

Abstract
Liver fibrosis, a consequence of unhealthy modern lifestyles, has a growing impact on human health, particularly in developed countries. Here, we have explored the anti‐fibrotic effects of propylene glycol alginate sodium sulphate (PSS), a natural extract from brown algae, in fibrotic mice and cell models. Thus, we established bile duct ligature and carbon tetrachloride mouse models and LX‐2 cell models with or without PSS treatment. Liver pathological sections and the relevant indicators in serum and liver tissues were examined. PSS prevented hepatic injury and fibrosis to a significant extent, and induced up‐regulation of matrix metalloproteinase‐2 and down‐regulation of tissue inhibitor of metalloproteinase‐1 through suppressing the transforming growth factor β1 (TGF‐β1)/Smad pathway. PSS additionally exerted an anti‐autophagy effect through suppressing the Janus kinase (JAK) 2/transducer and activator of transcription 3 (STAT3) pathway. In conclusion, PSS prevents hepatic fibrosis by suppressing inflammation, promoting extracellular matrix (ECM) decomposition and inactivating hepatic stellate cells through mechanisms involving the TGF‐β1/Smad2/3 and JAK2/STAT3 pathways in vivo and in vitro.
Keywords: autophagy, JAK2/STAT3, liver fibrosis, propylene glycol alginate sodium sulphate, TGF‐β1/Smad2/3
1. INTRODUCTION
Liver fibrosis or scarring, a damage‐induced reaction to heal wounds by encapsulating the injury, is a significant cause of morbidity worldwide and contributes to 45% mortality in developed countries. 1 , 2 Following chronic injury, the extracellular matrix (ECM) accumulates, mainly due to activation of hepatic stellate cells (HSC). 3 , 4 HSCs, normally quiescent cells that store vitamin A, can be proliferated in response to liver injury. This process goes along with release of the transforming growth factor β1 (TGF‐β1) from activated Kupffer cells and is characterized by the appearance of smooth muscle α‐actin (α‐SMA). 1 , 3 , 5 , 6 Collagen type I (Col‐1), a crucial constituent of the fibril‐forming matrix, increases gradually in a background of HSC activation and presents a relatively late event in hepatic fibrosis. 1 , 7 Matrix metalloproteinase‐2 (MMP‐2), a basement membrane protease and type IV collagenase, degrades the extracellular matrix and acts against fibrosis. Conversely, binding of tissue inhibitors of metalloproteinase‐1 (TIMP‐1) to interstitial collagenases suppresses degradation of the accumulating matrix and prevents clearance of HSCs. 8 Smad2 and Smad3 proteins are phosphorylated with release of TGF‐β1 and, in turn, promoting its activity and HSC activation. 1
Propylene glycol alginate sodium sulphate (PSS) is a heparinoid drug initially identified in brown algae by a Chinese scientist. 9 , 10 Studies have reported protective effects of PSS on concanavalin A and ischaemia reperfusion‐induced liver injury models that are attributed to its anti‐inflammatory ability and activity in reducing blood viscosity. 11 , 12 However, no literature has studied the efficiency of PSS in liver fibrosis. Interestingly, low–molecular‐weight heparin is reported to exert anti‐fibrotic effects, 13 bringing forward the hypothesis that PSS could prevent hepatic fibrosis in mice.
Autophagy is a well‐known dynamic cellular process that plays an important role in fibrosis. 14 Activation of HSCs has been shown to facilitate autophagic flux, 15 , 16 and involvement of specific molecular pathways, including Janus kinase (JAK) 2/signal transducer and activator of transcription 3 (STAT3), was in regulation of autophagy as reported. 17 , 18 Furthermore, several studies have investigated the roles of STAT3 in fibrosis, both in vitro and in vivo. 19 , 20 Based on the findings to date, JAK2/STAT3 pathway has been highlighted as a potential anti‐fibrosis target in clinical therapy.
Bile duct ligature (BDL) and carbon tetrachloride (CCl4) mice models and activated LX‐2 cells models are widely established for the assessment of liver fibrosis and cirrhosis. 4 , 21 As few effective treatments exist for hepatic cirrhosis, end stage of fibrosis or precancerous stages of hepatocellular carcinoma, more research focus is required on earlier fibrosis. 22 In this study, the above fibrotic models were employed to investigate the effects of PSS following chronic administration and the underlying mechanisms.
2. MATERIALS AND METHODS
2.1. Drugs and reagents
Propylene glycol alginate sodium sulphate was purchased from Dalian Tianyu Pharmaceuticals Co., Ltd and disposed in saline to obtain different drug doses of 12.5, 25 and 50 mg/kg, and stored at 4°C. 12 CCl4 was acquired from Sinopharm. Microplate test kits of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were obtained from Nanjing Jiancheng Bioengineering Institute (Jiancheng Biotech). Antibodies against IL‐6, Col‐1, α‐SMA, TGF‐β1, MMP‐2, TIMP‐1, Beclin‐1, p62, Smad2 and Smad3 were purchased from Proteintech, and those against p‐Smad2, p‐Smad3, JAK2, STAT3 and p‐STAT3 were purchased from Cell Signaling Technologies. The polymerase chain reaction (PCR) kit was acquired from Takara Biotechnology.
2.2. Animal experiments
All animal experiments were conducted following the institutional guidelines and approved by the Animal Care and Use Committee of Shanghai Tongji University, China. Healthy male C57 mice (20‐22 g) 6 weeks of age, provided by Shanghai Laboratory Animal Co., Ltd, were maintained in tidy cages with free access to food and water, and equilibrated for a week before experimental enrolment.
In the BDL experiment, 48 mice were divided into six groups: (1) normal control (NC), (2) sham, (3) BDL, (4) BDL + PSS (12.5), (5) BDL + PSS (25) and (6) BDL + PSS (50). The BDL operation was performed as described previously. 21 In groups (4), (5) and (6), mice were intraperitoneally injected with the indicated doses of PSS at 12.5, 25 and 50 mg/kg, respectively, and mice in groups (1), (2) and (3) were treated with the same volume of saline twice a week for two subsequent weeks. In the CCl4 experiment, 40 CCl4 model mice were correspondingly divided into five groups: (1) vehicle‐treated control, (2) CCl4, (3) CCl4 + PSS (12.5), (4) CCl4 + PSS (25) and (5) CCl4 + PSS (50). Mice were treated with CCl4 with or without PSS for a total of 12 weeks. In order to induce hepatic fibrosis, CCl4 dissolved in olive oil (10%) was injected intraperitoneally (1.0 mL/kg) twice a week. The dose of PSS in groups (3), (4) and (5) was 12.5, 25 and 50 mg/kg, respectively, in CCl4 experiment.
Following the experiment, mice were killed by CO2 inhalation. Blood samples were collected into 20 µL heparin (1000 U/mL), and serum samples were obtained by centrifugation for 10 minutes at 4500 g and 4°C. Liver samples were excised and processed after mice were killed, and stored at −80°C.
2.3. Cell culture
Human primary HSC cell line, LX‐2, was purchased from Cell Bank of Type Culture Collection of the Chinese Academy of Sciences and maintained in Dulbecco's modified Eagle Medium (DMEM) containing penicillin, streptomycin and foetal bovine serum as described. 4 HSC cells have been shown to be a primary effector of hepatic fibrosis, and in our study, LX‐2 cells were treated with 10 ng/mL TGF‐β1 (PeproTech) to be activated after 24 hours. 4 , 23
2.4. Biochemical measurements and enzyme‐linked immunosorbent assay (ELISA)
ALT and AST activities in serum were determined according to standard protocols using the test kits and an automated chemistry analyser (Olympus AU1000). Serum α‐SMA and Col‐1 were detected with commercial ELISA kits (R&D Systems).
2.5. Histological analysis
Liver specimens were washed with saline, followed by fixation in 10% formalin and embedding in paraffin. Specimens 5 µm thick were subjected to haematoxylin and eosin (HE), Sirius Red and Mason's trichrome staining to evaluate hepatic fibrosis under a light microscope. The degree of liver fibrosis was examined by a specialist blinded to sample information according to strict criteria. Fibrosis scores and related stages were as follows: 0, no fibrosis; 1, perisinusoidal or periportal fibrosis; 2, perisinusoidal and portal/periportal fibrosis; 3, bridging fibrosis; and 4, cirrhosis. 24
2.6. Immunohistochemistry
Immunostaining of liver sections embedded in paraffin was performed with primary antibodies against TGF‐β1, Col‐1, α‐SMA, MMP‐2, TIMP‐1, Beclin‐1, p62, p‐smad2, p‐smad3 and p‐STAT3 as reported. 25 Sections were subsequently incubated with the corresponding secondary antibodies. Bound antibodies were observed under a light microscope and images obtained with a digital camera (Leica Wetzlar) after incubation with a peroxidase substrate (DAB) kit (Vector).
2.7. Western blot analysis
Protein samples were obtained using standard protocols and the concentrations determined with a bicinchoninic acid protein assay kit (Solarbio). Western blot analysis of IL‐6, TGF‐β1, Col‐1, α‐SMA, MMP‐2, TIMP‐1, Beclin‐1, p62, p‐smad2, p‐smad3, p‐STAT3 and β‐actin (control) was conducted with standard methods. Different band quantities were detected via an Odyssey two‐colour infrared laser imaging system (LI‐COR Biosciences).
2.8. Reverse transcription (RT)‐PCR
Total RNA was obtained with TRIzol reagent (Takara) using standard protocols. After purity determination, single‐stranded cDNA was synthesized via reverse transcription using a ThermoScript RT‐PCR system (Invitrogen). Related gene expression was examined with a ViiA™ 7 Real‐Time PCR System (Applied Biosystems). All the detections were performed in duplicate, and experiments were repeated at least three times. The primers used are listed in Table 1.
Table 1.
The primers used in the study
| Gene | Primers sequence (5′–3′) | |
|---|---|---|
| α‐SMA | Forward | CCCAGACATCAGGGAGTAATGG |
| Reverse | TCTATCGGATACTTCAGCGTCA | |
| Col Ⅰ | Forward | CAATGGCACGGCTGTGTGCG |
| Reverse | AGCACTCGCCCTCCCGTCTT | |
| MMP‐2 | Forward | GGACAAGTGGTCCGTGTAAA |
| Reverse | CCGACCGTTGAACAGGAAGG | |
| TIMP‐1 | Forward | CGAGACCACCTTATACCAGCG |
| Reverse | ATGACTGGGGTGTAGGCGTA | |
| TGF‐β1 | Forward | CCACCTGCAAGACCATCGAC |
| Reverse | CTGGCGAGCCTTAGTTTGGAC | |
| Beclin‐1 | Forward | ATGGAGGGGTCTAAGGCGTC |
| Reverse | TGGGCTGTGGTAAGTAATGGA | |
| p62 | Forward | GAGGCACCCCGAAACATGG |
| Reverse | ACTTATAGCGAGTTCCCACCA | |
| β‐Actin | Forward | GGCTGTATTCCCCTCCATCG |
| Reverse | CCAGTTGGTAACAATGCCATGT |
2.9. Detection of cell viability and the TGF‐β/Smad and JAK/STAT pathways in vitro
We detected the viable cells before and after the activation of TGF‐β1 with the CCK8 assay (Dojindo Laboratories). LX‐2 cells were seeded into 96‐well plates for 48 hours, followed with the addition of PSS at concentration of 0, 1.0, 2.5, 5.0, 7.5, 10.0, 12.5 and 15.0 μg/mL for 24 hours. CCK8 assay was used to detect cell viability according to the manufacturer's instructions, assuming the absorbance of the cells with 0 μg/mL PSS as 100%. To further study whether TGF‐β/Smad and JAK/STAT pathways were involved in our study, LX‐2 cells, cultured in 6‐well plates, were divided into three groups in our vitro experiment. LX‐2 cells, cultured in 6‐well plates, were divided into three groups in our vitro experiment: (a) normal group: cells were treated with DMEM only; (b) TGF‐β1–treated group: cells were treated with TGF‐β1 without PSS administration; and (c) PSS group: cells were treated with TGF‐β1 and PSS at the half‐maximum inhibition concentration (IC50) that could be gained in accordance with the above studies.
2.10. Immunofluorescence (IF)
LX‐2 cells were cultured, and the IF staining against p‐Smad2 and p‐Stat3 was carried based on a recent study. 27
2.11. Statistical analysis
Results were presented as means ± standard deviation (SD) of at least three repeated independent experiments. Statistical analysis was conducted with the Student t test. P‐values < .05 were considered significant.
3. RESULTS
3.1. Protection of liver against the effects of BDL and CCl4 by PSS
Sham operation or oil injection led to no significant differences in HE staining when compared with the NC group, and it was consistent with a recent research. 21 The results of HE staining in the experimental models revealed considerably greater histological changes in the BDL group relative to the sham group. We observed a marked reduction in these changes after administration of different doses of PSS (Figure 1A). Injection of CCl4 for 12 weeks caused significant damage to liver, compared with the oil vehicle group, which was markedly prevented with simultaneous PSS treatment (Figure 1B). Next, we examined the expression of IL‐6, an important inflammatory factor in liver injury, via Western blot (Figure 1C) and immunohistochemistry analyses (Figure 1D). Notably, IL‐6 expression increased in the BDL and CCl4 groups was inhibited by PSS. To further confirm the protective function of PSS, ALT and AST activities in BDL operation and repeat CCl4 injection groups were examined. Notably, the significantly elevated activities of ALT and AST observed in both fibrosis models were reduced in the presence of PSS (Figure 1E).
Figure 1.
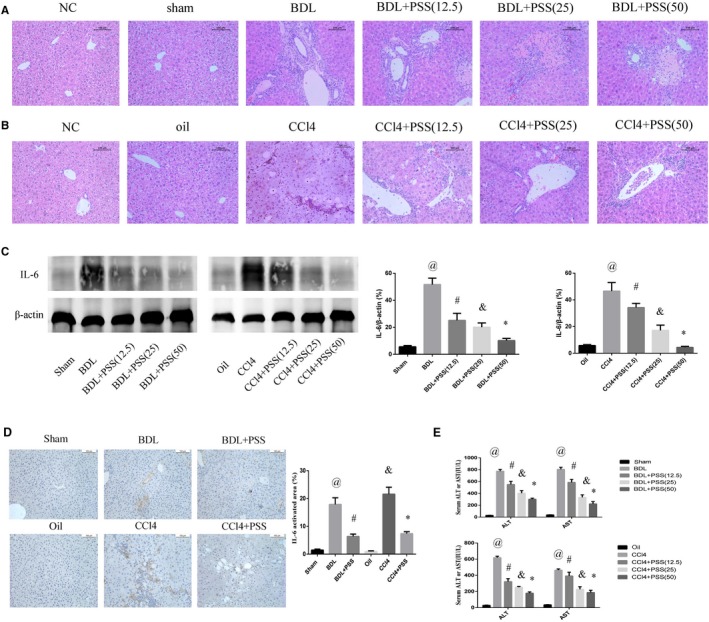
Protective effects of PSS on liver in BDL and CCl4 mouse models. A, HE staining of liver tissues in BDL experiment. Original magnification, 200×. B, HE staining of liver tissues in CCL4 experiment. Original magnification, 200×. C, IL‐6 protein expression was detected via Western blot. D, IHC staining of IL‐6 in liver tissues. We chose the PSS dose of 50 mg/kg as the representative dose in the subsequent experiment. Data were expressed as means ± SD (n = 8). @ P < .05 for BDL vs sham group, # P < .05 for BDL + PSS vs BDL group, & P < .05 for CCl4 vs oil group, *P < .05 for CCl4 + PSS vs CCl4 group. E, Suppression of serum ALT and AST levels. Above data are presented as means ± SD (n = 8). In BDL experiment, @ P < .05 for BDL vs sham group, # P < .05 for BDL + PSS (12.5) vs BDL group. & P < .05 for BDL + PSS (25) vs BDL group, *P < .05 for BDL + PSS (50) vs BDL group. In CCl4 experiment, @ P < .05 for CCl4 vs oil group, # P < .05 for CCl4 + PSS (12.5) vs CCl4 group. & P < .05 for CCl4 + PSS (25) vs CCl4 group, *P < .05 for CCl4 + PSS (50) vs CCl4 group
3.2. PSS prevents hepatic fibrosis induced by BDL and CCl4
To evaluate liver fibrosis, we examined deposition of Col‐1 with Sirius red staining and collagen fibre with Masson's trichrome staining. Hepatic fibrosis was markedly amplified after the BDL operation. In contrast, PSS‐treated mice displayed pronounced reduction in fibrosis (Figure 2A). In the CCl4 model, PSS consistently prevented fibrosis to a significant extent. After BDL operation or CCl4 injection, the expression of Col‐1 and α‐SMA was significantly increased. Up‐regulation of Col‐1 and α‐SMA was blocked upon PSS treatment, as observed from immunohistochemical staining data (Figure 2B‐D). Western blot analysis consistently indicated preventive functions of PSS against hepatic fibrosis (Figure 2E). The efficiency of PSS treatment was further validated via ELISA of serum α‐SMA and Col‐1 (Figure 2F) and quantitative PCR (Figure 2G). The serum level of α‐SMA and Col‐1 increased in BDL or CCl4 group, and they were reduced in PSS‐treated groups. The mRNA expression of α‐SMA and Col I showed consistent results.
Figure 2.
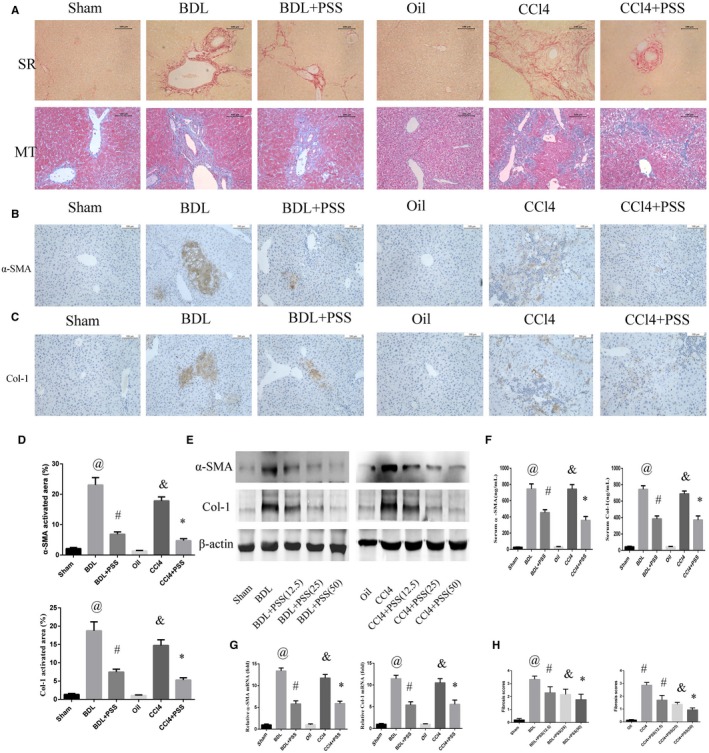
Propylene glycol alginate sodium sulphate prevents hepatic fibrosis induced by BDL and CCl4. A, Sirius red and Masson's trichrome staining in liver tissues. Original magnification, 200×. B, IHC staining of anti‐α‐SMA in liver tissues. C, IHC staining of anti‐Col I in liver tissues. D, Labelling index data for (B) and (C). E, Western blotting of α‐SMA and Col I. F, Serum α‐SMA and Col I levels. G, Expression of α‐SMA and Col I mRNA. Data of (D), (F) and (G) were presented as means ± SD (n = 8), @ P < .05 for BDL vs sham group, # P < .05 for BDL + PSS vs BDL group, & P < .05 for CCl4 vs oil group, *P < .05 for CCl4 + PSS vs CCl4 group. H, Fibrosis scores of different groups. Data were shown as means ± SD (n = 8). In BDL experiment, @ P < .05 for BDL vs sham group, # P < .05 for BDL + PSS (12.5) vs BDL group. & P < .05 for BDL + PSS (25) vs BDL group, *P < .05 for BDL + PSS (50) vs BDL group. In CCl4 experiment, @ P < .05 for CCl4 vs oil group, # P < .05 for CCl4 + PSS (12.5) vs CCl4 group. & P < .05 for CCl4 + PSS (25) vs CCl4 group, *P < .05 for CCl4 + PSS (50) vs CCl4 group
Fibrosis scores were obtained through rigorous evaluation by a specialist based on histological staining (Figure 2H). Notably, the high scores of BDL and CCl4 mice were reduced significantly following treatment with PSS.
3.3. PSS up‐regulates MMP‐2 and down‐regulates TIMP‐1 in hepatic fibrosis
Next, the expression patterns of MMP‐2 and TIMP‐1 were examined in our fibrosis models. MMP‐2 protein expression was significantly decreased after the BDL operation or CCl4 injection, which was enhanced upon PSS treatment. Conversely, high TIMP‐1 levels observed in the BDL and CCl4 models were down‐regulated upon PSS injection, indicating alleviation of fibrosis (Figure 3A). Moreover, immunostaining with anti‐MMP‐2 and TIMP‐1 antibodies confirmed the expression patterns of MMP‐2 and TIMP‐1 in tissue samples from PSS‐treated mice (Figure 3B). MMP‐2 and TIMP‐1 mRNA expression were consistent with those of the corresponding proteins (Figure 3C).
Figure 3.
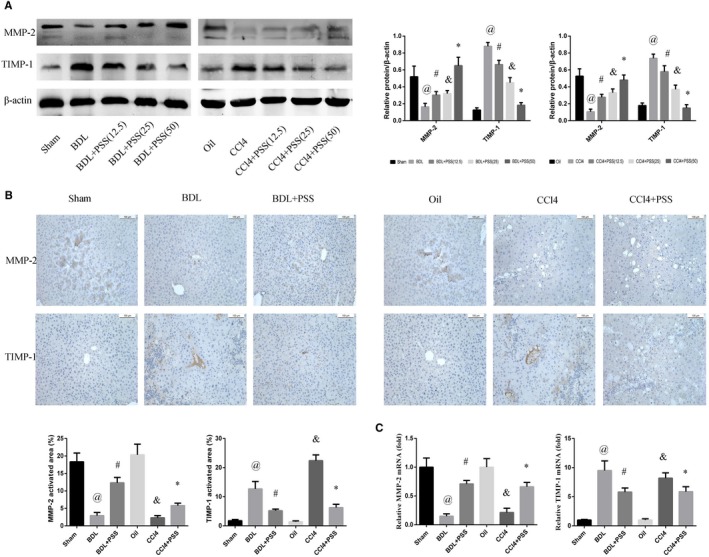
Propylene glycol alginate sodium sulphate up‐regulates MMP‐2 and down‐regulates TIMP‐1 in hepatic fibrosis. A, Western blots of MMP‐2 and TIMP‐1. Data are presented as means ± SD (n = 8). In BDL experiment, @ P < .05 for BDL vs sham, # P < .05 for BDL + PSS (12.5) vs BDL. & P < .05 for BDL + PSS (25) vs BDL, *P < .05 for BDL + PSS (50) vs BDL. In CCl4 experiment, @ P < .05 for CCl4 vs oil, # P (except the serum level of AST in CCl4 experiment) < .05 for CCl4 + PSS (12.5) vs CCl4. & P < .05 for CCl4 + PSS (25) vs CCl4, *P < .05 for CCl4 + PSS (50) vs CCl4. B, IHC staining of MMP‐2 and TIMP‐1 in liver tissues. C, The mRNA expression of MMP‐2 and TIMP‐1. Data of (B) and (C) were presented as means ± SD (n = 8). @ P < .05 for BDL vs sham group, # P < .05 for BDL + PSS vs BDL group, & P < .05 for CCl4 vs oil group, *P < .05 for CCl4 + PSS vs CCl4 group
3.4. PSS suppresses the TGF‐β1/Smad pathway in hepatic fibrosis
Phosphorylated Smad2 (p‐Smad2) and phosphorylated Smad3 (p‐Smad3) generated by TGF‐β1 lead to the activation of hepatic fibrosis. 1 Accordingly, we evaluated p‐Smad2, p‐Smad3 and TGF‐β1 expression to determine the mechanisms underlying the anti‐fibrotic effects of PSS. In the BDL and CCl4 model groups, TGF‐β1, p‐Smad2 and p‐Smad3 levels were remarkably increased, which were suppressed following PSS treatment (Figure 4A). We further examined these factors in liver tissues to confirm Western blot findings (Figure 4B). As expected, PSS effectively reduced the expression levels of TGF‐β1, p‐Smad2 and p‐Smad3 (Figure 4B). Notably, the p‐Smad2 and p‐Smad3 in the liver tissues of BDL and CCl4 group accumulated in the nuclei when compared with the sham or oil group. But this tendency could be mitigated with PSS treatment. Consistently, mRNA expression of TGF‐β1 was decreased upon treatment with PSS after BDL operation or repeated CCl4 injection (Figure 4C).
Figure 4.
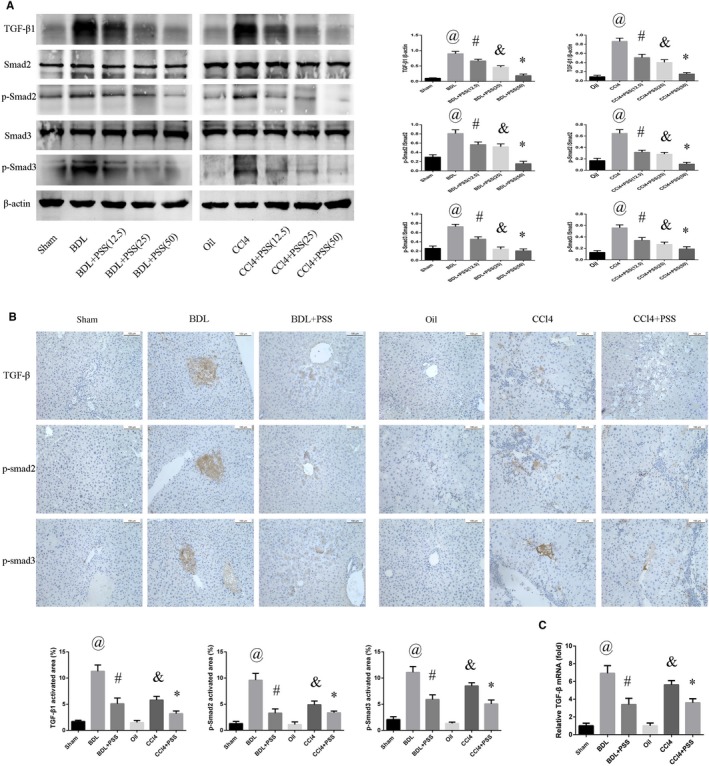
Propylene glycol alginate sodium sulphate suppresses hepatic fibrosis through the TGF‐β1/Smad pathway. A, Western blot analysis of TGF‐β1, Smad2, Smad3, p‐Smad2 and p‐Smad3 expression. Data were presented as means ± SD (n = 8). In BDL experiment, @ P < .05 for BDL vs sham group, # P < .05 for BDL + PSS (12.5) vs BDL group. & P < .05 for BDL + PSS (25) vs BDL group, *P < .05 for BDL + PSS (50) vs BDL group. In CCl4 experiment, @ P < .05 for CCl4 vs oil group, # P < .05 for CCl4 + PSS (12.5) vs CCl4 group. & P < .05 for CCl4 + PSS (25) vs CCl4 group, *P < .05 for CCl4 + PSS (50) vs CCl4 group. B, IHC staining of anti‐p‐Smad3, p‐Smad2 and TGF‐β1 antibodies. C, Expression of TGF‐β1 mRNA. Data of (B) and (C) were presented as means ± SD (n = 8), @ P < .05 for BDL vs sham group, # P < .05 for BDL + PSS vs BDL group, & P < .05 for CCl4 vs oil group, *P < .05 for CCl4 + PSS vs CCl4 group
3.5. PSS inhibits the JAK2/STAT3 pathway to exert anti‐autophagic effects in hepatic fibrosis
In view of the finding that autophagic death plays a significant role in progression of hepatic fibrosis, we examined the potential impact of the JAK2/STAT3 pathway on regulation of cellular autophagy. Suppression of JAK2/STAT3 pathway has been showed to ameliorate liver fibrosis in rat in a recent study. 28 JAK2 is a novel regulator of TGF‐β, and TGF‐β/JAK2/STAT3 signalling is regarded as a non‐canonical TGF‐β signalling in fibrosis. 28 , 29 , 30 Protein expression of JAK2 and total STAT3 was not significantly different among the groups, while BDL‐operated and CCl4‐treated mice showed enhanced expression of phosphorylated STAT3 (Figure 5A). STAT3 has been shown to be a major regulator of autophagy in some recent studies. 31 , 32 Beclin‐1, a key regulator of autophagy, tended to show a significant increase in both BDL and CCl4 models, which was markedly suppressed by PSS. Conversely, p62, inhibited in BDL‐operated and CCl4‐treated groups, was promoted upon synchronous PSS injection. Significant immunohistochemical staining of p‐STAT3 in liver tissues was observed in hepatic fibrosis model mice, and in particular, the expression of p‐STAT3 was highlighted in the nuclei. Meanwhile, PSS administration had a marked suppression effect (Figure 5B). The results of Beclin‐1 and p62 staining and RT‐PCR were consistent with Western blot data (Figure 5C).
Figure 5.
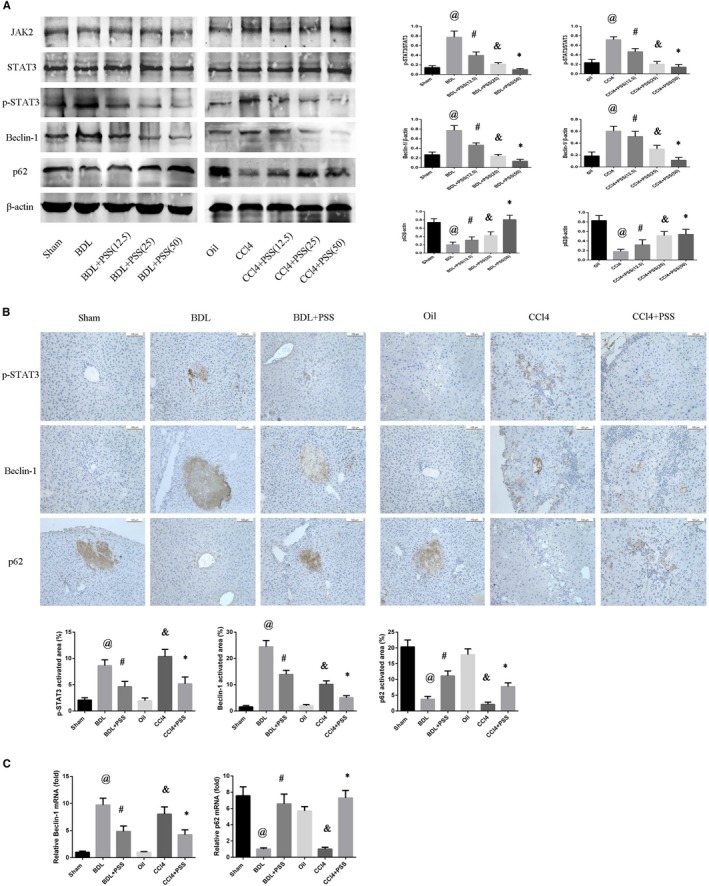
Propylene glycol alginate sodium sulphate inhibits the JAK2/STAT3 pathway to exert anti‐autophagic effects in hepatic fibrosis. A, Western blot analysis of JAK2, STAT3, p‐STAT3, Beclin‐1 and p62 expression. Data are presented as means ± SD (n = 8). In BDL experiment, @ P < .05 for BDL vs sham group, # P < .05 for BDL + PSS (12.5) vs BDL group. & P < .05 for BDL + PSS (25) vs BDL group, *P < .05 for BDL + PSS (50) vs BDL group. In CCl4 experiment, @ P < .05 for CCl4 vs oil group, # P < .05 for CCl4 + PSS (12.5) vs CCl4 group. & P < .05 for CCl4 + PSS (25) vs CCl4 group, *P < .05 for CCl4 + PSS (50) vs CCl4 group. B, IHC staining of anti‐p62, Beclin‐1 and p‐STAT3 in liver tissues. C, Expression of Beclin‐1 and p62 mRNA. Data of (B) and (C) were presented as means ± SD (n = 8), @ P < .05 for BDL vs sham group, # P < .05 for BDL + PSS vs BDL group, & P < .05 for CCl4 vs oil group, *P < .05 for CCl4 + PSS vs CCl4 group
3.6. PSS suppressed the activation of HSCs down‐regulated the activation of TGF‐β/Smad and JAK/STAT pathways in vitro
To further reveal the inhibitory effect of PSS on hepatic fibrosis, we then used LX‐2 cells to perform in vitro studies. As shown in Figure 6A, PSS showed no significant cytotoxicity in normal HSCs. However, the viability of TGF‐β1–activated LX‐2 cells was significantly suppressed with PSS administration in a dose‐dependent way in this study, and the IC50 of PSS was 5.8 mg/mL according to the data (Figure 6B). Therefore, PSS of 5.8 mg/mL was used in subsequent experiments. As expected, enhanced expression of protein Col‐1 and α‐SMA after TGF‐β1 activation was reduced in the presence of PSS (Figure 6C) in LX2 cells. Then, we examined the mRNA expression of Col‐1 and α‐SMA, and the study showed that the mounting expression of Col‐1 and α‐SMA mRNA after activated by TGF‐β1 was down‐regulated in the PSS group (Figure 6D). We therefore confirmed PSS also possessed anti‐fibrosis function by suppressing the activation of HSCs in vitro.
Figure 6.
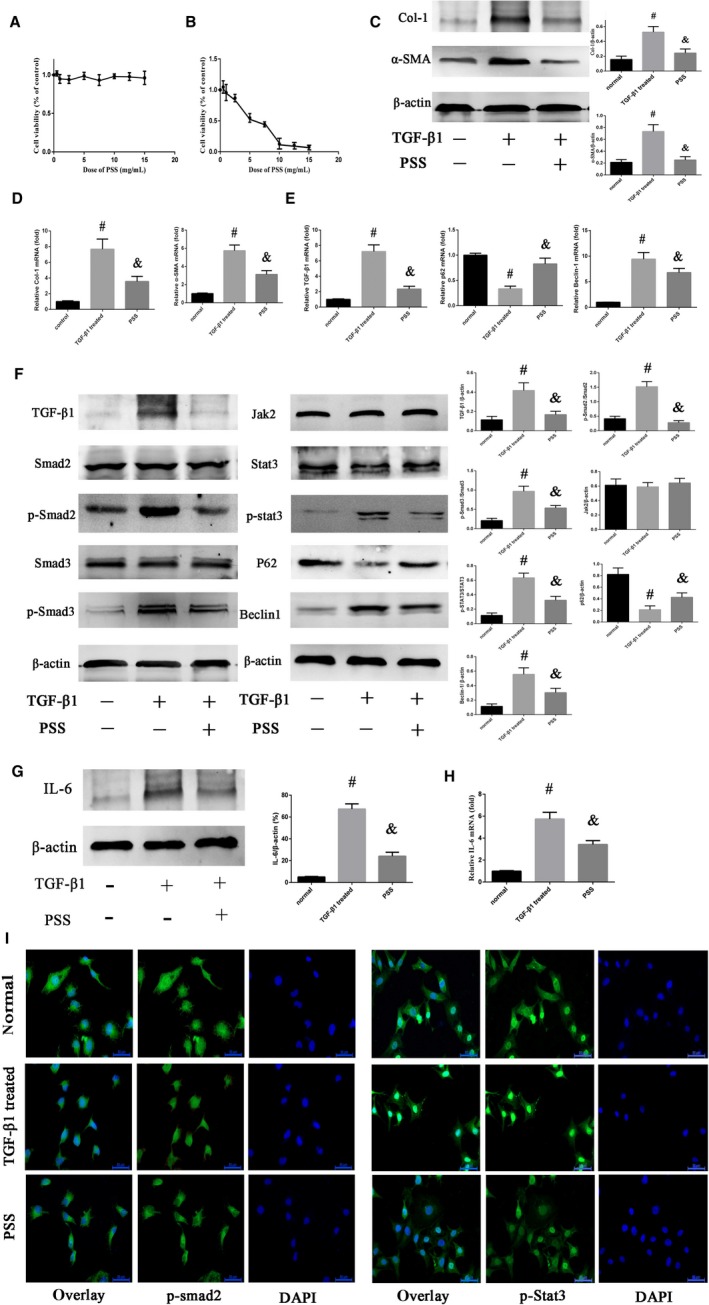
Inhibitory effects of PSS on HSC activation and TGF‐β/Smad and JAK/STAT pathways in vitro. A, The cytotoxicity examination of PSS in normal HSC cells. B, Cell viability after PSS administration in activated HSCs. C, The results of Western blot of Col‐1 and α‐SMA in normal, TGF‐β1–treated and PSS groups. D, mRNA expression of Col‐1 and α‐SMA. E, mRNA expression of TGF‐β1, P62 and Beclin‐1. F, Protein expression of TGF‐β1, Smad2, p‐Smad2, Smad3, p‐Smad3, Jak2, STAT3, p‐STAT3, P62 and Beclin‐1 was detected by Western blotting. G, Protein expression of IL‐6. H, mRNA expression of IL‐6. I, IF staining of p‐Smad2 and p‐STAT3 in LX‐2 cells (original magnification, 400×). Above experiments were all repeated three times. Data are presented as means ± SD. # P < .05 for TGF‐β1–treated group vs normal group; & P < .05 for PSS group vs TGF‐β1–treated group
Then, our study managed to investigate the effect of PSS on TGF‐β/Smad and JAK/STAT pathways in vitro. In Figure 6E, the mRNA expression of TGF‐β1 and Beclin‐1 was up‐regulated after the HSCs were activated, and PSS administration could down‐regulate TGF‐β1 and Beclin‐1 mRNA significantly. On the contrary, the decreasing p62 mRNA expression was prevented by PSS. The result of Western blot assay showed that the expression of TGF‐β1, p‐Smad2, p‐Smad3 and p‐STAT3 was up‐regulated in TGF‐β–treated group (Figure 6F), indicating TGF‐β/Smad and JAK/STAT pathways were activated after TGF‐β1 treatment in LX2 cells. In addition to TGF‐β1, IL‐6, a upstream of JAK2, can also directly stimulate the JAK2/STAT3 signalling. 33 , 34 In the result of Western blots, as the direct upstream factor of JAK2/STAT3, IL‐6 was up‐regulated in activated HSCs and the increasing level of IL‐6 was prevented by PSS significantly (Figure 6G). The examination of mRNA expression showed consistent results (Figure 6H). Two downstream proteins of p62 and Beclin‐1, known as the autophagic regulators, were also detected in this study. Consistently, p62 was restrained and Beclin‐1 promoted after the activation of HSCs. But these effects could be obviously suppressed in the presence of PSS. Besides, the IF results of p‐Smad2 and p‐Stat3 were consistent with the above experiments (Figure 6I). p‐smad2 accumulated in nucleus from cytoplasm in TGF‐β1–treated group when compared with the normal group. However, this effect was significantly mitigated in the PSS group. Similar results could also be detected in the IF staining of p‐Stat3. To sum up, PSS could suppress the activation of HSCs through down‐regulating the activation of TGF‐β/Smad and JAK2/STAT3 pathways to show its anti‐fibrosis function in vitro in our study.
4. DISCUSSION
In this study, we demonstrated the anti‐fibrotic activity of PSS and examined the mechanisms underlying PSS‐mediated hepatic fibrosis amelioration in our models (Figure 7). PSS suppressed inflammation and synthesis of ECM and inhibited activation of HSCs through a mechanism involving the TGF‐β1/Smad2/3 pathway. Furthermore, JAK2/STAT3 pathway–related autophagy was also identified as a potential therapeutic target for hepatic fibrosis in our study.
Figure 7.
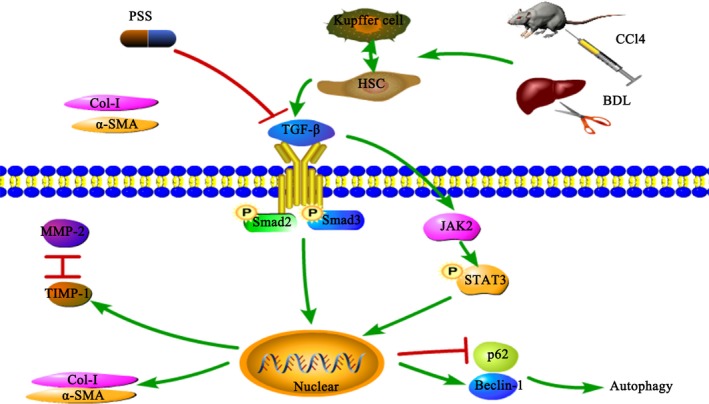
Potential mechanism of action of PSS against hepatic fibrosis in our models. After BDL operation or CCl4 injection, Kupffer cells are activated to secrete IL‐6 and HSCs were activated to produce TGF‐β. Through the TGF‐β1/Smad2/3 pathway, higher levels of Col I and α‐SMA are produced that induce excessive ECM accumulation. Higher Beclin‐1 and lower p62 levels are secreted through the JAK2/STAT3 pathway to induce cellular autophagy. Up‐regulation of TIMP‐1 is associated with hepatic fibrosis, and the TIMP‐1/MMP‐2 balance is altered to regulate production and elimination of the extracellular matrix. PSS significantly suppresses the production of TGF‐β1 and exerts anti‐fibrosis effects
Chronic liver injury leads to hepatic fibrosis, which usually progresses to cirrhosis, hepatocellular carcinoma and liver failure. 22 While significant progress has been made in clarifying the underlying mechanisms of hepatic fibrosis, no effective drugs or treatment methods are available at present, 36 highlighting the urgent medical requirement for novel anti‐fibrosis strategies. PSS, an oral heparinoid with good anti‐coagulative, hypotensive and anti‐inflammation activities, has been used to treat patients with cerebrovascular, cardiovascular and other diseases in China for the past 30 years. 10 The low cost and high efficacy of this compound make it an ideal choice as a long‐term therapeutic drug. 37
Here, we used the widely accepted BDL and CCl4 mouse models and LX‐2 cell models to verify the protective effects of PSS. Inflammation is a critical regulator in hepatic fibrosis. 6 Hepatic injury triggers inflammatory activity and releases cytokines to synthesize ECM through activating HSCs. 38 And high level of TGF‐β1 and IL‐6 produced by Kupffer cells has been investigated to be important pro‐inflammatory factors in HSC activation. 6 Activated HSCs, in turn, regulate inflammation and generate ECM and collagen. 38 In our in vivo study, significant histological changes and high IL‐6 expression were reduced in the presence of PSS, which was further confirmed by Col‐1 and collagen fibres detections with Sirius red and Masson's trichrome staining. Increasing α‐SMA expression is also a crucial character of HSC activation. 6 However, this expression pattern could be inhibited upon administration of PSS. Together with our in vitro study, these results supported the theory that PSS suppresses inflammation and HSC activation and promotes degradation of Col‐1 to inhibit hepatic fibrosis development.
TGF‐β1 secreted from HSC and Kupffer cells is the principal isoform of the TGF‐β family and a key profibrogenic cytokine in the development of hepatic fibrosis. 3 , 38 , 39 Numerous signalling pathways are regulated by TGF‐β, and drugs that can intervene those pathways may have the potential anti‐fibrosis pharmacological effects. 41 TGF‐β1 binds with TGF‐β type Ⅱ receptor, then activates the TGF‐β type Ⅰ receptor and eventually phosphorylates Smad2 and Smad3. 41 , 42 After phosphorylation, Smad2 and Smad3 can bind with Smad4 to form oligomeric complexes, which accumulate in the nucleus and affect the transcription of specific genes and their downstream factors. 43 , 44 In our study, we observed a significant increase in TGF‐β1, p‐Smad3, and p‐Smad2 levels in fibrotic mice, which were suppressed upon PSS administration. Recent studies advocate that TGF‐β1 is required to alter the balance between MMPs and TIMPs to affect matrix degradation. 38 TGF‐β1 and its latency‐associated peptide (LAP) can bind to the latency TGF‐β1–binding protein‐1 (LTBP‐1) to form part of ECM. 46 Moreover, latent TGF‐β1 is closely associated with MMP‐2 and MMPs are fibrolysis of proteolytic enzymes that can counterbalance fibrogenesis. 17 , 46 In contrast to MMP2, TIMP‐1 produced by TGF‐β1 can inhibit ECM degradation through the activation of Smad3. 48 In our animal model, MMP‐2 protein and mRNA levels were suppressed, while those of TIMP‐1 were enhanced in fibrotic mice. Notably, altered expression patterns of these marker molecules were significantly abrogated upon PSS injection. Our results collectively indicate that the TGF‐β1 signalling pathway is involved in the anti‐fibrotic activity of PSS.
Accumulating evidence has established that autophagy is a major participant in numerous liver diseases, such as autoimmune hepatitis, hepatic ischaemia reperfusion injury and hepatocellular carcinoma. 49 Although the autophagy of HSCs may moderate fibrogenesis in some studies, it saves energy for activated HSCs. 47 Increased autophagic flux during HSC activation was recently shown to be involved in the pathogenesis of hepatic fibrosis. 15 , 49 Beclin‐1, together with autophagy‐related genes (ATGs) and class III phosphatidylinositol 3‐kinase (VPS34), forms a complex, which is necessary to the formation of autophagosome. 51 As one of the autophagy adaptors, p62 sequesters the autophagosome by selectively targeting cargo to the autophagosome membrane. 51 In our study, Beclin‐1 displayed significantly higher expression in the fibrotic model groups and p62 was simultaneously inhibited. Interestingly, PSS administration led to suppression of autophagy conditions, clearly indicative of anti‐autophagic activity.
We further demonstrated an important role of JAK2/STAT3 in fibrotic autophagy in our study. Both TGF‐β1 and IL‐6 have been shown to be regulators of JAK2/STAT3 pathway. 30 , 34 STAT3 is a potential transcription factor, and phosphorylated STAT3 may dimerize, translocate to the nucleus and mediate the transcription for extracellular signals after binding to DNA target sites. 52 The pro‐autophagic effect of nuclear p‐STAT3 is associated with the modulation of hypoxia‐inducible factor 1, α subunit (HIF1A) in hypoxia and BCL2/adenovirus E1B 19 kD interacting protein 3 (BNIP3). 52 HIF1A promotes the expression of BNIP3 and its ligands, which can strength the inductions of autophagosome. 52 While the expression of total STAT3 and JAK2 proteins among these groups was not significantly different in our study, the p‐STAT3 level was promoted in fibrotic mice and activated LX‐2 cells, which was suppressed by PSS, as expected. These findings clearly demonstrate that PSS administration markedly inhibits the activation of the JAK2/STAT3 pathway, in turn, preventing autophagy, which may at least partially explain the mechanisms by which PSS exerts anti‐fibrotic effects and inhibits HSC activation.
Our results collectively suggest that PSS prevents hepatic fibrosis by suppressing inflammation, promoting ECM decomposition and inactivating HSCs through the TGF‐β1/Smad2/3 and JAK2/STAT3 pathways. Because of its multiple therapeutic effects, large number of sources and low cost, the natural extract PSS may effectively serve as a clinical supplement to treat hepatic fibrosis.
5. CONCLUSIONS
Propylene glycol alginate sodium sulphate exerts anti‐fibrotic activity in BDL and CCl4 mouse models, and in vitro. TGF‐β1/Smad2/3 and JAK2/STAT3 pathways are closely associated with the therapeutic effects of PSS, supporting its potential as an effective treatment agent for hepatic fibrosis.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
S. Xu and Y. Mao designed the research; S. Xu, Y. Mao, J. Feng, J. Li and L. Wu performed the data analysis; S. Xu, Q. Yu, Y. Zhou, J. Zhang and J. Chen perform the animal experiments; Y. Mao, J. Ji, K. Chen, F. Wang and W. Dai conducted some other experiments; S. Xu wrote the manuscript; X. Fan and C. Guo edited the manuscript.
ACKNOWLEDGEMENTS
This research was supported by Natural Science Foundation of Shanghai (grant number: 19ZR1447700), National Natural Science Foundation of China (grant number: 81800538), the Yangfan Project of Shanghai Science and Technology Commission (grant number: 18YF1420000), the Health System Innovation Project of Shanghai Putuo Science and Technology Commission (grant number: ptkwws201901), and the WBN liver disease research fund of Chinese Foundation for Hepatitis Prevention and Control (grant number: 2019031). We thank all the staff in the Central Laboratory of Shanghai Tenth People's Hospital for helping with our research and International Science Editing (http://www.internationalscienceediting.com) for editing this manuscript for us.
Xu S, Mao Y, Wu J, et al. TGF‐β/Smad and JAK/STAT pathways are involved in the anti‐fibrotic effects of propylene glycol alginate sodium sulphate on hepatic fibrosis. J Cell Mol Med. 2020;24:5224–5237. 10.1111/jcmm.15175
Shizan Xu and Yuqing Mao contributed equally to this work.
Contributor Information
Xiaoming Fan, Email: xiaomingfan57@hotmail.com.
Chuanyong Guo, Email: guochuanyong@hotmail.com.
DATA AVAILABILITY STATEMENT
Readers with reasonable requests can access the data supporting the conclusions of the study from the corresponding author or other authors.
REFERENCES
- 1. Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134:1655‐1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Karsdal MA, Genovese F, Madsen EA, Manon‐Jensen T, Schuppan D. Collagen and tissue turnover as a function of age: implications for fibrosis. J Hepatol. 2016;64:103‐109. [DOI] [PubMed] [Google Scholar]
- 3. Lee JH, Jang EJ, Seo HL, et al. Sauchinone attenuates liver fibrosis and hepatic stellate cell activation through TGF‐beta/Smad signaling pathway. Chem Biol Interact. 2014;224:58‐67. [DOI] [PubMed] [Google Scholar]
- 4. Feng J, Wang C, Liu T, et al. Procyanidin B2 inhibits the activation of hepatic stellate cells and angiogenesis via the Hedgehog pathway during liver fibrosis. J Cell Mol Med. 2019;23:6479‐6493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Balta C, Herman H, Boldura OM, et al. Chrysin attenuates liver fibrosis and hepatic stellate cell activation through TGF‐beta/Smad signaling pathway. Chem Biol Interact. 2015;240:94‐101. [DOI] [PubMed] [Google Scholar]
- 6. Mimche PN, Brady LM, Bray CF, et al. The receptor tyrosine kinase EphB2 promotes hepatic fibrosis in mice. Hepatology. 2015;62:900‐914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Guha IN, Myers RP, Patel K, Talwalkar JA. Biomarkers of liver fibrosis: what lies beneath the receiver operating characteristic curve? Hepatology. 2011;54:1454‐1462. [DOI] [PubMed] [Google Scholar]
- 8. Parsons CJ, Bradford BU, Pan CQ, et al. Antifibrotic effects of a tissue inhibitor of metalloproteinase‐1 antibody on established liver fibrosis in rats. Hepatology. 2004;40:1106‐1115. [DOI] [PubMed] [Google Scholar]
- 9. Xin M, Ren L, Sun Y, et al. Anticoagulant and antithrombotic activities of low‐molecular‐weight propylene glycol alginate sodium sulfate (PSS). Eur J Med Chem. 2016;114:33‐40. [DOI] [PubMed] [Google Scholar]
- 10. Xue YT, Ren L, Li S, et al. Study on quality control of sulfated polysaccharide drug, propylene glycol alginate sodium sulfate (PSS). Carbohydr Polym. 2016;144:330‐337. [DOI] [PubMed] [Google Scholar]
- 11. Xu S, Wu L, Zhang Q, et al. Pretreatment with propylene glycol alginate sodium sulfate ameliorated concanavalin A‐induced liver injury by regulating the PI3K/Akt pathway in mice. Life Sci. 2017;185:103‐113. [DOI] [PubMed] [Google Scholar]
- 12. Xu S, Niu P, Chen K, et al. The liver protection of propylene glycol alginate sodium sulfate preconditioning against ischemia reperfusion injury: focusing MAPK pathway activity. Sci Rep. 2017;7:15175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lee JH, Lee H, Joung YK, et al. The use of low molecular weight heparin‐pluronic nanogels to impede liver fibrosis by inhibition the TGF‐beta/Smad signaling pathway. Biomaterials. 2011;32:1438‐1445. [DOI] [PubMed] [Google Scholar]
- 14. Liu T, Xu L, Wang C, et al. Alleviation of hepatic fibrosis and autophagy via inhibition of transforming growth factor‐beta1/Smads pathway through shikonin. J Gastroenterol Hepatol. 2019;34:263‐276. [DOI] [PubMed] [Google Scholar]
- 15. Thoen LF, Guimaraes EL, Dolle L, et al. A role for autophagy during hepatic stellate cell activation. J Hepatol. 2011;55:1353‐1360. [DOI] [PubMed] [Google Scholar]
- 16. Kim KM, Han CY, Kim JY, et al. Galpha12 overexpression induced by miR‐16 dysregulation contributes to liver fibrosis by promoting autophagy in hepatic stellate cells. J Hepatol. 2018;68:493‐504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li XM, Peng JH, Sun ZL, et al. Chinese medicine CGA formula ameliorates DMN‐induced liver fibrosis in rats via inhibiting MMP2/9, TIMP1/2 and the TGF‐beta/Smad signaling pathways. Acta Pharmacol Sin. 2016;37:783‐793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wu L, Li J, Liu T, et al. Quercetin shows anti‐tumor effect in hepatocellular carcinoma LM3 cells by abrogating JAK2/STAT3 signaling pathway. Cancer Med. 2019;8:4806‐4820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jeong WI, Park O, Radaeva S, Gao B. STAT1 inhibits liver fibrosis in mice by inhibiting stellate cell proliferation and stimulating NK cell cytotoxicity. Hepatology. 2006;44:1441‐1451. [DOI] [PubMed] [Google Scholar]
- 20. Kong X, Feng D, Wang H, et al. Interleukin‐22 induces hepatic stellate cell senescence and restricts liver fibrosis in mice. Hepatology. 2012;56:1150‐1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wu L, Zhang Q, Mo W, et al. Quercetin prevents hepatic fibrosis by inhibiting hepatic stellate cell activation and reducing autophagy via the TGF‐beta1/Smads and PI3K/Akt pathways. Sci Rep. 2017;7:9289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fuchs BC, Hoshida Y, Fujii T, et al. Epidermal growth factor receptor inhibition attenuates liver fibrosis and development of hepatocellular carcinoma. Hepatology. 2014;59:1577‐1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tomita K, Teratani T, Suzuki T, et al. Acyl‐CoA:cholesterol acyltransferase 1 mediates liver fibrosis by regulating free cholesterol accumulation in hepatic stellate cells. J Hepatol. 2014;61:98‐106. [DOI] [PubMed] [Google Scholar]
- 24. Kleiner DE, Brunt EM, Van Natta M, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313‐1321. [DOI] [PubMed] [Google Scholar]
- 25. Wang W, Chen K, Xia Y, et al. The hepatoprotection by oleanolic acid preconditioning: focusing on PPARalpha activation. PPAR Res. 2018;2018:3180396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Feng J, Wu L, Ji J, et al. PKM2 is the target of proanthocyanidin B2 during the inhibition of hepatocellular carcinoma. J Exp Clin Cancer Res. 2019;38:204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yang YZ, Zhao XJ, Xu HJ, et al. Magnesium isoglycyrrhizinate ameliorates high fructose‐induced liver fibrosis in rat by increasing miR‐375‐3p to suppress JAK2/STAT3 pathway and TGF‐beta1/Smad signaling. Acta Pharmacol Sin. 2019;40:879‐894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dees C, Tomcik M, Palumbo‐Zerr K, et al. JAK‐2 as a novel mediator of the profibrotic effects of transforming growth factor beta in systemic sclerosis. Arthritis Rheum. 2012;64:3006‐3015. [DOI] [PubMed] [Google Scholar]
- 29. Liu Y, Liu H, Meyer C, et al. Transforming growth factor‐beta (TGF‐beta)‐mediated connective tissue growth factor (CTGF) expression in hepatic stellate cells requires Stat3 signaling activation. J Biol Chem. 2013;288:30708‐30719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Finnson KW, Almadani Y, Philip A. Non‐canonical (non‐SMAD2/3) TGF‐beta signaling in fibrosis: mechanisms and targets. Semin Cell Dev Biol. 2019. [DOI] [PubMed] [Google Scholar]
- 31. Larrue C, Heydt Q, Saland E, et al. Oncogenic KIT mutations induce STAT3‐dependent autophagy to support cell proliferation in acute myeloid leukemia. Oncogenesis. 2019;8:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liu Y, Zhang H, Wang Z, Wu P, Gong W. 5‐Hydroxytryptamine1a receptors on tumour cells induce immune evasion in lung adenocarcinoma patients with depression via autophagy/pSTAT3. Eur J Cancer. 2019;114:8‐24. [DOI] [PubMed] [Google Scholar]
- 33. Shi J, Feng J, Xie J, et al. Targeted blockade of TGF‐beta and IL‐6/JAK2/STAT3 pathways inhibits lung cancer growth promoted by bone marrow‐derived myofibroblasts. Sci Rep. 2017;7:8660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shi J, Li J, Yang S, et al. LncRNA SNHG3 is activated by E2F1 and promotes proliferation and migration of non‐small‐cell lung cancer cells through activating TGF‐beta pathway and IL‐6/JAK2/STAT3 pathway. J Cell Physiol. 2020;235:2891‐2900. [DOI] [PubMed] [Google Scholar]
- 35. Zhang Z, Guo M, Zhao S, Shao J, Zheng S. ROS‐JNK1/2‐dependent activation of autophagy is required for the induction of anti‐inflammatory effect of dihydroartemisinin in liver fibrosis. Free Radic Biol Med. 2016;101:272‐283. [DOI] [PubMed] [Google Scholar]
- 36. Zeng Y, Yang D, Qiu P, et al. Efficacy of heparinoid PSS in treating cardiovascular diseases and beyond‐a review of 27 years clinical experiences in China. Clin Appl Thromb Hemost. 2016;22:222‐229. [DOI] [PubMed] [Google Scholar]
- 37. Chiu YS, Wei CC, Lin YJ, Hsu YH, Chang MS. IL‐20 and IL‐20R1 antibodies protect against liver fibrosis. Hepatology. 2014;60:1003‐1014. [DOI] [PubMed] [Google Scholar]
- 38. Yang JH, Kim SC, Kim KM, et al. Isorhamnetin attenuates liver fibrosis by inhibiting TGF‐beta/Smad signaling and relieving oxidative stress. Eur J Pharmacol. 2016;783:92‐102. [DOI] [PubMed] [Google Scholar]
- 39. Choi YJ, Kim DH, Kim SJ, et al. Decursin attenuates hepatic fibrogenesis through interrupting TGF‐beta‐mediated NAD(P)H oxidase activation and Smad signaling in vivo and in vitro. Life Sci. 2014;108:94‐103. [DOI] [PubMed] [Google Scholar]
- 40. Gyorfi AH, Matei AE, Distler JHW. Targeting TGF‐beta signaling for the treatment of fibrosis. Matrix Biol. 2018;68–69:8‐27. [DOI] [PubMed] [Google Scholar]
- 41. Liu T, Feng XH. Regulation of TGF‐beta signalling by protein phosphatases. Biochem J. 2010;430:191‐198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wrighton KH, Lin X, Feng XH. Phospho‐control of TGF‐beta superfamily signaling. Cell Res. 2009;19:8‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dooley S, Hamzavi J, Breitkopf K, et al. Smad7 prevents activation of hepatic stellate cells and liver fibrosis in rats. Gastroenterology. 2003;125:178‐191. [DOI] [PubMed] [Google Scholar]
- 44. Hill CS. Nucleocytoplasmic shuttling of Smad proteins. Cell Res. 2009;19:36‐46. [DOI] [PubMed] [Google Scholar]
- 45. Buscemi L, Ramonet D, Klingberg F, et al. The single‐molecule mechanics of the latent TGF‐beta1 complex. Curr Biol. 2011;21:2046‐2054. [DOI] [PubMed] [Google Scholar]
- 46. Mao Y, Zhang S, Yu F, Li H, Guo C, Fan X. Ghrelin attenuates liver fibrosis through regulation of TGF‐beta1 expression and autophagy. Int J Mol Sci. 2015;16:21911‐21930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Xu F, Liu C, Zhou D, Zhang L. TGF‐beta/SMAD pathway and its regulation in hepatic fibrosis. J Histochem Cytochem. 2016;64:157‐167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rautou PE, Mansouri A, Lebrec D, Durand F, Valla D, Moreau R. Autophagy in liver diseases. J Hepatol. 2010;53:1123‐1134. [DOI] [PubMed] [Google Scholar]
- 49. Hernandez‐Gea V, Ghiassi‐Nejad Z, Rozenfeld R, et al. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology. 2012;142:938‐946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hazari Y, Bravo‐San Pedro JM, Hetz C, Galluzzi L, Kroemer G. Autophagy in hepatic adaptation to stress. J Hepatol. 2020;72:183‐196. [DOI] [PubMed] [Google Scholar]
- 51. You L, Wang Z, Li H, et al. The role of STAT3 in autophagy. Autophagy. 2015;11:729‐739. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Readers with reasonable requests can access the data supporting the conclusions of the study from the corresponding author or other authors.