes critically require the microbiota as an environmental factor for disease initiation(Dianda et al., 1997; Taurog et al., 1994). For example, many rodent models of colitis including rats transgenic for the human HLA B27 allele and mice lacking TCRbeta develop intestinal inflammation that is dependent on the presence of intestinal microbiota(Dianda et al., 1997; Taurog et al., 1994). In some of these animal models, disease severity is determined by gut microbiota composition and this phenotype can be transferred to non-genetically modified mice via microbiota transplant(Elinav et al., 2011; Garrett et al., 2007).
Specific bacteria appear to have a disproportionate ability to influence host immunity, via their localization close to host tissue and/or production of metabolites influencing the immune system, as illustrated in gnotobiotic and conventional mouse models(Ahern et al., 2014; Faith et al., 2014; Geva-Zatorsky et al., 2017). Segmented Filamentous Bacteria (SFB) are intimately host associated via attachments to intestinal epithelial cells in the ileum leading to a potent Th17 and IgA response that in turn, limits its expansion(Ivanov et al., 2009; Klaasen et al., 1993; Lécuyer et al., 2014; Suzuki et al., 2004). SFB appear to exist in many mammalian species, and may colonize humans at low frequency, but their importance in contributing to human immune responses remains to be clarified(Chen et al., 2018; Ericsson et al., 2014; Yin et al., 2013). Clostridial species are able to promote mouse CD4+ Treg induction via high production of SCFAs(Arpaia et al., 2013; Atarashi et al., 2011; Furusawa et al., 2013). Recent research identified bacterial species that reside in the mouse colonic mucus able to promote CD8+ T cell responses that protect against infection and augment anti-tumor immunity(Tanoue et al., 2019). A growing repertoire of commensal specific T cell transgenics and MHC-peptide tetramer based technology now allow for mechanistic studies of the role of anti-commensal T cell responses in regulating aspects of host-microbe interactions(Chai et al., 2017; Cong et al., 2009; Xu et al., 2018; Yang et al., 2014). Using these tools, it was found that the Bacteroides thetaiotaomicron (Bt) antigen driving T cell responses is regulated by dietary nutrients, highlighting the dynamic and interconnected nature of diet-host-microbe interactions(Wegorzewska et al., 2019).
Microbial metabolites are a key means by which the gut microbiota communicates with the immune system both in the intestine and systemically(Cohen et al., 2017; Rooks and Garrett, 2016). The host senses microbial metabolites via cell surface and intracellular receptors, such as the G protein coupled receptors (GPCR) and nuclear receptors (Figure 1). Through the processing of dietary fibers by bacteria, the short chain fatty acids (SCFA), acetate, propionate, and butyrate are potent and diverse mediators of diet-host-microbe interactions(Koh et al., 2016; Maslowski et al., 2009; Trompette et al., 2014). SCFAs can signal via the GPCR, GPR41 (FFAR3), GPR43 (FFAR2), and GPR109A expressed on a diversity of immune cells and on intestinal epithelial cells(McKenzie et al., 2017; Rooks and Garrett, 2016). As well, SCFA can inhibit histone deacetylases (HDACs) to mediate their effects through epigenetic modifications as well as integrating into host metabolic pathways(McKenzie et al., 2017; Rooks and Garrett, 2016). Mice deficient in GPR43 fail to resolve colitis and have more severe arthritis and asthma(Maslowski et al., 2009). SCFA’s can be found in biologically significant levels circulating in the blood and influence distal sites, regulating lung inflammation, autoimmunity and hematopoiesis(Mariño et al., 2017; Trompette et al., 2018; Trompette et al., 2014). The relevance of most of these findings to humans remains to be tested, but some promising data has begun to emerge: humans given oral butyrate had detectable anti-inflammatory signature in monocytes(Cleophas et al., 2019).
With these data in mind, several murine studies have supported the “Diet-microbiota hypothesis”: that industrialized diets are leading to increased inflammatory disorders via alterations in gut microbiota composition and metabolite production(Burkitt et al., 1972; Devereux, 2006; Maslowski and Mackay, 2011; Sonnenburg and Sonnenburg, 2014; Thorburn et al., 2014). The amount of fiber consumed in industrialized societies is ~16g/day, well below the recommended level of 25 to 38 g/day and increased fiber in the diet is associated with lower risk of death in human longitudinal studies(King et al., 2012; Park et al., 2011). Accordingly, a diet deficient in dietary fiber leads to more severe colitis and heightened pathogen susceptibility in mice, likely via both decreased SCFA production and mucus degradation by bacteria lacking a dietary source of complex carbohydrates(Desai et al., 2016; Earle et al., 2015; Hryckowian et al., 2018).
In addition to SCFAs, a growing number of microbial metabolites can influence the immune system directly, or indirectly via signaling in epithelial, stromal or adipose cells(Dodd et al., 2017; Virtue et al., 2019). Tryptophan metabolites serve as ligands for host receptors including the aryl hydrocarbon receptor (AhR) that promotes IL-22 transcription and mediate mucosal barrier protection(Zelante et al., 2013). In settings of abundant tryptophan, Lactobacillus reuteri, generates the AhR ligand indole-3-aldehyde. Trypamine is another microbial derived tryptophan metabolite that is abundant in human stool and signals via the GPCR serotonin receptor-4 (5-HT4R) on colonic epithelial cells to promote colonic motility(Bhattarai et al., 2018).
A recent study utilized a library of 144 strains, generated from inflammatory bowel disease (IBD) patients to perform a high throughput screen of metabolite signaling via human G Protein Coupled Receptors (GPCRs)(Chen et al., 2019; Palm et al., 2014). A wide range of signaling potential from this diverse set of strains was observed, with metabolites likely activating >75 GPCRs. Specifically, Morganella morganii and Lactobacillus reuteri strains produce histamine and promote colonic motility, likely via the GPCR histamine receptors (H1–H4). Additionally, phenylalanine (Phe) produced by a Bacteroides thetaiotaomicron strain (C34) signaled via GPR56/AGRG1 and participated in a metabolic network with M. morganii to produce the neuroactive chemical phenethylamine. Throughout this study, strain level variation in metabolite production was observed, highlighting the importance of attaining strain level resolution in microbiota analysis when pursuing functional insights.
Bile acids synthesized and secreted into the intestine by the host (primary bile acids) and microbial metabolites of host derived bile acids (secondary bile acids) are emerging as important mediators of metabolic health and immune status(Fiorucci et al., 2018; Fiorucci and Distrutti, 2015; Ridlon et al., 2006). The majority of primary bile acids secreted into the small intestine are absorbed in the terminal ileum (~95%) with the remaining bile acids reaching the large intestine where they are processed by the microbiota into a wide array of metabolites that signal to host receptors, including the GPCR TGR5, the farnesoid X receptor (FXR), the liver X receptor (LXR), and the vitamin D receptor (VDR)(Fiorucci et al., 2018). Microbial produced secondary bile acids appear to be generally anti-inflammatory, with TGR5/FXR signaling inhibiting NLRP3 activation, while promoting increased IL-10 production in macrophages and decreased TNF-α and IL-12 from dendritic cells (recently reviewed(Fiorucci et al., 2018)). The role of secondary bile acid-signaling on host immunity will likely be a significant area of focus going forward.
One key in connecting a microbial metabolite to a host receptor or pathway is to combine molecular genetics of host and microbe in the context of gnotobiotic mouse models with highly defined colonization states. Indolepropionic acid (IPA) produced by Clostridium sporogenes from the substrate tryptophan signals via the pregnane X receptor (PXR) on epithelial cells to promote barrier function(Dodd et al., 2017; Venkatesh et al., 2014). Colonizing gnotobiotic mice in mono-association with a genetically modified strain of C. sporogenes unable to produce IPA results in increased gut permeability and systemic inflammation(Dodd et al., 2017). Germ-free mice have also been widely used as recipients of intact complex microbial communities from other mouse models or humans with a particular disease state (known as “humanization”). Transfer of a host phenotype or disease state by microbiota transplant serves to provide support of microbiota causation, and also the utility of mice as a pre-clinical experimental model(Faith et al., 2010; Turnbaugh et al., 2009). Microbiota has been shown to transfer the disease states of obesity, malnutrition and colitis(Blanton et al., 2016b; Britton et al., 2019; Ridaura et al., 2013). As well gnotobiotic mice with humanized flora can be used to assess the role of dietary interventions(Faith et al., 2011). These approaches have been particularly enlightening in demonstrating a causal role for the microbiota in perpetuating host malnutrition despite nutritional intervention and led to a successful therapeutic approach(Blanton et al., 2016a; Blanton et al., 2016b; Trehan et al., 2013). The use of mice colonized with human-derived strains and communities will be an important vehicle for testing whether candidate host-microbe pathways identified in human-based studies can be validated and investigated using an animal model.
With 100 million years of evolutionary distance between humans and mice, the rapid positive selection of many immune genes coupled with co-evolving microbiome—many of the specific host-microbe interactions in mice are likely different than humans(Nielsen et al., 2005). With a bottleneck in translation of studies to humans, a means to prioritize pathways and findings relevant to human health is badly needed. A critical step to both interpret and inform studies in animal models will be investing in human-based studies and interventional trials.
Prioritization of microbiota-immune relationships: lessons to learn from human intervention studies (“Bedside to Bench”)
Studies in animal models discussed above have provided critical understanding and evidence for the microbiota’s causal role in immune health and pathology, environmental factors that influence the microbiota such as diet, and illustrate the specificity of interactions that occur between the microbiota and the immune system. However, as is the case in many fields, extending these principles to human health and disease has been a challenge, largely due to differences in biology that arise when moving from animal models to human(Nguyen et al., 2015). Compounding typical challenges in this translational leap, is humans differing from one another in their genetics, and in the complexity, individuality, and dynamics of the their gut microbiota. In addition to the translational hurdle, these features can make the prospect of human studies seem challenging, particularly when considering the response of each person’s microbiota to intervention is also individualized. We argue here that human-based studies are essential if we want to identify microbiota-immune wiring relevant for human health (Figure 2A, Top Panel).
Figure 2: Human-based approaches for studying microbiota-immune interactions.
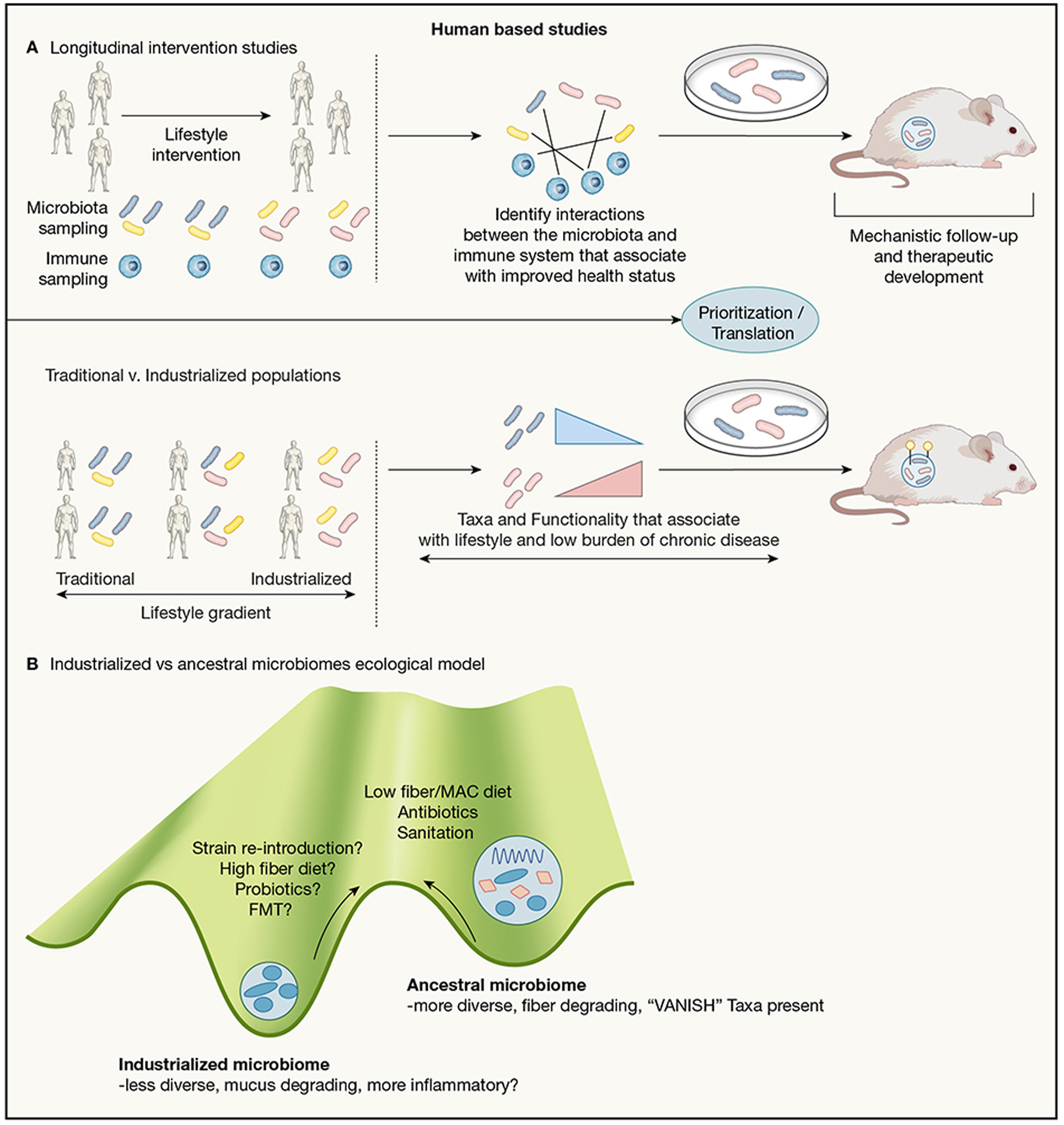
2A, Top panel: Longitudinal intervention studies in humans. A suggested model for microbiota-immune studies: starting with a cohort of participants that undergo a lifestyle intervention that perturbs the microbiota (diet, weight loss, antibiotic use, etc.). Participants are monitored over time, donating samples for both microbiota and immune system profiling. Candidate interactions between microbiota and immune features are identified using machine learning or other means. These interactions can then be studied in a mouse model and in vitro to elucidate their mechanistic underpinnings.
2A, Bottom panel: Traditional v. industrialized population studies. A suggested model for identifying microbes/microbial functionality relevant to lifestyle-related immune disorders: comparing the microbiome of individuals along a gradient of industrialized lifestyles. Focus should be placed on microbial elements that are shared across geographically distinct populations of similar lifestyles, but vary along a gradient of industrialization rather than geography. Once identified, those microbes along with their microbial genetic elements (Yellow markers in figure) or metabolites can be studied mechanistically in murine and in vitro models for their role in immune health or dysregulation.
2B: Ecological concepts applied to microbiota states(Costello et al., 2012). A suggested model for understanding microbial communities: Ancestral microbiomes represent stable communities different from those found in industrialized settings, with changes seen in overall diversity and in carbohydrate utilization capacity. Proposed drivers for this change over time are: dietary changes, more widespread use of antibiotics, increased sanitation contribute to microbiome configuration seen in industrialized settings. To restore beneficial aspects of our microbiota absent in western guts, we propose dietary changes, strain re-introduction, or possibly even Fecal Microbiota Transplant (FMT).
To date, the majority of microbiome studies have been cross-sectional, sampling at a single timepoint, while longitudinal studies with a defined intervention have been limited. Significant efforts have been made to perform cross-sectional microbiota profiling on large cohorts of individuals including the Human Microbiome Project and the American Gut project(Lloyd-Price et al., 2017; McDonald et al., 2018). These projects have provided data and insight that serve as a tremendous resources for the microbiome community, for example revealing the level of variation and diversity found across individuals. Due to the large number of relevant factors to consider, many of which correlate, relating aspects of the microbiota to other host and environmental factors has been a challenge. With the ultimate goal of establishing causality, very large numbers of individuals with high quality metadata are required to disentangle factors that commonly co-vary for such observational studies. Longitudinal intervention studies offer several advantages including establishing causation and, because each individual can serve as their own control (i.e., baseline state), being able to drastically decrease the number of subjects required to elucidate relationships between the microbiota and host parameters. Thus, responses measured in subjects to an intervention over time can be matched to alterations of the microbiome or microbial metabolites to increase the likelihood of identifying mechanistic/causal interactions.
Several longitudinal dietary intervention studies point to the power of diet for altering the microbiota as well as host health(Cotillard et al., 2013; Lewis et al., 2017; Zeevi et al., 2015; Zhao et al., 2018). It is unclear what mixture of general, population-wide interventions and personalized approaches will be the path forward for successful diet-microbiota based interventions. The introduction of a high-fiber diet and prebiotic supplementation showed increases in short-chain fatty acid production by the microbiota and improvements in markers for type-2 diabetes across the cohort(Zhao et al., 2018). In contrast, personalized diets based on microbiota signatures were identified that improved post-prandial glycemic responses to a greater extent than the universal dietary recommendations(Zeevi et al., 2015). Sub-groups additionally have been identified based on gene-richness of the microbiota in obese individuals that reflected to the extent to which dietary intervention decreased inflammatory signatures(Cotillard et al., 2013; Le Chatelier et al., 2013). Likely a combination of population-wide and personalized interventions will be useful in leveraging microbiota and immune interactions for improved human health, whether preventative or therapeutic.
In order to identify microbiota and immune interactions in studies with limited starting constraints, high-dimensional measurements (a “multi-omic” approach) should be used to capture an in-depth profile of both the microbiota and the immune system(Schirmer et al., 2016; Thomas et al., 2015). Several tools for high-dimensional profiling of the microbiota were discussed earlier in this review including sequencing and culture based methods as well as high-throughput metabolomics. Complementary tools are available for immune system profiling, including cytometry by time of flight (CyTOF), RNAseq, antibody repertoire sequencing, and high-dimensional serum proteomics(Hasin et al., 2017; Landhuis, 2018; Olsen et al., 2019; Spitzer and Nolan, 2016). A significant challenge for the field is to create user-friendly intuitive approaches for integration and analysis of multi-omic data which is likely to drive the application and discoveries of the approach, analogous to the democratization of 16S rRNA amplicon sequencing analysis by QIIME(Caporaso et al., 2010).
Other approaches to probe microbiota-host interactions include examining which components of the microbiota are coated with IgA (IgA-Seq), or to examine systemic antibody responses to commensal bacteria(Kau et al., 2015; Palm et al., 2014; Paun et al., 2019; Planer et al., 2016). Both of these antibody-based approaches identify bacteria that have interacted closely with the host immune system, leading to an adaptive immune response. Although some component of IgA coating will be T cell independent, IgA-Seq based studies suggest the majority of bacterial coating identified is T cell dependent(Palm et al., 2014). Used in combination, these tools enable the generation of a large set of immune and microbiota features that can then be used as inputs for machine learning models to identify otherwise non-obvious patterns in the microbiota and the immune system in response to perturbation, such as co-varying elements or states predictive of specific outcomes(Zmora et al., 2018).
Human-based studies help identify candidate interventions for improvement of microbiota and immune health, as well as a series of possible microbiota-immune interactions that occur in humans. However, the capacity for further testing and mechanistic insight is limited within the constraints of working with humans. Moving to animal and in vitro models as a second step, to test hypothesis informed by human data and perhaps incorporating human samples, offers a progression toward mechanism that is informed by human-relevant findings. This approach can include fecal microbiota transfer experiments (FMTs) from a human subject into gnotobiotic mice or colonization with a single microbe that was identified in a candidate interaction. In parallel with colonization, the diet provided to the mice, or other aspects of environmental conditions, can be tailored to test a given hypothesis or to replicate a prior human-based trial (i.e. high fat, low fiber, geographically-based)(Blanton et al., 2016b; Sonnenburg et al., 2016). In addition, should certain microbial metabolites be identified as immune modulators, those can be introduced either genetically or as substrates delivered to the gut to assess the effect on the murine immune system, or applied directly as stimuli to human immune cells in vitro(Dodd et al., 2017; Levy et al., 2015; Skelly et al., 2019). A limitation in this pipeline is that human specific microbiota-immune interactions may not be recapitulated in mice due to biological differences between the host organisms. While failing to reverse translate from humans into a laboratory model hinders mechanistic study, this progression offers advantages over studies started in mice that fail to translate to humans, which often serve as dead-ends(Davis and Brodin, 2018). The proposed human to mouse pipeline will likely contribute to understanding mechanistic causation of the microbiota’s influence on immune parameters and can be used iteratively to better establish candidate microbiota-immune interactions, establishing the foundation for refined interventions and precision therapeutics.
Prioritization of microbiota-immune relationships: lessons to learn from the ancestral microbiota
Elucidating important human microbiota-immune interactions is likely to be propelled by incorporating an evolutionary perspective. The majority of studies, including large government funded and international efforts like the Human Microbiome Project (HMP) and Metagenomes of the Human Intestinal Tract (MetaHIT), which collected 16S rRNA and/or metagenomic data for 300 and 124 individuals, respectively, have provided a detailed view of industrialized populations’ gut microbiota(Consortium, 2012; Qin et al., 2010). In 2010, a study of 14 children living in a rural agricultural region of Burkina Faso revealed a microbiota distinct in composition from Western microbiotas, providing a hint that mapping the human microbiota would require sampling diverse populations living diverse lifestyles(De Filippo et al., 2010). Ensuing studies of traditional populations around the world including Africa, South America, Papua New Guinea, Madagascar, and Asia have expanded our understanding of the variety of forms the human microbiome can take in healthy individuals(Ayeni et al., 2018; Clemente et al., 2015; Jha et al., 2018; Martínez et al., 2015; Morton et al., 2015; Obregon-Tito et al., 2015; Schnorr et al., 2014; Smits et al., 2017; Sonnenburg and Sonnenburg, 2019; Suzuki and Worobey, 2014; Yatsunenko et al., 2012; Zhang et al., 2014). More importantly, many of these studies reveal common features that are specific to non-industrialized microbiomes(Sonnenburg and Sonnenburg, 2019). Notably, many of the same taxa, known as VANISH (Volatile and/or Associated Negatively with Industrialized Societies of Humans) taxa have been identified in other comparative studies revealing a global pattern of microbiota change that is dictated by urbanization(Smits et al., 2017; Vangay et al., 2018). Similar analyses performed using metagenomic data focusing on functional attributes of the community reveal a similar pattern(Pasolli et al., 2019). One possibility is that shifts in lifestyle including diet, antibiotic use, and sanitation have altered the industrialized microbiota to a state that is distinct from what shaped our human genome(Blaser and Falkow, 2009; Sonnenburg and Sonnenburg, 2014).
Notably, the global burden of disease is, over time, shifting from infectious to non-communicable chronic diseases (NCCD), many of which are rooted in immune dysregulation and chronic inflammation(Chan, 2017; Lozano et al., 2012). These conditions, including diabetes, obesity, cardiovascular disease, and cancer, disproportionately affect industrialized societies, though are becoming more prevalent in developing countries as well, likely due to rapid changes in diet, lifestyle, and infections(Chan, 2017). In non-industrialized, hunter-gather, populations, the life expectancy once adulthood has been reached is ~72 and the burden of obesity and cardiovascular disease is estimated to be <5%(Pontzer et al., 2018). Along with the stark contrast in rates of NCCD, differences in microbial diversity and specific changes in taxa presence/absence can be found between industrialized and non-industrialized societies(Fragiadakis et al., 2018; Jha et al., 2018).
While the features of industrialized society have provided many benefits, it leads to the possibility that one unforeseen cost may be the selection of an “industrialized” microbiota that is not well-suited to promote human health. We can look to populations with traditional lifestyles, such as hunter-gatherers and rural agrarians, as a window into the state of the microbiome with fewer of the effects of industrialization(Fragiadakis et al., 2018; Gomez et al., 2016; Morton et al., 2015; Smits et al., 2017). Looking across a lifestyle gradient can highlight specific microbes and functionality that track with lifestyle. A study of Nepalese populations, some of which were hunter-gatherers, and others at various points of transition to subsistence farming, identified gradients of microbiota composition and identified specific taxa whose decrease in prevalence and relative abundance mirror lifestyle change(Jha et al., 2018). A tantalizing hypothesis is that the loss of microbial taxa and associated functions integral to proper immune regulation results in a dysregulated immune system in industrialized populations and predisposition to inflammatory diseases. The VANISH taxa and under-represented functional elements serve as candidate drivers of immune health or dysregulation to be explored in mice, in vitro, and, ultimately, in humans (Figure 2A, Bottom panel).
Concluding remarks
Industrialization has led to a series of advancements in human health through sanitation, antibiotics, vaccination, food production, and medical technology. Accordingly, industrialization has resulted in a dramatic reduction in infectious diseases (diarrheal infectious agents, parasitic infections and other chronic infections), resulting in reduced infant mortality and prolonged lifespan that are of clear overall benefit. However, coincident with these improvements has been a dysregulation of the immune system of unclear etiology, that can be seen in the global rise of non-communicable chronic diseases (obesity, diabetes, heart disease etc). The source of immune dysregulation in industrialized societies remains unclear and alterations in the microbiota may be both a result of, as well as a contributor to immune dysregulation. However, as the microbiota is a malleable and potent contributor to immune status, it offers a path forward to recalibrate our immune set points to mitigate these conditions.
Both the intestinal microbiota and host immune system are complex. In this review, we discuss the various levels of complexities and considerations of the microbiota including spatial organization, taxonomic diversity, functional capacity, and metabolic output. These levels of complexity also offer a variety of access points to target the microbiota for improvement of host immunity.
There are key questions and challenges for understanding and capitalizing on microbiome-immune interactions. An important challenge lies in distilling the guiding principles for host-microbe dialogue. To effectively parse through the most important of a seemingly infinite list of interactions requires a prioritization method to identify those that are relevant for understanding the nature of these relationships and are also relevant for human health. We presented two strategies in pursuit of this challenge that are both human-based “bedside-to-bench” approaches, i.e., that start with observations in human to be followed up with mouse and in vitro models. Both lifestyle intervention studies and analyses that compare human populations with diverse lifestyles can provide candidate interactions that are relevant to human health, and can be explored mechanistically as avenues for therapeutics.
The degree to which we can stably manipulate the microbiota in humans and through what means remains an open question. Promising data in mice and some human studies suggest diet and other lifestyle interventions can be important modulators of the microbiota, but further work must be done to establish our degree of control over therapeutic interventions as well as the extent to which an established adult microbiota is resilient to change. There may be additional requirements for introducing change into the microbiota, including adjunct targeting the host immune environment. Other tools such as engineered microbes or delivery of microbially-derived metabolites may also be viable approaches (Figure 2B).
Finally, further work must be done to identify which disease processes will be amenable to microbiota-targeted therapeutic intervention. While links have been established to a large number of conditions, it is unclear which will be the most fruitful for treatment benefit. Recent progress in understanding how the microbiota influence cancer therapy, or alter the severity of autoimmune diseases suggest hopeful avenues. The microbiota at sites outside of the gut, including the skin, lung and vagina offer additional points of interaction between resident microbes and human immunity. There is great potential for leveraging the microbiome as a tool in recalibrating immune system health. We envision a future in which we create an arsenal of microbial, diet-based, microbiota- and diet-derived tools for elucidating specific immune responses, trajectories, and alterations in immune set point. This likely will include monitoring of microbiome colonization and immune status during critical periods of development in infancy and early childhood with precision approaches used to guide therapy. The microbiome can become a powerful tool in precision health efforts, tailoring therapies to individualized microbiomes and circumstances. These approaches should be considered in conjunction with immune modulation and therapeutics, as may soon become the case in checkpoint-inhibitor based cancer therapy (Gong et al., 2019). Monitoring of both the microbiota and the immune system, possibly at-home or in real-time, will be a key step to propel precision and personalized interventions. Ultimately, as we begin to appreciate the vast complicated network of immune-microbiota interaction, it is important to think carefully about how we can most efficiently mobilize resources and time to improve understanding of these complex systems towards realizing the tremendous potential of harnessing these interactions to improve human health.
Acknowledgments:
We would like to thank members of the Sonnenburg labs for helpful discussions. National Science Foundation for their funding support (grant number 1648230) and the NIH (R01-DK085025 and DP1-AT00989201 to J.L.S.). JLS is a Chan Zuckerberg Biohub Investigator. S.P.S. is supported by postdoctoral training grant (NIH 5T32DK007056-44).
Footnotes
Declaration of Interests:
The authors declare no conflicts of interest
References:
- Ahern PP, Faith JJ, and Gordon JI (2014). Mining the human gut microbiota for effector strains that shape the immune system. Immunity 40, 815–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, Liu H, Cross JR, Pfeffer K, Coffer PJ, et al. (2013). Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 504, 451–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, Cheng G, Yamasaki S, Saito T, Ohba Y, et al. (2011). Induction of colonic regulatory T cells by indigenous Clostridium species. Science 331, 337–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayeni FA, Biagi E, Rampelli S, Fiori J, Soverini M, Audu HJ, Cristino S, Caporali L, Schnorr SL, Carelli V, et al. (2018). Infant and Adult Gut Microbiome and Metabolome in Rural Bassa and Urban Settlers from Nigeria. Cell Rep 23, 3056–3067. [DOI] [PubMed] [Google Scholar]
- Bach JF (2002). The effect of infections on susceptibility to autoimmune and allergic diseases. N Engl J Med 347, 911–920. [DOI] [PubMed] [Google Scholar]
- Bacher P, Hohnstein T, Beerbaum E, Röcker M, Blango MG, Kaufmann S, Röhmel J, Eschenhagen P, Grehn C, Seidel K, et al. (2019). Human Anti-fungal Th17 Immunity and Pathology Rely on Cross-Reactivity against Candida albicans. Cell 176, 1340–1355.e1315. [DOI] [PubMed] [Google Scholar]
- Bashiardes S, Godneva A, Elinav E, and Segal E (2018). Towards utilization of the human genome and microbiome for personalized nutrition. Curr Opin Biotechnol 51, 57–63. [DOI] [PubMed] [Google Scholar]
- Belkaid Y, and Hand TW (2014). Role of the microbiota in immunity and inflammation. Cell 157, 121–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattarai Y, Williams BB, Battaglioli EJ, Whitaker WR, Till L, Grover M, Linden DR, Akiba Y, Kandimalla KK, Zachos NC, et al. (2018). Gut Microbiota-Produced Tryptamine Activates an Epithelial G-Protein-Coupled Receptor to Increase Colonic Secretion. Cell Host Microbe 23, 775–785.e775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilen M, Dufour JC, Lagier JC, Cadoret F, Daoud Z, Dubourg G, and Raoult D (2018). The contribution of culturomics to the repertoire of isolated human bacterial and archaeal species. Microbiome 6, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanton LV, Barratt MJ, Charbonneau MR, Ahmed T, and Gordon JI (2016a). Childhood undernutrition, the gut microbiota, and microbiota-directed therapeutics. Science 352, 1533. [DOI] [PubMed] [Google Scholar]
- Blanton LV, Charbonneau MR, Salih T, Barratt MJ, Venkatesh S, Ilkaveya O, Subramanian S, Manary MJ, Trehan I, Jorgensen JM, et al. (2016b). Gut bacteria that prevent growth impairments transmitted by microbiota from malnourished children. Science 351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaser MJ, and Falkow S (2009). What are the consequences of the disappearing human microbiota? Nat Rev Microbiol 7, 887–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britton GJ, Contijoch EJ, Mogno I, Vennaro OH, Llewellyn SR, Ng R, Li Z, Mortha A, Merad M, Das A, et al. (2019). Microbiotas from Humans with Inflammatory Bowel Disease Alter the Balance of Gut Th17 and RORγt. Immunity 50, 212–224.e214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buffie CG, Bucci V, Stein RR, McKenney PT, Ling L, Gobourne A, No D, Liu H, Kinnebrew M, Viale A, et al. (2015). Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature 517, 205–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkitt DP, Walker AR, and Painter NS (1972). Effect of dietary fibre on stools and the transit-times, and its role in the causation of disease. Lancet 2, 1408–1412. [DOI] [PubMed] [Google Scholar]
- Bushman FD (2019). De-Discovery of the Placenta Microbiome. Am J Obstet Gynecol 220, 213–214. [DOI] [PubMed] [Google Scholar]
- Callahan BJ, McMurdie PJ, and Holmes SP (2017). Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J 11, 2639–2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell C, Dikiy S, Bhattarai SK, Chinen T, Matheis F, Calafiore M, Hoyos B, Hanash A, Mucida D, Bucci V, et al. (2018). Extrathymically Generated Regulatory T Cells Establish a Niche for Intestinal Border-Dwelling Bacteria and Affect Physiologic Metabolite Balance. Immunity 48, 1245–1257.e1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7, 335–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Lauber CL, Costello EK, Berg-Lyons D, Gonzalez A, Stombaugh J, Knights D, Gajer P, Ravel J, Fierer N, et al. (2011). Moving pictures of the human microbiome. Genome Biol 12, R50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmody RN, Gerber GK, Luevano JM, Gatti DM, Somes L, Svenson KL, and Turnbaugh PJ (2015). Diet dominates host genotype in shaping the murine gut microbiota. Cell Host Microbe 17, 72–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cebra JJ (1999). Influences of microbiota on intestinal immune system development. Am J Clin Nutr 69, 1046S–1051S. [DOI] [PubMed] [Google Scholar]
- Chai JN, Peng Y, Rengarajan S, Solomon BD, Ai TL, Shen Z, Perry JSA, Knoop KA, Tanoue T, Narushima S, et al. (2017). Helicobacter species are potent drivers of colonic T cell responses in homeostasis and inflammation. Sci Immunol 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan M (2017). Noncommunicable diseases: the slow-motion disaster In Ten years in public health, 2007–2017: report by Dr Margaret Chan, Director-General, World Health Organization (World Health Organization; ), pp. 89–105. [Google Scholar]
- Chen B, Chen H, Shu X, Yin Y, Li J, Qin J, Chen L, Peng K, Xu F, Gu W, et al. (2018). Presence of Segmented Filamentous Bacteria in Human Children and Its Potential Role in the Modulation of Human Gut Immunity. Front Microbiol 9, 1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Nwe PK, Yang Y, Rosen CE, Bielecka AA, Kuchroo M, Cline GW, Kruse AC, Ring AM, Crawford JM, et al. (2019). A Forward Chemical Genetic Screen Reveals Gut Microbiota Metabolites That Modulate Host Physiology. Cell 177, 1217–1231.e1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemente JC, Pehrsson EC, Blaser MJ, Sandhu K, Gao Z, Wang B, Magris M, Hidalgo G, Contreras M, Noya-Alarcón Ó, et al. (2015). The microbiome of uncontacted Amerindians. Sci Adv 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleophas MCP, Ratter JM, Bekkering S, Quintin J, Schraa K, Stroes ES, Netea MG, and Joosten LAB (2019). Effects of oral butyrate supplementation on inflammatory potential of circulating peripheral blood mononuclear cells in healthy and obese males. Sci Rep 9, 775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen LJ, Esterhazy D, Kim SH, Lemetre C, Aguilar RR, Gordon EA, Pickard AJ, Cross JR, Emiliano AB, Han SM, et al. (2017). Commensal bacteria make GPCR ligands that mimic human signalling molecules. Nature 549, 48–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen LJ, Kang HS, Chu J, Huang YH, Gordon EA, Reddy BV, Ternei MA, Craig JW, and Brady SF (2015). Functional metagenomic discovery of bacterial effectors in the human microbiome and isolation of commendamide, a GPCR G2A/132 agonist. Proc Natl Acad Sci U S A 112, E4825–4834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong Y, Feng T, Fujihashi K, Schoeb TR, and Elson CO (2009). A dominant, coordinated T regulatory cell-IgA response to the intestinal microbiota. Proc Natl Acad Sci U S A 106, 19256–19261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costello EK, Stagaman K, Dethlefsen L, Bohannan BJ, and Relman DA (2012). The application of ecological theory toward an understanding of the human microbiome. Science 336, 1255–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotillard A, Kennedy SP, Kong LC, Prifti E, Pons N, Le Chatelier E, Almeida M, Quinquis B, Levenez F, Galleron N, et al. (2013). Dietary intervention impact on gut microbial gene richness. Nature 500, 585–588. [DOI] [PubMed] [Google Scholar]
- Crabbé PA, Bazin H, Eyssen H, and Heremans JF (1968). The normal microbial flora as a major stimulus for proliferation of plasma cells synthesizing IgA in the gut. The germ-free intestinal tract. Int Arch Allergy Appl Immunol 34, 362–375. [DOI] [PubMed] [Google Scholar]
- Davenport ER, Sanders JG, Song SJ, Amato KR, Clark AG, and Knight R (2017). The human microbiome in evolution. BMC Biol 15, 127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, Ling AV, Devlin AS, Varma Y, Fischbach MA, et al. (2014). Diet rapidly and reproducibly alters the human gut microbiome. Nature 505, 559–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MM, and Brodin P (2018). Rebooting Human Immunology. Annu Rev Immunol 36, 843–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Filippis F, Pasolli E, Tett A, Tarallo S, Naccarati A, De Angelis M, Neviani E, Cocolin L, Gobbetti M, Segata N, et al. (2019). Distinct Genetic and Functional Traits of Human Intestinal Prevotella copri Strains Are Associated with Different Habitual Diets. Cell Host Microbe 25, 444–453.e443. [DOI] [PubMed] [Google Scholar]
- De Filippo C, Cavalieri D, Di Paola M, Ramazzotti M, Poullet JB, Massart S, Collini S, Pieraccini G, and Lionetti P (2010). Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci U S A 107, 14691–14696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debelius J, Song SJ, Vazquez-Baeza Y, Xu ZZ, Gonzalez A, and Knight R (2016). Tiny microbes, enormous impacts: what matters in gut microbiome studies? Genome Biol 17, 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejea CM, Fathi P, Craig JM, Boleij A, Taddese R, Geis AL, Wu X, DeStefano Shields CE, Hechenbleikner EM, Huso DL, et al. (2018). Patients with familial adenomatous polyposis harbor colonic biofilms containing tumorigenic bacteria. Science 359, 592–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai MS, Seekatz AM, Koropatkin NM, Kamada N, Hickey CA, Wolter M, Pudlo NA, Kitamoto S, Terrapon N, Muller A, et al. (2016). A Dietary Fiber-Deprived Gut Microbiota Degrades the Colonic Mucus Barrier and Enhances Pathogen Susceptibility. Cell 167, 1339–1353.e1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dethlefsen L, Huse S, Sogin ML, and Relman DA (2008). The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol 6, e280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dethlefsen L, McFall-Ngai M, and Relman DA (2007). An ecological and evolutionary perspective on human-microbe mutualism and disease. Nature 449, 811–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dethlefsen L, and Relman DA (2011). Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc Natl Acad Sci U S A 108 Suppl 1, 4554–4561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devereux G (2006). The increase in the prevalence of asthma and allergy: food for thought. Nat Rev Immunol 6, 869–874. [DOI] [PubMed] [Google Scholar]
- Dianda L, Hanby AM, Wright NA, Sebesteny A, Hayday AC, and Owen MJ (1997). T cell receptor-alpha beta-deficient mice fail to develop colitis in the absence of a microbial environment. Am J Pathol 150, 91–97. [PMC free article] [PubMed] [Google Scholar]
- Dodd D, Spitzer MH, Van Treuren W, Merrill BD, Hryckowian AJ, Higginbottom SK, Le A, Cowan TM, Nolan GP, Fischbach MA, et al. (2017). A gut bacterial pathway metabolizes aromatic amino acids into nine circulating metabolites. Nature 551, 648–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson GP, Ladinsky MS, Yu KB, Sanders JG, Yoo BB, Chou WC, Conner ME, Earl AM, Knight R, Bjorkman PJ, et al. (2018). Gut microbiota utilize immunoglobulin A for mucosal colonization. Science 360, 795–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson GP, Lee SM, and Mazmanian SK (2016). Gut biogeography of the bacterial microbiota. Nat Rev Microbiol 14, 20–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorrestein PC, Mazmanian SK, and Knight R (2014). Finding the missing links among metabolites, microbes, and the host. Immunity 40, 824–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earle KA, Billings G, Sigal M, Lichtman JS, Hansson GC, Elias JE, Amieva MR, Huang KC, and Sonnenburg JL (2015). Quantitative Imaging of Gut Microbiota Spatial Organization. Cell Host Microbe 18, 478–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE, and Relman DA (2005). Diversity of the human intestinal microbial flora. Science 308, 1635–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Kaoutari A, Armougom F, Gordon JI, Raoult D, and Henrissat B (2013). The abundance and variety of carbohydrate-active enzymes in the human gut microbiota. Nat Rev Microbiol 11, 497–504. [DOI] [PubMed] [Google Scholar]
- Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, Peaper DR, Bertin J, Eisenbarth SC, Gordon JI, et al. (2011). NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 145, 745–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ericsson AC, Hagan CE, Davis DJ, and Franklin CL (2014). Segmented filamentous bacteria: commensal microbes with potential effects on research. Comp Med 64, 90–98. [PMC free article] [PubMed] [Google Scholar]
- Faith JJ, Ahern PP, Ridaura VK, Cheng J, and Gordon JI (2014). Identifying gut microbe-host phenotype relationships using combinatorial communities in gnotobiotic mice. Sci Transl Med 6, 220ra211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faith JJ, Guruge JL, Charbonneau M, Subramanian S, Seedorf H, Goodman AL, Clemente JC, Knight R, Heath AC, Leibel RL, et al. (2013). The long-term stability of the human gut microbiota. Science 341, 1237439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faith JJ, McNulty NP, Rey FE, and Gordon JI (2011). Predicting a human gut microbiota’s response to diet in gnotobiotic mice. Science 333, 101–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faith JJ, Rey FE, O’Donnell D, Karlsson M, McNulty NP, Kallstrom G, Goodman AL, and Gordon JI (2010). Creating and characterizing communities of human gut microbes in gnotobiotic mice. ISME J 4, 1094–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falush D, Wirth T, Linz B, Pritchard JK, Stephens M, Kidd M, Blaser MJ, Graham DY, Vacher S, Perez-Perez GI, et al. (2003). Traces of human migrations in Helicobacter pylori populations. Science 299, 1582–1585. [DOI] [PubMed] [Google Scholar]
- Fiorucci S, Biagioli M, Zampella A, and Distrutti E (2018). Bile Acids Activated Receptors Regulate Innate Immunity. Front Immunol 9, 1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorucci S, and Distrutti E (2015). Bile Acid-Activated Receptors, Intestinal Microbiota, and the Treatment of Metabolic Disorders. Trends Mol Med 21, 702–714. [DOI] [PubMed] [Google Scholar]
- Forster SC, Kumar N, Anonye BO, Almeida A, Viciani E, Stares MD, Dunn M, Mkandawire TT, Zhu A, Shao Y, et al. (2019). A human gut bacterial genome and culture collection for improved metagenomic analyses. Nat Biotechnol 37, 186–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster KR, Schluter J, Coyte KZ, and Rakoff-Nahoum S (2017). The evolution of the host microbiome as an ecosystem on a leash. Nature 548, 43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox GE, Magrum LJ, Balch WE, Wolfe RS, and Woese CR (1977). Classification of methanogenic bacteria by 16S ribosomal RNA characterization. Proc Natl Acad Sci U S A 74, 4537–4541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fragiadakis GK, Smits SA, Sonnenburg ED, Van Treuren W, Reid G, Knight R, Manjurano A, Changalucha J, Dominguez-Bello MG, Leach J, et al. (2018). Links between environment, diet, and the hunter-gatherer microbiome. Gut Microbes, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N, and Pace NR (2007). Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci U S A 104, 13780–13785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimura KE, Sitarik AR, Havstad S, Lin DL, Levan S, Fadrosh D, Panzer AR, LaMere B, Rackaityte E, Lukacs NW, et al. (2016). Neonatal gut microbiota associates with childhood multisensitized atopy and T cell differentiation. Nat Med 22, 1187–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, Nakanishi Y, Uetake C, Kato K, Kato T, et al. (2013). Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 504, 446–450. [DOI] [PubMed] [Google Scholar]
- Garrett WS, Lord GM, Punit S, Lugo-Villarino G, Mazmanian SK, Ito S, Glickman JN, and Glimcher LH (2007). Communicable ulcerative colitis induced by T-bet deficiency in the innate immune system. Cell 131, 33–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geva-Zatorsky N, Sefik E, Kua L, Pasman L, Tan TG, Ortiz-Lopez A, Yanortsang TB, Yang L, Jupp R, Mathis D, et al. (2017). Mining the Human Gut Microbiota for Immunomodulatory Organisms. Cell 168, 928–943.e911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert JA, Blaser MJ, Caporaso JG, Jansson JK, Lynch SV, and Knight R (2018). Current understanding of the human microbiome. Nat Med 24, 392–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez A, Petrzelkova KJ, Burns MB, Yeoman CJ, Amato KR, Vlckova K, Modry D, Todd A, Jost Robinson CA, Remis MJ, et al. (2016). Gut Microbiome of Coexisting BaAka Pygmies and Bantu Reflects Gradients of Traditional Subsistence Patterns. Cell Rep 14, 2142–2153. [DOI] [PubMed] [Google Scholar]
- Gong J, Chehrazi-Raffle A, Placencio-Hickok V, Guan M, Hendifar A, and Salgia R (2019). The gut microbiome and response to immune checkpoint inhibitors: preclinical and clinical strategies. Clin Transl Med 8, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon HA, and Pesti L (1971). The gnotobiotic animal as a tool in the study of host microbial relationships. Bacteriol Rev 35, 390–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- H.M.P. Consortium (2012). Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halfvarson J, Brislawn CJ, Lamendella R, Vázquez-Baeza Y, Walters WA, Bramer LM, D’Amato M, Bonfiglio F, McDonald D, Gonzalez A, et al. (2017). Dynamics of the human gut microbiome in inflammatory bowel disease. Nat Microbiol 2, 17004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasin Y, Seldin M, and Lusis A (2017). Multi-omics approaches to disease. Genome Biol 18, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmink BA, Khan MAW, Hermann A, Gopalakrishnan V, and Wargo JA (2019). The microbiome, cancer, and cancer therapy. Nat Med 25, 377–388. [DOI] [PubMed] [Google Scholar]
- Hold GL, Pryde SE, Russell VJ, Furrie E, and Flint HJ (2002). Assessment of microbial diversity in human colonic samples by 16S rDNA sequence analysis. FEMS Microbiol Ecol 39, 33–39. [DOI] [PubMed] [Google Scholar]
- Hryckowian AJ, Van Treuren W, Smits SA, Davis NM, Gardner JO, Bouley DM, and Sonnenburg JL (2018). Microbiota-accessible carbohydrates suppress Clostridium difficile infection in a murine model. Nat Microbiol 3, 662–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugenholtz P, and Pace NR (1996). Identifying microbial diversity in the natural environment: a molecular phylogenetic approach. Trends Biotechnol 14, 190–197. [DOI] [PubMed] [Google Scholar]
- Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, Wei D, Goldfarb KC, Santee CA, Lynch SV, et al. (2009). Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139, 485–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha AR, Davenport ER, Gautam Y, Bhandari D, Tandukar S, Ng KM, Fragiadakis GK, Holmes S, Gautam GP, Leach J, et al. (2018). Gut microbiome transition across a lifestyle gradient in Himalaya. PLoS Biol 16, e2005396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kau AL, Planer JD, Liu J, Rao S, Yatsunenko T, Trehan I, Manary MJ, Liu TC, Stappenbeck TS, Maleta KM, et al. (2015). Functional characterization of IgA-targeted bacterial taxa from undernourished Malawian children that produce diet-dependent enteropathy. Sci Transl Med 7, 276ra224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King DE, Mainous AG, and Lambourne CA (2012). Trends in dietary fiber intake in the United States, 1999–2008. J Acad Nutr Diet 112, 642–648. [DOI] [PubMed] [Google Scholar]
- Klaasen HL, Van der Heijden PJ, Stok W, Poelma FG, Koopman JP, Van den Brink ME, Bakker MH, Eling WM, and Beynen AC (1993). Apathogenic, intestinal, segmented, filamentous bacteria stimulate the mucosal immune system of mice. Infect Immun 61, 303–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight R, Vrbanac A, Taylor BC, Aksenov A, Callewaert C, Debelius J, Gonzalez A, Kosciolek T, McCall LI, McDonald D, et al. (2018). Best practices for analysing microbiomes. Nat Rev Microbiol 16, 410–422. [DOI] [PubMed] [Google Scholar]
- Koh A, De Vadder F, Kovatcheva-Datchary P, and Bäckhed F (2016). From Dietary Fiber to Host Physiology: Short-Chain Fatty Acids as Key Bacterial Metabolites. Cell 165, 1332–1345. [DOI] [PubMed] [Google Scholar]
- Korpela K, Salonen A, Virta LJ, Kekkonen RA, Forslund K, Bork P, and de Vos WM (2016). Intestinal microbiome is related to lifetime antibiotic use in Finnish pre-school children. Nat Commun 7, 10410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundu P, Blacher E, Elinav E, and Pettersson S (2017). Our Gut Microbiome: The Evolving Inner Self. Cell 171, 1481–1493. [DOI] [PubMed] [Google Scholar]
- Laforest-Lapointe I, and Arrieta MC (2018). Microbial Eukaryotes: a Missing Link in Gut Microbiome Studies. mSystems 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagier JC, Dubourg G, Million M, Cadoret F, Bilen M, Fenollar F, Levasseur A, Rolain JM, Fournier PE, and Raoult D (2018). Culturing the human microbiota and culturomics. Nat Rev Microbiol, 540–550. [DOI] [PubMed] [Google Scholar]
- Lagier JC, Khelaifia S, Alou MT, Ndongo S, Dione N, Hugon P, Caputo A, Cadoret F, Traore SI, Seck EH, et al. (2016). Culture of previously uncultured members of the human gut microbiota by culturomics. Nat Microbiol 1, 16203. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Landhuis E (2018). Single-cell approaches to immune profiling. Nature 557, 595–597. [DOI] [PubMed] [Google Scholar]
- Le Chatelier E, Nielsen T, Qin J, Prifti E, Hildebrand F, Falony G, Almeida M, Arumugam M, Batto JM, Kennedy S, et al. (2013). Richness of human gut microbiome correlates with metabolic markers. Nature 500, 541–546. [DOI] [PubMed] [Google Scholar]
- Levy M, Kolodziejczyk AA, Thaiss CA, and Elinav E (2017). Dysbiosis and the immune system. Nat Rev Immunol 17, 219–232. [DOI] [PubMed] [Google Scholar]
- Levy M, Thaiss CA, Zeevi D, Dohnalová L, Zilberman-Schapira G, Mahdi JA, David E, Savidor A, Korem T, Herzig Y, et al. (2015). Microbiota-Modulated Metabolites Shape the Intestinal Microenvironment by Regulating NLRP6 Inflammasome Signaling. Cell 163, 1428–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis JD, Albenberg L, Lee D, Kratz M, Gottlieb K, and Reinisch W (2017). The Importance and Challenges of Dietary Intervention Trials for Inflammatory Bowel Disease. Inflamm Bowel Dis 23, 181–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley RE, Hamady M, Lozupone C, Turnbaugh PJ, Ramey RR, Bircher JS, Schlegel ML, Tucker TA, Schrenzel MD, Knight R, et al. (2008). Evolution of mammals and their gut microbes. Science 320, 1647–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley RE, Turnbaugh PJ, Klein S, and Gordon JI (2006). Microbial ecology: human gut microbes associated with obesity. Nature 444, 1022–1023. [DOI] [PubMed] [Google Scholar]
- Lloyd-Price J, Mahurkar A, Rahnavard G, Crabtree J, Orvis J, Hall AB, Brady A, Creasy HH, McCracken C, Giglio MG, et al. (2017). Strains, functions and dynamics in the expanded Human Microbiome Project. Nature 550, 61–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, Abraham J, Adair T, Aggarwal R, Ahn SY, et al. (2012). Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 380, 2095–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch SV, and Pedersen O (2016). The Human Intestinal Microbiome in Health and Disease. N Engl J Med 375, 2369–2379. [DOI] [PubMed] [Google Scholar]
- Lécuyer E, Rakotobe S, Lengliné-Garnier H, Lebreton C, Picard M, Juste C, Fritzen R, Eberl G, McCoy KD, Macpherson AJ, et al. (2014). Segmented filamentous bacterium uses secondary and tertiary lymphoid tissues to induce gut IgA and specific T helper 17 cell responses. Immunity 40, 608–620. [DOI] [PubMed] [Google Scholar]
- Magurran AE (1988). Ecological diversity and its measurement (London: Croom Helm; ). [Google Scholar]
- Mariño E, Richards JL, McLeod KH, Stanley D, Yap YA, Knight J, McKenzie C, Kranich J, Oliveira AC, Rossello FJ, et al. (2017). Gut microbial metabolites limit the frequency of autoimmune T cells and protect against type 1 diabetes. Nat Immunol 18, 552–562. [DOI] [PubMed] [Google Scholar]
- Martínez I, Stegen JC, Maldonado-Gómez MX, Eren AM, Siba PM, Greenhill AR, and Walter J (2015). The gut microbiota of rural papua new guineans: composition, diversity patterns, and ecological processes. Cell Rep 11, 527–538. [DOI] [PubMed] [Google Scholar]
- Maslowski KM, and Mackay CR (2011). Diet, gut microbiota and immune responses. Nat Immunol 12, 5–9. [DOI] [PubMed] [Google Scholar]
- Maslowski KM, Vieira AT, Ng A, Kranich J, Sierro F, Yu D, Schilter HC, Rolph MS, Mackay F, Artis D, et al. (2009). Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 461, 1282–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald D, Hyde E, Debelius JW, Morton JT, Gonzalez A, Ackermann G, Aksenov AA, Behsaz B, Brennan C, Chen Y, et al. (2018). American Gut: an Open Platform for Citizen Science Microbiome Research. mSystems 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie C, Tan J, Macia L, and Mackay CR (2017). The nutrition-gut microbiome-physiology axis and allergic diseases. Immunol Rev 278, 277–295. [DOI] [PubMed] [Google Scholar]
- Miller ET, Svanbäck R, and Bohannan BJM (2018). Microbiomes as Metacommunities: Understanding Host-Associated Microbes through Metacommunity Ecology. Trends Ecol Evol 33, 926–935. [DOI] [PubMed] [Google Scholar]
- Moeller AH, Caro-Quintero A, Mjungu D, Georgiev AV, Lonsdorf EV, Muller MN, Pusey AE, Peeters M, Hahn BH, and Ochman H (2016). Cospeciation of gut microbiota with hominids. Science 353, 380–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moeller AH, Li Y, Mpoudi Ngole E, Ahuka-Mundeke S, Lonsdorf EV, Pusey AE, Peeters M, Hahn BH, and Ochman H (2014). Rapid changes in the gut microbiome during human evolution. Proc Natl Acad Sci U S A 111, 16431–16435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morowitz MJ, Denef VJ, Costello EK, Thomas BC, Poroyko V, Relman DA, and Banfield JF (2011). Strain-resolved community genomic analysis of gut microbial colonization in a premature infant. Proc Natl Acad Sci U S A 108, 1128–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton ER, Lynch J, Froment A, Lafosse S, Heyer E, Przeworski M, Blekhman R, and Ségurel L (2015). Variation in Rural African Gut Microbiota Is Strongly Correlated with Colonization by Entamoeba and Subsistence. PLoS Genet 11, e1005658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nash AK, Auchtung TA, Wong MC, Smith DP, Gesell JR, Ross MC, Stewart CJ, Metcalf GA, Muzny DM, Gibbs RA, et al. (2017). The gut mycobiome of the Human Microbiome Project healthy cohort. Microbiome 5, 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TL, Vieira-Silva S, Liston A, and Raes J (2015). How informative is the mouse for human gut microbiota research? Dis Model Mech 8, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen R, Bustamante C, Clark AG, Glanowski S, Sackton TB, Hubisz MJ, Fledel-Alon A, Tanenbaum DM, Civello D, White TJ, et al. (2005). A scan for positively selected genes in the genomes of humans and chimpanzees. PLoS Biol 3, e170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obregon-Tito AJ, Tito RY, Metcalf J, Sankaranarayanan K, Clemente JC, Ursell LK, Zech Xu Z, Van Treuren W, Knight R, Gaffney PM, et al. (2015). Subsistence strategies in traditional societies distinguish gut microbiomes. Nat Commun 6, 6505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen LR, Pedersen CB, Leipold MD, and Maecker HT (2019). Getting the Most from Your High-Dimensional Cytometry Data. Immunity 50, 535–536. [DOI] [PubMed] [Google Scholar]
- Palm NW, de Zoete MR, Cullen TW, Barry NA, Stefanowski J, Hao L, Degnan PH, Hu J, Peter I, Zhang W, et al. (2014). Immunoglobulin A coating identifies colitogenic bacteria in inflammatory bowel disease. Cell 158, 1000–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer C, Bik EM, DiGiulio DB, Relman DA, and Brown PO (2007). Development of the human infant intestinal microbiota. PLoS Biol 5, e177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park Y, Subar AF, Hollenbeck A, and Schatzkin A (2011). Dietary fiber intake and mortality in the NIH-AARP diet and health study. Arch Intern Med 171, 1061–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasolli E, Asnicar F, Manara S, Zolfo M, Karcher N, Armanini F, Beghini F, Manghi P, Tett A, Ghensi P, et al. (2019). Extensive Unexplored Human Microbiome Diversity Revealed by Over 150,000 Genomes from Metagenomes Spanning Age, Geography, and Lifestyle. Cell 176, 649–662.e620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paun A, Yau C, Meshkibaf S, Daigneault MC, Marandi L, Mortin-Toth S, Bar-Or A, Allen-Vercoe E, Poussier P, and Danska JS (2019). Association of HLA-dependent islet autoimmunity with systemic antibody responses to intestinal commensal bacteria in children. Sci Immunol 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planer JD, Peng Y, Kau AL, Blanton LV, Ndao IM, Tarr PI, Warner BB, and Gordon JI (2016). Development of the gut microbiota and mucosal IgA responses in twins and gnotobiotic mice. Nature 534, 263–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plunkett CH, and Nagler CR (2017). The Influence of the Microbiome on Allergic Sensitization to Food. J Immunol 198, 581–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pontzer H, Wood BM, and Raichlen DA (2018). Hunter-gatherers as models in public health. Obes Rev 19 Suppl 1, 24–35. [DOI] [PubMed] [Google Scholar]
- Pédron T, Mulet C, Dauga C, Frangeul L, Chervaux C, Grompone G, and Sansonetti PJ (2012). A crypt-specific core microbiota resides in the mouse colon. MBio 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, et al. (2010). A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464, 59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, and Medzhitov R (2004). Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell 118, 229–241. [DOI] [PubMed] [Google Scholar]
- Reese AT, and Dunn RR (2018). Drivers of Microbiome Biodiversity: A Review of General Rules, Feces, and Ignorance. MBio 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Relman DA (2012). The human microbiome: ecosystem resilience and health. Nutr Rev 70 Suppl 1, S2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL, Griffin NW, Lombard V, Henrissat B, Bain JR, et al. (2013). Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 341, 1241214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridlon JM, Kang DJ, and Hylemon PB (2006). Bile salt biotransformations by human intestinal bacteria. J Lipid Res 47, 241–259. [DOI] [PubMed] [Google Scholar]
- Rooks MG, and Garrett WS (2016). Gut microbiota, metabolites and host immunity. Nat Rev Immunol 16, 341–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothschild D, Weissbrod O, Barkan E, Kurilshikov A, Korem T, Zeevi D, Costea PI, Godneva A, Kalka IN, Bar N, et al. (2018). Environment dominates over host genetics in shaping human gut microbiota. Nature 555, 210–215. [DOI] [PubMed] [Google Scholar]
- Schirmer M, Denson L, Vlamakis H, Franzosa EA, Thomas S, Gotman NM, Rufo P, Baker SS, Sauer C, Markowitz J, et al. (2018). Compositional and Temporal Changes in the Gut Microbiome of Pediatric Ulcerative Colitis Patients Are Linked to Disease Course. Cell Host Microbe 24, 600–610.e604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirmer M, Smeekens SP, Vlamakis H, Jaeger M, Oosting M, Franzosa EA, Ter Horst R, Jansen T, Jacobs L, Bonder MJ, et al. (2016). Linking the Human Gut Microbiome to Inflammatory Cytokine Production Capacity. Cell 167, 1125–1136.e1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnorr SL, Candela M, Rampelli S, Centanni M, Consolandi C, Basaglia G, Turroni S, Biagi E, Peano C, Severgnini M, et al. (2014). Gut microbiome of the Hadza hunter-gatherers. Nat Commun 5, 3654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder BO, and Bäckhed F (2016). Signals from the gut microbiota to distant organs in physiology and disease. Nat Med 22, 1079–1089. [DOI] [PubMed] [Google Scholar]
- Schulfer AF, Battaglia T, Alvarez Y, Bijnens L, Ruiz VE, Ho M, Robinson S, Ward T, Cox LM, Rogers AB, et al. (2018). Intergenerational transfer of antibiotic-perturbed microbiota enhances colitis in susceptible mice. Nat Microbiol 3, 234–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao TY, Ang WXG, Jiang TT, Huang FS, Andersen H, Kinder JM, Pham G, Burg AR, Ruff B, Gonzalez T, et al. (2019). Commensal Candida albicans Positively Calibrates Systemic Th17 Immunological Responses. Cell Host Microbe 25, 404–417.e406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpton TJ (2014). An introduction to the analysis of shotgun metagenomic data. Front Plant Sci 5, 209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd ES, DeLoache WC, Pruss KM, Whitaker WR, and Sonnenburg JL (2018). An exclusive metabolic niche enables strain engraftment in the gut microbiota. Nature 557, 434–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheth RU, Li M, Jiang W, Sims PA, Leong KW, and Wang HH Spatial metagenomic characterization of microbial biogeography in the gut. [DOI] [PMC free article] [PubMed]
- Skelly AN, Sato Y, Kearney S, and Honda K (2019). Mining the microbiota for microbial and metabolite-based immunotherapies. Nat Rev Immunol. [DOI] [PubMed] [Google Scholar]
- Smillie CS, Sauk J, Gevers D, Friedman J, Sung J, Youngster I, Hohmann EL, Staley C, Khoruts A, Sadowsky MJ, et al. (2018). Strain Tracking Reveals the Determinants of Bacterial Engraftment in the Human Gut Following Fecal Microbiota Transplantation. Cell Host Microbe 23, 229–240.e225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith K, McCoy KD, and Macpherson AJ (2007). Use of axenic animals in studying the adaptation of mammals to their commensal intestinal microbiota. Semin Immunol 19, 59–69. [DOI] [PubMed] [Google Scholar]
- Smits SA, Leach J, Sonnenburg ED, Gonzalez CG, Lichtman JS, Reid G, Knight R, Manjurano A, Changalucha J, Elias JE, et al. (2017). Seasonal cycling in the gut microbiome of the Hadza hunter-gatherers of Tanzania. Science 357, 802–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenburg ED, Smits SA, Tikhonov M, Higginbottom SK, Wingreen NS, and Sonnenburg JL (2016). Diet-induced extinctions in the gut microbiota compound over generations. Nature 529, 212–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenburg ED, and Sonnenburg JL (2014). Starving our microbial self: the deleterious consequences of a diet deficient in microbiota-accessible carbohydrates. Cell Metab 20, 779–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenburg ED, and Sonnenburg JL (2019). The ancestral and industrialized gut microbiota and implications for human health. Nat Rev Microbiol 17, 383–390. [DOI] [PubMed] [Google Scholar]
- Spitzer MH, and Nolan GP (2016). Mass Cytometry: Single Cells, Many Features. Cell 165, 780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprockett D, Fukami T, and Relman DA (2018). Role of priority effects in the early-life assembly of the gut microbiota. Nat Rev Gastroenterol Hepatol 15, 197–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staley C, Kelly CR, Brandt LJ, Khoruts A, and Sadowsky MJ (2016). Complete Microbiota Engraftment Is Not Essential for Recovery from Recurrent Clostridium difficile Infection following Fecal Microbiota Transplantation. MBio 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Meek B, Doi Y, Muramatsu M, Chiba T, Honjo T, and Fagarasan S (2004). Aberrant expansion of segmented filamentous bacteria in IgA-deficient gut. Proc Natl Acad Sci U S A 101, 1981–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki TA, and Worobey M (2014). Geographical variation of human gut microbial composition. Biol Lett 10, 20131037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swidsinski A, Weber J, Loening-Baucke V, Hale LP, and Lochs H (2005). Spatial organization and composition of the mucosal flora in patients with inflammatory bowel disease. J Clin Microbiol 43, 3380–3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanoue T, Morita S, Plichta DR, Skelly AN, Suda W, Sugiura Y, Narushima S, Vlamakis H, Motoo I, Sugita K, et al. (2019). A defined commensal consortium elicits CD8 T cells and anti-cancer immunity. Nature 565, 600–605. [DOI] [PubMed] [Google Scholar]
- Taurog JD, Richardson JA, Croft JT, Simmons WA, Zhou M, Fernández-Sueiro JL, Balish E, and Hammer RE (1994). The germfree state prevents development of gut and joint inflammatory disease in HLA-B27 transgenic rats. J Exp Med 180, 2359–2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas S, Rouilly V, Patin E, Alanio C, Dubois A, Delval C, Marquier LG, Fauchoux N, Sayegrih S, Vray M, et al. (2015). The Milieu Intérieur study - an integrative approach for study of human immunological variance. Clin Immunol 157, 277–293. [DOI] [PubMed] [Google Scholar]
- Thorburn AN, Macia L, and Mackay CR (2014). Diet, metabolites, and “western-lifestyle” inflammatory diseases. Immunity 40, 833–842. [DOI] [PubMed] [Google Scholar]
- Trehan I, Goldbach HS, LaGrone LN, Meuli GJ, Wang RJ, Maleta KM, and Manary MJ (2013). Antibiotics as part of the management of severe acute malnutrition. N Engl J Med 368, 425–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trompette A, Gollwitzer ES, Pattaroni C, Lopez-Mejia IC, Riva E, Pernot J, Ubags N, Fajas L, Nicod LP, and Marsland BJ (2018). Dietary Fiber Confers Protection against Flu by Shaping Ly6c. Immunity 48, 992–1005.e1008. [DOI] [PubMed] [Google Scholar]
- Trompette A, Gollwitzer ES, Yadava K, Sichelstiel AK, Sprenger N, Ngom-Bru C, Blanchard C, Junt T, Nicod LP, Harris NL, et al. (2014). Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat Med 20, 159–166. [DOI] [PubMed] [Google Scholar]
- Tropini C, Earle KA, Huang KC, and Sonnenburg JL (2017). The Gut Microbiome: Connecting Spatial Organization to Function. Cell Host Microbe 21, 433–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, et al. (2009). A core gut microbiome in obese and lean twins. Nature 457, 480–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vangay P, Johnson AJ, Ward TL, Al-Ghalith GA, Shields-Cutler RR, Hillmann BM, Lucas SK, Beura LK, Thompson EA, Till LM, et al. (2018). US Immigration Westernizes the Human Gut Microbiome. Cell 175, 962–972.e910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesh M, Mukherjee S, Wang H, Li H, Sun K, Benechet AP, Qiu Z, Maher L, Redinbo MR, Phillips RS, et al. (2014). Symbiotic bacterial metabolites regulate gastrointestinal barrier function via the xenobiotic sensor PXR and Toll-like receptor 4. Immunity 41, 296–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernocchi P, Del Chierico F, and Putignani L (2016). Gut Microbiota Profiling: Metabolomics Based Approach to Unravel Compounds Affecting Human Health. Front Microbiol 7, 1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virtue AT, McCright SJ, Wright JM, Jimenez MT, Mowel WK, Kotzin JJ, Joannas L, Basavappa MG, Spencer SP, Clark ML, et al. (2019). The gut microbiota regulates white adipose tissue inflammation and obesity via a family of microRNAs. Sci Transl Med 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner M, and Haider S (2012). New trends in fluorescence in situ hybridization for identification and functional analyses of microbes. Curr Opin Biotechnol 23, 96–102. [DOI] [PubMed] [Google Scholar]
- Wegorzewska MM, Glowacki RWP, Hsieh SA, Donermeyer DL, Hickey CA, Horvath SC, Martens EC, Stappenbeck TS, and Allen PM (2019). Diet modulates colonic T cell responses by regulating the expression of a Bacteroides thetaiotaomicron antigen. Science Immunology 4, eaau9079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittaker RJ, Willis KJ, and Field R (2001). Scale and species richness: towards a general, hierarchical theory of species diversity (Journal of biogeography: Wiley Online Library), pp. 453–470.
- Woese CR (1987). Bacterial evolution. Microbiol Rev 51, 221–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu GD, Chen J, Hoffmann C, Bittinger K, Chen YY, Keilbaugh SA, Bewtra M, Knights D, Walters WA, Knight R, et al. (2011). Linking long-term dietary patterns with gut microbial enterotypes. Science 334, 105–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Pokrovskii M, Ding Y, Yi R, Au C, Harrison OJ, Galan C, Belkaid Y, Bonneau R, and Littman DR (2018). c-MAF-dependent regulatory T cells mediate immunological tolerance to a gut pathobiont. Nature 554, 373–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Torchinsky MB, Gobert M, Xiong H, Xu M, Linehan JL, Alonzo F, Ng C, Chen A, Lin X, et al. (2014). Focused specificity of intestinal TH17 cells towards commensal bacterial antigens. Nature 510, 152–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yassour M, Jason E, Hogstrom LJ, Arthur TD, Tripathi S, Siljander H, Selvenius J, Oikarinen S, Hyöty H, Virtanen SM, et al. (2018). Strain-Level Analysis of Mother-to-Child Bacterial Transmission during the First Few Months of Life. Cell Host Microbe 24, 146–154.e144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, Magris M, Hidalgo G, Baldassano RN, Anokhin AP, et al. (2012). Human gut microbiome viewed across age and geography. Nature 486, 222–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y, Wang Y, Zhu L, Liu W, Liao N, Jiang M, Zhu B, Yu HD, Xiang C, and Wang X (2013). Comparative analysis of the distribution of segmented filamentous bacteria in humans, mice and chickens. ISME J 7, 615–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeevi D, Korem T, Zmora N, Israeli D, Rothschild D, Weinberger A, Ben-Yacov O, Lador D, Avnit-Sagi T, Lotan-Pompan M, et al. (2015). Personalized Nutrition by Prediction of Glycemic Responses. Cell 163, 1079–1094. [DOI] [PubMed] [Google Scholar]
- Zelante T, Iannitti RG, Cunha C, De Luca A, Giovannini G, Pieraccini G, Zecchi R, D’Angelo C, Massi-Benedetti C, Fallarino F, et al. (2013). Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity 39, 372–385. [DOI] [PubMed] [Google Scholar]
- Zhang J, Guo Z, Lim AA, Zheng Y, Koh EY, Ho D, Qiao J, Huo D, Hou Q, Huang W, et al. (2014). Mongolians core gut microbiota and its correlation with seasonal dietary changes. Sci Rep 4, 5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Zhang F, Ding X, Wu G, Lam YY, Wang X, Fu H, Xue X, Lu C, Ma J, et al. (2018). Gut bacteria selectively promoted by dietary fibers alleviate type 2 diabetes. Science 359, 1151–1156. [DOI] [PubMed] [Google Scholar]
- Zierer J, Jackson MA, Kastenmüller G, Mangino M, Long T, Telenti A, Mohney RP, Small KS, Bell JT, Steves CJ, et al. (2018). The fecal metabolome as a functional readout of the gut microbiome. Nat Genet 50, 790–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zmora N, Suez J, and Elinav E (2019). You are what you eat: diet, health and the gut microbiota. Nat Rev Gastroenterol Hepatol 16, 35–56. [DOI] [PubMed] [Google Scholar]
- Zmora N, Zilberman-Schapira G, Suez J, Mor U, Dori-Bachash M, Bashiardes S, Kotler E, Zur M, Regev-Lehavi D, Brik RB, et al. (2018). Personalized Gut Mucosal Colonization Resistance to Empiric Probiotics Is Associated with Unique Host and Microbiome Features. Cell 174, 1388–1405.e1321. [DOI] [PubMed] [Google Scholar]