

Abstract
Autophagy allows for cellular material to be delivered to lysosomes for degradation resulting in basal or stress-induced turnover of cell components that provide energy and macromolecular precursors. These activities are thought to be particularly important in cancer where both tumor-promoting and tumor-inhibiting functions of autophagy have been described. Autophagy has also been intricately linked to apoptosis and programmed cell death, and understanding these interactions is becoming increasingly important in improving cancer therapy and patient outcomes. In this review, we consider how recent discoveries about how autophagy manipulation elicits its effects on cancer cell behavior can be leveraged to improve therapeutic responses.
Subject terms: Cancer, Cancer
▓
Facts
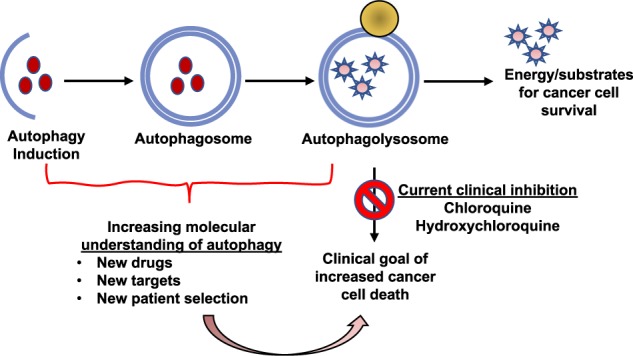
Autophagy has complicated and often competing roles in cancer.
There are five distinct stages: initiation, nucleation of the autophagosome, expansion and elongation of the autophagosome membrane, closure and fusion with the lysosome, and degradation of intravesicular products. Each stage has a subset of potential therapeutic targets for inhibiting autophagy in humans.
Inhibition of autophagy using chloroquine or hydroxychloroquine has already shown promise in clinical trials and numerous other clinical trials are ongoing to determine the optimal tumors to treat and chemotherapy combinations.
Markers of autophagy dependence including BRAF alterations, KRAS mutations, and EGFRvIII mutations may provide a way to select patients that will have a maximal response to autophagy inhibition therapy.
Autophagy interacts with apoptosis to define a tumor cell’s apoptotic threshold.
Open Questions
How should we target autophagy to maximize benefit—early vs. late stage targets; inhibition or stimulation?
How do we best understand the molecular mechanisms underlying autophagy’s effects on tumor behavior and how our deliberate interventions to target the pathway actually work?
How do we develop rationally based interventions to manipulate autophagy to improve cancer therapy?
Will more potent and selective autophagy inhibitors improve on the clinical benefits that we have seen with chloroquine and hydroxychloroquine?
Introduction
In 2016, Yoshinori Ohsumi was awarded the Nobel Prize for Physiology or Medicine for his work on autophagy and its impact in the study of human health and disease [1]. Its role in cancer therapy is particularly important. Indeed, although it is well accepted that autophagy is important in many diseases, until now, the majority of clinical studies that involve deliberate attempts to manipulate autophagy are in cancer therapy, almost always in patients with advanced disease. It is felt that autophagy is an important mechanism to prevent cancer development in both cell autonomous and non-cell autonomous methods. Autophagy maintains normal cell homeostasis through the removal of oncogenic protein substrates, toxic unfolded proteins and damaged organelles. This helps prevent chronic cellular damage and transition into a cancer-initiating cell [2]. Autophagy also works with immunosurveillance to provide a non-cellular autonomous cancer prevention method. For example, decreased autophagy is associated with infiltration of regulatory T cells that suppress the immune system and decrease effective immunosurveillance allowing for increased tumor initiation [3]. Once malignant cancers are fully established, increased autophagy enables tumor cell survival and growth [2, 4]. Thus, in premalignant lesions, enhancing autophagy might prevent cancer [5]. Conversely, in advanced cancers, while both enhancing autophagy and inhibiting it have been proposed as therapeutic strategies [4, 6, 7], clinical interventions to deliberately manipulate autophagy in cancer therapy are already underway [7] with the vast majority focused on inhibiting autophagy. These studies focus on blocking the recycling mechanism of autophagy to prevent the renewal of cellular proteins and other molecules that help cancer cells survive under stressful conditions such as hypoxia [8–10], nutrient deprivation [11, 12] and to enhance other cancer treatments including chemotherapy [13] and radiation [14]. Autophagy can have both tumor cell autonomous and non-autonomous promoting effects on tumor growth [15] and both the degradative process of autophagy itself related but distinct degradative processes as well as non-degradative activities of the autophagy machinery can affect tumor cell behavior [16]. Additionally, we now have a better mechanistic understanding of how autophagy interacts with cell death pathways to alter therapeutic responses to cancer treatments. In this review, we focus on how we may be able to leverage our understanding of these interactions and mechanisms to better harness the power of autophagy manipulation in cancer care.
Autophagy as a therapeutically targetable process
Macroautophagy (referred to hereafter as autophagy) is a highly conserved catabolic process with the formation of double membrane vesicles called autophagosomes that engulf cellular proteins and organelles for delivery to the lysosome [17, 18] (Fig. 1). Autophagy occurs at a basal level in all cells and can be induced by various signals and cellular stresses [7]. Autophagy related (ATG) genes are evolutionarily conserved and tightly regulate the production of autophagosomes [18], which is divided into five distinct stages: initiation, nucleation of the autophagosome, expansion and elongation of the autophagosome membrane, closure and fusion with the lysosome, and degradation of intravesicular products. This breakdown of stages is important as each stage has a subset of potential therapeutic targets for inhibiting autophagy in humans. Importantly, while blocking autophagy at any stage should be similarly effective at preventing degradation of autophagic cargos, recent work raises the possibility that biological effects on tumor cell behavior may be different when the autophagy pathway is blocked at different stages.
Fig. 1. Clinical and pre-clinical targets of inhibition along the five stages of autophagy.

The process of autophagy is divided into five distinct stages: initiation, nucleation, expansion and elongation, closure and fusion, and cargo degradation. Each stage has potential clinical targets. The ULK/FIP200/ATG13 complex induces vesicle nucleation, which continues with involvement of the BECN1 complex which includes VPS34. Multiple ULK1 and VPS34 inhibitors are in pre-clinical evaluation. Expansion and elongation are mediated by ubiquitin-like conjugation systems with conjugation of phosphatidylethanolamine (PE) to LC3-II facilitated by the protease, ATG4B, and the E1-like enzyme, ATG7, resulting in LC3-II incorporation into the growing membrane. Multiple ATG4B inhibitors are in pre-clinical development. Finally, fusion of the autophagosome and lysosome as well as degradation of intravesicular products can be inhibited through the use of lysosomal targeted agents such as CQ and HCQ, which are already in clinical use and under investigation in clinical trials. Additionally, more potent drugs are in preclinical development that target the lysosome and the lysosomal enzyme PPT1.
Initiation can be impeded at the activation complex which involves potential targets including the protein kinases ULK1and ULK2 [19, 20] and a scaffolding protein FIP200 [21]. A class III PI3K complex, also important for initiation, includes the potential targets VPS34 [22–24] (also known as PIK3C3) and Beclin 1, a putative tumor suppressor [25]. Expansion of the autophagosome proceeds as the LC3 and GABARAP families of proteins are conjugated to phosphatidylethanolamine (PE) and added to the growing membrane. This part of the process can be interrupted by targeting the ATG4 family of proteases [26], which cleave LC3/GABARAP family members to allow them to undergo a lipid conjugation reaction that requires an E1-like enzyme ATG7. It is also theoretically possible to target the conjugation machinery with an ATG7 inhibitor. Genetic targeting of ATG7 has shown the potential of blocking this enzyme in cancer cells [27, 28]. Conjugation of LC3-I and PE form LC3-II (MAP1LC3B), a common autophagosome marker [29].
The contents of the autophagosome, including cargo proteins and organelles, are eventually degraded following fusion with a lysosome and the creation of the autophagolysosome. This is where currently clinically available drugs including chloroquine (CQ) and hydroxychloroquine (HCQ) inhibit autophagy by decreasing autophagosome/lysosome fusion [30]. Newer drugs are also in development to target this stage including lys05 [31], a bisaminoquinoline, and DQ661, a unique inhibitor of autophagy and mTOR that is much more lysosome-specific than the current widely used drugs like CQ [32].
Alternatives to macroautophagy
In most discussions, autophagy is synonymous for macroautophagy (as it is throughout this article). But, there are other important forms of autophagy that should be understood and considered when thinking of development of new cancer therapies. Chaperone-mediated autophagy (CMA) occurs when cytosolic proteins with motifs related to the pentapeptide KFERQ are recognized by Heat Shock cognate 70 kDa Protein (HSC70; also known as HSPA8). The chaperone complex translocates into the lysosome via the lysosomal-associated membrane protein 2A, LAMP2A [33, 34]. CMA has been implicated in cancer with inhibition of LAMP2A resulting in decreased tumor growth and metastasis [35]. However, as with macroautophagy, CMA’s effects in cancer also appear to be context dependent. For example, CMA has also been shown to reduce MYC by regulating its proteasomal degradation, suggesting a tumor suppressive role of CMA in normal tissues [36]. Importantly, some of the currently proposed therapeutic interventions designed to inhibit macroautophagy (those that target the lysosome) might also affect CMA, whereas others (e.g., those affecting the ULK1/2 initiation complex) should not.
Selective macroautophagy is used to remove specific cellular components [37] and targeting specific selective autophagy processes may represent a way to more selectively achieve therapeutic effects with autophagy manipulations. Cargo adaptors such as Sequestosome 1 (p62/SQSTM1) target specific substrates to autophagosomes, including specific proteins and complexes of proteins, organelles including peroxisomes, mitochondria, lysosomes, endoplasmic reticulum and other organisms such as bacteria. These cargos are often selected by adaptor recognition of polyubiquitinated molecules followed by binding of the adaptor to LC3 via LC3-interacting regions (LIRs) that bind hydrophobic pockets on LC3 (LIR-docking sites). Iron homeostasis is in part controlled by selective autophagy as well [38], but through a somewhat different mechanism. When cells sense that they are deficient in iron, ferritin is degraded by autophagy. The co-activator 4 (NCOA4) binds ferritin heavy chain (FTH1) molecules resulting in delivery to autophagosomes and release of iron into the cell [38]. Interestingly, however, NCOA4 does not contain a conventional LIR [37].
Identification of alternative LC3-binding proteins (also known as ATG8-binding proteins in lower eukaryotes) has grown substantially in recent years [39]. Ubiquitin-interacting motifs (UIMs) have been shown to have high-affinity binding to ATG8 through recently described UIM-docking sites (UDS), a receptor binding patch on ATG8. This is important in the degradation of targets such as CDC48/p97, an essential segregase that extracts proteins from membranes or multi-subunit complexes [39]. Mutations within CDC48/p97 have been implicated in diseases such as Charcot–Marie–Tooth and familial amyotrophic lateral sclerosis [40] and recycling of this protein through autophagy potentially influences the development or severity of such diseases [39]. A better understanding of the mechanisms behind such targeted autophagy could enable us to make inhibitors that would block specific cargo from being taken in to autophagosomes in cancer cells, as well as other diseases, and could provide new ways to more selectively target autophagy for therapeutic benefit (Table 1).
Table 1.
Pre-clinical and clinical inhibitors of autophagy.
| Stage of autophagy | Target | Drug | Status |
|---|---|---|---|
| Initiation | ULK1/2 | SBI-0206965 | Pre-clinical |
| ULK1/2 | MRT67307 | Pre-clinical | |
| ULK1/2 | MRT68921 | Pre-clinical | |
| Nucleation | VPS34 | VPS34-IN1 | Pre-clinical |
| VPS34 | SB02024 | Pre-clinical | |
| VPS34 | PIK-III | Pre-clinical | |
| PIK3C3/VPS34 | SAR405 | Pre-clinical | |
| Expansion/elongation | ATG4B | NSC185058 | Pre-clinical |
| ATG4B | UAMC-2526 | Pre-clinical | |
| ATG4B | S130 | Pre-clinical | |
| Fusion | Lysosome | CQ | Clinical trial |
| Lysosome | HCQ | Clinical trial | |
| Lysosome | Lys05 | Pre-clinical | |
| Lysosome/mTORC1 | DQ661 | Pre-clinical | |
| Degradation | PPT1 | HCQ | Clinical trial |
| PPT1 | Lys05 | Pre-clinical | |
| PPT1/mTORC1 | DQ661 | Pre-clinical |
Targeting the early stages of autophagy
One potential approach of targeting autophagy is blocking the early stages of initiation and elongation through regulators such as ULK1 [19, 41], VPS34 [22, 23, 42] and ATG4B [26]. ULK1 and ULK2 are the only serine/threonine kinases in the autophagy pathway making them a prime potential target [43]. There are various ATP-competitive inhibitors against ULK1 kinase [44] including the selective SBI-0206965 (SBI) [43]. SBI inhibits autophagy and synergizes with mTOR inhibition [43] and other standard chemotherapies [20]. SBI also effectively induces a cytotoxic apoptotic response in lung cancer during nutrient starvation [19]. This is important as solid tumors often have hypoxic and nutrient-deficient centers [45]. Similarly, inhibitors of VPS34, such as VPS34-IN1 [22] and SB02024 [46], have been developed. SB02024 was shown to successfully block autophagy as well as reduce xenograft models of breast cancer as well as synergize with other therapies in vitro [46]. Both ULK1 and VPS34 inhibitors have also been shown to be effective in autophagy-dependent CNS tumor cells [47]. ATG4B is a cysteine protease that is important for the processing of LC3B and inhibition of this enzyme through targeted agents such as NSC185058 can limit osteosarcoma tumor cell in vitro and in vivo [26]. Additional ATG4B inhibitors UAMC-2526 [48] and S130 [49] are effective at blocking autophagy as well and have anti-cancer effects in xenograft colorectal tumors. While the preliminary data for these early stage inhibitors is encouraging, these compounds are still in early pre-clinical studies and additional research is ongoing. Potential issues with selectivity and the need for higher concentrations may limit clinical utility. Further work to optimize lead drugs will be needed before moving to clinical trials [50].
Targeting the late stages of autophagy—our current clinical focus
Current clinical efforts in cancer therapy focus on the use of the already FDA-approved CQ or HCQ due to their affordability and long-term history of use in a variety of illnesses. CQ was initially developed and pioneered in the treatment of malaria [51]. HCQ was developed by adding a hydroxyl group to CQ which decreased potential toxicities but maintained its overall effectiveness [52]. While CQ continued to be used to treat malaria around the world, HCQ was recognized for an additional benefit of controlling inflammatory conditions such as systemic lupus erythematosus (SLE) [53] and rheumatoid arthritis (RA) [54]. Importantly, these drugs were adopted early in pediatric patients with SLE and RA [55]. The routine use of these drugs in both adult and pediatric patients for over 50 years has provided a wealth of dosing and safety data that has been vital in early investigations of autophagy manipulation in cancer therapy.
The first indication of CQ as a potential cancer therapy was by Victor Bedoya in 1970 who noted CQ was toxic against lymphoma and melanoma cells, although the identification of autophagic vesicles, which accumulate in response to these drugs because autophagosome fusion and degradation is blocked, was more of a side note than a focus of his study [56]. Murakami et. al. were the first to clearly identify CQ’s ability to inhibit autophagy and its relation to the inhibition of lysosomal degradation of cellular contents [57]. The first clinical trial of CQ in the treatment of glioblastoma (GBM) patients demonstrated significantly improved clinical outcomes for patients [14]. These findings were the first in the fast-paced advancement of the study of CQ and HCQ in cancer therapy.
In 2007, Amaravadi and colleagues [58] showed that in Myc-induced p53 negative lymphomas, where tumor cells were naturally resistant to apoptosis, re-expression of p53 lead to apoptosis that was further enhanced with the inhibition of autophagy, while, importantly, surviving cells demonstrated increased autophagy. Subsequent inhibition of autophagy in these cells along with alkylating agents lead to increased apoptosis and cell death. Following this first demonstration that autophagy inhibition could enhance the tumor killing activity of other anti-cancer agents, many in vitro studies, genetically engineered mouse models (GEMMs) and patient-derived xenograft (PDX) mouse models provided abundant pre-clinical evidence that inhibition of autophagy with various anti-cancer drugs can, at least under some circumstances (see below), produce improved tumor cell killing. This large body of studies lead to both veterinary and human clinical trials [4, 6, 59], currently 61 such trials are listed on clinicaltrials.gov.
Briceno et al.’s initial small study of GBM patients treated with CQ in conjunction with standard of care radiation and temozolomide showed a median survival of 33 months compared with 11 months for control patients [14]. Follow-up clinical trials and retrospective data from this group supported these initial findings [60, 61] suggesting a role for CQ in GBM therapy. Combining CQ with whole-brain radiation for brain metastasis also demonstrated improved intracranial tumor control [62, 63].
Later clinical trials made an effort to correlate depth of autophagy inhibition by HCQ and changes in therapy response [13, 64–69]. Human trials included a broad range of tumors including GBM [67], advanced solid tumors and melanoma [64–66] and myeloma [68]. Consistent with the idea that autophagy inhibition alters cellular responses in both normal cells and tumor cells to other treatments, the maximum tolerated dose (MTD) of HCQ varied in relation to the concomitant therapy. Studies performed with HCQ in combination with targeted therapies such as vorinostat were able to reach an MTD of 600 mg twice daily [64] while HCQ in combination with cytotoxic chemotherapy such as temozolamide and radiation in GBM required a smaller dose of 400 mg twice daily [67]. Common dose-limiting toxicities included gastrointestinal toxicity and fatigue [64–66]. As a single agent, the MTD of HCQ is not known and 600 mg twice daily in adult patients is the highest dose tested in combination therapy studies to date [65].
Measurable responses to autophagy inhibition have varied by tumor type, therapy combinations and measured outcome (Table 2). The initial GBM studies of CQ in combination with temozolomide and radiation demonstrated more than a doubling of median survival [14, 60, 61]. A follow-up phase I/II trial with HCQ in place of CQ found no significant improvement in survival [67]. This difference may be related to dose limitations of HCQ due to myelosuppression not reported in the CQ studies. More intensive pharmacokinetic and pharmacodynamic studies in the HCQ trial also demonstrated these patients had inconsistent inhibition of autophagy [67]. Pharmacokinetic studies in canine (dog) lymphoma patients using HCQ and doxorubicin demonstrated a 100-fold increase in HCQ in the tumor compared to plasma concentrations demonstrating a disconnect between tumor exposure and measuring autophagy inhibition in peripheral blood mononuclear cells (PBMCs) as a surrogate for effectiveness [13]. There is also an inverse relationship between tumor extracellular pH and the level of HCQ uptake [70], which may result in different levels of autophagy inhibition at the center of a tumor compared to peripheral areas. A recently developed physiologically based pharmacokinetic model of HCQ offers a possible way to understand these differences and predict individual patient responses to HCQ [71]. Developed and validated in both mice and human samples, the physiologically based pharmacokinetic model of HCQ is able to predict blood and tissue exposures and could potentially be used to guide future clinical trials.
Table 2.
Human clinical autophagy inhibition trials.
| Published clinical trials | ||||||
|---|---|---|---|---|---|---|
| Tumor type | Inhibitor | Phase | Additional therapy | Clinical response | Autophagy evaluation | Ref. |
| Solid tumors | HCQ | I | Rapamycin, Metronomic cyproterone, docetaxel |
PR 40% SD 44% |
Not evaluated | [134] |
| Solid tumors | HCQ | I | Vorinostat | 1 patient (renal cell carcinoma) durable PR | EM of PBMC for AVs | [64] |
| 2 patients (colorectal cancer) prolonged SD | IHC for LC3II | |||||
| Solid tumors and melanoma | HCQ | I | Temsirolimus |
Solid tumor: SD 67% Melanoma: SD 74% |
EM of PBMC for AVs | [65] |
| Solid tumors and melanoma | HCQ | I | TMZ |
Solid tumor: PR 10% SD 27% Metastatic melanoma: PR 14% SD 27% |
EM of PBMC for AVs | [66] |
| Sarcoma | HCQ | Case series | Rapamycin |
6 PR 3 SD 1 PD |
Evaluation of 18FDG-PET as measure of tumor response after two weeks | [135] |
| Glioblastoma | CQ | III | TMZ and radiation | Median survival 24 mo (controls 11 mo) | Not evaluated | [61] |
| Relapsed glioblastoma | CQ | Case series (n = 5) | Radiation |
2 mo response: 2 PR 1 SD |
Not evaluated | [136] |
| Glioblastoma | HCQ | I/II | TMZ and radiation | Median survival 15.6 months |
Plasma concentrations of HCQ EM of PBMC for mean AVs PBMC LC3-II:Actin ratio |
[67] |
| Glioblastoma | CQ | III | TMZ and radiation | Median survival: 33 months (controls 11 months) | Not evaluated | [14] |
| Brain metastases: NSCLC, SCLC, breast, ovarian | CQ | Pilot | Radiation |
Median OS: 5.7 months PFS of brain metastasis at 1 year 55% |
Not evaluated | [62] |
| Brain metastases: NSCLS, breast cancer | CQ | II | Radiation |
ORR 54% (control 55%) PFS (brain metastasis) at 1 year: 83.9% (control 55.1%) |
Not evaluated | [63] |
| Refractory myeloma | HCQ | I | Bortezomib |
14% very good PR 45% period of SD |
Plasma concentrations of HCQ EM of PBMC and bone marrow plasma cells for mean AVs PBMC LC3-II:Actin ratio |
[68] |
| Metastatic PDAC | HCQ | II | None |
Median PFS 1.5 mo OS: 2.3 mo |
PBMC LC3-II:β-Actin ratio | [69] |
| PDAC | HCQ | I/II | Gemcitabine | 61% decreased CA19-9 | LC3-II in PBMC If >51% increase, OS 34.83 months v 10.83 months | [74] |
| NSCLC | HCQ | I | Erlotinib | ORR 5% | Plasma concentrations of HCQ | [137] |
| Ongoing clinical trials | ||||||
|---|---|---|---|---|---|---|
| Tumor type | Inhibitor | Phase | Additional therapy | Clinical response | Autophagy marker | Ref. |
| Colorectal | HCQ | I/II | FOLFOX + Bevacizumab | Ongoing | JNK1 | [138] |
| Glioblastoma | CQ | I/II | Temozolomide + radiation | Ongoing | EGFRvIII | [133] |
| Glioblastoma, HGG, DIPG | CQ | III |
Temozolomide Valproic acid |
Ongoing | None | [139] |
| Pancreatic | HCQ, nab-Paclitaxel | I/II | Gemcitabine, nab-Paclitaxel | Ongoing | Mutant RAS | [128] |
| Pancreatic | HCQ, nab-Paclitaxel | II | Gemcitabine, nab-Paclitaxel | Ongoing | Mutant RAS | [140] |
| Pancreatic | HCQ | I/II | Gemcitabine | Ongoing | Mutant RAS | [141] |
| Pancreatic | HCQ | I | Trametinib | Ongoing | Mutant RAS | [128] |
| BRAF Mutant Melanoma | HCQ | I | Vemurafenib | Ongoing | Mutant BRAF | [142] |
| BRAF Mutant Melanoma | HCQ | I/II | Dabrafenib, Trametinib | Ongoing | Mutant BRAF | [143] |
| NRAS Mutant Melanoma | HCQ | I | Trametinib | Ongoing | Mutant NRAS | [125] |
| Hepatocelluar | HCQ | II | Sorafenib | Ongoing | None | [144] |
| Osteosarcoma | HCQ | I/11 | Gemcitabine, Docetaxel | Ongoing | None | [145] |
| Breast cancer | HCQ | I/II | Palbociclib Letrozole | Ongoing |
ER positive HER2 negative |
[146] |
| Non-small cell lung cancer | HCQ | II | Erlotinib | Ongoing | EGFR | [132] |
Potential alternatives to CQ and HCQ
More potent and selective next- generation lysosomal targeted inhibitors are in development including Lys05, a bisaminoquinoline and DQ661, a dimeric quinacrine with the additional benefit of parallel lysosome and mTOR inhibition [32] (Table 1). Lys05, which is approximately tenfold more potent than CQ [72], has been successful in limiting melanoma and colorectal adenocarcinoma growth as a single agent in mouse models [72]. DQ661 has also shown impressive in vivo single-agent activity in melanoma and colorectal cancer and in vivo efficacy in combination with gemcitabine in pancreatic ductal adenocarcinoma (PDAC) [32] This is in contrast to HCQ, which has shown limited clinical responses as a single agent in cancer such as advanced metastatic pancreatic cancer [69]. Additional pre-clinical data in PDX studies of less advanced pancreatic cancer suggests HCQ may have some limited single-agent response [73] but combination therapy is more likely to produce clinically relevant responses [74].
It is often assumed that any agent that inhibits autophagy has the same consequence irrespective of the inhibitor used. The developing studies on these agents suggest that this assumption is wrong even if we limit the analysis only to drugs that target the lysosome. Lys05 is a more potent autophagy inhibitor than HCQ due to a greater accumulation in and deacidification of the lysosome [72]. DQ661 was also shown to have improved deacidification of the lysosome. Additionally, this agent allowed identification of the molecular target for the drug in the lysosome. DQ661 inhibition of autophagy is associated with its ability to inhibit palmitoyl-protein thioesterase 1 (PPT1), a glycoprotein important in the catabolism of lipid-modified protein during lysosomal degradation [32]. When DQ661 binds PPT1, it blocks its ability to cleave thioester bonds from palmitoylated proteins. This results in lysosomal displacement of v-ATPase subunits and also causes impaired lysosomal recruitment of mTOR. This is important because in amino-acid rich environments, mTOR is recruited to the lysosome and activated by its master regulator RHEB [32]. Concurrent inhibition of both the lysosomal component of autophagy and mTOR results in superior nullification of protein translation and potentially improved activity in cancer therapy [32]. Further investigation determined that CQ derivatives such as HCQ and Lys05 are also able to inhibit PPT1, but only DQ661 maintains its activity in acidic media. This is particularly important due to limiting of CQ and HCQ uptake into tumor tissue with decreasing pH, making it difficult for these drugs to effectively block autophagy in more acidic tumors [70]. Interestingly, recent data have shown that CQ itself inhibits autophagy by impairing autophagosome fusion with lysosomes, rather than affecting lysosomal acidity like Lys05 [30]. This results in additional cellular changes and CQ is known to have other autophagy-independent anti-cancer effects [75, 76] and to be able to sensitize cancer cells to therapy through autophagy-independent mechanisms [77]. The mechanistic differences between how CQ and CQ-derivatives such as HCQ and Lys05 inhibit autophagy may explain some of the clinical responses and uses for these drugs, such as HCQ’s anti-inflammatory and autoimmune benefits in SLE [53, 78] and RA [54, 79]. Further understanding of the mechanistic differences between the various CQ-like molecules may allow improved treatment strategies and help us take advantage of both the autophagy-dependent and autophagy-independent effects of these drugs.
Additional methods are available to influence autophagy including the use of epigenetic modifiers. The epigenetic control of autophagy has been demonstrated through acetylation of histones, hyper-methylation of CpG islands and the disruption of mRNA function due to cytoplasmic non-coding RNAs [80]. Epigenetic targeted therapy can affect this control. For example, the use of histone deacetylase inhibitors to treat malignant peripheral nerve sheath tumors led to an increase in autophagy and a resistance to therapy [81]. Vidoni et al. recently reviewed natural products that have been shown to alter gene expression via epigenetic effects resulting in changes in autophagy [80]. For example, curcumin is able to restore expression of miR-143 which in turn inhibits autophagy via ATG2B and has been shown to sensitize prostate cancer cells to apoptosis following radiation [82]. Numerous other natural products have been shown to epigenetically upregulate autophagy [80].
The interplay between autophagy and apoptosis in cancer therapy
Autophagy is a known survival mechanism in several tumor types [10, 83–85]. This can be due to protection against nutrient starvation and providing substrates for basic cell survival [12]. But it can also be related to its role in protecting cells from undergoing programmed cell death such as apoptosis [86]. Anti-cancer treatments usually work by inducing apoptosis taking advantage of the closer proximity of cancer cells to their apoptotic threshold than is the case for normal cells [87]. The role of autophagy in the control and modulation of apoptosis and apoptotic thresholds is important to understand in developing cancer therapies.
Autophagy is known to interact with and control canonical apoptosis. In a normal cell, pro and anti-apoptotic signals work together to maintain a balance between the life and death of the cell. Intrinsic stimuli that tip this scale toward a pro-apoptotic path drive mitochondria to become permeabilized, known as mitochondrial outer membrane permeabilization (MOMP). This results in a cascade of activity that starts with the release cytochrome C into the cytosol where it can bind APAF-1. This allows APAF-1 to oligomerize to form a scaffold, called the apoptosome, for caspase-9 activation and ultimately a full caspase cascade including the effector caspases, caspase-3, 6 and 7, leading to cell death. Extrinsic apoptosis results from death receptor activation through receptors such as Fas (CD95) and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and caspase-8 activation. Caspase-8 can lead directly to cell death through activation of the effector caspases or, more commonly, by cleaving the BCL-2 homology 3 (BH3)-only protein BID and activating the intrinsic mitochondrial pathway by inducing MOMP [88].
A historic understanding of apoptosis held that once MOMP has occurred, cells were committed to death. This belief has recently been questioned with the concepts of incomplete [89] or minority [90] MOMP. These incomplete forms of MOMP lead to limited caspase activation and a pool of mitochondria that survive and support cell survival. Autophagy within cancer cells can lead to inefficient MOMP, slowing the rate of cell death and presenting an opportunity for the cell to recover and regain the ability to grow [91]. More concerning, cells that survive minority MOMP can develop genomic instability, cellular transformation, and enhanced tumorigenesis [90, 92].
This has led to the concept that autophagy controls apoptosis, and vice versa and, that this relationship may control a cell’s ability to reach its apoptotic threshold. BCL family proteins control the balance of pro and anti-apoptotic signals and set the apoptotic threshold of a cell [93]. These proteins are also involved in the initiation of autophagy [86]. For example, the production of autophagosomes is stimulated when the Beclin 1/BCL-2 complex is disrupted by BH3-only proteins (PUMA, NOXA, NIX, BID, BNIP3) [94]. This allows Beclin 1 to complex with VPS34 and stimulate autophagy. In turn, BH3 proteins such as PUMA suppress BCL-2 function resulting in increased apoptosis. This function has also been shown in reverse with autophagy regulating the level of PUMA [91]. The mechanism underlying this effect (Fig. 2) has recently been described to involve the transcription factor Forkhead box O3 (FOXO3), which is itself known to regulate autophagy, e.g., in stem cells [95]. A decrease in autophagy results in the increased transcription of PUMA by FOXO3 through a single forkhead response element (FHRE). Loss of this FHRE was sufficient to block apoptosis when an autophagy inhibitor was combined with the MDM2 inhibitor Nutlin 3 [96]. Interestingly, this relationship arises because FOXO3a is itself degraded by autophagy [96] creating a potential homeostatic feedback loop whereby a transcription factor that is itself regulated by autophagy controls the expression of autophagy regulators and simultaneously regulates the apoptotic threshold by controlling a key apoptotic regulator. Thus, we suggest that a cell can correct short periods of autophagy inhibition by increasing FOXO3a levels and subsequently activating downstream autophagy targets. However, with prolonged autophagy inhibition, this will activate pro-apoptotic genes as well, such as PUMA, sensitizing the cell to death [97].
Fig. 2. The interplay between autophagy and apoptosis.

Autophagy and apoptosis interact through the control of multiple proteins. BH3-only proteins disrupt the interaction of Bcl-2 and Beclin 1. This allows Beclin 1 to stimulate autophagy through Vps34 and Bcl-2 to inhibit apoptosis through interaction with Bax/Bax. Autophagy controls cellular levels of FOXO3. Increased FOXO3 stimulates autophagy regulation genes to induce autophagy and reduce overall FOXO3 levels. If there is continued presence of elevated FOXO3, it results in increased transcription of PUMA, which is then free to block with interaction between Bcl-2 and Bax/Bak. Once Bax/Bak are released, they can permeabilize mitochondria resulting in effector caspase activation and apoptotic cell death.
Therapies targeting BH3-only proteins are already in clinical use, with the above data suggesting manipulation of these proteins will influence not only apoptosis but may also affect autophagy. Venetoclax, a BH3-mimetic inhibitor of BCL-2, was approved by the FDA in 2015 for the treatment of chronic lymphocytic leukemia (CLL) and small lymphocytic lymphoma. Newer generation of BH3-mimetics are also in development targeting not only BCL-2 but also MCL-1 and BCL-XL. While there are no current clinical trials investigating the combination of such BH3-mimetics and autophagy inhibition, there is strong evidence to suggest their combined potential for clinical benefit. For example, it has been shown in acute myeloid leukemia (AML) that there is an increase in vacuole membrane protein (VMP1), an autophagy-associated protein, and that cells overexpressing this protein resulted in increased autophagy and was correlated with increased mitochondrial health and increased protection against oxidative stress. It also decreased AML response to venetoclax-induced MOMP [98], an important consideration as it is being actively investigated in over 30 active adult and pediatric AML clinical trials (www.clinicaltrials.gov).
Autophagy manipulation and the tumor immune response
The importance of the immune system in cancer therapy cannot be understated. Therefore, understanding how autophagy manipulation affects tumor immune response is vital. It has been reported that autophagy in dying tumor cells is needed for immunogenic cell death and efficient tumor recognition by the immune system [99, 100]. In melanoma, an effective tumor immune response was shown to require intact autophagy [101]. A further study found that calorie restriction mimetics increased autophagy and could boost anti-tumor immune responses [102]. Autophagy can also stimulate tumor antigen cross-presentation supporting improved tumor immune responses; [103] autophagy inhibition could potentially interfere with process. These studies would suggest it would be better to increase, not decrease, autophagy during cancer therapy. This is also supported by some correlative clinical data where breast cancer patients with higher LC3 puncta (a surrogate for autophagosomes) and the presence of nuclear HMGB1 had improved overall and metastatic-free survival [104] and increased immune infiltration of the tumor [105].
However, as with most aspects of the immune system, its interaction with autophagy is complex with activation of autophagy being associated with immunological promotion as well as inhibition of tumors [106]. Some anti-tumor immune responses are enhanced by autophagy inhibition [107–109]. Furthermore, a recent study found equivalent T-cell immune responses with both genetically inhibited autophagy in tumors and following treatment with CQ to block autophagy [110]. Thus, there are arguments both for and against autophagy inhibition considering the effects on anti-tumor immune responses. It will therefore be important to ensure that clinical studies incorporate analysis of anti-tumor immune responses when autophagy is being manipulated.
Potential reasons for failure of autophagy inhibition as a therapeutic strategy
Current methods of clinical autophagy manipulation could have potential toxicity from global autophagy inhibition. Acute whole-body knockout of an essential autophagy gene (Atg7) in adult mice had a profound anti-tumor effect on KRAS-driven lung cancer. However, it also led the death of the mice due to severe neuronal toxicity, disruption of glucose homeostasis, and increased susceptibility to infection [111]. Importantly, however, the chronic use of HCQ for rheumatologic disorders and treatment of some cancer patients with CQ for years [112, 113] suggests incomplete autophagy inhibition (as is seen with these agents) can be done safely. Furthermore, patients could be taken off the drug for periods of time to allow for therapy breaks.
Difficulties obtaining efficient autophagy inhibition within solid tumors may also limit clinical effectiveness. As noted above, HCQ uptake may differ based on pH and from outer to the inner areas of a tumor, resulting in unequal inhibition between tumor cells [70]. Inconsistent inhibition has been shown in clinical trials that did not show significant effects [67, 69]. Finally, proper patient selection may limit positive findings in clinical trials. Without a clear understanding of which patients should receive such therapy, a potential response in one subset of patients may be overshadowed by non-responders in a larger subset.
How do we maximize the therapeutic benefit of autophagy manipulation in cancer?
The published results of autophagy inhibition clinical trials support the safe use of CQ and HCQ in the clinic. Positive clinical results such as those seen in CQ in the treatment of GBM [14, 61] and HCQ in select pancreatic cancer patients [74] are encouraging. Unfortunately, conflicting results with less positive or no evidence of clinical improvements make it difficult to know how to best utilize autophagy inhibition. Correspondingly, preclinical evidence would suggest, in select cases, autophagy inhibition may be contraindicated. One recent study demonstrated that inhbition of autophagy led to reactivation of aberrant expression of 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3) in dormant murine breast cancer stem cells (BCSCs) allowing them to reactivate and proliferate resulting in recurrent metastatic disease [114]. These results highlight that proper patient and drug selection are needed to maximize the benefits of autophagy inhibition and limit potential adverse outcomes.
The drugs currently available including CQ and HCQ that are being actively used in clinic are not truly autophagy specific. Even the newer drugs such as ULK1 and VPS34 inhibitors affect other pathways. Additionally, the majority of clinical trials have been in patients with no selection criteria beyond tumor type on which to think autophagy inhibition will work well (Table 2). There has also been little rationale for which therapies to combine with autophagy inhibition beyond adding CQ or HCQ to current standard therapies, leading to a variety of combinations with little basis or testing whether some combinations will be better than others. However, one set of tumors, those driven by RAS-RAF-MEK pathway, may demonstrate a way forward.
The concept of autophagy addiction, also known as autophagy-dependence, is important because in at least some cases, has been shown that only autophagy-dependent tumors respond well to autophagy inhibition even when the same combination of drugs with CQ are tested [115]. Indeed, synergy between autophagy inhibitors and other drugs may occur in autophagy-dependent tumor cells while being antagonistic in autophagy-independent tumors [115, 116]. Some mechanisms of autophagy addiction are now known and are driving new clinical trials, particularly in tumors driven by the RAS-RAF-MEK pathway. Mouse models of lung cancer and melanoma driven by the BrafV600E were amongst the first tumors to demonstrate autophagy-dependence and were highly sensitive to Atg7 deletion [27, 117]. Further studies have demonstrated that autophagy inhibition is sufficient to kill BrafV600E-positive CNS tumor cells, but not their wildtype BRAF-expressing counterparts [116]. This has led to multiple clinical trials in melanoma and CNS tumors with patients selected for the presence of BrafV600E (Table 2).
Tumors driven by mutations in KRAS increase autophagy to maintain cell metabolism [118–120]. PDAC has high rates of KRAS mutation and this is thought to contribute to PDAC being autophagy dependent [119, 121, 122]. Additionally increased activity of transcription factors promote autophagy [123] and pancreatic stellate cells in the tumor microenvironment use autophagy to fuel tumor metabolism [124]. Early in vivo xenograft trials of PDAC treated with autophagy inhibition demonstrated decreased tumor growth [73]. Follow-up clinical trials found improved surgical outcomes in PDAC patients treated with gemcitabine, nab-paclitaxel and HCQ pre-operatively [74, 114]. More recent studies have demonstrated the benefit of autophagy inhibition in combination with targeting the RAS pathway itself through inhibition of MEK [122] or ERK [52] directly. These encouraging results have led to the development of clinical trials inhibiting MEK and autophagy in metastatic NRAS melanoma (NCT03979651 [125]) and PDAC (NCT03825289 [126]) (Table 2).
Mutations outside the RAS pathway have also been implicated as markers of autophagy addiction. Functional JUN N-terminal kinase 1 (JNK1) is required for hypoxia-induced autophagy [127] and is being studied as a potential predictor of response to autophagy inhibition in PDAC (NCT01506973 [128]). Epidermal growth factor receptor (EGFR) mutations or amplifications have also been utilized to identify autophagy-dependence. EGFR regulates numerous downstream pathways that influence autophagy, including PI3K-AKT-mTOR, STAT3 and RAS family signaling [129]. EGFR mutations and amplifications are associated with radioresistance in GBM and head and neck squamous cell carcinomas. These tumors are highly autophagy-dependent and responsive to pharmacologic inhibition of autophagy [130]. GBM tumors expressing EGFR variant III (EGFRvIII), a common mutation in the extracellular domain of EGFR, have been also been shown to be autophagy dependent [131]. These studies have resulted in selection of patients with mutant or high expressing EGFR tumors for autophagy inhibition clinical trials in non-small cell lung cancer (NCT00977470 [132]) and GBM (NCT02432417 [133]).
Summary and future directions
Even just the limited discussion above shows that autophagy has complicated and often competing roles in cancer. Thus, it has sometimes been suggested that it is unwise to consider targeting autophagy in cancer. We think that such a position is unduly pessimistic. Moreover, we believe that since many of the interventions that are currently used in patients as well as the physiological stimuli that affect tumors such as nutrient deprivation affect autophagy, we do not have a choice—we have always been altering autophagy in cancer patients. The real question is not whether we should or should not target autophagy. Instead, we should focus on how to do so in a way that will maximize the benefit. This could include improved drugs to manipulate autophagy and improved clinical trial design and patient selection. By understanding the molecular mechanisms by which autophagy elicits its effects on tumor behavior and combining this knowledge with a better understanding of how our deliberate interventions to target the pathway actually work, it may be possible to build on the examples described above and develop rationally based interventions to control and manipulate this process to improve treatment.
Acknowledgements
Work in our laboratories is supported by NIH/NCI (K08CA193982), NIH/NINDS (R01NS107313), a Webb-Waring Biomedical Research Award and the Morgan Adams Foundation (JML). NIH CA150925, CA190170 and CA197887 (AT). We thank Christina Towers, PhD, for her assistance with the development of Fig. 1.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Edited by F. Pentimalli
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Assembly TN. The Nobel Assembly at Karolinska Instiutet has today decided to award the 2016 Nobel Prize in Physiology or Medicine to Yoshinori Ohsumi. 2016. https://www.nobelprize.org/nobel_prizes/medicine/laureates/2016/press.html. Accessed 25 May 2017.
- 2.White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer. 2012;12:401–10. doi: 10.1038/nrc3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rao S, Tortola L, Perlot T, Wirnsberger G, Novatchkova M, Nitsch R, et al. A dual role for autophagy in a murine model of lung cancer. Nat Commun. 2014;5:3056. doi: 10.1038/ncomms4056. [DOI] [PubMed] [Google Scholar]
- 4.Amaravadi R, Kimmelman AC, White E. Recent insights into the function of autophagy in cancer. Genes Dev. 2016;30:1913–30. doi: 10.1101/gad.287524.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Galluzzi Lorenzo, Pietrocola Federico, Bravo‐San Pedro José Manuel, Amaravadi Ravi K, Baehrecke Eric H, Cecconi Francesco, Codogno Patrice, Debnath Jayanta, Gewirtz David A, Karantza Vassiliki, Kimmelman Alec, Kumar Sharad, Levine Beth, Maiuri Maria Chiara, Martin Seamus J, Penninger Josef, Piacentini Mauro, Rubinsztein David C, Simon Hans‐Uwe, Simonsen Anne, Thorburn Andrew M, Velasco Guillermo, Ryan Kevin M, Kroemer Guido. Autophagy in malignant transformation and cancer progression. The EMBO Journal. 2015;34(7):856–880. doi: 10.15252/embj.201490784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Levy JM, Thorburn A. Targeting autophagy during cancer therapy to improve clinical outcomes. Pharmcol Ther. 2011;131:130–41. doi: 10.1016/j.pharmthera.2011.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Towers Christina G., Thorburn Andrew. Therapeutic Targeting of Autophagy. EBioMedicine. 2016;14:15–23. doi: 10.1016/j.ebiom.2016.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blagosklonny MV. Hypoxia, MTOR and autophagy: converging on senescence or quiescence. Autophagy. 2013;9:260–2. doi: 10.4161/auto.22783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu HM, Jiang ZF, Ding PS, Shao LJ, Liu RY. Hypoxia-induced autophagy mediates cisplatin resistance in lung cancer cells. Sci Rep. 2015;5:12291. doi: 10.1038/srep12291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tan Q, Wang M, Yu M, Zhang J, Bristow RG, Hill RP, et al. Role of autophagy as a survival mechanism for hypoxic cells in tumors. Neoplasia. 2016;18:347–55. doi: 10.1016/j.neo.2016.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 11.Guo XL, Hu F, Wang H, Fang JM, Zhu Z, Wei L, et al. Inhibition of autophagy in hepatocarcinoma cells promotes chemotherapeutic agent-induced apoptosis during nutrient deprivation. Oncol Rep. 2018;39:773–83. doi: 10.3892/or.2017.6115. [DOI] [PubMed] [Google Scholar]
- 12.Sato K, Tsuchihara K, Fujii S, Sugiyama M, Goya T, Atomi Y, et al. Autophagy is activated in colorectal cancer cells and contributes to the tolerance to nutrient deprivation. Cancer Res. 2007;67:9677–84. doi: 10.1158/0008-5472.CAN-07-1462. [DOI] [PubMed] [Google Scholar]
- 13.Barnard RA, Wittenburg LA, Amaravadi RK, Gustafson DL, Thorburn A, Thamm DH. Phase I clinical trial and pharmacodynamic evaluation of combination hydroxychloroquine and doxorubicin treatment in pet dogs treated for spontaneously occurring lymphoma. Autophagy. 2014;10:1415–25. doi: 10.4161/auto.29165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Briceno E, Reyes S, Sotelo J. Therapy of glioblastoma multiforme improved by the antimutagenic chloroquine. Neurosurgical FOCUS. 2003;14:e3. doi: 10.3171/foc.2003.14.2.4. [DOI] [PubMed] [Google Scholar]
- 15.Yang A, Herter-Sprie G, Zhang H, Lin EY, Biancur D, Wang X, et al. Autophagy sustains pancreatic cancer growth through both cell-autonomous and nonautonomous mechanisms. Cancer Discov. 2018;8:276–87. doi: 10.1158/2159-8290.CD-17-0952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Galluzzi L, Green DR. Autophagy-independent functions of the autophagy machinery. Cell. 2019;177:1682–99. doi: 10.1016/j.cell.2019.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mizushima N. Autophagy: process and function. Genes Dev. 2007;21:2861–73. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- 18.Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27:107–32. doi: 10.1146/annurev-cellbio-092910-154005. [DOI] [PubMed] [Google Scholar]
- 19.Egan DF, Chun MGH, Vamos M, Zou H, Rong J, Miller CJ, et al. Small molecule inhibition of the autophagy kinase ULK1 and identification of ULK1 substrates. Mol Cell. 2015;59:285–97. doi: 10.1016/j.molcel.2015.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tang F, Hu P, Yang Z, Xue C, Gong J, Sun S, et al. SBI0206965, a novel inhibitor of Ulk1, suppresses non-small cell lung cancer cell growth by modulating both autophagy and apoptosis pathways. Oncol Rep. 2017;37:3449–58. doi: 10.3892/or.2017.5635. [DOI] [PubMed] [Google Scholar]
- 21.Bae H, Guan JL. Suppression of autophagy by FIP200 deletion impairs DNA damage repair and increases cell death upon treatments with anticancer agents. Mol Cancer Res. 2011;9:1232–41. doi: 10.1158/1541-7786.MCR-11-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bago R, Malik N, Munson MJ, Prescott AR, Davies P, Sommer E, et al. Characterization of VPS34-IN1, a selective inhibitor of Vps34, reveals that the phosphatidylinositol 3-phosphate-binding SGK3 protein kinase is a downstream target of class III phosphoinositide 3-kinase. Biochemical J. 2014;463:413–27. doi: 10.1042/BJ20140889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dowdle WE, Nyfeler B, Nagel J, Elling RA, Liu S, Triantafellow E, et al. Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat Cell Biol. 2014;16:1069–79. doi: 10.1038/ncb3053. [DOI] [PubMed] [Google Scholar]
- 24.Pasquier B. SAR405, a PIK3C3/Vps34 inhibitor that prevents autophagy and synergizes with MTOR inhibition in tumor cells. Autophagy. 2015;11:725–6. doi: 10.1080/15548627.2015.1033601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–6. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- 26.Akin D, Wang SK, Habibzadegah-Tari P, Law B, Ostrov D, Li M, et al. A novel ATG4B antagonist inhibits autophagy and has a negative impact on osteosarcoma tumors. Autophagy. 2014;10:2021–35. doi: 10.4161/auto.32229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xie X, Koh JY, Price S, White E, Mehnert JM. Atg7 overcomes senescence and promotes growth of BrafV600E-driven melanoma. Cancer Discov. 2015;5:410–23. doi: 10.1158/2159-8290.CD-14-1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karvela M, Baquero P, Kuntz EM, Mukhopadhyay A, Mitchell R, Allan EK, et al. ATG7 regulates energy metabolism, differentiation and survival of Philadelphia-chromosome-positive cells. Autophagy. 2016;12:936–48. doi: 10.1080/15548627.2016.1162359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12:1–222. [DOI] [PMC free article] [PubMed]
- 30.Mauthe M, Orhon I, Rocchi C, Zhou X, Luhr M, Hijlkema KJ, et al. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy. 2018;14:1435–55. doi: 10.1080/15548627.2018.1474314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Amaravadi RK, Winkler JD. Lys05: a new lysosomal autophagy inhibitor. Autophagy. 2012;8:1383–4. doi: 10.4161/auto.20958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rebecca VW, Nicastri MC, McLaughlin N, Fennelly C, McAfee Q, Ronghe A, et al. A unified approach to targeting the lysosome's degradative and growth signaling roles. Cancer Discov. 2017;7:1266–83. doi: 10.1158/2159-8290.CD-17-0741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arias E, Cuervo AM. Chaperone-mediated autophagy in protein quality control. Curr Opin Cell Biol. 2011;23:184–9. doi: 10.1016/j.ceb.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaushik S, Bandyopadhyay U, Sridhar S, Kiffin R, Martinez-Vicente M, Kon M, et al. Chaperone-mediated autophagy at a glance. J Cell Sci. 2011;124:495–9. doi: 10.1242/jcs.073874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kon M, Kiffin R, Koga H, Chapochnick J, Macian F, Varticovski L, et al. Chaperone-mediated autophagy is required for tumor growth. Sci Transl Med. 2011;3:109ra117. doi: 10.1126/scitranslmed.3003182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gomes LR, Menck CFM, Cuervo AM. Chaperone-mediated autophagy prevents cellular transformation by regulating MYC proteasomal degradation. Autophagy. 2017;13:928–40. doi: 10.1080/15548627.2017.1293767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gatica D, Lahiri V, Klionsky DJ. Cargo recognition and degradation by selective autophagy. Nat Cell Biol. 2018;20:233–42. doi: 10.1038/s41556-018-0037-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature. 2014;509:105–9. doi: 10.1038/nature13148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marshall RS, Hua Z, Mali S, McLoughlin F, Vierstra RD. ATG8-binding UIM proteins define a new class of autophagy adaptors and receptors. Cell. 2019;177:766–81.e24. doi: 10.1016/j.cell.2019.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tang WK, Xia D. Mutations in the human AAA(+) chaperone p97 and related diseases. Front Mol Biosci. 2016;3:79. doi: 10.3389/fmolb.2016.00079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Petherick KJ, Conway OJL, Mpamhanga C, Osborne SA, Kamal A, Saxty B, et al. Pharmacological inhibition of ULK1 kinase blocks mammalian target of rapamycin (mTOR)-dependent autophagy. J Biol Chem. 2015;290:11376–83. doi: 10.1074/jbc.C114.627778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ronan B, Flamand O, Vescovi L, Dureuil C, Durand L, Fassy F, et al. A highly potent and selective Vps34 inhibitor alters vesicle trafficking and autophagy. Nat Chem Biol. 2014;10:1013–9. doi: 10.1038/nchembio.1681. [DOI] [PubMed] [Google Scholar]
- 43.Egan DF, Chun MG, Vamos M, Zou H, Rong J, Miller CJ, et al. Small molecule inhibition of the autophagy kinase ULK1 and identification of ULK1 substrates. Mol Cell. 2015;59:285–97. doi: 10.1016/j.molcel.2015.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Petherick KJ, Conway OJ, Mpamhanga C, Osborne SA, Kamal A, Saxty B, et al. Pharmacological inhibition of ULK1 kinase blocks mammalian target of rapamycin (mTOR)-dependent autophagy. J Biol Chem. 2015;290:28726. doi: 10.1074/jbc.A114.627778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lin TW, Chen MT, Lin LT, Huang PI, Lo WL, Yang YP, et al. TDP-43/HDAC6 axis promoted tumor progression and regulated nutrient deprivation-induced autophagy in glioblastoma. Oncotarget. 2017;8:56612–25. [DOI] [PMC free article] [PubMed]
- 46.Dyczynski M, Yu Y, Otrocka M, Parpal S, Braga T, Henley AB, et al. Targeting autophagy by small molecule inhibitors of vacuolar protein sorting 34 (Vps34) improves the sensitivity of breast cancer cells to Sunitinib. Cancer Lett. 2018;435:32–43. doi: 10.1016/j.canlet.2018.07.028. [DOI] [PubMed] [Google Scholar]
- 47.Zahedi SF, Fitzwalter BE, Morin A, Grob S, Desmarais M, Nellan A, et al. Effect of early stage autophagy inhibition in BRAFV600E autophagy dependent brain tumor cells. Cell Death Dis. 2019;10:679. [DOI] [PMC free article] [PubMed]
- 48.Kurdi A, Cleenewerck M, Vangestel C, Lyssens S, Declercq W, Timmermans JP, et al. ATG4B inhibitors with a benzotropolone core structure block autophagy and augment efficiency of chemotherapy in mice. Biochem Pharm. 2017;138:150–62. doi: 10.1016/j.bcp.2017.06.119. [DOI] [PubMed] [Google Scholar]
- 49.Fu Y, Hong L, Xu J, Zhong G, Gu Q, Gu Q, et al. Discovery of a small molecule targeting autophagy via ATG4B inhibition and cell death of colorectal cancer cells in vitro and in vivo. Autophagy. 2019;15:295–311. doi: 10.1080/15548627.2018.1517073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pasquier B. Autophagy inhibitors. Cell Mol life Sci. 2016;73:985–1001. doi: 10.1007/s00018-015-2104-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jensen M, Mehlhorn H. Seventy-five years of Resochin in the fight against malaria. Parasitol Res. 2009;105:609–27. doi: 10.1007/s00436-009-1524-8. [DOI] [PubMed] [Google Scholar]
- 52.Finbloom DS, Silver K, Newsome DA, Gunkel R. Comparison of hydroxychloroquine and chloroquine use and the development of retinal toxicity. J Rheumatol. 1985;12:692–4. [PubMed] [Google Scholar]
- 53.Canadian Hydroxychloroquine Study Group. A randomized study of the effect of withdrawing hydroxychloroquine sulfate in systemic lupus erythematosus. N Engl J Med. 1991;324:150–4. [DOI] [PubMed]
- 54.Bell CL. Hydroxychloroquine sulfate in rheumatoid arthritis: long-term response rate and predictive parameters. Am J Med. 1983;75:46–51. doi: 10.1016/0002-9343(83)91270-6. [DOI] [PubMed] [Google Scholar]
- 55.Laaksonen AL, Koskiahde V, Juva K. Dosage of antimalarial drugs for children with juvenile rheumatoid arthritis and systemic lupus erythematosus. A clinical study with determination of serum concentrations of chloroquine and hydroxychloroquine. Scand J Rheumatol. 1974;3:103–8. doi: 10.3109/03009747409115809. [DOI] [PubMed] [Google Scholar]
- 56.Bedoya V. Effect of chloroquine on malignant lymphoreticular and pigmented cells in vitro. Cancer Res. 1970;30:1262–75. [PubMed] [Google Scholar]
- 57.Murakami N, Oyama F, Gu Y, McLennan IS, Nonaka I, Ihara Y. Accumulation of tau in autophagic vacuoles in chloroquine myopathy. J Neuropathol Exp Neurol. 1998;57:664–73. doi: 10.1097/00005072-199807000-00003. [DOI] [PubMed] [Google Scholar]
- 58.Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI, et al. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest. 2007;117:326–36. doi: 10.1172/JCI28833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Thorburn A, Thamm DH, Gustafson DL. Autophagy and cancer therapy. Mol Pharmacol. 2014;85:830–8. doi: 10.1124/mol.114.091850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Briceno E, Calderon A, Sotelo J. Institutional experience with chloroquine as an adjuvant to the therapy for glioblastoma multiforme. Surg. Neurol. 2007;67:388–91. doi: 10.1016/j.surneu.2006.08.080. [DOI] [PubMed] [Google Scholar]
- 61.Sotelo J, Briceno E, Lopez-Gonzalez MA. Adding chloroquine to conventional treatment for glioblastoma multiforme: a randomized, double-blind, placebo-controlled trial. Ann Intern Med. 2006;144:337–43. doi: 10.7326/0003-4819-144-5-200603070-00008. [DOI] [PubMed] [Google Scholar]
- 62.Eldredge Harriet B., DeNittis Albert, DuHadaway James B., Chernick Michael, Metz Richard, Prendergast George C. Concurrent whole brain radiotherapy and short-course chloroquine in patients with brain metastases: a pilot trial. Journal of Radiation Oncology. 2013;2(3):315–321. doi: 10.1007/s13566-013-0111-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rojas-Puentes LL, Gonzalez-Pinedo M, Crismatt A, Ortega-Gomez A, Gamboa-Vignolle C, Nunez-Gomez R, et al. Phase II randomized, double-blind, placebo-controlled study of whole-brain irradiation with concomitant chloroquine for brain metastases. Radiat Oncol. 2013;8:209. doi: 10.1186/1748-717X-8-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mahalingam D, Mita M, Sarantopoulos J, Wood L, Amaravadi R, Davis LE, et al. Combined autophagy and HDAC inhibition: a phase I safety, tolerability, pharmacokinetic, and pharmacodynamic analysis of hydroxychloroquine in combination with the HDAC inhibitor vorinostat in patients with advanced solid tumors. Autophagy. 2014;10:1403–14. doi: 10.4161/auto.29231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rangwala R, Chang YC, Hu J, Algazy K, Evans T, Fecher L, et al. Combined MTOR and autophagy inhibition: Phase I trial of hydroxychloroquine and temsirolimus in patients with advanced solid tumors and melanoma. Autophagy. 2014;10:1391–402. doi: 10.4161/auto.29119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rangwala R, Leone R, Chang YC, Fecher L, Schuchter L, Kramer A, et al. Phase I trial of hydroxychloroquine with dose-intense temozolomide in patients with advanced solid tumors and melanoma. Autophagy. 2014;10:1369–79. doi: 10.4161/auto.29118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rosenfeld MR, Ye X, Supko JG, Desideri S, Grossman SA, Brem S, et al. A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy. 2014;10:1359–68. doi: 10.4161/auto.28984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vogl DT, Stadtmauer EA, Tan K-S, Heitjan DF, Davis LE, Pontiggia L, et al. Combined autophagy and proteasome inhibition: a phase 1 trial of hydroxychloroquine and bortezomib in patients with relapsed/refractory myeloma. Autophagy. 2014;10:1380–90. doi: 10.4161/auto.29264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wolpin BM, Rubinson DA, Wang X, Chan JA, Cleary JM, Enzinger PC, et al. Phase II and pharmacodynamic study of autophagy inhibition using hydroxychloroquine in patients with metastatic pancreatic adenocarcinoma. Oncologist. 2014;19:637–8. doi: 10.1634/theoncologist.2014-0086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pellegrini P, Strambi A, Zipoli C, Hägg-Olofsson M, Buoncervello M, Linder S, et al. Acidic extracellular pH neutralizes the autophagy-inhibiting activity of chloroquine: implications for cancer therapies. Autophagy. 2014;10:562–71. doi: 10.4161/auto.27901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Collins KP, Jackson KM, Gustafson DL. Hydroxychloroquine: a physiologically-based pharmacokinetic model in the context of cancer-related autophagy modulation. J Pharmacol Exp Ther. 2018;365:447–59. doi: 10.1124/jpet.117.245639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.McAfee Q, Zhang Z, Samanta A, Levi SM, Ma XH, Piao S, et al. Autophagy inhibitor Lys05 has single-agent antitumor activity and reproduces the phenotype of a genetic autophagy deficiency. Proc Natl Acad Sci USA. 2012;109:8253–8. doi: 10.1073/pnas.1118193109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yang A, Rajeshkumar NV, Wang X, Yabuuchi S, Alexander BM, Chu GC, et al. Autophagy is critical for pancreatic tumor growth and progression in tumors with p53 alterations. Cancer Discov. 2014;4:905–13. doi: 10.1158/2159-8290.CD-14-0362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Boone BA, Bahary N, Zureikat AH, Moser AJ, Normolle DP, Wu WC, et al. Safety and biologic response of pre-operative autophagy inhibition in combination with gemcitabine in patients with pancreatic adenocarcinoma. Ann Surg Oncol. 2015;22:4402–10. doi: 10.1245/s10434-015-4566-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Eng CH, Wang Z, Tkach D, Toral-Barza L, Ugwonali S, Liu S, et al. Macroautophagy is dispensable for growth of KRAS mutant tumors and chloroquine efficacy. Proc Natl Acad Sci USA. 2016;113:182–7. doi: 10.1073/pnas.1515617113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Maes H, Kuchnio A, Peric A, Moens S, Nys K, De Bock K, et al. Tumor vessel normalization by chloroquine independent of autophagy. Cancer Cell. 2014;26:190–206. doi: 10.1016/j.ccr.2014.06.025. [DOI] [PubMed] [Google Scholar]
- 77.Maycotte P, Aryal S, Cummings CT, Thorburn J, Morgan MJ, Thorburn A. Chloroquine sensitizes breast cancer cells to chemotherapy independent of autophagy. Autophagy. 2012;8:200–12. doi: 10.4161/auto.8.2.18554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rainsford KD, Parke AL, Clifford-Rashotte M, Kean WF. Therapy and pharmacological properties of hydroxychloroquine and chloroquine in treatment of systemic lupus erythematosus, rheumatoid arthritis and related diseases. Inflammopharmacology. 2015;23:231–69. doi: 10.1007/s10787-015-0239-y. [DOI] [PubMed] [Google Scholar]
- 79.Das SK, Pareek A, Mathur DS, Wanchu A, Srivastava R, Agarwal GG, et al. Efficacy and safety of hydroxychloroquine sulphate in rheumatoid arthritis: a randomized, double-blind, placebo controlled clinical trial-an Indian experience. Curr Med Res Opin. 2007;23:2227–34. doi: 10.1185/030079907X219634. [DOI] [PubMed] [Google Scholar]
- 80.Vidoni C, Ferraresi A, Secomandi E, Vallino L, Dhanasekaran DN, Isidoro C. Epigenetic targeting of autophagy for cancer prevention and treatment by natural compounds. Semin Cancer Biol. 2019. 10.1016/j.semcancer.2019.04.006. [DOI] [PubMed]
- 81.Lopez G, Torres K, Liu J, Hernandez B, Young E, Belousov R, et al. Autophagic survival in resistance to histone deacetylase inhibitors: novel strategies to treat malignant peripheral nerve sheath tumors. Cancer Res. 2011;71:185–96. doi: 10.1158/0008-5472.CAN-10-2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Liu J, Li M, Wang Y, Luo J. Curcumin sensitizes prostate cancer cells to radiation partly via epigenetic activation of miR-143 and miR-143 mediated autophagy inhibition. J Drug Target. 2017;25:645–52. doi: 10.1080/1061186X.2017.1315686. [DOI] [PubMed] [Google Scholar]
- 83.Altman JK, Szilard A, Goussetis DJ, Sassano A, Colamonici M, Gounaris E, et al. Autophagy is a survival mechanism of acute myelogenous leukemia precursors during dual mTORC2/mTORC1 targeting. Clin Cancer Res. 2014;20:2400–9. doi: 10.1158/1078-0432.CCR-13-3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kun Zhuang, Hanqing Guo, Hailing Tang, Yuan Yan, Jun Zhang, Lingxia Zhang, Kun Han, Xin Zhang. Gastrin Enhances Autophagy and Promotes Gastric Carcinoma Proliferation via Inducing AMPKα. Oncology Research Featuring Preclinical and Clinical Cancer Therapeutics. 2017;25(8):1399–1407. doi: 10.3727/096504016X14823648620870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Masui A, Hamada M, Kameyama H, Wakabayashi K, Takasu A, Imai T, et al. Autophagy as a survival mechanism for squamous cell carcinoma cells in endonuclease G-mediated apoptosis. PLoS ONE. 2016;11:e0162786. doi: 10.1371/journal.pone.0162786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fitzwalter BE, Thorburn A. Recent insights into cell death and autophagy. FEBS J. 2015;282:4279–88. doi: 10.1111/febs.13515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Singh R, Letai A, Sarosiek K. Regulation of apoptosis in health and disease: the balancing act of BCL-2 family proteins. Nat Rev Mol Cell Biol. 2019;20:175–93. doi: 10.1038/s41580-018-0089-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Marino G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. 2014;15:81–94. doi: 10.1038/nrm3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tait SW, Parsons MJ, Llambi F, Bouchier-Hayes L, Connell S, Munoz-Pinedo C, et al. Resistance to caspase-independent cell death requires persistence of intact mitochondria. Dev Cell. 2010;18:802–13. doi: 10.1016/j.devcel.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ichim G, Lopez J, Ahmed SU, Muthalagu N, Giampazolias E, Delgado ME, et al. Limited mitochondrial permeabilization causes DNA damage and genomic instability in the absence of cell death. Mol Cell. 2015;57:860–72. doi: 10.1016/j.molcel.2015.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Thorburn J, Andrysik Z, Staskiewicz L, Gump J, Maycotte P, Oberst A, et al. Autophagy controls the kinetics and extent of mitochondrial apoptosis by regulating PUMA levels. Cell Rep. 2014;7:45–52. doi: 10.1016/j.celrep.2014.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Liu X, He Y, Li F, Huang Q, Kato TA, Hall RP, et al. Caspase-3 promotes genetic instability and carcinogenesis. Mol Cell. 2015;58:284–96. doi: 10.1016/j.molcel.2015.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. 2014;15:49–63. doi: 10.1038/nrm3722. [DOI] [PubMed] [Google Scholar]
- 94.Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–39. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 95.Warr Matthew R., Binnewies Mikhail, Flach Johanna, Reynaud Damien, Garg Trit, Malhotra Ritu, Debnath Jayanta, Passegué Emmanuelle. FOXO3A directs a protective autophagy program in haematopoietic stem cells. Nature. 2013;494(7437):323–327. doi: 10.1038/nature11895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Fitzwalter BE, Towers CG, Sullivan KD, Andrysik Z, Hoh M, Ludwig M, et al. Autophagy inhibition mediates apoptosis sensitization in cancer therapy by relieving FOXO3a turnover. Dev Cell. 2018;44:555–65.e3. doi: 10.1016/j.devcel.2018.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fitzwalter BE, Thorburn A. FOXO3 links autophagy to apoptosis. Autophagy. 2018;14:1467–8. doi: 10.1080/15548627.2018.1475819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Folkerts H, Wierenga AT, van den Heuvel FA, Woldhuis RR, Kluit DS, Jaques J, et al. Elevated VMP1 expression in acute myeloid leukemia amplifies autophagy and is protective against venetoclax-induced apoptosis. Cell Death Dis. 2019;10:421. doi: 10.1038/s41419-019-1648-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ko A, Kanehisa A, Martins I, Senovilla L, Chargari C, Dugue D, et al. Autophagy inhibition radiosensitizes in vitro, yet reduces radioresponses in vivo due to deficient immunogenic signalling. Cell Death Differ. 2014;21:92–99. doi: 10.1038/cdd.2013.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P, et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science. 2011;334:1573–7. doi: 10.1126/science.1208347. [DOI] [PubMed] [Google Scholar]
- 101.Michaud M, Xie X, Bravo-San Pedro JM, Zitvogel L, White E, Kroemer G. An autophagy-dependent anticancer immune response determines the efficacy of melanoma chemotherapy. Oncoimmunology. 2014;3:e944047. doi: 10.4161/21624011.2014.944047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pietrocola F, Pol J, Vacchelli E, Rao S, Enot DP, Baracco EE, et al. Caloric Restriction mimetics enhance anticancer immunosurveillance. Cancer Cell. 2016;30:147–60. doi: 10.1016/j.ccell.2016.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Li Y, Hahn T, Garrison K, Cui ZH, Thorburn A, Thorburn J, et al. The vitamin E analogue alpha-TEA stimulates tumor autophagy and enhances antigen cross-presentation. Cancer Res. 2012;72:3535–45. doi: 10.1158/0008-5472.CAN-11-3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ladoire S, Penault-Llorca F, Senovilla L, Dalban C, Enot D, Locher C, et al. Combined evaluation of LC3B puncta and HMGB1 expression predicts residual risk of relapse after adjuvant chemotherapy in breast cancer. Autophagy. 2015;11:1878–90. doi: 10.1080/15548627.2015.1082022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ladoire S, Enot D, Senovilla L, Ghiringhelli F, Poirier-Colame V, Chaba K, et al. The presence of LC3B puncta and HMGB1 expression in malignant cells correlate with the immune infiltrate in breast cancer. Autophagy. 2016;12:864–75. doi: 10.1080/15548627.2016.1154244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Jiang GM, Tan Y, Wang H, Peng L, Chen HT, Meng XJ, et al. The relationship between autophagy and the immune system and its applications for tumor immunotherapy. Mol Cancer. 2019;18:17. doi: 10.1186/s12943-019-0944-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Baginska J, Viry E, Berchem G, Poli A, Noman MZ, van Moer K, et al. Granzyme B degradation by autophagy decreases tumor cell susceptibility to natural killer-mediated lysis under hypoxia. Proc Natl Acad Sci USA. 2013;110:17450–5. doi: 10.1073/pnas.1304790110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.DeVorkin L, Pavey N, Carleton G, Comber A, Ho C, Lim J, et al. Autophagy regulation of metabolism is required for CD8(+) T cell anti-tumor immunity. Cell Rep. 2019;27:502–13 e505. doi: 10.1016/j.celrep.2019.03.037. [DOI] [PubMed] [Google Scholar]
- 109.Liang X, De Vera ME, Buchser WJ, Romo de Vivar Chavez A, Loughran P, Beer Stolz D, et al. Inhibiting systemic autophagy during interleukin 2 immunotherapy promotes long-term tumor regression. Cancer Res. 2012;72:2791–801. doi: 10.1158/0008-5472.CAN-12-0320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Starobinets H, Ye J, Broz M, Barry K, Goldsmith J, Marsh T, et al. Antitumor adaptive immunity remains intact following inhibition of autophagy and antimalarial treatment. J Clin Invest. 2016;126:4417–29. doi: 10.1172/JCI85705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Karsli-Uzunbas G, Guo JY, Price S, Teng X, Laddha SV, Khor S, et al. Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov. 2014;4:914–27. doi: 10.1158/2159-8290.CD-14-0363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Levy JM, Thompson JC, Griesinger AM, Amani V, Donson AM, Birks DK, et al. Autophagy inhibition improves chemosensitivity in BRAF(V600E) brain tumors. Cancer Discov. 2014;4:773–80. doi: 10.1158/2159-8290.CD-14-0049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Mulcahy Levy JM, Zahedi S, Griesinger AM, Morin A, Davies KD, Aisner DL, et al. Autophagy inhibition overcomes multiple mechanisms of resistance to BRAF inhibition in brain tumors. Elife. 2017:6 pii: e19671. 10.7554/eLife.19671. [DOI] [PMC free article] [PubMed]
- 114.La Belle Flynn A, Calhoun BC, Sharma A, Chang JC, Almasan A, Schiemann WP. Autophagy inhibition elicits emergence from metastatic dormancy by inducing and stabilizing Pfkfb3 expression. Nat Commun. 2019;10:3668. doi: 10.1038/s41467-019-11640-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Maycotte P, Gearheart CM, Barnard R, Aryal S, Mulcahy Levy JM, Fosmire SP, et al. STAT3-mediated autophagy dependence identifies subtypes of breast cancer where autophagy inhibition can be efficacious. Cancer Res. 2014;74:2579–90. doi: 10.1158/0008-5472.CAN-13-3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Levy JMM, Thompson JC, Griesinger AM, Amani V, Donson AM, Birks DK, et al. Autophagy inhibition improves chemosensitivity in BRAFV600E brain tumors. Cancer Discov. 2014;4:773–80. doi: 10.1158/2159-8290.CD-14-0049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Strohecker AM, Guo JY, Karsli-Uzunbas G, Price SM, Chen GJ, Mathew R, et al. Autophagy sustains mitochondrial glutamine metabolism and growth of BrafV600E-driven lung tumors. Cancer Discov. 2013;3:1272–85. doi: 10.1158/2159-8290.CD-13-0397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Guo JY, Chen H-Y, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011;25:460–70. doi: 10.1101/gad.2016311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lock R, Roy S, Kenific CM, Su JS, Salas E, Ronen SM, et al. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol Biol Cell. 2011;22:165–78. doi: 10.1091/mbc.E10-06-0500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011;25:717–29. doi: 10.1101/gad.2016111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bryant KL, Stalnecker CA, Zeitouni D, Klomp JE, Peng S, Tikunov AP, et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat Med. 2019;25:628–40. doi: 10.1038/s41591-019-0368-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kinsey CG, Camolotto SA, Boespflug AM, Guillen KP, Foth M, Truong A, et al. Protective autophagy elicited by RAF->MEK->ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat Med. 2019;25:620–7. doi: 10.1038/s41591-019-0367-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Perera RM, Stoykova S, Nicolay BN, Ross KN, Fitamant J, Boukhali M, et al. Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature. 2015;524:361–5. doi: 10.1038/nature14587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Sousa CM, Biancur DE, Wang X, Halbrook CJ, Sherman MH, Zhang L, et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature. 2016;536:479–83. doi: 10.1038/nature19084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.MEK and autophagy inhibition in metastatic/locally advanced, unresectable neuroblastoma RAS (NRAS) melanoma. https://ClinicalTrials.gov/show/NCT03979651.
- 126.Trametinib and hydroxychloroquine in treating patients with pancreatic cancer. https://ClinicalTrials.gov/show/NCT03825289.
- 127.Vasilevskaya IA, Selvakumaran M, Roberts D, O'Dwyer PJ. JNK1 inhibition attenuates hypoxia-induced autophagy and sensitizes to chemotherapy. Mol Cancer Res. 2016;14:753–63. doi: 10.1158/1541-7786.MCR-16-0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.A phase I/II/pharmacodynamic study of hydroxychloroquine in combination with gemcitabine/abraxane to inhibit autophagy in pancreatic cancer. https://ClinicalTrials.gov/show/NCT01506973.
- 129.Jutten B, Rouschop KM. EGFR signaling and autophagy dependence for growth, survival, and therapy resistance. Cell Cycle. 2014;13:42–51. doi: 10.4161/cc.27518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Jutten B, Keulers TG, Schaaf MB, Savelkouls K, Theys J, Span PN, et al. EGFR overexpressing cells and tumors are dependent on autophagy for growth and survival. Radiother Oncol. 2013;108:479–83. doi: 10.1016/j.radonc.2013.06.033. [DOI] [PubMed] [Google Scholar]
- 131.Jutten B, Keulers TG, Peeters HJM, Schaaf MBE, Savelkouls KGM, Compter I, et al. EGFRvIII expression triggers a metabolic dependency and therapeutic vulnerability sensitive to autophagy inhibition. Autophagy. 2018;14:283–95. doi: 10.1080/15548627.2017.1409926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Erlotinib with or without hydroxychloroquine in chemo-naive advanced NSCLC and (EGFR) mutations. https://ClinicalTrials.gov/show/NCT00977470.
- 133.The addition of chloroquine to chemoradiation for glioblastoma. https://ClinicalTrials.gov/show/NCT02378532.
- 134.Chi KH, Ko HL, Yang KL, Lee CY, Chi MS, Kao SJ. Addition of rapamycin and hydroxychloroquine to metronomic chemotherapy as a second line treatment results in high salvage rates for refractory metastatic solid tumors: a pilot safety and effectiveness analysis in a small patient cohort. Oncotarget. 2015;6:16735–45. doi: 10.18632/oncotarget.3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Chi MS, Lee CY, Huang SC, Yang KL, Ko HL, Chen YK, et al. Double autophagy modulators reduce 2-deoxyglucose uptake in sarcoma patients. Oncotarget. 2015;6:29808–17. doi: 10.18632/oncotarget.5060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Bilger A, Bittner MI, Grosu AL, Wiedenmann N, Meyer PT, Firat E, et al. FET-PET-based reirradiation and chloroquine in patients with recurrent glioblastoma: first tolerability and feasibility results. Strahlenther Onkol. 2014;190:957–61. doi: 10.1007/s00066-014-0693-2. [DOI] [PubMed] [Google Scholar]
- 137.Goldberg SB, Supko JG, Neal JW, Muzikansky A, Digumarthy S, Fidias P, et al. A phase I study of erlotinib and hydroxychloroquine in advanced non-small-cell lung cancer. J Thorac Oncol. 2012;7:1602–8. doi: 10.1097/JTO.0b013e318262de4a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.FOLFOX/Bevacizumab/Hydroxychloroquine (HCQ) in colorectal cancer. https://ClinicalTrials.gov/show/NCT01206530.
- 139.Göttingen Uo, Kinderkrebsstiftung D, School HM. International cooperative phase III trial of the HIT-HGG study group (HIT-HGG-2013). 2018. https://ClinicalTrials.gov/show/NCT03243461.
- 140.Randomized phase II trial of pre-operative gemcitabine and nab paclitacel with or with out hydroxychloroquine. https://ClinicalTrials.gov/show/NCT01978184.
- 141.Study of pre-surgery gemcitabine + hydroxychloroquine (GcHc) in stage IIb or III adenocarcinoma of the pancreas. https://ClinicalTrials.gov/show/NCT01128296.
- 142.A phase I trial of vemurafenib and hydroxychloroquine in patients with advanced BRAF mutant melanoma. https://ClinicalTrials.gov/show/NCT01897116.
- 143.The BAMM Trial: BRAF, autophagy and MEK inhibition in metastatic melanoma: a phase I/2 trial of dabrafenib, trametinib and hydroxychloroquine in patients with advanced BRAF mutant melanoma. https://ClinicalTrials.gov/show/NCT02257424. [DOI] [PMC free article] [PubMed]
- 144.Sorafenib induced autophagy using hydroxychloroquine in hepatocellular cancer. https://ClinicalTrials.gov/show/NCT03037437.
- 145.Gemcitabine, docetaxel, and hydroxychloroquine in treating participants with recurrent or refractory osteosarcoma. https://ClinicalTrials.gov/show/NCT03598595.
- 146.Hydroxychloroquine, palbociclib, and letrozole before surgery in treating participants with estrogen receptor positive, HER2 negative breast cancer. https://ClinicalTrials.gov/show/NCT03774472.