

Abstract
Individuals with diabetes are more susceptible to cerebral vascular aging. However, the underlying mechanisms are not well elucidated. The present study examined whether the myogenic response of the middle cerebral artery (MCA) is impaired in diabetic rats due to high glucose (HG)–induced cerebral vascular smooth muscle cell (CVSMC) dysfunction, and whether this is associated with ATP depletion and changes in mitochondrial dynamics and membrane potential. The diameters of the MCA of diabetic rats increased to 135.3 ± 11.3% when perfusion pressure was increased from 40 to 180 mmHg, while it fell to 85.1 ± 3.1% in non-diabetic controls. The production of ROS and mitochondrial-derived superoxide were enhanced in cerebral arteries of diabetic rats. Levels of mitochondrial superoxide were significantly elevated in HG-treated primary CVSMCs, which was associated with decreased ATP production, mitochondrial respiration, and membrane potential. The expression of OPA1 was reduced, and MFF was elevated in HG-treated CVSMCs in association with fragmented mitochondria. Moreover, HG-treated CVSMCs displayed lower contractile and proliferation capabilities. These results demonstrate that imbalanced mitochondrial dynamics (increased fission and decreased fusion) and membrane depolarization contribute to ATP depletion in HG-treated CVSMCs, which promotes CVSMC dysfunction and may play an essential role in exacerbating the impaired myogenic response in the cerebral circulation in diabetes and accelerating vascular aging.
Keywords: Hyperglycemia, ATP depletion, Mitochondria dynamics, Myogenic response, Cerebral vascular smooth muscle cells
Introduction
Diabetes mellitus (DM) is a complex and progressive metabolic disorder clinically characterized by hyperglycemia, dyslipidemia, inflammation, and insulin resistance. DM accelerates oxidative stress and cellular senescence as in aging (Ahn et al. 2018; Fulop et al. 2018; McDaniel 1999; Springo et al. 2015a; Zhang et al. 2013). These lead to many pathological outcomes, including age-related microvascular (nephropathy, retinopathy, and neuropathy) and macrovascular (stroke, coronary and peripheral arterial diseases) complications and end-organ damage (American Diabetes A 2014; Corriere et al. 2013). Moreover, DM is a leading risk factor for stroke, dementia, and accelerating vascular aging (Ergul et al. 2012; Hoyer et al. 1988; Samaras et al. 2014; Zhou et al. 2014), but the underlying mechanisms are still poorly understood.
It is well established that high glucose (HG) enhances the production of mitochondrial reactive oxygen species (ROS) and promotes endothelial dysfunction (Lagaud et al. 2001; Tabit et al. 2010). DM is also associated with impaired myogenic response (MR), an intrinsic property of vascular smooth muscle cells, in systemic microcirculatory beds (Hill and Meininger 1993; Walmsley and Wiles 1990). In the cerebral circulation, the MR regulates cerebral blood flow (CBF) by constriction of cerebral vasculature in response to elevations in blood pressure (Cipolla et al. 2002). CBF autoregulation maintains constant blood flow to the brain to meet the energy demands (Fan et al. 2017). The impaired MR and autoregulation have been implicated in playing an essential role in the pathogenesis of cerebrovascular diseases (Shekhar et al. 2017; Wang et al. 2016). However, it remains to be determined whether hyperglycemia impairs the MR in the cerebral circulation and if this is secondary to changes in ATP production in cerebral vascular smooth muscle cells (CVSMCs).
The present study investigated whether HG-induced elevation in ROS production and mitochondrial dysfunction in CVSMCs result from changes in mitochondrial dynamics and membrane potential that diminish cell contraction and proliferation, and contributes to the impaired MR of the middle cerebral artery (MCA) in diabetic rats.
Materials and methods
Animals
These experiments were performed using 24 age-matched Sprague Dawley (SD) and 25 diabetic T2DN rats. The rats were obtained from in house colonies maintained at the University of Mississippi Medical Center (UMMC). All animals were housed under standard laboratory animal conditions, including a 12-h light/dark cycle with free access to food and water. The UMMC animal care facility is approved by the American Association for the Accreditation of Laboratory Animal Care. All experiments and protocols involving animals were approved by the Institutional Animal Care and Use Committees (IACUC) of the UMMC and conformed to NIH guidelines.
Primary cell culture and treatments
Primary CVSMCs were isolated from the MCA of 3-week-old SD rats, as described previously (Fan et al. 2017). Briefly, the rat was sacrificed with isoflurane (2–3%, inhalation administration), and the brain was collected immediately. The MCAs were dissected from the brain and washed with ice-cold Tyrode’s solution, as we previously described (Fan et al. 2017; Fan et al. 2013). The MCAs were cut into small pieces and incubated with gentle rotation in Tyrode’s solution, including papain (22.5 u/mL, Sigma-Aldrich, St. Louis, MO) and dithiothreitol (2 mg/mL, Sigma-Aldrich) at 37 °C for 15 min. The vessels were pelleted at 1000 RPM and resuspended in fresh Tyrode’s solution containing collagenase (250 u/mL, Sigma-Aldrich), trypsin inhibitor (10,000 u/mL, Sigma-Aldrich) and elastase (2.4 u/mL, Sigma-Aldrich) and incubated for another 15 min at 37 °C with gentle rotation. The digested vessels were centrifuged at 1500 RPM for 10 min. Single CVSMCs were resuspended in Dulbecco’s modified Eagle’s medium (DMEM, Thermo Scientific, Waltham, MA) containing 5 mM glucose, 20% fetal bovine serum, and 1% penicillin/streptomycin. The cells were seeded into a 6-well plate precoated with CellTak (Thermo Scientific) and cultured in either normal glucose (NG, 5.5 mM) or HG (30 mM) media for 7 days prior to subsequent experiments. The same concentration of mannitol was used as an osmotic control. Early passages (P2–P4) of primary CVSMCs were used in all experiments.
The myogenic response of the MCA
The MCAs were isolated from 9- to 12-week-old SD and diabetic rats, and the myogenic response of the MCA was compared as previously described (Fan et al. 2015; Fan et al. 2014). A branch-free portion of the MCA with inner diameters (ID) ranging from 100 to 200 μm was cannulated with glass pipettes in a pressure myograph that was filled with physiological salt solution (PSS) (Fan et al. 2017; Fan et al. 2013). The bath solution was bubbled with 5% CO2, and 95% O2 at 37 °C to maintain adequate oxygenation and pH at 7.4. The MCAs were equilibrated for 30 min and preconditioned three times by raising intraluminal pressure from 40 to 140 mmHg. The MR was determined by measuring the ID of the MCA in response to an increase of intraluminal pressures from 40 to 180 mmHg in steps of 20 mmHg. The ID of the MCA was measured using a × 10 objective of an inverted microscope connected to an AmScope microscope camera (Olympus, Center Valley, PA). MR was calculated by the formula: ID at a given perfusion pressure/ID at 40 mmHg in PSS × 100% (Fan et al. 2017; Kelly-Cobbs et al. 2011). The passive pressure-diameter relationship was examined by replacing the bath solution with calcium-free PSS.
Determination of ROS production
ROS production in MCAs was measured with dihydroethidium (DHE, Thermo Scientific) staining. Freshly isolated MCAs were washed with phosphate-buffered saline (PBS, Thermo Scientific), placed in a 6-well plate and incubated with 10 μM DHE at room temperature for 10 min. MCAs were washed 3× with PBS and fixed with 3.7% paraformaldehyde (Thermo Scientific) for 10 min. Vessels were then mounted on coverslips and imaged with 405 nm excitation and 590 nm emission (Robinson et al. 2008) using the Nikon Eclipse 55i fluorescence microscope (Nikon, Melville, NY). Quantitation of ROS was determined by comparing the mean intensities of the red fluorescence using ImageJ software (https://imagej.nih.gov/ij/download.html).
Mitochondrial ROS production was measured using the MitoSOX™ Red Mitochondrial Superoxide Indicator kit (Thermo Scientific). CVSMCs were seeded into a 6-well plate and treated with NG or HG media for 7 days. The same concentration of mannitol was used as an osmotic control. The cells were washed with PBS (Thermo Scientific) and incubated with 5 μM MitoSOX™ reagent for 10 min at room temperature. The cells were then washed three times, and images were taken immediately (time 0) and 10, 20, and 30 min with 405-nm excitation and 590-nm emission using a Lionheart live-cell imager (BioTek Instruments, Inc., Winooski, VT). Fluorescence excitation at ~ 400 nm with emission at ~ 590 nm specifically detects mitochondrial superoxide without obfuscating the results from detecting ethidium at an excitation wavelength of ~ 510 nm (Robinson et al. 2008), which is generated by oxidation of DHE with oxygen species. Relative mitochondrial ROS production was also measured in freshly isolated MCAs of control and diabetic rats. Vessels were incubated with 10 μM MitoSOX™ reagent for 10 min at room temperature, and images were taken using Nikon Eclipse 55i fluorescence microscopy. Mitochondrial ROS production was compared by the mean red fluorescence intensities using ImageJ.
Western blotting
NG- or HG-treated CVSMCs were scraped into radioimmunoprecipitation assay (RIPA) buffer (Sigma-Aldrich) containing protease and phosphatase inhibitors (Thermo Scientific) on ice and sonicated. Homogenates were centrifuged at 9000g for 15 min at 4 °C, and supernatants were collected. Protein concentration was measured using the Bradford method (Bio-Rad Laboratories, Hercules, CA). Equal amounts of protein (30 μg) were suspended with 2× Laemmli buffer, including 2-mercaptoethanol and denatured at 100 °C for 10 min. Protein samples and a molecular marker were separated by 4–20% SDS-polyacrylamide gel electrophoresis and transferred to 0.2 μm nitrocellulose membranes by Trans-Blot Turbo Transfer System (Bio-Rad). Membranes were blocked with 5% non-fat milk in Tris-buffered saline with 0.1% Tween-20 (TBST) at room temperature for 1 h and incubated at 4 °C overnight with rabbit anti-Tomm20 antibody (42406, 1:1000, Cell Signaling, Danvers, MA), rabbit anti-MFF antibody (84580, 1:1000, Cell Signaling), or rabbit anti-OPA1 antibody (80471, 1:1000, Cell Signaling), followed by HRP-conjugated goat anti-rabbit secondary antibody (7074, 1:2000, Cell Signaling). The membranes were exposed to the SuperSignal West Dura substrate (Thermo Scientific), and the relative intensities of immunoreactive bands were visualized and analyzed using a ChemiDoc Imaging System (Bio-Rad).
Immunocytochemistry
CVSMCs were seeded on autoclaved glass coverslips placed in sterile 6-well plates. Cells were fixed with 3.7% paraformaldehyde (Thermo Scientific) for 10 min, permeabilized with 0.1% Triton-100 (Sigma-Aldrich) in PBS for 5 min, and blocked with 1% BSA for 30 min, followed by incubation with primary antibody Tomm20 (42406, 1:200, Cell Signaling) for 1 h and secondary antibody Alexa Fluor 555 (A-11036, 1:500, Thermo Scientific) for 1 h at room temperature. The slides were coverslipped using an anti-fade mounting medium with DAPI (Vector Laboratories, Burlingame, CA). Images were collected using a Nikon C2+ confocal microscope (Nikon) and the length of mitochondria was measured using ImageJ.
Comparison of mitochondria function in NG- and HG-treated CVSMCs
The oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) of CVSMCs were measured using the Seahorse XFe24 Extracellular Flux Analyzer (Agilent, Santa Clara, CA). CVSMCs (5 × 103/well) were seeded onto CellTak-precoated XFe24 plates and treated with NG or HG media for 7 days. The same concentration of mannitol was used as an osmotic control. On the day of the experiment, the cells were washed and replaced with fresh XF assay medium (XF base medium containing 10 mM glucose, 2 mM l-glutamine, and 1 mM sodium pyruvate; pH 7.4), and incubated in a non-CO2 environment at 37 °C for 1 h. OCR was compared under basal conditions and in response to sequential addition of 1 μM oligomycin (Sigma-Aldrich), 2 μM carbonyl cyanide-4-(trifluoromethoxy) phenylhydrazone (FCCP, Sigma-Aldrich), 0.5 μM rotenone (Sigma-Aldrich), and 0.5 μM antimycin A (Sigma-Aldrich) for three cycles. The results were normalized to protein levels in each well determined by the Bradford method (Bio-Rad). Glycolytic reserve capacity was calculated as oligomycin-induced ECAR-basal ECAR (Lee et al. 2014; Phong et al. 2017).
Comparison of mitochondrial ΔΨm in NG- and HG-treated CVSMCs
Mitochondrial ΔΨm was detected using a MitoProbe™ JC-1 Assay Kit (Thermo Scientific). Cells were seeded onto a 6-well plate and treated with HG for 7 days. After removing the medium, 5 μM JC-1 dye in PBS was added to wells, and the cells incubated for 20 min at 37 °C in a 5% CO2 humidified atmosphere. The cells were photographed using an excitation and emission wavelengths of ~ 545 nm (green) and ~ 595 nm (red) using Lionheart live-cell imager (BioTek). Quantitation was performed using ImageJ.
Comparison of cell proliferation in NG- and HG-treated CVSMCs
Cell proliferation in NG- and HG-treated CVSMCs was compared using an MTS Assay Kit (Abcam, Cambridge, MA). CVSMCs (2.5 × 104/well) were seeded onto a 96-well plate. After 72-h incubation, MTS reagent (20 μL/well) was added, and the cells were incubated for 3 h at 37 °C in a 5% CO2 humidified atmosphere. The formazan product was compared by measuring the absorbance at 490 nm using a plate reader (BioTek).
Comparison of the contractile capability of NG- and HG-treated CVSMCs
Cell contractile capability in NG- and HG-treated CVSMCs was compared using a collagen gel-based cell contraction assay kit (Cell Biolabs, San Diego, CA). The cells were trypsinized and resuspended in the culture medium at 2 × 106 cells/mL, mixed with collagen gel working solution on the ice at a ratio of 1:4. The mixture (0.5 mL) containing 2 × 105 cells were added to each well of 24-well plates, and 1 mL of culture medium was then dropped at the top of the gel after collagen polymerized at 37 °C for 1 h. The cells were incubated for another 48 h to develop contractile force. The stressed matrix was detached from the wall using a sterile needle to initiate cell contraction. Changes in the collagen gel size were imaged after stimulation and quantified with ImageJ.
Statistical analysis
Statistical differences of mean values between groups were determined using an unpaired t test. Analyses were performed using GraphPad Prism 8 (GraphPad Software, San Diego, CA), and P < 0.05 was considered statistically significant.
Results
The production of ROS and mitochondrial superoxide were enhanced in the MCA of diabetic rats
The production of ROS and mitochondria-derived superoxide were enhanced in the MCA of diabetic T2DN rats. As presented in Fig. 1, the fold change in the increase in DHE fluorescence intensity in freshly isolated MCA of diabetic T2DN rats (6.51 ± 0.53, n = 8) was significantly higher than in control SD rats (n = 7, Fig. 1 a and c). Similarly, the fold change of the increase in mean MitoSOX fluorescence intensity in MCA of diabetic rats (3.70 ± 0.39, n = 9) was significantly higher than in control SD rats (n = 9, Fig. 1 b and d).
Fig. 1.

The production of ROS and mitochondrial superoxide were enhanced in the MCA of diabetic rats. a Representative images of DHE staining in MCA freshly isolated from diabetic (DM) and control (Ctrl) rats. b Representative images of MitoSOX staining in MCA freshly isolated from DM and Ctrl rats. c Quantitative analysis of the fold change of DHE fluorescence. d Quantitative analysis of the fold change of MitoSOX fluorescence intensity. ROS, reactive oxygen species; MCA, middle cerebral artery; DHE, dihydroethidium. Mean values ± SE are presented as horizontal lines. Asterisk (*) indicates P < 0.05 from the corresponding values in DM versus Ctrl rats
The MR of the MCA was impaired in diabetic rats
The inner diameters of the MCA decreased to 85.08 ± 3.08% in control rats (n = 5) but increased to 135.30 ± 11.29% in diabetic rats (n = 5) when perfusion pressure was increased from 40 to 180 mmHg (Fig. 2), while the MCAs of control and diabetic rats were fully passive dilated in response to elevations in perfusion pressure (data not shown). These results demonstrate that the MR of MCA was impaired in diabetic rats.
Fig. 2.
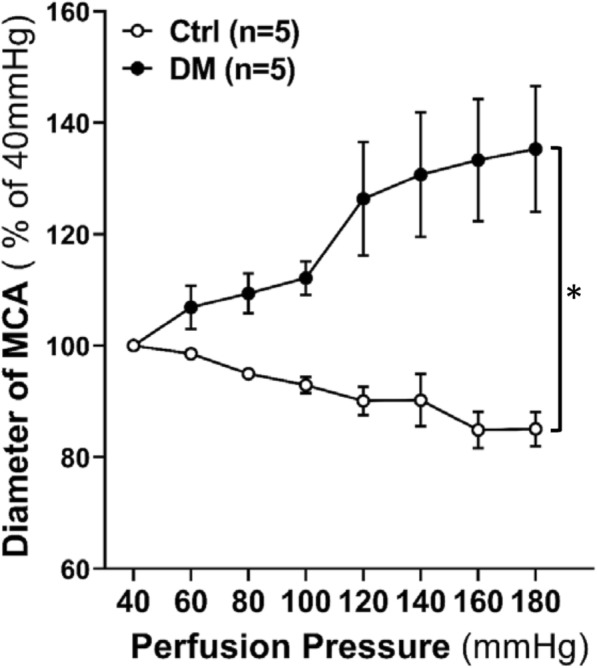
Comparison of the changes in the inner diameter of isolated perfused MCA of diabetic (DM) and non-diabetic control (Ctrl) rats in response to perfusion pressure increased from 40 to 180 mmHg. MR, myogenic response; MCA, middle cerebral artery. Mean values ± SE are presented. Asterisk (*) indicates P < 0.05 from the corresponding value in DM vs. Ctrl rats
The production of mitochondrial superoxide was increased in HG-treated CVSMCs
A comparison of mitochondrial superoxide in HG and NG-treated CVSMCs is presented in Fig. 3. The fold change of the increase in fluorescence intensity of HG-treated CVSMCs (1.09 ± 0.01) was significantly higher than that measured in NG-treated CVSMCs.
Fig. 3.

Comparison of the production of mitochondrial superoxide in HG- and NG-treated CVSMCs. a Representative images of MitoSOX staining. b Quantitative analysis of the fold change of MitoSOX fluorescence intensity in CVSMCs. Primary CVSMCs were isolated from five rats, and experiments were repeated three times. a.u., arbitrary unit. NG, normal glucose; HG, high glucose; CVSMCs, cerebral vascular smooth muscle cells. Mean values ± SE are presented. Asterisk (*) indicates P < 0.05 from the corresponding value in HG- versus NG-treated cells
ATP production and mitochondrial respiration were diminished in HG-treated CVSMCs
To further investigate the effects of HG on mitochondrial function in CVSMCs, we conducted a Seahorse XF Cell Mito Stress Test. As shown in Fig. 4, mitochondrial basal respiration and ATP production were significantly lower in HG-treated than NG-treated CVSMCs (1995 ± 86.99 vs. 1185 ± 87.67 pmol/min/mg, and 1561 ± 53.36 vs. 959.7 ± 61.01 pmol/min/mg, respectively). There was no significant difference in basal ECAR, oligomycin-induced ECAR, and glycolytic reserve capacity between HG- and NG-treated CVSMCs.
Fig. 4.

Determination of ATP production and mitochondrial respiration. OCR (a, b) and ECAR (c, d) Mito Stress Test Seahorse XF profiles in HG-treated and NG-treated CVSMCs. Oligomycin (1 μM, an inhibitor of Complex V), FCCP (2 μM, an uncoupling agent which promotes transport of hydrogen ions), rotenone (0.5 μM, an inhibitor of complex I), and antimycin A (0.5 μM, an inhibitor of complex III) were added at the indicated points. Primary CVSMCs were isolated from five rats, and experiments were repeated three times. OCR, oxygen consumption rate; ECAR, extracellular acidification rate; NG, normal glucose; HG, high glucose; CVSMCs, cerebral vascular smooth muscle cells; ATP, adenosine triphosphate; FCCP, carbonyl cyanide-4-(trifluoromethoxy) phenylhydrazone. Mean values ± SE are presented. Asterisk (*) indicates P < 0.05 from the corresponding values measured in HG- versus NG-treated cells
HG promoted mitochondrial fission and reduced fusion processes in CVSMCs
The effects of HG on mitochondrial dynamics of CVSMCs are depicted in Fig. 5a–c. An interconnected network and elongated mitochondria were observed in NG-treated CVSMCs. In contrast, dot-like clusters and short mitochondrial filaments and fragmentation were observed in HG-treated CVSMCs. The length of mitochondria was significantly shorter in HG-treated vs. NG-treated CVSMCs (0.71 ± 0.006 μm vs.1.51 ± 0.01 μm) (Fig. 5a). Mitochondrial fusion protein OPA1 (80–100 kDa) level was significantly lower, while the fission protein MFF (25, 27, 30, 35 kDa) (Shen et al. 2014) level was markedly higher in HG-treated than NG-treated CVSMCs (Fig. 5 b and c).
Fig. 5.

Determination of mitochondrial fission and fusion processes and ΔΨm in CVSMCs. a Representative images (× 1000 magnification) of the morphology of mitochondria and comparison of the mitochondria length in HG- vs. NG-treated CVSMCs. Red fluorescence represents Tomm20, and blue fluorescence represents Hoechst. b Representative images of Western blots and quantification of the ratio of OPA1 to Tomm20 in HG- vs. NG-treated cells. c Representative images of Western blots and quantification of the ratio of MFF to Tomm20 in HG- vs. NG-treated cells. d Representative images of JC1 staining and quantitative analysis of the ratio of red to green fluorescence intensity in HG vs. NG-treated CVSMCs. Primary CVSMCs were isolated from five rats, and experiments were all repeated three times. NG, normal glucose; HG, high glucose; CVSMCs, cerebral vascular smooth muscle cells
HG promoted depolarization of mitochondrial ΔΨm in CVSMCs
The effects of HG on ΔΨm of CVSMCs are shown in Fig. 5d. Mitochondrial membrane potentials in HG-treated CVSMCs were compared by JC-1 dye staining. JC-1 is a positively charged dye that accumulates in the electronegative interior of the mitochondria, which can be detected at an emission ~ 595 nm by red fluorescent aggregates. When ΔΨm decreases or depolarizes, the potential-sensitive color shifts from red to green (emission ~ 545 nm). We found that JC-1 strained HG-treated CVSMCs exhibited a change in the emission spectrum from red to green compared with NG-treated cells indicating more depolarization of ΔΨm.
HG decreased cell proliferation in CVSMCs
The effects of HG on cell proliferation of CVSMCs are described in Fig. 6a. The MTS assay is a reliable and sensitive indicator for detecting cell proliferation and cytotoxicity in response to many factors (Cory et al. 1991). After treatment with HG, cell viability was significantly decreased in HG-treated than NG-treated CVSMCs (0.86 ± 0.007 vs. 1.00 ± 0.009, respectively).
Fig. 6.

Determination of cell proliferation and contractile capability of CVSMCs. a Comparison of relative viability in HG- vs. NG-treated CVSMCs using MTS assay. b Comparison of the contractile capability of HG- vs. NG-treated CVSMCs. Representative images are presented at the left panel. Quantification of % constriction relative to the initial size of the CVSMCs is presented at the right panel. The white dotted circle represented the gel area. Primary CVSMCs were isolated from five rats, and experiments were repeated three times. NG, normal glucose; HG, high glucose; CVSMCs, cerebral vascular smooth muscle cells. Mean values ± SE are presented. Asterisk (*) indicates P < 0.05 from HG- vs. NG-treated cells
HG reduced the contractile capability of CVSMCs
The effects of HG on the ability of CVSMCs to contract were studied using a collagen gel-based n assay. The results of these experiments are presented in Fig. 6b. Cell contraction developed much slower in HG-treated CVSMCs, and the gel size was reduced by 27.2% vs. 48.7% in HG- vs. NG-treated CVSMCs. These findings indicate that HG diminishes the contractile capability of CVSMCs, which may contribute to the impairment of MR of MCA in diabetic rats.
Discussion
DM is a complex and progressive metabolic disorder characterized by a hallmark of hyperglycemia that accelerates vascular aging. Aging exacerbates DM-related vascular damage, especially in highly vascularized organs such as the kidney, heart, and brain and promotes the development of acute kidney injury, coronary heart disease, and cerebrovascular diseases (Assar et al. 2016; Chao et al. 2018; Goldberg and Dansky 2006). The present study provides evidence that HG-induced dysregulation of mitochondrial dynamics in cerebral CVSMCs is associated with ATP depletion and elevated mitochondria-derived ROS production in MCA isolated from diabetic rats. These changes may contribute to the impaired MR of the MCA and cerebrovascular dysfunction in diabetes, which is similar to the changes previously reported with aging (Springo et al. 2015a; Springo et al. 2015b; Sure et al. 2018; Toth et al. 2013).
Chronic hyperglycemia can produce a wide range of serious complications; however, symptoms and organ damage only become apparent when plasma glucose exceeds 15–20 mM in humans (American Diabetes A 2014). In the present study, we first compared the myogenic response of the MCA in diabetic T2DN and non-diabetic SD rats. The T2DN rat model closely mimics changes seen in diabetic patients with hyperglycemia, hyperinsulinemia, and hyperlipidemia (Kojima et al. 2013; Nobrega et al. 2004). Plasma glucose levels in T2DN rats averaged 20–30 mM in comparison with 5 mM in SD rats (Kojima et al. 2013; Muroya et al. 2018; Nobrega et al. 2004). We found that the MCA exhibited impaired MR in association with elevated ROS and mitochondrial superoxide production in freshly isolated MCA of diabetic vs. non-diabetic rats. These results suggest that hyperglycemia-induced mitochondrial superoxide over-production is at least one of the factors that contribute to impaired cerebral vascular function in these animals.
The MR is an intrinsic property of vascular smooth muscle cells to regulate CBF by constriction of cerebral vasculature in response to elevations in blood pressure (Cipolla et al. 2002). To determine whether exposure to hyperglycemic conditions alone is sufficient to damage CVSMCs, we treated CVSMCs isolated from MCA of SD rats with a glucose concentration of 30 mM. This concentration mimics the plasma glucose levels in our diabetic rats (Kojima et al. 2013; Muroya et al. 2018; Nobrega et al. 2004) and is reported to damage endothelial, human skeletal muscle satellite cells, and mesenchymal stem cells (Kargl et al. 2019; Weil et al. 2009). We confirmed that mitochondrial superoxide production increased significantly in HG-treated primary CVSMCs, which is similar to our findings in intact MCA isolated from the diabetic rats. Oxidative phosphorylation in the mitochondria is the primary source of ATP by aerobic cellular respiration. ATP can also be synthesized by glycolysis, pyruvate oxidation, and Krebs cycle (Jose et al. 2011). Using a Seahorse XFe24 analyzer, OCR that indicates mitochondrial respiration and ECAR that indicates glycolysis were compared (Lee et al. 2014). We found that basal mitochondrial respiration and ATP production were significantly reduced, but ECAR was not changed in HG-treated compared with NG-treated CVSMCs. The results suggested that HG-induced decrease in ATP production in CVSMCs was mainly due to impaired mitochondrial function.
Mitochondria occupy a central role in energy production. They are one of the primary sources to produce ROS in disease. Mitochondrial function is regulated by the balance between mitochondrial biogenesis and degradation and is vital to maintain energy homeostasis. Changes in the size, shape, and subcellular distribution of mitochondria via fission and fusion, the proteolytic system, and mitophagy are essential to maintain mitochondrial function (Lemasters 2014; van der Bliek et al. 2013). Mitochondrial fusion is regulated by optic atrophy 1 (OPA1), mitofusin-1 and mitofusin-2 (MFN1 and MFN2). Mitochondrial fission is regulated by mitochondria fission factor (MFF), fission protein 1 (FIS1), and dynamin-related protein 1 (DRP1) proteins (Tilokani et al. 2018). Mitochondria have a bean-shaped structure with inner membrane forming mitochondrial cristae, and outer membrane. They form a tubular network in the endothelial cells, are ovoid or rod shape in vascular smooth muscle cells, are spherical and oval-shaped in hepatocytes, and aggregate into long filaments in fibroblasts (McCarron et al. 2013; Shenouda et al. 2011; Westermann 2010; Youle and van der Bliek 2012). Perturbations in the regulation of mitochondrial dynamics cause mitochondrial dysfunction, which promotes mitochondria-derived ROS production and induces cellular damage and causes diminished function. Imbalanced activation or inactivation of fusion or fission by genetic or other factors results in mitochondrial fragmentation or an increase in connectivity (Karbowski and Youle 2003). The depletion of mitochondrial DNA in human fibroblasts and 143B osteosarcoma cell lines were found to alter mitochondrial morphology and produced smaller individual mitochondria and functional insufficiency (Gilkerson et al. 2000). Treatment of HeLa cells with mitochondrial toxins dissipated the mitochondrial membrane potential (ΔΨm) and inhibited fusion (Legros et al. 2002). Transient increases in fusion were observed in human umbilical vein endothelial cells treated with low doses of hydrogen peroxide (Jendrach et al. 2008). Decreased plasma glucose levels associated with starvation promoted mitochondrial elongation and inhibited mitochondrial fission (Gomes et al. 2011). In type 2 diabetes, mitochondria in skeletal muscle exhibited a shorter and less internal structure with vacuolization (Kelley et al. 2002). This phenomenon was also reported in cumulus cells of type 1 diabetic mice that showed more fragmented mitochondria (Wang et al. 2010).
Mitochondria dynamics play a critical role in regulating mitochondrial homeostasis and superoxide production. These organelles are constantly changing their shape and distribution within the cell to accommodate evolving cellular energy demands via the processes of fission or fusion, leading to fragmentation or elongation of mitochondria (Yoon et al. 2011). The disruption of fission-fusion balance is considered a significant cause of mitochondrial ROS overproduction and loss of ATP production (Yoon et al. 2011). In the present study, we found that mitochondria in CVSMCs underwent fragmentation with shorter filaments after exposure to HG for 7 days compared with control cells. These results are consistent with previous reports in various tissues of diabetic patients and animal models (Kelley et al. 2002; Wang et al. 2010). The observation was further confirmed by detecting changes in the expression of recognized biomarkers of mitochondrial fission and fusion. We found the expression of OPA1 was reduced, and MFF was increased in HG-treated CVSMCs. OPA1, a dynamin-related guanosine triphosphatase localizing on mitochondria inner membrane, is required for the fusion of mitochondria (Song et al. 2007). MFF recruits DRP1 to promote mitochondrial fission (Otera et al. 2010). These results suggest that the fragmented or shorter mitochondria in HG-treated CVSMCs resulted from imbalanced mitochondrial dynamics by enhanced fission and reduced fusion.
ΔΨm is generated by proton pumps in complexes I, III, and IV by moving electrons through the energy gradient (Zorova et al. 2018). ΔΨm is essential for the regulation of a wide variety of mitochondrial functions, including, ATP synthesis, Ca2+ uptake and storage, ROS production, and opening of the mitochondrial permeability transition pore (MPTP) (Zorova et al. 2018). ΔΨm provides the driving force for non-proton ions passing through complex V to trigger the generation of ATP, whereas depolarization of mitochondria leads to the reduction of ATP production (Chalmers and McCarron 2008). Oxidative stress has been reported to open MPTP that induces mitochondrial membrane depolarization, ATP depletion, and mitochondrial matrix swelling. The subsequent rupture of the mitochondrial membrane leads to the release of cytochrome C, resulting in the reduction of ATP levels and the promotion of cell apoptosis (Lieven et al. 2003). Increases in fragmented mitochondria in diabetic mice were observed in association with reduced ΔΨm (Wang et al. 2010). In the present study, we detected mitochondrial depolarization in HG-treated CVSMCs. Reduced ΔΨm is an early detector of cell apoptosis or cell death, which was confirmed in a cell proliferation assay in our study, demonstrating that HG-treated CVSMCs exhibited more cell death.
Vascular smooth muscle cells have high rates of glucose metabolism, and approximately 30% ATP is generated in the mitochondria (Butler and Siegman 1985; Paul 1983). Abnormal mitochondrial dynamics, reduction of the synthesis of ATP, and overproduction of ROS in HG-treated cells were associated with mitochondrial dysfunction in our study. Our findings that the contractile capability of CVSMCs of MCA was markedly reduced in HG-treated cells strongly supports the view that HG-induced mitochondrial dysfunction plays a vital role in the loss of CVSMCs function. As a significant factor that initiates the MR, the diminished contractile capability in HG-treated CVSMCs fundamentally contributes to the impaired MR of MCA as we detected in diabetic rats. Indeed, there is a limitation of the present study, since we only focused on the smooth muscle cells and did not study the other cell types that comprise the vascular wall, such as endothelial cells, which also modulate cerebral vascular function. Our study provides another piece of information from a unique angle to dissect the underlying mechanisms of diabetes-related cerebral vascular dysfunction.
The underlying mechanisms by which DM-related vascular injury with aging may involve several signaling pathways. For example, knockout of a recently identified specific form of myosin light chain kinase (MLCK210) protected the functional integrity of the blood-brain barrier and prevented cerebral microhemorrhages and neuroinflammation and the development of vascular cognitive impairment and dementia in a mouse model (Braun et al. 2019). Although MLCK210 mainly expressed in the endothelium and deletion of MLCK210 had no effects on the constriction of aortic rings in response to vasoconstrictor and vasodilator agonists (Ohlmann et al. 2005), it remains to be determined whether the MLCK210 plays a role in the constriction of CVSMCs. Administration of nicotinamide mononucleotide, an important NAD+ intermediate, improved mitochondrial membrane potential, enhanced ATP and NO production, and reduced mitochondrial ROS in cerebral microvascular endothelial cells in aged mice (Tarantini et al. 2019a). NAD+, an oxidized form of nicotinamide adenine dinucleotide (NAD), is a substrate of poly (ADP-ribose) polymerase that regulates DNA damage and repair, and is a rate-limiting cosubstrate for silent information regulator-2-like enzymes that mediate mitochondrial biogenesis, energetics, and mitochondrial antioxidant defenses (Csiszar et al. 2019; Kiss et al. 2020; Pehar et al. 2018; Tarantini et al. 2019b). These findings suggest NAD+ plays an essential role in the cerebrovascular function. It is not clear whether NAD+ also affects the mitochondrial functions of CVSMCs in DM. Hence, examining the protection effects of NAD+ on CVSMCs under hyperglycemic conditions is needed in future studies.
In summary, HG promotes mitochondrial fission and attenuates fusion that contributes to elevated mitochondria-derived ROS production and mitochondrial membrane depolarization to reduce ATP production. These pathological subcellular events diminish CVSMCs contractile capability, which may contribute to the impairment of MR of MCA in diabetes (Fig. 7). The current study provides a novel insight into understanding the link between diabetes and vascular aging. Stabilization of mitochondrial dynamics and reduction of mitochondrial-derived ROS production may serve as a novel approach for the prevention and treatment of cerebrovascular diseases associated with diabetes and aging.
Fig. 7.
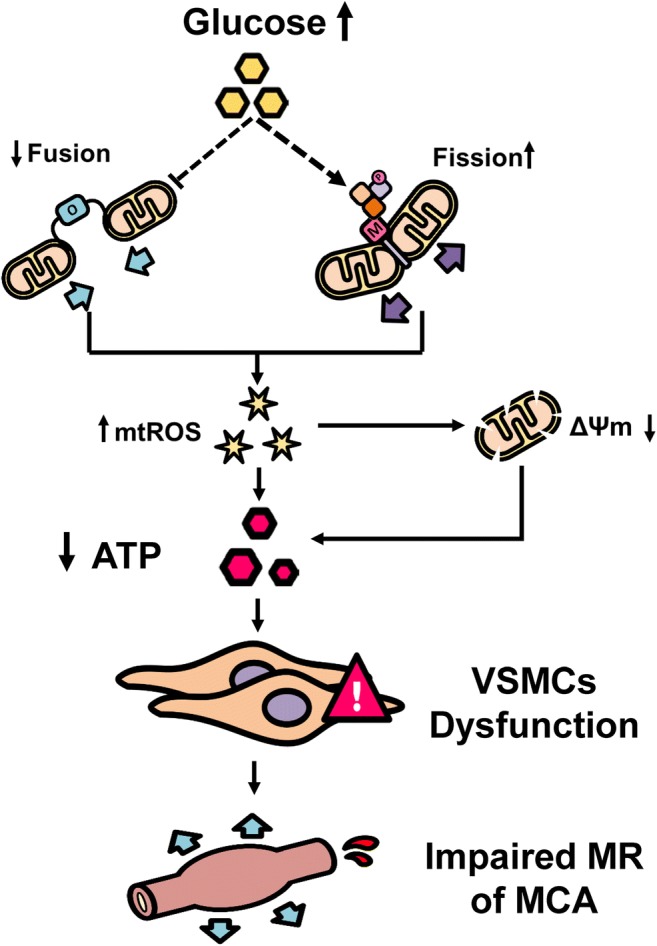
Summary of mechanisms by which hyperglycemia induces mitochondria dysfunction in CVSMVs. Exposure to high glucose promotes mitochondrial fission and inhibits fusion in CVSMCs, which elevates mitochondrial ROS (mtROS) production, and reduces mitochondrial ΔΨm and ATP production. These events diminish CVSMCs proliferation and contractile capabilities, which plays an essential role in impaired MR of MCAs in diabetes. Dash lines indicate multiple steps
Funding information
This work was supported by the National Institutes of Health (AG050049, AG057842, P20GM104357, DK104184, and HL138685) and the American Heart Association (16GRNT31200036 and 20PRE35210043).
Compliance with ethical standards
The UMMC animal care facility is approved by the American Association for the Accreditation of Laboratory Animal Care. All experiments and protocols involving animals were approved by the Institutional Animal Care and Use Committees (IACUC) of the UMMC and conformed to NIH guidelines.
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Ya Guo and Shaoxun Wang contributed equally to this work.
References
- Ahn B, et al. Nrf2 deficiency exacerbates age-related contractile dysfunction and loss of skeletal muscle mass. Redox Biol. 2018;17:47–58. doi: 10.1016/j.redox.2018.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Diabetes A Diagnosis and classification of diabetes mellitus. Diabetes Care. 2014;37(Suppl 1):S81–S90. doi: 10.2337/dc14-S081. [DOI] [PubMed] [Google Scholar]
- Assar ME, Angulo J, Rodriguez-Manas L. Diabetes and ageing-induced vascular inflammation. J Physiol. 2016;594:2125–2146. doi: 10.1113/JP270841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun DJ, Bachstetter AD, Sudduth TL, Wilcock DM, Watterson DM, Van Eldik LJ. Genetic knockout of myosin light chain kinase (MLCK210) prevents cerebral microhemorrhages and attenuates neuroinflammation in a mouse model of vascular cognitive impairment and dementia. Geroscience. 2019;41:671–679. doi: 10.1007/s11357-019-00072-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler TM, Siegman MJ. High-energy phosphate metabolism in vascular smooth muscle. Annu Rev Physiol. 1985;47:629–643. doi: 10.1146/annurev.ph.47.030185.003213. [DOI] [PubMed] [Google Scholar]
- Chalmers S, McCarron JG. The mitochondrial membrane potential and Ca<sup>2+</sup> oscillations in smooth muscle. J Cell Sci. 2008;121:75. doi: 10.1242/jcs.014522. [DOI] [PubMed] [Google Scholar]
- Chao CT, Wang J, Wu HY, Huang JW, Chien KL. Age modifies the risk factor profiles for acute kidney injury among recently diagnosed type 2 diabetic patients: a population-based study. Geroscience. 2018;40:201–217. doi: 10.1007/s11357-018-0013-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipolla MJ, Gokina NI, Osol G. Pressure-induced actin polymerization in vascular smooth muscle as a mechanism underlying myogenic behavior. FASEB J. 2002;16:72–76. doi: 10.1096/cj.01-0104hyp. [DOI] [PubMed] [Google Scholar]
- Corriere M, Rooparinesingh N, Kalyani RR. Epidemiology of diabetes and diabetes complications in the elderly: an emerging public health burden. Curr Diab Rep. 2013;13:805–813. doi: 10.1007/s11892-013-0425-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cory AH, Owen TC, Barltrop JA, Cory JG. Use of an aqueous soluble tetrazolium/formazan assay for cell growth assays in culture. Cancer Commun. 1991;3:207–212. doi: 10.3727/095535491820873191. [DOI] [PubMed] [Google Scholar]
- Csiszar A, et al. Role of endothelial NAD(+) deficiency in age-related vascular dysfunction. Am J Physiol Heart Circ Physiol. 2019;316:H1253–H1266. doi: 10.1152/ajpheart.00039.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ergul A, Kelly-Cobbs A, Abdalla M, Fagan SC. Cerebrovascular complications of diabetes: focus on stroke. Endocr Metab Immune Disord Drug Targets. 2012;12:148–158. doi: 10.2174/187153012800493477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan F, et al. 20-Hydroxyeicosatetraenoic acid contributes to the inhibition of K+ channel activity and vasoconstrictor response to angiotensin II in rat renal microvessels. PloS One. 2013;8:e82482. doi: 10.1371/journal.pone.0082482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan F, Geurts AM, Pabbidi MR, Smith SV, Harder DR, Jacob H, Roman RJ. Zinc-finger nuclease knockout of dual-specificity protein phosphatase-5 enhances the myogenic response and autoregulation of cerebral blood flow in FHH.1BN rats. PLoS One. 2014;9:e112878. doi: 10.1371/journal.pone.0112878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan F, Geurts AM, Murphy SR, Pabbidi MR, Jacob HJ, Roman RJ. Impaired myogenic response and autoregulation of cerebral blood flow is rescued in CYP4A1 transgenic Dahl salt-sensitive rat. Am J Physiol Regul Integr Comp Physiol. 2015;308:R379–R390. doi: 10.1152/ajpregu.00256.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan F, Pabbidi MR, Ge Y, Li L, Wang S, Mims PN, Roman RJ. Knockdown of Add3 impairs the myogenic response of renal afferent arterioles and middle cerebral arteries. Am J Physiol Renal Physiol. 2017;312:F971–F981. doi: 10.1152/ajprenal.00529.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulop GA, et al. Nrf2 deficiency in aged mice exacerbates cellular senescence promoting cerebrovascular inflammation. Geroscience. 2018;40:513–521. doi: 10.1007/s11357-018-0047-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilkerson RW, Margineantu DH, Capaldi RA, Selker JML. Mitochondrial DNA depletion causes morphological changes in the mitochondrial reticulum of cultured human cells. FEBS Lett. 2000;474:1–4. doi: 10.1016/S0014-5793(00)01527-1. [DOI] [PubMed] [Google Scholar]
- Goldberg IJ, Dansky HM. Diabetic vascular disease: an experimental objective. Arterioscler Thromb Vasc Biol. 2006;26:1693–1701. doi: 10.1161/01.ATV.0000231521.76545.f6. [DOI] [PubMed] [Google Scholar]
- Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol. 2011;13:589–598. doi: 10.1038/ncb2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill MA, Meininger GA. Impaired arteriolar myogenic reactivity in early experimental diabetes. Diabetes. 1993;42:1226–1232. doi: 10.2337/diab.42.9.1226. [DOI] [PubMed] [Google Scholar]
- Hoyer S, Oesterreich K, Wagner O. Glucose metabolism as the site of the primary abnormality in early-onset dementia of Alzheimer type? J Neurol. 1988;235:143–148. doi: 10.1007/bf00314304. [DOI] [PubMed] [Google Scholar]
- Jendrach M, Mai S, Pohl S, Voth M, Bereiter-Hahn J. Short- and long-term alterations of mitochondrial morphology, dynamics and mtDNA after transient oxidative stress. Mitochondrion. 2008;8:293–304. doi: 10.1016/j.mito.2008.06.001. [DOI] [PubMed] [Google Scholar]
- Jose C, Bellance N, Rossignol R. Choosing between glycolysis and oxidative phosphorylation: a tumor’s dilemma? Biochim Biophys Acta. 2011;1807:552–561. doi: 10.1016/j.bbabio.2010.10.012. [DOI] [PubMed] [Google Scholar]
- Karbowski M, Youle RJ. Dynamics of mitochondrial morphology in healthy cells and during apoptosis. Cell Death Differ. 2003;10:870–880. doi: 10.1038/sj.cdd.4401260. [DOI] [PubMed] [Google Scholar]
- Kargl CK, Nie Y, Evans S, Stout J, Shannahan JH, Kuang S, Gavin TP. Factors secreted from high glucose treated endothelial cells impair expansion and differentiation of human skeletal muscle satellite cells. J Physiol. 2019;597:5109–5124. doi: 10.1113/JP278165. [DOI] [PubMed] [Google Scholar]
- Kelley DE, He J, Menshikova EV, Ritov VB. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes. 2002;51:2944–2950. doi: 10.2337/diabetes.51.10.2944. [DOI] [PubMed] [Google Scholar]
- Kelly-Cobbs A, Elgebaly MM, Li W, Ergul A. Pressure-independent cerebrovascular remodelling and changes in myogenic reactivity in diabetic Goto-Kakizaki rat in response to glycaemic control. Acta Physiol (Oxf) 2011;203:245–251. doi: 10.1111/j.1748-1716.2010.02230.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss T, et al. Nicotinamide mononucleotide (NMN) supplementation promotes neurovascular rejuvenation in aged mice: transcriptional footprint of SIRT1 activation, mitochondrial protection, anti-inflammatory, and anti-apoptotic effects. Geroscience. 2020. 10.1007/s11357-020-00165-5. [DOI] [PMC free article] [PubMed]
- Kojima N, Williams JM, Takahashi T, Miyata N, Roman RJ. Effects of a new SGLT2 inhibitor, luseogliflozin, on diabetic nephropathy in T2DN rats. J Pharmacol Exp Ther. 2013;345:464–472. doi: 10.1124/jpet.113.203869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagaud GJ, Masih-Khan E, Kai S, van Breemen C, Dube GP. Influence of type II diabetes on arterial tone and endothelial function in murine mesenteric resistance arteries. J Vasc Res. 2001;38:578–589. doi: 10.1159/000051094. [DOI] [PubMed] [Google Scholar]
- Lee H, et al. Increased mitochondrial activity in renal proximal tubule cells from young spontaneously hypertensive rats. Kidney Int. 2014;85:561–569. doi: 10.1038/ki.2013.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legros F, Lombes A, Frachon P, Rojo M. Mitochondrial fusion in human cells is efficient, requires the inner membrane potential, and is mediated by mitofusins. Mol Biol Cell. 2002;13:4343–4354. doi: 10.1091/mbc.e02-06-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemasters JJ. Variants of mitochondrial autophagy: types 1 and 2 mitophagy and micromitophagy (type 3) Redox Biol. 2014;2:749–754. doi: 10.1016/j.redox.2014.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieven CJ, Vrabec JP, Levin LA. The effects of oxidative stress on mitochondrial transmembrane potential in retinal ganglion cells. Antioxid Redox Signal. 2003;5:641–646. doi: 10.1089/152308603770310310. [DOI] [PubMed] [Google Scholar]
- McCarron JG, Wilson C, Sandison ME, Olson ML, Girkin JM, Saunter C, Chalmers S. From structure to function: mitochondrial morphology, motion and shaping in vascular smooth muscle. J Vasc Res. 2013;50:357–371. doi: 10.1159/000353883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDaniel CF. Diabetes: a model of oxidative accelerated aging. Age (Omaha) 1999;22:145–148. doi: 10.1007/s11357-999-0016-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muroya Y, He X, Fan L, Wang S, Xu R, Fan F, Roman RJ. Enhanced renal ischemia-reperfusion injury in aging and diabetes. Am J Physiol Renal Physiol. 2018;315:F1843–F1854. doi: 10.1152/ajprenal.00184.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobrega MA, Fleming S, Roman RJ, Shiozawa M, Schlick N, Lazar J, Jacob HJ. Initial characterization of a rat model of diabetic nephropathy. Diabetes. 2004;53:735–742. doi: 10.2337/diabetes.53.3.735. [DOI] [PubMed] [Google Scholar]
- Ohlmann P, et al. Deletion of MLCK210 induces subtle changes in vascular reactivity but does not affect cardiac function. Am J Physiol Heart Circ Physiol. 2005;289:H2342–H2349. doi: 10.1152/ajpheart.00511.2004. [DOI] [PubMed] [Google Scholar]
- Otera H, Wang C, Cleland MM, Setoguchi K, Yokota S, Youle RJ, Mihara K. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J Cell Biol. 2010;191:1141. doi: 10.1083/jcb.201007152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul RJ. Functional compartmentalization of oxidative and glycolytic metabolism in vascular smooth muscle. Am J Physiol. 1983;244:C399–C409. doi: 10.1152/ajpcell.1983.244.5.C399. [DOI] [PubMed] [Google Scholar]
- Pehar M, Harlan BA, Killoy KM, Vargas MR. Nicotinamide adenine dinucleotide metabolism and neurodegeneration. Antioxid Redox Signal. 2018;28:1652–1668. doi: 10.1089/ars.2017.7145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phong B, Avery L, Menk AV, Delgoffe GM, Kane LP. Cutting edge: murine mast cells rapidly modulate metabolic pathways essential for distinct effector functions. J Immunol. 2017;198:640–644. doi: 10.4049/jimmunol.1601150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson KM, Janes MS, Beckman JS. The selective detection of mitochondrial superoxide by live cell imaging. Nat Protoc. 2008;3:941–947. doi: 10.1038/nprot.2008.56. [DOI] [PubMed] [Google Scholar]
- Samaras K, et al. The impact of glucose disorders on cognition and brain volumes in the elderly: the Sydney Memory and Ageing Study. Age (Dordr) 2014;36:977–993. doi: 10.1007/s11357-013-9613-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shekhar S, et al. Impaired cerebral autoregulation-a common neurovascular pathway in diabetes may play a critical role in diabetes-related Alzheimer’s disease. Curr Res Diabetes Obes J. 2017;2:555587. [PMC free article] [PubMed] [Google Scholar]
- Shen Q, et al. Mutations in Fis1 disrupt orderly disposal of defective mitochondria. Mol Biol Cell. 2014;25:145–159. doi: 10.1091/mbc.E13-09-0525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenouda SM, et al. Altered mitochondrial dynamics contributes to endothelial dysfunction in diabetes mellitus. Circulation. 2011;124:444–453. doi: 10.1161/CIRCULATIONAHA.110.014506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Z, Chen H, Fiket M, Alexander C, Chan DC. OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J Cell Biol. 2007;178:749. doi: 10.1083/jcb.200704110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springo Z, Tarantini S, Toth P, Tucsek Z, Koller A, Sonntag WE, Csiszar A, Ungvari Z. Aging exacerbates pressure-induced mitochondrial oxidative stress in mouse cerebral arteries. J Gerontol A Biol Sci Med Sci. 2015;70:1355–1359. doi: 10.1093/gerona/glu244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springo Z, Toth P, Tarantini S, Ashpole NM, Tucsek Z, Sonntag WE, Csiszar A, Koller A, Ungvari ZI. Aging impairs myogenic adaptation to pulsatile pressure in mouse cerebral arteries. J Cereb Blood Flow Metab. 2015;35:527–530. doi: 10.1038/jcbfm.2014.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sure VN, et al. A novel high-throughput assay for respiration in isolated brain microvessels reveals impaired mitochondrial function in the aged mice. Geroscience. 2018;40:365–375. doi: 10.1007/s11357-018-0037-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabit CE, Chung WB, Hamburg NM, Vita JA. Endothelial dysfunction in diabetes mellitus: molecular mechanisms and clinical implications. Rev Endocr Metab Disord. 2010;11:61–74. doi: 10.1007/s11154-010-9134-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarantini S, Valcarcel-Ares MN, Toth P, Yabluchanskiy A, Tucsek Z, Kiss T, Hertelendy P, Kinter M, Ballabh P, Süle Z, Farkas E, Baur JA, Sinclair DA, Csiszar A, Ungvari Z. Nicotinamide mononucleotide (NMN) supplementation rescues cerebromicrovascular endothelial function and neurovascular coupling responses and improves cognitive function in aged mice. Redox Biol. 2019;24:101192. doi: 10.1016/j.redox.2019.101192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarantini S, et al. Treatment with the poly(ADP-ribose) polymerase inhibitor PJ-34 improves cerebromicrovascular endothelial function, neurovascular coupling responses and cognitive performance in aged mice, supporting the NAD+ depletion hypothesis of neurovascular aging. Geroscience. 2019;41:533–542. doi: 10.1007/s11357-019-00101-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilokani L, Nagashima S, Paupe V, Prudent J. Mitochondrial dynamics: overview of molecular mechanisms. Essays Biochem. 2018;62:341–360. doi: 10.1042/EBC20170104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth P, et al. Age-related autoregulatory dysfunction and cerebromicrovascular injury in mice with angiotensin II-induced hypertension. J Cereb Blood Flow Metab. 2013;33:1732–1742. doi: 10.1038/jcbfm.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Bliek AM, Shen Q, Kawajiri S. Mechanisms of mitochondrial fission and fusion. Cold Spring Harb Perspect Biol. 2013;5. 10.1101/cshperspect.a011072. [DOI] [PMC free article] [PubMed]
- Walmsley D, Wiles PG. Myogenic microvascular responses are impaired in long-duration type 1 diabetes. Diabet Med. 1990;7:222–227. doi: 10.1111/j.1464-5491.1990.tb01374.x. [DOI] [PubMed] [Google Scholar]
- Wang Q, et al. Mitochondrial dysfunction and apoptosis in cumulus cells of type I diabetic mice. PLoS One. 2010;5:e15901. doi: 10.1371/journal.pone.0015901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Mims PN, Roman RJ, Fan F. Is beta-amyloid accumulation a cause or consequence of Alzheimer’s disease? J Alzheimers Parkinsonism Dement. 2016;1:007. [PMC free article] [PubMed] [Google Scholar]
- Weil BR, Abarbanell AM, Herrmann JL, Wang Y, Meldrum DR. High glucose concentration in cell culture medium does not acutely affect human mesenchymal stem cell growth factor production or proliferation. Am J Physiol Regul Integr Comp Physiol. 2009;296:R1735–R1743. doi: 10.1152/ajpregu.90876.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermann B. Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol. 2010;11:872–884. doi: 10.1038/nrm3013. [DOI] [PubMed] [Google Scholar]
- Yoon Y, Galloway CA, Jhun BS, Yu T. Mitochondrial dynamics in diabetes. Antioxid Redox Signal. 2011;14:439–457. doi: 10.1089/ars.2010.3286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012;337:1062–1065. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, et al. SIRT3 overexpression antagonizes high glucose accelerated cellular senescence in human diploid fibroblasts via the SIRT3-FOXO1 signaling pathway. Age (Dordr) 2013;35:2237–2253. doi: 10.1007/s11357-013-9520-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Zhang X, Lu J. Progress on diabetic cerebrovascular diseases. Bosn J Basic Med Sci. 2014;14:185–190. doi: 10.17305/bjbms.2014.4.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorova LD, et al. Mitochondrial membrane potential. Anal Biochem. 2018;552:50–59. doi: 10.1016/j.ab.2017.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]