

Abstract
Pancreatic cancer has one of the highest mortality rates (5‐year survival ~9%) among cancers. Pancreatic adenocarcinoma (PAAD) is the most common (>80%) and the most lethal type of pancreatic cancer. A need exists for new approaches to treat pancreatic adenocarcinoma. GPCRs, the largest family of cell‐surface receptors and drug targets, account for ~35% of approved drugs. Recent studies have revealed roles for GPCRs in PAAD cells and cells in the tumour micro‐environment. This review assesses current information regarding GPCRs in PAAD by summarizing omics data for GPCRs expression in PAAD. The PAAD “GPCRome” includes GPCRs with approved agents, thereby offering potential for their repurposing/repositioning. We then reviewed the evidence for functional roles of specific GPCRs in PAAD. We also highlight gaps in understanding the contribution of GPCRs to PAAD biology and identify several GPCRs that may be novel therapeutic targets for future work in search of GPCR‐targeted drugs to treat PAAD tumours.
Abbreviations
- CAF
cancer‐associated fibroblast
- CCLE
Cancer Cell Line Encyclopedia
- ECM
extracellular matrix
- EMT
epithelial‐to‐mesenchymal transformation
- GtoPdb
Guide to Pharmacology Database
- LPA
lysophosphatidic acid
- PAAD
pancreatic adenocarcinoma
- PDAC
pancreatic ductal adenocarcinoma
- PSC
pancreatic stellate cell
- TCGA
The Cancer Genome Atlas
- TPM
transcripts per million
1. INTRODUCTION
Pancreatic cancer accounted for ~430,000 deaths globally and ~4.5% of all cancer deaths in 2018 (Bray et al., 2018). The 5‐year survival (~9% in the United States) identifies pancreatic cancer as the most lethal major category of solid tumours (http://seer.cancer.gov/statfacts/html/pancreas.html). The most common pancreatic cancer (85%) is pancreatic adenocarcinoma (PAAD), which arises from the exocrine duct of the pancreas (Rawla, Sunkara, & Gaduputi, 2019). The incidence of pancreatic cancer is highest in the developed world: 7.7 and 7.6 cases per 100,000 people in Europe and North America, respectively (Rawla et al., 2019). Current trends indicate a sharp increase in number of cases and deaths in coming decades, with an anticipated additional ~350,000 deaths per year globally by 2040, in part because of the growing incidence of obesity, a risk factor for pancreatic adenocarcinoma (Ilic & Ilic, 2016; Rawla et al., 2019). Other risk factors include smoking, chronic pancreatitis, family history, late onset diabetes mellitus and rare genetic mutations (Ilic & Ilic, 2016).
Current drug therapy for pancreatic adenocarcinoma involves treatment with cytotoxic agents: https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4793/nab‐https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2770 or Folfirinox (a combination of https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4816 and three chemotherapeutic drugs, https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4789 [5FU], https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6823 and https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7433). These agents and others approved for treating pancreatic adenocarcinoma in recent decades (e.g. https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4920; Wang et al., 2015) typically only increase survival by several months. Surgical removal of the tumour, the most successful therapy, is possible in only a limited number of patients. Little improvement has occurred in pancreatic adenocarcinoma survival rates over the past 40 years (Siegel, Miller, & Jemal, 2019). Identification of new therapies for pancreatic adenocarcinoma is thus an important unmet medical need.
A key problem in treating pancreatic adenocarcinoma is the TME; a dense tumour stroma (termed desmoplasia) composed of extracellular matrix (ECM) derived from cancer‐associated fibroblasts (CAFs) and that also contains immune/inflammatory cells and vascular cells. This desmoplasia can impede the delivery of therapeutic agents to tumour cells. Pancreatic adenocarcinoma tumour cells and the extracellular matrix/stromal cells in the microenvironment are potential targets for the treatment of pancreatic adenocarcinoma. Immuno‐oncology approaches that have improved the outcome of other cancers have not been successful for pancreatic adenocarcinoma, a “cold (non‐T‐cell inflamed) cancer” (Upadhrasta & Zheng, 2019).
In this article, we review the potential role of GPCRs in the biology of pancreatic adenocarcinoma and of GPCR‐targeted drugs as potential therapeutics for pancreatic adenocarcinoma. GPCRs are the largest family of proteins on the cell surface, with >800 members in humans: ~400 are endoGPCRs that respond to endogenous agonists (e.g. hormones and neurotransmitters) in contrast with chemosensory (vision, taste and olfactory) GPCRs. EndoGPCRs are the largest family of targets for approved drugs, in part because of their cell‐surface expression, which facilitates access from the extracellular milieu. GPCRs are targets for ~35% of Food and Drug Administration and European Medicines Agency‐approved drugs and ~14% of all drug targets in the human proteome (Sriram & Insel, 2018).
The relatively low frequency of mutations of GPCRs has likely inhibited interest in them as targets in cancer beyond the treatment of certain endocrine and hormone‐responsive tumours. However, evolving evidence has revealed functional roles for GPCRs in cancers, including pancreatic adenocarcinoma—both in pancreatic adenocarcinoma cells and the TME. Cells in pancreatic adenocarcinoma tumours express a large repertoire of GPCRs (Insel et al., 2018; Sriram, Moyung, Corriden, Carter, & Insel, 2019; Wiley et al., 2018). In this review, we provide a perspective on GPCR biology in pancreatic adenocarcinoma and on specific GPCRs as potential therapeutic targets. Part 1 provides an overview of the GPCRome in pancreatic adenocarcinoma tumours. Part 2 reviews functional data for several GPCR families in the context of GPCR expression in pancreatic adenocarcinoma tumours, pancreatic adenocarcinoma cells, and CAFs. In the final section, we identify unresolved issues and directions for future efforts.
2. PART 1: THE GPCRome OF PANCREATIC ADENOCARCINOMA TUMOURS
2.1. Methods for mining GPCR expression data
RNA‐sequencing (RNA‐seq) data for pancreatic adenocarcinoma tumours [from The Cancer Genome Atlas (TCGA)] and for normal pancreas [from the Genotype‐Tissue Expression (GTEx) database] are hosted by the Xena project from the University of California, Santa Cruz (http://xena.ucsc.edu). These data consist of analysis of the TCGA and GTEx databases using the same bioinformatics pipeline (the TOIL pipeline, Vivian et al., 2017). Thus quantifying gene expression for further analyses, such as quantification of differential expression (DE) of mRNA between tumours and normal tissue. Methods for obtaining GPCR expression and differential expression analysis of these data are described in Sriram, Moyung, et al. (2019). Briefly, estimated counts for genes were used as input for analysis of differential expression via edgeR (Robinson, McCarthy, & Smyth, 2010); false discovery rates (FDRs) of <0.05 were used for statistical significance. Normalized gene expression in transcripts per million (TPM) define magnitude of expression of GPCRs. We also accessed TCGA data for mutation and copy number variation of GPCRs (Sriram, Moyung, et al., 2019).
RNA‐seq data from the Cancer Cell Line Encyclopedia (CCLE, Barretina et al., 2012) were used to calculate gene expression of cancer cell lines using Kallisto (Bray, Pimentel, Melsted, & Pachter, 2016; Tatlow & Piccolo, 2016), thus providing a database with gene expression in cancer cell lines. The analysed data (quantified in TPM) were queried by us for expression of GPCRs and other genes, such as receptor ligands.
Coupling of GPCRs to heterotrimeric G proteins was determined by using primary linkages identified in the International Union of Basic and Clinical Pharmacology (IUPHAR)/British Pharmacological Society (BPS) Guide to Pharmacology (GtoPdb) database (Alexander et al., 2019). The number of GPCRs that couple to each G protein is presented below. We include all G proteins to which a GPCR couples. For weighted analysis, we incorporated the magnitude of expression of each GPCR in TPM and summed the results to define the potential contribution to cell signalling of an overall GPCR‐G protein cohort.
We also present analysis of RNA‐seq data from microdissection of pancreatic adenocarcinoma tumours (Maurer et al., 2019), which yielded results for stromal and epithelial components. Those authors also provide data in raw counts (i.e. estimates of how many times each gene is encountered in each sequenced mRNA sample) for each sample at the Gene Expression Omnibus accession number: GSE93326. We analysed those counts data using the edgeR (Robinson et al., 2010) tool to generate differential expression data, quantifying differences in gene expression between stromal and epithelial regions and extracted results for GPCRs.
The omics data in Part 1 help place in context the findings in Part 2 regarding functional effects of specific GPCRs. Highly and widely expressed GPCRs in pancreatic adenocarcinoma tumours and cells that have functional effects are candidates for further study related to pancreatic adenocarcinoma biology and may be therapeutic targets.
Analysis of association of GPCR expression with markers for various cell types in pancreatic adenocarcinoma tumours was performed as previously described (Sriram, Moyung, et al., 2019). In brief, we used RNA‐seq data for pancreatic adenocarcinoma/pancreatic ductal adenocarcinoma (PDAC) tumours from TCGA (from http://xena.ucsc.edu, analysed via edgeR, as discussed above) to obtain gene expression in counts per million, thereby facilitating between‐sample comparisons of gene expression and analysis of co‐expressed genes. The Pearson correlation (Pearson's r) was calculated between expression of GPCRs and markers for specific cell types, for example E‐Cadherin or EpCAM (cancer cellshttps://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4899 (COL2A1; fibroblasts), https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6755 (endothelial cells) and https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1852 (RTP Type C; a general immune cell marker). From the Pearson's r, we calculated P values, followed by correction for multiple testing to yield false discovery rates, using the p.adjust function (in R software), to quantify the statistical significance of association of expression between GPCRs and cell‐type markers. Associations were considered significant if the false discovery rate was <0.001. For example, strong association of a particular GPCR with expression of CD45 implies that the GPCR is likely expressed in CD45‐expressing cells, especially if that GPCR is not associated with markers for CAFs or cancer cells. CD45 immune‐associated GPCRs can be further associated with markers for specific immune cell types, such as https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2601 (myeloid cells), https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2742 (T‐cells), https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=852 (monocytes) and so forth (Sriram, Moyung, et al., 2019).
The expressions of GPCRs and other genes in cancer cell lines are also available from the CCLE (Barretina et al., 2012) and Genentech (Klijn et al., 2015) databases, hosted by multiple sources, including the European Molecular Biology Laboratory (EMBL)‐European Bioinformatics Institute (EBI) expression atlas (Kapushesky et al., 2009). We have previously discussed these data (Sriram, Moyung, et al., 2019) and created a website (http://insellab.github.io) with GPCR expression data from these sources and data extracted from re‐analysis of the CCLE (Tatlow & Piccolo, 2016). Data from Witkiewicz et al. (2016), with RNA‐seq analysis of low‐passage “primary” pancreatic adenocarcinoma cells from 73 patients are available at Gene Expression Omnibus (accession number: GSE84023) and provide additional findings for GPCRs. GPCR expression data in pancreatic adenocarcinoma cells lines from these different sources are generally highly concordant (Sriram, Moyung, et al., 2019).
We have provided a downloadable table at https://insellab.github.io/gpcr_cells_exp of GPCR expression in all pancreatic adenocarcinoma cell lines, with expression of all ~400 GPCRs annotated by the GtoPdb (Alexander et al., 2019), quantified in TPM, based on analysis by Tatlow and Piccolo (2016). Expression of GPCRs differs among the different pancreatic adenocarcinoma cell lines; interested readers can explore the variability of expression of individual GPCRs across cell lines using these data.
2.2. Results
2.2.1. Mutations: Certain proteins but not GPCRs are frequently mutated in pancreatic adenocarcinoma
Analysis of somatic, non‐silent mutations of GPCRs reveals a low frequency of GPCR mutations in pancreatic adenocarcinoma tumours (e.g. O'Hayre et al., 2013; Sriram, Moyung, et al., 2019; Wu et al., 2019) and low expression levels of such mutants (Sriram, Moyung, et al., 2019). Analysis via MutsigCV and related tools (hosted at the Broad Firebrowse TCGA web portal) that assess mutations for all coding genes, including GPCRs, for possible functional relevance (based on accumulation of mutations at specific domains and if such regions are highly evolutionarily conserved) reveals that none of the GPCR mutations in pancreatic adenocarcinoma are “significant” or highly frequent. Mutated GPCRs are thus unlikely to be important oncogenes in pancreatic adenocarcinoma. Databases are available that include the mutational frequency of GPCRs and the significance of such mutations in pancreatic adenocarcinoma (Sriram, Moyung, et al., 2019 and accompanying online data). Changes in copy number of GPCR genes in pancreatic adenocarcinoma are relatively infrequent and also unlikely to meaningfully effect GPCR mRNA expression or cell function.
By contrast, the GNAS gene, which encodes the Gαs protein of the heterotrimeric (αβγ) Gs protein that stimulates adenylyl cyclase (AC), is mutated in ~5% of pancreatic adenocarcinoma tumours (TCGA data). Based on MutsigCV analysis, these mutations are predicted to have high significance. Such findings imply a potential role in pancreatic adenocarcinoma biology for Gs‐ and Gi‐coupled GPCRs that regulate the synthesis of cAMP. Modulating such GPCRs or signalling by Gs and Gi may influence the impact of mutated GNAS on tumour development and other features of pancreatic adenocarcinoma. Limited data exist regarding the potential synergy in pancreatic adenocarcinoma between GNAS mutations and activation of Gs‐coupled GPCRs. Other Gα proteins are not frequently mutated in pancreatic adenocarcinoma.
Mutated KRAS, a frequent driver gene in pancreatic adenocarcinoma, expressed in ~70% of pancreatic adenocarcinoma tumours in TCGA, shows crosstalk with GPCR pathways (Eibl, 2019), suggesting a role for GPCRs in modulating the impact of KRAS mutations. Hence, signalling by certain GPCRs may be oncogenic in pancreatic adenocarcinoma tumours, especially tumours with GNAS and KRAS mutations.
2.2.2. GPCRs expressed by pancreatic adenocarcinoma tumours
TCGA contains 179 pancreatic adenocarcinoma tumours, 147 of which are histologically identified as pancreatic ductal adenocarcinoma (Insel et al., 2018), while the others lack a more specific histological classification. These other pancreatic tumours in TCGA were mostly outliers in analysis of RNA‐seq data, and hence, we restricted our analysis to the 147 pancreatic ductal adenocarcinoma tumours. These tumours typically express ~100–150 GPCRs, many of which have elevated expression in pancreatic ductal adenocarcinoma compared to the normal pancreas (Sriram, Moyung, et al., 2019). Numerous GPCRs with increased expression in pancreatic ductal adenocarcinoma are found in virtually every tumour. Some of these GPCRs are targets for approved drugs, which potentially might be repurposed/repositioned to treat pancreatic ductal adenocarcinoma. GPCR expression in pancreatic ductal adenocarcinoma appears independent of tumour grade/stage, patient sex or KRAS mutation status (Sriram, Moyung, et al., 2019). A subset of ~30 GPCRs are consistently expressed in most (>80%) TCGA pancreatic ductal adenocarcinoma tumours, suggesting their potential biological importance and possible utility as therapeutic targets.
Pancreatic adenocarcinoma tumours possess dense stroma and generally large numbers of CAFs. Patient‐derived CAFs have a different GPCR repertoire than that of fibroblast precursor cells, pancreatic stellate cells (PSCs) and pancreatic fibroblasts (Wiley et al., 2018). Assessment of TCGA data using bioinformatics methods can predict GPCRs in pancreatic adenocarcinoma tumours that are likely expressed in resident (tumour‐infiltrating) immune cells (Sriram, Moyung, et al., 2019). Figure 1a–d shows GPCR expression in tumours (TCGA), pancreatic adenocarcinoma cell lines (CCLE) and CAFs and bioinformatically inferred expression in TCGA tumours of immune‐associated GPCRs.
FIGURE 1.
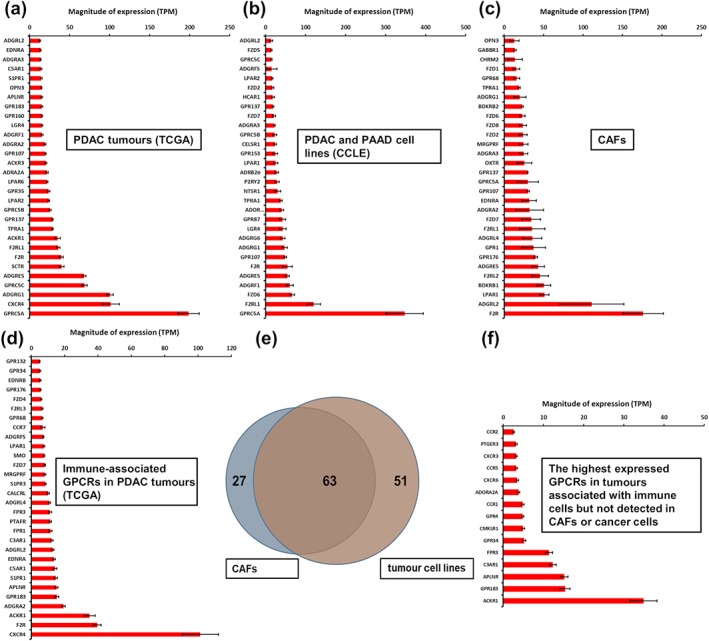
(a–c) The highest expressed GPCRs from RNA‐seq data in (a) pancreatic ductal adenocarcinoma (PDAC) tumours (in TCGA), (b) pancreatic adenocarcinoma (PAAD) cell lines (in CCLE, analysed by Tatlow & Piccolo, 2016), and (c) cancer‐associated fibroblast (CAFs) in culture. (d) GPCRs expressed in TCGA PDAC tumours, associated with the immune marker CD45. (e) Venn diagram that shows the number of GPCRs with average expression >1 TPM in pancreatic adenocarcinoma cell lines and CAFs. (f) The most highly expressed immune compartment‐associated GPCRs (expressed at >1 TPM) in TCGA pancreatic adenocarcinoma tumours but not expressed in pancreatic adenocarcinoma CAFs or tumour cells
Distinct GPCR profiles are predicted to localize in major cellular compartments in pancreatic adenocarcinoma tumours and together represent the overall GPCR repertoire. Figure 1e shows the overlap in GPCR expression, between CAFs (assessed in our laboratory via RNA‐seq, qPCR, microarrays and functional studies; Wiley et al., 2018) and pancreatic adenocarcinoma cell lines from the CCLE, based on whether a GPCR was detectable at >1 TPM (which likely predicts functional activity). These results highlight candidate GPCRs that may have cell‐selective effects in pancreatic adenocarcinoma tumours. Figure 1f shows immune‐associated GPCRs detected in pancreatic adenocarcinoma tumours but not substantially expressed (<1 TPM) in pancreatic adenocarcinoma tumour cells or CAFs.
Figure 2a–f shows the number of GPCRs in each cell compartment that couple to each of the four Gα protein families (Gs, Gi, Gq/11 and G12/13), based primarily on curated data from the GtoPdb (Alexander et al., 2019). Pancreatic adenocarcinoma tumours and each cell compartment express GPCRs that can couple to each of the Gα families. Also shown are those data weighted for magnitude of expression of each GPCR (see Section 2.1), thus quantifying the size of the “repertoire” of GPCRs that couple to each type of Gα protein. Gi‐/Go‐coupled GPCRs are most abundant and most highly expressed. Gs‐coupled GPCRs comprise only a small portion of the GPCRome, implying that pancreatic adenocarcinoma cells have fewer highly expressed Gs‐coupled GPCRs and that this category of GPCR may show the least redundancy of signalling. These GPCRomic data for pancreatic adenocarcinoma/pancreatic ductal adenocarcinoma tumours and cell lines help identify GPCRs for prioritization for further experiments and aid in evaluation of findings from functional studies, as discussed below.
FIGURE 2.
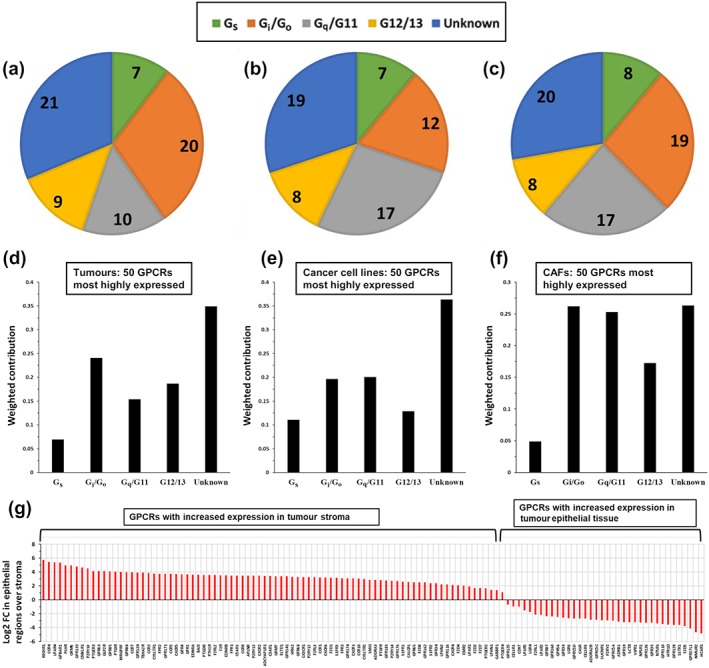
(a–c) Gα coupling of the 50 highest expression GPCRs in pancreatic ductal adenocarcinoma (PDAC) tumours, pancreatic adenocarcinoma (PAAD) cell lines, and CAF srespectively. As described in Section 2.1, the tumour data are from TCGA, pancreatic adenocarcinoma cell line are from CCLE and CAF data from Wiley et al. (2018). Analysis to define coupling to G proteins was performed as previously described (Sriram, Moyung, et al., 2019) and in Section 2.1. (d–f) The weighted contribution of GPCRs coupling to each type of Gα protein in PDAC tumours, pancreatic adenocarcinoma cell lines and CAFs (as in a–c), as described in Section 2.1. (g) Differential expression analysis of RNA‐seq data from tumour stroma compared to epithelial regions (Maurer et al., 2019) identifies GPCRs that are enriched in each compartment. Log2 FC indicates the Log2 fold‐change in expression between epithelial and stromal regions; positive values indicate higher expression in stroma compared to epithelium, vice versa for negative values
2.2.3. GPCR expression in stromal and epithelial compartments of pancreatic adenocarcinoma tumours
Using microdissection methods with pancreatic adenocarcinoma tumours, Maurer et al. (2019) isolated regions enriched for epithelial cells (i.e. mainly cancer cells) or stroma (mainly fibroblasts and immune cells) and then performed RNA‐seq to evaluate gene expression in these compartments. Mining of these data and differential expression analysis (Section 2.1) facilitate identification of the most highly enriched GPCRs in epithelial cells (i.e., expressed in cancer cells) and components of the tumour micro‐environment (i.e. likely expressed in CAFs and/or immune cells; Figure 2g). Such results suggest GPCR candidates for targeting of specific cellular compartments or possible targets in both pancreatic adenocarcinoma cells and other cells in the TME.
The data in Figure 2g are consistent with those in Figure 1: Stroma‐associated GPCRs are enriched in the immune and CAF compartments of pancreatic adenocarcinoma tumours (Sriram, Moyung, et al., 2019). Many highly expressed GPCRs in CAFs have elevated expression in the stromal compared to epithelial regions, while higher expressed GPCRs in epithelial tissue tend to be highly expressed in cancer cell lines (e.g. Figure 1b). CAFs and cancer cells in tissue culture generally retain the GPCR expression profiles detected in tumours in vivo.
Pancreatic adenocarcinoma cancer cell lines in CCLE typically have high expression of epithelial markers (e.g. EpCAM and E‐Cadherin), consistent with their epithelial origin. Pancreatic adenocarcinoma cell lines show considerable heterogeneity in GPCR expression, but in general, GPCRs strongly associated with the epithelial compartment in the analysis above are also highly expressed (on average) in pancreatic adenocarcinoma cell lines. Further, expression of GPCRs in cell lines analysed in CCLE is generally consistent with that in low‐passage tumour cell lines isolated from patients with pancreatic adenocarcinoma (Witkiewicz et al., 2016), implying that the expression of GPCRs in cancer cell lines tends to be maintained, despite passage in culture. GPCR expression data in pancreatic adenocarcinoma cell lines are accessible at https://insellab.github.io/gpcr_cells_exp.
3. PART 2: FUNCTIONAL DATA ON GPCRs IN PANCREATIC ADENOCARCINOMA
In Part 2, we critically evaluate published data, based on the pancreatic adenocarcinoma GPCRome, for certain GPCR families and GPCRs implicated in pancreatic adenocarcinoma cells and tumours. Experimental studies may report findings for a particular GPCR (e.g., in a cell line) that is not widely expressed in pancreatic adenocarcinoma tumours or pancreatic adenocarcinoma cell lines. We advise skepticism regarding a potential role for such GPCRs in pancreatic adenocarcinoma biology or as candidate drug targets. Conversely, we advocate greater confidence for those GPCRs that omics data reveal that are widely expressed in pancreatic adenocarcinoma tumours and cells.
No lower limit has been established regarding the level of mRNA expression needed for a GPCR to be a functional protein. However, it is difficult to reconcile findings of a strong functional response for an mRNA that is expressed below or near detection thresholds by highly sensitive methods (e.g, RNA‐seq). For several GPCRs discussed below, we note reports of functional effects, typically in pancreatic adenocarcinoma cell lines, which contrast with data for the same cell lines (from CCLE) that indicate near‐zero mRNA expression. Such contradictory results may reflect hitherto unknown relationships between mRNA and protein expression, or perhaps more likely, challenges associated with reproducibility of data related to reagents, protocols and cell lines among different laboratories.
In addition, we identify endogenous agonist‐GPCR pairs. In some cases, GPCR activation may be constitutive (agonist‐independent), but for other GPCRs, agonists (e.g. peptides, proteins [such as proteases] and “small” molecules [e.g. biogenic amines, https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2844 and ATP sphingosine‐1‐phosphate]) may be derived from the circulation or as autocrine/paracrine entities within the tumour itself, a relationship recently termed “oncocrine” (Wu et al., 2019).
3.1. Smoothened GPCR (SMO)
Numerous studies have shown a role for smoothened (https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=239), a member of the frizzled class of GPCRs, which are primarily activated by Wnt proteins. SMO is believed to be constitutively active, signalling via Gi/Go and Gq/G11 mechanisms and hedgehog (Alexander et al., 2019). SMO is highly expressed in pancreatic adenocarcinoma in CAFs and pancreatic adenocarcinoma cells; tumourigenic effects of SMO have been shown in cultured cells and animal models (Ji, Mei, Xie, & Cheng, 2007; Lee et al., 2014; Li et al., 2012; Onishi et al., 2011; Thayer et al., 2003). Unfortunately, clinical trials with SMO inhibitors in pancreatic ductal adenocarcinoma patients were unsuccessful (Catenacci et al., 2015). A large literature exists on SMO in pancreatic adenocarcinoma, including multiple review articles. Interested readers can obtain further information regarding SMO in the references above and a review by Gu et al. (Gu, Schlotman, & Xie, 2016).
3.2. https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=14
Figure 3a–c summarizes the expression of chemokine receptors in pancreatic adenocarcinoma tumours, tumour cells and CAFs. TCGA data indicate that C–X–C motif chemokine receptor 4 (https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=71) is the highest expressed chemokine receptor in pancreatic adenocarcinoma/pancreatic ductal adenocarcinoma tumours (~100 TPM). The atypical chemokine receptors https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=316 and https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=80 are also expressed at relatively high levels. Pancreatic adenocarcinoma cell lines assessed by CCLE express relatively few chemokine receptors; C–C motif chemokine receptor‐like 2 (https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=78) and CXCR4 are most highly expressed. CAFs prominently express ACKR3 and https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=315. Chemokines can be synthesized within pancreatic adenocarcinoma tumours. Figure 3d–f shows expression of chemokine ligand genes in pancreatic adenocarcinoma tumours and cell types.
FIGURE 3.
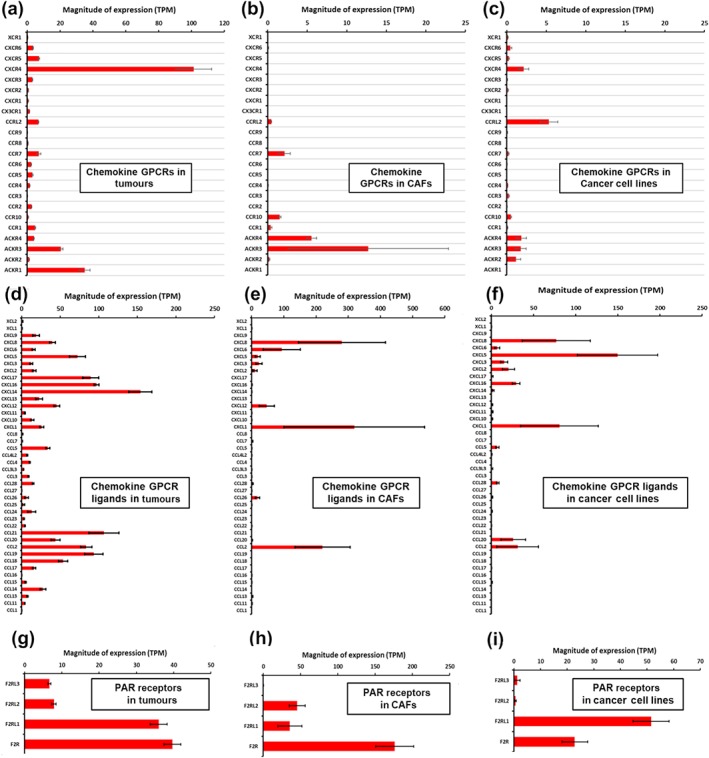
mRNA expression in tumour tissue, cancer‐associated fibroblasts (CAFs) and tumour cell lines, respectively, for (a–c): chemokine GPCRs; (d–f): chemokine ligands and (g–i) Protease‐activated receptor (PAR) receptors. Data were obtained as described in Section 2.1
Pancreatic ductal adenocarcinoma tumours (in TCGA) express numerous chemokine receptors: 16 of the 23 chemokine receptors are expressed at levels >1 TPM. In general, cell lines from pancreatic adenocarcinoma and primary CAFs show very low expression of chemokine receptors; only CXCR4, CCRL2, ACKR3/CXCR7 and ACKR4 are expressed at >1 TPM. Bioinformatic analysis of TCGA tumours supports these findings, suggesting a strong association of chemokine receptors with the immune compartment (Figure 1d,f). Most published functional data on chemokine receptors in pancreatic adenocarcinoma are from studies of pancreatic adenocarcinoma cell lines. Relatively little such information is available regarding these receptors in immune cells in pancreatic adenocarcinoma tumours.
Among the first studies indicating a role for CXCR4 and its ligand, C–X–C motif chemokine ligand 12 (https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4358; aka stromal cell‐derived factor [SDF]‐1) was work by Koshiba et al. (2000), who showed that proteins for the ligand and receptor are highly expressed in pancreatic adenocarcinoma tumours and cells. CXCL12–CXCR4 signalling increased pancreatic adenocarcinoma cell migration, which was blocked by a CXCR4 antagonist. Subsequent work (Wehler et al., 2006) suggested that higher CXCR4 protein expression is associated with more advanced tumours and high rates of metastasis. Further work (Heinrich, Lee, Lu, Lowy, & Kim, 2012) showed that CXCR4 and ACKR3/CXCR7 are expressed in numerous pancreatic adenocarcinoma cell lines and mediate responses, including extracellular signal–regulated kinase (https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=514) phosphorylation by CXCL12 and tumour cell proliferation. The authors also found that KRAS can drive CXCL12 signalling. Overall, the results suggest an autocrine mechanism by which pancreatic adenocarcinoma tumour cells drive their proliferation via chemokine receptors.
Work by Cui et al. (2011) provided additional support for the relevance of the CXCL12–CXCR4 axis. The authors showed that increased ligand and receptor protein expression in pancreatic adenocarcinoma tumour is indicative of more advanced, de‐differentiated tumours and higher levels of angiogenesis. CXCR4 signalling via CXCL12 promoted invasion and epithelial–mesenchymal transition (EMT) in tumour cells via non‐canonical activation of the hedgehog pathway. Others have observed crosstalk between SMO and CXCR4 signalling (Li et al., 2012) and that CXCR4 can promote tumour cell proliferation by canonical GPCR signalling pathways, via ERK and PKB/AKT (Shen et al., 2010).
Together, these findings suggest that CXCR4 antagonists may be therapeutics for pancreatic adenocarcinoma tumours. Indeed, Morimoto et al. (2016) found that CXCR4 enhances the resistance of pancreatic adenocarcinoma cells to treatment with gemcitabine and that CXCR4 antagonists reduce the growth of drug‐resistant cells. Data supporting the possible combination of CXCR4 antagonists and cytotoxic agents to treat pancreatic adenocarcinoma are provided by findings that indicate that the antimalarial drugs https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5535 and https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7198 are CXCR4 antagonists and can reduce CXCR4‐mediated proliferation of pancreatic adenocarcinoma tumour cells (Kim et al., 2012).
However, concerns exist regarding the potential use of CXCR4 antagonists for pancreatic adenocarcinoma. One concern is that CXCL12–CXCR4 may be tumour suppressive (Roy et al., 2014), which contradicts the data above showing promotion of tumour progression by CXCR4. Further, while CXCR4 is expressed by tumour cells, most CXCR4 expression in pancreatic adenocarcinoma tumours is likely in immune cells (Figure 1d,f). CXCR4 is highly expressed in T‐cell subtypes and has been studied in many tumour types (Susek, Karvouni, Alici, & Lundqvist, 2018). CXCR4 can mediate tumour cell‐immune cell communication, perhaps altering the balance between numbers of T‐regulatory and natural killer cells. It is unclear if this occurs in pancreatic adenocarcinoma. Understanding potential impacts of CXCR4 on immune cell types in pancreatic adenocarcinoma tumours will be crucial for determining its utility as a potential target for pancreatic adenocarcinoma.
Similar ideas have been suggested for the https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=70, https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=69 and https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=68 receptors in tumour cell lines: a signalling axis involving factors such as https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=819, https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=829 and https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=821 (Chen et al., 2014, 2012). These chemokine receptors are not significantly expressed in most pancreatic adenocarcinoma tumour cell lines and have generally low expression in pancreatic adenocarcinoma tumours. This suggests potential heterogeneity of cell lines in different laboratories. Also, certain chemokine ligands can activate multiple receptors. The roles of CXCR1–3 appear to be less functionally important than CXCR4 but require further clarification.
A role for https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=64 and https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=66 receptors has also been suggested. Immunohistochemistry has revealed that CCR7 is expressed in pancreatic adenocarcinoma tumours, perhaps associated with lymph node metastasis and poorer survival (Nakata et al., 2008). It was further shown (Zhang, Wang et al., 2016) that CCR7, activated by https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=811, promotes metastatic and EMT transformation phenotypes in pancreatic adenocarcinoma tumour cells, via ERK and NF‐κB. These results are somewhat challenging to reconcile with the negligible mRNA expression of CCR7 in tumour cell lines (Figure 3c), including in those used in the studies by (Zhang, Wang, et al., 2016). Immunohistochemistry of pancreatic adenocarcinoma tumours also revealed that CCR9 expression is elevated in tumour sections and CCR9 activation ex vivo promoted proliferation of pancreatic adenocarcinoma cancer cells (Shen et al., 2009). However, there is negligible CCR9 mRNA expression in tumours or tumour cell lines from TCGA or CCLE, respectively. Thus, there are striking contrasts for RNA and reported protein/functional data for CCR7 and CCR9 from different sources. These inconsistencies highlight the challenges with studying GPCRs, for example, ensuring the specificity of GPCR antibodies and of the action of chemokine (and other) ligands at particular receptors. Further efforts are needed to define the role for CCR7 and CCR9 in pancreatic adenocarcinoma, given the incongruities between the very low mRNA expression and suggested relevance in pancreatic adenocarcinoma.
3.3. Proteinase‐activated receptors
The https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=59 (PAR) family consists of four receptor (PAR1–4, aka https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=347 or PAR1; ([Coagulation Factor II Thrombin Receptor]) and F2RL1–3 [Coagulation Factor II (Thrombin) Receptor‐Like 1–3]) activated by thrombin and other proteases. All four receptors are expressed in pancreatic adenocarcinoma tumours, especially F2R (PAR1) and https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=348 (PAR2). F2RL1 is the highest expressed PAR receptor in pancreatic adenocarcinoma cancer cell lines, while F2R is most highly expressed in CAFs (>150 TPM; Figure 3g–i). Functional effects of F2R and F2RL1 have been shown in pancreatic adenocarcinoma cells. Hence, both F2R and F2RL1 are potential regulators of features of the malignant phenotype in pancreatic adenocarcinoma since these receptors are present on both major cell types in pancreatic adenocarcinoma tumours.
Functional data for F2R in pancreatic adenocarcinoma have been primarily derived from studies on tumour cell lines. For example, early studies with MIA PaCa‐2 (pancreatic adenocarcinoma) cells suggested that high F2R expression was correlated with de‐differentiation, enhanced DNA synthesis and calcium signalling (Rudroff et al., 1998, 2002). More recent work (e.g., Huang et al., 2018) identified a feedback loop that includes https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1628, https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2098 and https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=360, which together promote a migratory, invasive phenotype, thereby likely enhancing metastasis. The authors also showed that F2R increases perineural invasion in ex vivo and in vivo models. Other studies (Tekin et al., 2018) implicated F2R in promotion of pancreatic adenocarcinoma metastasis by maintaining a mesenchymal phenotype in pancreatic adenocarcinoma cells. F2R depletion reduced tumour growth but increased metastasis in an orthotopic mouse model of pancreatic adenocarcinoma, thus suggesting that in vivo, F2R signalling in pancreatic adenocarcinoma tumour cells can slow tumour growth but increase metastasis.
These results contrast with those from Yang et al. (2019). Studies with cell lines from wild‐type (WT) and KPC mice (which have mutations in KRAS and p53 and spontaneously develop pancreatic adenocarcinoma tumours) or cell lines with F2R knockout (KO) revealed that F2R KO cells were incapable of forming tumours in orthotopic models in WT mice but formed such tumours in immune‐deficient mice. These findings suggested that F2R is critical for tumour cells to evade tumour suppression by immune cells.
Resolving the differing results with respect to how F2R drives tumour growth in vivo will be important for defining its potential as a target for treating pancreatic adenocarcinoma. Work by Queiroz et al. (2014) used a genetic mouse model with F2R KO to study orthotopic xenografts of human pancreatic tumours in mouse pancreas. The authors showed that compared to WT mice, F2R KO mice had lower tumour growth rates, less angiogenesis and improved drug response. F2R KO did not change the degree of fibrosis but appeared to impact on macrophage recruitment and enhanced resistance to chemotherapy. Che, Park, and Stokol (2017) showed effects of F2R in endothelial cells, treated with pancreatic adenocarcinoma cell line‐derived extracellular vesicles containing tissue factor and Factor X. Extracellular vesicles may be a means to engage F2R/GPCR signalling in the TME.
F2R thus has numerous pro‐oncogenic effects that involve multiple cell types in pancreatic adenocarcinoma tumours, including not only pancreatic adenocarcinoma cells but also endothelial cells, monocytes and fibroblasts. Of note, https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?tab=biology&ligandId=4047, an approved antagonist of F2R, is a drug that might be repurposed for the treatment of pancreatic adenocarcinoma tumours.
As with F2R, functional data for F2RL1 are primarily from studies in pancreatic adenocarcinoma cells. F2RL1 activation by trypsin (Ohta et al., 2003; Xie, Duan, Liu, Zheng, & Zhou, 2015) or an activating peptide, Ser‐Leu‐Ile‐Gly‐Lys‐Val (SLIGKV; Xie et al., 2015) enhances proliferation in high F2RL1‐expressing pancreatic adenocarcinoma cell lines. Using the same agonist peptide with different cell lines, Shi, Queiroz, Stap, Richel, and Spek (2013) reported that F2RL1 activation promotes migration, but not proliferation, likely via activation of ERK, https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1499 (https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=519) and https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2206 (SRC Proto‐Oncogene, Non‐Receptor Tyrosine Kinase). Work by the same group also demonstrated a role for F2RL1 in the tumour micro‐environment: Using orthotopic xenografts of human pancreatic adenocarcinoma tumour cells in mice with or without F2RL1 KO, the authors found that F2RL1 expression in the tumour micro‐environment promotes tumour growth but may reduce lymphangiogenesis and metastasis to lymph nodes, thus, perhaps playing a dual role, akin to that suggested above for F2R.
F2R and F2RL1 are activated by thrombin and trypsin. The F2 gene, which codes for pro‐thrombin (the precursor for https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4453) is not highly expressed (<1 TPM) in pancreatic adenocarcinoma tumours (TCGA data) and not meaningfully expressed in pancreatic adenocarcinoma cells or CAFs. Thrombin may thus not be the agonist for F2R and F2RL1 in pancreatic adenocarcinoma tumours. F2R is activated by other proteases, including MMP1, which is highly expressed in pancreatic adenocarcinoma tumours (TCGA data) and pancreatic adenocarcinoma cells (from CCLE), suggesting paracrine signalling between cancer cells and CAFs and perhaps autocrine signalling in cancer cells. Moreover, the exocrine pancreas forms and secretes trypsin, which may be available within the microenvironment of pancreatic adenocarcinoma tumours in vivo.
3.4. EDG (S1P and LPA) receptors
https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=135 (EDG) receptors are activated by sphingosine 1‐phosphate (https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=135), an agonist for S1P receptors1–5 (S1PR1–5) and lysophosphatidic acid (https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=36), which activates LPA receptor (LPAR)1–6. Both groups of receptors primarily signal via Gi/Go or Gq/G11, though exceptions exist (e.g. https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=277 receptor also signals via G12/ 13; Alexander et al., 2019). Figure 4a–c summarize the expression of S1P and LPA receptors in pancreatic adenocarcinoma tumours, tumour cells and CAFs.
FIGURE 4.
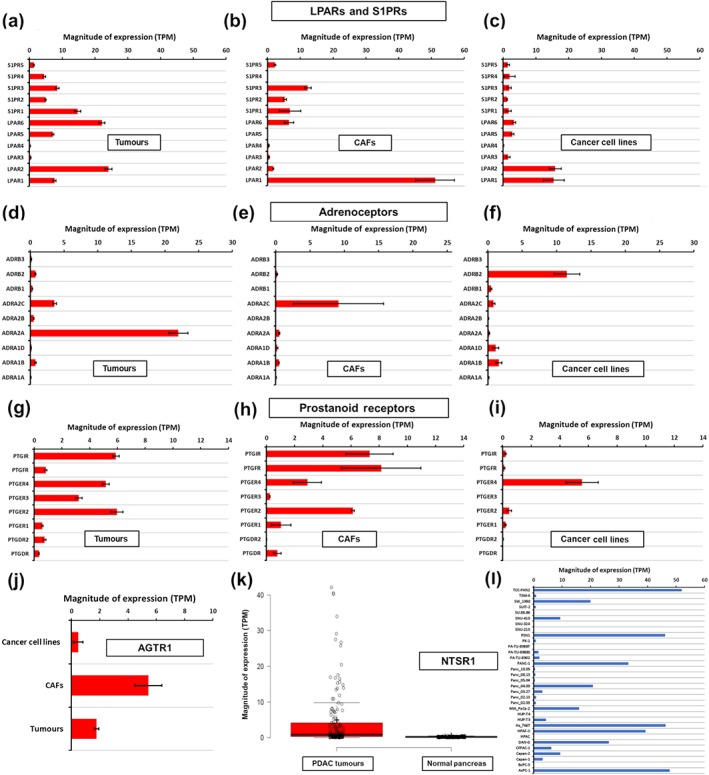
mRNA expression in tumour tissue, cancer‐associated fibroblasts (CAFs), and tumour cell lines for LPA and S1P receptors (a–c); adrenoceptors (d–f); prostanoid receptors (g–i). (j) mRNA expression in tumour tissue, CAFs, and tumour cell lines for AT1 receptors. Expression of NTSR1 is shown comparing tumour tissue with normal tissue (k) and in pancreatic adenocarcinoma (PAAD) cell lines from CCLE (l). Data were obtained as described in Section 2.1
Multiple EDG receptors are expressed in pancreatic ductal adenocarcinoma tumours. https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=272 receptor is among the highest expressed GPCRs in CAFs and is also expressed by pancreatic adenocarcinoma cancer cells; https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=273 receptor is primarily expressed in pancreatic adenocarcinoma cancer cells. S1P receptors are present in CAFs and likely other cells in the microenvironment but with relatively low expression in tumour cells. ENPP2 (ectonucleotide pyrophosphatase/PDE family member 2, https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2901), the gene for https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2901, which catalyses LPA synthesis from lysophosphatidylcholine (LPC), is highly expressed (~20 TPM) in pancreatic adenocarcinoma tumours (TCGA) and CAFs (Wiley et al., 2018) but is not significantly expressed in pancreatic adenocarcinoma cell lines (CCLE). LPA synthesis thus likely occurs in the TME and may activate pancreatic adenocarcinoma cells in a paracrine manner.
With respect to functional data for LPA receptors, LPAR1, 2 and 3 promote migration in pancreatic adenocarcinoma cell lines (PANC‐1 and BxPC‐3) in a pertussis‐toxin dependent manner, implying that this is Gi‐/Go‐mediated effect (Stähle et al., 2003). The authors also showed post‐Gi/Go responses: ERK1/2 phosphorylation and translocation of activated ERK1/2 to the leading edge of cells, thereby promoting cell polarization and migration. The effects on migration were shown to depend on Ras, Rac 1 (Ras‐related C3 botulinum toxin substrate 1), MEK (MAPK kinase), JNK and RhoA (Ras homolog gene family member A). Komachi et al. (2009) demonstrated that in PANC‐1 cells LPA2 receptors acting via G12/13, inhibited EGF‐promoted migration. Those results are surprising, because LPA‐induced G12/13‐Rho activation is generally pro‐migratory (e.g. Gardner, Ha, Jayaraman, & Dhanasekaran, 2013). Tevteraas et al. (2016) reported that LPA induces DNA synthesis and migration in multiple pancreatic adenocarcinoma cell lines, including via transactivation of the EGFR (EGF receptor). Komachi et al. (2009) also identified LPA in ascites, potentially accessible to pancreatic adenocarcinoma tumours. In ensuing work, the same group showed that the LPA1/https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=274 receptor antagonist Ki16198 reduced migration and invasion in pancreatic adenocarcinoma cell lines and that orally administered Ki16198 decreased tumour size and metastasis in nude mice, in which the YAPC pancreatic adenocarcinoma cell line was placed into the abdominal cavity (Komachi et al., 2012).
Other studies showed that LPA1,2 and 3 receptors are active in PANC‐1 cells and stimulate the activation/phosphorylation of https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2180 (focal adhesion kinase) and https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2309, resulting in their localization to focal adhesions, whereas Ki16198 inhibition of LPA1 and LPA3 receptors induced a more diffuse, cytosolic distribution (Liao et al., 2013). Activation of these focal adhesion proteins promoted cell migration. Knockdown of LPA1 and LPA3 receptor in PANC‐1 cells confirmed their role as drivers of migration and invasion, mediated at least in part, by their stimulation of MMP activity (Fukushima et al., 2017). The authors also found that cisplatin treatment of PANC‐1 cells increased expression of ENPP2/ATX and the migratory and invasive properties of the cells, effects abrogated by knockdown of LPA1 and LPA3 receptors.
https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=94 ,5 and 6 receptors have also been reported to impact on PANC‐1 cells (Ishii et al., 2015). Knockdown of https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=163 receptor reduced cell motility, but LPA4 and https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=124 receptor knockdown had the opposite effect. Knockdown of LPA4 and LPA5 receptors also increased, but LPA6 receptor knockdown decreased colony formation.
A few studies have assessed S1P receptors in pancreatic adenocarcinoma. https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2407 (FTY720), an approved S1P receptor functional inhibitor (i.e. fingolimod drives internalization and subsequent loss of active S1P receptors; see GtoPdb entry on S1P receptors/fingolimod), reduced proliferation, migration, colony formation and invasion, decreased levels of phosphorylated Akt, https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2844 (B‐cell lymphoma 2) and https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1619, while up‐regulating https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1625 in three pancreatic adenocarcinoma cells lines (Shen et al., 2007). Lankadasari et al. (2018) observed that Fingolimod treatment of pancreatic adenocarcinoma cells reduced growth rates and EMT, increased apoptosis and inhibited signalling of https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=275 to https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2994, a possible mechanism for the functional effects. Fingolimod treatment of orthotopic tumours (MIA PaCa‐2 and PAN 02 cell lines) in mice decreased tumour growth and increased efficacy of gemcitabine. Bi et al. (2014) found effects of S1P on PSCs: https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2204 (SPHK1), the enzyme that generates S1P, is up‐regulated in pancreatic adenocarcinoma tumours compared to normal tissue and present in pancreatic adenocarcinoma cells. S1P treatment induced ERK and AKT phosphorylation, fibrotic gene expression, MMP9 expression and proliferation of PSCs. Conditioned media from S1P‐stimulated PSCs increased PANC‐1 cell migration and invasion, effects dependent on https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=276. Moreover, S1PR2 knockdown in PSCs reduced growth of PSCs and tumour cells orthotopically xenografted into mice. These findings imply interaction of pancreatic adenocarcinoma cells with PSCs/CAFs via S1P receptors.
Considerable evidence thus exists for functional effects of LPA and S1P receptors in pancreatic adenocarcinoma, especially for the LPA1–3 receptors. Much of the data derives from studies with PANC‐1 cells. Further work is needed to determine if such functional effects occur in other pancreatic adenocarcinoma cell lines and pancreatic adenocarcinoma tumours in vivo. Given the high expression of LPA1 receptor in CAFs, studies assessing effects of LPA in CAFs would be of interest, especially to determine if blocking LPA receptors is a useful therapeutic approach for pancreatic adenocarcinoma tumours.
3.5. Adrenoceptors
https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=4 are activated by endogenous catecholamines, that is, https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=509 (epinephrine) and https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=484 (norepinephrine) released from chromaffin tissue (e.g. the adrenal medulla) and for noradrenaline, at post‐ganglionic sympathetic nerve endings. Epinephrine and norepinephrine can activate: (a) α1‐adrenoceptors (https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=22, https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=23 and https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=24) which signal primarily via Gq/G11; (b) α2‐adrenoceptors (https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=25, https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=26 and https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=27), which signal primarily via Gi/Go; (c) β‐adrenoceptors (https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=28, https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=29 and https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=30) which signal primarily via Gs.
With respect to adrenoceptor expression (Figure 4d–f), pancreatic adenocarcinoma cell lines express much more ß2 (~10 TPM) than do pancreatic adenocarcinoma tumours (~1 TPM). This discrepancy may be a consequence of cell culture, but alternatively, ß2‐adrenoceptors may be expressed by other cell types in pancreatic adenocarcinoma tumours. α2A‐adrenoceptors is the highest expressed adrenoceptor in tumours and is not expressed in pancreatic adenocarcinoma cell lines or CAFs but is associated with immune cell markers and thus may be present on immune cells in pancreatic adenocarcinoma tumours.
The impact of β‐adrenoceptors in pancreatic adenocarcinoma tumours has been studied, but other adrenoceptors are largely unexplored. Evidence for protein expression and a functional role of β‐adrenoceptors in pancreatic adenocarcinoma cell lines (PANC‐1 and BxPC‐3) was shown by Weddle, Tithoff, Williams, and Schuller (2001), who used radioligand binding and found that β2‐adrenoceptors were more abundant than β1‐adrenoceptors. In addition, β2‐adrenoceptors activation of cell lines with mutant KRAS increased DNA synthesis. Noradrenaline, via β‐adrenoceptors, reportedly stimulates proliferation and migration in PANC‐1 cells, perhaps via p38 phosphorylation (Huang, Wang, Yuan, Huang, & Zheng, 2011). Zhang, Yong Ma, Hu, and Zhang (2010) demonstrated a role for β1‐ and β2‐adrenoceptors in MIA PaCa‐2 and BxPC‐3 (pancreatic adenocarcinoma) cells, driving cAMP response element‐binding (CREB), https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1376, NF‐κB, and activator protein 1 (AP‐1) signalling, effects that were blocked by β2‐adrenoceptor antagonists. β2‐adrenoceptor antagonists reduced invasion of tumour cells via multiple signalling mechanisms, while antagonism of β1‐adrenoceptors suppressed invasion. These effects of β‐adrenoceptor antagonism occurred without addition of agonists, suggesting constitutive activity of these receptors in the cell lines. Zhang et al. (2011) have suggested that β2‐adrenoceptor antagonists induce G1/S cell cycle phase arrest and apoptotic cell death, perhaps via AKT and NF‐κB. The combined use of β2‐adrenoceptor antagonist with gemcitabine amplified the efficacy of gemcitabine in killing pancreatic adenocarcinoma cell lines (Shan et al., 2011). The same group subsequently showed a role for β‐adrenoceptors in up‐regulating https://www.guidetopharmacology.org/GRAC/FamilyIntroductionForward?familyId=900 1α (HIF‐1α), in response to the tobacco‐specific NNK (nicotine‐derived nitrosamine ketone), which reportedly activates β2‐adrenoceptors (Zhang et al., 2016).
A role for β‐adrenoceptors in the pancreatic adenocarcinoma microenvironment has been suggested. Kim‐Fuchs et al. (2014) tested communication between the sympathetic nervous system and pancreatic adenocarcinoma tumours using an orthotopic mouse model. The authors showed that exposure of mice to chronic stress (achieved by restraining mice in confined spaces) increased tumour progression and metastasis, effects blocked by administration of https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=564, a non‐selective β‐adrenoceptor antagonist. These results suggest that β‐adrenoceptors may mediate tumour sensing of and response to stress, an idea further explored by Renz et al. (2018), who showed that catecholamines produced by restraint‐induced stress promote pancreatic adenocarcinoma tumour progression in KPC mice. The authors found that adrenoceptor signalling induces secretion by tumour cells of neurotrophins, which in turn increased innervation of tumours and local concentrations of catecholamines, creating a positive feedback loop. Using a retrospective analysis, these authors suggested a pro‐survival effect of non‐selective β‐adrenoceptor antagonists in patients with Stage II and III pancreatic adenocarcinoma/pancreatic ductal adenocarcinoma. Immunostaining indicated epithelial expression of adrenoceptors in human tumours. Further, Pu et al. (2017) found that pancreatic adenocarcinoma patients have higher plasma epinephrine concentrations than do non‐pancreatic adenocarcinoma patients; higher concentrations in pancreatic adenocarcinoma patients were associated with shorter survival times. The authors also demonstrated that epinephrine enhances migration of PANC‐1 cells via TGF‐β and human antigen receptor (HuR) signalling. Epidemiological evidence for a role of β‐adrenoceptors in pancreatic adenocarcinoma tumours has been provided by Udumyan et al. (2017), who analysed the Swedish Cancer Register for effects of β‐adrenoceptor antagonist usage on survival rates in pancreatic adenocarcinoma patients. The authors found that β‐adrenoceptor antagonists were associated with lower mortality rates (adjusted hazard rate = 0.79, P < 0.001) and patients who received higher doses of β‐adrenoceptors antagonist or who had localized tumours had a larger effect.
Evidence thus exists from ex vivo cellular, in vivo experiments in mice and data from patients that β‐adrenoceptors antagonist, most likely β2‐adrenoceptors, play a role in pancreatic adenocarcinoma. However, low mRNA expression of β2‐adrenoceptor is found in pancreatic adenocarcinoma tumours in TCGA (Figure 4d–f), raising the concern that the high levels of expression of β‐adrenoceptors seen in certain pancreatic adenocarcinoma cell lines may not be observed in vivo. The epidemiological data and some of the in vivo data cited above may relate to other systemic effects of β‐adrenoceptors antagonists. Further study is needed to determine if β‐adrenoceptors are expressed in pancreatic adenocarcinoma tumours and help drive tumourigenic phenotypes. Such data could help provide evidence to test β‐adrenoceptors antagonists in clinical trials.
3.6. Prostanoid receptors
Pancreatic adenocarcinoma tumours express https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=58 (EP2–4 receptors), for which https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1883 is the primary endogenous agonist. Numerous approved https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=269 inhibitors blunt prostaglandin (PG) synthesis; agonists (e.g. https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1882, https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1883 and https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1915 analogues) are also approved drugs. Pancreatic adenocarcinoma tumours also express the https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=345), which is preferentially activated by PGI2 (prostacyclin). https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=341, https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=343 and https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=345 all signal via Gs, whereas https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=342 signals via Gq/G11.
Figure 4g–i shows that pancreatic adenocarcinoma tumours and cells express multiple types of prostanoid receptors. IP and EP2–4 receptors are expressed, on average, between 1 and 10 TPM. CAFs show high expression of IP, EP2, and PGF receptors, while pancreatic adenocarcinoma cells have high expression of EP4 (PTGER4). Consistent with the expression data, most functional evidence for prostanoid receptors in pancreatic adenocarcinoma relates to EP4 in pancreatic adenocarcinoma cells and implies that EP4 activity is tumourigenic and EP4 antagonism may have therapeutic benefit.
Studies with several pancreatic adenocarcinoma cell lines that express https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1376 and increase PGE2 production in response to added arachidonic acid imply that pancreatic adenocarcinoma tumour cells can produce PGE2, which has been shown to increase https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=324 synthesis and migration of tumour cells, effects blocked by COX‐2 inhibitors or EP2 antagonists (Eibl et al., 2003). In other studies, PGE2 increased COX‐2 expression in pancreatic adenocarcinoma cell lines via cAMP, https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=284 and CREB signalling, effects blocked by EP2 antagonists (Pino et al., 2005).
Evidence for function of EP4 includes data from PSCs and pancreatic adenocarcinoma cells. Charo et al. (2013) found that PGE2 stimulated proliferation, migration and expression of extracellular matrix proteins of human PSCs, effects blunted by EP4 antagonists, thereby providing evidence for PGE2 and prostanoid receptors in the TME. Takahashi et al. (2015) detected EP2 and/or EP4 activity in pancreatic adenocarcinoma cell lines; both receptors showed evidence for crosstalk with https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4971 (IGF‐1) receptors. EP2 and EP4 receptor antagonists abrogated the ability of IGF‐1 to stimulate growth in tumour cells and increased phosphorylation of PKC‐θ, suggesting that EP2 and EP4 signalling may reduce PKC activity. Administration of receptor antagonists in vivo to mice with orthotopic xenografts of IGF‐1‐expressing tumour cells reduced tumour growth and increased PKC‐θ phosphorylation. Further studies indicated that proliferative effects of EP2/EP4 receptors and IGF‐1 are associated with metabolic changes, including elevated levels of https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1067 and 5‐oxoproline and increased expression of the 5‐oxoproline‐catalysing enzyme, https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=272#1392 (Takahashi, Ichikawa, Morimoto, Tsuneyama, & Hijikata, 2019).
Other work on EP4 signalling in PANC‐1 cells confirmed a synergistic effect of PGE2 on IGF‐1 signalling, promoting mammalian target of rapamycin complex 1 (mTORC‐1) phosphorylation and activation via cAMP and PKA (Chang et al., 2015). Activation of EP1 receptors synergistically enhanced mTORC‐1‐promoted Ca2+ signalling. The authors hypothesized an additive/synergistic effect of https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4971 and PGE2, via IGF‐1 receptors and EP1/EP4 receptors, respectively, each contributing to mTORC‐1 phosphorylation. Schmidt et al. (2017) reported an opposite effect of PGE2‐EP4 signalling in PANC‐1 cells. PGE2 treatment reduced ERK1/2 phosphorylation via PKA/CREB, colony formation/growth and DNA synthesis, implying an anti‐cancer effect of EP4 receptors. Reconciling these contradictory data is needed to formulate more precise hypotheses regarding the role of those prostanoid receptors in pancreatic adenocarcinoma.
Prostacyclin (PGI2) is among the highest expressed Gs‐coupled GPCRs in CAFs and is also highly expressed in pancreatic adenocarcinoma tumours (Figure 4g). Based on the evidence above that implicate a role for Gs‐coupled EP4 receptors in PSCs, PGI2 receptors may have similar effects. Other prostanoid receptors, in particular, EP3 receptors are likely expressed in other cell types in the TME, as they are moderately expressed in tumours but not in CAFs or tumour cells.
3.7. Neurotensin receptor 1 (NTS1 receptor)
The 13 amino acid peptide https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1579) are synthesized by the NTS gene. NT activates neurotensin receptor 1 (https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=309 receptor), which signals via Gq/G11, increasing inositol trisphosphate (IP3)‐dependent calcium increase and PKC signalling. Expression of NTS1 receptor is elevated >10‐fold in pancreatic adenocarcinoma tumours compared to normal tissue , with high statistical significance (FDR<0.05) but is not highly expressed in magnitude (i.e. <1 TPM) in most pancreatic adenocarcinoma tumours (Figure 4k; Sriram, Moyung, et al., 2019). However, a subset (~25%) of patients have substantially higher NTS1 receptor expression (>1 TPM, in some cases >10 TPM). Several pancreatic ductal adenocarcinoma cell lines from CCLE also show high expression of NTS1 receptor (>10 TPM; Figure 4l). NTS1 receptor is not highly expressed in CAFs.
Neurotensin can enhance MIA PaCa‐2 (pancreatic adenocarcinoma) cell proliferation, primarily via stimulation of calcium signalling (Ishizuka, Townsend, & Thompson, 1993), and is expressed in pancreatic adenocarcinoma/pancreatic ductal adenocarcinoma but not in normal pancreas, endocrine tumours, or pancreatitis (Körner, Waser, Strobel, Büchler, & Reubi, 2015; Reubi, Waser, Friess, Büchler, & Laissue, 1998). NTS1 receptor activates https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=514, https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=518 and AP‐1 and, in turn, can increase IL‐8 secretion, DNA synthesis, proliferation and migration of pancreatic adenocarcinoma cells (Ehlers, Zhang, Hellmich, & Evers, 2000). NTS1 receptor can act synergistically with EGF to activate ERK via PKC‐dependent mechanisms (via NTS1 receptor) and PKC‐independent mechanisms (via EGF; Kisfalvi, Guha, & Rozengurt, 2005). NTS1 receptor may also regulate pH as NTS1 receptor expression appears to increase with lowering of pH in tumour cells (Olszewski‐Hamilton & Hamilton, 2011) and to promote intracellular alkalinization by activation of the Na+/H+ exchanger (Olszewski, Hlozek, & Hamilton, 2010). The NTS1 receptor antagonist https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1582 (SR‐48692) reduces growth of pancreatic adenocarcinoma cells (Wang, Li, Li, Cui, & Wang, 2011). NTS1 receptor is among the most highly expressed Gq‐coupled GPCRs with ability to increase intracellular calcium and promote migration of AsPC‐1 cells (Sriram et al., 2019). Neurotensin was effective at the low nM range, suggesting that in pancreatic adenocarcinoma tumours with high NTS1 receptor expression, neurotensin is likely able to activate Gq‐mediated responses.
NTS receptor has also been tested as a candidate for peptides conjugated with radionuclides for imaging and therapy of pancreatic adenocarcinoma. de Visser et al. (2003) first demonstrated this approach, using neurotensin analogues conjugated to the chelators Pentetic acid (DTPA) or 1,4,7,10‐tetraazacyclododecane‐1,4,7,10‐tetraacetic acid (DOTA) to facilitate labelling with indium‐111 (111In), lutetium‐177 (177Lu), or yttrium‐90. A 111In‐labelled analogue was stable in serum and bound with high affinity to neurotensin receptors in pancreatic adenocarcinoma tumour sections. The authors also demonstrated excellent uptake in mice injected with HT‐29 tumour cells but minimal accumulation in neurotensin receptor‐null tissues. Injection of a technetium (99Tc)‐labelled neurotensin analogue was well‐tolerated in four patients with pancreatic adenocarcinoma tumours (Buchegger et al., 2003), but only one had significant neurotensin receptor expression. This patient had high uptake of the radiolabelled neurotensin in the tumour, providing the first direct evidence for uptake of radiolabelled neurotensin in a patient's tumour.
Work on NTS1 receptor as an imaging target by Yin et al. (2017) used neurotensin analogues conjugated with Cu‐64 or IRDye800 to label pancreatic adenocarcinoma cell lines. Cu‐64 NT was able to detect AsPC‐1 orthotopic xenografts in mice by PET and of IRDye800‐labelled neurotensin for in vivo fluorescent imaging. Wang et al. (2018) reported that [18F]AlF‐labelled neurotensin can be a PET probe in mice with subcutaneous pancreatic adenocarcinoma xenografts.
Radiolabelled neurotensin targeted compounds may be potential therapeutics. Baum et al. (2018) treated six pancreatic adenocarcinoma patients with a NTS1 receptor antagonist, 3bp‐227, labelled with 177Lu. The compound was well tolerated. A positive response occurred in one patient, who lived nearly 1 year following treatment with 177Lu‐3bp‐227. A clinical trial (Identifier: NCT03525392) with this compound is in progress and has an enrollment target of 320 subjects. Since NTS1 receptor is expressed in only a subset of pancreatic adenocarcinoma patients, it will be important to confirm the presence of NTS1 receptor prior to administration of the NTS1 receptor radiotherapeutic.
3.8. Angiotensin receptors (AT1 and AT2)
The https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=6 1 and 2 (AT1 and AT2 ) are activated by angiotensin peptides, are synthesized from the AGT gene, and are part of the renin–angiotensin system. Angiotensin acts via angiotensin receptors in numerous tissues and cell types. Several AT1 antagonists are approved for use in patients, especially those with cardiovascular disorders, making such drugs potentially attractive for repurposing.
Multiple studies with pancreatic adenocarcinoma cell lines have implicated AT receptors as drivers of an oncogenic phenotype and possible targets for inhibition of pancreatic adenocarcinoma. Data from the TCGA, CCLE, and Genentech databases Klijn et al. (2015) indicate that AT2 receptor is not expressed in pancreatic adenocarcinoma tumours per cells or CAFs. AT1 receptor is, generally, also not highly expressed in pancreatic aessed at >5 TPM in CAFs (Figure 4j), consistent with data (discussed below) indicating that AT1 receptor is functional in CAFs and that AT1 receptor antagonists may blunt fibrosis in pancreatic adenocarcinoma.
A role for AT2 receptors in tumour cells and cell lines has been suggested via effects on migration or proliferation, which are blunted by AT receptor antagonists (e.g. Anandanadesan et al., 2008; Fujimoto, Sasaki, Tsuchida, & Chayama, 2001; Gong, Davis, Chipitsyna, Yeo, & Arafat, 2010; Ishiguro et al., 2015; McGhee et al., 2011). It is difficult to reconcile these findings with expression data from databases on cancer cells, showing very low expression of AT1 receptor in pancreatic adenocarcinoma cell lines and virtually no expression of AT2 receptor in tumour tissue, cancer cells or CAFs. Moreover, mRNA expression data (in CCLE) for pancreatic adenocarcinoma cell lines used in studies above showed negligible AT1 and AT2 receptor expression, implying the unlikely presence of functional AT receptor receptors in pancreatic adenocarcinoma tumour cell lines. Data from TCGA, CCLE, and other sources discussed in Part 1 indicate that AT2 receptor mRNA is not expressed in pancreatic adenocarcinoma. Thus, it is unclear how functional effects might occur for AT2 receptor, especially AT2 receptor, in pancreatic adenocarcinoma cells/cell lines.
Evidence for a functional role for AT1 receptor in the pancreatic adenocarcinoma stroma was provided by Masamune et al. (2013), who used immortalized PSCs and AsPC‐1 tumour cells for subcutaneous xenografts in nude (immunodeficient) mice. Co‐injection of PSCs with AsPC‐1 cells resulted in formation of larger tumours than AsPC‐1 cells alone. Treatment with https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=591, an AT1 receptor antagonist, reduced the growth of PSCs + AsPC‐1 tumours and the expression of fibrotic markers. Olmesartan treatment ofPSCs ex vivo confirmed these results, reducing proliferation and collagen‐I expression. Arnold et al. (2012) tested the AT1 receptor antagonist losartan in mice with orthotopic grafts of murine Pan02 pancreatic adenocarcinoma tumour cells in mice with a KO of the osteonectin (ON/SPARC) gene. Losartan treatment reduced tumour growth, perhaps via effects on PSCs.
Retrospective epidemiological study of pancreatic adenocarcinoma patients treated with angiotensin–renin pathway inhibitors has revealed that patients with hypertension who receive angiotensin‐converting‐enzyme (ACE) inhibitors or AT1 antagonists have improved survival compared to patients not receiving such drugs (Nakai et al., 2010). Independent of the presence of hypertension as a potential confounder, pancreatic adenocarcinoma patients who received ACE or angiotensin receptor inhibitors had improved survival. A large U.K. study subsequently revealed that the use of AT1 antagonists and ACE inhibitor was not associated with an overall decrease in risk of pancreatic adenocarcinoma (Mandilaras, Bouganim, Yin, Asselah, & Azoulay, 2017).
Limited evidence thus exists for AT1 antagonists inhibition as a way to target the TME. Additional studies are needed to strengthen this finding and for the possible repurposing of approved ACE inhibitors and AT1 receptor antagonists for treating pancreatic adenocarcinoma.
3.9. GPRC5A (GPCR class C group 5 member A)
https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=258, an orphan receptor, is the highest expressed GPCR in pancreatic adenocarcinoma tumours and is highly expressed in pancreatic adenocarcinoma cells (Sriram, Moyung, et al., 2019). Bioinformatics analysis reveals a strong correlation of GPCR5A mRNA expression with that of other pancreatic adenocarcinoma cell‐associated GPCRs. Together, these multiple receptor mRNAs form a composite GPCR signature for pancreatic adenocarcinoma. High expression of GPRC5A is an adverse predictor of survival in pancreatic adenocarcinoma/pancreatic ductal adenocarcinoma and its expression is associated with that of multiple oncogenic pathways, for example, those linked with KRAS (Sriram, Moyung, et al., 2019). GPRC5A protein expression has been documented in pancreatic adenocarcinoma cells and tumours (Insel et al., 2018; Jahny et al., 2017; Zhou et al., 2016). GPRC5A is also expressed in CAFs, though its function in these cells is unknown. Work by Zhou et al. (2016) showed that GPRC5A expression increases following treatment with gemcitabine and may contribute to drug resistance. Knockdown of GPRC5A reduces oncogenic phenotypes such as proliferation, migration, and colony formation. Data regarding GPRC5A as an adverse prognostic factor have also been reported by Jahny et al. (2017), who, in addition, provided data suggesting that GPRC5A drives the STAT3 pathway, which is known to be oncogenic. https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2030 (GSK‐3B), another potential signalling protein in cancer, is phosphorylated as a consequence of GPRC5A signalling, though the precise Gα signalling mechanism remains unknown (Liu, Yang, Pilarsky, & Weber, 2018). Together, these findings identify GPRC5A as a GPCR for deorphanization efforts and perhaps as a therapeutic target for pancreatic adenocarcinoma.
3.10. Additional GPCRs
Table 1 lists gene IDs, citations, and brief details for additional GPCRs that have been implicated in pancreatic adenocarcinoma in small numbers of studies, and thus, the available information is less than for the GPCRs discussed above. We focus on GPCRs discussed in >1 publication. Single publications have suggested a role in pancreatic adenocarcinoma for several GPCRs. This review does not include those GPCRs as they require replication and/or bioinformatics data in support of the findings.
TABLE 1.
Evidence in the literature for a role of additional GPCRs in pancreatic adenocarcinoma, not discussed in the text
| GPCR | Context | Findings and comments | Source |
|---|---|---|---|
| https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=222 and https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=223 | MIA PaCa‐2 cells and orthotopic mouse grafts | FPR1 and FPR2 (PAR1 and 2) are both expressed in these cells, but annexin1‐mediated effects on driving tumour metastasis are likely independent of FPRs. | Belvedere et al. (2014) |
| Various human cell lines |
Annexin‐1 activates both FPRs in the cell lines, increases calcium signalling and migration/invasion. Comment: Results from this study and the one above are difficult to reconcile with data from CCLE that indicate negligible expression of FPR mRNAs in these cell lines. |
Belvedere et al. (2014) | |
| Human pancreatic adenocarcinoma cancer stem cells and KPC mice | The antimicrobial peptide 18/leucine leucine‐37 (hCAP‐18/LL‐37) is produced in pancreatic adenocarcinoma stroma by macrophages, activates FPR2, and promotes tumour growth, which can be blocked by an FPR2 antagonist. | Sainz et al. (2015) | |
| https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=324 | Human cell lines, tissue sections, and several mouse models | P2Y2 is overexpressed in pancreatic adenocarcinoma and is a negative prognostic marker. Inhibition of P2Y2 reduces tumour growth and increases the effect of gemcitabine, improving survival of mice with pancreatic adenocarcinoma tumours. | Hu et al. (2019) |
| PANC‐1 cells | P2Y2 agonists enhance proliferation, via mechanisms including Src, CaM kinase II, and AKT, effects blocked by PKC inhibition. | Choi, Ji, and Lee (2013) | |
| PANC‐1 cells |
Multiple P2Y receptors, including https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=323, P2Y2 and https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=326 are expressed in these cells. ADP and UDP increase proliferation, which is blocked by antagonists of P2Y1 and P2Y6, respectively. Comment: CCLE data indicate, P2Y2 receptor is expressed, but P2Y1 and P2Y6 receptors are not expressed in PANC‐1 cells. |
Ko, An, Ji, Kim, and Lee (2012) | |
| https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=215 | Human tissue samples, multiple cell lines, and orthotopic xenografts in mice | Protein expression of dopamine receptor D2 and Gαi2 is elevated in pancreatic adenocarcinoma. D2 antagonism or knockdown reduces proliferation and migration of cell lines. D2 antagonism via haloperidol reduces tumour size and metastasis in orthotopic xenografts. | Jandaghi et al. (2016) |
| Review article |
Data combined from multiple cancer types suggest D2 receptor may be a target in pancreatic adenocarcinoma. Comment: D2 receptor is very low expressed in most human pancreatic adenocarcinoma cell lines and TCGA tumours. |
Bakadlag, Jandaghi, Hoheisel, and Riazalhosseini, (2019) | |
| https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=61 | Human cell lines; mouse subcutaneous xenografts | Somatostatin2–5 receptors can be targeted with peptides derived from somatostatin. Conjugation of peptides with anti‐cancer agents are used for targeted delivery, which is shown in grafted tumours. | Ragozin et al. (2018) |
| Human cell lines; mouse subcutaneous xenografts | SST1 expression is lost in pancreatic adenocarcinoma cells but re‐expressing it reduces cell growth by inducing cell‐cycle arrest. In vivo results: reduced tumour growth with SST1 receptor overexpression in grafted tumours. | Li et al. (2008) | |
| Capan‐1 cells and subcutaneous xenografts in mice; allografts using PC1.0 cells in hamsters | Gene transfer in vivo to restore SST2 receptor expression in pancreatic adenocarcinoma tumours inhibits vascularization, VEGF expression, and growth of tumours but induces somatostatin production and SST3 receptor expression. | Carrere et al. (2005) | |
| Human CAFs and orthotopic xenografts in mice with human CAFs+MIA PaCa‐2 cells | https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2018 (SOM230), an approved somatostatin analogue reduces tumorigenic properties and contents of the secretome from CAFs. IL‐6 drives CAF‐cancer cell communication; its secretion is reduced by pasireotide. In vivo, metastases are decreased, but tumour growth is not changed. | Moatassim‐Billah et al. (2016) | |
| https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=114 | Human CAFs | The proton‐sensing receptor, GPR68, is more highly expressed in human CAFs than in normal precursors. GPR68 signalling impacts the fibrotic phenotype and secretome of CAFs via cAMP/PKA/CREB increasing IL‐6 mRNA/protein. In low pH, CAFs secrete factors that promote pancreatic ductal adenocarcinoma cell growth, an effect blocked by GPR68 knockdown in CAFs and by IL‐6 neutralizing antibody treatment of CAFs conditioned media. | Wiley et al. (2018) |
| In silico analysis of TCGA data | GPR68 is ~10‐fold higher expressed in pancreatic adenocarcinoma tumours than normal pancreatic tissue. Expression of GPR68 is strongly associated with the expression of fibrotic markers and is associated with fibrotic pathways. | Sriram, Moyung, et al. (2019) | |
| Review + meta‐analysis | Review of GPR68 functional data and expression indicates a role for GPR68 in the TME in a range of tumour types (including pancreatic adenocarcinoma); expression in various cell types including CAFs, monocytes, and T‐cells. | Wiley, Sriram, Salmerón, and Insel (2019) |
4. DISCUSSION AND PERSPECTIVE
Other than their role in certain hormone‐responsive tumours, GPCRs have not been considered high priority targets in cancer, but studies, including in pancreatic adenocarcinoma, have increasingly explored this possibility, especially in the past 10 years. Numerous GPCRs are potential therapeutic targets but understanding of the regulation of pancreatic adenocarcinoma tumours by GPCRs is still evolving.
Figure 5 shows a workflow to illustrate the steps involved in target/drug discovery efforts to identify GPCR targets in pancreatic adenocarcinoma. These steps involve methods based on generation of omics data as well as mining of publicly available bioinformatics data on pancreatic adenocarcinoma, from which GPCR expression can be ascertained. Subsequent to such efforts, a variety of validation and cell‐based methods in vitro facilitate prioritization of GPCRs for subsequent study, ultimately in vivo, typically using mouse models, followed by eventual clinical studies using novel compounds or repurposing of existing drugs. This general schema also provides a means to organize data regarding specific GPCRs, in order to evaluate progress towards their potential realization as targets in pancreatic adenocarcinoma. At present, the NTS1 receptor is the most well‐developed candidate GPCR target in pancreatic adenocarcinoma, with multiple studies furnishing evidence for every step in the schema in Figure 5, leading to current clinical trials.
FIGURE 5.
A flowchart that describes a sequence of steps from omics and bioinformatics to early clinical studies, as applied to evaluate candidate GPCR drug targets in pancreatic adenocarcinoma. ADME, absorption, distribution, metabolism and excretion; KD/KO, knockdown/knockout; KPC mice, KRAS and TP53 mutant mice, which spontaneously develop pancreatic adenocarcinoma (PAAD)
Most studies regarding GPCR function in pancreatic adenocarcinoma have been on cancer cells, typically commercially available cell lines. Cancer cells in vivo may have GPCRomes and functional responses that differ from those of pancreatic adenocarcinoma cell lines studied in vitro. Mining of data on low‐passage, patient‐derived cancer cell lines generated by Witkiewicz et al. (2016) allays some of these concerns: GPCR expression appears relatively consistent between the low‐passage cancer cells and established cell lines. Further, analysis of epithelial‐ and stromal‐enriched regions of pancreatic adenocarcinoma tumours (from Maurer et al., 2019) discussed in Section 1 indicates that GPCRs highly expressed in cancer cells ex vivo are also highly expressed in cancer cells in vivo. However, direct evidence is needed to show if cell lines accurately reproduce the functional responses of cells in vivo. In this regard, the use of patient‐derived organoids, perhaps in co‐culture with other pancreatic adenocarcinoma tumour cell types, may prove informative (Baker, Tiriac, Clevers, & Tuveson, 2016; Tuveson & Clevers, 2019).
A sizable GPCRome exists in immune cells present in the TME (Section 1; Figure 1d,f), but little is known regarding roles for these receptors or the GPCRs expressed in subsets of immune cells. Current single‐cell RNA‐seq analysis of pancreatic adenocarcinoma tumours (e.g. Elyada et al., 2019; Lytle et al., 2019) yields limited data for GPCRs: they are often not detected consistently, as a result of their low expression and the current state of single‐cell sequencing technology. With improved cell isolation protocols that can yield larger numbers of individual cell types and access to primary tissue, methods such as fluorescent‐activated cell sorting (FACS) may aid in separating immune cell populations for study of their function and GPCR expression. Such an approach with pancreatic adenocarcinoma tumours from mice has provided novel information regarding CAFs and cancer stem cells (Elyada et al., 2019; Lytle et al., 2019). In particular, since immune‐oncology therapeutic approaches for pancreatic adenocarcinoma have not been successful (Upadhrasta & Zheng, 2019), such efforts in tumour immune cells might reveal GPCRs that could be useful targets for such approaches.
Our laboratory has described the GPCRome in pancreatic adenocarcinoma patient‐derived CAFs and the identification of a novel GPCR target, https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=114, that is activated by acidic pH (Wiley et al., 2018; Table 1). Studies by others have identified additional GPCRs, as noted in Section 2 and Table 1, that influence the function ofCAFs. However, the precise biological roles played by the many GPCRs expressed in CAFs is unknown. Of particular interest are highly expressed GPCRs, for example, F2R (PAR1), F2RL1 (PAR2), the https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=369 and the https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=41, for some of which approved drugs might be repurposed. GPCRs in sub‐populations of CAFs (Elyada et al., 2019) need to be further studied. In this review, we note several examples of paracrine communication and regulation between CAFs and pancreatic adenocarcinoma cells, which appear to involve ligand–receptor pairs that can modulate function of pancreatic adenocarcinoma tumours.
Methodological concerns exist regarding the identification of GPCRs and efforts to define their functions in pancreatic adenocarcinoma cells and tumours. One problem relates to GPCRs that have been suggested as functional in certain pancreatic adenocarcinoma cell lines despite their very low (or lack of) expression in tumours (from TCGA) and/or cell lines (from CCLE). Reasons for such difficulties include: (a) unreliable GPCR antibodies, resulting in false positive results for GPCR proteins; (b) issues related to validation/consistency of cell lines; and (c) the non‐selectivity of drugs that may interact with multiple GPCRs or other non‐GPCR targets.
The lack of knowledge regarding how well human and mouse pancreatic adenocarcinoma models align with respect to GPCR expression and function is a further challenge. Data comparing GPCR expression of different cell types in pancreatic adenocarcinoma tumours between the species may help clarify how well models (e.g. KPC mice) phenocopy human pancreatic adenocarcinoma and the potential for translating results for efficacy data of GPCR‐targeted drug from such models.
In summary, we draw the following general conclusions regarding the current status of GPCRs in pancreatic adenocarcinoma:
The GPCRome in pancreatic adenocarcinoma tumours comprise >100 GPCRs expressed at the mRNA level. Several GPCRs, for example, F2R (PAR1), GPRC5A, and F2RL1 (PAR2), are expressed in multiple cell types (e.g. pancreatic adenocarcinoma cells and CAFs), but other GPCRs are uniquely expressed in a particular cell compartment/type. Very limited data exist regarding GPCRs in pancreatic adenocarcinoma tumour endothelial cells. Most functional data in pancreatic adenocarcinoma have been derived from studies using established pancreatic adenocarcinoma cell lines.
Numerous highly expressed GPCRs, in particular, CXCR4, F2R (PAR1), F2RL1(PAR2), NTS1 receptor, E4 receptor, LPA1/2 receptor, SMO and GPRC5A, have functional (generally pro‐oncogenic) effects in pancreatic adenocarcinoma cells. Such findings imply that inhibition of one or more of those receptors may be a therapeutic strategy.
In vivo results for many of these GPCRs, including with pharmacological antagonists, provide evidence consistent with data from pancreatic adenocarcinoma cells; that is, GPCR antagonists may reduce pancreatic adenocarcinoma tumour growth and metastasis. Small molecules and peptides targeting GPCRs can enter the TME, but additional data demonstrating target engagement are needed.
Based on the regulation of immune cell types by GPCRs, including https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=19 (Lämmermann & Kastenmüller, 2019; Leone & Emens, 2018), GPCRs in immune cells in the pancreatic adenocarcinoma micro‐environment merit further study as potential targets to mitigate immune suppression.
As cell‐surface receptors, GPCRs may be useful pancreatic adenocarcinoma biomarkers, perhaps to detect or monitor patients, ideally by non‐invasive techniques (e.g. imaging of tumours). Recent data with NTS1 receptor encourages further such efforts for personalized/precision medicine approaches.
Studies are needed to define the mechanisms by which GPCR expression is altered and GPCRs signal in cell types within pancreatic adenocarcinoma tumours. Little is known about biased signalling of GPCRs or signalling via β‐arrestins, Gβγ, and canonical/non‐canonical downstream components in pancreatic adenocarcinoma.
GPCRs whose expression is reduced in pancreatic adenocarcinoma tumours/cells may provide valuable insight into how GPCR regulation occurs, as well as pointing to potential mechanisms for tumour suppression that can be exploited. GPCRs with reduced expression may be therapeutic targets, perhaps by approaches to enhance their expression in patients with pancreatic adenocarcinoma.
The greatest potential for GPCR drugs is likely their use in combination, especially with cytotoxic agents. GPCR activation may alter response to chemotherapy and contribute to drug resistance.
From a drug discovery perspective in the targeting of highly/differentially expressed GPCRs, inhibitors might be antagonists, inverse agonists, or negative allosteric modulators. In addition to small molecules targeting GPCRs, biologics such as antibodies, gene therapy, and gene editing may offer ways to modulate expression and activities mediated by GPCRs. It is currently unclear if there are GPCRs in pancreatic adenocarcinoma for which agonists may be beneficial.
This review provides evidence for the relevance of GPCRs in pancreatic adenocarcinoma biology and as possible targets for treating pancreatic adenocarcinoma. Many GPCRs have increased expression in pancreatic adenocarcinoma, but their contribution in this highly lethal cancer has not yet been elucidated. We believe that the potential impact of GPCRs in oncology and pharmacology—and most importantly for pancreatic adenocarcinoma patients—is yet to be realized. We urge readers to share our enthusiasm for the abundant opportunities to undertake what we believe may be highly impactful basic and translational research studies related to GPCRs in pancreatic adenocarcinoma.
4.1. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the concise Guide to PHARMACOLOGY 2019/20 (Alexander, Kelly, et al., 2019) and the chapter on GPCRs (Alexander, Christopoulos, et al., 2019).
CONFLICT OF INTEREST
The authors have no conflicts of interest.
ACKNOWLEDGEMENTS
This work was supported in part by a David Lehr Research Award (to P.A.I.) by the American Society for Pharmacology and Experimental Therapeutics (ASPET). We thank Drs Andrew M. Lowy and Randall French for providing pancreatic cancer patient‐derived cancer‐associated fibroblasts and cancer cells.
Sriram K, Salmerón C, Wiley SZ, Insel PA. GPCRs in pancreatic adenocarcinoma: Contributors to tumour biology and novel therapeutic targets. Br J Pharmacol. 2020;177:2434–2455. 10.1111/bph.15028
REFERENCES
- Alexander, S. P. , Christopoulos, A. , Davenport, A. P. , Kelly, E. , Mathie, A. , Peters, J. A. , … Pawson, A. J. (2019). The concise guide to pharmacology 2019/20: G protein‐coupled receptors. British Journal of Pharmacology, 176, S21–S141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. , Kelly, E. , Mathie, A. , Peters, J. A. , Veale, E. L. , Armstrong, J. F. , et al. (2019). The concise guide to pharmacology 2019/20: Introduction and other protein targets. British Journal of Pharmacology, 176, S1–S20. 10.1111/bph.14747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anandanadesan, R. , Gong, Q. , Chipitsyna, G. , Witkiewicz, A. , Yeo, C. J. , & Arafat, H. A. (2008). Angiotensin II induces vascular endothelial growth factor in pancreatic cancer cells through an angiotensin II type 1 receptor and ERK1/2 signaling. Journal of Gastrointestinal Surgery, 12, 57–66. 10.1007/s11605-007-0403-9 [DOI] [PubMed] [Google Scholar]
- Arnold, S. A. , Rivera, L. B. , Carbon, J. G. , Toombs, J. E. , Chang, C.‐L. , Bradshaw, A. D. , & Brekken, R. A. (2012). Losartan slows pancreatic tumor progression and extends survival of SPARC‐null mice by abrogating aberrant TGFβ activation. PLoS ONE, 7, e31384–e31384. 10.1371/journal.pone.0031384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakadlag, R. , Jandaghi, P. , Hoheisel, J. D. , & Riazalhosseini, Y. (2019). The potential of dopamine receptor D2 (DRD2) as a therapeutic target for tackling pancreatic cancer. Expert Opinion on Therapeutic Targets, 23, 365–367. [DOI] [PubMed] [Google Scholar]
- Baker, L. A. , Tiriac, H. , Clevers, H. , & Tuveson, D. A. (2016). Modeling pancreatic cancer with organoids. Trends in Cancer, 2, 176–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretina, J. , Caponigro, G. , Stransky, N. , Venkatesan, K. , Margolin, A. A. , Kim, S. , … Garraway, L. A. (2012). The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature, 483, 603–607. 10.1038/nature11003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum, R. P. , Singh, A. , Schuchardt, C. , Kulkarni, H. R. , Klette, I. , Wiessalla, S. , … Smerling, C. (2018). 177Lu‐3BP‐227 for neurotensin receptor 1–targeted therapy of metastatic pancreatic adenocarcinoma: First clinical results. Journal of Nuclear Medicine, 59, 809–814. 10.2967/jnumed.117.193847 [DOI] [PubMed] [Google Scholar]
- Belvedere, R. , Bizzarro, V. , Popolo, A. , Dal Piaz, F. , Vasaturo, M. , Picardi, P. , … Petrella, A. (2014). Role of intracellular and extracellular annexin A1 in migration and invasion of human pancreatic carcinoma cells. BMC Cancer, 14, 961 10.1186/1471-2407-14-961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi, Y. , Li, J. , Ji, B. , Kang, N. , Yang, L. , Simonetto, D. A. , … Shah, V. (2014). Sphingosine‐1‐phosphate mediates a reciprocal signaling pathway between stellate cells and cancer cells that promotes pancreatic cancer growth. The American Journal of Pathology, 184, 2791–2802. 10.1016/j.ajpath.2014.06.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray, F. , Ferlay, J. , Soerjomataram, I. , Siegel, R. L. , Torre, L. A. , & Jemal, A. (2018). Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer Journal for Clinicians, 68, 394–424. [DOI] [PubMed] [Google Scholar]
- Bray, N. L. , Pimentel, H. , Melsted, P. , & Pachter, L. (2016). Near‐optimal probabilistic RNA‐seq quantification. Nature Biotechnology, 34, 525–527. 10.1038/nbt.3519 [DOI] [PubMed] [Google Scholar]
- Buchegger, F. , Bonvin, F. , Kosinski, M. , Schaffland, A. O. , Prior, J. , Reubi, J. C. , … Bischof Delaloye, A. (2003). Radiolabeled neurotensin analog, 99mTc‐NT‐XI, evaluated in ductal pancreatic adenocarcinoma patients. Journal of Nuclear Medicine, 44, 1649–1654. [PubMed] [Google Scholar]
- Carrere, N. , Vernejoul, F. , Souque, A. , Asnacios, A. , Vaysse, N. , Pradayrol, L. , … Cordelier, P. (2005). Characterization of the bystander effect of somatostatin receptor sst2 after in vivo gene transfer into human pancreatic cancer cells. Human Gene Therapy, 16, 1175–1193. 10.1089/hum.2005.16.1175 [DOI] [PubMed] [Google Scholar]
- Catenacci, D. V. T. , Junttila, M. R. , Karrison, T. , Bahary, N. , Horiba, M. N. , Nattam, S. R. , … Kindler, H. L. (2015). Randomized phase Ib/II study of gemcitabine plus placebo or vismodegib, a hedgehog pathway inhibitor, in patients with metastatic pancreatic cancer. Journal of Clinical Oncology, 33, 4284–4292. 10.1200/JCO.2015.62.8719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, H.‐H. , Young, S. H. , Sinnett‐Smith, J. , Chou, C. E. N. , Moro, A. , Hertzer, K. M. , … Eibl, G. (2015). Prostaglandin E2 activates the mTORC1 pathway through an EP4/cAMP/PKA‐ and EP1/Ca2+‐mediated mechanism in the human pancreatic carcinoma cell line PANC‐1. American Journal of Physiology. Cell Physiology, 309, C639–C649. 10.1152/ajpcell.00417.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charo, C. , Holla, V. , Arumugam, T. , Hwang, R. , Yang, P. , Dubois, R. N. , … Ramachandran, V. (2013). Prostaglandin E2 regulates pancreatic stellate cell activity via the EP4 receptor. Pancreas, 42, 467–474. 10.1097/MPA.0b013e318264d0f8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Che, S. P. Y. , Park, J. Y. , & Stokol, T. (2017). Tissue factor‐expressing tumor‐derived extracellular vesicles activate quiescent endothelial cells via protease‐activated receptor‐1. Frontiers in Oncology, 7, 261 eCollection 2017. 10.3389/fonc.2017.00261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, L. , Fan, J. , Chen, H. , Meng, Z. , Chen, Z. , Wang, P. , & Liu, L. (2014). The IL‐8/CXCR1 axis is associated with cancer stem cell‐like properties and correlates with clinical prognosis in human pancreatic cancer cases. Scientific Reports, 4, 5911 10.1038/srep05911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Y. , Shi, M. , Yu, G.‐Z. , Qin, X.‐R. , Jin, G. , Chen, P. , & Zhu, M. H. (2012). Interleukin‐8, a promising predictor for prognosis of pancreatic cancer. World Journal of Gastroenterology, 18, 1123–1129. 10.3748/wjg.v18.i10.1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, J. H. , Ji, Y. G. , & Lee, D. H. (2013). Uridine triphosphate increases proliferation of human cancerous pancreatic duct epithelial cells by activating P2Y2 receptor. Pancreas, 42, 680–686. 10.1097/MPA.0b013e318271bb4b [DOI] [PubMed] [Google Scholar]
- Cui, K. , Zhao, W. , Wang, C. , Wang, A. , Zhang, B. , Zhou, W. , … Li, S. (2011). The CXCR4‐CXCL12 pathway facilitates the progression of pancreatic cancer via induction of angiogenesis and lymphangiogenesis. The Journal of Surgical Research, 171, 143–150. [DOI] [PubMed] [Google Scholar]
- de Visser, M. , Janssen, P. J. J. M. , Srinivasan, A. , Reubi, J. C. , Waser, B. , Erion, J. L. , … de Jong, M. (2003). Stabilised 111In‐labelled DTPA‐ and DOTA‐conjugated neurotensin analogues for imaging and therapy of exocrine pancreatic cancer. European Journal of Nuclear Medicine and Molecular Imaging, 30, 1134–1139. [DOI] [PubMed] [Google Scholar]
- Ehlers, R. A. , Zhang, Y. , Hellmich, M. R. , & Evers, B. M. (2000). Neurotensin‐mediated activation of MAPK pathways and AP‐1 binding in the human pancreatic cancer cell line, MIA PaCa‐2. Biochemical and Biophysical Research Communications, 269, 704–708. [DOI] [PubMed] [Google Scholar]
- Eibl, G. , Bruemmer, D. , Okada, Y. , Duffy, J. P. , Law, R. E. , Reber, H. A. , & Hines, O. J. (2003). PGE2 is generated by specific COX‐2 activity and increases VEGF production in COX‐2‐expressing human pancreatic cancer cells. Biochemical and Biophysical Research Communications, 306, 887–897. 10.1016/S0006-291X(03)01079-9 [DOI] [PubMed] [Google Scholar]
- Eibl, G. , & Rozengurt, E. (2019). KRAS, YAP, and obesity in pancreatic cancer: A signaling network with multiple loops. Seminars in Cancer Biology, 54, 50–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elyada, E. , Bolisetty, M. , Laise, P. , Flynn, W. F. , Courtois, E. T. , Burkhart, R. A. , … Tuveson, D. A. (2019). Cross‐species single‐cell analysis of pancreatic ductal adenocarcinoma reveals antigen‐presenting cancer‐associated fibroblasts. Cancer Discovery, 9, 1102–1123. 10.1158/2159-8290.CD-19-0094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto, Y. , Sasaki, T. , Tsuchida, A. , & Chayama, K. (2001). Angiotensin II type 1 receptor expression in human pancreatic cancer and growth inhibition by angiotensin II type 1 receptor antagonist. FEBS Letters, 495, 197–200. [DOI] [PubMed] [Google Scholar]
- Fukushima, K. , Takahashi, K. , Yamasaki, E. , Onishi, Y. , Fukushima, N. , Honoki, K. , & Tsujiuchi, T. (2017). Lysophosphatidic acid signaling via LPA1 and LPA3 regulates cellular functions during tumor progression in pancreatic cancer cells. Experimental Cell Research, 352, 139–145. 10.1016/j.yexcr.2017.02.007 [DOI] [PubMed] [Google Scholar]
- Gardner, J. A. , Ha, J. H. , Jayaraman, M. , & Dhanasekaran, D. N. (2013). The gep proto‐oncogene Gα13 mediates lysophosphatidic acid‐mediated migration of pancreatic cancer cells. Pancreas, 42, 819–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong, Q. , Davis, M. , Chipitsyna, G. , Yeo, C. J. , & Arafat, H. A. (2010). Blocking angiotensin II type 1 receptor triggers apoptotic cell death in human pancreatic cancer cells. Pancreas, 39, 581–594. 10.1097/MPA.0b013e3181c314cd [DOI] [PubMed] [Google Scholar]
- Gu, D. , Schlotman, K. E. , & Xie, J. (2016). Deciphering the role of hedgehog signaling in pancreatic cancer. Journal of Biomedical Research, 30, 353–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR . (2018). The IUPHAR/BPS guide to pharmacology in 2018: Updates and expansion to encompass the new guide to immunopharmacology. Nucleic Acids Research, 46, D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinrich, E. L. , Lee, W. , Lu, J. , Lowy, A. M. , & Kim, J. (2012). Chemokine CXCL12 activates dual CXCR4 and CXCR7‐mediated signaling pathways in pancreatic cancer cells. Journal of Translational Medicine, 10, 68 https://doi/org/10.1186/1479-5876-10-68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, L.‐P. , Zhang, X.‐X. , Jiang, S.‐H. , Tao, L.‐Y. , Li, Q. , Zhu, L.‐L. , … Cao, X. Y. (2019). Targeting purinergic receptor P2Y2 prevents the growth of pancreatic ductal adenocarcinoma by inhibiting cancer cell glycolysis. Clinical Cancer Research. 25(4), 1318–1330. 10.1158/1078-0432.CCR-18-2297 [DOI] [PubMed] [Google Scholar]
- Huang, C. , Li, Y. , Guo, Y. , Zhang, Z. , Lian, G. , Chen, Y. , … Zeng, L. (2018). MMP1/PAR1/SP/NK1R paracrine loop modulates early perineural invasion of pancreatic cancer cells. Theranostics, 8, 3074–3086. 10.7150/thno.24281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, X. , Wang, H.‐C. , Yuan, Z. , Huang, J. , & Zheng, Q. (2011). Norepinephrine stimulates pancreatic cancer cell proliferation, migration and invasion via β‐adrenergic receptor‐dependent activation of P38/MAPK pathway. Hepato‐Gastroenterology, 59, 889–893. [DOI] [PubMed] [Google Scholar]
- Ilic, M. , & Ilic, I. (2016). Epidemiology of pancreatic cancer. World Journal of Gastroenterology, 22, 9694–9705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insel, P. A. , Sriram, K. , Wiley, S. Z. , Wilderman, A. , Katakia, T. , McCann, T. , … Murray, F. (2018). GPCRomics: GPCR expression in cancer cells and tumors identifies new, potential biomarkers and therapeutic targets. Frontiers in Pharmacology, 9, 431 10.3389/fphar.2018.00431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiguro, S. , Yoshimura, K. , Tsunedomi, R. , Oka, M. , Takao, S. , Inui, M. , … Tamura, M. (2015). Involvement of angiotensin II type 2 receptor (AT2R) signaling in human pancreatic ductal adenocarcinoma (PDAC): A novel AT2R agonist effectively attenuates growth of PDAC grafts in mice. Cancer Biology & Therapy, 16, 307–316. 10.1080/15384047.2014.1002357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii, S. , Hirane, M. , Fukushima, K. , Tomimatsu, A. , Fukushima, N. , & Tsujiuchi, T. (2015). Diverse effects of LPA4, LPA5 and LPA6 on the activation of tumor progression in pancreatic cancer cells. Biochemical and Biophysical Research Communications, 461, 59–64. [DOI] [PubMed] [Google Scholar]
- Ishizuka, J. , Townsend, C. M. Jr. , & Thompson, J. C. (1993). Neurotensin regulates growth of human pancreatic cancer. Annals of Surgery, 217, 439–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahny, E. , Yang, H. , Liu, B. , Jahnke, B. , Lademann, F. , Knösel, T. , … Denz, A. (2017). The G protein‐coupled receptor RAI3 is an independent prognostic factor for pancreatic cancer survival and regulates proliferation via STAT3 phosphorylation. PLoS ONE, 12, e0170390–e0170390. 10.1371/journal.pone.0170390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jandaghi, P. , Najafabadi, H. S. , Bauer, A. S. , Papadakis, A. I. , Fassan, M. , Hall, A. , … Hoheisel, J. D. (2016). Expression of DRD2 is increased in human pancreatic ductal adenocarcinoma and inhibitors slow tumor growth in mice. Gastroenterology, 151, 1218–1231. 10.1053/j.gastro.2016.08.040 [DOI] [PubMed] [Google Scholar]
- Ji, Z. , Mei, F. C. , Xie, J. , & Cheng, X. (2007). Oncogenic KRAS activates hedgehog signaling pathway in pancreatic cancer cells. The Journal of Biological Chemistry, 282, 14048–14055. [DOI] [PubMed] [Google Scholar]
- Kapushesky, M. , Emam, I. , Holloway, E. , Kurnosov, P. , Zorin, A. , Malone, J. , … Brazma, A. (2009). Gene Expression Atlas at the European Bioinformatics Institute. Nucleic Acids Research, 38, D690–D698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, J. , Yip, M. L. R. , Shen, X. , Li, H. , Hsin, L.‐Y. C. , Labarge, S. , … Vaidehi, N. (2012). Identification of anti‐malarial compounds as novel antagonists to chemokine receptor CXCR4 in pancreatic cancer cells. PLoS ONE, 7, e31004–e31004. 10.1371/journal.pone.0031004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim‐Fuchs, C. , Le, C. P. , Pimentel, M. A. , Shackleford, D. , Ferrari, D. , Angst, E. , … Sloan, E. K. (2014). Chronic stress accelerates pancreatic cancer growth and invasion: A critical role for beta‐adrenergic signaling in the pancreatic microenvironment. Brain, Behavior, and Immunity, 40, 40–47. 10.1016/j.bbi.2014.02.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisfalvi, K. , Guha, S. , & Rozengurt, E. (2005). Neurotensin and EGF induce synergistic stimulation of DNA synthesis by increasing the duration of ERK signaling in ductal pancreatic cancer cells. Journal of Cellular Physiology, 202, 880–890. [DOI] [PubMed] [Google Scholar]
- Klijn, C. , Durinck, S. , Stawiski, E. W. , Haverty, P. M. , Jiang, Z. , Liu, H. , … Pau, G. (2015). A comprehensive transcriptional portrait of human cancer cell lines. Nature Biotechnology, 33(3), 306–312. https://doi.org.10.1038/nbt.3080. [DOI] [PubMed] [Google Scholar]
- Ko, T. , An, H. J. , Ji, Y. G. , Kim, O. J. , & Lee, D. H. (2012). P2Y receptors regulate proliferation of human pancreatic duct epithelial cells. Pancreas, 41, 797–803. [DOI] [PubMed] [Google Scholar]
- Komachi, M. , Sato, K. , Tobo, M. , Mogi, C. , Yamada, T. , Ohta, H. , … Okajima, F. (2012). Orally active lysophosphatidic acid receptor antagonist attenuates pancreatic cancer invasion and metastasis in vivo. Cancer Science, 103, 1099–1104. 10.1111/j.1349-7006.2012.02246.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komachi, M. , Tomura, H. , Malchinkhuu, E. , Tobo, M. , Mogi, C. , Yamada, T. , … Okajima, F. (2009). LPA 1 receptors mediate stimulation, whereas LPA 2 receptors mediate inhibition, of migration of pancreatic cancer cells in response to lysophosphatidic acid and malignant ascites. Carcinogenesis, 30, 457–465. 10.1093/carcin/bgp011 [DOI] [PubMed] [Google Scholar]
- Körner, M. , Waser, B. , Strobel, O. , Büchler, M. , & Reubi, J. C. (2015). Neurotensin receptors in pancreatic ductal carcinomas. EJNMMI Research, 5, 17 10.1186/s13550-015-0094-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshiba, T. , Hosotani, R. , Miyamoto, Y. , Ida, J. , Tsuji, S. , Nakajima, S. , … Fujii, N. (2000). Expression of stromal cell‐derived factor 1 and CXCR4 ligand receptor system in pancreatic cancer: A possible role for tumor progression. Clinical Cancer Research, 6, 3530–3535. [PubMed] [Google Scholar]
- Lämmermann, T. , & Kastenmüller, W. (2019). Concepts of GPCR‐controlled navigation in the immune system. Immunological Reviews, 289, 205–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lankadasari, M. B. , Aparna, J. S. , Mohammed, S. , James, S. , Aoki, K. , Binu, V. S. , … Harikumar, K. B. (2018). Targeting S1PR1/STAT3 loop abrogates desmoplasia and chemosensitizes pancreatic cancer to gemcitabine. Theranostics, 8, 3824–3840. 10.7150/thno.25308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, J. J. , Perera, R. M. , Wang, H. , Wu, D.‐C. , Liu, X. S. , Han, S. , … Beachy, P. A. (2014). Stromal response to hedgehog signaling restrains pancreatic cancer progression. Proceedings of the National Academy of Sciences of the United States of America, 111, E3091–E3100. 10.1073/pnas.1411679111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leone, R. D. , & Emens, L. A. (2018). Targeting adenosine for cancer immunotherapy. Journal for Immunotherapy of Cancer, 6, 57 10.1186/s40425-018-0360-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, M. , Wang, X. , Li, W. , Li, F. , Yang, H. , Wang, H. , … Fisher, W. E. (2008). Somatostatin receptor‐1 induces cell cycle arrest and inhibits tumor growth in pancreatic cancer. Cancer Science, 99, 2218–2223. 10.1111/j.1349-7006.2008.00940.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, X. , Ma, Q. , Xu, Q. , Liu, H. , Lei, J. , Duan, W. , … Wang, Z. (2012). SDF‐1/CXCR4 signaling induces pancreatic cancer cell invasion and epithelial‐mesenchymal transition in vitro through non‐canonical activation of hedgehog pathway. Cancer Letters, 322, 169–176. 10.1016/j.canlet.2012.02.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao, Y. , Mu, G. , Zhang, L. , Zhou, W. , Zhang, J. , & Yu, H. (2013). Lysophosphatidic acid stimulates activation of focal adhesion kinase and paxillin and promotes cell motility, via LPA1–3, in human pancreatic cancer. Digestive Diseases and Sciences, 58, 3524–3533. 10.1007/s10620-013-2878-4 [DOI] [PubMed] [Google Scholar]
- Liu, B. , Yang, H. , Pilarsky, C. , & Weber, G. F. (2018). The effect of GPRC5a on the proliferation, migration ability, chemotherapy resistance, and phosphorylation of GSK‐3β in pancreatic cancer. International Journal of Molecular Sciences, 19(7). pii: E1870. 10.3390/ijms19071870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lytle, N. K. , Ferguson, L. P. , Rajbhandari, N. , Gilroy, K. , Fox, R. G. , Deshpande, A. , … Reya, T. (2019). A multiscale map of the stem cell state in pancreatic adenocarcinoma. Cell, 177, 572, e22–586. 10.1016/j.cell.2019.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandilaras, V. , Bouganim, N. , Yin, H. , Asselah, J. , & Azoulay, L. (2017). The use of drugs acting on the renin‐angiotensin system and the incidence of pancreatic cancer. British Journal of Cancer, 116, 103–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masamune, A. , Hamada, S. , Kikuta, K. , Takikawa, T. , Miura, S. , Nakano, E. , & Shimosegawa, T. (2013). The angiotensin II type I receptor blocker olmesartan inhibits the growth of pancreatic cancer by targeting stellate cell activities in mice. Scandinavian Journal of Gastroenterology, 48, 602–609. 10.3109/00365521.2013.777776 [DOI] [PubMed] [Google Scholar]
- Maurer, C. , Holmstrom, S. R. , He, J. , Laise, P. , Su, T. , Ahmed, A. , … Olive, K. P. (2019). Experimental microdissection enables functional harmonisation of pancreatic cancer subtypes. Gut, 68, 1034–1043. 10.1136/gutjnl-2018-317706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGhee, A. , Sivarajah, M. , Gong, Q. , Lim, S. , Chipitsyna, G. , Yeo, C. J. , & Arafat, H. A. (2011). Angiotensin II type 2 receptor blockade inhibits fatty acid synthase production through activation of AMP‐activated protein kinase in pancreatic cancer cells. Surgery, 150, 284–298. 10.1016/j.surg.2011.06.002 [DOI] [PubMed] [Google Scholar]
- Moatassim‐Billah, S. , Duluc, C. , Samain, R. , Jean, C. , Perraud, A. , Decaup, E. , … Bousquet, C. (2016). Anti‐metastatic potential of somatostatin analog SOM230: Indirect pharmacological targeting of pancreatic cancer‐associated fibroblasts. Oncotarget, 7, 41584–41598. 10.18632/oncotarget.9296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto, M. , Matsuo, Y. , Koide, S. , Tsuboi, K. , Shamoto, T. , Sato, T. , … Takeyama, H. (2016). Enhancement of the CXCL12/CXCR4 axis due to acquisition of gemcitabine resistance in pancreatic cancer: Effect of CXCR4 antagonists. BMC Cancer, 16, 305 10.1186/s12885-016-2340-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakai, Y. , Isayama, H. , Ijichi, H. , Sasaki, T. , Sasahira, N. , Hirano, K. , … Koike, K. (2010). Inhibition of renin‐angiotensin system affects prognosis of advanced pancreatic cancer receiving gemcitabine. British Journal of Cancer, 103, 1644–1648. 10.1038/sj.bjc.6605955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakata, B. , Fukunaga, S. , Noda, E. , Amano, R. , Yamada, N. , & Hirakawa, K. (2008). Chemokine receptor CCR7 expression correlates with lymph node metastasis in pancreatic cancer. Oncology, 74, 69–75. [DOI] [PubMed] [Google Scholar]
- O'Hayre, M. , Vázquez‐Prado, J. , Kufareva, I. , Stawiski, E. W. , Handel, T. M. , Seshagiri, S. , & Gutkind, J. S. (2013). The emerging mutational landscape of G proteins and G‐protein‐coupled receptors in cancer. Nature Reviews. Cancer, 13, 412–424. 10.1038/nrc3521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta, T. , Shimizu, K. , Yi, S. , Takamura, H. , Amaya, K. , Kitagawa, H. , … Miwa, K. (2003). Protease‐activated receptor‐2 expression and the role of trypsin in cell proliferation in human pancreatic cancers. International Journal of Oncology, 23, 61–66. [PubMed] [Google Scholar]
- Olszewski, U. , Hlozek, M. , & Hamilton, G. (2010). Activation of Na+/H+ exchanger 1 by neurotensin signaling in pancreatic cancer cell lines. Biochemical and Biophysical Research Communications, 393, 414–419. [DOI] [PubMed] [Google Scholar]
- Olszewski‐Hamilton, U. , & Hamilton, G. (2011). Dependence of relative expression of NTR1 and EGFR on cell density and extracellular pH in human pancreatic cancer cell lines. Cancers (Basel)., 3, 182–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onishi, H. , Kai, M. , Odate, S. , Iwasaki, H. , Morifuji, Y. , Ogino, T. , … Katano, M. (2011). Hypoxia activates the hedgehog signaling pathway in a ligand‐independent manner by upregulation of SMO transcription in pancreatic cancer. Cancer Science, 102, 1144–1150. 10.1111/j.1349-7006.2011.01912.x [DOI] [PubMed] [Google Scholar]
- Pino, M. S. , Nawrocki, S. T. , Cognetti, F. , Abruzzese, J. L. , Xiong, H. , & McConkey, D. J. (2005). Prostaglandin E2 drives cyclooxygenase 2 expression via cyclic AMP response element activation in human pancreatic cancer cells. Cancer Biology & Therapy, 4, 1263–1269. [DOI] [PubMed] [Google Scholar]
- Pu, J. , Zhang, X. , Luo, H. , Xu, L. , Lu, X. , & Lu, J. (2017). Adrenaline promotes epithelial‐to‐mesenchymal transition via HuR‐TGFβ regulatory axis in pancreatic cancer cells and the implication in cancer prognosis. Biochemical and Biophysical Research Communications, 493, 1273–1279. [DOI] [PubMed] [Google Scholar]
- Queiroz, K. C. S. , Shi, K. , Duitman, J. , Aberson, H. L. , Wilmink, J. W. , van Noesel, C. J. M. , … Spek, C. A. (2014). Protease‐activated receptor‐1 drives pancreatic cancer progression and chemoresistance. International Journal of Cancer, 135, 2294–2304. 10.1002/ijc.28726 [DOI] [PubMed] [Google Scholar]
- Ragozin, E. , Hesin, A. , Bazylevich, A. , Tuchinsky, H. , Bovina, A. , Shekhter Zahavi, T. , … Gellerman, G. (2018). New somatostatin‐drug conjugates for effective targeting pancreatic cancer. Bioorganic & Medicinal Chemistry, 26, 3825–3836. 10.1016/j.bmc.2018.06.032 [DOI] [PubMed] [Google Scholar]
- Rawla, P. , Sunkara, T. , & Gaduputi, V. (2019). Epidemiology of pancreatic cancer: Global trends, etiology and risk factors. World Journal of Oncology, 10, 10–27. 10.14740/wjon1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renz, B. W. , Takahashi, R. , Tanaka, T. , Macchini, M. , Hayakawa, Y. , Dantes, Z. , … Wang, T. C. (2018). β2 adrenergic‐neurotrophin feedforward loop promotes pancreatic cancer. Cancer Cell, 33, 75, e7–90. 10.1016/j.ccell.2017.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reubi, J. C. , Waser, B. , Friess, H. , Büchler, M. , & Laissue, J. (1998). Neurotensin receptors: A new marker for human ductal pancreatic adenocarcinoma. Gut, 42, 546–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson, M. D. , McCarthy, D. J. , & Smyth, G. K. (2010). edgeR: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics, 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy, I. , Zimmerman, N. P. , Mackinnon, A. C. , Tsai, S. , Evans, D. B. , & Dwinell, M. B. (2014). CXCL12 chemokine expression suppresses human pancreatic cancer growth and metastasis. PLoS ONE, 9, e90400–e90400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudroff, C. , Schafberg, H. , Nowak, G. , Weinel, R. , Scheele, J. , & Kaufmann, R. (1998). Characterization of functional thrombin receptors in human pancreatic tumor cells (MIA PACA‐2). Pancreas, 16, 189–194. [DOI] [PubMed] [Google Scholar]
- Rudroff, C. , Seibold, S. , Kaufmann, R. , Cu Zetina, C. , Reise, K. , Schäfer, U. , … Neugebauer, E. A. M. (2002). Expression of the thrombin receptor PAR‐1 correlates with tumour cell differentiation of pancreatic adenocarcinoma in vitro. Clinical & Experimental Metastasis, 19, 181–189. 10.1023/A:1014598904644 [DOI] [PubMed] [Google Scholar]
- Sainz, B. , Alcala, S. , Garcia, E. , Sanchez‐Ripoll, Y. , Azevedo, M. M. , Cioffi, M. , … Cañamero, M. (2015). Microenvironmental hCAP‐18/LL‐37 promotes pancreatic ductal adenocarcinoma by activating its cancer stem cell compartment. Gut, 64, 1921–1935. 10.1136/gutjnl-2014-308935 [DOI] [PubMed] [Google Scholar]
- Schmidt, A. , Sinnett‐Smith, J. , Young, S. , Chang, H.‐H. , Hines, O. J. , Dawson, D. W. , … Eibl, G. (2017). Direct growth‐inhibitory effects of prostaglandin E2 in pancreatic cancer cells in vitro through an EP4/PKA‐mediated mechanism. Surgery, 161, 1570–1578. 10.1016/j.surg.2016.12.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan, T. , Ma, Q. , Zhang, D. , Guo, K. , Liu, H. , Wang, F. , & Wu, E. (2011). β2‐Adrenoceptor blocker synergizes with gemcitabine to inhibit the proliferation of pancreatic cancer cells via apoptosis induction. European Journal of Pharmacology, 665, 1–7. 10.1016/j.ejphar.2011.04.055 [DOI] [PubMed] [Google Scholar]
- Shen, X. , Artinyan, A. , Jackson, D. , Thomas, R. M. , Lowy, A. M. , & Kim, J. (2010). Chemokine receptor CXCR4 enhances proliferation in pancreatic cancer cells through AKT and ERK dependent pathways. Pancreas, 39, 81–87. 10.1097/MPA.0b013e3181bb2ab7 [DOI] [PubMed] [Google Scholar]
- Shen, X. , Mailey, B. , Ellenhorn, J. D. I. , Chu, P. G. , Lowy, A. M. , & Kim, J. (2009). CC chemokine receptor 9 enhances proliferation in pancreatic intraepithelial neoplasia and pancreatic cancer cells. Journal of Gastrointestinal Surgery, 13, 1955–1962. 10.1007/s11605-009-1002-8 [DOI] [PubMed] [Google Scholar]
- Shen, Y. , Cai, M. , Xia, W. , Liu, J. , Zhang, Q. , Xie, H. , … Zheng, S. (2007). FTY720, a synthetic compound from Isaria sinclairii, inhibits proliferation and induces apoptosis in pancreatic cancer cells. Cancer Letters, 254, 288–297. 10.1016/j.canlet.2007.03.013 [DOI] [PubMed] [Google Scholar]
- Shi, K. , Queiroz, K. C. S. , Stap, J. , Richel, D. J. , & Spek, C. A. (2013). Protease‐activated receptor‐2 induces migration of pancreatic cancer cells in an extracellular ATP‐dependent manner. Journal of Thrombosis and Haemostasis, 11, 1892–1902. [DOI] [PubMed] [Google Scholar]
- Siegel, R. L. , Miller, K. D. , & Jemal, A. (2019). Cancer statistics, 2019. CA: A Cancer Journal for Clinicians, 69, 7–34. [DOI] [PubMed] [Google Scholar]
- Sriram, K. , & Insel, P. A. (2018). G protein‐coupled receptors as targets for approved drugs: How many targets and how many drugs? Molecular Pharmacology, 93, 251–258. 10.1124/mol.117.111062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriram, K. , Moyung, K. , Corriden, R. , Carter, H. , & Insel, P. A. (2019). GPCRs show widespread differential mRNA expression and frequent mutation and copy number variation in solid tumors. PLoS Biol, 17, e3000434 https://doi.org10.1371/journal.pbio.3000434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriram, K. , Wiley, S. Z. , Moyung, K. , Gorr, M. W. , Salmerón, C. , Marucut, J. , … Insel, P. A. (2019). Detection and quantification of GPCR mRNA: An assessment and implications of data from high‐content methods. ACS Omega, 4, 17048–17059. 10.1021/acsomega.9b02811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stähle, M. , Veit, C. , Bachfischer, U. , Schierling, K. , Skripczynski, B. , Hall, A. , … Giehl, K. (2003). Mechanisms in LPA‐induced tumor cell migration: Critical role of phosphorylated ERK. Journal of Cell Science, 116, 3835–3846. 10.1242/jcs.00679 [DOI] [PubMed] [Google Scholar]
- Susek, K. H. , Karvouni, M. , Alici, E. , & Lundqvist, A. (2018). The role of CXC chemokine receptors 1–4 on immune cells in the tumor microenvironment. Frontiers in Immunology, 9, 2159 eCollection 2018. 10.3389/fimmu.2018.02159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi, T. , Ichikawa, H. , Morimoto, Y. , Tsuneyama, K. , & Hijikata, T. (2019). Inhibition of EP2/EP4 prostanoid receptor‐mediated signaling suppresses IGF‐1‐induced proliferation of pancreatic cancer BxPC‐3 cells via upregulating γ‐glutamyl cyclotransferase expression. Biochemical and Biophysical Research Communications, 516, 388–396. [DOI] [PubMed] [Google Scholar]
- Takahashi, T. , Uehara, H. , Ogawa, H. , Umemoto, H. , Bando, Y. , & Izumi, K. (2015). Inhibition of EP2/EP4 signaling abrogates IGF‐1R‐mediated cancer cell growth: Involvement of protein kinase C‐θ activation. Oncotarget, 6, 4829–4844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatlow, P. J. , & Piccolo, S. R. (2016). A cloud‐based workflow to quantify transcript‐expression levels in public cancer compendia. Scientific Reports, 6, 39259 10.1038/srep39259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tekin, C. , Shi, K. , Daalhuisen, J. B. , Ten Brink, M. S. , Bijlsma, M. F. , & Spek, C. A. (2018). PAR1 signaling on tumor cells limits tumor growth by maintaining a mesenchymal phenotype in pancreatic cancer. Oncotarget, 9, 32010–32023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thayer, S. P. , di Magliano, M. P. , Heiser, P. W. , Nielsen, C. M. , Roberts, D. J. , Lauwers, G. Y. , … Hebrok, M. (2003). Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature, 425, 851–856. 10.1038/nature02009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuveson, D. , & Clevers, H. (2019). Cancer modeling meets human organoid technology. Science, 80(364), 952–955. [DOI] [PubMed] [Google Scholar]
- Tveteraas, I. H. , Aasrum, M. , Brusevold, I. J. , Ødegård, J. , Christoffersen, T. , & Sandnes, D. (2016). Lysophosphatidic acid induces both EGFR‐dependent and EGFR‐independent effects on DNA synthesis and migration in pancreatic and colorectal carcinoma cells. Tumor Biology, 37, 2519–2526. [DOI] [PubMed] [Google Scholar]
- Udumyan, R. , Montgomery, S. , Fang, F. , Almroth, H. , Valdimarsdottir, U. , Ekbom, A. , … Fall, K. (2017). Beta‐blocker drug use and survival among patients with pancreatic adenocarcinoma. Cancer Research, 77, 3700–3707. 10.1158/0008-5472.CAN-17-0108 [DOI] [PubMed] [Google Scholar]
- Upadhrasta, S. , & Zheng, L. (2019). Strategies in developing immunotherapy for pancreatic cancer: Recognizing and correcting multiple immune ‘defects’ in the tumor microenvironment. Journal of Clinical Medicine, 8 10.3390/jcm8091472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivian, J. , Rao, A. A. , Nothaft, F. A. , Ketchum, C. , Armstrong, J. , Novak, A. , … Paten, B. (2017). Toil enables reproducible, open source, big biomedical data analyses. Nature Biotechnology, 35, 314–316. 10.1038/nbt.3772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J.‐G. , Li, N.‐N. , Li, H.‐N. , Cui, L. , & Wang, P. (2011). Pancreatic cancer bears overexpression of neurotensin and neurotensin receptor subtype‐1 and SR 48692 counteracts neurotensin induced cell proliferation in human pancreatic ductal carcinoma cell line PANC‐1. Neuropeptides, 45, 151–156. [DOI] [PubMed] [Google Scholar]
- Wang, J. P. , Wu, C.‐Y. , Yeh, Y.‐C. , Shyr, Y.‐M. , Wu, Y.‐Y. , Kuo, C.‐Y. , … Chao, Y. (2015). Erlotinib is effective in pancreatic cancer with epidermal growth factor receptor mutations: A randomized, open‐label, prospective trial. Oncotarget, 6, 18162–18173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, M. , Zhang, H. , Wang, H. , Feng, H. , Deng, H. , Wu, Z. , … Li, Z. (2018). Development of [18F]AlF‐NOTA‐NT as PET agents of neurotensin receptor‐1 positive pancreatic cancer. Molecular Pharmaceutics, 15, 3093–3100. 10.1021/acs.molpharmaceut.8b00192 [DOI] [PubMed] [Google Scholar]
- Weddle, D. L. , Tithoff, P. , Williams, M. , & Schuller, H. M. (2001). β‐Adrenergic growth regulation of human cancer cell lines derived from pancreatic ductal carcinomas. Carcinogenesis, 22, 473–479. [DOI] [PubMed] [Google Scholar]
- Wehler, T. , Wolfert, F. , Schimanski, C. C. , Gockel, I. , Herr, W. , Biesterfeld, S. , … Moehler, M. (2006). Strong expression of chemokine receptor CXCR4 by pancreatic cancer correlates with advanced disease. Oncology Reports, 16, 1159–1164. [PubMed] [Google Scholar]
- Wiley, S. Z. , Sriram, K. , Liang, W. , Chang, S. E. , French, R. , McCann, T. , … Insel, P. A. (2018). GPR68, a proton‐sensing GPCR, mediates interaction of cancer‐associated fibroblasts and cancer cells. The FASEB Journal, 32, 1170–1183. 10.1096/fj.201700834R [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley, S. Z. , Sriram, K. , Salmerón, C. , & Insel, P. A. (2019). GPR68: An emerging drug target in cancer. International Journal of Molecular Sciences, 20 10.3390/ijms20030559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witkiewicz, A. K. , Balaji, U. , Eslinger, C. , McMillan, E. , Conway, W. , Posner, B. , … Knudsen, E. S. (2016). Integrated patient‐derived models delineate individualized therapeutic vulnerabilities of pancreatic cancer. Cell Reports, 16, 2017–2031. 10.1016/j.celrep.2016.07.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, V. , Yeerna, H. , Nohata, N. , Chiou, J. , Harismendy, O. , Raimondi, F. , … Gutkind, J. S. (2019). Illuminating the Onco‐GPCRome: Novel G protein‐coupled receptor‐driven oncocrine networks and targets for cancer immunotherapy. The Journal of Biological Chemistry, 294, 11062–11086. 10.1074/jbc.REV119.005601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie, L. , Duan, Z. , Liu, C. , Zheng, Y. , & Zhou, J. (2015). Protease‐activated receptor 2 agonist increases cell proliferation and invasion of human pancreatic cancer cells. Experimental and Therapeutic Medicine, 9, 239–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Y. , Stang, A. , Schweickert, P. G. , Lanman, N. A. , Paul, E. N. , Monia, B. P. , … Flick, M. J. (2019). Thrombin signaling promotes pancreatic adenocarcinoma through PAR‐1‐dependent immune evasion. Cancer Research, 79, 3417–3430. 10.1158/0008-5472.CAN-18-3206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin, X. , Wang, M. , Wang, H. , Deng, H. , He, T. , Tan, Y. , … Li, Z. (2017). Evaluation of neurotensin receptor 1 as a potential imaging target in pancreatic ductal adenocarcinoma. Amino Acids, 49, 1325–1335. 10.1007/s00726-017-2430-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, D. , Lei, J. , Ma, J. , Chen, X. , Sheng, L. , Jiang, Z. , … Huo, X. (2016). β2‐Adrenogenic signaling regulates NNK‐induced pancreatic cancer progression via upregulation of HIF‐1α. Oncotarget, 7, 17760–17772. 10.18632/oncotarget.5677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, D. , Ma, Q. , Wang, Z. , Zhang, M. , Guo, K. , Wang, F. , & Wu, E. (2011). β2‐Adrenoceptor blockage induces G1/S phase arrest and apoptosis in pancreatic cancer cells via Ras/Akt/NFκB pathway. Molecular Cancer, 10, 146 10.1186/1476-4598-10-146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, D. , Yong Ma, Q. , Hu, H.‐T. , & Zhang, M. (2010). β2‐Adrenergic antagonists suppress pancreatic cancer cell invasion by inhibiting CREB, NF‐κB and AP‐1. Cancer Biology & Therapy, 10, 19–29. 10.4161/cbt.10.1.11944 [DOI] [PubMed] [Google Scholar]
- Zhang, L. , Wang, D. , Li, Y. , Liu, Y. , Xie, X. , Wu, Y. , … Su, Z. (2016). CCL21/CCR7 axis contributed to CD133+ pancreatic cancer stem‐like cell metastasis via EMT and Erk/NF‐κB pathway. PLoS ONE, 11, e0158529–e0158529. 10.1371/journal.pone.0158529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, H. , Telonis, A. G. , Jing, Y. , Xia, N. L. , Biederman, L. , Jimbo, M. , … Rigoutsos, I. (2016). GPRC5A is a potential oncogene in pancreatic ductal adenocarcinoma cells that is upregulated by gemcitabine with help from HuR. Cell Death & Disease, 7, e2294–e2294. 10.1038/cddis.2016.169 [DOI] [PMC free article] [PubMed] [Google Scholar]