

Abstract
Viruses are a constant threat to global health as highlighted by the current COVID-19 pandemic. Currently, lack of data underlying how the human host interacts with viruses, including the SARS-CoV-2 virus, limits effective therapeutic intervention. We introduce Viral-Track, a computational method that globally scans unmapped single-cell RNA sequencing (scRNA-seq) data for the presence of viral RNA, enabling transcriptional cell sorting of infected versus bystander cells. We demonstrate the sensitivity and specificity of Viral-Track to systematically detect viruses from multiple models of infection, including hepatitis B virus, in an unsupervised manner. Applying Viral-Track to bronchoalveloar-lavage samples from severe and mild COVID-19 patients reveals a dramatic impact of the virus on the immune system of severe patients compared to mild cases. Viral-Track detects an unexpected co-infection of the human metapneumovirus, present mainly in monocytes perturbed in type-I interferon (IFN)-signaling. Viral-Track provides a robust technology for dissecting the mechanisms of viral-infection and pathology.
Keywords: virus host interactions, COVID-19, single-cell RNA-seq, Viral-Track
Graphical Abstract
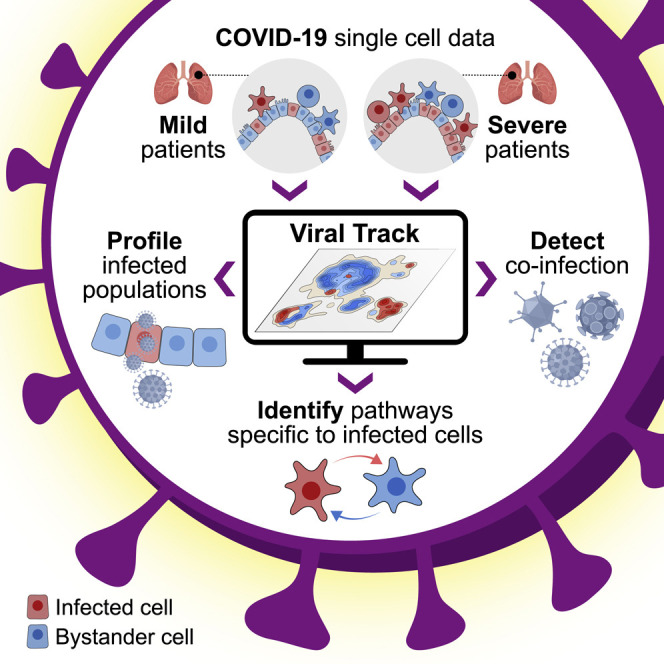
A computational framework that allows for the identification and characterization of virus-infected cells as well as bystander cell responses reveals how SARS-CoV-2 alters the immune responses of patients.
Introduction
The development of efficient vaccines against viral pathogens is considered one of the biggest achievements of modern medicine and has significantly contributed to the increase in life expectancy worldwide. However, no vaccines exist for many life-threatening viruses such as HIV (Burton, 2019), Zika virus (Pierson and Diamond, 2018), or hepatitis C virus (HCV) (Bailey et al., 2019). Additionally, efficient broad-spectrum antiviral drugs are still missing, making infectious diseases a significant challenge for modern health systems. Viruses can also trigger or fuel non-infectious diseases such as cancer (Young and Rickinson, 2004) and are suspected to contribute to various other chronic diseases such as Alzheimer disease (Itzhaki, 2018) and various auto-immune disorders (Münz et al., 2009). The recent emergence of highly pathogenic viruses such as the Ebola virus and the emerging SARS-CoV-2 pandemic recalls the constant threat that viruses represent to global health. So far, the SARS-CoV-2 pandemic has caused a global financial and social catastrophe and is expected to make a significant long-lasting impact on human health (Zhu et al., 2020). Despite intensive research efforts, little is known thus far regarding the interaction of the SARS-CoV-2 virus with the human host and, as a consequence, no efficient treatment has been designed so far (Chen et al., 2020). Moreover, only few therapeutic targets have been identified, highlighting the urgency to develop additional strategies to dissect the virus-host interactions.
Single-cell RNA sequencing (scRNA-seq) is an emerging technology that has been extensively used to study several complex diseases, including cancer (Li et al., 2019), neurodegeneration (Keren-Shaul et al., 2017), and auto-immune (Zhang et al., 2019) and metabolic diseases (Jaitin et al., 2019), providing new insights and revealing new therapeutic targets and strategies (Yofe et al., 2020). In the context of infectious diseases, scRNA-seq studies identified the underlying cells and pathways interacting with various pathogens (Drayman et al., 2019, Shnayder et al., 2018, Steuerman et al., 2018, Zanini et al., 2018). During the immune response to a pathogen, a limited number of antigen-positive or infected cells initiate and modulate the host immune response (Blecher-Gonen et al., 2019), while most of the tissue response is propagated through cytokines, such as type I interferon (IFN) signaling, to bystander, uninfected cells. It is therefore essential to develop new analytical tools to identify the rare infected cells in order to better understand complex host-virus interactions underlying these pathologies. Multiple experimental tools have been developed over the years to track virus-infected cells in vivo, characterize the cellular state of the infected cells, and differentiate them from their bystander neighbors. These include fluorescently labeled pathogens or pathogens expressing fluorescent proteins (De Baets et al., 2015, Blecher-Gonen et al., 2019), as well as reporter mice (Lienenklaus et al., 2009). However, in the case of human clinical samples, these tools are limited, making the pathogen-infected cells and viral reservoir cell types hard to detect.
Viruses exploit their host cells to first express viral genes, optimize the cellular environment, and then fully activate the viral replication program. Because scRNA-seq technologies rely on polyadenylated RNA isolation and amplification, current scRNA-seq methods can, in theory, detect these viral RNA programs and therefore enable accurate identification of the bona fide infected cells and their unique properties at single-cell resolution. While such an approach has already been used to study both in vitro (Drayman et al., 2019, Shnayder et al., 2018) and in vivo infection models (Steuerman et al., 2018), no general computational framework has been developed to detect viruses and analyze host-viral maps in clinical samples. Here, we present a new computational tool, called Viral-Track, that is designed to systematically scan for viral RNA in scRNA-seq data of physiological viral infections using a direct mapping strategy. Viral-Track performs comprehensive mapping of scRNA-seq data onto a large database of known viral genomes, providing precise annotation of the cell types associated with viral infections. Integrating these data with the host transcriptome enables transcriptional sorting and differential profiling of the viral-infected cells compared to bystander cells. Using a new statistical approach for differential gene expression between infected and bystander cells, we are able to recover virus-induced programs and reveal key host factors required for viral replication. Viral-Track is able to annotate the viral program with high accuracy and sensitivity, as we demonstrate in several in vivo mouse models of infection, as well as human samples of hepatitis B virus (HBV) infection. Applying Viral-Track on bronchoalveolar lavage (BAL) samples from moderate and severe COVID-19 patients, we reveal the infection landscape of SARS-CoV-2 and its interaction with the host tissue. Our analysis shows a dramatic impact of the SARS-CoV-2 virus on the immune system of severe patients, compared to mild cases, including replacement of the tissue-resident alveolar macrophages with recruited inflammatory monocytes, neutrophils, and macrophages and an altered CD8+ T cell cytotoxic response. We find that SARS-CoV-2 mainly infects the epithelial and macrophage subsets. In addition, Viral-Track detects an unexpected co-infection of the human metapneumovirus in one of the severe patients. This study establishes Viral-Track as a broadly applicable tool for dissecting mechanisms of viral infections, including identification of the cellular and molecular signatures involved in virus-induced pathologies.
Results
Viral-Track: An Unsupervised Pipeline for Characterization of Viral Infections in scRNA-Seq Data
All scRNA-seq computational packages implement a pipeline that initially aligns the sequenced reads to the expressed part of a reference host genome of the relevant profiled organism. Irrelevant reads, representing other organisms, primers, adaptors, template switching oligonucleotides, and other contaminants are then commonly discarded. We reasoned that during infection, and likely many other pathological processes, these reads can potentially carry valuable information about viral RNA that is discarded in this filtering step. In order to efficiently detect viral reads from raw scRNA-seq data in an unsupervised manner, we developed Viral-Track, an R-based computational pipeline (Figure 1 A; STAR Methods). Briefly, Viral-Track relies on the STAR aligner (Dobin et al., 2013) to map the reads of scRNA-seq data to both the host reference genome and an extensive list of high-quality viral genomes (Stano et al., 2016). Because viral reads are highly repetitive and generate substantial sequencing artifacts, the viral genomes identified in Viral-Track with a sufficient number of mapped reads are then filtered, based on read mapping quality, nucleotide composition, sequence complexity, and genome coverage, to limit the occurrence of false-positives (STAR Methods). Due to the lack of high-quality viral genome annotations, Viral-Track includes de novo transcriptome assembly of the identified viruses using StringTie (Pertea et al., 2015). Finally, viral reads are demultiplexed, quantified using unique molecular identifiers (UMI), and assigned to unique viral transcripts and cells (Figures 1A and S1 A). The Viral-Track algorithm has been designed to robustly handle various types of scRNA-seq datasets, as illustrated below, and is publicly accessible at https://github.com/PierreBSC/Viral-Track.
Figure 1.

Viral-Track Retrieves Viral Reads in a Variety of Tissues, Viral Strains, and Sequencing Platforms
(A) Schematics of the Viral-Track approach. Single-cell sequencing data of cells from an infected tissue, containing infected and bystander cells are analyzed by Viral-Track. Viral-Track maps the sequenced reads to both the host reference genome and a database of viral genomes, overlaying infection status on top of the host transcriptional landscape.
(B) Results of Viral-Track analysis on scRNA-seq data from influenza A PR8-infected mouse lungs. For each viral segment, represented by a dot, the complexity of the sequences (measured by entropy, i.e., how repetitive are the mapped sequences) and the percentage of the segment that is mapped are plotted. Dark red dots correspond to viral segments of the influenza A PR8 strain and yellow dots to segments belonging to other H1N1 influenza strains. Viral segments with more than 50 mapped reads are plotted.
(C) Coverage plot of the influenza A segment NC_002016 (influenza A PR8 segment 7), M2 transcript location estimated using StringTie is shown below with the splicing site position.
(D) Quantification of the number of reads assigned to influenza viral segments across experimental settings. Each dot corresponds to a technical replicate (384-well plate). Two-tailed Welch’s t test was used to compare viral load betwen CD45− and CD45+ cells (p = 0.039).
(E) Quantification of the number of reads assigned to LCMV viral segments in the different zones of the spleen. Each dot corresponds to a technical replicate (384-well plate). Two-tailed Welch’s t test was used to compare viral load between cells from the infected marginal zone to cells from the B zone or the whole speen (p = 0.0067 and 0.0083 respectively).
(F) Result of Viral-Track analysis on scRNA-seq data from a HBV patient. For each viral segment, represented by a dot, the entropy of the sequence and the percentage of the segment that is mapped is plotted. Green dots correspond to viral segments that passed quality control. Viral segments with more than 50 mapped reads are plotted.
(G) Coverage plot of the HBV genome. Locations of the different viral genes from NCBI database are depicted at the bottom.
(H) Enrichment of infected cells across hepatic cell subsets (left panel); red line corresponds to an enrichment of one. Distribution of the number of HBV UMIs per cell in each cell subset (right panel).
See also Figure S1.
Figure S1.

Benchmarking of Viral-Track on Diverse Infection Models, Related to Figure 1
A. Graph chart representing the different steps of the Viral-Track pipeline. B-D. Results of Viral-Track analysis performed on LCMV spleen, LCMV lymph node and VSV lymph node datasets, respectively. Viral segments with more than 50 mapped reads are plotted. (E). Number of detected LCMV (left panel) and VSV (right panel) reads in the different samples from the lymph node experiment. F. Results of Viral-Track analysis performed on the in-vitro HSV-1 data. G. Quantification of the number of HSV-1 reads in HSV-1 infected and control samples. (H). Results of Viral-Track analysis performed on the in-vitro HIV data. I. Quantification of the number of HIV reads in HIV infected and control samples. J. UMAP plot of the liver HBV data, dots are colored by cell subset assignment based on Louvain clustering. K. UMAP plot of the liver HBV data. infected cells are colored in orange and bystander cells in gray.
In order to evaluate the specificity and sensitivity of Viral-Track, we benchmarked Viral-Track on several scRNA-seq datasets (Table S1). These datasets include a large number of experiments we conducted, as well as published studies, that span several tissues (lung, spleen, liver, and lymph node) and a wide range of viruses: influenza A, lymphocytic choriomeningitis virus (LCMV), vesicular stomatitis virus (VSV), herpes simplex virus 1 (HSV-1), human immunodeficiency virus (HIV), and HBV. We first evaluated mouse lungs infected in vivo by influenza A virus and sequenced using MARS-seq2.0 (Keren-Shaul et al., 2019, Steuerman et al., 2018). Viral-Track analysis specifically detected the 8 distinct influenza A viral segments (NC_002016 to NC_002023 Refseq nucleotide sequences) from the specific infecting strain (H1N1 Puerto Rico 8 strain) (Figure 1B). We performed transcriptome assembly to test the feasibility of reconstructing the viral transcriptome from 3′-enriched scRNA-seq data. The results were highly coherent with the current knowledge of influenza A transcriptome, exemplified by Viral-Track’s ability to identify documented spliced transcript structures with single-nucleotide precision. For instance, we identified the exact location of the key splicing site on segment 7 that gives rise to M2 transcript and links nucleotides 51 and 740 (Dubois et al., 2014) (Figure 1C). Quantification of the number of viral reads across different experimental conditions was consistent with current knowledge of the disease, with lung stomal cells of non-immune lineages (CD45−) exhibiting a significantly higher viral load compared to immune cells (CD45+) (p = 0.039, two-tailed Welch’s t test) (Figure 1D).
As inbred mice lack the influenza-specific restriction factor Mx1, influenza A infection is extremely virulent in inbred mice (Haller et al., 1980). Moreover, all influenza A mRNA are capped and polyadenylated, making them an optimal substrate for scRNA-seq isolation and amplification protocols. We therefore evaluated the sensitivity and specificity of Viral-Track in a more challenging dataset. In this model, photoactivatable-GFP (PA-GFP) mice were infected with LCMV (Armstrong acute strain), a virus lacking strong poly(A) mRNA signals (Burrell et al., 2017), via injection to the footpad. 72 h post-infection, CD45+ splenic immune cells from different spatial niches (T zone, B zone, marginal zone, and total spleen) were profiled using the NICHE-seq technology (Medaglia et al., 2017). Even though the LCMV viral mRNAs are not polyadenylated, we detected mRNA molecules that converted to cDNA through priming of the MARS-seq oligo(dt) RT primer, and Viral-Track successfully identified the two viral segments (LCMV segment L [NC_004291] and S [NC_004294]) (Figure S1B), albeit the number of detected reads was an order of magnitude lower than the number observed in influenza A infection (Figure 1E). We detected viral reads in samples from the marginal zone, B zone, and the total spleen, but not in T zone samples, and marginal zone samples exhibited significantly higher viral load compared to B zone and total spleen samples (Figure 1E; p = 0.0067 and 0.0083 respectively, two-tailed Welch’s t test). This observation is in line with the biology of LCMV, which primarily infects macrophages and lymphocytes from the marginal zone of the spleen (Müller et al., 2002).
We next evaluated whether Viral-Track is sensitive to barcode swapping during Illumina-based scRNA-seq (Griffiths et al., 2018), which, in the case of viral RNA detection, can lead to the false assignment of viral reads to uninfected cells. To this end, we infected mice with one of two different viruses, LCMV and VSV, and performed MARS-seq2.0 on CD45+CD19−CD3− non-B/T cells from the auricular draining lymph node 1 day after infection (STAR Methods). All samples were sequenced concurrently to test for cross-sample viral read contamination. For both viruses, Viral-Track was able to identify the correct viral segments (Figures S1C and S1D), with no cross-contamination, evident by the absence of VSV reads detected in the LCMV-infected cells and vice versa (Figure S1E). We further generalized Viral-Track for commonly used scRNA-seq technologies and non-RNA viruses. We applied Viral-Track to scRNA-seq data from a recently publication of human primary cells infected ex vivo with HSV-1, a linear double-stranded DNA virus, generated by the Drop-seq platform (Drayman et al., 2019, Macosko et al., 2015). We found that Viral-Track detected and identified correctly HSV-1 RNA specifically in the infected samples but not in the controls (NC_001806 Refseq nucleotide sequences) (Figures S1F and S1G). Finally, we analyzed scRNA-seq data of CD4+ T cells infected ex vivo with HIV-1 (Bradley et al., 2018), generated using the droplet-based chromium platform (Zheng et al., 2017). Viral-Track successfully identified HIV as the unique virus present in the infected samples (Figures S1H and S1I), but detected significant amounts of HIV-1 viral reads in one control samples probably due to ambient contamination (Yang et al., 2020).
Defining the Host Viral Interactions of HBV Using Viral-Track
We further tested Viral-Track’s applicability for detecting viral reads in human clinical samples. For this purpose, we generated scRNA-seq data from a liver biopsy of an untreated hepatitis B patient and analyzed the data using Viral-Track. Viral-Track successfully identified HBV as the only virus present in the sample (Figure 1F) with 18,420 reads assigned to the HBV genome (NC_003977 Refseq sequence). Coverage analysis revealed a strong peak located at the 5′ end of the C gene, encoding for the main core protein, suggesting that the HBV virus is actively producing virions (Figure 1G). We then overlaid the viral data on the host transcriptome to identify infected and bystander populations. A total of 13,803 cells passed a lenient quality control, permitting apoptotic signals that may arise from viral infection. We identified several non-immune cell types (Figure S1J), including hepatocytes (expressing ALB and APOA2), as well as hepatocytes showing apoptotic signatures (ALB with high expression of mitochondrial genes), sinusoidal endothelial cells (FCN2), and epithelial cells (KRT7). We also observed several subsets of immune cells such as B cells (MS4A1), plasma cells (MZB1), conventional dendritic cells 1 (cDC1; XCR1), plasmacytoid dendritic cells (pDCs) (TCF4), and three different macrophage subsets (expressing TREM2, CD163, and FCN1, respectively). We observed a large diversity among the lymphocyte compartment with CD8+ T cells (CD8A), Th17 cells (CCR6, IL23A), γδ T cells (TRGC1), activated CD4 T cells (LEF1, OX40), natural killer (NK) cells (NKG7), and a distinct cluster of activated CD8+ T cells (CSF2 and TOX2). We analyzed infected cells using automated thresholding over the viral signal (Figure S1J; STAR Methods). As expected, hepatocytes and apoptotic hepatocytes were strongly enriched among the infected cells (Figures 1H and S1K). Interestingly, we also detected viral reads in non-hepatocyte clusters, including two subsets of macrophages (CD163+ and TREM2+ populations, respectively), the cDC1 subset (XCR1+), as well as endothelial (OIT3+ cells) and epithelial cells (KRT7+) (Figures 1H and S1K). Infection of non-hepatocyte clusters, although with relatively low viral load, is coherent with several studies, reporting active infection of macrophages (Faure-Dupuy et al., 2019).
Together, this extensive list of validations demonstrate that Viral-Track is a sensitive and accurate method to detect and identify, in an unsupervised manner, virus strains in diverse scRNA-seq samples, in different tissues, and at varying viral types and loads. Importantly, Viral-Track can be applied to human clinical samples to extract valuable insight into the biology of the host-virus interactions.
Viral-Track Identifies Infected versus Bystander Cells and Uncovers Virus-Induced Pathways
To further evaluate the accuracy of Viral-Track against a well-established model for tracking infection in single cells, we infected mice with a GFP-expressing LCMV virus (LCMV-GFP virus) (Medaglia et al., 2017). We performed MARS-seq on GFP+ splenocytes and total spleen cells 72 h post-infection and analyzed the sequenced cells (Figures S2 A and S2B; STAR Methods). GFP+ cells were enriched for vUMI+ cells compared to total spleen (Figure S2A). We then calculated whether the cells positive for the LCMV-GFP signal (GFP+ cells) were similar to the ones designated by Viral-Track as containing viral UMIs (vUMI+). Following clustering and annotation, we observed similar proportions of GFP+ and vUMI+ cells across cell clusters (Figures 2 A and S2C; R = 0.95, p = 9.0 ∗ 10−12), with monocytes, marginal zone B cells (MZBs), and macrophages being the major infected cell types. We then evaluated the transcriptional signatures within these two sets of cells by computing the Pearson correlation between each pair of cells. We observed similar distribution of Pearson correlation within the GFP+ and vUMI+ monocyte cells (Figure 2B) that was significantly higher (median correlation of 0.65, 0.64, and 0.51, respectively) than the correlation observed between GFP− vUMI− bystander monocytes. We conclude that Viral-Track correctly identifies a homogeneous set of infected cells from in vivo scRNA-seq samples similar to the one identified by conventional reporter viruses, even in the more difficult scenario in which viral transcripts are poorly polyadenylated.
Figure S2.

Comparison of Viral-Track Performance to Fluorescence Tagging Techniques, Related to Figure 2
A. Proportion of vUMI+ cells from total spleen and the LCMV-GFP+ population B. UMAP plot of the spleen LCMV data, spots are colored based on Louvain clustering. C. UMAP plot of the spleen LCMV data, bystander cells are colored in gray, vUMI+ cells are colored in red and GFP+ cells in green. D. Mean gene expression in bystander and infected MZB cells. Genes with a log2FC bigger than 1 or lower than −1 and a corrected p value lower than 0.01 are colored in orange.
Figure 2.

Viral-Track Identifies Virus-Modified Transcription in Infected Cell Subsets
(A) Distribution of vUMI+ and GFP+ cells across cells types found in the spleen.
(B) Distribution of the Pearson Correlation between GFP+ cells, vUMI+, and bystander (GFP−vUMI−) cells. Two-tailed Kruskal-Wallis test.
(C) Number of differentially expressed genes between bystander and infected cells in MZB cells, monocytes, and macrophages.
(D) Top 10 enriched terms identified by Gene Ontology enrichment analysis.
(E) Mean expression of four top differentially expressed genes in bystander and infected MZB cells.
See also Figure S2.
We next evaluated the ability of Viral-Track to detect host factors associated with virus replication. For this purpose, we developed a statistical method that detects differentially expressed genes based on data binarization and complementary log-log regression (STAR Methods; Methods S1). We used this approach to test for transcriptional differences between bystander and infected cells during spleen LCMV infection across the three main infected cell types: macrophages, MZB cells, and monocytes. We observed that MZB cells were the most influenced by the viral infection, compared to monocytes and macrophages (107, 42, and 3 genes upregulated, respectively, Z score >3) (Figure 2C). We performed Gene Ontology enrichment analysis on the upregulated genes in MZB cells and observed a significant enrichment in several pathways, including “chromosome organization,” “DNA replication,” and “cell cycle,” suggesting that LCMV triggers cell division in MZB cells (Figure 2D). Indeed, LCMV-infected MZB cells exhibited higher levels of cell cycle-related genes such as Smc2 (required for chromatin condensation), Cdc6 (regulator of DNA replication), and Stmn1 (regulator of mitotic spindle) (Figures 2E and S2D), but also fibrillarin (Fbl), a host factor whose expression is required by several viruses (Deffrasnes et al., 2016) (Figure 2E). This is in line with a previous report highlighting the ability of LCMV to trigger an abortive form of cell division blocked in the G1 phase (Beier et al., 2015). Altogether, our results show that Viral-Track is sufficient to detect infected cells in in vivo scRNA-seq data and infer the differential gene expression in infected versus bystander cells.
A Single-Cell Map of SARS-CoV-2 Infection in Mild and Severe Patients
COVID-19 is a viral disease caused by SARS-CoV-2 infection, which has recently been recognized as the cause for a pandemic (Wang et al., 2020a). Little is currently known about the course of the disease and how the virus interacts with the host immune system in its mild and severe manifestations. To gain insights on the infection course in humans, we performed scRNA-seq and Viral-Track analysis on BALF samples from three mild and six severe COVID-19 patients (Liao et al., 2020). In total, 50,615 cells passed quality control and were analyzed using the MetaCell algorithm (Baran et al., 2019) (Figure 3 A; STAR Methods). Metacell analysis coarsely grouped the metacells into the myeloid, lymphoid, and epithelial lineages, and each lineage was further subdivided into smaller subsets (Figures 3A, 3B and S3 A). Among epithelial cells, we identified epithelial progenitors (expressing SOX4), type II alveolar cells (AT2, expressing SFTPB), ciliated cells (FOXJ1), ionocytes (CFTR), goblet cells (MUC5B), and club cells (SCGB1A1; Figure S3B). Lymphoid cells consisted several subtypes of CD4+ T cells, including naive CD4+ T cells (expressing CCR7), regulatory T cells (Treg, expressing FOXP3), and T follicular helper cells (Tfh, expressing CXCL13 and PDCD1), but also diverse CD8+ subsets, such as NK cells (NCAM1), resident memory CD8+ T cells (Trm , CD8A, and ZNF683), effector CD8+ T cells (GZMA and GZMK), and cytotoxic CD8+ T cells (GNLY, PRF1), as well as B cells (CD79A; Figure S3C). The myeloid compartment exhibited a high diversity of cell states, including neutrophils (FCGR3B), mast cells (CPA3), alveolar macrophages (FABP4), dendritic cells (DCs; FSCN1), and plasmacytoid DCs (pDC; TCF4) as well as a large diversity of monocytes (FCN1) and monocyte-derived macrophages (SPP1) sub-populations (Figure S3D). These results were robust across different analysis platforms (Liao et al., 2020).
Figure 3.

scRNA-Seq of 6 COVID-19 Samples Reveals Myeloid Remodeling in Severe Patients
(A) A 2-dimensional visualization of 50,615 single cells from three mild and six severe COVID-19 patients, generated by the MetaCell algorithm. Colors indicate grouping of cells into 27 subsets, based on transcriptional similarity (Figure S3A).
(B) Quantification of the three main compartments, myeloid, lymphoid, and epithelial, across the three mild (M1–M3) and six severe (S1–S6) patients.
(C) Density plots depicting projection of cells from the mild (left) and severe (right) patients on the 2D map shown in (A).
(D–F) Quantification of the frequency of specific cell subsets in the myeloid (D), lymphoid (E), and epithelial (F) compartments, across the nine patients. Diamond marks patient S1, co-infected with the human metapneumovirus (Figures 4D–4H). Horizontal lines indicate mean frequency.
(G) Percentage of proliferating cells (determined by thresholding over a cell-cycle-related gene module, detailed in Table S3) in each of 455 metacells, projected on the 2D map shown in (A).
(H) Quantification of the type I interferon response gene module across 455 metacells, projected on the 2D map shown in (A). Color scale represents log2 fold change over the median expression of the module across all metacells.
(I) Differential gene expression analysis. Each panel compares pooled gene expression between naive and non-naive CD4+ T cells (left) and effector and cytotoxic CD8+ T cells (right) cell subsets.
(J) Differential gene expression analysis between cells belonging to AM (left) and SPP1hiC1Qhi macrophages (right) from mild (x axis) and severe (y axis) patients. (I and J) Values represent log2 size-normalized expression (transcripts per 1,000 UMI).
See also Figure S3.
Figure S3.

Detailed Molecular and Cellular Profiling of COVID-19 BAL Samples, Related to Figure 3
A. The confusion matrix of the MetaCell model shown in Figure 3A. Entries denote for each pair of metacells the propensity of cells from both metacells to be clustered together in a bootstrap analysis. B-D. Gene expression profiles of cells belonging to the epithelial (B), lymphoid (C), and myeloid (D). In A-D, color bars indicate association to 27 cell subsets depicted in Figure 3A. E-G. Quantification of the frequency of specific cell subsets in the myeloid (E), lymphoid (F), and epithelial (G) compartments, across the nine patients. Diamond marks patient S1, co-infected with the human Metapneumovirus (Figures 4D-4H). Horizontal lines indicate mean frequency. (H). Projection of IL6 and IL8 (CXCL8) expression on the 2D map shown in Figure 4A. Colors represent expression quantiles.
Comparison of the cellular landscape of mild and severe patients revealed key differences in the composition of BAL samples (Figures 3B and 3C). We found changes to each of the three compartments (Figures 3D–3F and S3E–S3G). While alveolar macrophages and pDC where enriched in the myeloid compartment in the mild patients, the severe patients’ myeloid cells were characterized by a patient-specific diversity associated with accumulation of neutrophils, FCN1+ monocytes, and monocyte-derived SPP1+ macrophages (Figures 3D and S3E). Additionally, NK cells and naive CCR7+ CD4+ T cells were consistently enriched across severe patients BAL, while ZNF683hi CD8+ Trm cells were specific to mild patients (Figures 3E and S3F). We also observed changes in the epithelial compartment, as severe patients exhibited higher numbers of club cells and AT2 cells (Figures 3F and S3G). By investigating expression patterns of shared gene expression programs, we observed that cytotoxic CD8+ cells and the CD4+ Tfh cells are the most proliferative compartments (Figure 3G), while a broad interferon type I response, a hallmark of viral response, is mainly expressed by neutrophils and, to a lesser extent, FCN1+ monocytes (Figure 3H). We next performed in-depth differential gene expression analysis between subsets characteristic of mild or severe patients. We found that CD4+ T cells in the severe patients exhibit a more naive phenotype, expressing higher levels of IL7R, CCR7, S1PR1, and LTB. The CD8+ Trm cells signatures are restricted to the mild patients and have higher levels of the effector molecules XCL1, ITGAE, CXCR6, and ZNF683 (Figure 3I). Comparing gene expression differences in myeloid types between severe and mild patients revealed disease severity-associated upregulation of inflammatory chemokine genes in SPP1+ monocyte-derived macrophages populations (CCL2, CCL3, CCL4, CCL7, and CCL8; Figure 3J), as well as genes associated with hypoxia or oxidative stress (HMOX1 and HIF1A), and downregulation of MHC class II (HLA-A and HLA-DQA1) and type I IFN genes (IFIT1 and OAS1). Alveolar macrophages displayed a severity-associated signature, including upregulation of the chemokines CCL18 and CCL4L2 and the cathepsins CTSL and CTSB (Figure 3J). Together, we identified dramatic differences between the mild and severe COVID-19 patients, including an inflammatory signature and a perturbed immune response associated with the severe manifestation of the COVID-19 disease. These also highlight potential immunotherapy treatment of the severe patients by targeting the hyper inflammatory response that is activated by inflammatory cytokines such as interleukin (IL)-6 and IL-8 (Liu et al., 2019) (Figure S3H).
Viral-Track Identifies Co-infection of SARS-CoV-2 with the Human Metapneumovirus
To characterize the in vivo crosstalk of SARS-CoV-2 with its human host, we applied Viral-Track on the data generated from the nine SARS-CoV-2 patients and the rich cellular landscape we identified. SARS-CoV-2 transcripts were detected in all six severe samples in variable amounts, ranging from less than 400 transcripts to more than 15,000 (Figures 4 A and S4 A). In contrast, no viral reads were detected in the three mild patients (Figure 4A). Coverage analysis revealed that the majority of the viral reads mapped to the 3′ end of the viral segment and corresponded to positive-stranded RNA (Figure 4B). This is in agreement with the coronavirus transcription: due to a nested transcription process all genomic and subgenomic RNA molecules share the same 3′ end (Masters, 2006). We then analyzed the enrichment of vUMIs in the cell populations represented in the BAL samples. We observed a strong enrichment of viral reads in the ciliated and epithelial progenitor population, two known cellular targets of the virus, which express the main receptor of the SARS-CoV-2 virus ACE2, as well as TMPRSS2, a protease essential for SARS-CoV-2 entry (Figures 4C and S4B; Table S2) (Hoffmann et al., 2020). We also observed enrichment of SARS-CoV-2 reads in the SPP1+ macrophage population, suggesting either that SARS-CoV-2 can infect immune cells from the myeloid compartment or that SPP1+ macrophages phagocytose infected cells or viral particles. Differential gene expression analysis between vUMI+ infected and vUMI− bystander SPP1+ macrophages in the patients with the highest viral load, revealed that infected macrophages have a higher expression of chemokines (CCL7, CCL8, and CCL18) and APOE, and a lower expression of TAOK1, a serine/threonine-protein kinase in the p38 MAPK cascade (Figure S4C). Interestingly, CD147 (also known as BSG), a potential new SARS-CoV-2 receptor (Wang et al., 2020b), is expressed by all cell types, including immune cells, suggesting alternative routes for the virus to infect these cells.
Figure 4.

Viral-Track Reveals Infection Specificity and a Co-infection in Severe COVID-19
(A) Total number of viral reads mapped to the SARS-CoV-2 viral genome in the profiled COVID-19 patients.
(B) Coverage plot of the SARS-CoV-2 viral genome.
(C) Enrichment of viral UMIs over expected values across 361 metacells, projected on the 2D map shown in Figure 4A. Color scale indicates log2 observed/expected vUMIs. Only metacells with more than one expected UMI are plotted.
(D) Result of Viral-Track analysis on patient S1. For each viral segment, represented by a dot, the entropy of the sequence (how repetitive are the mapped sequences) and the percentage of the segment that is mapped is plotted. Green dots correspond to viral segments that have passed quality control. Viral segments with more than 50 mapped reads are plotted.
(E) Coverage plot of the human metapneumovirus (hMPV) genome.
(F) Distribution of hMPV UMIs across patient S1 sequenced cells. Red dashed line indicates automatic thresholding of vUMI+ cells.
(G) Enrichment of vUMI+ cells over expected values across 297 metacells, projected on the 2D map shown in Figure 4A. Color scale indicates log2 observed/expected. Only metacells with more than one expected vUMI+ cell are plotted.
(H) Volcano plot showing the relative expression between infected and bystander monocytes of patient S1. Differentially expressed (>1 log2 fold change) and statistically significant (p value <0.01) are colored in orange.
See also Figure S4.
Figure S4.

Viral-Track Performance on COVID-19 BAL Samples, Related to Figure 4
A. Results of Viral-Track analysis performed on samples with highest viral load (patients S2 and S3). B. Mean normalized expression of ACE2, TMPRSS2 and BSG across the 27 cell subsets C. Log2 fold change between vUMI+ and vUMI- SPP1+ monocyte-derived macrophages in patient S2 (x axis) and patient S3 (y axis). D. Relation between total human and viral UMIs in cells from patient S1. E. Projection of cells from patient S1, co-infected with hMPV, on the metacell map from Figure 3A. F. Enrichment analysis of the downregulated genes in hMPV infected monocytes. G. Number of hMPV UMIs in cells producing type I IFN or not. P value was computed by fitting a logistic regression predicting if a cell would produce type I IFN using total host and viral UMIs.
Often in cases of infectious diseases, the specific infecting virus is not known, or may be accompanied by co-infection with additional unknown viruses. Viral-Track applies an unsupervised mapping strategy and is optimally designed to systematically profile the source of infection or co-infections in human clinical samples. To our surprise, Viral-Track analysis of data from one of the severe patients (S1) revealed the presence of a second virus, the human metapneumovirus (hMPV) (NC_039199 Refseq sequence, Figure 4D) with more than one million reads mapped to hMPV in this specific patient. hMPV is a non-segmented, single-stranded, and negative-sense RNA virus that is responsible for upper and lower respiratory tract infections in mostly young (<5 years) children but can also target elderly as well as immuno-compromised patients (Panda et al., 2014). hMPV has been implicated as a possible source of co-infection with the original SARS-CoV virus (Chan et al., 2003).
Coverage analysis revealed that most reads fall into the N, P, M, F, M2, SH, G, but not L, genes of hMPV (Figure 4E). We observed a typical pattern of biased scRNA-seq coverage, indicating that the N, P, M, F, M2, SH, and G genes are actively transcribed, and suggesting that the hMPV was active and replicating at the time of sample collection. Analysis of the viral UMI distribution across cells revealed a substantial viral load in a large subset of the cells, spanning hundreds to thousands vUMIs per infected cell (Figure 4F), independently of the total host UMIs in that cell (Figure S4D). We mapped the infected cells and characterized their distribution across cell types. The infected patient is characterized by high levels of monocytes and CD4+ T cells (Figure S4E). Unlike the SARS-CoV-2 virus infection map, hMPV-infected cells were highly enriched in the monocyte compartment but not in the epithelial and SPP1+ macrophage compartments (Figure 4G).
We tested whether the hMPV could alter the function of the infected monocytes, and therefore influence the course of the disease. Using Viral-Track, we detected a large number of up- and downregulated genes in infected monocytes compared to bystander monocytes (Figure 4H). Interestingly, several key receptor genes required for monocyte activation such as CD16 (FCGR3B), G-CSF receptor (CSF3R), and the formyl peptide receptor (FRP1) were downregulated in the infected compared to the bystander cells. Moreover, we observed a dramatic downregulation of type I Interferon signaling and interferon stimulated genes (ISGs), including viral restriction factors, (e.g., IFIT3). A gene set enrichment analysis (Figure S4F) revealed a strong enrichment of interferon response genes in the downregulated gene set, suggesting that the hMPV is strongly downregulating the IFN response pathway. Several anti-inflammatory genes were upregulated, including LILRB4 (a potent inhibitor of monocyte activation) (Lu et al., 2009) and MITF, a transcription factor known to be a critical suppressor of innate immunity (Harris et al., 2018). Last, we observed a positive and significant association between total number of hMPV UMIs and production of type I IFN, highlighting that while hMPV dampens the response to type I IFN, production of this signal is highly restricted to a rare (~1%) population of cells with a high viral load (Figure S4G). Altogether, our analysis described the distribution of SARS-CoV-2-infected cells in patient’s BAL and revealed the presence of a viral co-infection by the hMPV that dampens the immune activation of the monocyte compartment in the infected patient. Further large-scale analyses of mild versus severe patients need to be conducted to better understand if the co-infection is correlated or even causative in SARS-CoV-2 pathology.
Discussion
The virosphere contains hundreds of thousands of species that constantly interact with their host cells. Over the years, several genomic techniques have been developed to detect virus-derived sequences in human samples. For instance, deep sequencing assays are unbiased and sensitive in their ability to detect extremely rare viral sequences (Moustafa et al., 2017), but do not provide information about the infected cells and the cellular changes induced by the infection. Alternatively, it is possible to combine DNA probes with scRNA-seq to enrich for viral sequences and increase the sensitivity of the assay, but this requires prior knowledge of the viruses present in each sample (Zanini et al., 2018). Here, we present Viral-Track, a robust and unsupervised computational pipeline that can detect viral RNA in any scRNA-seq dataset without the need for experimental modifications or prior knowledge of the infecting agent. Viral-Track was benchmarked on data originating from various tissues, infected by viruses with marked differences in their RNA properties, and generated with different scRNA-seq platforms. We demonstrate that Viral-Track can readily provide essential information on infection status in clinical samples, identify infected cells, probe viral-induced transcriptional alterations, and reveal cases of co-infection.
In practice, only 70%–85% of scRNA-seq reads map to the host genome and represent polyadenylated exonic host transcripts, whereas the remainder of the data is usually overlooked in analysis. We show that these unmapped scRNA-seq reads, in pathological human samples, potentially contain valuable information on viral infection and can be effectively used for viral genome assembly. Viral-Track can resolve complex cellular ecosystems perturbed by viral infection and provide an unbiased map of the infected cells, as well as the transcriptional perturbations induced by the virus at the single cell level. We combine Viral-Track with a novel statistical approach to detect differentially expressed genes from scRNA-seq data, therefore allowing the detection of gene expression changes triggered by viral infection and differentiating them from the more abundant bystander effects, such as type I IFN signaling, at the single cell level. Further advances will focus on applying Viral-Track on largescale datasets containing scRNA-seq data from dozens of samples, leading to robust single-cell viral metagenomic studies that characterize the viral evolution and interactions of virus-induced disease mechanisms with host genetics.
Here, we applied scRNA-seq and Viral-Track analysis to COVID-19 patient-derived samples to provide a cellular and viral atlas of the BAL lung cells from COVID-19 patients. This analysis revealed the diversity of the immune responses across COVID-19 patients and between mild and severe patients. We expect that as the pandemic keeps spreading and global research efforts grow, additional scRNA-seq samples from COVID-19 patients will be generated, including patients treated with emerging immunotherapies (Liu et al., 2019). Such an approach might help to solve key questions including the contribution of the humoral response (Iwasaki and Yang, 2020), the role of the IL6 pathway (Herold et al., 2020), and the immune memory induced by the virus (Prompetchara et al., 2020). Viral-Track can contribute to the global effort to identify the different cellular compartments that are targeted and affected by COVID-19 and other viruses and to detect possible co-infection by unexpected viruses. Co-infections are gaining recognition in the scientific and medical community as critical factors in disease prognosis (Zhang et al., 2020). So far, research focused mainly on co-infections of bacterial sources or of well-known viruses such as influenza A (Wu et al., 2020). Understanding the diversity of viral co-infections and their mechanisms of immune suppression at the cellular and molecular level could therefore provide highly valuable information and lead toward possible therapeutic targets, especially for severe patients, whose treatment options are limited.
Limitations
Viral-Track is a new and powerful tool to decipher host-viral interactions. However, its impact is dependent on several factors, the most critical one being the biochemical and pathophysiological properties of the virus. The absence of a poly(A) tail at the end of viral RNA molecules can significantly decrease their capture rate efficiency in current scRNA-seq techniques, as shown by the LCMV example. This may hinder Viral-Track’s ability to robustly identify infected cells or discern differential expression between infected and bystander cells in such viruses. Other properties of the viral RNA molecules, absence/presence of 5′ capping, nucleotide composition, or dependence on RNA binding proteins, may also affect capture efficiency, and as the technology develops, further research will focus on the classification of molecular features that facilitate or prevent virus identification by scRNA-seq. Notably, non poly(A)-based scRNA-seq techniques, such as RamDA-seq (Hayashi et al., 2018), can be potentially used when profiling these datasets.
Another limiting factor for Viral-Track’s applicability is the potential scarcity of viral reads and infected cells in the sample. As shown in our analysis of SARS-CoV-2-infected samples, only a limited number of viral reads are detected in some of the samples. This may be due to the specific stage of the disease (He et al., 2020), or sampling biases favoring mainly the lung immune populations, with lower representation of non-immune cells that are the primary targets of the virus. Therefore, future COVID-19 scRNA-seq studies should consider this limitation in their experimental design and aim for a better representation of the upper respiratory tissue and the lung parenchyma. Alternative approaches may rely on index sorting and single-cell transcriptome-trained sorting to design optimal gating strategies for capturing and enriching the stromal populations.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| santi-mouse TCRb biotin (clone H57-597) | Biolegend | Cat#:109203; RRID:AB_313426 |
| anti-mouse CD3 biotin (clone 17A2) | Biolegend | Cat#:100243; RRID:AB_2563946 |
| anti-mouse CD19 biotin (clone 6D5) | Biolegend | Cat#:115503; RRID:AB_313638 |
| Bacterial and Virus Strains | ||
| Vesicular Stomatitis Virus (VSV) Indiana Strain | In house | N/A |
| Lymphocytic choriomeningitis virus (LCMV)- Armstrong (Arm) strain | In house | N/A |
| LCMV-Arm-eGFP | In house | N/A |
| Biological Samples | ||
| COVID-19 BAL samples | Shenzhen Third People’s Hospital | N/A |
| HBV liver sample | Shenzhen Third People’s Hospital | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Liberase TL | Roche | Cat#:5401020001 |
| Dnase I, grade II | Roche | Cat#:10104159001 |
| Critical Commercial Assays | ||
| Chromium Single Cell 3ʹ Reagent Kit (v3 chemistry) | 10X Genomics | 1000075 |
| Chromium Single Cell V(D)J Reagent Kits (v1 Chemistry) | 10X Genomics | 1000006 |
| Deposited Data | ||
| Raw data files for the 10X COVID-19 and HBV patients | This paper | GEO: GSE145926 |
| Raw data files for the LCMV/VSV single-cell RNA-seq | This paper | GEO: GSE149443 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6 WT | Jackson laboratories | RRID:IMSR_JAX:000664 |
| Software and Algorithms | ||
| R (3.5.0) | The R project | https://www.r-project.org |
| Python (3.6.5) | Python software foundation | https://www.python.org |
| STAR (2.7.0) | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| Samtools (1.4.0) | Li et al., 2009 | http://www.htslib.org/download/ |
| StringTie (1.3.5) | Pertea et al., 2015 | https://ccb.jhu.edu/software/stringtie/ |
| UMI-tools (1.0.0) | Smith et al., 2017 | https://umi-tools.readthedocs.io/en/latest/ |
| Pagoda2 (0.1.0) | Lake et al., 2018 | https://github.com/hms-dbmi/pagoda2/ |
| MetaCell (0.3.41) | Baran et al., 2019 | https://github.com/tanaylab/metacell |
| Cell Ranger (3.1.0) | N/A | https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/what-is-cell-ranger |
| Other | ||
| MARS-seq reagents | Jaitin et al., 2014 | N/A |
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Ido Amit (ido.amit@weizmann.ac.il).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The whole Viral-Track pipeline is freely available at https://github.com/PierreBSC/Viral-Track. The datasets generated during this study were deposited to the Gene Expression Omnibus (GEO) repository with accession codes GEO: GSE145926 and GSE149443.
Experimental Model and Subject Details
Mice
C57BL/6 mice were purchased from Jackson Laboratories and bred and housed at the Weizmann Institute of Science animal facility, under specific pathogen-free conditions. Female mice, 6-8 weeks of age, were used for all experiments. Experimental protocols were approved by the Weizmann Institute of Science Ethics Committee and were performed according to institutional guidelines.
LCMV/VSV infections
For LCMV infection, 1x105 Focus-Forming Units (FFUs) of the LCMV-Arm strain were injected. For VSV, 1x105 Plaque-Forming Units (PFUs) of the VSV Indiana strain were used. Mice were anesthetized and viruses administered by intradermal injection into the ear pinna. 24h later, mice were sacrificed and auricular LN were harvested.
Subjects
This study was conducted according to the principles expressed in the Declaration of Helsinki. Ethical approval was obtained from the Research Ethics Committee of Shenzhen Third People’s Hospital. All participants provided written informed consent for sample collection and subsequent analyses.
Method Details
Lymph Node MARS-seq data generation
To prepare single cell suspensions for MARS-seq and flow cytometry, auricular LNs were digested in IMDM containing 100mg/mL Liberase TL and 100mg/mL DNase I (both from Roche, Germany) for 20 minutes at 37C. In the last 5 minutes of incubation, EDTA was added at a final concentration of 10mM. Cells were collected, filtered through a 70mm cell strainer, washed with IMDM and maintained strictly at 4C. Cells were sorted with FACSARIA-FUSION (BD Biosciences, San Jose, CA). Prior to sorting, all samples were filtered through a 70-mm nylon mesh. Isolated cells were single cell sorted into 384-well cell capture plates containing 2 mL of lysis solution and barcoded poly(T) reversetranscription (RT) primers for single-cell RNA-seq (Jaitin et al., 2014). Four empty wells were kept in each 384-well plate as a no-cell control for data analysis. Immediately after sorting, each plate was spun down to ensure cell immersion into the lysis solution, snap frozen on dry ice, and stored at –80C until processing Single-cell RNA-seq libraries were prepared as previously described (Jaitin et al., 2014). In brief, mRNA from single cells sorted into capture plates were barcoded and converted into cDNA and then pooled using an automated pipeline. The pooled sample was linearly amplified by T7 in vitro transcription, and the resulting RNA was fragmented and converted into a sequencing-ready library by tagging the samples with pool barcodes and Illumina sequences during ligation, RT, and PCR. Each pool of cells was tested for library quality and concentration as described previously (Jaitin et al., 2014).
Influenza MARS-seq data generation
Full description of the protocol used to generate the Influenza A lung data can be found in Steuerman et al. (2018). Influenza PR8 H1N1 influenza virus (A/Puerto Rico/8/34) was cultivated in hen egg anion. 40μL of diluted virus (6x103 PFU per mouse) were inoculated intranasaly to the mice, or 40μL of PBS for the control mice. Mice were killed 48 or 72h post infection and the lung perfused. Immune and none-immune cells were then extracted using two different extraction protocols before being single-cell sorted in 384-well plates and sequenced using the original MARS-seq protocol (Jaitin et al., 2014).
LCMV spleen MARS-seq data generation
Description for the full protocol used to generate the NICHE-Seq spleen data can be found in Medaglia et al. (2017). Briefly female mice received 1x106 FFU of LCMV-Arm or LCMV-Arm-eGFP in the footpad. 72 hours after injection, spleens were harvested and forced through a 70μm mesh to form a single-cell suspension. Cells were then single-cell sorted using a SORP-aria into 384-well plates containing lysis buffer before processing the plate according to the MARS-seq protocol (Jaitin et al., 2014). All infectious work was performed in designated Biosafety Level 2 (BSL-2) and BSL-3 workspaces in accordance with institutional guidelines
10X HBV liver data generation
The approximately 1 cm long Liver biopsy was homogenized by mincing with scissors into smaller pieces (~0.5 mm2 per piece). Then the tissue was transferred into 10 mL of enzyme mix consisting of 0.3 mg/ml collagenase type IV (Sigma, C9891) and DNase I (Sigma, D5025) for mild enzymatic digestion for 1 h at 37°C while shaking. 5 mL of Dulbecco’s phosphate-buffered saline (DPBS, Thermo, 14190250) supplemented with 5% FBS was added to interrupt digestion and dissociated cells in suspension were passed through a 40 μm strainer and centrifuged at 300 g for 5 min at 4°C. Erythrocytes were lysed using Ammonium-Chloride-Potassium (ACK, Thermo, A1049201), and finally cells were re-suspended in DPBS supplemented with 1% FBS at the concentration of 2, 000 cells/μl for scRNA-Seq. The single-cell capturing and downstream library constructions were performed using the Chromium Single Cell 3′ V3 library preparation kit according to the manufacturer’s protocol (10x Genomics). Full-length cDNA along with cell-barcode identifiers were PCR-amplified and sequencing libraries were prepared and normalized to 3 nM. The constructed library was sequenced on BGI MGISEQ-2000 platform. The Cell Ranger Software Suite (Version 3.1.0) was then used to perform sample de-multiplexing, barcode processing and single-cell 3′ UMI counting with human GRCh38 as the reference genome
10X COVID-19 data generation
20 mL of BALF was obtained and placed on ice. BALF was processed within 2 hours and all operations were performed in BSL-3 laboratory. By passing BALF through a 100 μm nylon cell strainer to filter out lumps, the supernatant was centrifuged and the cells were re-suspended in the cooled RPMI 1640 complete medium. Then the cells were counted in 0.4% trypan blued, centrifuged and re-suspended at the concentration of 2 × 106 /ml for further use. Total 11 μl of single cell suspension and 40 μl barcoded Gel Beads were loaded to Chromium Chip A to generate single-cell gel bead-in-emulsion (GEM). The poly-adenylated transcripts were reverse-transcribed later. The single-cell capturing and downstream library constructions were performed using the Chromium Single Cell 5′ library preparation kit according to the manufacturer’s protocol (10x Genomics). Full-length cDNA along with cell-barcode identifiers were PCR-amplified and sequencing libraries were prepared and normalized to 3 nM. The constructed library was sequenced on BGI MGISEQ-2000 platform. Each sample was sequenced on a different sequencing run to avoid contamination between samples. The Cell Ranger Software Suite (Version 3.1.0) was then used to perform sample de-multiplexing, barcode processing and single-cell 5′ UMI counting with human GRCh38 as the reference genome. A more extensive description of the data generation process can be found in Liao et al. (2020).
Quantification and Statistical Analysis
Read mapping/alignment
Reads were aligned using STAR 2.7.0 (Dobin et al., 2013) in the two-pass mode using the following parameters:–runThreadN was set to 14,–outSAMattributes to ‘NH HI AS nM NM XS’,–outSAMtype to ‘BAM SortedByCoordinate’,–outFilterScoreMinOverLread to 0.6,–outFilterMatchNminPverLread to 0.6, and–twopassMode to ‘Basic’.
Viral database and STAR Index building
As STAR performance drastically dropped when the reference index contains more than 10.000 scaffold/chromosomes, we decided to base our analysis on the limited, but high-quality, viruSITE database (Stano et al., 2016), derived from the NCBI Refseq database. The corresponding FASTA file was downloaded from the viruSITE website (http://www.virusite.org/archive/2019.1/genomes.fasta.zip). STAR indexes were build for both human and mouse samples using respectively the GRCh38 (hg38) and GRCm38 (mm10) reference genomes in addition with the whole viruSITE database. Both reference genomes were downloaded at http://www.ensembl.org//useast.ensembl.org/info/data/ftp/index.html?redirectsrc=//www.ensembl.org%2Finfo%2Fdata%2Fftp%2Findex.html. For the analysis of COVID-19 patients we added the official SARS-CoV-2 reference genome from the Refseq database (NC_045512.2) as it has not been added to the viruSITE database yet. In total this database contains 11988 viral segments from 9431 different viruses.
Processing and filtering of the BAM files
We empirically observed that viral genome sequences can contain highly repetitive subsequences and can therefore create false positive signal. Moreover, some viral genes can share a significant similarity with host genes and also generate mapping artifacts. To remove those, we implemented a strict filtering approach where for each viral segment, a list of mapping features are measured and used to estimate the quality of the mapping.
Following the alignment, the resulting BAM files were processed using the samtools toolbox (Li et al., 2009): first the BAM files were indexed using the samtools index command. Virus segment with more than 50 mapped reads were detected using the samtools idxstats command and a unique bam file was then created for each of the viral segment using the samtools view command.
Each viral bam files were then loaded into an R environment using the readGAlignments() function from the GenomicAlignments package. Various features were then extracted to assess the quality of the mapping:
-
•
The length of the longest mapped contig computed using the coverage() function.
-
•
The percentage of the viral segment that is mapped, also computed using the coverage() function.
-
•
The mean sequencing quality of the mapped reads.
-
•
The number and percentage of uniquely mapped reads.
-
•
The mean sequenced entropy of the mapped reads defined as follows: for each mapped read each nucleotide frequency was extracted using the alphabetFrequency() function of the Biostring package and averaged over the reads. Then the corresponding Shanon entropy was computed using napierian logarithm.
Empirically we determined that a mean sequence entropy bigger than 1.2, a coverage bigger than 5% and the longest contig bigger than three times the mean read length is sufficient to consider a viral segment to be present. This filter configuration eliminated all manually identified artifacts in the various benchmarked datasets and was used unchanged in the HBV and COVID-19 patient data analysis.
When using this strategy, we observed two different kinds of ‘contamination’:
-
•
- the first one consists of the detection of retroviruses specific to the sequenced host species: this is likely due to the expression of host endogenous retro-viral elements that highly similar to ‘real’ retroviruses.
-
•
- the second is the presence of a plant virus, the Tomato brown rugose fruit virus: this is an emerging virus that infects tomatoes and peppers and is endemic in Israel and Jordan. It is highly contagious and spreads easily. We detected this virus only in samples sequenced in Rehovot (Israel) suggesting that it was due to an airborne contamination.
To improve computation speed, this step was parallelised using the doParallel R package.
Transcript reconstruction
As viral genomes are poorly annotated, we decided to systemically reconstruct the transcriptome of each viral segment detected using the transcript assembler StringTie (Pertea et al., 2015). StringTie was used with default parameter except the minimum isoform abundance parameter -f which was set to 0.01 to detect lowly abundant transcripts and the minimal distance between two transcript -g set to 5.
MARS-seq data demultiplexing and UMI count
In order to have a UMI-counting procedure adapted to viral genomes, i.e that distinguish spliced and un-spliced RNA molecule, we developed an in-house R script based on the GenomicRanges, GenomicAlignments and GenomicFeatures packages that used the same strategy as the commercial CellRanger toolkit. Briefly cell barcodes were extracted and compared with a cell barcode whitelist provided by the MARS-seq2 demultiplexing pipeline (Keren-Shaul et al., 2019): cell barcode that belong to the whitelist were kept while cell barcodes that did not belong to the whitelist but that has a highly similar barcode (Hamming distance equal to one, computed using the stringdist() function from the stringdist package) were corrected and kept. UMIs were also extracted and mono-nucleotide UMIs filtered out. Hamming distances between UMIs assigned to the same cell and the same gene were then computed similarly to cell barcodes and UMIs with a Hamming distance equal to one were aggregated and considered as redundant UMIs. Lastly the mapping file was loaded using the readGAlignments() function from the GenomicAlignments package and reads were assigned to a specific viral gene using the findOverlaps() function from the same package. In case the read mapped to a given viral transcript but was not assigned to any viral gene, it was considered as coming from an un-spliced viral RNA molecule.
Drop-seq and 10X data download, pre-processing and demultiplexing
Fastq files were downloaded through the SRA Explorer tool (https://sra-explorer.info/#). Identification and correction of cellular barcode, as well as UMI demultiplexing was performed using UMI-tools 1.0.0 (Smith et al., 2017). First, cell barcodes were extracted and a putative whitelist computed using the umi_tools whitelist command with the parameters ‘–stdin —bc-pattern = CCCCCCCCCCCCCCCCNNNNNNNNNN–log2stderr ’ for the 10X data. For Drop-Seq data the same command is used except the–bc-pattern option set to CCCCCCCCCCCCNNNNNNNN. Collapsing of the UMIs is performed using the command umi_tools extract with parameters ‘—bc-pattern = CCCCCCCCCCCCCCCCNNNNNNNNNN —stdin —filter-cell-barcode’ on the 10X data and with the same command for Drop-seq data except for the–bc-pattern option set to ‘CCCCCCCCCCCCNNNNNNNN’. Following the mapping of the reads to viral genomes and transcript assembly, the mapped reads were assigned to transcripts using the R package Rsubread through the function featureCounts() with default parameters. The command umi_tools count is then used to compute the final expression table with the following parameters:–per-gene–gene-tag = XT–assigned-status-tag = XS–per-cell.
Analysis of the MARS-seq spleen LCMV dataset
High-level analysis were performed using the R-based Pagoda2 pipeline (https://github.com/hms-dbmi/pagoda2/) (Lake et al., 2018) in addition to an in-house R script. Briefly UMI table were loaded and cells with less than 350 UMIs were removed. Lowly abundant genes (less than 100 UMIs) were also removed from analysis. Analysis of the filtered dataset was then performed similarly to our previous paper (Blecher-Gonen et al., 2019) by using the 1500 most variant genes and 100 PCs for dimensionality reduction. kNN graph was build with a parameter K equal to 30 and Louvain’s method used for clustering. Cluster marker genes were computed by using the getdiffGenes function with default parameters. Data were visualized using UMAP (McInnes et al., 2018) implemented by the uwot package.
Analysis of the 10X HBV liver dataset
High-level analysis were performed using the R-based Pagoda2 pipeline (https://github.com/hms-dbmi/pagoda2/ ) (Lake et al., 2018) in addition to an in-house R script. Briefly UMI table were loaded and cells with less than 1000 UMIs were removed. Lowly abundant genes (less than 50 UMIs) were also removed from analysis. Analysis of the filtered dataset was then performed similarly to our previous paper (Blecher-Gonen et al., 2019) by using the 1000 most variant genes and 100 PCs for dimensionality reduction. kNN graph was build with a parameter K equal to 30 and Louvain’s method used for clustering. Cluster marker genes were computed by using the getdiffGenes function with default parameters. Data were visualized using UMAP (McInnes et al., 2018) implemented by the uwot package.
Analysis of the COVID-19 BAL dataset
Upstream processing of reads was done with the CellRanger toolkit, resulting in a UMI table of 75,790 cells with a median UMI count of 2,442, and a median of 868 genes per cell. Cells with less than 500 UMI, or more than 50% mitochondrial genes were excluded.
We used the MetaCell package (Baran et al., 2019) to group single cells from all patients into groups of transcriptionally homogeneous groups, termed metacells . We first removed mitochondrial genes, ERCC, and the diverse immunoglobulin genes (IGH, IGK, and IGL).
Gene features for metacell covers were selected using the parameter Tvm = 0.4, total umi > 30, and more than 4 UMI in at least 3 cells (using the functions mcell_gset_filter_varmean, and mcell_gset_filter_cov). We excluded gene features associated with the cell cycle, stress response, type I interferon, and batch-specific genes via a clustering approach (using the functions mcell_mat_rpt_cor_anchors and mcell_gset_split_by_dsmat). To this end we first identified all genes with a correlation coefficient of at least 0.1 for one of the anchor genes TOP2A, MKI67, PCNA, MCM4, UBE2C, STMN1 (cell cycle), HSPA1B, HSPA1A, DNAJB1, HSPB1, HSPA6, FOS, JUN, CCL4, CCL4L2, MT1E, MT1X, MT1F, TYMS, GADPH, DUT, HMGB2 (stress and batch effect), IFIT1, IFIT3, OASL, IRF7, IRF1, STAT1, and STAT3 (type I IFN). We then hierarchically clustered the correlation matrix between these genes (filtering genes with low coverage and computing correlation using a down-sampled UMI matrix) and selected the gene clusters that contained the above anchor genes. We thus retained 402 genes as features (Table S3). We used metacell to build a kNN graph, perform boot-strapped co-clustering (500 iterations; resampling 70% of the cells in each iteration), and derive a cover of the co-clustering kNN graph (K = 100). Outlier cells featuring gene expresssion higher than 4-fold than the geometric mean in the metacells in at least one gene were discarded.
Annotation of the metacell model was done using the metacell confusion matrix and analysis of marker genes. Detailed annotation within the myeloid, lymphoid and epithelial compartments was performed using hierarchical clustering of the metacell confusion matrix (Figure S3A) and supervised analysis of enriched genes. Metacells enriched for markers from more than one lineage (either T (TRBC2), myeloid (S100A8, C1QB), epithel (KRT18), and plasma cells (XBP1)) were marked as doublets and discarded from further analysis. We additionally discarded metacells of erythrocytes or plasma cells from further analysis.
To derive cell cycle and type I interferon response co-expressed gene modules, we used a clustering-approach as described in the previous paragraphs (using the functions mcell_mat_rpt_cor_anchors and mcell_gset_split_by_dsmat) on a set of cell cycle and interferon genes. We clustered, and manually inspected the resulting clusters, retrieving 72 cell-cycle related and 65 interferon related genes (Table S3).
To extract proportion of proliferating cells (Figure 3G), we calculated for each cells the number of cell-cycle related transcripts per 1,000 UMI. Cells with more than 8 transcripts were determined proliferating.
Testing for infection specificity in COVID-19 BAL dataset
To test for SARS-CoV-2 infection specificity in different cell populations, we computed for each metacell the total number of host UMIs (hUMI) and viral UMIs (vUMI) in the three severe patients (S1-3). We then computed for each metacell its expected vUMI cout, based on its total UMI count (hUMI + vUMI) and the total vUMI proportion across all cells. Figure 4C shows log2 fold change between the observed and expected UMI in each metacell, after adding a regularization factor ( = 5) for each factor. Log2 fold change for the 27 subsets in Figure 3A, and calculated for each severe patient separately is shown in Table S2.
Testing for hMPV infection specificity was done in a similar manner. However, since UMI distribution across cells was abundant and heavy-tailed, we computed for each metacell the expected number of vUMI+ cells instead of its total vUMI count. A cell was determined vUMI+ if it had more than 10 viral UMI, as determined by automatic thresholding (Figure 4F). Figure 4G shows log2 fold change between the observed and expected vUMI+ cells in each metacell, after adding a regularization factor ( = 5) for each factor.
Dichotomized differential gene expression analysis
ScRNA-seq data are intrinsically noisy data with a large proportion of zeros values (previously called dropouts) due to limited sampling of the initial mRNA molecule pool. In addition, cell library size is a major cofounder variable, even after common normalization procedures such as TPM, especially for lowly expressed genes (Hafemeister and Satija, 2019). We therefore improved the method used in our former paper (Blecher-Gonen et al., 2019) that was based on logistic regression.
Briefly our method is based on the global trend of the field that consists in sequencing large amounts of cells but with a limited sequencing depth. Such approach will produce mostly ‘binary’ data and seem to be represent the best compromise on a cost/efficiency point of view (Svensson et al., 2019). So far, several statistical models have been used to model and analyze scRNA-seq count data, most of them being based on the zero-inflated negative-binomial (ZINB) distribution (Finak et al., 2015, Kharchenko et al., 2014). However, recent studies suggested that those models are too complex and introduce artificial complexity (Silverman et al., 2018, Svensson, 2020, Townes et al., 2019). We hypothesize that with such binary data, current models will not fit properly and more suited ones need to be developed.
We therefore developed a new approach based on the binomial complementary Log-log regression (cloglog model): once a given group of cells has been isolated, through Louvain’s clustering for instance (Blondel et al., 2008), we first dichotomized gene expression (if the normalized expression is bigger than 0 the gene is considered as expressed) and then computed a binomial Generalized Linear Model (GLM) with a complementary log log link function (cloglog) using the glm() R function. To mitigate the variation of the library size as well as the global effect of the infection (bystander effect), we include both variables in the regression model. The corresponding p value are then computed using a Likelihood Ratio Test (LRT) and then corrected using Benjamini Hochberg correction (Benjamini and Hochberg, 1995).
For a more comprehensive description of the approach please see Methods S1.
Automate thresholding to detect HBV and hMPV infected cells
In the case of the HBV and hMPV infections, we observed that cells could contain from one to several thousands UMIs. In order to know which cells were really infected and which one contain viral UMIs due to ambient contamination, we decided to apply Otsu’s thresholding after logarithmic transformation. Otsu’s method was implemented using an in-house R script (Otsu, 1979).
Gene set enrichment analysis
Gene set enrichment analysis was performed using the online GSEA tool https://www.gsea-msigdb.org/gsea/index.jsp (Liberzon et al., 2015, Subramanian et al., 2005). The enrichment analysis was performed using the Hallmark and Gene Ontology biological process databases. False detection rate was set to 0.05. Only the top 10 most enriched terms were reported.
Acknowledgments
We thank Dr. Noam Stern Ginossar and Dr. Yoav Golan for careful evaluation of the manuscript; Dr. Etienne Simon-Loriere, Thomas Jacquemont, and Alice Balfourier for valuable advices; Tali Wiesel from the Scientific Illustration unit of the Weizmann Institute for artwork; and members of the Amit laboratory for discussions. I.A. is an Eden and Steven Romick Professorial Chair and supported by Merck KGaA (Darmstadt, Germany), the Chan Zuckerberg Initiative (CZI), an HHMI International Scholar award, the European Research Council Consolidator Grant (ERC-COG) 724471-HemTree2.0, an SCA award from the Wolfson Foundation and Family Charitable Trust, the Thompson Family Foundation, an MRA Established Investigator Award (509044), the Israel Science Foundation (703/15), the Ernest and Bonnie Beutler Research Program for Excellence in Genomic Medicine, the Helen and Martin Kimmel award for innovative investigation, a NeuroMac DFG/Transregional Collaborative Research Center grant, an International Progressive MS Alliance/NMSS (PA-1604 08459), and an Adelis Foundation grant. P.B. is supported by a PhD scholarship from the Ecole Normale Supérieure, Paris. B.S. has received funding from the French Government’s Investissement d’Avenir program, Laboratoire d’Excellence “Integrative Biology of Emerging Infectious Diseases” (ANR-10-LABX-62-IBEID). Z.Z. and Y.L. were supported by fundings from the National Natural Science Foundation of China (91442127 to Z.Z.;81700540 to Y.L.).
Author Contributions
P.B. designed and developed Viral-Track, performed various computational analyses, and wrote the manuscript. A.G. performed computational analyses and wrote the manuscript. Y.L., G.X., and S.Z. designed and performed experiments. Y.B. developed Viral-Track. E.D. contributed to data processing and analysis. R.B.-G., M.C., and C.M. designed and performed experiments. H.L. contributed to data analysis. A.D. contributed to data analysis, manuscript writing, and scientific communication. B.S. contributed to development of computational methods and bioinformatic analysis and wrote the manuscript. Z.Z. conceived, designed, and analyzed experiments and wrote the manuscript. I.A. directed the project, conceived, designed, and analyzed experiments, and wrote the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: May 8, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.cell.2020.05.006.
Supplemental Information
Important gene lists used for analyzing COVID-19 BAL dataset.
References
- Bailey J.R., Barnes E., Cox A.L. Approaches, Progress, and Challenges to Hepatitis C Vaccine Development. Gastroenterology. 2019;156:418–430. doi: 10.1053/j.gastro.2018.08.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baran Y., Bercovich A., Sebe-Pedros A., Lubling Y., Giladi A., Chomsky E., Meir Z., Hoichman M., Lifshitz A., Tanay A. MetaCell: analysis of single-cell RNA-seq data using K-nn graph partitions. Genome Biol. 2019;20:206. doi: 10.1186/s13059-019-1812-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beier J.I., Jokinen J.D., Holz G.E., Whang P.S., Martin A.M., Warner N.L., Arteel G.E., Lukashevich I.S. Novel mechanism of arenavirus-induced liver pathology. PLoS ONE. 2015;10:e0122839. doi: 10.1371/journal.pone.0122839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y., Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B. 1995;57:289–300. [Google Scholar]
- Blecher-Gonen R., Bost P., Hilligan K.L., David E., Salame T.M., Roussel E., Connor L.M., Mayer J.U., Bahar Halpern K., Tóth B. Single-Cell Analysis of Diverse Pathogen Responses Defines a Molecular Roadmap for Generating Antigen-Specific Immunity. Cell Syst. 2019;8:109–121. doi: 10.1016/j.cels.2019.01.001. [DOI] [PubMed] [Google Scholar]
- Blondel V.D., Guillaume J.-L., Lambiotte R., Lefebvre E. Fast unfolding of communities in large networks. J. Stat. Mech. Theory Exp. 2008;2008:P10008. [Google Scholar]
- Bradley T., Ferrari G., Haynes B.F., Margolis D.M., Browne E.P. Single-Cell Analysis of Quiescent HIV Infection Reveals Host Transcriptional Profiles that Regulate Proviral Latency. Cell Rep. 2018;25:107–117. doi: 10.1016/j.celrep.2018.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrell C.J., Howard C.R., Murphy F.A. Academic Press; 2017. Fenner and White’s Medical Virology. [Google Scholar]
- Burton D.R. Advancing an HIV vaccine; advancing vaccinology. Nat. Rev. Immunol. 2019;19:77–78. doi: 10.1038/s41577-018-0103-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan P.K.S., Tam J.S., Lam C.-W., Chan E., Wu A., Li C.-K., Buckley T.A., Ng K.-C., Joynt G.M., Cheng F.W.T. Human metapneumovirus detection in patients with severe acute respiratory syndrome. Emerg. Infect. Dis. 2003;9:1058–1063. doi: 10.3201/eid0909.030304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Baets S., Verhelst J., Van den Hoecke S., Smet A., Schotsaert M., Job E.R., Roose K., Schepens B., Fiers W., Saelens X. A GFP expressing influenza A virus to report in vivo tropism and protection by a matrix protein 2 ectodomain-specific monoclonal antibody. PLoS ONE. 2015;10:e0121491. doi: 10.1371/journal.pone.0121491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deffrasnes C., Marsh G.A., Foo C.H., Rootes C.L., Gould C.M., Grusovin J., Monaghan P., Lo M.K., Tompkins S.M., Adams T.E. Genome-wide siRNA screening at biosafety level 4 reveals a crucial role for fibrillarin in henipavirus infection. PLoS Pathog. 2016;12:e1005478. doi: 10.1371/journal.ppat.1005478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drayman N., Patel P., Vistain L., Tay S. HSV-1 single-cell analysis reveals the activation of anti-viral and developmental programs in distinct sub-populations. eLife. 2019;8:e46339. doi: 10.7554/eLife.46339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois J., Terrier O., Rosa-Calatrava M. Influenza viruses and mRNA splicing: doing more with less. MBio. 2014;5:e00070-14. doi: 10.1128/mBio.00070-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faure-Dupuy S., Delphin M., Aillot L., Dimier L., Lebossé F., Fresquet J., Parent R., Matter M.S., Rivoire M., Bendriss-Vermare N. Hepatitis B virus-induced modulation of liver macrophage function promotes hepatocyte infection. J. Hepatol. 2019;71:1086–1098. doi: 10.1016/j.jhep.2019.06.032. [DOI] [PubMed] [Google Scholar]
- Finak G., McDavid A., Yajima M., Deng J., Gersuk V., Shalek A.K., Slichter C.K., Miller H.W., McElrath M.J., Prlic M. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 2015;16:278. doi: 10.1186/s13059-015-0844-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths J.A., Richard A.C., Bach K., Lun A.T.L., Marioni J.C. Detection and removal of barcode swapping in single-cell RNA-seq data. Nat. Commun. 2018;9:2667. doi: 10.1038/s41467-018-05083-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafemeister C., Satija R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 2019;20:296. doi: 10.1186/s13059-019-1874-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haller O., Arnheiter H., Lindenmann J., Gresser I. Host gene influences sensitivity to interferon action selectively for influenza virus. Nature. 1980;283:660–662. doi: 10.1038/283660a0. [DOI] [PubMed] [Google Scholar]
- Harris M.L., Fufa T.D., Palmer J.W., Joshi S.S., Larson D.M., Incao A., Gildea D.E., Trivedi N.S., Lee A.N., Day C.-P., NISC Comparative Sequencing Program A direct link between MITF, innate immunity, and hair graying. PLoS Biol. 2018;16:e2003648. doi: 10.1371/journal.pbio.2003648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T., Ozaki H., Sasagawa Y., Umeda M., Danno H., Nikaido I. Single-cell full-length total RNA sequencing uncovers dynamics of recursive splicing and enhancer RNAs. Nat. Commun. 2018;9:619. doi: 10.1038/s41467-018-02866-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X., Lau E.H.Y., Wu P., Deng X., Wang J., Hao X., Lau Y.C., Wong J.Y., Guan Y., Tan X. Temporal dynamics in viral shedding and transmissibility of COVID-19. Nat. Med. 2020;26:672–675. doi: 10.1038/s41591-020-0869-5. [DOI] [PubMed] [Google Scholar]
- Herold T., Jurinovic V., Arnreich C., Hellmuth J.C., von Bergwelt-Baildon M., Klein M., Weinberger T. Level of IL-6 predicts respiratory failure in hospitalized symptomatic COVID-19 patients. MedRxiv. 2020 doi: 10.1101/2020.04.01.20047381. [DOI] [Google Scholar]
- Hoffmann M., Kleine-Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S., Schiergens T.S., Herrler G., Wu N.-H., Nitsche A. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell. 2020;181:271–280. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itzhaki R.F. Corroboration of a Major Role for Herpes Simplex Virus Type 1 in Alzheimer’s Disease. Front. Aging Neurosci. 2018;10:324. doi: 10.3389/fnagi.2018.00324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki A., Yang Y. The potential danger of suboptimal antibody responses in COVID-19. Nat. Rev. Immunol. 2020 doi: 10.1038/s41577-020-0321-6. Published online April 21, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaitin D.A., Kenigsberg E., Keren-Shaul H., Elefant N., Paul F., Zaretsky I., Mildner A., Cohen N., Jung S., Tanay A., Amit I. Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science. 2014;343:776–779. doi: 10.1126/science.1247651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaitin D.A., Adlung L., Thaiss C.A., Weiner A., Li B., Descamps H., Lundgren P., Bleriot C., Liu Z., Deczkowska A. Lipid-Associated Macrophages Control Metabolic Homeostasis in a Trem2-Dependent Manner. Cell. 2019;178:686–698.. doi: 10.1016/j.cell.2019.05.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J., Liu D., Liu L., Liu P., Xu Q., Xia L., Ling Y., Huang D., Song S., Zhang D. A pilot study of hydroxychloroquine in treatment of patients with common coronavirus disease-19 (COVID-19). J. Zhejiang Univ. Medical Sci. 2020;49 doi: 10.3785/j.issn.1008-9292.2020.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren-Shaul H., Spinrad A., Weiner A., Matcovitch-Natan O., Dvir-Szternfeld R., Ulland T.K., David E., Baruch K., Lara-Astaiso D., Toth B. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell. 2017;169:1276–1290. doi: 10.1016/j.cell.2017.05.018. [DOI] [PubMed] [Google Scholar]
- Keren-Shaul H., Kenigsberg E., Jaitin D.A., David E., Paul F., Tanay A., Amit I. MARS-seq2.0: an experimental and analytical pipeline for indexed sorting combined with single-cell RNA sequencing. Nat. Protoc. 2019;14:1841–1862. doi: 10.1038/s41596-019-0164-4. [DOI] [PubMed] [Google Scholar]
- Kharchenko P.V., Silberstein L., Scadden D.T. Bayesian approach to single-cell differential expression analysis. Nat. Methods. 2014;11:740–742. doi: 10.1038/nmeth.2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lake B.B., Chen S., Sos B.C., Fan J., Kaeser G.E., Yung Y.C., Duong T.E., Gao D., Chun J., Kharchenko P.V., Zhang K. Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nat. Biotechnol. 2018;36:70–80. doi: 10.1038/nbt.4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., van der Leun A.M., Yofe I., Lubling Y., Gelbard-Solodkin D., van Akkooi A.C.J., van den Braber M., Rozeman E.A., Haanen J.B.A.G., Blank C.U. Dysfunctional CD8 T Cells Form a Proliferative, Dynamically Regulated Compartment within Human Melanoma. Cell. 2019;176:775–789. doi: 10.1016/j.cell.2018.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao M., Liu Y., Yuan J., Wen Y., Xu G., Zhao J., Cheng L., Li J., Wang X., Wang F. The landscape of lung bronchoalveolar immune cells in COVID-19 revealed by single-cell RNA sequencing. medRxiv. 2020 doi: 10.1101/2020.02.23.20026690. [DOI] [Google Scholar]
- Liberzon A., Birger C., Thorvaldsdóttir H., Ghandi M., Mesirov J.P., Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1:417–425. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lienenklaus S., Cornitescu M., Ziętara N., Łyszkiewicz M., Gekara N., Jabłónska J., Edenhofer F., Rajewsky K., Bruder D., Hafner M. Novel reporter mouse reveals constitutive and inflammatory expression of IFN-β in vivo. J. Immunol. 2009;183:3229–3236. doi: 10.4049/jimmunol.0804277. [DOI] [PubMed] [Google Scholar]
- Liu L., Wei Q., Lin Q., Fang J., Wang H., Kwok H., Tang H., Nishiura K., Peng J., Tan Z. Anti-spike IgG causes severe acute lung injury by skewing macrophage responses during acute SARS-CoV infection. JCI Insight. 2019;4:123158. doi: 10.1172/jci.insight.123158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H.K., Rentero C., Raftery M.J., Borges L., Bryant K., Tedla N. Leukocyte Ig-like receptor B4 (LILRB4) is a potent inhibitor of FcgammaRI-mediated monocyte activation via dephosphorylation of multiple kinases. J. Biol. Chem. 2009;284:34839–34848. doi: 10.1074/jbc.M109.035683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macosko E.Z., Basu A., Satija R., Nemesh J., Shekhar K., Goldman M., Tirosh I., Bialas A.R., Kamitaki N., Martersteck E.M. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell. 2015;161:1202–1214. doi: 10.1016/j.cell.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masters P.S. The molecular biology of coronaviruses. Adv. Virus Res. 2006;66:193–292. doi: 10.1016/S0065-3527(06)66005-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McInnes L., Healy J., Melville J. Umap: Uniform manifold approximation and projection for dimension reduction. ArXiv. 2018 ArXiv1802.03426. [Google Scholar]
- Medaglia C., Giladi A., Stoler-Barak L., De Giovanni M., Salame T.M., Biram A., David E., Li H., Iannacone M., Shulman Z. Spatial reconstruction of immune niches by combining photoactivatable reporters and scRNA-seq. Science. 2017;358:1622–1626. doi: 10.1126/science.aao4277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moustafa A., Xie C., Kirkness E., Biggs W., Wong E., Turpaz Y., Bloom K., Delwart E., Nelson K.E., Venter J.C., Telenti A. The blood DNA virome in 8,000 humans. PLoS Pathog. 2017;13:e1006292. doi: 10.1371/journal.ppat.1006292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller S., Hunziker L., Enzler S., Bühler-Jungo M., Di Santo J.P., Zinkernagel R.M., Mueller C. Role of an intact splenic microarchitecture in early lymphocytic choriomeningitis virus production. J. Virol. 2002;76:2375–2383. doi: 10.1128/jvi.76.5.2375-2383.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Münz C., Lünemann J.D., Getts M.T., Miller S.D. Antiviral immune responses: triggers of or triggered by autoimmunity? Nat. Rev. Immunol. 2009;9:246–258. doi: 10.1038/nri2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsu N. A Threshold Selection Method from Gray-Level Histograms. IEEE Trans. Syst. Man Cybern. 1979;9:62–66. [Google Scholar]
- Panda S., Mohakud N.K., Pena L., Kumar S. Human metapneumovirus: review of an important respiratory pathogen. Int. J. Infect. Dis. 2014;25:45–52. doi: 10.1016/j.ijid.2014.03.1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea M., Pertea G.M., Antonescu C.M., Chang T.-C., Mendell J.T., Salzberg S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015;33:290–295. doi: 10.1038/nbt.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierson T.C., Diamond M.S. The emergence of Zika virus and its new clinical syndromes. Nature. 2018;560:573–581. doi: 10.1038/s41586-018-0446-y. [DOI] [PubMed] [Google Scholar]
- Prompetchara E., Ketloy C., Palaga T. Immune responses in COVID-19 and potential vaccines: Lessons learned from SARS and MERS epidemic. Asian Pac. J. Allergy Immunol. 2020;38:1–9. doi: 10.12932/AP-200220-0772. [DOI] [PubMed] [Google Scholar]
- Shnayder M., Nachshon A., Krishna B., Poole E., Boshkov A., Binyamin A., Maza I., Sinclair J., Schwartz M., Stern-Ginossar N. Defining the Transcriptional Landscape during Cytomegalovirus Latency with Single-Cell RNA Sequencing. MBio. 2018;9:e00013-18. doi: 10.1128/mBio.00013-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman J.D., Roche K., Mukherjee S., David L.A. Naught all zeros in sequence count data are the same. bioRxiv. 2018 doi: 10.1101/477794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith T., Heger A., Sudbery I. UMI-tools: modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res. 2017;27:491–499. doi: 10.1101/gr.209601.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stano M., Beke G., Klucar L. viruSITE-integrated database for viral genomics. Database J. Biol. Databases Curation 2016. 2016 doi: 10.1093/database/baw162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steuerman Y., Cohen M., Peshes-Yaloz N., Valadarsky L., Cohn O., David E., Frishberg A., Mayo L., Bacharach E., Amit I., Gat-Viks I. Dissection of Influenza Infection In Vivo by Single-Cell RNA Sequencing. Cell Syst. 2018;6:679–691. doi: 10.1016/j.cels.2018.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson V. Droplet scRNA-seq is not zero-inflated. Nat. Biotechnol. 2020;38:147–150. doi: 10.1038/s41587-019-0379-5. [DOI] [PubMed] [Google Scholar]
- Svensson V., da Veiga Beltrame E., Pachter L. Quantifying the tradeoff between sequencing depth and cell number in single-cell RNA-seq. bioRxiv. 2019 doi: 10.1101/762773. [DOI] [Google Scholar]
- Townes F.W., Hicks S.C., Aryee M.J., Irizarry R.A. Feature selection and dimension reduction for single-cell RNA-Seq based on a multinomial model. Genome Biol. 2019;20:295. doi: 10.1186/s13059-019-1861-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C., Horby P.W., Hayden F.G., Gao G.F. A novel coronavirus outbreak of global health concern. Lancet. 2020;395:470–473. doi: 10.1016/S0140-6736(20)30185-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K., Chen W., Zhou Y.-S., Lian J.-Q., Zhang Z., Du P., Gong L., Zhang Y., Cui H.-Y., Geng J.-J. SARS-CoV-2 invades host cells via a novel route: CD147-spike protein. BioRxiv. 2020 doi: 10.1101/2020.03.14.988345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X., Cai Y., Huang X., Yu X., Zhao L., Wang F., Li Q., Gu S., Xu T., Li Y. Co-infection with SARS-CoV-2 and Influenza A Virus in Patient with Pneumonia, China. Emerg. Infect. Dis. 2020;26 doi: 10.3201/eid2606.200299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S., Corbett S.E., Koga Y., Wang Z., Johnson W.E., Yajima M., Campbell J.D. Decontamination of ambient RNA in single-cell RNA-seq with DecontX. Genome Biol. 2020;21:57. doi: 10.1186/s13059-020-1950-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yofe I., Dahan R., Amit I. Single-cell genomic approaches for developing the next generation of immunotherapies. Nat. Med. 2020;26:171–177. doi: 10.1038/s41591-019-0736-4. [DOI] [PubMed] [Google Scholar]
- Young L.S., Rickinson A.B. Epstein-Barr virus: 40 years on. Nat. Rev. Cancer. 2004;4:757–768. doi: 10.1038/nrc1452. [DOI] [PubMed] [Google Scholar]
- Zanini F., Robinson M.L., Croote D., Sahoo M.K., Sanz A.M., Ortiz-Lasso E., Albornoz L.L., Rosso F., Montoya J.G., Goo L. Virus-inclusive single-cell RNA sequencing reveals the molecular signature of progression to severe dengue. Proc. Natl. Acad. Sci. USA. 2018;115:E12363–E12369. doi: 10.1073/pnas.1813819115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F., Wei K., Slowikowski K., Fonseka C.Y., Rao D.A., Kelly S., Goodman S.M., Tabechian D., Hughes L.B., Salomon-Escoto K., Accelerating Medicines Partnership Rheumatoid Arthritis and Systemic Lupus Erythematosus (AMP RA/SLE) Consortium Defining inflammatory cell states in rheumatoid arthritis joint synovial tissues by integrating single-cell transcriptomics and mass cytometry. Nat. Immunol. 2019;20:928–942. doi: 10.1038/s41590-019-0378-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H., Wang X., Fu Z., Luo M., Zhang Z., Zhang K., He Y., Wan D., Zhang L., Wang J. Potential Factors for Prediction of Disease Severity of COVID-19 Patients. MedRxiv. 2020 doi: 10.1101/2020.03.20.20039818. [DOI] [Google Scholar]
- Zheng G.X.Y., Terry J.M., Belgrader P., Ryvkin P., Bent Z.W., Wilson R., Ziraldo S.B., Wheeler T.D., McDermott G.P., Zhu J. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017;8:14049. doi: 10.1038/ncomms14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu N., Zhang D., Wang W., Li X., Yang B., Song J., Zhao X., Huang B., Shi W., Lu R., China Novel Coronavirus Investigating and Research Team A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020;382:727–733. doi: 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Important gene lists used for analyzing COVID-19 BAL dataset.
Data Availability Statement
The whole Viral-Track pipeline is freely available at https://github.com/PierreBSC/Viral-Track. The datasets generated during this study were deposited to the Gene Expression Omnibus (GEO) repository with accession codes GEO: GSE145926 and GSE149443.