

Abstract
The Siva protein, named after the Hindu God of Destruction, plays important roles in apoptosis in various contexts, including downstream of death receptor activation or p53 tumor suppressor engagement. The function of Siva in organismal development and homeostasis, however, has remained uncharacterized. Here, we generate Siva knockout mice to characterize the physiological function of Siva in vivo. Interestingly, we find that Siva deficiency causes early embryonic lethality accompanied by multiple phenotypes, including developmental delay, abnormal neural tube closure, and defective placenta and yolk sac formation. Examination of Siva expression during embryogenesis shows that Siva is expressed in both embryonic and extra-embryonic tissues, including within the mesoderm, which may explain the vascular defects observed in the placenta and yolk sac. The embryonic phenotypes caused by Siva loss are not rescued by p53 deficiency, nor do they resemble those of p53 null embryos, suggesting that the embryonic function of Siva is not related to the p53 pathway. Moreover, loss of the Ripk3 necroptosis protein does not rescue the observed lethality or developmental defects, suggesting that Siva may play a non-apoptotic role in development. Collectively, these studies reveal a key role for Siva in proper embryonic development.
Subject terms: Cell biology, Development
Introduction
Siva is a death domain homology region-containing protein that promotes apoptosis in a wide variety of cellular contexts. Originally identified as an interacting partner of the TNF-superfamily member CD27 [1], subsequent studies found that Siva also interacts with other TNF receptors, including GITR, CD40, and OX40 [2]. SIVA loss was also found to render T-cells resistant to anti-CD3-triggered apoptosis, revealing a key role for SIVA in T cell receptor-mediated activation-induced cell death [3, 4]. These pro-apoptotic effects are likely mediated through the NF-кB pathway, as SIVA loss increases NF-кB activity, resulting in enhanced expression of NF-кB-responsive anti-apoptotic proteins, such as BCL-xL and c-FLIP [3, 4]. This effect on NF-кB may be mediated at least in part through formation of a ternary complex between SIVA, XIAP1 (X-linked inhibitor of apoptosis), and TAK1 (TGF-Beta Activated Kinase-1) that both dampens NF-кB survival signals and enhances pro-apoptotic JNK activity [5]. Beyond its role in membrane signaling events, Siva has also been proposed to promote apoptosis through interaction with the BCL-2 family of proteins [6, 7]. Roles for Siva in apoptosis have since been observed in numerous different cell types and in response to various triggers [8–12], demonstrating that Siva acts in a broad range of contexts.
One of the cellular contexts in which Siva functions is apoptotic cell death induced by the p53 tumor suppressor protein, which directly activates Siva expression [13, 14]. p53 restricts cellular proliferation through either inhibition of cell cycle progression or induction of apoptosis [15, 16], and Siva is specifically upregulated during apoptosis but not during p53-induced cell-cycle arrest in a mouse embryonic fibroblast system [14]. Siva is also highly induced in a p53-dependent manner by DNA damage and ischemia in other cell types, such as neurons and kidney cells, both in vitro and in vivo [12–14]. Genetic perturbation studies demonstrate that Siva is critically important for p53-mediated apoptosis in cerebellar granule neurons in response to DNA damage and in mouse kidney following ischemia/reperfusion injury [12, 14]. Overexpression of Siva in mouse embryonic fibroblasts and lymphocytes triggers apoptosis through the intrinsic mitochondrial apoptotic pathway involving localization of Siva to the plasma membrane and signaling through Bak/Bax, Bid, cytochrome C, and Caspase 3 [14, 17]. Collectively, these findings support a role for Siva in the p53 apoptotic pathway in addition to the varied contexts of apoptosis described above.
Although Siva has a well-established role as an activator of apoptosis in diverse contexts, its physiological function in vivo has heretofore not been determined. To better understand the physiological functions of Siva in vivo, we generated Siva knockout mice using gene-trapped embryonic stem (ES) cells carrying a β-galactosidase reporter. Analysis of reporter expression during early embryogenesis revealed that Siva expression is enriched in the heart, forebrain, and extra-embryonic tissues. We found further that Siva deficiency results in mid-gestational embryonic lethality associated with defects in extra-embryonic vasculature and nervous system development. Together, these findings reveal an essential role for Siva during embryonic development.
Results
Generation of Siva-deficient mice
To examine the function of Siva in vivo, we generated Siva knockout mice using ES cells in which the Siva gene is inactivated through gene trapping. In the Siva gene-trapped allele, a β-galactosidase-neomycin resistance fusion (β-geo) gene and an SV40 polyadenylation sequence are inserted into the Siva locus, thereby both eliminating Siva gene expression and providing a marker for cells in which the Siva promoter is active (Fig. 1a) [18]. We first confirmed that the Siva locus is disrupted in these ES cells by Southern blot analysis (Fig. 1b). Sequencing of the transcript produced by this insertion revealed that the gene trap vector is inserted near the 5′-end of exon 1, as the transcript comprises Siva 5′-untranslated sequences and the first three codons fused to β-geo sequences, suggesting that the insertion completely disrupts the locus (Fig. 1a; data not shown). We next used these ES cells to generate knockout mice through blastocyst injection. Once knockout mice were established (Fig. 1c), Northern blot analysis on wild-type, Siva+/-, and Siva−/− embryos was used to verify reduced Siva RNA in heterozygous animals and a complete absence of Siva transcript in Siva−/− embryos (Fig. 5b). Importantly, we did not detect transcripts of alternate sizes, suggesting that this mutation creates a null allele.
Fig. 1.

Generation of Siva-knockout mice. a Schematic representation of Siva gene-trapped allele and transcript. The gene trap vector consists of Engrailed 2 (En2) intronic sequences followed by a splice acceptor (SA) and coding sequences for the β-galactosidase-neomycin (β-geo) gene. An SV40 polyA sequence acts as a transcriptional stop element. The gene-trapped transcript consists of the Siva 5’UTR and sequences encoding the first 3 amino acids from Siva exon1 fused to β-geo. BamHI sites and PCR primers used for genotyping are indicated. b Southern blot analysis of J1 (WT) and RRR467 (Siva gene trap) ES cell DNA demonstrates disruption of the Siva locus in the gene-trapped ES cells. Following DNA digestion with BamHI, Siva exon 2 sequences were used as a probe. (c) Genotyping PCR used to identify Siva+/+, Siva+/−, and Siva−/− mice and embryos. The forward primer recognizes a portion of 5′-UTR from Siva exon 1 (primer “a”) and is used in combination with reverse primers to a region of intron 1 (primer “b”) and the gene trap vector (primer “c”) The expected band sizes for the wild-type and gene-trapped alleles are 415 and 550 bp, respectively
Fig. 5.

Siva-knockout yolk sacs fail to undergo vascular remodeling. a Whole-mount examination of wild-type and Siva−/− e10.5 yolk sacs. Inset shows embryo associated with Siva−/− yolk sac. b Northern blot examining expression of the hypoxia marker Slc2a1 in Siva-null embryos. c–e Whole-mount CD31 immunohistochemistry on wild-type and Siva−/− yolk sacs. The Siva−/− yolk sac shown in (e) is the same as shown in (a). f–h Cross-sections of CD31-immunostained yolk sacs shown in (c–e). i–k H&E staining of CD31-immunostained yolk sacs. (c–k) Arrows (black) indicate large vitelline vessels in wild-type yolk sac. Arrowheads (red) indicate small capillaries. l–n Higher magnification of H&E-stained yolk sacs. Arrowheads indicate mesoderm cells. Note the loosely attached mesoderm cells in the Siva−/− yolk sac shown in (n) and the apparent absence of mesoderm cells in the Siva−/− yolk sac shown in (m). Asterisks denote endothelial cell-lined blood vessels. o–q Picrosirius Red staining of yolk sac collagen (arrowheads) reveals a disruption in extracellular matrix production in the absence of Siva. end, endoderm; mes, mesoderm
Loss of Siva induces embryonic lethality
To define the physiological role of Siva, Siva heterozygous mice from a mixed 129P2/OlaHsd and C57BL/6 J background were intercrossed. No viable Siva−/− mice were obtained at weaning or neonatally, suggesting that Siva deficiency causes embryonic lethality. To delineate the stage of development at which Siva loss induces lethality, we examined embryos at embryonic days (e) 8.5 through 13.5. As early as e8.5, the number of Siva−/− embryos is lower than the expected Mendelian ratio (Table 1). Similarly, Siva−/− embryos are underrepresented at all stages through e11.5, and no Siva−/− embryos are observed at later stages. At e11.5, approximately half of the Siva−/− embryos isolated are dead, as determined by lack of heartbeat, or are in the process of resorption. Thus, Siva deficiency results in embryonic lethality between e8.5 and e12.5. The lethality associated with Siva loss was confirmed in a second Siva knockout model generated by Cre-mediated deletion of a previously described conditional Siva allele (data not shown) [19].
Table 1.
Siva deficiency induces mid-gestational embryonic lethality
| Age | Litters | Conceptuses | Genotype: actual (expected) | P value | |||
|---|---|---|---|---|---|---|---|
| Siva+/+ | Siva+/− | Siva−/− | ND | ||||
| e8-8.5 | 12 | 92 | 22 (23) | 46 (46) | 11 (23) | 13 | P < 0.05 |
| e9-9.5 | 32 | 277 | 69 (69.25) | 135 (138.5) | 28 (69.25) | 44 | P < 0.0001 |
| e10-10.5 | 36 | 298 | 97 (74.5) | 127 (149) | 35 (74.5) | 39 | P < 0.0001 |
| e11-11.5 | 15 | 120 | 32 (30) | 57 (60) | 9 (30) | 16 | P < 0.001 |
| e12.5+ | 6 | 42 | 14 (10.5) | 20 (21) | 0 (10.5) | 8 | P < 0.005 |
| P21 | 53 | 334 | 100 (83.5) | 234 (167) | 0 (83.5) | N/A | P < 0.0001 |
The actual (and expected) number of embryos and pups recovered from Siva-heterozygous intercrosses at the indicated embryonic (e) and postnatal (P) ages. ND includes decidua from which embryos could not be isolated and embryos for which genotype could not be determined. P values denote significant loss of Siva−/− embryos as determined by Χ2 test
Siva is expressed in embryonic and extra-embryonic tissues
To uncover the basis for the lethality observed in the absence of Siva, we examined the expression pattern of Siva during early- to mid-gestation embryonic development, using the β-galactosidase (β-gal) reporter driven by the Siva promoter as a marker of Siva expression. During gastrulation (e7–e7.5), we find β-gal staining in heterozygous embryos within the ectoderm and mesoderm, but not in the endoderm (Fig. 2a, b). We also observe expression in extra-embryonic tissues, including the yolk sac and allantois. During early organogenesis (e8.5–e11.5), the strongest Siva expression is observed in the developing heart, with ongoing expression in the allantois and umbilical cord – a derivative of the allantois – and lesser expression in the forebrain (Fig. 2c–f). Interestingly, the Siva expression pattern, as determined by β-gal staining of e11.5 Siva heterozygous embryos, is independent of p53 status (Supplementary Fig. 1). Because Siva has also been shown to be regulated by the p53-related transcription factor p73 in cultured cells [20], we also examined whether Siva expression is dependent on the p53 family members p73 or p63 during embryogenesis (Supplementary Fig. 1). We found that the Siva expression pattern does not change when p63 or p73 are deleted, or even when p53 and p63 are both deleted, suggesting that Siva expression is not controlled by the p53 family during development. Together, these data show that Siva is expressed in multiple embryonic and extra-embryonic regions that are critical for proper morphogenesis and survival, and that embryonic expression is driven independently of p53 family members.
Fig. 2.
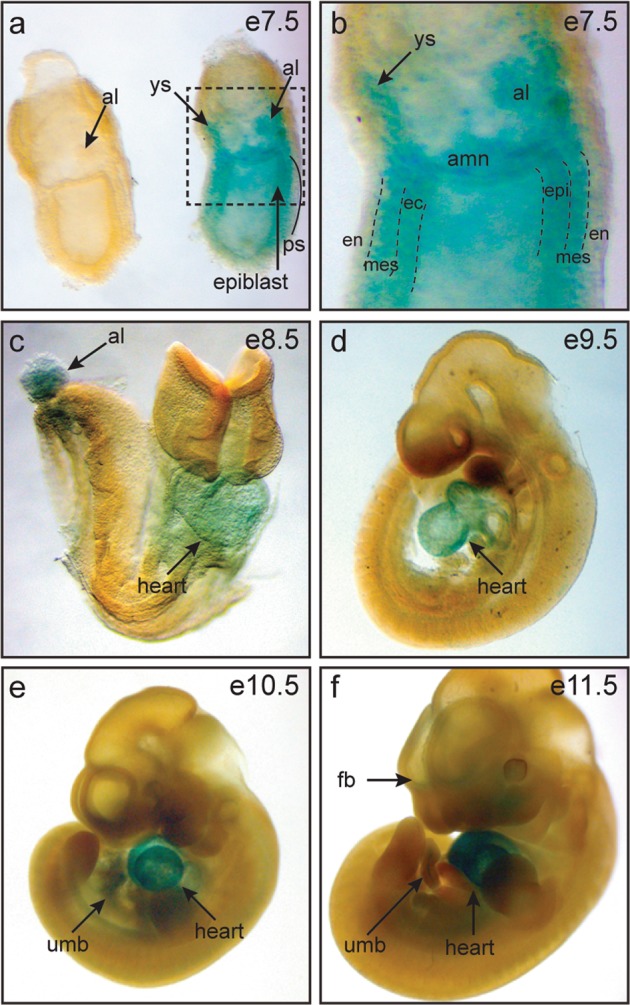
Siva is expressed in embryonic and extra-embryonic tissues. a, b Whole-mount β-galactosidase staining of wild-type and Siva-heterozygous embryos at embryonic day e7.5 reveals Siva expression in the primitive streak, allantois, and yolk sac. b Higher magnification of box in (a) showing β-galactosidase expression in the primitive streak. (c–f) Whole-mount β-galactosidase staining of embryos aged between e8.5 and e11.5 reveals continuing expression of Siva in the allantois and umbilical cord as well as in the heart and forebrain. al allantois, amn amnion, en endoderm, epi epiblast, fb forebrain, mes mesoderm, ec ectoderm, ps primitive streak, ys yolk sac, umb umbilical cord
Siva deficiency results in developmental delay and neural tube closure defects
Morphological examination of embryos between e8.5 and e11.5 revealed that Siva deficiency results in a variety of developmental defects. At e8.5, Siva−/− embryos have yet to undergo somitogenesis, while their wild-type littermates have 8–12 somites already formed (Fig. 3a). At e9.5, Siva-null embryos continue to display developmental delay, resembling wild-type e8.5 embryos in size and developmental features: they are unturned, have 8–12 somites, and have not yet completed neural tube closure or begun limb development (Fig. 3b, c, f, g). This apparent 24-hour developmental delay continues to be a prominent feature of Siva deficiency in embryos between e9.5 and e11.5 (95%; 54/57 embryos; Fig. 3d, e, h–k).
Fig. 3.

Siva deficiency results in developmental delay and neurulation defects. a–e Lateral view of morphology of wild-type and Siva−/− embryos from e8.5 through e11.5 reveals a developmental delay in the absence of Siva. Note that Siva−/− embryos are smaller and less developed than their littermates at all ages examined. Examination of limb development (arrows) demonstrates that Siva−/− embryos are developmentally delayed by ~1 day. In addition, quantitation of somite number in e9.5 Siva−/− embryos suggests a 1 day developmental delay. In (c), a side-by-side comparison of an e8.5 wild-type embryo with an e9.5 Siva−/− embryo demonstrates the similarity in size and developmental features, including the extent of neural tube closure, somite number, turning stage, and heart tube development. 54/57 e9.5–e11.5 Siva−/− embryos exhibited developmental delay. f–k Morphological examination of neurulation defects (arrows) in Siva−/− embryos. Ventral and dorsal views of wild-type and Siva−/− embryos at e9.5 (f, g), e10.5 (h, i), and e11.5 (j, k). At all ages examined, Siva−/− embryos exhibit a failure to complete cranial neural tube closure (NT), kinked spines, and abnormally compacted hindbrains (HB)
In addition to overall developmental delay, Siva-deficient embryos have defects in nervous system development, most notably exhibiting exencephaly, a condition in which the cranial neural folds are persistently open. By e10.5, 94% of Siva−/− embryos (48/51) fail to complete neural tube closure (Fig. 3d, h), although apposition of the neural folds does ultimately occur in these embryos (Fig. 3i, inset). Loss of Siva also induces defects in spinal cord neurulation, resulting in kinked spines. Although the vast majority of e9.5–e11.5 embryos (97%; 43/44) show some phenotype (Fig. 3g, i, k), the severity of the spinal kinks is variable, ranging from mild to severe. Furthermore, the hindbrain roof plate appears smaller and more compact in e10.5 and e11.5 Siva-null embryos than in controls (Fig. 3i, k). Together, these findings suggest an important role for Siva in proper nervous system development. Although Siva-null embryos display neurological phenotypes, this is not a likely cause of lethality as mouse embryos do not typically die in utero from abnormal brain development [21].
Siva deficiency does not dramatically affect heart development or the embryonic vasculature
As congenital defects in the cardiovascular system are detected in nearly half of the embryonic lethal mouse knockouts [22] and whole-mount expression analysis revealed high levels of Siva in the embryonic heart beginning at e8.5 (Fig. 2c), we sought to establish whether Siva-null embryos exhibit a defect in heart development. Whole-mount examination of e9.5 and e10.5 Siva−/− embryos reveals that the initial steps of heart tube formation and looping occur normally in Siva−/− embryos, albeit with delayed kinetics consistent with the observed developmental delay (Fig. 3b, d). Furthermore, Siva deficiency does not affect the onset of myocardial contractility, as we routinely observed heartbeats in Siva−/− embryos up to e10.5. By histological analysis, major hallmarks of heart development are evident in e10.5 Siva−/− embryos, including myocardial trabeculation, epithelial to mesenchymal transition to form endocardial cushions, the appearance of mesenchymal cells in the outflow tract, and development of the epicardium (Supplementary Fig. 2). The main phenotype observed is that heart development in e10.5 Siva–/– embryos is developmentally delayed relative to wild-type littermates. For example, while the intraventricular septum is beginning to form e10.5 wild-type embryos, it is not yet present in e10.5 Siva–/– embryos (Supplementary Fig. 2). Together, these data suggest that heart development is delayed but not dramatically disrupted by Siva deficiency and that defects in heart development are unlikely to be the cause of lethality of Siva–/– embryos.
As disruption of embryonic vasculature formation can also lead to growth retardation and lethality, we examined the early embryonic vasculature using a marker of endothelial cells, CD31, by whole-mount immunohistochemistry. Visual inspection of the initial formation of the embryonic vascular system, in particular the dorsal aorta, by whole-mount analysis of stage-matched e8.5 wild-type and e9.5 Siva−/− embryos, suggests no obvious differences with Siva loss (Supplementary Fig. 3a). Similarly, the vascular network of e10.5 Siva−/− embryos appears well developed and grossly normal (Supplementary Fig. 3b–d), suggesting the observed developmental phenotypes and lethality are not due to a failure in embryonic vasculature formation.
Siva is required for vascularization of the placental labyrinth
We next sought to ascertain whether defects in extra-embryonic structures might explain the phenotypes associated with Siva deficiency [23]. The placenta is a highly-vascularized extra-embryonic tissue that functions as a key site of nutrient and gas exchange between mother and embryo, and disruption of placental development results in mid-gestation embryonic growth retardation and lethality around e10.5 [23, 24]. Vascularization of the placenta begins when the allantoic mesoderm, a site of Siva expression (Fig. 2a, c), fuses with the ectodermal chorionic plate around e8.5. Although chorioallantoic fusion is detected in most Siva−/− embryos, it does not occur until at least e9.5, which is consistent with the observed developmental delay phenotypes. Importantly, while fusion occurs by e10.5 in the majority of Siva null embryos, the attachment is notably weak and easily disrupted, in contrast to the strong connection that develops in wild-type and heterozygous embryos (data not shown). Additionally, in three e10.5 Siva−/− embryos, concomitant with a failure to fuse to the chorion, the allantois formed a bulbous sac attached to the embryonic gut in place of an umbilical cord (Fig. 4a, b).
Fig. 4.

Siva is required for chorioallantoic fusion and placental labyrinth formation. a Whole-mount image of e10.5 Siva−/− embryo in which chorioallantoic fusion failed to occur. The allantois (arrow) can be seen as a sac protruding from the embryonic gut. b H&E staining of embryo in (a), with arrow indicating allantois. c–e Upper panels: Histological analysis of placentas from e9.5 and e10.5 embryos reveals defective labyrinth formation in the absence of Siva. H&E staining of placentas, in which chorioallantoic attachment occurred, shows that the Siva−/− allantois attaches but fails to penetrate the chorion. Lower panels: Higher magnification of boxed area of labyrinth layer. Embryonic and maternal blood vessels can be identified by the presence of nucleated erythroblasts (yellow arrowheads) and enucleated red blood cells (black arrows), respectively. Note the absence of embryonic blood vessels in the labyrinth from Siva−/− placentas (d). de decidua, gi trophoblast giant cells, sp spongiotrophoblast, lb labyrinth, ch chorion
The placental labyrinth consists of closely intermingled maternal and embryonic blood vessels, and results from penetration of embryonic blood vessels from the allantois into the chorion, followed by branching morphogenesis of the vasculature [23]. We examined the architecture of placentas associated with e10.5 Siva−/− embryos in which chorioallantoic fusion had occurred. Both wild-type and Siva-deficient placentas have identifiable trophoblast giant cell (gi), spongiotrophoblast (sp), labyrinth (lb), and chorion (ch) layers (Fig. 4c–e). However, whereas wild-type or Siva+/− placentas have a dense vascular network comprising both maternal and embryonic blood vessels, 5 of the 8 Siva−/− placentas analyzed at e10.5 have no embryonic blood vessels present in the labyrinth, as determined by the lack of nucleated erythroblasts (Fig. 4c–e). Notably, several of the Siva−/− placentas display large embryonic blood vessels at the interface between the chorion and allantois (Fig. 4d, asterisk), indicating that although chorioallantoic attachment had occurred, the embryonic vessels failed to penetrate the chorion and undergo branching morphogenesis. The absence of proper labyrinth formation in Siva−/− placentas is not attributable to the developmental delay since wild-type placentas from e9.5 embryos have begun branching morphogenesis (Fig. 4e). These findings suggest that Siva deficiency perturbs normal placental labyrinth formation and establishment of a functional umbilical circulation.
Siva-deficient yolk sacs exhibit disrupted vascular remodeling and mesoderm function
As with the placenta, the yolk sac is an extra-embryonic structure in which proper vascularization is essential for supporting embryonic growth and survival, and failure to form and maintain a functioning yolk sac circulation results in embryonic lethality [25, 26]. In contrast to wild-type and Siva-heterozygous yolk sacs, which have a smooth appearance and in which blood vessels are readily visible, yolk sacs from Siva-deficient embryos are abnormal, often having a pale and wrinkled appearance (Fig. 5a). We examined yolk sac vasculature by whole-mount CD31 immunohistochemistry (Fig. 5c–h). Unlike wild-type yolk sacs, which have undergone extensive remodeling to form a highly organized vascular network consisting of both large vitelline vessels and small capillaries, the e10.5 Siva−/− yolk sac vasculature retains a honeycomb-like appearance and lacks large vitelline vessels, phenotypes consistent with a failure to undergo angiogenic remodeling [26]. Additionally, we noted a relationship between the severity of the yolk sac vascular defect and the severity of embryonic phenotypes, as illustrated by the dramatically affected embryo and yolk sac in Fig. 5a, e.
To gain insight into the underlying cause of the abnormal yolk sac vasculature, we examined yolk sac structure by histology. The yolk sac is a bilaminar membrane consisting of an outer visceral endoderm and an inner mesoderm surrounding endothelial-cell-lined blood vessels. In contrast to the tightly attached mesodermal layer in wild-type yolk sac, the mesoderm in the Siva-null yolk sacs is poorly attached to the visceral endoderm layer (Fig. 5i–n), and, in some instances, completely absent (not shown). As the yolk sac mesoderm is responsible for production of extracellular matrix (ECM), we examined ECM deposition in Siva−/− yolk sacs by staining for collagen with picrosirius red (Fig. 5o–q). Indeed, we observed a reduction in collagen fibers in the absence of Siva. Vascular defects such as those observed in the Siva-null placenta and yolk sac would constrain embryonic growth and development by not allowing for sufficient nutrient and gas exchange. Consistent with this notion, Siva−/− embryos express high levels of the hypoxia marker Slc2a1 (Fig. 5b), implicating the lack of proper extra-embryonic vasculature as a contributor to embryonic lethality [27].
Phenotypes resulting from Siva deficiency are p53 independent
We next sought to determine if the role of Siva in development relates to its function in the p53 pathway. Although our expression analyses suggest that the developmental role of Siva does not reflect it being a p53 target gene (Supplementary Fig. 1), it remained a possibility that Siva acts upstream of p53. Specifically, previous studies have suggested that Siva can negatively regulate p53 both by stabilizing the interaction of the Mdm2 ubiquitin ligase with p53 to promote p53 degradation and by promoting degradation of the p53 activator, p19Arf [28, 29]. Thus, Siva loss could result in p53 hyperactivation, which is a known cause of early embryonic lethality [30–32]. We interrogated whether Siva deficiency induces embryonic lethality through activation of p53 by examining Siva−/−;p53−/− compound mutant embryos. Loss of p53 does not rescue any of the phenotypes associated with Siva deficiency, including the lethality, developmental delay, exencephaly, and yolk sac vasculature defects (Table 2, Supplementary Fig. 4). These observations suggest that Siva deficiency does not trigger phenotypes by activating the p53 pathway.
Table 2.
Loss of p53 does not rescue lethality induced by Siva deficiency
| Siva | +/+ | +/+ | +/+ | +/− | +/− | +/− | −/− | −/− | −/− | ND | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| p53 | +/+ | +/− | −/− | +/+ | +/− | −/− | +/+ | +/− | −/− | ||||
| Age | Litters | Total | P value | ||||||||||
| e10.5 | 10 | 84 | 6 (5.25) | 12 (10.5) | 8 (5.25) | 14 (10.5) | 25 (21) | 11 (10.5) | 1 (5.25) | 3 (10.5) | 0 (5.25) | 6 | <2 × 10−3 |
| P1.5 | 10 | 71 | 7 (4.4375) | 11 (8.875) | 4 (4.4375) | 10 (8.875) | 24 (17.75) | 13 (8.875) | 0 (4.4375) | 0 (8.875) | 0 (4.4375) | 2 | <1 × 10−5 |
| P21 | 9 | 54 | 5 (3.375) | 7 (6.75) | 4 (3.375) | 9 (6.75) | 20 (13.5) | 9 (6.75) | 0 (3.375) | 0 (6.75) | 0 (3.375) | NA | <2 × 10−5 |
The actual (and expected) numbers of embryos and pups recovered from Siva;p53-heterozygous intercrosses at the indicated embryonic (e) and postnatal (P) ages. ND includes decidua from which embryos could not be isolated and embryos for which genotype could not be determined. P values denote significant loss of Siva−/−;p53−/− embryos and pups as determined by Χ2 test
Siva−/− embryonic phenotypes are not rescued by loss of necroptosis factor Ripk3
Intriguingly, mice deficient for the extrinsic cell death pathway components Caspase-8 or Fadd display embryonic lethality with phenotypes similar to those observed with Siva deficiency, including developmental delay, yolk sac vasculature defects, and neural tube defects [33, 34]. Notably, loss of the necroptotic factors Ripk1 or Ripk3 rescues the embryonic lethality of Caspase-8 and Fadd knockout mice by preventing necroptosis, a form of cell death that is specifically coordinated by Ripk proteins upon deficiency of extrinsic apoptotic signaling pathways [35–37]. As Siva can act in the extrinsic cell death pathway, we investigated whether the Siva-deficient embryonic phenotypes and lethality are similarly due to upregulation of necroptosis. We crossed Siva knockout mice to Ripk3 knockout mice and assessed mouse viability at postnatal day 21 and embryo viability at e10.5. No viable Siva;Ripk3 double-knockout mice are observed at P21, indicating that Ripk3 loss is not able to rescue the lethality associated with Siva deficiency (Table 3). Furthermore, at e10.5, Siva;Ripk3 double-knockout embryos show developmental delay and display exencephaly and kinked spine phenotypes similar to those observed with Siva loss (Supplementary Fig. 5). Together, these observations indicate that Siva deficiency does not provoke lethality through activation of necroptosis pathways.
Table 3.
Rip3 loss does not rescue lethality induced by Siva deficiency
| Siva | +/+ | +/+ | +/+ | +/− | +/− | +/− | −/− | −/− | −/− | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Rip3 | +/+ | +/− | −/− | +/+ | +/− | −/− | +/+ | +/− | −/− | |||
| Age | Litters | Total | P value | |||||||||
| e10.5 | 9 | 62 | 5 (3.875) | 15 (7.75) | 7 (3.875) | 9 (7.75) | 15 (15.5) | 8 (7.75) | 1 (3.875) | 1 (7.75) | 1 (3.875) | <0.01 |
| P21 | 15 | 92 | 6 (5.75) | 14 (11.5) | 11 (5.75) | 17 (11.5) | 31 (23) | 13 (11.5) | 0 (5.75) | 0 (23) | 0 (5.75) | <4× 10−5 |
The actual (and expected) numbers of embryos and pups recovered from Siva;Rip3-heterozygous intercrosses at embryonic (e) day 10.5 and postnatal (P) day 21. P values denote significant loss of Siva−/−;Rip3−/− embryos and pups as determined by Chi-square test
Discussion
Here we examine the physiological function of Siva in vivo by generating and analyzing Siva knockout mice. Siva deficiency induces mid-gestational embryonic lethality, associated with a variety of developmental abnormalities, including defects in extra-embryonic tissues, improper neural tube closure and developmental delay. Whereas the exencephaly and kinked spines reveal a key role for Siva in nervous system development, the observed developmental delay, growth retardation and lethality indicate essential functions for Siva in embryonic growth and survival.
Developmental defects that lead to embryonic lethality generally compromise either the cardiovascular system of the embryo proper or the ability of the embryo to carry out nutrient and gas exchange with the mother through the yolk sac and placenta [38, 39]. The yolk sac is the first site of vasculogenesis and hematopoiesis, providing nourishment by acting as a circulatory system before the intra-embryonic circulation begins [40–42]. When the allantois fuses with the chorionic plate at e8.5, embryonic blood vessels undergo a branching morphogenesis program to form a labyrinth, which allows maternal-fetal gas exchange. Notably, abnormal vascularization and labyrinth formation in the placenta commonly triggers growth retardation and lethality at ~e10.5 [23]. The specific vascular problems identified in the yolk sac and placenta of Siva-null embryos suggest that these defects are primary contributors to the lethality induced by Siva loss. Indeed, we detect Siva expression in the mesodermal components of the yolk sac and the allantois [43]. In mouse models, inactivating mutations in genes important for mesoderm development and function – such as Brachyury, TGFβ signaling molecules (e.g., both Bmp5 and Bmp7 or Tak1), or extracellular matrix components and adhesion molecules (e.g., Fibronectin, α5 integrin) – result in embryonic lethality associated with a variety of developmental phenotypes similar to those observed in Siva-null embryos, including developmental delay, kinked spines, and defective yolk sac vascularization, chorioallantoic fusion, and neural tube closure [44, 45]. Interestingly, Siva has been previously reported to interact with TAK1, a key component of TGFβ-induced, SMAD-independent signaling to NFκB, p38, and JNK [5], suggesting that these components may act in the same pathway during embryonic development.
A major unanswered question – and important area of investigation for future studies – is the cellular function of Siva important for its role in development. Programmed cell death has essential roles in many developmental processes, including neural tube closure, remodeling of blood vessels, and sculpting of the heart and limbs [46–48]. It is unclear, however, whether the Siva-knockout phenotypes reflect loss of its capacity as a pro-apoptotic protein. Knockout mice lacking genes encoding key pro-apoptotic proteins – such as Apaf-1, Caspase-3, and Caspase-9 – do not exhibit developmental delay, kinked spines, and early-mid-gestational embryonic lethality as is observed with loss of Siva [49–51]. A notable exception to this are Caspase-8 knockout mice, which display mid-gestational embryonic lethality accompanied by vascular defects in the yolk sac, cardiac malformations, and kinked neural tubes [33, 52]. Interestingly, the neural tube and heart defects are secondary to Caspase-8 deficiency in the yolk sac, implicating yolk sac vasculature defects as the primary event leading to embryonic lethality [33]. Further investigation into the mechanism established that activation of Ripk3-dependent necroptosis, an alternate cell death pathway, contributes to the embryonic lethality in Caspase-8-deficient mice [35]. Although the Caspase-8-mediated, death receptor pathway has been implicated in Siva apoptotic function [14], our observation that Siva-null phenotypes are not rescued by Ripk3 deficiency suggests that Siva function during embryonic development may rely on an alternate pathway – potentially a non-apoptotic one. Indeed, Caspase-8 is proposed to promote proper development not only through apoptosis-dependent but also through apoptosis-independent functions [53]. Thus, Siva deficiency may promote defects in the placenta and yolk sac through non-apoptotic cellular mechanisms.
Beyond its role in apoptosis, Siva has been reported to regulate various other cellular pathways and processes. For example, Siva can: suppress migration and invasion through phosphorylation of STATHMIN [54]; enhance mTorc1 activity, inhibit autophagy, and stimulate mitochondrial output to promote proliferation [19]; and translocate into the nucleus to interact with PCNA and regulate translesion repair through RAD18-mediated monoubiquitination of PCNA [55]. However, in contrast to the embryonic lethality associated with Siva inactivation, Stathmin deficiency does not cause embryonic lethality [56] and loss of mTor, Raptor, or Pcna induces pre-implantation lethality [57, 58]. Notably, loss of either Lst8 or Rictor – both of which encode mTorc2 complex members – results in lethality at e10.5 with yolk sac and embryonic vasculature defects due to impaired Akt-Foxo signaling [57]. This unexplored potential link between Siva and mTorc2 during embryonic development appears distinct from Siva’s previously reported role in mTorc1 signaling during tumorigenesis [19], but may represent an additional connection between Siva and mTor signaling. Thus, despite Siva’s reported actions in various signaling pathways, the mechanisms through which Siva loss results in embryonic lethality remain unclear and may be through an as-yet-unexplored pathway in which Siva functions. Interestingly, Siva deficiency impedes lung cancer development, irrespective of p53 status, suggesting a p53-independent role for Siva in enabling tumorigenesis [19]. As the established roles of Siva are becoming increasingly complex, it will be key to elucidate which cellular functions of Siva are important for each of its essential physiological functions, including during embryonic development. Many of the signaling pathways essential for developmental processes during embryogenesis also have important functions during tumorigenesis [59]. Therefore, further investigation into the p53-independent role and mechanism of action of Siva during embryonic development may also illuminate roles for Siva in tumorigenesis.
Materials and methods
Generation of Siva Gene-Trapped Mice
We obtained the RRR467 gene-trapped ES cell line, which was derived on a 129P2/OlaHsd background, from Baygenomics [18]. This ES cell line was generated by insertional mutation of the pGT0lxf gene trap vector, which comprises intron 1 sequences and a splice acceptor from the mouse Engrailed 2 gene followed by β-galactosidase-neomycin (β-geo) fusion-encoding sequences and an SV40 polyadenylation transcriptional stop element, into the Siva locus. ES cells were cultured using standard methods. Chimeric mice were generated by blastocyst injection into pseudopregnant C57BL/6 mothers by the transgenic facility at Stanford University. Chimeras were backcrossed onto a C57BL/6 J background and mice genotyped by PCR analysis of tail or yolk sac DNA. The primers used include a forward primer in exon 1 (5′-CGCTGCTGAAGGCTGTGTCTGG-3′), a wild-type reverse primer (5′-CCATCACTGCAAGGCAATGCTC-3′), and a gene-trap reverse primer (5′-CTTATCCACAACCAACGCAC-3′). All mice studied were on a mixed 129P2/OlaHsd and C57BL/6 J background.
Embryo isolation and analysis
Siva-heterozygous mice were intercrossed and timed matings were performed. Females were checked for vaginal plugs in the morning and the day of plug was counted as embryonic day 0.5. Embryos were isolated, and yolk sacs, placentas, and allantois/umbilical cords were examined upon dissection. A small piece of yolk sac or embryo was used for PCR-based genotyping. Age of embryos in each litter was confirmed at time of dissection by examination of general morphology. For better visualization of embryonic morphology in Fig. 3, embryos were soaked briefly in methanol/PBS containing ethidium bromide. Embryos were photographed under ultraviolet light, and images were then converted to grayscale.
Northern Blot Analysis
RNA was prepared from embryos using Trizol reagent (Invitrogen). Northern blotting was performed using standard methods [27]. cDNAs for murine Siva and Gapdh and human SLC2A1 served as Northern blot probes [27].
LacZ staining
For all LacZ staining, embryos were isolated from matings of Siva-heterozygous males crossed to wild-type females to eliminate maternal background. Whole-mount LacZ staining of embryos was done according to standard protocols. Briefly, embryos were fixed in 0.1 M phosphate buffer with 0.2% glutaraldehyde, 2 mM MgCl2, 5 mM EGTA for 5–15 min and then washed with Wash Buffer (0.1 M phosphate buffer pH 7.3, 2 mM MgCl2, 0.01% sodium deoxycholate, 0.02% NP-40). Embryos were stained in X-gal staining solution (wash buffer supplemented with 5 mM potassium ferrocyanide, 5 mM potassium ferricyanide, and 1 mg/ml 5-bromo-4-chloro-3-indolyl-β-D-galactoside) for 24–48 h at 37 °C. Embryos were washed and stored at 4 °C to allow stain to intensify prior to post-fixing with formalin overnight. Embryos were cleared into 75% glycerol prior to photographing. Wild-type embryos were used as negative controls in all experiments.
Histology & Immunohistochemistry
For histological analyses, embryos, placentas, and yolk sacs were fixed in formalin (3.7% formaldehyde) overnight and then embedded in paraffin. Hematoxylin and eosin (H&E) staining was performed by the Pathology core at Stanford University School of Medicine. For CD31 whole-mount immunohistochemistry (IHC), embryos and yolk sacs were fixed in 4% PFA at 4 °C for 30 min to overnight and then dehydrated into methanol. IHC was performed essentially as described previously [60]. Briefly, endogenous peroxidase activity was quenched with 5% H202 for 5 h and then tissue was rehydrated in PBS. Tissue was blocked in PBS containing 3% milk and 0.1% Triton X-100 (PBSMT) for 2 h prior to overnight incubation with α-CD31 (BD Pharmingen, MEC 13.3 clone, 1:50) at 4 °C. Following 5 h of washing in PBSMT, tissue was incubated with HRP-conjugated secondary antibody (Jackson, 1:200) overnight at 4 °C. After washing, peroxidase was developed using DAB (Vector). Tissue was post-fixed with 2% PFA/0.1% glutaraldehyde prior to clearing into 75% glycerol for photographing. Following photography, whole-mount stained tissues were returned to PBS and then processed for paraffin embedding. Sections of whole-mount IHC stained tissues were examined for CD31 IHC or stained for H&E or collagen expression. For Picrosirius Red staining, deparaffinized tissue was incubated for 30 s in Mayer’s Hematoxylin and then washed under running water for 30 s prior to staining in Picrosirius Red solution for 30 min. Tissue was then washed with acidified water, dehydrated, and mounted.
Supplementary information
Acknowledgements
We are grateful to Ching-Pin Chang, Kryn Stanukas, and Jill Helms for helpful comments. We thank Jacqueline Benjamin, Tiara Kawahara, Samantha Brugmann, Elora Majumder, and Sean Massa for technical assistance.
Funding
This work was supported by the Giannini Family Foundation to SBRJ, the NSF and NIH (1F31CA167917-01) to JLVN, and the Damon Runyon Cancer Research Foundation, the Donald E. and Delia B. Baxter Foundation, and the NIH (5R03NS05618002 and R35 CA197591) to LDA.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Edited by G. Melino
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version of this article (10.1038/s41418-019-0358-x) contains supplementary material, which is available to authorized users.
References
- 1.Prasad KV, Ao Z, Yoon Y, Wu MX, Rizk M, Jacquot S, et al. CD27, a member of the tumor necrosis factor receptor family, induces apoptosis and binds to Siva, a proapoptotic protein. Proc Natl Acad Sci USA. 1997;94:6346–51. doi: 10.1073/pnas.94.12.6346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Spinicelli S, Nocentini G, Ronchetti S, Krausz LT, Bianchini R, Riccardi C. GITR interacts with the pro-apoptotic protein Siva and induces apoptosis. Cell Death Differ. 2002;9:1382–4. doi: 10.1038/sj.cdd.4401140. [DOI] [PubMed] [Google Scholar]
- 3.Gudi R, Barkinge J, Hawkins S, Chu F, Manicassamy S, Sun Z, et al. Siva-1 negatively regulates NF-kappaB activity: effect on T-cell receptor-mediated activation-induced cell death (AICD) Oncogene. 2006;25:3458–62. doi: 10.1038/sj.onc.1209381. [DOI] [PubMed] [Google Scholar]
- 4.Gudi R, Barkinge J, Hawkins S, Prabhakar B, Kanteti P. Siva-1 promotes K-48 polyubiquitination of TRAF2 and inhibits TCR-mediated activation of NF-kappaB. J Environ Pathol Toxicol Oncol. 2009;28:25–38. doi: 10.1615/jenvironpatholtoxicoloncol.v28.i1.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Resch U, Schichl YM, Winsauer G, Gudi R, Prasad K, de Martin R. Siva1 is a XIAP-interacting protein that balances NFkappaB and JNK signalling to promote apoptosis. J Cell Sci. 2009;122(Pt 15):2651–61. doi: 10.1242/jcs.049940. [DOI] [PubMed] [Google Scholar]
- 6.Chu F, Borthakur A, Sun X, Barkinge J, Gudi R, Hawkins S, et al. The Siva-1 putative amphipathic helical region (SAH) is sufficient to bind to BCL-XL and sensitize cells to UV radiation induced apoptosis. Apoptosis. 2004;9:83–95. doi: 10.1023/B:APPT.0000012125.01799.4c. [DOI] [PubMed] [Google Scholar]
- 7.Xue L, Chu F, Cheng Y, Sun X, Borthakur A, Ramarao M, et al. Siva-1 binds to and inhibits BCL-X(L)-mediated protection against UV radiation-induced apoptosis. Proc Natl Acad Sci USA. 2002;99:6925–30. doi: 10.1073/pnas.102182299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zins K, Pomyje J, Hofer E, Abraham D, Lucas T, Aharinejad S. Egr-1 upregulates Siva-1 expression and induces cardiac fibroblast apoptosis. Int J Mol Sci. 2014;15:1538–53. doi: 10.3390/ijms15011538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cao C, Ren X, Kharbanda S, Koleske AJ, Prasad KV, Kufe D. The ARG tyrosine kinase interacts with Siva-1 in the apoptotic response to oxidative stress. J Biol Chem. 2001;276:11465–8. doi: 10.1074/jbc.C100050200. [DOI] [PubMed] [Google Scholar]
- 10.Py B, Bouchet J, Jacquot G, Sol-Foulon N, Basmaciogullari S, Schwartz O, et al. The Siva protein is a novel intracellular ligand of the CD4 receptor that promotes HIV-1 envelope-induced apoptosis in T-lymphoid cells. Apoptosis. 2007;12:1879–92. doi: 10.1007/s10495-007-0106-4. [DOI] [PubMed] [Google Scholar]
- 11.Shiozaki T, Iwai A, Kawaoka Y, Takada A, Kida H, Miyazaki T. Requirement for Siva-1 for replication of influenza A virus through apoptosis induction. J Gen Virol. 2011;92(Pt 2):315–25. doi: 10.1099/vir.0.028316-0. [DOI] [PubMed] [Google Scholar]
- 12.Singaravelu K, Padanilam BJ. p53 target Siva regulates apoptosis in ischemic kidneys. Am J Physiol Ren Physiol. 2011;300:F1130–41. doi: 10.1152/ajprenal.00591.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fortin A, MacLaurin JG, Arbour N, Cregan SP, Kushwaha N, Callaghan SM, et al. The proapoptotic gene SIVA is a direct transcriptional target for the tumor suppressors p53 and E2F1. J Biol Chem. 2004;279:28706–14. doi: 10.1074/jbc.M400376200. [DOI] [PubMed] [Google Scholar]
- 14.Jacobs SB, Basak S, Murray JI, Pathak N, Attardi LD. Siva is an apoptosis-selective p53 target gene important for neuronal cell death. Cell Death Differ. 2007;14:1374–85. doi: 10.1038/sj.cdd.4402128. [DOI] [PubMed] [Google Scholar]
- 15.Bieging KT, Mello SS, Attardi LD. Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev Cancer. 2014;14:359–70. doi: 10.1038/nrc3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mello SS, Attardi LD. Deciphering p53 signaling in tumor suppression. Curr Opin Cell Biol. 2018;51:65–72. doi: 10.1016/j.ceb.2017.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Py B, Slomianny C, Auberger P, Petit PX, Benichou S. Siva-1 and an alternative splice form lacking the death domain, Siva-2, similarly induce apoptosis in T lymphocytes via a caspase-dependent mitochondrial pathway. J Immunol. 2004;172:4008–17. doi: 10.4049/jimmunol.172.7.4008. [DOI] [PubMed] [Google Scholar]
- 18.Stryke D, Kawamoto M, Huang CC, Johns SJ, King LA, Harper CA, et al. BayGenomics: a resource of insertional mutations in mouse embryonic stem cells. Nucleic Acids Res. 2003;31:278–81. doi: 10.1093/nar/gkg064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Van Nostrand JL, Brisac A, Mello SS, Jacobs SB, Luong R, Attardi LD. The p53 target gene siva enables non-small cell lung cancer development. Cancer Discov. 2015;5:622–35. doi: 10.1158/2159-8290.CD-14-0921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ray RM, Bhattacharya S, Johnson LR. Mdm2 inhibition induces apoptosis in p53 deficient human colon cancer cells by activating p73- and E2F1-mediated expression of PUMA and Siva-1. Apoptosis. 2011;16:35–44. doi: 10.1007/s10495-010-0538-0. [DOI] [PubMed] [Google Scholar]
- 21.Copp AJ, Brook FA, Estibeiro JP, Shum AS, Cockroft DL. The embryonic development of mammalian neural tube defects. Prog Neurobiol. 1990;35:363–403. doi: 10.1016/0301-0082(90)90037-h. [DOI] [PubMed] [Google Scholar]
- 22.Conway SJ, Kruzynska-Frejtag A, Kneer PL, Machnicki M, Koushik SV. What cardiovascular defect does my prenatal mouse mutant have, and why? Genesis. 2003;35:1–21. doi: 10.1002/gene.10152. [DOI] [PubMed] [Google Scholar]
- 23.Perez-Garcia V, Fineberg E, Wilson R, Murray A, Mazzeo CI, Tudor C, et al. Placentation defects are highly prevalent in embryonic lethal mouse mutants. Nature. 2018;555:463–8. doi: 10.1038/nature26002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cross JC, Werb Z, Fisher SJ. Implantation and the placenta: key pieces of the development puzzle. Science. 1994;266:1508–18. doi: 10.1126/science.7985020. [DOI] [PubMed] [Google Scholar]
- 25.Conway EM, Collen D, Carmeliet P. Molecular mechanisms of blood vessel growth. Cardiovasc Res. 2001;49:507–21. doi: 10.1016/s0008-6363(00)00281-9. [DOI] [PubMed] [Google Scholar]
- 26.Garcia MD. Larina IV. Vascular development and hemodynamic force in the mouse yolk sac. Front Physiol. 2014;5:308. doi: 10.3389/fphys.2014.00308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnson TM, Hammond EM, Giaccia A, Attardi LD. The p53QS transactivation-deficient mutant shows stress-specific apoptotic activity and induces embryonic lethality. Nat Genet. 2005;37:145–52. doi: 10.1038/ng1498. [DOI] [PubMed] [Google Scholar]
- 28.Du W, Jiang P, Li N, Mei Y, Wang X, Wen L, et al. Suppression of p53 activity by Siva1. Cell Death Differ. 2009;16:1493–504. doi: 10.1038/cdd.2009.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang X, Zha M, Zhao X, Jiang P, Du W, Tam AY, et al. Siva1 inhibits p53 function by acting as an ARF E3 ubiquitin ligase. Nat Commun. 2013;4:1551. doi: 10.1038/ncomms2533. [DOI] [PubMed] [Google Scholar]
- 30.Jones SN, Roe AE, Donehower LA, Bradley A. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature. 1995;378:206–8. doi: 10.1038/378206a0. [DOI] [PubMed] [Google Scholar]
- 31.Montes de Oca Luna R, Wagner DS, Lozano G. Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature. 1995;378:203–6. doi: 10.1038/378203a0. [DOI] [PubMed] [Google Scholar]
- 32.Bowen Margot E, Attardi Laura D. The role of p53 in developmental syndromes. Journal of Molecular Cell Biology. 2019;11(3):200–211. doi: 10.1093/jmcb/mjy087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sakamaki K, Inoue T, Asano M, Sudo K, Kazama H, Sakagami J, et al. Ex vivo whole-embryo culture of caspase-8-deficient embryos normalize their aberrant phenotypes in the developing neural tube and heart. Cell Death Differ. 2002;9:1196–206. doi: 10.1038/sj.cdd.4401090. [DOI] [PubMed] [Google Scholar]
- 34.Yeh WC, de la Pompa JL, McCurrach ME, Shu HB, Elia AJ, Shahinian A, et al. FADD: essential for embryo development and signaling from some, but not all, inducers of apoptosis. Science. 1998;279:1954–8. doi: 10.1126/science.279.5358.1954. [DOI] [PubMed] [Google Scholar]
- 35.Kaiser WJ, Upton JW, Long AB, Livingston-Rosanoff D, Daley-Bauer LP, Hakem R, et al. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature. 2011;471:368–72. doi: 10.1038/nature09857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang H, Zhou X, McQuade T, Li J, Chan FK, Zhang J. Functional complementation between FADD and RIP1 in embryos and lymphocytes. Nature. 2011;471:373–6. doi: 10.1038/nature09878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dillon CP, Oberst A, Weinlich R, Janke LJ, Kang TB, Ben-Moshe T, et al. Survival function of the FADD-CASPASE-8-cFLIP(L) complex. Cell Rep. 2012;1:401–7. doi: 10.1016/j.celrep.2012.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Copp AJ. Death before birth: clues from gene knockouts and mutations. Trends Genet. 1995;11:87–93. doi: 10.1016/S0168-9525(00)89008-3. [DOI] [PubMed] [Google Scholar]
- 39.Poelmann RE, Molin D, Wisse LJ, Gittenberger-de Groot AC. Apoptosis in cardiac development. Cell Tissue Res. 2000;301:43–52. doi: 10.1007/s004410000227. [DOI] [PubMed] [Google Scholar]
- 40.Flamme I, Frolich T, Risau W. Molecular mechanisms of vasculogenesis and embryonic angiogenesis. J Cell Physiol. 1997;173:206–10. doi: 10.1002/(SICI)1097-4652(199711)173:2<206::AID-JCP22>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 41.Baron MH. Embryonic origins of mammalian hematopoiesis. Exp Hematol. 2003;31:1160–9. doi: 10.1016/j.exphem.2003.08.019. [DOI] [PubMed] [Google Scholar]
- 42.Eichmann A, Yuan L, Moyon D, Lenoble F, Pardanaud L, Breant C. Vascular development: from precursor cells to branched arterial and venous networks. Int J Dev Biol. 2005;49:259–67. doi: 10.1387/ijdb.041941ae. [DOI] [PubMed] [Google Scholar]
- 43.Arora R, Papaioannou VE. The murine allantois: a model system for the study of blood vessel formation. Blood. 2012;120:2562–72. doi: 10.1182/blood-2012-03-390070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.George EL, Georges-Labouesse EN, Patel-King RS, Rayburn H, Hynes RO. Defects in mesoderm, neural tube and vascular development in mouse embryos lacking fibronectin. Development. 1993;119:1079–91. doi: 10.1242/dev.119.4.1079. [DOI] [PubMed] [Google Scholar]
- 45.Goumans MJ, Mummery C. Functional analysis of the TGFbeta receptor/Smad pathway through gene ablation in mice. Int J Dev Biol. 2000;44:253–65. [PubMed] [Google Scholar]
- 46.Fuchs Y, Steller H. Programmed cell death in animal development and disease. Cell. 2011;147:742–58. doi: 10.1016/j.cell.2011.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Clarke M, Bennett M, Littlewood T. Cell death in the cardiovascular system. Heart. 2007;93:659–64. doi: 10.1136/hrt.2006.088203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mallat Z, Tedgui A. Apoptosis in the vasculature: mechanisms and functional importance. Br J Pharmacol. 2000;130:947–62. doi: 10.1038/sj.bjp.0703407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cecconi F, Alvarez-Bolado G, Meyer BI, Roth KA, Gruss P. Apaf1 (CED-4 homolog) regulates programmed cell death in mammalian development. Cell. 1998;94:727–37. doi: 10.1016/s0092-8674(00)81732-8. [DOI] [PubMed] [Google Scholar]
- 50.Kuida K, Haydar TF, Kuan CY, Gu Y, Taya C, Karasuyama H, et al. Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell. 1998;94:325–37. doi: 10.1016/s0092-8674(00)81476-2. [DOI] [PubMed] [Google Scholar]
- 51.Woo M, Hakem R, Soengas MS, Duncan GS, Shahinian A, Kagi D, et al. Essential contribution of caspase 3/CPP32 to apoptosis and its associated nuclear changes. Genes Dev. 1998;12:806–19. doi: 10.1101/gad.12.6.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Varfolomeev EE, Schuchmann M, Luria V, Chiannilkulchai N, Beckmann JS, Mett IL, et al. Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity. 1998;9:267–76. doi: 10.1016/s1074-7613(00)80609-3. [DOI] [PubMed] [Google Scholar]
- 53.Kang TB, Ben-Moshe T, Varfolomeev EE, Pewzner-Jung Y, Yogev N, Jurewicz A, et al. Caspase-8 serves both apoptotic and nonapoptotic roles. J Immunol. 2004;173:2976–84. doi: 10.4049/jimmunol.173.5.2976. [DOI] [PubMed] [Google Scholar]
- 54.Li N, Jiang P, Du W, Wu Z, Li C, Qiao M, et al. Siva1 suppresses epithelial-mesenchymal transition and metastasis of tumor cells by inhibiting stathmin and stabilizing microtubules. Proc Natl Acad Sci USA. 2011;108:12851–6. doi: 10.1073/pnas.1017372108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Han J, Liu T, Huen MS, Hu L, Chen Z, Huang J. SIVA1 directs the E3 ubiquitin ligase RAD18 for PCNA monoubiquitination. J Cell Biol. 2014;205:811–27. doi: 10.1083/jcb.201311007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liedtke W, Leman EE, Fyffe RE, Raine CS, Schubart UK. Stathmin-deficient mice develop an age-dependent axonopathy of the central and peripheral nervous systems. Am J Pathol. 2002;160:469–80. doi: 10.1016/S0002-9440(10)64866-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Guertin DA, Stevens DM, Thoreen CC, Burds AA, Kalaany NY, Moffat J, et al. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev Cell. 2006;11:859–71. doi: 10.1016/j.devcel.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 58.Roa S, Avdievich E, Peled JU, Maccarthy T, Werling U, Kuang FL, et al. Ubiquitylated PCNA plays a role in somatic hypermutation and class-switch recombination and is required for meiotic progression. Proc Natl Acad Sci USA. 2008;105:16248–53. doi: 10.1073/pnas.0808182105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Aiello NM, Stanger BZ. Echoes of the embryo: using the developmental biology toolkit to study cancer. Dis Model Mech. 2016;9:105–14. doi: 10.1242/dmm.023184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Van Nostrand JL, Brady CA, Jung H, Fuentes DR, Kozak MM, Johnson TM, et al. Inappropriate p53 activation during development induces features of CHARGE syndrome. Nature. 2014;514:228–32. doi: 10.1038/nature13585. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.