

Abstract
Hypomagnesemia is a significant risk factor for critically ill patients to develop sepsis, a life-threatening disease with a mortality rate over 25%. Our clinic data analysis showed that hypomagnesemia is associated with a decreased monocyte count in septic patients. At the cellular level, we found that Mg2+ inhibits pyroptosis. Specifically, Mg2+ limits the oligomerization and membrane localization of gasdermin D N-terminal (GSDMD-NT) upon the activation of either the canonical or noncanonical pyroptotic pathway. Mechanistically, we demonstrated that Ca2+ influx is a prerequisite for the function of GSDMD-NT. Mg2+ blocks Ca2+ influx by inhibiting the ATP-gated Ca2+ channel P2X7, thereby impeding the function of GSDMD-NT and inhibiting lipopolysaccharide (LPS)-induced noncanonical pyroptosis. Furthermore, Mg2+ administration protects mice from LPS-induced lethal septic shock. Together, our data reveal the underlying mechanism of how Mg2+ inhibits pyroptosis and suggest potential clinic applications of magnesium supplementation for sepsis prevention and treatment.
Subject terms: Immune cell death, Inflammasome
Introduction
Hypomagnesemia is a common feature in critically ill patients and is associated with poor clinical outcomes [1]. Patients with hypomagnesemia appear to have a longer intensive care unit (ICU) stay, longer duration of mechanical ventilation, higher risk of developing sepsis and septic shock, and higher mortality rate [2–5]. Nevertheless, due to our poor understanding of the underlying mechanisms of hypomagnesemia and the lack of pathognomonic symptoms, it is often overlooked in clinical practice [2, 6].
Mg2+ has anti-inflammatory properties and acts as a Ca2+ antagonist [7]. Whereas magnesium supplementation reduces the inflammatory response, its depletion increases susceptibility to inflammation in experimental models [8, 9]. In particular, magnesium supplementation affords significant protection against lipopolysaccharide (LPS)-induced cardiotoxicity, lung injury, and liver injury [10–12]. Although these could be attributed to the inhibitory effect of Mg2+ against the release of inflammatory cytokines and oxidative stress, the detailed mechanisms involved remain to be determined.
Recent studies indicated that caspase-11-mediated noncanonical pyroptosis plays a key role in LPS-induced sepsis [13]. Pyroptosis is a cell-intrinsic death mechanism activated by cytosolic danger signals that aims to remove the infected cells and simultaneously trigger an inflammatory response [14]. However, an excessive inflammatory response can damage the host organism and even cause lethal septic shock. LPS is a strong activator of pyroptosis. Intraperitoneal injection with high-dose LPS in mice results in acute septic shock and death, while the deletion of genes involved in the LPS-induced pyroptotic death pathway (e.g., caspase-11, gasdermin D (Gsdmd)) significantly enhances survival [13, 15]. Intriguingly, ablation of Toll-like receptor 4 fails to protect mice primed with Toll-like receptor 3 ligand and then challenged with LPS, whereas ablation of caspase-11 succeeds [16, 17]. Given the extensive protective effect of magnesium in endotoxaemia, it is worthwhile to investigate whether Mg2+ regulates LPS-stimulated pyroptosis.
LPS-stimulated pyroptosis is mainly mediated by mouse caspase-11 and its human counterparts, caspase-4/5 [18]. Direct binding of these caspases with intracellular LPS leads to the cleavage of GSDMD and the release of N-terminal (GSDMD-NT), which is an executor of cell pyroptosis [15, 19]. GSDMD-NT then oligomerizes and translocates to the plasma membrane and forms a pore with an inner diameter of 10–14 nm that causes cell swelling and pyroptosis [20–23]. Notably, in addition to GSDMD, caspase-4/11 also functions upstream of a canonical Nod-like receptor protein 3 (NLRP3) inflammasome; upon activation, NLRP3 recruits the adaptor protein ASC and triggers caspase-1 activation, which in turn processes the maturation of proinflammatory cytokines interleukin-1β (IL-1β) and IL-18 [13, 24, 25]. Furthermore, canonical pyroptosis, which is mediated by canonical inflammasomes, caspase-1, and GSDMD, plays a critical role in polymicrobial sepsis. Blocking this process protects mice against caecum ligation and puncture (CLP)-induced sepsis [26, 27].
In this work, we found that Mg2+ restrains both canonical and noncanonical pyroptosis by blocking GSDMD-NT oligomerization and membrane localization. Its potential mechanism in noncanonical pyroptosis was also investigated. Furthermore, mice intraperitoneally injected with Mg2+ had significantly reduced susceptibility to lethal septic shock. Collectively, these findings reflect that magnesium, as a critical macroelement, is responsible for the regulation of the immune response during infection. Correction of hypomagnesemia and proper magnesium supplementation should be beneficial to reducing the sepsis morbidity of critically ill patients.
Results
Hypomagnesemia is associated with decreased monocyte levels in septic patients
Hypomagnesemia in critically ill patients has been linked to poor clinical outcomes [1]. To confirm this in Chinese patients, clinical data were collected from the surgical ICU of Jinling Hospital, Nanjing, China. From a total of 311 patients, 32 were excluded according to the exclusion criteria. Of the 279 patients studied, 136 (48.7%) were traumatic patients, and 96 (34.4%) were surgical patients. At admission, 97 (34.8%) patients had hypomagnesemia and 182 (65.2%) had normomagnesemia (Table S1). There was no significant difference in age, gender, or primary disease between the two groups of patients, but hypomagnesemic patients had a higher SOFA score and APACHE II score at admission than patients with normomagnesemia (P = 0.075 for both scores) (Table S1). Furthermore, patients with hypomagnesemia had a significantly longer ICU length of stay, higher risk of developing sepsis, and higher mortality rate than patients with normomagnesemia (Table S1).
To further investigate the impact of hypomagnesemia on sepsis development, 87 patients diagnosed with sepsis during their ICU stay were analyzed separately. Among these patients, 39 (44.8%) had hypomagnesemia at admission. Compared to the others, these patients did not have a significantly different white blood cell (WBC) count or WBC differential at admission (Fig. 1a). However, when sepsis struck, these patients had a significantly lower monocyte percentage (P < 0.01); WBC count, neutrophil percentage, and lymphocyte percentage were not significantly different from the other patients (Fig. 1a). To look for a linear relationship between the serum magnesium level at admission and the monocyte percentage on the day of sepsis diagnosis, a Pearson’s correlation analysis was conducted. Importantly, a significant positive correlation was found between the two parameters (P < 0.001, R(85) = 0.381) (Fig. 1b), indicating that patients with lower serum magnesium at admission might have a lower monocyte percentage when sepsis strikes.
Fig. 1.

Hypomagnesemia is associated with decreased monocyte level when sepsis strikes. a Patients developing sepsis during their ICU stay were divided into two groups according to their serum magnesium at admission. Of the 87 septic patients, 48 were normomagnesemic (0.7–1.0 mmol/L) and 39 were hypomagnesemic (<0.7 mmol/L). Box plots of white blood cell (WBC) count, neutrophil percentages, monocyte percentages, and lymphocyte percentages in these two groups of patients are shown. In the upper panels, parameters were collected at admission; in the lower panels, parameters were collected on the day of sepsis diagnosis. The center line represents the median value, the limits represent the 25th and 75th percentile, and the whiskers represent the minimum and maximum value of the distribution. **P < 0.01. b Scatter plot and regression line for the regression between serum magnesium at admission and monocyte percentage on the day of sepsis diagnosis in septic patients (n = 87). See also Table S1. ICU intensive care unit
Mg2+ inhibits noncanonical pyroptosis in macrophages
The above analyses implied that magnesium might be protective to the death of monocytes during sepsis. Recently, noncanonical pyroptosis was reported to play a critical role in the development of LPS-induced sepsis [13]. To examine whether Mg2+ is involved in the regulation of noncanonical pyroptosis, mouse immortalized bone marrow-derived macrophages (iBMDMs) were primed with Pam3CSK4 and then electroporated with LPS. We showed that cells stimulated with intracellular LPS presented robust lactate dehydrogenase (LDH) release, which was attenuated by Mg2+ in a concentration-dependent manner, with as low as 5 mM being effective (Fig. 2a). Furthermore, cells incubated with 20 mM Mg2+ did not develop the typical pyroptotic morphology with cell swelling and membrane blebbing (Fig. 2b), and propidium iodide (PI) uptake and IL-1β release were also significantly decreased (Fig. 2c, d). By contrast, 5 mM glycine, an antilytic reagent, did not prevent the membrane blebbing, PI uptake, or IL-1β release in iBMDMs, albeit it significantly reduced the LDH release (Fig. 2a–d). These data indicated that Mg2+ and glycine act differently in preventing pyroptosis.
Fig. 2.

Mg2+ inhibits noncanonical pyroptosis in iBMDMs downstream of GSDMD cleavage. a−f Mouse immortalized bone marrow-derived macrophages (iBMDMs) primed overnight with Pam3CSK4 in advance were electroporated with LPS for 2 h, in the absence or presence of MgCl2, MgSO4, MgGluc2, or 5 mM glycine as indicated. Cells electroporated without LPS (Ctrl) are used as controls. a Percentage of LDH release (used to measure cytotoxicity). b Representative brightfield cell images. Representative pyroptotic cells were indicated with white arrows. Bar = 20 μm. c Representative images of propidium iodide (red) uptake. Bar = 20 μm. d IL-1β release. e Immunoblots for pro-caspase-1 (Casp-1 p45) and its cleavage product (Casp-1 p20), and full-length gasdermin D (GSDMD-FL) and its N-terminal (GSDMD-NT) in whole cell lysate. α-tubulin is used as a loading control. f Immunoblots for GSDMD-NT in the cytosolic and membrane fractions. Cytosolic and membrane-bound proteins were separated by using a Plasma membrane protein isolation kit (see Methods). α-tubulin and transferrin receptor 1 (TFR1) are used as loading controls respectively. For panels (a) and (d), data are presented as mean ± SD; *P < 0.05, **P < 0.01, compared to the cells electroporated with LPS in the absence of Mg2+ and glycine. Each panel is a representative experiment of at least three replicates. See also Figs. S1 and S2. LPS lipopolysaccharide, LDH lactate dehydrogenase
Caspase-11-mediated GSDMD cleavage is considered as a critical step in noncanonical pyroptosis [15, 19]; thus, we next examined whether GSDMD cleavage is blocked by Mg2+ treatment. Notably, the cleavage of GSDMD (i.e. the expression of GSDMD-NT) was not impeded (Fig. 2e). Instead, the membrane binding of GSDMD-NT was significantly blocked by Mg2+ (Fig. 2f). To further confirm this notion, we used different methods to separate the cytosolic and membrane fractions, and the two results were in complete agreement. In addition, Mg2+ did not block the activation of caspase-1, implying that the release but not maturation of IL-1β was inhibited by Mg2+ (Fig. 2e).
To examine whether the protective effect of Mg2+ on pyroptosis is cell-line specific, a similar set of experiments were conducted in human THP-1 cells which were transfected with LPS by FuGENE (Fig. S2A−E). Except that 5 mM glycine failed to reduce LDH release from THP-1 cells (Fig. S2A), the other results were in total agreement with the data in mouse iBMDMs. Together, these data indicate that Mg2+ inhibits noncanonical pyroptosis by impeding the membrane localization of GSDMD-NT, without affecting the activation of NLRP3 inflammasome.
Mg2+ inhibits canonical pyroptosis in macrophages
Given that GSDMD-NT is indispensable for both noncanonical and canonical pyroptosis [14, 15, 19, 28] while Mg2+ impedes the function of GSDMD-NT, we assumed that Mg2+ also restrained canonical pyroptosis. We first used LPS plus ATP to trigger canonical pyroptosis in iBMDMs, and the inhibitory effect of Mg2+ was confirmed by both LDH assay and morphological observation (Fig. S3A and S3B). However, since Mg2+ binds to ATP as evidenced by the earlier studies [29, 30], it functioned upstream of NLRP3 activation and thus blocked ASC oligomerization, caspase-1 activation, GSDMD cleavage, and IL-1β maturation and release (Fig. S3C−E). Therefore, we cannot observe the effect of Mg2+ on GSDMD-NT in this case.
Nigericin is a K+ ionophore that activates the NLRP3 inflammasome by depleting cytosolic K+ [31]. We showed that Mg2+ also inhibited nigericin-induced pyroptosis in a concentration-dependent manner as measured by LDH release, and that 20 mM Mg2+ significantly prevented the membrane blebbing and PI uptake in iBMDMs in response to nigericin stimulation (Fig. 3a–c). Importantly, IL-1β release was decreased while ASC oligomerization, caspase-1 activation, or GSDMD cleavage were not affected by Mg2+ (Fig. 3d–f). Further analysis indicated that Mg2+ blocked the membrane binding of GSDMD-NT (Fig. 3g and Fig. S4), consistent with the results in noncanonical pyroptosis. Together, these data confirm that the inhibitory effect of Mg2+ on the membrane binding of GSDMD-NT exists not only in noncanonical pyroptosis but also in canonical pyroptosis.
Fig. 3.

Mg2+ inhibits nigericin-induced pyroptosis in iBMDMs downstream of GSDMD cleavage. a−g Mouse iBMDMs primed with LPS for 4 h in advance were stimulated with (or without) 1 μM nigericin for 1 h, in the absence or presence of MgCl2, MgSO4, MgGluc2, or 5 mM glycine as indicated. a Percentage of LDH release. b Representative brightfield cell images. Representative pyroptotic cells were indicated with white arrows. Bar = 20 μm. c Representative images of propidium iodide (red) uptake. Bar = 20 μm. d IL-1β release. e Representative confocal immunofluorescence images of endogenous ASC specks (green) and DAPI (blue) in cells. Bar = 20 μm. f Immunoblots for Casp-1 p45, Casp-1 p20, GSDMD-FL, and GSDMD-NT in whole cell lysate. α-tubulin is used as a loading control. g Immunoblots for GSDMD-NT in the cytosolic and membrane fractions. Cytosolic and membrane-bound proteins were separated by using a Plasma membrane protein isolation kit. α-tubulin and TFR1 are used as loading controls respectively. For panels (a) and (d), data are presented as mean ± SD; **P < 0.01, compared to the cells treated with LPS + nigericin in the absence of Mg2+ and glycine. Each panel is a representative experiment of at least three replicates. See also Figs. S3 and S4. iBMDM immortalized bone marrow-derived macrophage, LDH lactate dehydrogenase
Mg2+ inhibits GSDMD-NT membrane binding and oligomerization in HEK293T cells
GSDMD-NT oligomerizes on the plasma membrane during pyroptosis [20, 22]; thus, we used HEK293T cells to determine whether GSDMD-NT oligomerization is blocked by Mg2+. HEK293T cells transfected with human GSDMD-NT cDNA were largely killed, displaying pyroptotic morphology and robust LDH release, but these were significantly abrogated by Mg2+ (Fig. 4a and Fig. S5). Further analysis indicated that the membrane localization, but not the expression, of GSDMD-NT was inhibited (Fig. 4b), and these results were visualized in HEK293T cells expressing enhanced green fluorescent protein (eGFP)-labeled GSDMD-NT (Fig. 4c). To detect oligomer formation, HEK293T cells expressing Flag-GSDMD-NT were lysed under nonreducing conditions, and Flag immunoblot was performed. Consistent with a previous report [22], GSDMD-NT oligomers were detected at >180 kDa on a nonreducing gel, and the band disappeared in the presence of Mg2+, suggesting that Mg2+ inhibits GSDMD-NT oligomerization (Fig. 4d). To further confirm this, GSDMD-NT was labeled with cyan fluorescent protein (CFP) or yellow fluorescent protein (YFP) to record fluorescence resonance energy transfer (FRET). We first demonstrated that these tagged proteins were still functional in the absence but not presence of Mg2+ (Fig. S6A and S6B). FRET experiments were then performed to examine their interactions. Whereas the FRET signals could barely be determined in the presence of Mg2+, robust signals emerged after a brief washout (Fig. 4e). The quantification of these signals indicated a significant increase in FRET efficiency after the washout, confirming that GSDMD-NT oligomerization is blocked by Mg2+ (Fig. 4f). Taken together, our results demonstrate that both membrane localization and oligomerization of GSDMD-NT are blocked by Mg2+.
Fig. 4.

Mg2+ inhibits GSDMD-NT membrane localization and oligomerization in HEK293T cells. a−f HEK293T cells transfected with indicated cDNA were cultured for 16 h, in the absence or presence of MgCl2, MgSO4, or MgGluc2 as indicated. a Percentage of LDH release. b Immunoblots for Flag in the whole cell lysate, cytosolic fraction, and membrane fraction. Cytosolic and membrane-bound proteins were separated by using a Plasma membrane protein isolation kit. α-tubulin and TFR1 are used as loading controls respectively. c Representative confocal immunofluorescence images of cells. Bar = 5 μm. d Oligomer formation assessed by Flag immunoblot of nonreducing gel (left) and reducing gel (right). e, f Cells transfected with CFP-GSDMD-NT and YFP-GSDMD-NT were cultured for 16 h in the presence of 10 mM MgSO4. The fluorescence resonance energy transfer (FRET) signal was then detected before and after a washout of MgSO4. Shown are representative images (e) and quantification of FRET efficiency (f). Bar = 10 μm. For panels (a) and (f), data are presented as mean ± SD; *P < 0.05, **P < 0.01, compared to the cells transfected with Flag-GSDMD-NT in the absence of Mg2+ (a) or the cells before the washout (f). Each panel is a representative experiment of at least three replicates. See also Figs. S5 and S6. LDH lactate dehydrogenase
Mg2+ inhibits GSDMD-NT-induced pyroptosis by blocking Ca2+ influx
Mg2+ is a powerful Ca2+ antagonist physiologically [7], and we next investigated whether the inhibitory effect of Mg2+ on GSDMD-NT was associated with its role as a Ca2+ antagonist. Interestingly, EGTA, a selective chelator of extracellular Ca2+, and BAPTA-AM, a strong chelator of intracellular Ca2+, both significantly inhibited GSDMD-NT-induced pyroptosis in HEK293T cells (Fig. 5a). Similar effects were observed in cells treated with lanthanides (La3+ and Gd3+), which are broad-spectrum inhibitors of cation channels, and in cells incubated with Ca2+/Mg2+-free medium or Ca2+-free medium (Ca2+/Mg2+-free medium supplemented with 0.8 mM Mg2+) (Fig. 5a). These data indicate that blocking Ca2+ influx could also rescue GSDMD-NT-induced pyroptosis.
Fig. 5.

Mg2+ inhibits GSDMD-NT-induced pyroptosis by blocking Ca2+ influx. a, e HEK293T cells transfected with Flag-GSDMD-NT (or empty vector) were treated as indicated. Percentage of LDH release was measured (a), and immunoblots for GSDMD-NT in the cytosolic and membrane fractions were performed (e). Cytosolic and membrane-bound proteins were separated by using a Plasma membrane protein isolation kit. α-tubulin and TFR1 are used as loading controls respectively. b−d HEK293T cells transduced with a doxycycline- (DOX-) inducible human GSDMD-NT cDNA were transfected with GCaMP6 plasmid, a genetically encoded Ca2+ indicator. Cells were treated (or untreated) with DOX 16 h later, in the absence or presence of 10 mM MgSO4. b Representative time-lapse cell images (brightfield and fluorescence) taken from 0 to 210 min after DOX addition. Bar = 100 μm. c Scatter plot of the fluorescence intensity of GCaMP6. d LDH release measured from 35 to 350 min after DOX addition. f iBMDMs primed with Pam3CSK4 overnight were electroporated with (or without) LPS and incubated in the indicated media for 2 h. LDH (left) and IL-1β (right) release were measured. Cells electroporated without LPS (Ctrl) were used as controls. g iBMDMs primed with LPS for 4 h were treated (or untreated) with 1 μM nigericin in the indicated media for 1 h. LDH (left) and IL-1β (right) release were measured. For panels (a, f, and g), data are presented as mean ± SD; **P < 0.01, compared to the cells transfected with Flag-GSDMD-NT (a), electroporated with LPS (f), or treated with nigericin (g) in normal DMEM. Each panel is a representative experiment of at least three replicates. See also Video S1. LPS lipopolysaccharide, iBMDM immortalized bone marrow-derived macrophage, DMEM Dulbecco’s modified Eagle’s medium
To directly demonstrate that Mg2+ inhibits GSDMD-NT-induced pyroptosis via blocking Ca2+ influx, human GSDMD-NT was inducibly expressed in HEK293T cells, and GCaMP6 plasmid, a genetically encoded Ca2+ indicator, was used to monitor the Ca2+ level in live cells. We noticed that the fluorescence intensity of GCaMP6 was markedly increased in the absence, but not presence, of Mg2+ (Fig. 5b and Video S1). Further analysis revealed that the Ca2+ signal emerged much earlier than LDH release (Fig. 5c, d), consistent with the previous report [32]. Additionally, the membrane localization of GSDMD-NT was impeded when Ca2+ was removed (Fig. 5e), and both intracellular LPS- and nigericin-induced pyroptosis were blocked when cells were cultured in Ca2+/Mg2+-free or Ca2+-free medium (Fig. 5f, g). Collectively, these data indicate that Ca2+ influx is essential for GSDMD-NT-induced pyroptosis and that Mg2+ inhibits Ca2+ influx and thereby blocks pyroptosis.
The ATP-gated Ca2+ channel P2X7 is essential for noncanonical pyroptosis
To determine which type of Ca2+ channel is involved in noncanonical pyroptosis, iBMDMs electroporated with LPS were treated with various Ca2+ channel blockers. Four types of voltage-dependent Ca2+ channel blockers, including nifedipine, mibefradil, amlodipine, and manidipine, all failed to rescue the cells (Fig. 6a). By contrast, two types of broad-spectrum Ca2+ channel blockers, La3+ and Gd3+, succeeded (Fig. 6a).
Fig. 6.

The ATP-gated Ca2+ channel P2X7 is essential for noncanonical pyroptosis. a, c−g iBMDMs primed overnight with Pam3CSK4 in advance were electroporated with LPS for 2 h. Cells electroporated without LPS (Ctrl) are used as controls. Ca2+ channel inhibitors (i.e. nifedipine, mibefradil, amlodipine, manidipine, LaCl3, GdCl3), pannexin-1 inhibitors (i.e., carbonoxolone, probenecid, and trovafloxacin), and P2X7 inhibitors (i.e., A438079, A740003, and A804598) were added 30 min before the electroporation, while MgCl2, CuCl2, ZnCl2, and NiCl2 were added right after the electroporation. The concentration of the inhibitors were all 0.1 mM. a Percentage of LDH release from cells treated as indicated. b Immunoblot for P2X7 in whole-cell lysate of WT iBMDMs and P2X7 knockout (KO) iBMDMs transfected with empty vector or P2X7 cDNA. α-tubulin is used as a loading control. c WT iBMDMs and P2X7 KO iBMDMs transfected with empty vector or P2X7 cDNA were primed and electroporated as described above. Shown are LDH (upper) and IL-1β (lower) release.**P < 0.01. d Percentage of LDH release from cells treated as indicated. e, g Immunoblots for GSDMD-FL, GSDMD-NT, Casp-1 p45, Casp-1 p20 in the whole-cell lysate, cytosolic fraction, and membrane fraction of cells treated as indicated. Cytosolic and membrane-bound proteins were separated through the use of OptiPrepTM (f) or by using a Plasma membrane protein isolation kit (g). α-tubulin, glyceraldehyde 3-phosphate dehydrogenase (GAPDH), and TFR1 are used as loading controls respectively. f LDH (left) and IL-1β (right) released from cells treated as indicated. Data are presented as mean ± SD. For panels (a, d, and f), **P < 0.01, compared to the cells electroporated with LPS and then left untreated or treated with DMSO. Each panel is a representative experiment of at least three replicates. See also Fig. S7. iBMDM immortalized bone marrow-derived macrophage, LPS lipopolysaccharide, LDH lactate dehydrogenase
P2X7 is a Ca2+ channel sensitive to La3+ and Gd3+ and has been reported to play a key role in noncanonical pyroptosis [33–35]. Ablation of P2X7 protects BMDMs against intracellular LPS stimulation [33]. Therefore, we assumed that P2X7-mediated Ca2+ influx is critical for the normal function of GSDMD-NT. As P2X7 is expressed in iBMDMs, we used CRISPR/Cas9 technology to knockout (KO) P2X7 gene. Consistent with the previous study [33], P2X7 KO iBMDMs were resistant to LPS electroporation while WT iBMDMs and P2X7 KO iBMDMs reconstituted with plasmid producing P2X7 remained susceptible (Fig. 6b, c). Furthermore, IL-1β release was partly blocked by P2X7 KO (Fig. 6c). These data were further confirmed by P2X7 shRNA knockdown in iBMDMs (Fig. S7A and S7B).
P2X7 is activated by ATP released from pannexin-1 channel in noncanonical pyroptosis [33], and Mg2+ is an ATP chelator which can block the activation of P2X7 [29, 30]. As other divalent metal cations, such as Cu2+, Zn2+, Ni2+, have been reported to have much higher potency than Mg2+ to reduce the affinity of ATP binding to P2X7 [30], we treated iBMDMs undergoing noncanonical pyroptosis with 1 mM Mg2+, Cu2+, Zn2+, and Ni2+. Whereas 1 mM Mg2+ basically had no effect on pyroptosis, 1 mM Cu2+, Zn2+, and Ni2+ significantly reduced the LDH release and impeded the membrane binding of GSDMD-NT (Fig. 6d, e), similar to the effect of 20 mM Mg2+.
Furthermore, consistent with the previous report [33], both pannexin-1 inhibitors (carbonoxolone, probenecid, and trovafloxacin) and P2X7 inhibitors (A438079, A740003, and A804598) rescued iBMDMs electroporated with LPS, while only pannexin-1 inhibitors completely blocked the cleavage of caspase-1 and the release of IL-1β (Fig. 6f, g). Notably, blockade of either pannexin-1 or P2X7 had no impact on the cleavage of GSDMD but impeded the membrane localization of GSDMD-NT (Fig. 6g). These data suggest that P2X7-mediated Ca2+ influx is crucial for the membrane localization of GSDMD-NT to induce noncanonical pyroptosis.
Mg2+ protects against LPS-induced septic shock
To extend our in vitro findings, we next examined whether Mg2+ solutions could protect mice from LPS-induced septic shock. Mice were primed with 0.4 mg/kg LPS and then challenged with 10 mg/kg LPS 6 h later. In agreement with a previous study [33], wild-type mice that were treated with 0.9% NaCl (LPS group) succumbed within 24 h; whereas over 50% of mice that were treated with 1 mmol/kg Mg2+ survived beyond 72 h (Fig. 7a). Under the protection of Mg2+, the amount of serum IL-1β and IL-6 at 4 and 8 h after LPS challenge was significantly reduced, confirming the anti-inflammatory effects of Mg2+ (Fig. 7b). Other cytokines (e.g., TNF-α, IL-2, IL-4, IL-5, IL-10, IL-12p70, and IFN-γ), however, were not significantly different between the two groups (Fig. 7b and Fig. S8A). By routine blood test, we found that the WBC levels and WBC differential count in mice treated with Mg2+ were significantly higher than in the LPS group after LPS challenge (Fig. 7c). In particular, the monocyte count in mice treated with Mg2+ was twice that in the LPS group (Fig. 7c). Consistent with this finding, flow cytometry analysis showed that mice treated with Mg2+ had apparently higher monocyte and macrophage percentages than those in the LPS group (Fig. 7d), whereas T cell, B cell, and nature killer cell percentages were comparable between the two groups (Fig. S8B). LPS-induced endothelial pyroptosis is the leading cause of death in endotoxaemic mouse models [36]. By H&E staining, we observed that Mg2+ markedly alleviated the inflammation and lung injury caused by LPS challenge in lung (Fig. 7e), which is in accord with a previous report [10]. Collectively, these results suggest that Mg2+ might be protective in LPS-induced septic shock; the underlying mechanisms might be associated with its inhibitory effect on pyroptosis and on the release of proinflammatory cytokines.
Fig. 7.

Mg2+ is protective in LPS-induced endotoxic shock. a−e C57BL/6 male mice were left untreated (Ctrl group) or primed with 0.4 mg/kg E. coli O111:B4 LPS intraperitoneally and then challenged 6 h later with 10 mg/kg E. coli O111:B4 LPS intraperitoneally. Upon the secondary challenge, LPS was mixed with 1 mmol/kg MgSO4, MgCl2 or MgGluc2 as indicated, or an equal volume of 0.9% NaCl (LPS group). N = 8 per group. a Survival of mice after LPS challenge. **P < 0.01, compared to the LPS group. b IL-1β, IL-6, and TNF-α levels in mouse plasma measured at the indicated time points after LPS challenge. Mice treated with MgSO4 were compared to the LPS group at each time point. c Cell counts of white blood cell (WBC), neutrophil, monocyte, and lymphocyte in mouse whole blood measured in a normal state (Ctrl) or at 12 h after LPS challenge. d Representative flow cytometry plots (left) and quantification (right) of monocytes (CD11b+LY6C+LY6G−) and macrophages (CD11b+MHC-II+F4/80+) in mouse whole blood measured in a normal state (Ctrl) or at 12 h after LPS challenge. e Representative H&E-stained lung sections obtained from mice in a normal state (Ctrl) or at 0 or 12 h after LPS challenge. Bar = 100 μm. Data are presented as mean ± SD. *P < 0.05, **P < 0.01, ns = not significant. Each panel is a representative experiment of at least two replicates. See also Fig. S8
Discussion
Magnesium homeostasis is essential for the physiological function of multiple organs, and magnesium supplementation has been considered as a treatment for several major diseases, such as preeclampsia, migraine, depression, and asthma [7]. However, modest hypomagnesemia is often neglected in clinical practice due to the lack of characteristic clinical features [7]. Several studies, along with our current work, indicate that critically ill patients with hypomagnesemia have a higher risk of developing sepsis than patients with normomagnesemia [2–5]. This is probably because hypomagnesemia makes cells more susceptible to pyroptosis. When infection occurs, cells are excessively activated, resulting in extensive pyroptosis and robust inflammatory response. Our data indicate that Mg2+ is essential for protecting cells from pyroptosis, and the magnesium level is an important index for critically ill patients.
Mg2+ acts as a physiological Ca2+ antagonist, and changes in intracellular and extracellular Mg2+ concentration often cause disturbed Ca2+ signaling [7]. In this study, we proposed that Mg2+ impeded pyroptosis by blocking Ca2+ influx. In noncanonical pyroptosis, caspase-11-dependent cleavage of pannexin-1 leads to ATP release, which in turn activates P2X7, a Ca2+ channel, to mediate pyroptosis [33]. We showed that extracellular Mg2+ bound to ATP and thereby impeded the activation of P2X7 as well as P2X7-mediated Ca2+ influx, and that the ATP-gated Ca2+ channel P2X7 is indispensable for noncanonical pyroptosis (Fig. 8). For regulating canonical pyroptosis elicited by nigericin, however, the specific Ca2+ channel remains to be determined. We know this specific channel can be inhibited by Mg2+, La3+, and Gd3+, and the channel has nothing to do with pannexin-1 or P2X7 [37, 38]. Furthermore, as P2X7 is not expressed in HEK293T cells [39], the Ca2+ influx triggered by GSDMD-NT in HEK293T cells must be mediated by other channels, probably TMEM16F [40]. However, it is still an open question as to whether TMEM16F is essential for pyroptosis in macrophages. These results indicate that although Ca2+ influx is essential for the function of GSDMD-NT, the specific channel may differ among various cell types and various pathways.
Fig. 8.
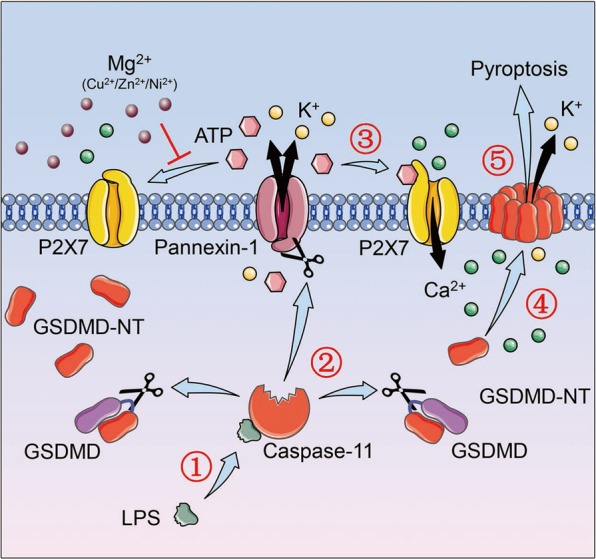
A model regarding how Mg2+ impedes noncanonical pyroptosis. Upon intracellular LPS stimulation, activated caspase-11 cleaves and activates the K+ channel pannexin-1 on the one hand, and cleaves GSDMD to form GSDMD-NT on the other. Subsequently, activated pannexin-1 releases ATP to elicit Ca2+ channel P2X7, which licenses the Ca2+ influx. With the help of Ca2+ influx, either directly or indirectly, GSDMD-NT then binds to the plasma membrane and oligomerizes to form pores which result in membrane rupture and pyroptosis. Meanwhile, the K+ efflux mediated by pannexin-1 and GSDMD-NT pore, and probably the mitochondrial reactive oxygen species driven by GSDMD-NT, all contribute to the activation of NLRP3 inflammasome (not shown in the figure). Nevertheless, divalent metal cations such as Mg2+, Cu2+, Zn2+, Ni2+, may disrupt the pyroptotic process by binding to ATP. In this case, the ATP released by pannexin-1 cannot activate P2X7, and thus Ca2+ influx is blocked. Without Ca2+ influx, GSDMD-NT cannot bind to the membrane and form pores, and pyroptosis is therefore inhibited. LPS lipopolysaccharide
Notably, previous studies have showed that GSDMD pores can be assembled in synthetic liposomes in the absence of Ca2+ [22]. We reasoned that this discrepancy may be attributed to the different systems used. Specifically, cell is much more complicated than synthetic liposome. Indeed, it is still unclear whether Ca2+ assists the assembly of GSDMD-NT pore directly or indirectly, and we cannot rule out the possibility that Ca2+ may regulate other proteins in cells which license the assembly of GSDMD-NT. Hence, the underlying mechanism concerning how Ca2+ regulates GSDMD-NT pore assembly must be further investigated in the future.
Many previous studies have indicated that the ability of noncanonical inflammasome activation to secondarily activate NLRP3/caspase-1 is mediated by K+ efflux, and that GSDMD is indispensable in this process. Nevertheless, no study, as far as we are concerned, has demonstrated that K+ efflux is necessarily via the GSDMD-NT pore. Notably, a previous study showed that activated caspase-11 triggers the opening of the pannexin-1 channel through cleavage, which induces the K+ efflux required for NLRP3 inflammasome activation [33]. Our data also demonstrated that pannexin-1 inhibitors completely impeded caspase-1 activation and IL-1β release. In addition, another study proposed that GSDMD-NT-induced mitochondrial reactive oxygen species (ROS) also contributed to the activation of NLRP3 inflammasome in caspase-4-mediated noncanonical pyroptosis [41]. Pretreating cells with antioxidant N-acetylcysteine impeded caspase-1 cleavage and IL-1β production without affecting caspase-4 cleavage [41]. Taken together, it appears that the K+ efflux mediated by pannexin-1 and GSDMD-NT pore, as well as the mitochondrial ROS elicited by GSDMD-NT, all contribute to NLRP3 inflammasome activation in noncanonical pyroptosis. Impeding any of these (e.g. ablation of GSDMD, inhibition of pannexin-1, blockade of the K+ efflux, elimination of mitochondrial ROS) may affect the normal activation of NLRP3 inflammasome. In this study, we showed that Mg2+ impeded the membrane binding of GSDMD-NT without affecting caspase-1 activation. This may be attributed to the K+ efflux mediated by pannexin-1 and the mitochondrial ROS mediated by GSDMD-NT. In addition, we cannot rule out the possibility that a small amount of GSDMD-NT pores generated and contributed to K+ efflux even in the presence of 20 mM Mg2+.
In summary, our current work reveals that Mg2+, as a Ca2+ antagonist, regulates the oligomerization and membrane localization of GSDMD-NT and thereby protects the cell from pyroptosis. The role of Ca2+ influx as a prerequisite for GSDMD-NT to function was proposed, and the relationship between caspase-11-pannexin-1-P2X7 signaling and caspase-11-GSDMD signaling was determined. Furthermore, our in vivo studies have demonstrated the protective effects of Mg2+ in an endotoxaemic mouse model. Further investigation is needed to determine whether Mg2+ supplementation improves the outcomes of critically ill patients.
Methods
Clinical data
Clinical data was abstracted from the medical records in surgical ICU of Jinling Hospital, Nanjing, China. All adult patients (>18 years old) admitted consecutively to the surgical ICU from January 2015 to August 2015 with available admission serum magnesium levels were primarily enrolled. Exclusion criteria were patients who presented with hypermagnesemia (serum magnesium > 1.0 mmol/L) or diagnosed with sepsis at admission, and who received magnesium supplementation during their ICU stay. The Acute Physiology And Chronic Health Evaluation (APACHE) II score and the Sequential Organ Failure Assessment (SOFA) score were determined at admission. Sepsis was diagnosed according to the diagnostic criteria of the American College of Chest Physicians/Society of Critical Care Medicine [42].
Cell culture
Human embryonic kidney (HEK) 293T cells and 293FT cells were obtained from ATCC and were cultured in Dulbecco’s modified Eagle’s medium (DMEM). C57BL/6 mice-derived immortalized BMDM (iBMDM) cells were kindly provided by F. Shao (National Institute of Biological Sciences, China) and THP-1 (human monocyte) cells were obtained from Shanghai Institute of Cell Biology (Shanghai, China). These two cell lines were grown in RPMI 1640 medium. In some cases, DMEM without Ca2+/Mg2+ (Cat#SH30262.01, Hyclone) was used, and DMEM was used as control. All mediums mentioned above were supplemented with 10% fetal bovine serum (Cat#P30-3302, PANBiotech, Germany) and 1% penicillin/streptomycin. Cells of passages fewer than 15 were used for experiments. All cells were cultured at 37 ℃ and 5% CO2.
Plasmids and transfection
Complementary DNA (cDNA) for human gasdermin D (GSDMD) was amplified from reverse-transcribed cDNA from THP-1 cells, and cDNA for its N-terminal (GSDMD-NT) (1–275) was derived by PCR. For transient expression in 293T cells, these two cDNAs were inserted into the NotI and XbaI sites of a pCS2 vector with an N-terminal 3× Flag epitope tag respectively; GSDMD-NT cDNA was additionally inserted into the XhoI and EcoRI sites of a pEGFP-N1 vector with a C-terminal eGFP epitope tag. For FRET experiment, CFP or YFP were inserted into the N-terminal of GSDMD-NT cDNA by homologous recombination. For Tet-On expression in 293T cells, GSDMD-NT cDNA was inserted into a pTRIPZ vector. For the plasmid harboring the GSDMD-NT gene was then cotransfected with the packing plasmids pSPAX2 and pMD2G into 293FT cells. The lentivirus particles were harvested, filtered, and concentrated by ultracentrifugation 48 h later and were used to infect 293T cells for another 48 h. 293T cells that are resistant to puromycin were selected. cDNA for mouse P2X7 was amplified from reverse-transcribed cDNA from BMDM cells, and was inserted into the EcoRI and XbaI sites of the PCS2 vector. All plasmids were verified by DNA sequencing. Lipofectamine 2000 Transfection Reagent (Invitrogen) was used for transfection.
CRISPR knockout and shRNA knockdown
To knockout P2X7 in iBMDMs, the sgRNA sequences of P2X7 (AATGGAGAGGGGCCGTTAAT and CAAAATAGCACCGCGCGTCC) were inserted into pSpCas9(BB)-2A-Puro (PX459) V2.0 (#62988, Addgene). These two plasmids were electroporated using the Neon Transfection System (Invitrogen). To transiently knockdown P2X7 in iBMDMs, pLKO-1 plasmids targeting mouse P2X7 were electroporated using the Neon Transfection System (Invitrogen). The targeting sequences of P2X7 shRNA1 and P2X7 shRNA2 are CCCGGCTACAACTTCAGATAT and GCCACAACTATACCACGAGAA, respectively.
Activation of canonical and noncanonical pyroptosis
To stimulate caspase-1-mediated canonical pyroptosis, iBMDMs were primed with 1 μg/mL LPS for 4 h followed by 5 mM ATP or 1 μM nigericin treatment for 1 h. To stimulate caspase-4/11-mediated noncanonical pyroptosis, THP-1 cells or iBMDMs were primed with 1 μg/mL Pam3CSK4 overnight and then either transfected with 2 μg/mL LPS by using 0.25% v/v FuGENE HD (Promega) or electroporated with 50 ng/mL LPS by using the Neon Transfection System (Invitrogen) following the manufacturer’s instructions.
Measurement of LDH release
LDH activity in the supernatant was used to evaluate cell death. Cellular debris was removed by centrifuging the supernatant at 300 × g for 5 min. LDH measurement was performed using a CytoTox 96 Non-Radioactive Cytotoxicity Assay kit (Promega) according to the manufacturer’s protocol. All values represent the percentage of LDH release by a maximum lysis control (1% Triton X-100-lysed cells).
Immunoblotting
Total cell protein was extracted on ice using RIPA lysis buffer (50 mmol/L Tris-HCl (PH 7.4), 150 mmol/L NaCl, 1% NP-40, 0.1 mmol/L ethylenediaminetetraacetic acid (EDTA), 1 mmol/L DTT with 0.4 mmol/L phenylmethanesulfonyl fluoride, 0.1 mmol/L Na3VO4, 0.1 mmol/L NaF and cocktail protein inhibitor). To separate cytosolic and membrane protein, two different methods were used as indicated in the relative figure legends. The first method is through the use of a plasma membrane protein isolation kit (SM-005, Invent Biotechnologies, Inc.), according to the manufacturer’s instructions. The second method is through the use of OptiPrepTM Density Gradient Medium (D1556, Sigma-Aldrich) according to the protocol (Application Sheet S35; eight edition, February 2018) obtained from the official website of Axis-Shield Density Gradient Media, a brand of Alere Technologies AS (https://www.axis-shield-density-gradient-media.com/organelleindexes.htm), and the link of the protocol is: https://www.axis-shield-density-gradient-media.com/S35.pdf. For nonreducing gels, cells were lysed in lysis buffer without DTT and cell lysates were prepared with 2ME-free SDS loading buffer. Protein concentrations were determined using a Bradford protein assay kit (Sangon, Shanghai, China). The proteins were run in ExpressPlus™ PAGE gels (M42015C, GenScript, China) and then transferred to polyvinylidene difluoride membranes (Amersham Bioscience). After blocking with 5% fat-free milk at room temperature for 1 h, blots were incubated with primary antibody overnight at 4 °C. The next day, the blots were washed with Tris buffered saline Tween 20 and then incubated with secondary antibody for 1 h at room temperature. Proteins were visualized using enhanced chemiluminescence substrate (Tanon, Shanghai, China). Antibodies used in this work included anti-Flag (F1804, Sigma-Aldrich), anti-tubulin (BS1699, Bioworld), anti-GAPDH (G8795, Sigma-Aldrich) anti-GSDMD (ab209845, abcam), anti-caspase-1 (SC-515, Santa Cruz), anti-TfR1 (ab84036, abcam), and anti-P2X7 (#13809, Cell Signaling Technology). Secondary antibodies including anti-rat IgG, anti-mouse IgG, and anti-rabbit IgG were all purchased from Sigma-Aldrich.
Immunostaining
Cells grown on coverslips were first fixed in PBS with 4% paraformaldehyde (PFA) for 15 min, and then permeabilized in 0.1% Triton X-100 in PBS for 15 min. After blocking with 5% BSA for 1 h, cells were stained with ASC antibody (ab175449, abcam) and then Alexa Fluor 488-conjugated secondary antibody. Nuclei were counterstained with DAPI (Cell Signaling). Images were captured at room temperature using a confocal microscope (FV-1000, Olympus).
FRET assay
HEK293T cells cultured on poly-lysine-coated (50 ng/mL) confocal dish were transfected with CFP- and YFP-GSDMD in a ratio of 3 to 1. To accumulate adequate fluorescence, 10 mM MgSO4 was added immediately after the transfection. FRET signal was detected and analyzed by the FRET sensitized emission approach (Leica Microsystems Inc.) 16 h later, before and after a washout of Mg2+.
Live-cell imaging
To examine cell death morphology, cells were treated as indicated in the six-well plates. Static brightfield images of pyroptotic cells were captured using an Olympus IX71 microscope.
For PI staining, 1 μg/mL PI dye obtained from an Annexin V-FITC/PI Apoptosis Detection Kit (catalog no. A211-01, Vazyme) was added right after the electroporation or nigericin stimulation. Images were captured using a confocal microscope (FV-1000, Olympus) 60 min later.
For Ca2+ signaling detection, cells were transfected with Ca2+ indicator GCaMP6 plasmid 24 h before imaging. The GCaMP6 fluorescence was excited using a 488 nm argon laser, and the images were recorded every 30 s using a confocal microscope (FV-1000, Olympus). Cells were incubated in a chamber with a 5% CO2 atmosphere at 37 °C throughout the experiment. The average fluorescence intensity of the ROI was calculated using SV10-ASW software (Olympus). Image acquisition started immediately after doxycline addition (100 ng/mL).
ELISA
The amount of IL-1β in cell culture supernatants was measured by ELISA according to the manufacturer’s protocol. For iBMDMs, a Mouse IL-1 beta/IL-1F2 Quantikine ELISA Kit (Cat#MLB00C, R&D Systems) was used. For THP-1 cells, a Human IL-1 beta/IL-1F2 Quantikine ELISA Kit (Cat#DLB50, R&D Systems) was used.
Animal studies
C57BL/6 mice were provided by the Model Animal Research Center (MARC) of Nanjing University (Nanjing, Jiangsu Province, China) and were housed in a specific pathogen-free facility accredited by Association for Assessment and Accreditation of Laboratory Animal Care International.
6- to 8-week-old male mice were primed with 0.4 mg/kg E. coli O111:B4 LPS intraperitoneally (i.p.) and then challenged 6 h later with 10 mg/kg E. coli O111:B4 LPS i.p. Upon the secondary challenge, LPS was mingled with 1 mmol/kg MgSO4, MgCl2, MgGluc2, or equal volume 0.9% NaCl as indicated. At indicated time points, mice were euthanized and whole blood was collected by cardiac puncture using the EDTA-2K tubes, and lung tissue was collected and fixed in 4% PFA solution. Subsequently, whole blood was used for hematological analysis, flow cytometry, and was centrifuged to provide plasma for the measurement of cytokines. Lung tissue was embedded in paraffin and sectioned, and H&E staining was performed according to the manufacturer’s protocol (Cat#G1005, Servicebio).
Hematological analysis, flow cytometry, and measurement of plasma cytokines
Hematological analysis was performed using a Hemavet 950FS Hematology Analyzer (Drew Scientific, Miami Lakes, FL, USA) following the manufacturer’s manual.
For flow cytometry, red blood cells (RBCs) were lysed using an RBC Lysis Buffer (Cat#00-4300, Invitrogen) and live cells/dead cells were discriminated using SYTOX Blue (Cat#S34857, Invitrogen). To assess cell types, CD11b-PE-CF594 (BD), Ly6G-BV421 (BD), Ly6C-FITC (BD), MHCII-APC-Cy7 (Biolegend), and F4/80-BV605 (Biolegend) antibodies were used for panel 1; CD3-FITC (BD), CD335-AF-647 (BD), CD4-APC-H7 (BD), CD8a-PE-CF594 (BD), and CD19-AF700 (Biolegend) antibodies were used for panel 2. Cells that stained were analyzed by flow cytometry (BD LSRFortessa™) and data were analyzed using FlowJo software.
For measurement of plasma cytokines (IL-1β, IL-6, TNF-α, IL-2, IL-4, IL-5, IL-10, IL-12p70, IFN-γ, KC/GRO), a V-PLEX Proinflammatory Panel 1 (mouse) Kit (Cat#K15048D-1, Meso Scale Diagnostics) was used and data were collected using a multiplex imager (QuickPlex SQ120, Meso Scale Diagnostics). However, the values of IL-6 and KC/GRO were excluded for exceeding the upper limits; thus, IL-6 was re-measured using a Mouse IL-6 Quantikine ELISA Kit (Cat#M6000B, R&D Systems) according to the manufacturer’s protocol.
Statistics
Data are shown as mean ± SD using GraphPad Prism 7 Software (GraphPad Software, San Diego, CA). Unless otherwise specified, statistical analysis was performed using t test. The relationship between serum magnesium and monocyte percentage was assessed using a Pearson’s correlation and then a linear regression analysis. Statistical analysis for survival was performed with log-rank (Mantel−Cox) test. *P < 0.05. **P < 0.01.
Study approval
Animal studies were approved by the MARC Animal Care and Use Committee. The clinical study was retrospective and did not involve interference with routine patient management. The institutional review board of Jinling Hospital indicated that ethical approval is not necessary and they indicated no objection for conducting this study.
Supplementary information
Acknowledgements
We thank XN Zhang, J. Liang, YY Dai, and Nanjing Biomedical Research Institute of Nanjing University for providing technical support. This work was supported by grants from the Ministry of Science and Technology of China (2015BAI08B02), the National Natural Science Foundation of China (31772550 and 31301217), the Key Project of Jiangsu Social Development (BE2016752), Natural Science Foundation in Jiangsu Province (BK20181260) and the Innovation Project of Military Medicine (16CXZ007).
Author contributions
DW, JZ, and ZL conceived this study, designed the experiments and wrote the paper. DW performed most of the experiments, assisted by JZ, Qianyue C, PS, Qin C, YZ, DZ, QL, and JP. JZ, QH, and CZ collected and analyzed the clinical data, supervised by XW and JR ZL and XG supervised all research.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Edited by J. SIlke
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Dingyu Wang, Jiashuo Zheng
Contributor Information
Xiang Gao, Phone: +8613815856111, Email: gaoxiang@nju.edu.cn.
Jianan Ren, Phone: +8613605169808, Email: jiananr@gmail.com.
Zhaoyu Lin, Phone: +8618114717776, Email: linzy@nju.edu.cn.
Supplementary information
The online version of this article (10.1038/s41418-019-0366-x) contains supplementary material, which is available to authorized users.
References
- 1.Velissaris D, Karamouzos V, Pierrakos C, Aretha D, Karanikolas M. Hypomagnesemia in critically ill sepsis patients. J Clin Med Res. 2015;7:911–8. doi: 10.14740/jocmr2351w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jiang P, Lv Q, Lai T, Xu F. Does hypomagnesemia impact on the outcome of patients admitted to the intensive care unit? A systematic review and meta-analysis. Shock. 2017;47:288–95. doi: 10.1097/SHK.0000000000000769. [DOI] [PubMed] [Google Scholar]
- 3.Limaye CS, Londhey VA, Nadkart MY, Borges NE. Hypomagnesemia in critically ill medical patients. J Assoc Physicians India. 2011;59:19–22. [PubMed] [Google Scholar]
- 4.Soliman HM, Mercan D, Lobo SS, Melot C, Vincent JL. Development of ionized hypomagnesemia is associated with higher mortality rates. Crit Care Med. 2003;31:1082–7. doi: 10.1097/01.CCM.0000060867.17556.A0. [DOI] [PubMed] [Google Scholar]
- 5.Thongprayoon C, Cheungpasitporn W, Erickson SB. Admission hypomagnesemia linked to septic shock in patients with systemic inflammatory response syndrome. Ren Fail. 2015;37:1518–21. doi: 10.3109/0886022X.2015.1074519. [DOI] [PubMed] [Google Scholar]
- 6.Weglicki WB. Hypomagnesemia and inflammation: clinical and basic aspects. Annu Rev Nutr. 2012;32:55–71. doi: 10.1146/annurev-nutr-071811-150656. [DOI] [PubMed] [Google Scholar]
- 7.de Baaij JH, Hoenderop JG, Bindels RJ. Magnesium in man: implications for health and disease. Physiol Rev. 2015;95:1–46. doi: 10.1152/physrev.00012.2014. [DOI] [PubMed] [Google Scholar]
- 8.Mazur A, Maier JA, Rock E, Gueux E, Nowacki W, Rayssiguier Y. Magnesium and the inflammatory response: potential physiopathological implications. Arch Biochem Biophys. 2007;458:48–56. doi: 10.1016/j.abb.2006.03.031. [DOI] [PubMed] [Google Scholar]
- 9.Schick V, Scheiber JA, Mooren FC, Turi S, Ceyhan GO, Schnekenburger J, et al. Effect of magnesium supplementation and depletion on the onset and course of acute experimental pancreatitis. Gut. 2014;63:1469–80. doi: 10.1136/gutjnl-2012-304274. [DOI] [PubMed] [Google Scholar]
- 10.Lee CY, Jan WC, Tsai PS, Huang CJ. Magnesium sulfate mitigates acute lung injury in endotoxemia rats. J Trauma. 2011;70:1177–85. doi: 10.1097/TA.0b013e31820ca695. [DOI] [PubMed] [Google Scholar]
- 11.Ahmed LA. Protective effects of magnesium supplementation on metabolic energy derangements in lipopolysaccharide-induced cardiotoxicity in mice. Eur J Pharm. 2012;694:75–81. doi: 10.1016/j.ejphar.2012.07.036. [DOI] [PubMed] [Google Scholar]
- 12.Eltanbouly DM, Abdelsalam RM, Attia AS, Abdelaziz MT. Pretreatment with magnesium ameliorates lipopolysaccharide-induced liver injury in mice. Pharmacol Rep Pr. 2015;67:914–20. doi: 10.1016/j.pharep.2015.02.004. [DOI] [PubMed] [Google Scholar]
- 13.Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, et al. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479:117–21. doi: 10.1038/nature10558. [DOI] [PubMed] [Google Scholar]
- 14.Kovacs SB, Miao EA. Gasdermins: effectors of pyroptosis. Trends Cell Biol. 2017;27:673–84. doi: 10.1016/j.tcb.2017.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526:666–71. doi: 10.1038/nature15541. [DOI] [PubMed] [Google Scholar]
- 16.Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA. Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science. 2013;341:1250–3. doi: 10.1126/science.1240988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science. 2013;341:1246–9. doi: 10.1126/science.1240248. [DOI] [PubMed] [Google Scholar]
- 18.Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. 2014;514:187–92. doi: 10.1038/nature13683. [DOI] [PubMed] [Google Scholar]
- 19.Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–5. doi: 10.1038/nature15514. [DOI] [PubMed] [Google Scholar]
- 20.Mulvihill E, Sborgi L, Mari SA, Pfreundschuh M, Hiller S, Muller DJ. Mechanism of membrane pore formation by human gasdermin-D. EMBO J. 2018;37:14. doi: 10.15252/embj.201798321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;535:111. doi: 10.1038/nature18590. [DOI] [PubMed] [Google Scholar]
- 22.Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535:153–8. doi: 10.1038/nature18629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aglietti RA, Estevez A, Gupta A, Ramirez MG, Liu PS, Kayagaki N, et al. GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc Natl Acad Sci USA. 2016;113:7858–63. doi: 10.1073/pnas.1607769113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schmid-Burgk JL, Gaidt MM, Schmidt T, Ebert TS, Bartok E, Hornung V. Caspase-4 mediates non-canonical activation of the NLRP3 inflammasome in human myeloid cells. Eur J Immunol. 2015;45:2911–7. doi: 10.1002/eji.201545523. [DOI] [PubMed] [Google Scholar]
- 25.Ruhl S, Broz P. Caspase-11 activates a canonical NLRP3 inflammasome by promoting K(+) efflux. Eur J Immunol. 2015;45:2927–36. doi: 10.1002/eji.201545772. [DOI] [PubMed] [Google Scholar]
- 26.Wu Y, Ren J, Zhou B, Ding C, Chen J, Wang G, et al. Gene silencing of non-obese diabetic receptor family (NLRP3) protects against the sepsis-induced hyper-bile acidaemia in a rat model. Clin Exp Immunol. 2015;179:277–93. doi: 10.1111/cei.12457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee S, Nakahira K, Dalli J, Siempos II, Norris PC, Colas RA, et al. NLRP3 inflammasome deficiency protects against microbial sepsis via increased lipoxin B4 synthesis. Am J Respir Crit Care Med. 2017;196:713–26. doi: 10.1164/rccm.201604-0892OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.He WT, Wan H, Hu L, Chen P, Xin W, Zhe H, et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015;25:1285–98. doi: 10.1038/cr.2015.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ross PE, Ehring GR, Cahalan MD. Dynamics of ATP-induced calcium signaling in single mouse thymocytes. J Cell Biol. 1997;138:987–98. doi: 10.1083/jcb.138.5.987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Virginio C, Church D, North RA, Surprenant A. Effects of divalent cations, protons and calmidazolium at the rat P2X7 receptor. Neuropharmacology. 1997;36:1285–94. doi: 10.1016/s0028-3908(97)00141-x. [DOI] [PubMed] [Google Scholar]
- 31.Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228–32. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- 32.de Vasconcelos Nathalia M., Van Opdenbosch Nina, Van Gorp Hanne, Parthoens Eef, Lamkanfi Mohamed. Single-cell analysis of pyroptosis dynamics reveals conserved GSDMD-mediated subcellular events that precede plasma membrane rupture. Cell Death & Differentiation. 2018;26(1):146–161. doi: 10.1038/s41418-018-0106-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang D, He Y, Munoz-Planillo R, Liu Q, Nunez G. Caspase-11 requires the Pannexin-1 channel and the purinergic P2X7 pore to mediate pyroptosis and endotoxic shock. Immunity. 2015;43:923–32. doi: 10.1016/j.immuni.2015.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bruzzone S, Basile G, Chothi MP, Nobbio L, Usai C, Jacchetti E, et al. Diadenosine homodinucleotide products of ADP-ribosyl cyclases behave as modulators of the purinergic receptor P2X7. J Biol Chem. 2010;285:21165–74. doi: 10.1074/jbc.M109.097964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hansen DB, Ye ZC, Calloe K, Braunstein TH, Hofgaard JP, Ransom BR, et al. Activation, permeability, and inhibition of astrocytic and neuronal large pore (hemi)channels. J Biol Chem. 2014;289:26058–73. doi: 10.1074/jbc.M114.582155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cheng KT, Xiong S, Ye Z, Hong Z, Di A, Tsang KM, et al. Caspase-11-mediated endothelial pyroptosis underlies endotoxemia-induced lung injury. J Clin Invest. 2017;127:4124–35. doi: 10.1172/JCI94495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Russo HM, Rathkey J, Boydtressler A, Katsnelson MA, Abbott DW, Dubyak GR. Active caspase-1 induces plasma membrane pores that precede pyroptotic lysis and are blocked by lanthanides. J Immunol. 2016;197:1353. doi: 10.4049/jimmunol.1600699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Solle M, Labasi J, Perregaux DG, Stam E, Petrushova N, Koller BH, et al. Altered cytokine production in mice lacking P2X(7) receptors. J Biol Chem. 2001;276:125–32. doi: 10.1074/jbc.M006781200. [DOI] [PubMed] [Google Scholar]
- 39.Morelli A, Chiozzi P, Chiesa A, Ferrari D, Sanz JM, Falzoni S, et al. Extracellular ATP causes ROCK I-dependent bleb formation in P2X7-transfected HEK293 cells. Mol Biol Cell. 2003;14:2655–64. doi: 10.1091/mbc.02-04-0061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ousingsawat J, Wanitchakool P, Schreiber R, Kunzelmann K. Contribution of TMEM16F to pyroptotic cell death. Cell Death Dis. 2018;9:300. doi: 10.1038/s41419-018-0373-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Platnich JM, Chung H, Lau A, Sandall CF, Bondzi-Simpson A, Chen HM, et al. Shiga toxin/lipopolysaccharide activates caspase-4 and gasdermin D to trigger mitochondrial reactive oxygen species upstream of the NLRP3 inflammasome. Cell Rep. 2018;25:1525–36 e7. doi: 10.1016/j.celrep.2018.09.071. [DOI] [PubMed] [Google Scholar]
- 42.American College of Chest Physicians/Society of Critical Care Medicine Consensus Conference. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Crit Care Med. 1992;20:864–74. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.