

Abstract
Gynaecological cancer is a main subtype of cancer in women, and acquired chemoresistance is a major contributor to the poor prognosis of gynaecological cancer, but its underlying mechanism remains ill-defined. JOSD1 has been recognized as a deubiquitinase, but its biological functions remain largely unknown, especially in the context of cancer. Here we established a chemoresistant xenograft model and acquired chemoresistant cell lines to mimic the establishment of acquired chemoresistance. We identified that JOSD1 is the most upregulated DUB during the development of chemoresistance. JOSD1 depletion led to severe apoptosis in gynaecological cancer cells both in vivo and in vitro. Mechanistically, we showed that JOSD1 deubiquitinated and stabilized MCL1 to suppress mitochondrial apoptotic signalling. JOSD1 overexpression caused chemoresistance in gynaecological cancer by upregulating the MCL1 protein. Importantly, high JOSD1 expression was correlated with poor prognosis among ovarian cancer patients, and serum JOSD1 levels could be a marker for clinical diagnosis. Our study showed that JOSD1 is a novel and critical oncogene that contributes to the acquisition of chemoresistance by inhibiting mitochondrial apoptotic signalling via MCL1 stabilization. We also suggest that JOSD1 is an ideal therapeutic target and a promising diagnostic marker.
Subject terms: Oncogenes, Deubiquitylating enzymes, Tumour biomarkers, Translational research
Introduction
Gynaecologic malignancy is the primary cancer subtype occurring in women, among which ovarian and cervical cancers are the most common. Ovarian cancer is the leading cause of death among gynaecologic malignancies, with a 5-year survival rate of <50% in women diagnosed with late-stage ovarian cancer [1, 2]. Despite the high response rate for chemotherapy, the majority of patients develop resistance to first-line agents, and the resultant prognosis of these patients is particularly poor [3, 4]. Cervical cancer is the second most common cancer among women [5], accounting for 528,000 new cases and 266,000 deaths worldwide each year [6]. Although the development of early diagnostic methods has improved the diagnosis of cervical cancer, patients at the progressive stage still suffer from a poor prognosis, and novel strategies for the treatment of progressive gynaecologic cancer are urgently needed [7, 8].
The ability to escape apoptosis is one of the hallmarks of cancer cells [9, 10]. Apoptosis can be triggered by extracellular or intracellular stimuli. The extrinsic pathway is activated by the binding of death-inducing ligands to death receptors expressed on the cell surface, while intrinsic apoptotic stimuli include DNA damage, growth factor deprivation and oxidative stress [11]. BCL-2 family members contain up to four conserved BCL-2 homology (BH) regions (BH1–4). The antiapoptotic proteins BCL-2, BCL-XL, BCL-W, myeloid cell leukaemia 1 (MCL1) and BCL-2A1 antagonize proapoptotic BH3-only proteins to inhibit the essential apoptosis effectors BCL-2 antagonist killer 1 (BAK) and BCL-2-associated X protein (BAX) [12]. Inhibition of apoptosis is considered to be a main contributor to chemoresistance in human cancer [13]; thus targeting the BCL-2 family is thought to be a promising therapeutic strategy for overcoming chemoresistance [14, 15].
Ubiquitination is one of the common posttranslational modifications, by which the C terminus of the 76 amino acid protein ubiquitin is covalently bound to ɛ-amino groups of lysine (K) residues (or, less commonly, to amino termini) of the substrate proteins. Substrates linked with polyubiquitin chains are directed to the 26S proteasome and, in most circumstances, subsequently undergo degradation through the ubiquitin–proteasome system (UPS) [16]. The UPS plays a critical role in regulating a wide variety of cellular events, such as cell growth, proliferation, apoptosis and DNA repair [17, 18]. Emerging studies have revealed the key functions of UPS in human diseases [19, 20]. Similar to other posttranslational modifications, ubiquitination could be reversed by peptidases termed deubiquitinating enzymes (also known as deubiquitinases (DUBs)) that cleave ubiquitin from the substrates, modify ubiquitin chains and process ubiquitin precursors [21]. DUBs are classified into six families: USPs (ubiquitin-specific proteases), UCHs (ubiquitin carboxy-terminal hydrolases), MJDs (Machado–Josephin domain-containing proteases), OTUs (ovarian tumour proteases), MINDYs (motif-interacting with ubiquitin-containing novel DUB family) and JAMMs (the JAB1, MPN, MOV34 family) [22]. The USP family comprises a large part of DUBs and is the most reviewed DUB family in cancer [23–26], while the roles of other families in cancer development are less studied.
Here we first established an in vivo chemoresistance model and identified that JOSD1 is a contributor to chemoresistance. JOSD1 depletion in gynaecological cancer cells triggered severe apoptosis and inhibited tumour growth both in vitro and in vivo. Moreover, adeno-associated virus (AAV)-mediated short hairpin RNA (shRNA) interference mimicking clinical gene therapy significantly inhibited the growth of patient-derived xenografts (PDXs) comprising ovarian cancer cells. Mechanically, JOSD1 deubiquitinated and stabilized the antiapoptotic protein MCL1, which then protected cells from apoptosis by inhibiting the mitochondrial apoptosis pathway. Clinical samples showed a strong relationship between JOSD1/MCL1 expression and the chemoresistance status of ovarian cancer patients. Most importantly, the serum JOSD1 levels were positively correlated with the intracellular JOSD1 expression levels, suggesting that serum JOSD1 is a promising prognostic indicator in gynaecological cancer patients. Our study identifies JOSD1 as a novel DUB that plays a key role in the antiapoptotic process of cancer and provides a potential therapeutic opportunity for the clinical treatment of gynaecological cancer.
Materials and methods
Cell culture and plasmids
HeLa, 293T and ES2 cell lines were purchased from the American Type Culture Collection (ATCC; Manassas, VA, USA), A2780 cells were derived from the National Experimental Cell Resource Sharing Platform (Beijing, China), and 3AO cells were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). HeLa and 293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% foetal bovine serum (FBS), and ES2, A2780 and 3AO cells were maintained in RPMI 1640 supplemented with 10% FBS. All cell lines were routinely authenticated using short tandem repeat (STR) DNA fingerprinting and were tested with a MycoBlue Mycoplasma Detector (Vazyme Biotech, Nanjing, China) to exclude mycoplasma contamination before cell use in any experiments. The acquired chemoresistant cell lines 3AO/CBP and A2780/CBP were established from parental lines. Briefly, parental cells were exposed to high doses of carboplatin (CBP) for 2 h and then cultured in complete medium; once the cell growth reached the logarithmic phase, the cells were re-exposed to high doses of CBP. After 6–10 cycles, the acquired chemoresistant cell lines were established. Ovarian cancer primary cell lines were established via sterile processing of fresh tumour biopsy specimens from patients at the time of primary resection. Tumours were mechanically disrupted, enzymatically digested and filtered through a 100-μm filter to obtain single-cell suspensions with the Cell Suspension Isolation Kit from Fresh/Frozen Tissues (Invent, Plymouth, MN, USA) according to the manufacturer’s protocol. Primary cells were cultured in DMEM supplemented with 20% FBS and were used within 10 passages. (Cell line characteristics and tissue sources are described in Supplementary Table 1.).
Full-length or truncated cDNAs were cloned into the pLVX-IRES-neo vector using a PCR-based approach. The mutants (JOSD1C36A, K0, K48 and K63) were generated using a Q5® Site-Directed Mutagenesis Kit (New England Biolabs, Ipswich, MA, USA) according to the manufacturer’s instructions.
Antibodies and reagents
The antibodies used for immunoblotting (IB), immunoprecipitation (IP), immunohistochemistry (IHC) and immunofluorescence (IF) in this study targeted the following proteins and were used at the dilutions listed: JOSD1, 1:1000 (IB; Abcam, ab118221), 1:100 (IHC, Sigma-Aldrich, HPA001168); MCL1, 1:1000 (IB; Cell Signaling Technology, #94296), 1:100 (IP; Cell Signaling Technology, #94296) and 1:400 (IHC; Sigma-Aldrich, HPA008455); V5-Tag, 1:1000 (IB; Cell Signaling Technology, #13202), 1:100 (IP; Cell Signaling Technology, #13202); Flag-Tag, 1:1000 (IB; Cell Signaling Technology, #8146); Myc-Tag, 1:1000 (IB; Cell Signaling Technology, #2276); HA-Tag, 1:5000 (IB; Abcam, ab9110); Ubiquitin, 1:1000 (IB; Cell Signaling Technology, #3936); Cleaved caspase-3, 1:1000 (IB; Cell Signaling Technology, #9664); Caspase-9, 1:1000 (IB; Cell Signaling Technology, #9502); PARP, 1:1000 (IB; Cell Signaling Technology, #9542); β-actin, 1:4000 (IB; Sigma-Aldrich, A5316); and normal rabbit IgG (IP; Santa Cruz, sc-2027). Cycloheximide (CHX; HY-12320) was obtained from MedChemExpress (MCE, NJ, USA), MG132 (C2211) was purchased from Sigma-Aldrich (St. Louis, MO, USA), and Z-VAD(OMe)-FMK was purchased from TargetMol (MA, USA).
Quantitative real-time PCR (qRT-PCR)
Cellular RNA was extracted from cells using TRIzol reagent (Thermo Scientific, Grand Island, NY, USA) and then reverse transcribed to cDNA using a Quantscript RT Kit (Tiangen, Beijing, China). The qRT-PCR analysis was performed on a StepOnePlus Real-Time PCR system (Applied Biosystems (ABI), Foster City, CA, USA) using standard procedures. The relative expression levels of the target genes were normalized to those of the housekeeping gene GAPDH. For screening the distinctly expressed DUBs, primers targeting 45 DUBs in the SENPs, OUTs, MJDs, JAMMs and UCHs families were used. The MCL1 (HQP010025) primers used in the qRT-PCR analyses were purchased from FulenGen (Guangzhou, China). The rest primers used for the qRT-PCR analyses are listed in Supplementary Table 2.
Immunoprecipitation and western blotting
Cell samples were lysed with RIPA lysis buffer supplemented with a protease inhibitor cocktail (Roche, Basel, Switzerland) for 20 min on ice and were then centrifuged at 14,000 × g for 10 min. The supernatants were collected, and the total protein concentrations were measured using a BCA Kit (Thermo Scientific). For immunoprecipitation, equal amounts of lysate were incubated with anti-FLAG M2 affinity gels (A2220, Sigma) or anti-V5-tag pAb-agarose (D291-8, MBL, Japan) or with protein A/G magnetic beads (HY-K0202, MCE) and anti-MCL1 antibody overnight at 4 °C. Thereafter, the beads were washed three times with cell lysis buffer, and the immunoprecipitated proteins were analysed by western blotting. The samples were separated on 10–15% gels depending on the molecular weights of the proteins, and then the proteins were transferred onto polyvinylidene difluoride membranes (Merck Millipore, Billerica, MA, USA).
Silver staining and mass spectrometry
Cellular extracts from ES2 cells stably expressing empty vector or Flag-JOSD1 were incubated with anti-FLAG M2 affinity gel (A2220, Sigma) and eluted with 1× loading buffer. The eluted proteins were collected and resolved on 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Gel was silver-stained using PierceTM Silver Stain for Mass Spectrometry (24600, Thermo Scientific) according to the manufacturer’s protocol, and bands were excised and subjected to a gel-based liquid chromatography–tandem mass spectrometric analysis.
In vivo deubiquitination assay
The in vivo deubiquitination assay was carried out in 293T and gynaecological cancer cells. In 293T cells, HA-Ub and Flag-MCL1 were co-transfected with empty vector, Myc-JOSD1 or Myc-JOSD1C36A plasmid as indicated. The cells were treated for 6 h with 20 mM MG132 before they were harvested. Proteins in the cell lysate were immunoprecipitated to isolate ubiquitinated MCL1, which was detected with an anti-HA antibody. In the gynaecological cancer cells, the experimental procedures were performed as described above, except for the isolation of ubiquitinated MCL1 with an anti-MCL1 antibody and the detection of endogenous ubiquitin chains on MCL1.
In vitro deubiquitination assay
293T cells were transfected with empty vector or HA-Ub and Flag-MCL1 plasmids. Forty-eight hours after transfection, Flag-MCL1 was pulled down by anti-FLAG M2 affinity gels (A2220, Sigma) and then incubated with bacterially purified GST-JOSD1 protein (wild type or C36A). The reaction was carried out at 37 °C for 3 h in deubiquitination buffer (50 mM Tris-HCl, pH 8.0, 50 mM NaCl, 10 mM dithiothreitol, 1 mM EDTA, 5% glycerol). After the reaction, the beads were washed three times with deubiquitination buffer. The proteins bound to beads were released by boiling in 1× loading buffer. The samples were then subjected to western blot analysis with the indicated antibodies.
IF staining
Cells were seeded into a μ-Slide V1 (ibidi, Germany) and cultured until they adhered to the surface. To promote cell adherence, plates were pretreated with the Enhanced Cell Adhesion Kit-3 (ECAK-3) (C028, Beijing Xigong Biological Technology co., LTD., Beijing, China) for 1 h before use. Then the cells were fixed in 4% paraformaldehyde and permeabilized with Triton X-100, after which they were blocked with 5% bovine serum albumin and incubated with primary antibodies overnight. Proteins were visualized by incubation with anti-mouse IgG conjugated to Alexa Fluor 488 (#4412, Cell Signaling Technology) or anti-rabbit IgG conjugated to Alexa Fluor 594 (#9854, Cell Signaling Technology), and nuclei were stained with Hoechst 33342 (Thermo Scientific) for 15 min at room temperature.
Flow cytometry
Flow cytometry was performed using an apoptosis detection kit (Dojindo, Tokyo, Japan) according to the manufacturer’s protocol, as previously described [27].
Terminal deoxynucleotidyl transferase-mediated dUTP-fluorescein nick end labelling (TUNEL) staining
Cell apoptosis was determined using the TUNEL Andy Fluor 488 Apoptosis Detection Kit (GeneCopoeia, MD, USA) in accordance with the manufacturer’s protocol, as previously described [27].
CCK-8 cell proliferation and cell viability assays
CCK-8 reagent (Dojindo) was added to the cell culture medium at a ratio of 1:10, and after the cells were incubated for 1 h at 37 °C, the absorbance was measured at 450 nm. For the cell proliferation assay, 2 × 103 cells were seeded into 96-well microplates, and one plate was measured each day for 5 consecutive days. The relative cell proliferation rate was calculated by normalizing the absorbance at 450 nm on days 2–5 to the absorbance measured on the first day. For the cell viability assay, 1 × 104 ovarian cancer cells were seeded into 96-well microplates, drugs were added after cell adhesion and cell viability was detected 24 h later as described in the proliferation assay. Cell viability was calculated by normalizing the absorbance at 450 nm of the experimental groups to that of the negative control group.
Animal experiments
All animal protocols were approved by the Animal Care and Use Committee of the Chinese Academy of Medical Sciences Cancer Hospital. For subcutaneous xenografting, 1 × 106 gynaecological cells were subcutaneously implanted into 6-week-old female BALB/c nude mice (Nanjing Biomedical Research Institute of Nanjing University, Nanjing, China). Mice were fed under normal conditions and administered saline or CBP, as indicated in the figure legends. The tumour lengths and widths were measured using a caliper, and the volume was calculated with the formula 0.52 × length × width2. After tumours had grown for the designated time, all the mice were euthanized, and the tumours were harvested.
For the establishment of PDX models, fresh tumour samples obtained during surgical resection were transferred immediately into RPMI 1640 medium supplemented with penicillin/streptomycin and kept on ice. Within 12 h after excision, the tumour sample was cut into 60 mm3 pieces and subcutaneously embedded into flanks of 6-week-old female NCG mice (Nanjing Biomedical Research Institute of Nanjing University, Nanjing, China). Once the xenografts reached approximately 1 cm3 in volume, tumours were dissected and regrafted into a new generation of mice.
RNA interference and AAV-mediated gene therapy
Small interfering RNAs (siRNAs) targeting JOSD1 and negative control siRNA were purchased from Invitrogen. The siRNA sequences targeting JOSD1 are as follows: siJOSD1#1, GGCAGCACUUCAGACCAAAGGCUAU, siJOSD1#2, GGGCCUACUACAACCUCGACUCCAA, and siJOSD1#3, CCUGCUGGUGGUACCAGAAGAGGUA. The shRNA sequences were as follows: JOSD1, sh1, GAGCGAGCTCAGGAAGTTTCT, and sh2, GGTGGTACCAGAAGAGGTAGA; and MCL1, sh1, GCCTTTGTGGCTAAACACTTG, and sh2, GCTGGTTTGGCATATCTAATA. The following nontargeting shRNA sequence was used as a negative control: AGTCTTAATCGCGTATAAGGC. Vector construction and lentivirus production were performed as previously described [27].
The PDX models were treated with AAV produced by ViGene Biosciences (Shandong, China). Briefly, once the ovarian cancer xenografts reached approximately 10 mm3, 2.5 × 1011 vg/mouse AAV9-shJOSD1 or AAV9-shCtrl was directly injected via the tail vein. The tumour volumes were measured every week, and the mice were sacrificed once the tumour volumes reached approximately 400 mm3.
Patient samples and IHC
Paraffin-embedded in situ tumour tissue blocks were collected from 150 patients with ovarian serous adenocarcinoma during their initial treatment at the Cancer Hospital, Chinese Academy of Medical Sciences [27]. Written informed consent was obtained from all patients. Patients with incomplete standard treatment, including cytoreductive surgery or tumour staging surgery followed by platinum-based adjuvant chemotherapy either in combination with neoadjuvant chemotherapy for 6–8 cycles or without, were excluded. Progression-free survival (PFS) was defined as the time from diagnosis to the first instance of recurrence/progression. Overall survival (OS) was calculated from the date of surgery to the date of death or the end of follow-up. Patients with recurrent ovarian cancer were categorized as either platinum-sensitive or platinum-resistant based on a platinum-free interval of greater than or less than 6 months, respectively. IHC experiments were performed as previously described [27]. All slides were scanned using an Aperio scanning system (Aperio, San Diego, CA, USA) and quantified as an H-score as follows: H-score = Σ(pi × i), where pi represents the percentage of positively stained cells (0–100%) and i represents the staining intensity (0: negative; 1: weak; 2: moderate; 3: strong). The H-score was evaluated by two independent observers.
Statistics
For all statistical analyses, differences for which p ≤ 0.05 were considered statistically significant, and at least three biologically independent experiments with similar results are reported. The data analysis was performed using GraphPad Prism version 6.01 (San Diego, CA, USA).
Results
JOSD1 depletion triggers gynaecological tumour cell apoptosis
To identify DUBs that play a key role in the emergence of chemoresistance in gynaecological cancer, we established an in vivo model by subcutaneously xenografting A2780 cells into nude mice. One week later, those mice were randomly divided into two groups and administered either CBP or saline every other day for 10 days and then monitored for 2 weeks under normal conditions to allow tumour growth (Fig. 1a). At the 2-week time point, tumours that formed in CBP-treated mice were significantly smaller than those that formed in the saline-treated group (Supplementary Fig. 1a). We then extracted total RNA from five xenografts of each group and performed qRT-PCR to identify differentially expressed genes. Saline or CBP was administered to another 10 mice in each group 3 times in 10 days to confirm their chemoresistant status (Supplementary Fig. 1b). Among the 45 DUBs we detected, JOSD1 was the most significantly upregulated gene (Fig. 1b). To further confirm the change in JOSD1 expression during the generation of acquired chemoresistance, we performed IB to detect the protein level of JOSD1 in two paired parental and chemoresistant ovarian cancer cell lines. In line with the in vivo screening results, the expression of both JOSD1 and its monoubiquitinated form were remarkably increased in both chemoresistant cell lines compared with their respective parental cell lines (Fig. 1c and Supplementary Fig. 1c). These results illustrated that JOSD1 might be a key factor that contributes to the acquired chemoresistance of ovarian cancer.
Fig. 1.

JOSD1 depletion causes severe apoptosis of gynaecological cancer cells. a Schematic showing the screening of the in vivo chemoresistant model. b Quantitative real-time PCR detection of gene expression in carboplatin-treated tumours relative to that in saline-treated tumours. c Immunoblotting for JOSD1 expression in acquired chemoresistant cell lines and the corresponding sensitive parental cell lines. d Immunoblotting for JOSD1 expression in A2780 and HeLa cells expressing either short hairpin RNAs (shRNAs) targeting JOSD1 or nontargeting shRNA. e Representative images of apoptotic HeLa cells after JOSD1 depletion. f In vitro growth of A2780 and HeLa cells expressing either shRNAs targeting JOSD1 or nontargeting shRNA. g Representative images and fraction of TUNEL-positive apoptotic cells in HeLa cells after JOSD1 depletion. Scale bars, 60 μm. Two-tailed Student’s t test; ****p < 0.0001. The data are presented as the mean ± SD, and representative results of three biological replicates are shown
To further study the function of JOSD1 in gynaecological cancer, we first knocked down JOSD1 in A2780 and HeLa cells using two independent shRNA sequences (Fig. 1d) and found that JOSD1 depletion triggered severe cell death under normal culture conditions (Fig. 1e; Supplementary Fig. 1d). The results of the CCK-8 assay also showed decreased cell growth in JOSD1-depleted A2780 and HeLa cells compared to that in the respective control cells (Fig. 1f). JOSD1 knockdown using siRNA pools also rescued the chemoresistance status of both chemoresistant cell lines (Supplementary Fig. 1e–h). To validate that the increased cell death and decreased cell growth were caused by activation of apoptosis, we performed TUNEL staining of control and JOSD1-depleted HeLa cells and found a significantly increased fraction of TUNEL-positive cells after JOSD1 depletion (Fig. 1g, h). These results demonstrated that JOSD1 is a critical gene that has an antiapoptotic function.
JOSD1 depletion inhibits gynaecological cancer in vivo
To investigate the effect of JOSD1 depletion in vivo, we subcutaneously xenografted JOSD1-depleted and control A2780 or HeLa cells into nude mice. Weeks later, the mice injected with the control A2780 or HeLa cells formed tumour masses, while the mice injected with the JOSD1-depleted cells barely formed tumours (Fig. 2a–d; Supplementary Fig. 2a, b). To further confirm the effect of JOSD1 depletion, we treated eight ovarian cancer primary cell lines with siRNA pools targeting JOSD1 (Supplementary Fig. 3a). siRNA-mediated JOSD1 depletion also significantly inhibited cell growth in these primary cell lines (Fig. 2e), which was in accordance with our previous in vivo results (Fig. 2a–d). To evaluate the potential toxicity of JOSD1 depletion in normal tissues, we knocked down JOSD1 in two normal cell lines, NE3 and RWPE-1 (Supplementary Fig. 3b). JOSD1 depletion showed no obvious effect on the proliferation of either normal cell line (Supplementary Fig. 3c). Subsequently, we sought to determine whether targeting JOSD1 could be a promising strategy for the gene therapy of gynaecological cancer. To confirm our hypothesis, we established four ovarian cancer PDX models. Each model was randomly distributed into two groups, and then AAV-shControl or AAV-shJOSD1 was administered via tail vein injections. The groups treated with AAV-shJOSD1 showed significantly inhibited growth of the xenografts compared with that of the AAV-shControl groups (Fig. 2f and Supplementary Fig. 3d).
Fig. 2.

Targeting JOSD1 significantly suppresses gynaecological cancer growth in vivo. a–d In vivo growth and tumour weights of xenografts generated from A2780 (n = 6 for each group) and HeLa (n = 8 for each group) cells expressing either short hairpin RNAs (shRNAs) targeting JOSD1 or nontargeting shRNA. e In vitro growth of eight gynaecological cancer primary cell lines transfected with either small interfering RNAs targeting JOSD1 or negative control siRNA. f Tumour weights of ovarian cancer patient-derived xenograft models administered adeno-associated virus (AAV) targeting JOSD1 or AAV with a nontargeting sequence (n = 5 per group). Two-tailed Student’s t test; *p < 0.05, **p < 0.01 and ****p < 0.0001. For a–d and f, the data are presented as the mean ± SEM. For e, the data are presented as the mean ± SD, and representative results of three biological replicates are shown
JOSD1 interacts with and deubiquitinates MCL1
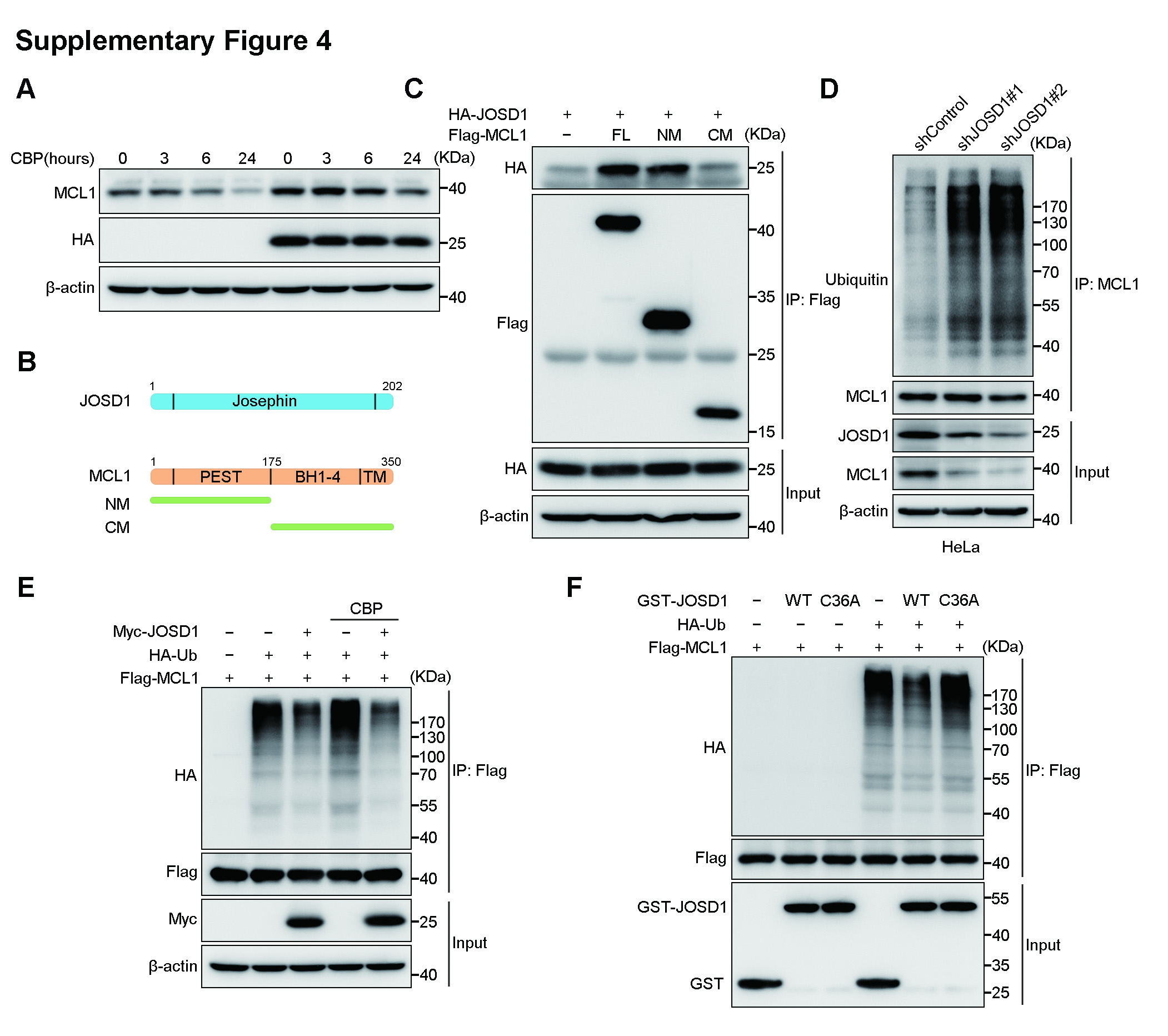
To illustrate the regulatory mechanism of JOSD1, we performed immunoprecipitation followed by mass spectrometric analysis. Among the proteins that pulled down together with JOSD1, MCL1, which is an essential antiapoptotic protein of the BCL-2 family, was our primary focus (Fig. 3a, b). Notably, no other antiapoptotic members of the BCL-2 family were detected, illustrating the specific association between JOSD1 and MCL1. To further confirm this association between JOSD1 and MCL1, a coimmunoprecipitation (co-IP) assay was performed. V5-tagged MCL1 was pulled down by Flag-tagged JOSD1, and vice versa (Fig. 3c). We next investigated the interaction between JOSD1 and MCL1 in an existing chemotherapy drug environment. HA-JOSD1 and Flag-MCL1 were co-transfected into 293T cells, which were then treated with CBP. The association between HA-JOSD1 and Flag-MCL1 was detected by co-IP 0, 3, 6 or 24 h after the CBP treatment. As shown in Fig. 3d, the interaction intensity between JOSD1 and MCL1 after the CBP treatment increased in a time-dependent manner, indicating that JOSD1 might exert its antiapoptotic function by binding MCL1. Notably, although JOSD1 maintained a more stable level of MCL1 protein compared with that in the control cells upon CBP treatment, JOSD1 could not completely inhibit the degradation of MCL1 when severe cell apoptosis occurred as the time of CBP treatment reached 24 h (Supplementary Fig. 4a). JOSD1 is a 202 amino acid small protein containing only a Josephin domain (Supplementary Fig. 4b) [28]. MCL1 consists of two PEST (proline, glutamate, serine and threonine) domains, four BH (BCL-2 homology) domains and a carboxyl-terminal transmembrane domain (Supplementary Fig. 4b) [29]. To map the specific binding region of MCL1 on JOSD1, we constructed two truncated MCL1 mutants (N terminus and C terminus) and performed a co-IP assay. The results indicated that only the N terminus is associated with JOSD1 (Supplementary Fig. 4c). The confocal imaging results showed co-localization of JOSD1 and MCL1 in the cytoplasm of the A2780 cells, further confirming the association between these two proteins in gynaecological cancer cells (Fig. 3e).
Fig. 3.

JOSD1 associates with and deubiquitinates myeloid cell leukaemia 1 (MCL1). a Immunoprecipitation was performed using an anti-Flag antibody in ES2 cells expressing empty vector or Flag-JOSD1 and then conducting sliver staining. b Number of unique peptide hits for JOSD1 and MCL1 are shown. c Immunoblotting for V5-MCL1 among proteins pulled down with anti-FLAG M2 affinity gels (left) and for Flag-JOSD1 among proteins pulled down with an anti-V5 antibody (right). d Immunoblotting for the association between HA-JOSD1 and Flag-MCL1 in 293T cells exposed to 0.5 mg/ml carboplatin for 0, 3, 6 and 24 h. e Confocal microscopy of the co-localization of Myc-JOSD1 (green) and MCL1 (red) in ES2 cells. Nuclei were stained with Hoechst 33342 (blue). Scale bars, 60 μm. f Immunoblotting to detect the ubiquitination of MCL1 in 293T cells co-transfected with Myc-JOSD1, HA-Ubiquitin and Flag-MCL1 (wild type or C36A). g Immunoblotting to detect the ubiquitination of MCL1 in A2780 cells expressing either short hairpin RNAs (shRNAs) targeting JOSD1 or nontargeting shRNA. h Immunoblotting to detect the ubiquitination of MCL1 in 293T cells co-transfected with Flag-MCL1, Myc-JOSD1 and HA-Ubiquitin mutant (K0 indicates that all lysines were replaced by arginines, and K48 and K63 indicate that all lysines, except for K48 or K63, were mutated to arginines). Representative results of three biological replicates are shown
We next performed a ubiquitination assay to validate whether MCL1 is indeed a substrate of JOSD1. The overexpression of wild-type JOSD1 significantly decreased the level of polyubiquitin chains on MCL1, while the overexpression of the enzymatically dead JOSD1C36A mutant resulted in the loss of this decrease (Fig. 3f), illustrating that JOSD1 could deubiquitinate MCL1 as a bona fide DUB. Consistently, JOSD1 depletion in A2780 and HeLa cells increased the level of ubiquitinated endogenous MCL1, and the protein level of MCL1 declined (Fig. 3g; Supplementary Fig. 4d). Supporting our former conclusion that JOSD1 maintains MCL1 stability after CBP-induced apoptosis, the deubiquitination of MCL1 by JOSD1 was increased upon CBP treatment (Supplementary Fig. 4e). Since the monoubiquitinated type of JOSD1 was also increased in the chemoresistant cell lines (Fig. 1c), we next performed an in vitro ubiquitination assay to confirm whether JOSD1 could deubiquitinate MCL1 directly. We showed that JOSD1 alone was sufficient to deubiquitinate MCL1 in vitro (Supplementary Fig. 4f). This result indicated that JOSD1 interacts with and deubiquitinates MCL1 in the cytoplasm, which is consistent with a previous study showing that monoubiquitinated JOSD1 is located mainly on the cell membrane [28]. To further specify the type of modification JOSD1 exerts on MCL1, we co-expressed Flag-MCL1 and Myc-JOSD1 along with wild-type or mutant HA-Ubiquitin. The results showed that JOSD1 only cleaved the K48 ubiquitin chains linked on MCL1 (Fig. 3h).
JOSD1 stabilizes MCL1 to inhibit the mitochondrial apoptosis pathway
Based on the above results, we next sought to clarify whether JOSD1 stabilization of MCL1 is depending on the enzymatic activity of JOSD1. Increasing amounts of Myc-tagged wild-type JOSD1 or the mutant Myc-JOSD1C36A were transfected into 293T cells. As shown in Fig. 4a, the protein level of MCL1 was increased in a dose-dependent manner only when wild-type JOSD1 was overexpressed, while JOSD1C36A had no effect on the stabilization of MCL1. Additionally, the qRT-PCR results indicated that JOSD1 did not affect the mRNA expression of MCL1 (Supplementary Fig. 5a), suggesting that the stabilizing effect of JOSD1 on MCL1 is dependent on its DUB activity and that this regulation functions at the posttranslational level. The CHX pulse-chase assay showed that the ectopic expression of wild-type but not mutant JOSD1 prolonged the half-life of endogenous MCL1 (Fig. 4b). Consistent with this observation, siRNA-mediated depletion of JOSD1 led to increased MCL1 instability (Fig. 4c). Similar results were also obtained in A2780 and HeLa cells using shRNAs (Supplementary Fig. 5b). Supporting the positive regulation of MCL1 by JOSD1, the expression level of MCL1 was also significantly upregulated in the chemoresistant xenografts and cell lines (Fig. 4d and Supplementary Fig. 5c). MCL1 is a key antiapoptotic factor involved in blocking the mitochondrial apoptosis pathway [30–32]. We hypothesized that JOSD1 inhibits apoptosis by suppressing the mitochondrial apoptosis pathway. To verify our hypothesis, we performed IB to detect several critical factors in this pathway. The results showed that the decrease in MCL1 and increase in cleaved PARP, cleaved caspase-9 and cleaved caspase-3 occurred in both the A2780 and HeLa cells following the JOSD1 depletion, suggesting that the apoptosis pathway is activated (Fig. 4e). Moreover, the overexpression of wild-type but not mutant JOSD1 inhibited the activation of the mitochondrial apoptosis pathway in ES2 cells subjected to CBP treatment (Fig. 4f). To further validate that the ability of JOSD1 to induce apoptosis is dependent on the mitochondrial apoptosis pathway, we pretreated A2780 cells with a pan-caspase inhibitor, Z-VAD(OMe)-FMK, and then knocked down JOSD1 using siRNAs. We found that Z-VAD(OMe)-FMK treatment could completely abolish the effect of JOSD1 depletion (Fig. 4g). In conclusion, these results demonstrated that JOSD1 stabilizes MCL1 to suppress the mitochondrial apoptosis pathway in gynaecological cancer cells and that this stabilization is dependent on its enzymatic activity.
Fig. 4.

JOSD1 stabilizes myeloid cell leukaemia 1 (MCL1) to suppress the mitochondrial apoptosis pathway. a Immunoblotting for MCL1 expression in 293T cells transfected with increasing amounts of JOSD1 or JOSD1C36A. b Immunoblotting for MCL1 expression in 293T cells transfected with JOSD1 or JOSD1C36A and subjected to the cycloheximide (CHX) pulse-chase assay. Quantification of MCL1 expression relative to β-actin expression is shown. c Immunoblotting for MCL1 expression in 293T cells transfected with either JOSD1-targeting or nontargeting siRNAs and subjected to the CHX pulse-chase assay. Quantification of MCL1 relative to β-actin expression is shown. d H-score and representative images of JOSD1 and MCL1 staining in chemoresistant and parental xenografts. Scale bars, 100 μm. e Immunoblotting for apoptosis markers in A2780 and HeLa cells expressing either JOSD1-targeting or nontargeting shRNAs. f Immunoblotting for apoptosis markers in ES2 cells expressing empty vector, JOSD1 or JOSD1C36A and were treated with different concentrations of carboplatin. g In vitro growth of A2780 cells expressing either siRNAs targeting JOSD1 or negative control siRNA and treated with Z-VAD(OMe)-FMK (50 μM) or dimethyl sulfoxide (DMSO). Z-VAD(OMe)-FMK (50 μM) or DMSO was added 2 h before small interfering RNA transfection. Two-tailed Student’s t test; ****p < 0.0001; ns, no significant difference. In b–d, the data are presented as the mean ± SEM; in g, the data are presented as the mean ± SD. Representative results of three biological replicates are shown
JOSD1 contributes to chemoresistance and is dependent on MCL1
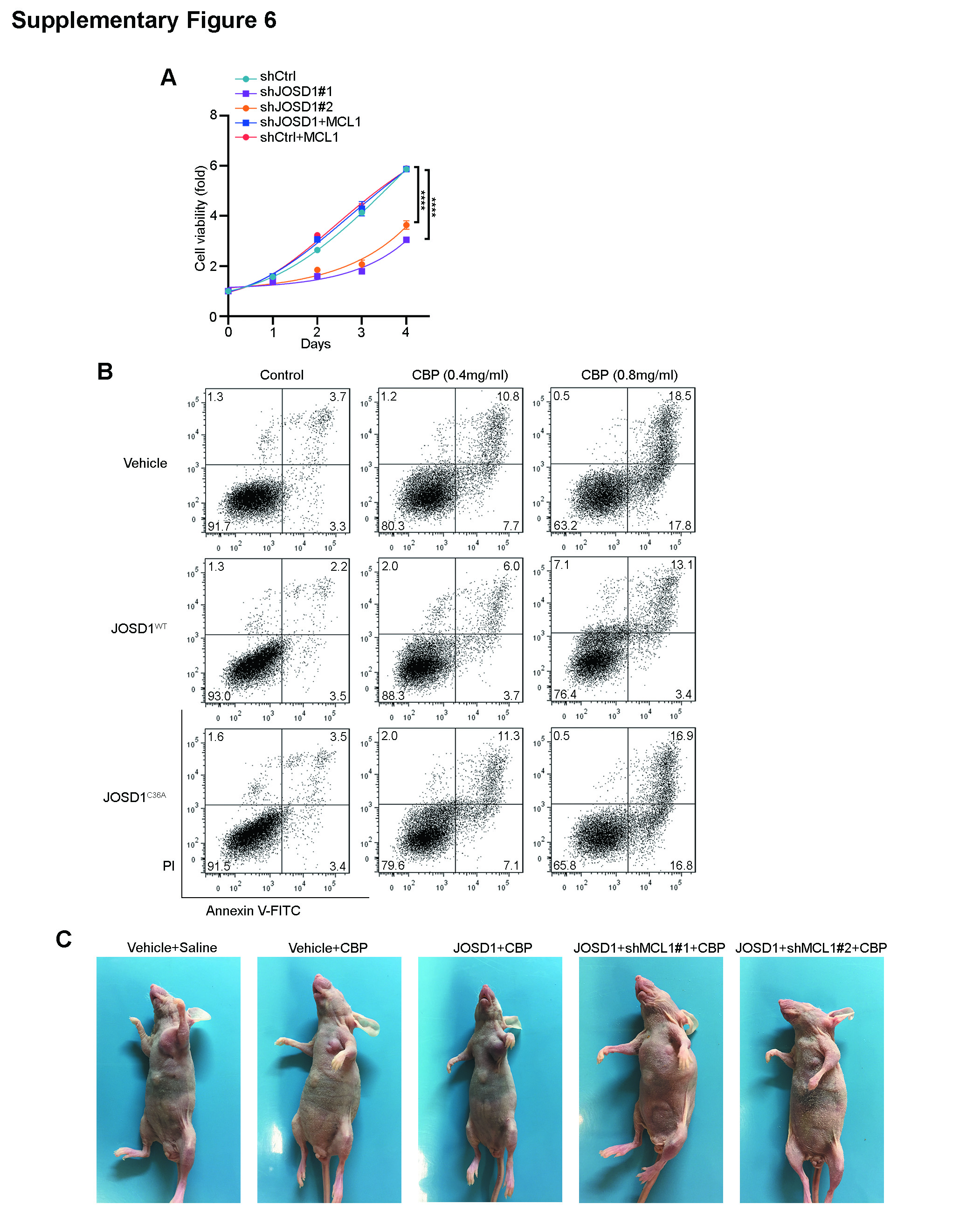
To investigate the function of JOSD1 in chemoresistance, JOSD1 was stably overexpressed in the ovarian cancer cell lines ES2 and 3AO. As shown in Fig. 5a, cells with wild-type JOSD1 overexpression developed resistance against CBP treatment, while cells overexpressing mutant JOSD1 showed no such effect. To further validate that the chemoresistance caused by JOSD1 overexpression is mediated by MCL1, we knocked down MCL1 in JOSD1-overexpressing cells and found that MCL1 depletion dramatically abolished the chemoresistance triggered by JOSD1 overexpression (Fig. 5b). In consistent with these findings, overexpression of MCL1 rescued the proapoptotic effect of JOSD1 depletion in A2780 cells (Supplementary Fig. 6a). Annexin V/fluorescein isothiocyanate double staining showed a significantly reduced apoptosis rate in JOSD1-overexpressing ES2 cells compared with empty vector- or JOSD1C36A-overexpressing ES2 cells (Fig. 5c; Supplementary Fig. 6b).
Fig. 5.

JOSD1 causes chemoresistance by stabilizing myeloid cell leukaemia 1 (MCL1). a Cell viability of ES2 and 3AO cells expressing empty vector, JOSD1 or JOSD1C36A and treated with different doses of carboplatin (CBP). b Cell viability of ES2 and 3AO cells expressing empty vector, JOSD1 or JOSD1 and MCL1-targeting short hairpin RNAs (shRNAs) and treated with different doses of CBP. c Quantitative analysis of the apoptosis rate of ES2 cells expressing empty vector, JOSD1 or JOSDC36A and treated with different doses of CBP. d–f In vivo growth curves and tumour weights of xenografts generated from ES2 cells expressing an empty vector, JOSD1 or JOSD1 and MCL1-targeting shRNAs and treated with CBP (50 mg/kg, three times a week). g, h H-score and representative images of immunohistochemical staining for JOSD1, MCL1 and cleaved caspase-3 in Fig. 5e. Scale bars, 250 μm. Two-tailed Student’s t test; **p < 0.01, ***p < 0.001 and ****p < 0.0001. In a and b, the data are presented as the mean ± SD, and representative results of three biological replicates are shown. In c, d, f and g, the data are presented as the mean ± SEM (n = 10)
To confirm the antiapoptotic function of JOSD1 in vivo, ES2 cells overexpressing an empty vector, JOSD1 or shRNAs targeting MCL1 were xenografted into nude mice. The CBP administration significantly reduced the tumour growth in the control ES2 cells, while the JOSD1-overexpressing ES2 cells grew much more rapidly than the control cells after the treatment with the same dose of CBP. Furthermore, knockdown of MCL1 reversed the chemoresistance effect caused by JOSD1 overexpression (Fig. 5d–f; Supplementary Fig. 6c). Meanwhile, IHC staining showed increased MCL1 and decreased cleaved caspase-3 expression levels in JOSD1-overexpressing xenografts, while xenografts with MCL1 depletion showed remarkably increased cleaved caspase-3 expression (Fig. 5g, h). Taken together, these data showed that JOSD1 is a crucial factor associated with chemoresistance that exerts its effects on drug resistance by stabilizing MCL1.
JOSD1 expression level predicts outcomes of ovarian cancer patients
To further validate the positive relationship of JOSD1 and MCL1 with chemoresistance in human samples, we stained for both JOSD1 and MCL1 in tissues from 150 ovarian cancer patients. Both JOSD1 and MCL1 were highly expressed in the chemoresistant ovarian cancer samples, while they were expressed at low levels in the chemosensitive samples (Fig. 6a and Supplementary Table 3). Correlation analysis showed that JOSD1 and MCL1 expression levels were tightly correlated with each other in these samples (Fig. 6b). High JOSD1/MCL1 expression levels were also associated with poor OS and PFS of ovarian cancer patients (Fig. 6c, d).
Fig. 6.

JOSD1 and myeloid cell leukaemia 1 (MCL1) expression correlates with chemoresistance and poor prognosis of ovarian cancer patients. a Representative images of JOSD1 and MCL1 staining in platinum-sensitive and platinum-resistant ovarian cancer samples. Scale bars, 250 μm. b Correlation analysis of JOSD1 and MCL1 expression levels in ovarian cancer samples (n = 150). Pearson correlation coefficients are shown. c, d Correlation between JOSD1 expression (c) or MCL1 expression (d) and overall survival or progression-free survival in ovarian cancer patients (n = 150). Kaplan–Meier survival plots are shown. e Immunoblotting for JOSD1 in the supernatants of chemoresistant cell lines and their parental cell lines. f Correlation between serum JOSD1 levels and intratumour JOSD1 expression in 20 paired serum and tumour tissue samples. Pearson correlation coefficients are shown
Because JOSD1 is also expressed in cell membranes [28], we speculated whether it could be secreted into serum and represent intracellular expression levels, which reflect chemosensitivity in ovarian cancer patients. To test this hypothesis, we first detected the existence of secreted JOSD1 in culture supernatants of parental and acquired chemoresistant 3AO and A2780 cells. Excitingly, the secreted JOSD1 levels in the supernatant of the chemoresistant cell lines were extremely higher than those in the supernatant of the parental cell lines (Fig. 6e). To further confirm this result in clinical samples, we performed both IB and IHC experiments to detect the serum and intracellular JOSD1 expression levels in 20 ovarian cancer patients and identified that the serum JOSD1 levels were positively correlated with intracellular JOSD1 expression (Fig. 6f and Supplementary Fig. 7). In summary, these results revealed that the intracellular JOSD1 expression levels could be adopted as a marker for the chemosensitivity of ovarian cancer patients and that the serum JOSD1 levels, which is easily detected, is an accurate indicator for intracellular JOSD1 expression.
Discussion
Ovarian and cervical cancers are two dominant types of gynaecological malignancy. Ovarian cancer patients suffer an extremely poor prognosis, mainly due to the emergence of acquired chemoresistance during treatment [33, 34]. The current standard of care for cervical cancer patients is pelvic radiotherapy (RT) and concurrent weekly cisplatin chemotherapy (RTCT), followed by brachytherapy [35]. Although 50–60% of patients are cured with this approach, recurrence and metastases after treatment remain major problems. There are very few effective treatments for patients with recurrent cervical cancer after RTCT, and virtually all patients die often with pain, bleeding and other debilitating symptoms [36]. Previous studies have revealed several factors that contribute to the chemoresistance of gynaecological cancer, but the specific factors that contribute to the occurrence of acquired chemoresistance remain ill-defined. JOSD1 is primarily characterized as a DUB [37], but a later study showed that JOSD1 might possess the ability to modulate gene transcription [38]. Moreover, the role of JOSD1 in the cancer context is still ambiguous. In this study, we established an in vivo chemoresistance model of ovarian cancer and identified that JOSD1 is the most strikingly upregulated DUB in chemoresistant tumours compared with the control group. Both in vitro and in vivo studies confirmed that JOSD1 is a key modulator of cell viability, while JOSD1 depletion significantly triggered gynaecological cancer cell death. Most excitingly, AAV-mediated JOSD1 shRNA could effectively restrict the growth of PDX models of ovarian cancer, providing a promising therapeutic option for gene therapy targeting gynaecological malignancy.
MCL1 is a critical antiapoptotic member of the BCL-2 family. The only selective inhibitor of MCL1 is S63845, which has ideal effects on blood malignancies but lesser effects on solid tumours [39–41]. Previously, two DUBs, USP9X and USP13, had been reported as modulators of MCL1 stabilization [29, 42]. However, USP9X exhibits tissue-specific expression primarily in the brain and immune system [43] and sometimes functions as a tumour suppressor [44, 45]. Additionally, USP13 has been reported to act as a tumour suppressor by stabilizing PTEN in breast cancer and colorectal cancer [26]. The controversial context-dependent functions of USP9X and USP13 make them less promising as gene therapeutic targets in cancer treatment owing to the probability of triggering side effects. Therefore, there is a need to develop other more specific and stronger regulators. Through a mass spectrometric analysis, we identified that MCL1 is a substrate of JOSD1. JOSD1 acts as a bona fide DUB that cleaves the K48 ubiquitin chains linked on MCL1. By stabilizing MCL1, JOSD1 inhibits the mitochondrial apoptosis pathway and exerts antiapoptosis effects (Fig. 7). Though the monoubiquitinated type of JOSD1 was also increased in the chemoresistant cell lines, our data showed that JOSD1 could directly deubiquitinate MCL1 in vitro, which illustrates that the increased amount of monoubiquitinated JOSD1 might just be a result of increased JOSD1 expression. Additionally, this result is also in accordance with the conclusion drawn from a previous study that monoubiquitinated JOSD1 is mainly located on the cell membrane [28]. Another point worth further discussed is that, although JOSD1 overexpression could not completely prevent the degradation of MCL1 after CBP treatment for >6 h, CBP treatment still increased the interaction between JOSD1 and MCL1 and increased the deubiquitination of MCL1 by JOSD1. Former studies showed that the elimination of MCL1 is required for the initiation of apoptosis [46-48]. Hence, the decreased MCL1 protein level after 6 h of CBP treatment resulted from a cell apoptosis event upon the chemical drug inducement rather than the loss of deubiquitination by JOSD1.
Fig. 7.

JOSD1 drives chemoresistance by stabilizing myeloid cell leukaemia 1 (MCL1) and suppressing mitochondrial apoptotic signalling. JOSD1 interacts with and deubiquitinates MCL1. JOSD1 stabilizes MCL1 protein in the cytoplasm, which leads to the inhibition of mitochondrial apoptotic signalling. The cytoplasmic JOSD1 expression level also correlates with the serum JOSD1 expression level
IHC analysis of 150 human ovarian cancer samples illustrated a strong correlation between JOSD1 and MCL1 expression levels; it was also found that high JOSD1/MCL1 expression is correlated with chemoresistance and poor prognosis of ovarian cancer patients. To explore more convenient methods for the detection of JOSD1 levels, we validated that the serum JOSD1 levels in patients directly corresponded to the intracellular JOSD1 expression levels, suggesting that serum JOSD1 is a predictor of chemosensitivity and prognosis in ovarian cancer patients. Taken together, these data comprise the first study to identify JOSD1 as an oncogene that inhibits mitochondrial apoptotic signalling and contributes to the emergence of acquired chemoresistance in gynaecological cancer. Most importantly, AAV-mediated JOSD1 depletion might be a promising therapeutic approach for the treatment of gynaecological cancer, and serum JOSD1 levels could be a diagnostic and prognostic indicator for gynaecological cancer patients.
Supplementary information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
This work was supported by the National Key R&D Program of China (2016YFC1302100), the CAMS Innovation Fund for Medical Sciences (2016-I2M-1-001) and the National Science Foundation of China (81420108025).
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Edited by H. Ichijo
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Xiaowei Wu, Qingyu Luo.
Supplementary information
The online version of this article (10.1038/s41418-019-0339-0) contains supplementary material, which is available to authorized users.
References
- 1.Vaughan S, Coward JI, Bast RC, Jr., Berchuck A, Berek JS, Brenton JD, et al. Rethinking ovarian cancer: recommendations for improving outcomes. Nat Rev Cancer. 2011;11:719–25. doi: 10.1038/nrc3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bast RC, Jr., Hennessy B, Mills GB. The biology of ovarian cancer: new opportunities for translation. Nat Rev Cancer. 2009;9:415–28. doi: 10.1038/nrc2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ozols RF, Bundy BN, Greer BE, Fowler JM, Clarke-Pearson D, Burger RA, et al. Phase III trial of carboplatin and paclitaxel compared with cisplatin and paclitaxel in patients with optimally resected stage III ovarian cancer: a Gynecologic Oncology Group study. J Clin Oncol. 2003;21:3194–3200. doi: 10.1200/JCO.2003.02.153. [DOI] [PubMed] [Google Scholar]
- 4.Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, Kepp O, et al. Molecular mechanisms of cisplatin resistance. Oncogene. 2012;31:1869–83. doi: 10.1038/onc.2011.384. [DOI] [PubMed] [Google Scholar]
- 5.Hou T, Liang D, Xu L, Huang X, Huang Y, Zhang Y. Atypical chemokine receptors predict lymph node metastasis and prognosis in patients with cervical squamous cell cancer. Gynecol Oncol. 2013;130:181–7. doi: 10.1016/j.ygyno.2013.04.015. [DOI] [PubMed] [Google Scholar]
- 6.Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359–86. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 7.Uyar D, Rader J. Genomics of cervical cancer and the role of human papillomavirus pathobiology. Clin Chem. 2014;60:144–6. doi: 10.1373/clinchem.2013.212985. [DOI] [PubMed] [Google Scholar]
- 8.Matulonis UA, Sood AK, Fallowfield L, Howitt BE, Sehouli J, Karlan BY. Ovarian cancer. Nat Rev Dis Prim. 2016;2:16061. doi: 10.1038/nrdp.2016.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 10.Schmitt CA. Senescence, apoptosis and therapy--cutting the lifelines of cancer. Nat Rev Cancer. 2003;3:286–95. doi: 10.1038/nrc1044. [DOI] [PubMed] [Google Scholar]
- 11.Ashkenazi A, Fairbrother WJ, Leverson JD, Souers AJ. From basic apoptosis discoveries to advanced selective BCL-2 family inhibitors. Nat Rev Drug Discov. 2017;16:273–84. doi: 10.1038/nrd.2016.253. [DOI] [PubMed] [Google Scholar]
- 12.O’Neill KL, Huang K, Zhang J, Chen Y, Luo X. Inactivation of prosurvival Bcl-2 proteins activates Bax/Bak through the outer mitochondrial membrane. Genes Dev. 2016;30:973–88. doi: 10.1101/gad.276725.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Amundson SA, Myers TG, Scudiero D, Kitada S, Reed JC, Fornace AJ., Jr. An informatics approach identifying markers of chemosensitivity in human cancer cell lines. Cancer Res. 2000;60:6101–10. [PubMed] [Google Scholar]
- 14.Cory S, Adams JM. The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer. 2002;2:647–56. doi: 10.1038/nrc883. [DOI] [PubMed] [Google Scholar]
- 15.Delbridge AR, Grabow S, Strasser A, Vaux DL. Thirty years of BCL-2: translating cell death discoveries into novel cancer therapies. Nat Rev Cancer. 2016;16:99–109. doi: 10.1038/nrc.2015.17. [DOI] [PubMed] [Google Scholar]
- 16.Ciechanover A. The unravelling of the ubiquitin system. Nat Rev Mol Cell Biol. 2015;16:322–4. doi: 10.1038/nrm3982. [DOI] [PubMed] [Google Scholar]
- 17.Witt A, Vucic D. Diverse ubiquitin linkages regulate RIP kinases-mediated inflammatory and cell death signaling. Cell Death Differ. 2017;24:1160–71. doi: 10.1038/cdd.2017.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jiang X, Chen ZJ. The role of ubiquitylation in immune defence and pathogen evasion. Nat Rev Immunol. 2011;12:35–48. doi: 10.1038/nri3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rape M. Ubiquitylation at the crossroads of development and disease. Nat Rev Mol Cell Biol. 2018;19:59–70. doi: 10.1038/nrm.2017.83. [DOI] [PubMed] [Google Scholar]
- 20.Rousseau A, Bertolotti A. Regulation of proteasome assembly and activity in health and disease. Nat Rev Mol Cell Biol. 2018;19:697–712. doi: 10.1038/s41580-018-0040-z. [DOI] [PubMed] [Google Scholar]
- 21.Komander D, Clague MJ, Urbe S. Breaking the chains: structure and function of the deubiquitinases. Nat Rev Mol Cell Biol. 2009;10:550–63. doi: 10.1038/nrm2731. [DOI] [PubMed] [Google Scholar]
- 22.Harrigan JA, Jacq X, Martin NM, Jackson SP. Deubiquitylating enzymes and drug discovery: emerging opportunities. Nat Rev Drug Discov. 2018;17:57–78. doi: 10.1038/nrd.2017.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang X, Liu Z, Zhang L, Yang Z, Chen X, Luo J, et al. Targeting deubiquitinase USP28 for cancer therapy. Cell Death Dis. 2018;9:186. doi: 10.1038/s41419-017-0208-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu C, Luo K, Zhao F, Yin P, Song Y, Deng M, et al. USP20 positively regulates tumorigenesis and chemoresistance through beta-catenin stabilization. Cell Death Differ. 2018;25:1855–69. doi: 10.1038/s41418-018-0138-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pringle LM, Young R, Quick L, Riquelme DN, Oliveira AM, May MJ, et al. Atypical mechanism of NF-kappaB activation by TRE17/ubiquitin-specific protease 6 (USP6) oncogene and its requirement in tumorigenesis. Oncogene. 2012;31:3525–35. doi: 10.1038/onc.2011.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang J, Zhang P, Wei Y, Piao HL, Wang W, Maddika S, et al. Deubiquitylation and stabilization of PTEN by USP13. Nat Cell Biol. 2013;15:1486–94. doi: 10.1038/ncb2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luo Q, Wu X, Zhang Y, Shu T, Ding F, Chen H, et al. ARID1A ablation leads to multiple drug resistance in ovarian cancer via transcriptional activation of MRP2. Cancer Lett. 2018;427:9–17. doi: 10.1016/j.canlet.2018.04.013. [DOI] [PubMed] [Google Scholar]
- 28.Seki T, Gong L, Williams AJ, Sakai N, Todi SV, Paulson HL. JosD1, a membrane-targeted deubiquitinating enzyme, is activated by ubiquitination and regulates membrane dynamics, cell motility, and endocytosis. J Biol Chem. 2013;288:17145–55. doi: 10.1074/jbc.M113.463406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang S, Zhang M, Jing Y, Yin X, Ma P, Zhang Z, et al. Deubiquitinase USP13 dictates MCL1 stability and sensitivity to BH3 mimetic inhibitors. Nat Commun. 2018;9:215. doi: 10.1038/s41467-017-02693-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13:714–26. doi: 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]
- 31.Juin P, Geneste O, Gautier F, Depil S, Campone M. Decoding and unlocking the BCL-2 dependency of cancer cells. Nat Rev Cancer. 2013;13:455–65. doi: 10.1038/nrc3538. [DOI] [PubMed] [Google Scholar]
- 32.Certo M, Del Gaizo Moore V, Nishino M, Wei G, Korsmeyer S, Armstrong SA, et al. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell. 2006;9:351–65. doi: 10.1016/j.ccr.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 33.Coleman RL, Monk BJ, Sood AK, Herzog TJ. Latest research and treatment of advanced-stage epithelial ovarian cancer. Nat Rev Clin Oncol. 2013;10:211–24. doi: 10.1038/nrclinonc.2013.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Burger RA, Brady MF, Bookman MA, Fleming GF, Monk BJ, Huang H, et al. Incorporation of bevacizumab in the primary treatment of ovarian cancer. N Engl J Med. 2011;365:2473–83. doi: 10.1056/NEJMoa1104390. [DOI] [PubMed] [Google Scholar]
- 35.Chemoradiotherapy for Cervical Cancer Meta-Analysis Collaboration. Reducing uncertainties about the effects of chemoradiotherapy for cervical cancer: a systematic review and meta-analysis of individual patient data from 18 randomized trials. J Clin Oncol. 2008;26:5802–12. doi: 10.1200/JCO.2008.16.4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chaudary N, Pintilie M, Jelveh S, Lindsay P, Hill RP, Milosevic M. Plerixafor improves primary tumor response and reduces metastases in cervical cancer treated with radio-chemotherapy. Clin Cancer Res. 2017;23:1242–9. doi: 10.1158/1078-0432.CCR-16-1730. [DOI] [PubMed] [Google Scholar]
- 37.Orcutt SJ, Wu J, Eddins MJ, Leach CA, Strickler JE. Bioluminescence assay platform for selective and sensitive detection of Ub/Ubl proteases. Biochim Biophys Acta. 2012;1823:2079–86. doi: 10.1016/j.bbamcr.2012.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sacco JJ, Yau TY, Darling S, Patel V, Liu H, Urbe S, et al. The deubiquitylase Ataxin-3 restricts PTEN transcription in lung cancer cells. Oncogene. 2014;33:4265–72. doi: 10.1038/onc.2013.512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kotschy A, Szlavik Z, Murray J, Davidson J, Maragno AL, Le Toumelin-Braizat G, et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature. 2016;538:477–82. doi: 10.1038/nature19830. [DOI] [PubMed] [Google Scholar]
- 40.Li Z, He S, Look AT. The MCL1-specific inhibitor S63845 acts synergistically with venetoclax/ABT-199 to induce apoptosis in T-cell acute lymphoblastic leukemia cells. Leukemia. 2018;33:262–6. doi: 10.1038/s41375-018-0201-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moujalled DM, Pomilio G, Ghiurau C, Ivey A, Salmon J, Rijal S, et al. Combining BH3-mimetics to target both BCL-2 and MCL1 has potent activity in pre-clinical models of acute myeloid leukemia. Leukemia. 2018;33:905–17. doi: 10.1038/s41375-018-0261-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schwickart M, Huang X, Lill JR, Liu J, Ferrando R, French DM, et al. Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival. Nature. 2010;463:103–7. doi: 10.1038/nature08646. [DOI] [PubMed] [Google Scholar]
- 43.Naik E, Webster JD, DeVoss J, Liu J, Suriben R, Dixit VM. Regulation of proximal T cell receptor signaling and tolerance induction by deubiquitinase Usp9X. J Exp Med. 2014;211:1947–55. doi: 10.1084/jem.20140860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Khan OM, Carvalho J, Spencer-Dene B, Mitter R, Frith D, Snijders AP, et al. The deubiquitinase USP9X regulates FBW7 stability and suppresses colorectal cancer. J Clin Invest. 2018;128:1326–37. doi: 10.1172/JCI97325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Toloczko A, Guo F, Yuen HF, Wen Q, Wood SA, Ong YS, et al. Deubiquitinating enzyme USP9X suppresses tumor growth via LATS kinase and core components of the Hippo pathway. Cancer Res. 2017;77:4921–33. doi: 10.1158/0008-5472.CAN-16-3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cuconati A, Mukherjee C, Perez D, White E. DNA damage response and MCL-1 destruction initiate apoptosis in adenovirus-infected cells. Genes Dev. 2003;17:2922–32. doi: 10.1101/gad.1156903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wertz IE, Kusam S, Lam C, Okamoto T, Sandoval W, Anderson DJ, et al. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature. 2011;471:110–4. doi: 10.1038/nature09779. [DOI] [PubMed] [Google Scholar]
- 48.Inuzuka H, Shaik S, Onoyama I, Gao D, Tseng A, Maser RS, et al. SCF(FBW7) regulates cellular apoptosis by targeting MCL1 for ubiquitylation and destruction. Nature. 2011;471:104–9. doi: 10.1038/nature09732. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.