

Abstract
Autophagy regulates the degradation of unnecessary or dysfunctional cellular components. This catabolic process requires the formation of a double-membrane vesicle, the autophagosome, that engulfs the cytosolic material and delivers it to the lysosome. Substrate specificity is achieved by autophagy receptors, which are characterized by the presence of at least one LC3-interaction region (LIR) or GABARAP-interaction motif (GIM). Only recently, several receptors that mediate the specific degradation of endoplasmic reticulum (ER) components via autophagy have been identified (the process known as ER-phagy or reticulophagy). Here, we give an update on the current knowledge about the role of ER-phagy receptors in health and disease.
Subject terms: Autophagy, Diseases
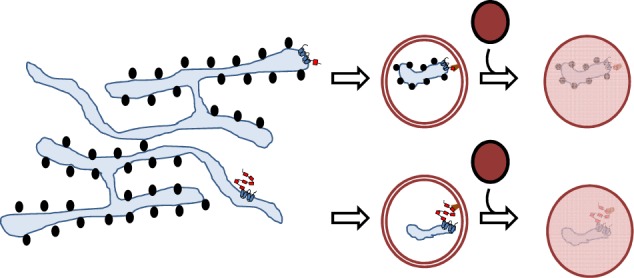
Different ER subdomains such as ER sheets and tubules can be degraded by ER-phagy via specific ER-phagy receptors.
Facts
ER-phagy plays a role in ER homeostasis
ER-phagy is involved in the recovery from ER stress
ER-phagy is subdomain specific
Multiple ER-phagy receptors exist in mammalian cells (six have been identified to date)
ER-phagy receptors can serve other functions than ER-phagy
Defects of FAM134B and ATL3 cause monogenic neurodegenerative disorders
FAM134B and SEC62 are associated with cancer
Open questions
How is ER-phagy regulated?
Does the regulation involve a posttranslational modification of ER-phagy receptors?
How does ER-phagy relate to ER functions?
What is the basis of ER-phagy defects in the pathogenesis of human diseases?
Is ER-phagy a target for therapy?
Is ER-phagy involved in responses to drug treatment via ER stress or disturbance of cellular homeostasis?
Structure and functions of the endoplasmic reticulum
A large amount of ER membranes is organized in tubular and lamellar structures, generating a complex and highly dynamic architecture that varies in response to functional requirements [1]. The ER is a continuum of membrane structures, from the nuclear envelope to a network of tubules and sheets, extended regions of parallel, flat membrane bilayers, spread throughout the cytoplasm (Fig. 1). The cisternal/sheet ER is densely packed with ribosomes (rough ER) and is the primary location of protein synthesis, protein translocation and protein modification, including N-glycosylation. Proteins such as p180, kinectin, and CLIMP-63 are thought to play a role in stacking and luminal spacing of ER sheets [2]. Improved spatial resolution uncovered that some of these sheets may rather be dense matrices of highly convoluted tubules [3]. Members of the Reticulon (RTN), FAM134 and REEP families, all of which contain hydrophobic hairpin domains, also known as Reticulon domains, are thought to promote curvature of ER sheets and tubules via scaffolding and hydrophobic wedging [4–7]. Tubular three-way junctions require members of the Atlastin (ATL) family of dynamin-related guanosine triphosphatases (GTPases) [2]. The functions of ER tubules are less well understood, but they may be the primary regions where lipid synthesis and signaling between the ER and other organelles occur. The ER also contains other specialized subcompartments, such as ER exit sites (ERES), where COPII-coated anterograde transport vesicles are generated. Lipids and native proteins are exported from the ER. In contrast, terminally misfolded proteins are retained in the ER and may trigger unfolded protein responses (UPR) [8]. Misfolded proteins may eventually be displaced across the ER membrane for ER-associated degradation (ERAD), or may be segregated in ER subdomains displaying ER-phagy receptors, which ensure lysosomal delivery by autophagic and nonautophagic pathways, collectively defined as ER-to-lysosome-associated degradation (ERLAD) [9–11].
Fig. 1.
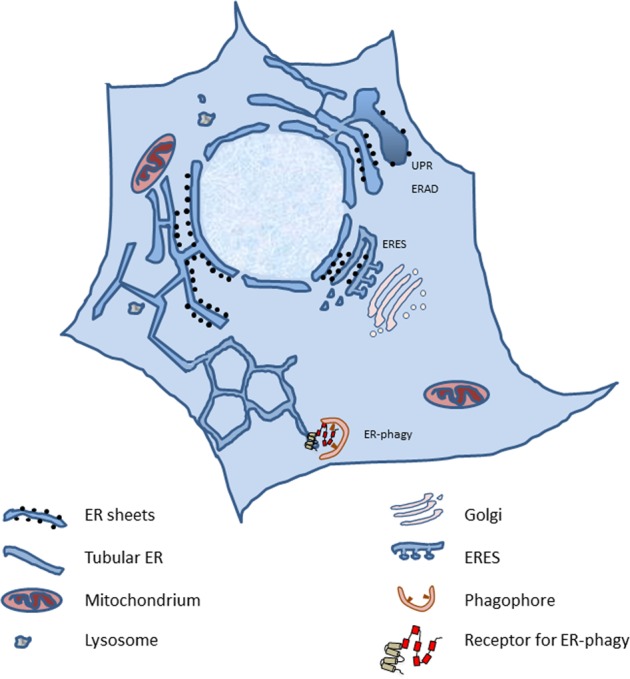
Gross architecture of an eukaryotic cell and its ER. ERES endoplasmic reticulum exit sites, UPR unfolded protein response, ERAD endoplasmic reticulum associated protein degradation
The ER is involved in the formation of junctional regions with essentially every other organelle, including the plasma membrane [12]. The cortical ER refers to the regions of the peripheral ER that are closely apposed and tethered to the plasma membrane. Contact sites between the ER and the plasma membrane allow the exchange of small molecules, such as lipids and signals [13]. Similarly, the contact sites between the ER and mitochondria form specific domains, termed mitochondria-ER associated membranes (MAMs), with their role ranging from the coordination of calcium transfer to the regulation of mitochondrial fission and inflammasome formation [12].
ER-phagy contributes to ER homeostasis
Dynamic remodeling of the ER is critical for cellular homeostasis and prevention of disease pathogenesis. Almost four decades ago, autophagy was found to be involved in controlling the dynamic changes of the ER during the recovery from ER stress [14]. Central to the process of autophagy is the formation of a double-membrane phagophore, also called the isolation membrane, which upon its elongation and closure forms an autophagosome that engulfs cellular material and delivers it to the lysosome (Fig. 2). This process can either be nonselective or targeted to selective cellular components [15, 16]. The latter relies on binding of cargos to specific autophagy receptors, which are in turn recognized by autophagy modifier proteins attached to autophagosomal membranes. Their genetic inactivation abolishes the turnover of the target proteins or organelles, but does not affect other forms of selective or nonselective autophagy. Originally, the turnover of ER components via autophagy was discovered as a back-up system for inefficient proteasomal degradation of ER proteins through the ERAD pathway [17]. Nevertheless, the insights into the mechanisms underlying ER-phagy have only been obtained recently by identifying receptors required for targeting ER fragments to the lysosome via a classical autophagy pathway [18]. Thus far, six mammalian ER-resident proteins have been identified to function as receptors for selective ER-phagy: FAM134B [19], SEC62 [20], RTN3L [21], CCPG1 [22], ATL3 [23], and TEX264 [24, 25] (Fig. 3). All of them contain at least one LIR or a GABARAP-interaction motif (GIM), a sequential peptide motif within the cytoplasmic region, which enables the binding to LC3/GABARAP proteins associated with phagophore membranes. The canonical consensus motif for LIRs and GIMs is characterized by [W/F/Y]xx[L/I/V], commonly adjacent to at least one acidic residue [26]. CCPG1 also contains a FIP200-interacting region [22]. ER-phagy receptors are selective for the ER, as well as for specific ER subdomains. Analogous systems have also been reported in yeast [27]. FAM134B was originally described as an ER-phagy receptor specific for ER sheets, while RTN3 and ATL3 preferentially target ER tubules for degradation [21, 23]. The most recently identified receptor, TEX264, is localized on ER three-way junctions and is involved in the regulation of a large proportion of the ER-phagy flux in stressed cells [24, 25]. The expression of ER-phagy receptors can differ between different cell types, and may thus allow cell- and tissue-specific responses to biological and stress stimuli. For example, SEC62, which is ubiquitously expressed, is involved in “recov-ER-phagy”, which reduces the ER size to a normal level after ER stress is resolved [20], whereas CCPG1 seems to be specific for gastric chief cells and pancreatic cells and is activated under secretory ER stress conditions [22]. ER-phagy receptors may also integrate other proteins or chaperones to select specific cargoes, as it was recently proposed for the role of FAM134B in a complex with Calnexin, resulting in removal of proteasome-resistant polymers of alpha1-antitrypsin Z (ATZ) [28] or misfolded procollagen [29]. The ER-phagy receptors may also be linked to components of the COPII complex to promote ER budding and fragmentation [30]. Moreover, ER-phagy pathways may be targeted by pathogens, such as viruses and bacteria [31, 32].
Fig. 2.

The process of ER-phagy. ER-phagy receptors are recognized by LC3/GABARAP proteins, which allows the formation of the phagophore around the ER fragments. As the phagophore seals, it forms an autophagosome, which delivers the enclosed ER fragments to the lysosome for degradation
Fig. 3.
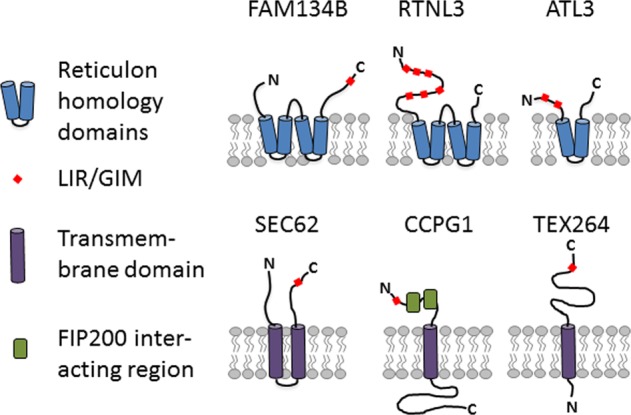
Structure and topology of known receptors for ER-phagy
FAM134B
FAM134B (also known as RETREG1) belongs to a family of three ER-resident proteins with sequence similarity (family with sequence similarity 134; FAM134A, -B, and -C) that share the hairpin domains, as described for Reticulons, and a cytoplasmic C-terminal LIR motif, which mediates the binding to LC3/GABARAP proteins (Fig. 3) [19]. FAM134B is the best studied family member. Depletion of FAM134B sensitizes cells to undergo apoptosis if challenged by starvation, ER stressors (such as thapsigargin and tunicamycin), staurosporine or carbonyl cyanide m-chlorophenyl hydrazone, an inhibitor of oxidative phosphorylation [19]. Since the overexpression of FAM134B increased the number of LC3-positive vesicles, also positive for some ER components and decreased the overall size of the ER, while its absence led to the expansion of the ER [19], FAM134B was characterized as a receptor for ER-phagy. Notably, the depletion of ATL2 repressed ER-phagy induced by the overexpression of FAM134B [33]. Therefore, it was proposed that ATL2 can remodel ER membranes to separate FAM134B-positive ER domains for efficient autophagosomal engulfment. Experimental data and modeling approaches further suggest that the clustering of FAM134B in autophagic puncta amplifies membrane deformation and thus supports membrane remodeling [34]. Consistent with its predominant localization at ER sheets, FAM134B is mainly involved in the remodeling and the degradation of ER sheets [21].
Recently, an involvement of FAM134B in lysosomal clearance of ER subdomains containing ERAD-resistant misfolded proteins has been shown for ATZ polymers [28] and for endogenous procollagen [29]. FAM134B-regulated clearance of ER-subdomains containing ATZ polymers involves LC3 lipidation, but not on the autophagosome biogenesis machinery [28]. For procollagen, both the LC3 lipidation and the autophagosome biogenesis machinery participate in the catabolic process [29]. In both cases, the transmembrane ER chaperone Calnexin controls the segregation of ERAD-resistant misfolded proteins in the ER subdomains displaying FAM134B. A similar mechanism may also apply for the Niemann-Pick type C disease protein, NPC1 [35]. Mutations in this multipass transmembrane glycoprotein, which is essential for intracellular lipid trafficking, result in a fatal, progressive neurodegenerative disorder. The most common disease-causing mutant, I1061T NPC1, is exported for degradation by either MARCH6-dependent ERAD or via FAM134B-directed ER-phagy. Analysis of samples obtained from Niemann-Pick patients showed alterations in key components of ER-phagy, including FAM134B [35]. Notably, a small quantity of wild-type (WT) NPC1 is also degraded through FAM134B, suggesting that a fraction of WT NPC1 can misfold similarly to the I1061T mutant [35]. However, it remains only partially understood how MARCH6 or FAM134B activities are coordinated in selecting substrates for degradation.
Several mutations in FAM134B have been reported to result in autosomal recessive hereditary sensory and autonomic neuropathy (HSAN2) [36]. The mutation spectrum includes nonsense, frameshift, and splice-site mutations [36–39], consistent with loss-of-function as the main pathophysiological event. This is further supported by knockout mice, which develop an age-dependent sensory deficit and a progressive loss of peripheral nerve fibers [19]. At the ultrastructural level, ER tubules were enlarged and the Golgi cisternae were distorted in dorsal root ganglia tissue sections from knockout mice. In agreement, knockdown of FAM134B in cultured dorsal root ganglia neurons resulted in apoptosis [36]. However, the complete series of events, which finally result in axon degeneration, remain elusive.
FAM134B has also been linked with cancer, since FAM134B (back then referred to as JK-1) is commonly found to be overexpressed in esophageal squamous carcinoma [40]. NIH 3T3 cells overexpressing FAM134B displayed accelerated cell growth and formed sarcomas when injected into athymic nude mice [40]. Adding support to this notion, FAM134B mutations were detected in lymph node metastases in patients suffering from esophageal squamous carcinoma [41], suggesting that a mutation in FAM134B may promote the progression. In addition, a significant correlation was found between FAM134B gene copy number alterations and clinical and pathological features in pre-invasive and invasive colorectal malignancies [42]. Overall, mutations in FAM134B were associated with biological aggressiveness in both esophageal and colorectal cancers [43].
Other reported disease associations involving FAM134B are allergic rhinitis [44], vascular diseases [45] and viral infections. Since the loss of FAM134B resulted in a much higher production of infectious Ebola virus and accumulation of nucleocapsid lattices in mouse embryonic fibroblasts [31], it was proposed that FAM134B-dependent ER-phagy limits Ebola virus replication in mouse cells and thus may be a target for the development of novel antiviral therapeutics. FAM134B was also shown to restrict both Dengue and ZIKA virus replication [46]. It is known that Flavi viruses utilize the virally-encoded NS3 proteases to cleave FAM134B at a single site within its reticulon homology domain and thus impair the formation of viral protein-enriched ER vesicles, suggesting that the cleavage of FAM134B serves to specifically suppress ER-phagy [46].
RTN3L
To date, four different Reticulons (RTN1–4) have been identified. Since there is a large number of different splicing isoforms for each RTN, the family structure of Reticulons is rather complex. All Reticulons share a reticulon domain, but only RTN3 has an extended N-terminal variant (RTN3L), which harbors six different functional LIR motifs (Fig. 3). Notably, the overexpression of this variant resulted in the fragmentation of ER tubules under starvation and drove the delivery of ER-derived tubular fragments to lysosomes [21]. Similar to FAM134B, the absence of RTN3L did not affect macroautophagy. It did not alter the ER sheets versus tubules ratio, while the ER turnover by starvation was reduced, indicating that both mechanisms contribute to ER homeostasis in an independent manner. In mice, the complete loss of RTN3 did not entail any obvious phenotypes [47] and no monogenic disorder has been linked with RTN3 so far. RTN3 may be relevant for Alzheimer’s disease, since it has been found to interact with β-site amyloid precursor protein cleaving enzyme 1 (BACE1). BACE1 initiates the generation of β-amyloid peptides from amyloid precursor protein and is the major protein in Alzheimer plaques. Mutant prohormones, such as the Akita proinsulin mutant, which causes the autosomal-dominant diabetic syndrome called mutant INS-gene-induced diabetes of the youth (MIDY), forms aggregates that are degraded by RTN3-dependent ER-phagy [48]. Interestingly, this function of RTN3 was independent of its LIR motifs, suggesting that ER-phagy may be mediated by another protein interacting with RTN3 [48].
ATL3
Members of the Atlastin family of dynamin-related GTPases play a critical role in the process to generate the three-way junctions between ER tubules [2, 49]. They are large, multimeric, integral membrane GTPases that localize predominantly to highly-curved ER membranes, including the tubular ER and the edges of ER sheets, which is a consequence of the hydrophobic segments predicted to form partially membrane-spanning hairpin structures (Fig. 3). ATLs can dimerize in cis and trans, and are thus believed to tether adjacent membranes and bring them closely together to allow fusion. In mammals, there are three closely related ATLs, each of which contains an N-terminal GTP-binding domain, a middle assembly domain, two very closely spaced hydrophobic segments near the C-terminus, and a C-terminal tail. Notably, ATL3 harbors two GIMs, thereby targeting ER tubules for lysosomal degradation [23, 50]. Overexpression of RTN3L could rescue defective ER-phagy in ATL3 KO COS-7 cells [23]. Importantly, mutations in ATL3 cause autosomal-dominant hereditary sensory neuropathy (HSN1F), which closely resembles the FAM134B-associated disorder [51–53]. In HeLa cells, the disease-associated variant ATL3 Y192C reduced the complexity of the tubular ER network, delayed ER export, impaired autophagy, promoted Golgi fragmentation and caused malformation of the nucleus [54]. In neurons, the expression of ATL3 Y192C protein variant was excluded from axons [54]. Moreover, it has been reported that the presence of HSAN-causing ATL3 mutations leads to increased formation of MAMs, with effects on mitochondrial function and motility [52]. The ATL3 Y192C mutation lies within the first GIM motif. ATL3 Y192C, as well as ATL3 P338R, which is another disease variant, both exhibit reduced binding to GABARAP, suggesting that impaired ER-phagy is involved in the pathogenesis of the disorder. To study the role of ATLs in membrane trafficking, ATL-depleted COS-7 cells have been used, which generally express reasonable levels of ATL2 and ATL3 [55]. Importantly, ATL depletion affected the formation of COPII-coated vesicles and cargo exit from the ER [55], suggesting that the tubular ER plays a key role in membrane trafficking.
TEX264
TEX264 was identified as an ER-phagy receptor in a differential interactome analysis for LC3 WT and a LIR-recognition defective LC3 mutant [25], as well as global analysis of cells after amino acid withdrawal or mTOR inhibition [24]. Under nutrient-rich conditions, TEX264 localizes to punctate structures on the ER, some of which represent three-way junctions. TEX264 also localizes to larger structures, which co-localize with autophagosomal proteins. Importantly, the C-terminal cytoplasmic part of TEX264 contains a long intrinsically disordered region near the LIR motif, which is flexible and is required for its activity as an ER-phagy receptor (Fig. 3). The interaction of ATG8 proteins with TEX264 may be analogous to a “zipper,” allowing the ER membrane to come into a close contact with the inner phagophore membrane through a trans interaction [24]. The ER-phagy receptors CCPG1 and TEX264 each have a single transmembrane domain and both of them require a LIR motif-driven interaction with ATG8 proteins for lysosomal degradation. Reticulon-type receptors (FAM134B and RTN3L) are able to induce ER-phagy by a simple overexpression in mammalian cells, whereas single membrane-passing receptors (CCPG1 and TEX264) are unable to promote ER fragmentation by overexpression in host cells. They seem to require another signal or an associated partner to promote the budding and fragmentation of the ER. The different types/topologies of the ER-phagy receptors suggest a possible cooperative/redundancy function, and possibly the formation of a coat complex, in order to generate ER fragments, which are consequently enclosed into an autophagosome and then degraded in the lysosome. The comparison of the relative contributions of FAM134B, CCPG1, RTN3L, and SEC62 to that of TEX264, by knockdown of each ER-phagy receptor, showed that the depletion of TEX264 most efficiently suppressed ER-phagy activity in HeLa cells under nutrient-rich and starvation conditions [25]. Proteomic studies [24], however, suggest that TEX264 likely targets specific ER contents for ER-phagy. To date, no pathologies associated with TEX264 have been identified.
SEC62
SEC62 is involved in the posttranslational insertion of polypeptides as a part of the translocon complex [56], which allows the nascent precursor polypeptides to enter the ER via the heterotrimeric polypeptide conducting SEC61 channel. As already mentioned, misfolded proteins, which accumulate within the ER, activate the UPR. This results in an expansion of the ER and an increase of chaperones and folding enzymes to restore ER homeostasis and adapt to stress conditions. If this is not possible, cells undergo apoptosis. The recovery from ER stress relies on ER-phagy via SEC62 [20], which contains a LIR motif in its cytosolic C-terminal part (Fig. 3). The resulting ER fragments contain ER-resident molecular chaperones and enzymes, but are devoid of other ER components, such as ERAD proteins, arguing in favor of a separation of ER functions within ER subdomains [9]. Notably, SEC62 does not participate in FAM134B-driven ATZ [28] and procollagen [29] proteostasis.
Mutations, amplifications and an overexpression of the SEC genes were linked to various disorders, such as kidney and liver diseases, diabetes, and cancer [57]. Increased SEC62 expression levels were reported in prostate cancer, lung cancer, head and neck squamous cell carcinoma, cervical lesions as well as hepatocellular carcinoma [58–62]. The expression of the obligatory interaction partners of SEC62 in protein translocation, such as SEC61 and SEC63, is not altered in tumors with elevated SEC62 expression [58, 61]. It has been suggested that enhanced ER-phagy via SEC62 may render the tumor cells more resistant to ER stress [63]. Consequently, it has been proposed that drugs blocking autophagy might be beneficial components in the therapy of selected tumor entities, characterized by the increased expression of SEC62.
CCPG1
CCPG1 is a vertebrate-specific protein, which is induced upon ER stress, leading to the activation of autophagic degradation of the peripheral ER [22]. CCPG1 consists of a cytosolic N-terminal region, a transmembrane domain that anchors it within the ER membrane, and an ER luminal C-terminal region [64]. In an unbiased affinity-purification mass spectrometry screen for the GABARAP interactome, CCPG1 was found to interact with GABARAP via its N-terminal LIR motif [22] (Fig. 3). The loss of CCPG1 blocked ER-phagy in response to starvation and DTT-induced ER stress. To mediate ER-phagy, CCPG1 requires two FIP200-binding regions, which bind to the C-terminal region of FIP200. Consistent with a role in ER-phagy, the ER is expanded in pancreatic acinar cells and in gastric chief cells of CCPG1 knockout mice [22]. In vivo, CCPG1 protects against ER luminal protein aggregation and consequent UPR hyper-activation, as well as tissue injury of the exocrine pancreas. To date, there are no reports that CCPG1 is linked to a human disease, but it has been proposed that the manipulation of CCPG1 functions might be beneficial to ameliorate ER stress in pancreatic inflammatory states or, conversely, to enhance ER stress and thus eliminate malignant cells in pancreatic cancers [22].
Insights into ER-phagy-related human diseases
ER-phagy receptors are associated with various human disorders (Table 1). Importantly, mutations in either FAM134B or ATL3 can lead to HSAN [36, 51], which is characterized by the predominant degeneration of afferent fibers and results in a sensory loss ranging from local numbness to the complete inability to feel pain [65]. Patients are typically susceptible to involuntary injuries and painless fractures. Notably, the FAM134B-associated disease is inherited in an autosomal recessive manner, while the ATL3-related disorder follows an autosomal-dominant trait. The latter may be explained by the fact that ATL can dimerize in cis and trans, thus potentially allowing ATL3 variants to act in a dominant-negative manner. Consistent with this notion, the clinical presentation for ATL3 patients appears to be less severe and usually excludes upper extremities, while this is common in FAM134B patients (Fig. 4). FAM134B patients can also suffer from autonomic alterations such as cardiac arrhythmia, an- or hypohydrosis and other symptoms of autonomic malfunction. In some patients, leg spasticity and weakness have also been reported [66], suggesting that axons of upper motoneurons can be affected as well. This latter manifestation is also typical for hereditary spastic paraplegia (HSP) [67]. In men, both sensory and motor circuits have to bridge distances of more than a meter, posing a permanent challenge for signaling, transport and supply to such exceptional cellular extents, thus appearing to be particularly sensitive to altered ER homeostasis [12]. Mutations in ATL1, a paralogue of ATL3, also manifest as an axonal disorder, either as autosomal-dominant spastic paraplegia (SPG3) [68] or as HSN1 [69]. Mutations in RTN2, a paralogue of RTN3L, have also been linked with HSP (SPG12) [70]. RTN3 is further associated with Alzheimer’s disease [71], since RTN3 deficiency facilitated amyloid deposition in Alzheimer’s mouse models [47]. Years ago, RTN3 expression was reported in dystrophic neurites, neuropil threads, granulovacuolar degeneration, glial cells, morphologically normal neurons in both hippocampal pyramidal cell layer and cerebral neocortex, and specifically in neurofibrillary tangles and Lewy bodies [72], suggesting that RTN3 expression may be a downstream stress response in Alzheimer’s disease. Further supporting this, screening of patients with sporadic early- and late-onset Alzheimer’s disease, as well as age-matched controls, resulted in the identification of several variants in the 5′ non-coding region and N-terminal domain of the RTN3 gene only in Alzheimer’s disease patients [71] and may hence represent genetic modifiers. To which extent this is related to the role of RTN3 in ER-phagy remains to be determined.
Table 1.
Known diseases linked to receptors for ER-phagy
| Known monogenic disorders | Related diseases | Known monogenic disorders linked with paralogues | |
|---|---|---|---|
| FAM134B | HSAN2B [36–39] autosomal recessive OMIM #613115 |
• Esophageal squamous carcinoma [40, 41] • Allergic rhinitis [44] • Vascular disease [45] |
|
| SEC62 | Cancer [58–62] | ||
| RTN3L | Alzheimer’s disease [71] | RTN2: hereditary spastic paraplegia (SPG12) [70] autosomal dominant | |
| ATL3 | HSN1F [51–53] autosomal-dominant OMIM #609369 | ATL1: hereditary spastic paraplegia (SPG3) [68] autosomal-dominant OMIM #182600 HMSN1D [69] autosomal-dominant OMIM #607678 |
HSAN2B: hereditary sensory and autonomic neuropathy 2B; HSN1F: hereditary sensory neuropathy 1F; HMSN1D: hereditary motor and sensory neuropathy 1D (Charcot-Marie-Tooth disease, type 1D); SPG: spastic paraplegia
Fig. 4.

Clinical presentation of a patient with the homozygous mutation p.S309X in FAM134B showing severe acro-osteolysis of fingers and toes and deformation of the skeleton of the foot because of recurrent painless bone fractures (HSAN2B)
Cancer is another recurrent link to ER-phagy, as already discussed for both FAM134B and SEC62. Since SEC62 amplifications have been reported in non-small cell lung cancer, prostate cancer, and thyroid cancers, as well as head and squamous cell carcinoma [61–63], it is tempting to assume that SEC62 amplification may render tumor cells more tolerant to ER stress, thus conferring a high metastatic and invasive potential. Similar mechanisms may also apply for CCPG1 [22]. In contrast, FAM134B may rather act as a tumor suppressor, since FAM134B mutations have been frequently found in colorectal adenocarcinoma, and esophageal squamous cell carcinoma [41, 43, 73]. In the particular case of glioma cells with isocitrate dehydrogenase 1 mutations [74], however, FAM134B was found to be upregulated and its knockdown sensitized the cells to undergo apoptosis. Taken together, ER-phagy receptors can play diverse roles in the pathogenesis of different cancer entities. Yet, further insights are needed to evaluate whether targeting ER-phagy may be considered relevant as an anticancer therapeutic strategy.
Open questions and concluding remarks
Considering that there are six different ER-phagy receptors identified thus far (and likely more to be identified in the future), the analysis of their physiological and pathophysiological roles will be a major challenge. They may either serve specific tasks or may act in concert during ER-phagy in more global responses. For example, the ER-phagy receptors RTN3L and ATL3 can trigger the degradation of ER tubules, while the degradation of ER sheets is promoted by FAM134B. Likely, the different receptors may also respond to different cellular cues. Indeed, it has been shown that SEC62 and CCPG1 are mainly involved in the recovery from ER stress to re-establish the pre-stress ER size, while the other receptors are related to starvation-induced ER-phagy. On the other hand, TEX264 and FAM134B have been found in the same ER vesicles and could thus act together with FAM134B inducing ER curvature, remodeling and finally fragmentation under starvation [24].
To resolve the molecular pathways responsible for activation of ER-phagy is another challenging topic. At present, for example, very little is known about the regulation of the expression levels of individual ER-phagy receptors. An increased transcription of the respective ER-phagy receptor gene may be the first trigger to initiate ER-phagy. The posttranslational modification of ER-phagy receptors during induction of ER-phagy is another attractive and likely hypothesis, but requires further studies. Such modifications may potentially modify the affinity to LC3 [75] or result in structural alterations that allow the oligomerization of the receptors, thus supporting membrane remodeling during ER-phagy. In addition, modifications may also allow interactions with other components of the ER-phagy machinery. To this end, the upregulation of the yeast ER-phagy receptor Atg40 induced its association with Lst1-Sec23 to package ER into autophagosomes, thus preventing the accumulation of an aggregation-prone protein within the ER [30]. Lst1 function appears to be conserved, since its mammalian homolog, SEC24C, is required for delivering ER sheets and tubules to lysosomes. Notably, the interactome of RTN3L includes SEC24C [21].
The importance of ER-phagy in the pathogenesis of different human disorders is currently underestimated. In addition to genetic mutations involving the ER-phagy receptors themselves, ER-phagy is also involved in the removal of disease-associated protein variants as demonstrated for Niemann-Pick type C disease [35] and may thus be relevant in the context of numerous monogenic disorders. Moreover, ER-phagy is also likely to play a role in immune competent cells and thus may have an impact on immune responses that are directly or indirectly implicated in the development of human diseases. Taken together, the increasing interest and research on ER-phagy will certainly provide a better understanding of ER-phagy in disease pathogenesis. This may ultimately promote the identification of novel therapeutic approaches targeting ER-phagy.
Acknowledgements
We are grateful to Paolo Grumati, Andrea Gubas, Ingo Kurth, and Antje-Kathrin Huebner for critical comments and insightful discussions. This work was partially supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)—Project number 259130777—SFB 1177 and DFG-funded Research Unit FOR 2625 on Mechanisms of Lysosomal Homeostasis, the European Research Council (ERC) grant 742720 UbBAC, Cardio-Pulmonary Institute (CPI), EXC 2026, Project ID: 390649896, and the LOEWE program on Ubiquitin Networks (Ub-Net), State of Hesse, Germany to ID and by German Research Foundation HU 800/6–2, HU 800/13–1, the DFG-funded Research Unit FOR 2625 on Mechanisms of Lysosomal Homeostasis and the SP6 within the DFG-funded research training group 1715 to CAH. CAH and ID envisioned and wrote the manuscript.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Edited by F. Pentimalli
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Christian A. Hübner, Email: Christian.Huebner@med.uni-jena.de
Ivan Dikic, Email: dikic@biochem2.uni-frankfurt.de.
References
- 1.Baumann O, Walz B. Endoplasmic reticulum of animal cells and its organization into structural and functional domains. Int Rev Cytol. 2001;205:149–214. doi: 10.1016/s0074-7696(01)05004-5. [DOI] [PubMed] [Google Scholar]
- 2.Goyal U, Blackstone C. Untangling the web: mechanisms underlying ER network formation. Biochim Biophys Acta. 2013;1833:2492–8. doi: 10.1016/j.bbamcr.2013.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nixon-Abell J, Obara CJ, Weigel AV, Li D, Legant WR, Xu CS, et al. Increased spatiotemporal resolution reveals highly dynamic dense tubular matrices in the peripheral ER. Science. 2016;354:aaf3928. doi: 10.1126/science.aaf3928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beetz C, Koch N, Khundadze M, Zimmer G, Nietzsche S, Hertel N, et al. A spastic paraplegia mouse model reveals REEP1-dependent ER shaping. J Clin Investig. 2013;123:4273–82. doi: 10.1172/JCI65665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hubner CA, Kurth I. Membrane-shaping disorders: a common pathway in axon degeneration. Brain. 2014;137:3109–21. doi: 10.1093/brain/awu287. [DOI] [PubMed] [Google Scholar]
- 6.Shibata Y, Shemesh T, Prinz WA, Palazzo AF, Kozlov MM, Rapoport TA. Mechanisms determining the morphology of the peripheral ER. Cell. 2010;143:774–88. doi: 10.1016/j.cell.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Voeltz GK, Prinz WA, Shibata Y, Rist JM, Rapoport TA. A class of membrane proteins shaping the tubular endoplasmic reticulum. Cell. 2006;124:573–86. doi: 10.1016/j.cell.2005.11.047. [DOI] [PubMed] [Google Scholar]
- 8.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–6. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 9.Fregno I, Molinari M. Endoplasmic reticulum turnover: ER-phagy and other flavors in selective and non-selective ER clearance. F1000 Res. 2018;7:454. doi: 10.12688/f1000research.13968.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fregno I, Molinari M. Proteasomal and lysosomal clearance of faulty secretory proteins: ER-associated degradation (ERAD) and ER-to-lysosome-associated degradation (ERLAD) pathways. Crit Rev Biochem Mol Biol. 2019;54:153–63. doi: 10.1080/10409238.2019.1610351. [DOI] [PubMed] [Google Scholar]
- 11.Sun Zhihao, Brodsky Jeffrey L. Protein quality control in the secretory pathway. The Journal of Cell Biology. 2019;218(10):3171–3187. doi: 10.1083/jcb.201906047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Helle SC, Kanfer G, Kolar K, Lang A, Michel AH, Kornmann B. Organization and function of membrane contact sites. Biochim Biophys Acta. 2013;1833:2526–41. doi: 10.1016/j.bbamcr.2013.01.028. [DOI] [PubMed] [Google Scholar]
- 13.Henne WM, Liou J, Emr SD. Molecular mechanisms of inter-organelle ER-PM contact sites. Curr Opin Cell Biol. 2015;35:123–30. doi: 10.1016/j.ceb.2015.05.001. [DOI] [PubMed] [Google Scholar]
- 14.Bolender RP, Weibel ER. A morphometric study of the removal of phenobarbital-induced membranes from hepatocytes after cessation of threatment. J Cell Biol. 1973;56:746–61. doi: 10.1083/jcb.56.3.746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rogov VV, Stolz A, Ravichandran AC, Rios-Szwed DO, Suzuki H, Kniss A, et al. Structural and functional analysis of the GABARAP interaction motif (GIM) EMBO Rep. 2018;19:e47268. doi: 10.15252/embr.201847268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stolz A, Ernst A, Dikic I. Cargo recognition and trafficking in selective autophagy. Nat Cell Biol. 2014;16:495–501. doi: 10.1038/ncb2979. [DOI] [PubMed] [Google Scholar]
- 17.Fujita E, Kouroku Y, Isoai A, Kumagai H, Misutani A, Matsuda C, et al. Two endoplasmic reticulum-associated degradation (ERAD) systems for the novel variant of the mutant dysferlin: ubiquitin/proteasome ERAD(I) and autophagy/lysosome ERAD(II) Hum Mol Genet. 2007;16:618–29. doi: 10.1093/hmg/ddm002. [DOI] [PubMed] [Google Scholar]
- 18.Dikic I. Open questions: why should we care about ER-phagy and ER remodelling? BMC Biol. 2018;16:131. doi: 10.1186/s12915-018-0603-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khaminets A, Heinrich T, Mari M, Grumati P, Huebner AK, Akutsu M, et al. Regulation of endoplasmic reticulum turnover by selective autophagy. Nature. 2015;522:354–8. doi: 10.1038/nature14498. [DOI] [PubMed] [Google Scholar]
- 20.Fumagalli F, Noack J, Bergmann TJ, Cebollero E, Pisoni GB, Fasana E, et al. Translocon component Sec62 acts in endoplasmic reticulum turnover during stress recovery. Nat Cell Biol. 2016;18:1173–84. doi: 10.1038/ncb3423. [DOI] [PubMed] [Google Scholar]
- 21.Grumati P, Morozzi G, Holper S, Mari M, Harwardt MI, Yan R, et al. Full length RTN3 regulates turnover of tubular endoplasmic reticulum via selective autophagy. Elife. 2017;6:e25555. doi: 10.7554/eLife.25555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smith MD, Harley ME, Kemp AJ, Wills J, Lee M, Arends M, et al. CCPG1 Is a Non-canonical Autophagy Cargo Receptor Essential for ER-Phagy and Pancreatic ER Proteostasis. Dev Cell. 2018;44:217–32 e211. doi: 10.1016/j.devcel.2017.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen Q, Xiao Y, Chai P, Zheng P, Teng J, Chen J. ATL3 Is a Tubular ER-Phagy Receptor for GABARAP-Mediated Selective Autophagy. Curr Biol. 2019;29:846–55 e846. doi: 10.1016/j.cub.2019.01.041. [DOI] [PubMed] [Google Scholar]
- 24.An H, Ordureau A, Paulo JA, Shoemaker CJ, Denic V, Harper JW. TEX264 is an endoplasmic reticulum-resident ATG8-interacting protein critical for ER remodeling during nutrient stress. Mol Cell. 2019;74:891–908 e810. doi: 10.1016/j.molcel.2019.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chino H, Hatta T, Natsume T, Mizushima N. Intrinsically disordered protein TEX264 mediates ER-phagy. Mol Cell. 2019;74:909–21 e906. doi: 10.1016/j.molcel.2019.03.033. [DOI] [PubMed] [Google Scholar]
- 26.Noda NN, Kumeta H, Nakatogawa H, Satoo K, Adachi W, Ishii J, et al. Structural basis of target recognition by Atg8/LC3 during selective autophagy. Genes Cells. 2008;13:1211–8. doi: 10.1111/j.1365-2443.2008.01238.x. [DOI] [PubMed] [Google Scholar]
- 27.Mochida K, Oikawa Y, Kimura Y, Kirisako H, Hirano H, Ohsumi Y, et al. Receptor-mediated selective autophagy degrades the endoplasmic reticulum and the nucleus. Nature. 2015;522:359–62. doi: 10.1038/nature14506. [DOI] [PubMed] [Google Scholar]
- 28.Fregno I, Fasana E, Bergmann TJ, Raimondi A, Loi M, Solda T, et al. ER-to-lysosome-associated degradation of proteasome-resistant ATZ polymers occurs via receptor-mediated vesicular transport. EMBO J. 2018;37:e99259. doi: 10.15252/embj.201899259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Forrester A, De Leonibus C, Grumati P, Fasana E, Piemontese M, Staiano L, et al. A selective ER-phagy exerts procollagen quality control via a Calnexin-FAM134B complex. EMBO J. 2019;38:e99847. doi: 10.15252/embj.201899847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cui Y, Parashar S, Zahoor M, Needham PG, Mari M, Zhu M, et al. A COPII subunit acts with an autophagy receptor to target endoplasmic reticulum for degradation. Science. 2019;365:53–60. doi: 10.1126/science.aau9263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chiramel AI, Dougherty JD, Nair V, Robertson SJ, Best SM. FAM134B, the selective autophagy receptor for endoplasmic reticulum turnover, inhibits replication of ebola virus strains makona and mayinga. J Infect Dis. 2016;214(suppl 3):S319–25. doi: 10.1093/infdis/jiw270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moretti J, Roy S, Bozec D, Martinez J, Chapman JR, Ueberheide B, et al. STING senses microbial viability to orchestrate stress-mediated autophagy of the endoplasmic reticulum. Cell. 2017;171:809–23 e813. doi: 10.1016/j.cell.2017.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liang JR, Lingeman E, Ahmed S, Corn JE. Atlastins remodel the endoplasmic reticulum for selective autophagy. J Cell Biol. 2018;217:3354–67. doi: 10.1083/jcb.201804185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bhaskara RM, Grumati P, Garcia-Pardo J, Kalayil S, Covarrubias-Pinto A, Chen W, et al. Curvature induction and membrane remodeling by FAM134B reticulon homology domain assist selective ER-phagy. Nat Commun. 2019;10:2370. doi: 10.1038/s41467-019-10345-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schultz ML, Krus KL, Kaushik S, Dang D, Chopra R, Qi L, et al. Coordinate regulation of mutant NPC1 degradation by selective ER autophagy and MARCH6-dependent ERAD. Nat Commun. 2018;9:3671. doi: 10.1038/s41467-018-06115-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kurth I, Pamminger T, Hennings JC, Soehendra D, Huebner AK, Rotthier A, et al. Mutations in FAM134B, encoding a newly identified Golgi protein, cause severe sensory and autonomic neuropathy. Nat Genet. 2009;41:1179–81. doi: 10.1038/ng.464. [DOI] [PubMed] [Google Scholar]
- 37.Davidson G, Murphy S, Polke J, Laura M, Salih M, Muntoni F, et al. Frequency of mutations in the genes associated with hereditary sensory and autonomic neuropathy in a UK cohort. J Neurol. 2012;259:1673–85. doi: 10.1007/s00415-011-6397-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ilgaz Aydinlar E, Rolfs A, Serteser M, Parman Y. Mutation in FAM134B causing hereditary sensory neuropathy with spasticity in a Turkish family. Muscle Nerve. 2014;49:774–5. doi: 10.1002/mus.24145. [DOI] [PubMed] [Google Scholar]
- 39.Murphy SM, Davidson GL, Brandner S, Houlden H, Reilly MM. Mutation in FAM134B causing severe hereditary sensory neuropathy. J Neurol Neurosurg Psychiatry. 2012;83:119–20. doi: 10.1136/jnnp.2010.228965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tang WK, Chui CH, Fatima S, Kok SH, Pak KC, Ou TM, et al. Oncogenic properties of a novel gene JK-1 located in chromosome 5p and its overexpression in human esophageal squamous cell carcinoma. Int J Mol Med. 2007;19:915–23.. [PubMed] [Google Scholar]
- 41.Haque MH, Gopalan V, Chan KW, Shiddiky MJ, Smith RA, Lam AK. Identification of Novel FAM134B (JK1) Mutations in Oesophageal Squamous Cell Carcinoma. Sci Rep. 2016;6:29173. doi: 10.1038/srep29173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kasem K, Gopalan V, Salajegheh A, Lu CT, Smith RA, Lam AK. The roles of JK-1 (FAM134B) expressions in colorectal cancer. Exp Cell Res. 2014;326:166–73. doi: 10.1016/j.yexcr.2014.06.013. [DOI] [PubMed] [Google Scholar]
- 43.Islam F, Gopalan V, Lam AK. RETREG1 (FAM134B): a new player in human diseases: 15 years after the discovery in cancer. J Cell Physiol. 2018;233:4479–89. doi: 10.1002/jcp.26384. [DOI] [PubMed] [Google Scholar]
- 44.Melchiotti R, Puan KJ, Andiappan AK, Poh TY, Starke M, Zhuang L, et al. Genetic analysis of an allergic rhinitis cohort reveals an intercellular epistasis between FAM134B and CD39. BMC Med Genet. 2014;15:73. doi: 10.1186/1471-2350-15-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kong M, Kim Y, Lee C. A strong synergistic epistasis between FAM134B and TNFRSF19 on the susceptibility to vascular dementia. Psychiatr Genet. 2011;21:37–41. doi: 10.1097/YPG.0b013e3283413496. [DOI] [PubMed] [Google Scholar]
- 46.Lennemann NJ, Coyne CB. Dengue and Zika viruses subvert reticulophagy by NS2B3-mediated cleavage of FAM134B. Autophagy. 2017;13:322–32. doi: 10.1080/15548627.2016.1265192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shi Q, Ge Y, Sharoar MG, He W, Xiang R, Zhang Z, et al. Impact of RTN3 deficiency on expression of BACE1 and amyloid deposition. J Neurosci. 2014;34:13954–62. doi: 10.1523/JNEUROSCI.1588-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cunningham CN, Williams JM, Knupp J, Arunagiri A, Arvan P, Tsai B. Cells deploy a two-pronged strategy to rectify misfolded proinsulin aggregates. Mol Cell. 2019;75:442–56 e444. doi: 10.1016/j.molcel.2019.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hu J, Shibata Y, Zhu PP, Voss C, Rismanchi N, Prinz WA, et al. A class of dynamin-like GTPases involved in the generation of the tubular ER network. Cell. 2009;138:549–61. doi: 10.1016/j.cell.2009.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen Q, Teng J, Chen J. ATL3, a cargo receptor for reticulophagy. Autophagy. 2019;15:1465–6. doi: 10.1080/15548627.2019.1609862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kornak U, Mademan I, Schinke M, Voigt M, Krawitz P, Hecht J, et al. Sensory neuropathy with bone destruction due to a mutation in the membrane-shaping atlastin GTPase 3. Brain. 2014;137:683–92. doi: 10.1093/brain/awt357. [DOI] [PubMed] [Google Scholar]
- 52.Krols M, Asselbergh B, De Rycke R, De Winter V, Seyer A, Muller FJ, et al. Sensory neuropathy-causing mutations in ATL3 affect ER-mitochondria contact sites and impair axonal mitochondrial distribution. Hum Mol Genet. 2019;28:615–27. doi: 10.1093/hmg/ddy352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xu H, Zhang C, Cao L, Song J, Xu X, Zhang B, et al. ATL3 gene mutation in a Chinese family with hereditary sensory neuropathy type 1F. J Peripher Nerv Syst. 2019;24:150–5. doi: 10.1111/jns.12309. [DOI] [PubMed] [Google Scholar]
- 54.Behrendt L, Kurth I, Kaether C. A disease causing ATLASTIN 3 mutation affects multiple endoplasmic reticulum-related pathways. Cell Mol Life Sci. 2019;76:1433–45. doi: 10.1007/s00018-019-03010-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Niu L, Ma T, Yang F, Yan B, Tang X, Yin H, et al. Atlastin-mediated membrane tethering is critical for cargo mobility and exit from the endoplasmic reticulum. Proc Natl Acad Sci USA. 2019;116:14029–38. doi: 10.1073/pnas.1908409116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Meyer HA, Grau H, Kraft R, Kostka S, Prehn S, Kalies KU, et al. Mammalian Sec61 is associated with Sec62 and Sec63. J Biol Chem. 2000;275:14550–7. doi: 10.1074/jbc.275.19.14550. [DOI] [PubMed] [Google Scholar]
- 57.Linxweiler M, Schick B, Zimmermann R. Let’s talk about Secs: Sec61, Sec62 and Sec63 in signal transduction, oncology and personalized medicine. Signal Transduct Target Ther. 2017;2:17002. doi: 10.1038/sigtrans.2017.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Greiner M, Kreutzer B, Jung V, Grobholz R, Hasenfus A, Stohr RF, et al. Silencing of the SEC62 gene inhibits migratory and invasive potential of various tumor cells. Int J Cancer. 2011;128:2284–95. doi: 10.1002/ijc.25580. [DOI] [PubMed] [Google Scholar]
- 59.Greiner M, Kreutzer B, Lang S, Jung V, Cavalie A, Unteregger G, et al. Sec62 protein level is crucial for the ER stress tolerance of prostate cancer. Prostate. 2011;71:1074–83. doi: 10.1002/pros.21324. [DOI] [PubMed] [Google Scholar]
- 60.Linxweiler M, Bochen F, Schick B, Wemmert S, Al Kadah B, Greiner M, et al. Identification of SEC62 as a potential marker for 3q amplification and cellular migration in dysplastic cervical lesions. BMC Cancer. 2016;16:676. doi: 10.1186/s12885-016-2739-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Linxweiler M, Linxweiler J, Barth M, Benedix J, Jung V, Kim YJ, et al. Sec62 bridges the gap from 3q amplification to molecular cell biology in non-small cell lung cancer. Am J Pathol. 2012;180:473–83. doi: 10.1016/j.ajpath.2011.10.039. [DOI] [PubMed] [Google Scholar]
- 62.Wemmert S, Lindner Y, Linxweiler J, Wagenpfeil S, Bohle R, Niewald M, et al. Initial evidence for Sec62 as a prognostic marker in advanced head and neck squamous cell carcinoma. Oncol Lett. 2016;11:1661–70. doi: 10.3892/ol.2016.4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bergmann TJ, Fumagalli F, Loi M, Molinari M. Role of SEC62 in ER maintenance: a link with ER stress tolerance in SEC62-overexpressing tumors? Mol Cell Oncol. 2017;4:e1264351. doi: 10.1080/23723556.2016.1264351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kostenko EV, Olabisi OO, Sahay S, Rodriguez PL, Whitehead IP. Ccpg1, a novel scaffold protein that regulates the activity of the Rho guanine nucleotide exchange factor Dbs. Mol Cell Biol. 2006;26:8964–75. doi: 10.1128/MCB.00670-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schwartzlow C, Kazamel M. Hereditary sensory and autonomic neuropathies: adding more to the classification. Curr Neurol Neurosci Rep. 2019;19:52. doi: 10.1007/s11910-019-0974-3. [DOI] [PubMed] [Google Scholar]
- 66.Eggermann K, Gess B, Hausler M, Weis J, Hahn A, Kurth I. Hereditary Neuropathies. Dtsch Arztebl Int. 2018;115:91–7. doi: 10.3238/arztebl.2018.0091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fink JK. Hereditary spastic paraplegia. Curr Neurol Neurosci Rep. 2006;6:65–76. doi: 10.1007/s11910-996-0011-1. [DOI] [PubMed] [Google Scholar]
- 68.Zhao X, Alvarado D, Rainier S, Lemons R, Hedera P, Weber CH, et al. Mutations in a newly identified GTPase gene cause autosomal dominant hereditary spastic paraplegia. Nat Genet. 2001;29:326–31. doi: 10.1038/ng758. [DOI] [PubMed] [Google Scholar]
- 69.Guelly C, Zhu PP, Leonardis L, Papic L, Zidar J, Schabhuttl M, et al. Targeted high-throughput sequencing identifies mutations in atlastin-1 as a cause of hereditary sensory neuropathy type I. Am J Hum Genet. 2011;88:99–105. doi: 10.1016/j.ajhg.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Montenegro G, Rebelo AP, Connell J, Allison R, Babalini C, D’Aloia M, et al. Mutations in the ER-shaping protein reticulon 2 cause the axon-degenerative disorder hereditary spastic paraplegia type 12. J Clin Investig. 2012;122:538–44. doi: 10.1172/JCI60560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zou Y, He W, Wang K, Han H, Xiao T, Chen X, et al. Identification of rare RTN3 variants in Alzheimer’s disease in Han Chinese. Hum Genet. 2018;137:141–50. doi: 10.1007/s00439-018-1868-1. [DOI] [PubMed] [Google Scholar]
- 72.Heath JE, Siedlak SL, Zhu X, Lee HG, Thakur A, Yan R, et al. Widespread distribution of reticulon-3 in various neurodegenerative diseases. Neuropathology. 2010;30:574–9. doi: 10.1111/j.1440-1789.2010.01107.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kasem K, Gopalan V, Salajegheh A, Lu CT, Smith RA, Lam AK. JK1 (FAM134B) gene and colorectal cancer: a pilot study on the gene copy number alterations and correlations with clinicopathological parameters. Exp Mol Pathol. 2014;97:31–36. doi: 10.1016/j.yexmp.2014.05.001. [DOI] [PubMed] [Google Scholar]
- 74.Viswanath P, Radoul M, Izquierdo-Garcia JL, Luchman HA, Gregory Cairncross J, Pieper RO, et al. Mutant IDH1 gliomas downregulate phosphocholine and phosphoethanolamine synthesis in a 2-hydroxyglutarate-dependent manner. Cancer Metabol. 2018;6:3. doi: 10.1186/s40170-018-0178-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science. 2011;333:228–33. doi: 10.1126/science.1205405. [DOI] [PMC free article] [PubMed] [Google Scholar]