

Abstract
Inflammation-resolution is a protective response that is mediated by specialized pro-resolving mediators (SPMs). The clearance of dead cells or efferocytosis is a critical cellular program of inflammation-resolution. Impaired efferocytosis can lead to tissue damage in prevalent human diseases, like atherosclerosis. Therefore understanding mechanisms associated with swift clearance of dead cells is of utmost clinical importance. Recently, the accumulation of necroptotic cells (NCs) was observed in human plaques and we postulated that this is due to defective clearance programs. Here we present evidence that NCs are inefficiently taken up by macrophages because they have increased surface expression of a well-known “don’t eat me” signal called CD47. High levels of CD47 on NCs stimulated RhoA-pMLC signaling in macrophages that promoted “nibbling”, rather than whole-cell engulfment of NCs. Anti-CD47 blocking antibodies limited RhoA-p-MLC signaling and promoted whole-cell NC engulfment. Treatment with anti-CD47 blocking antibodies to Ldlr−/− mice with established atherosclerosis decreased necrotic cores, limited the accumulation of plaque NCs and increased lesional SPMs, including Resolvin D1 (RvD1) compared with IgG controls. Mechanistically, RvD1 promoted whole-cell engulfment of NCs by decreasing RhoA signaling and activating CDC42. RvD1 specifically targeted NCs for engulfment by facilitating the release of the well-known “eat me signal” called calreticulin from macrophages in a CDC42 dependent manner. Lastly, RvD1 enhanced the clearance of NCs in advanced murine plaques. Together, these results suggest new molecules and signaling associated with the clearance of NCs, provide a new paradigm for the regulation of inflammation-resolution, and offer a potential treatment strategy for diseases where NCs underpin the pathology.
Subject terms: Cell death and immune response, Chronic inflammation
Introduction
Inflammation-resolution is a protective response that is largely controlled by specialized pro-resolving mediators (SPMs), such as lipoxins, resolvins, protectins, and maresins [1]. SPMs control inflammation and promote tissue repair, in a manner that does not compromise host defense [1–3]. Efferocytosis (or the clearance of dead cells) is a critical cellular process of inflammation-resolution. The uptake of apoptotic cells (ACs) enhances the biosynthesis of SPMs and SPMs, in a feed-forward manner, then facilitate a swift clearance of ACs [4–6].
Defective efferocytosis and impaired SPM biosynthesis are associated with non-resolving diseases like atherosclerosis [7, 8]. Specifically, defects in these programs result in atherosclerotic plaques that have large necrotic cores and are thus more prone to rupture [9]. Recently, the accumulation of necroptotic cells (NCs) has emerged as a major driver of atherosclerotic plaque necrosis [10–12]. Necroptosis is a pro-inflammatory form of cell death [13, 14] and so the accumulation of NCs can be damaging to tissues. Earlier work demonstrated that NCs were cleared in a manner that appears distinct and less efficient than ACs [12, 15]. However, detailed molecular mechanisms, quantification methods, and factors that augment the clearance of NCs are not known. Importantly, how the accumulation of NCs impacts inflammation-resolution is not known either.
Here, we demonstrate that NCs are less readily internalized by macrophages because they have increased levels of CD47. NCs were “nibbled” by macrophages due to a CD47-induced RhoA-p-myosin light chain 2 (MLC) signaling in macrophages. Blockade of CD47 enhanced the rate of NC uptake, prevented the “nibbling” response and promoted whole-cell engulfment. Treatment of anti-CD47 antibodies to Ldlr−/− mice with established atherosclerosis resulted in decreased lesional NCs and increased SPMs. Moreover, the SPM Resolvin D1 (RvD1) enhanced the clearance of NCs by limiting p-MLC and activating CDC42. Together, these results suggest new mechanisms associated with the recognition and clearance of NCs, links between NC clearance and inflammation-resolution, and a potential new treatment strategy for diseases that are associated with the accumulation of NCs.
Results
Necroptotic cells are not readily phagocytized by macrophages
NCs have a distinct engulfment process compared with ACs [15]. Recognition receptors, signaling molecules, and temporal aspects associated with this process are of interest. Here, we performed confocal z-stack live-cell imaging of primary murine bone-marrow-derived macrophages co-cultured with either ACs or NCs. Raw fluorescence intensity of representative images and videos are shown in Fig. S1a and in Videos 1,2 where the macrophages are green, the dead cells are red and ingested dead cells are yellow. Figure 1a displays representative images after 3D rendering in which macrophages are gray, dead cells are magenta, and engulfed cells are cyan (Fig. 1a, Video 3, 4). We observed that macrophages swiftly engulfed ACs (Fig. 1a, Videos 1 and 3), whereas macrophages did not readily ingest NCs and had significantly less engulfment compared with ACs over the same time period (Fig. 1a, Videos 2 and 4). Similar results were obtained using AMNIS image stream flow cytometry (Fig. S1b). Macrophages took small bites of the NCs, which is shown as small cyan spots within the gray macrophage (Fig. 1a, Video 4) or as small green spots from the representative AMNIS images (Fig. S1b). Moreover, the size of the engulfed dead ACs was significantly larger than internalized NCs (Fig. S1c). Macrophages briefly interacted with NCs, took a few nibbles, then swiftly retracted from NCs (Video 4). These results suggest that there are recognition molecule(s) on NCs that dictate inefficient clearance or there are a lack of molecule(s) on NCs that enable engulfment.
Fig. 1.

Elevated levels of CD47 on necroptotic cells promotes RhoA-p-MLC signaling in macrophages and result in inefficient clearance of NCs. ACs or NCs were co-cultured with live macrophages in a 3:1 ratio for 1 hr at 37 °C, 5% CO2. a Representative 3D-rendered images from live-cell imaging of macrophages (gray) and AC or NCs (magenta), ingested ACs or NCs (cyan) at 0, 15, and 30 min. Images were captured on a Zeiss LSM 880 microscope and rendered using Imaris software. Scale bar is 10 μm. Results are mean ± sem of n = 4–8 separate cells per group, **p < 0.01, Two-way ANOVA. b CD47 was determined by flow cytometry on live macrophages (Veh), ACs, or NCs and data were analyzed with FlowJo software. Results are represented as fold change and are shown as mean ± sem. Experiments were carried out in triplicate and repeated four separate times, **p < 0.01, One-way ANOVA, Tukey’s multiple comparisons test. c Live-cell confocal imaging was carried out as in panel a. NCs were incubated with 10 μg/mL of IgG or anti-CD47 and then added to macrophages. Images were acquired for 33 min and data was analyzed with Imaris software. Results are mean ± sem, of n = 8 separate cells per group, **p < 0.01 Two-way ANOVA. d Representative maximum intensity projection (MIP) images are shown. Macrophages were transfected with LifeAct-RFP and co-cultured with PKH67 (green) ACs or NCs as above for 40 mins at 37 °C, 5% CO2. Top panel is a merged image with both macrophages (red) and dead cells (green). Bottom panels display only the macrophages (red). Arrows denote the phagocytic synapse. e Quantification of p-MLC in macrophages from experiments carried out as in d. Results are mean ± sem, of n = 6 separate cells per group, **p < 0.01 student’s t-test. f Live-cell confocal imaging was carried out as in c. Results are mean ± sem of n = 8 separate cells per group, **p < 0.01, Two-way ANOVA
NCs have high levels of the “don’t eat me” signal CD47
Recognition molecules for NC internalization are not known. CD47 is a well-known “don’t eat me” signal and is upregulated during inflammation [16]. Since necroptosis is pro-inflammatory and because NCs are not readily internalized by macrophages, we questioned whether NCs had increased CD47. We found that NCs have significantly higher surface expression of CD47 compared with live macrophages or ACs (Fig. 1b). This increase in CD47 was dependent on NFkB signaling [16] (Fig. S2a). We also observed that CD47 is clustered on the surface of NCs (Fig. S2b). Macrophages did not internalize NCs in regions where CD47 was high (Fig. S2b, white arrow), whereas the macrophage “nibbled” NCs in regions devoid of CD47 (Fig. S2b, white asterisks). These results suggest that CD47 clustering promotes “nibbling.”
We next incubated NCs with an anti-CD47 antibody and observed significantly more uptake, compared with IgG controls (Fig. 1c). In contrast, AC uptake was not enhanced in the presence of anti-CD47 blocking antibodies (Fig. 1c). The size of the ingested NC with anti-CD47 treatment was significantly larger than IgG controls (Fig. S2c), which suggests that macrophages internalized whole NCs.
Mechanisms related to actin cytoskeletal changes during NC nibbling remain underexplored. We transfected macrophages with LifeAct-RFP, which illuminates F-actin in red. Macrophages internalize whole ACs and present “mouth-like” actin structures that resemble phagocytic cups (Fig. 1d, right panels). Macrophages that encountered IgG-NCs displayed “arm-like” structures (Fig. 1d, left panel, Fig. S3a). Interestingly, macrophages that internalized anti-CD47-NCs produced a “mouth-like” cup, similar to macrophage internalization of ACs (Fig. 1d middle and right panels). These results suggest that the distinct internalization process of NCs is dictated by aberrant CD47. We next developed a method to quantify and visualize the phagocytic synapse (i.e., the region in which macrophages encounter NCs). We 3D-rendered phagocytic synapses based on the fluorescence intensity of LifeAct-RFP and assessed volume (Fig. S3b left panel and see methods). We observed that macrophages produced significantly larger synapses when eating NCs with anti-CD47, compared with IgG controls (Fig. S3b, right panel). Representative 3D-rendered images also revealed actin branching in the synapse of the anti-CD47 group (Fig. S3b). Together, these results suggest that high levels of CD47 on NCs promote inefficient nibbling by macrophages.
GTPases control the internalization of ACs [17], therefore we questioned which GTPase promotes NC engulfment. We observed stress fibers that appear as linear bright red regions on macrophages that extend toward NCs (Figs. S2b, S3a). Stress fibers can be formed upon activation of RhoA and phospho-MLC (p-MLC) signaling. Live cells co-cultured with macrophages did not stimulate p-MLC, which suggests that homeostatic levels of CD47 did not activate p-MLC in macrophages (Fig. 1e). NCs significantly increased macrophage p-MLC, which was blocked by anti-CD47 treatment (Fig. 1e). Together, these results suggest that aberrant CD47 on NCs promote RhoA signaling.
Next, we treated macrophages with a RhoA inhibitor and performed live-cell imaging of macrophages co-cultured with NCs coated with IgG or an anti-CD47 antibody. First, we observed that RhoA inhibition significantly enhanced the uptake of NCs by macrophages (Fig. 1f), which further suggests that NCs activate RhoA signaling in macrophages. Importantly, RhoA inhibition and anti-CD47 treatment were not additive (Fig. 1f), which suggests that CD47 acts through RhoA signaling to promote uptake of NCs. RhoA inhibition also promoted a branched phagocytic synapse (Fig. S4a) that was similar to the anti-CD47 group (Fig. S3b). These results suggest that aberrant CD47 levels on NCs induce a RhoA-p-MLC signaling pathway on macrophages to promote nibbling versus whole-cell engulfment.
Anti-CD47 antibody treatment during established atherosclerosis decreases lesional NCs and promotes SPM
Aberrant expression of CD47 occurs in certain cancers, atherosclerosis, and myocardial infarction [16, 18, 19]. Treatment with anti-CD47 blocking antibodies to apoE−/− mice at the initiation of atherosclerosis resulted in decreased atheroprogression [16]. This treatment was given at the onset of atherosclerosis in apoE−/− mice that received Angiotensin II and Western Diet (WD) feeding for 4 weeks. We first questioned whether anti-CD47 antibody treatment reduced lesional p-MLKL (a marker for NCs) in this context. Indeed, we found that anti-CD47 antibody treatment significantly reduced lesional p-MLKL+ cells in apoE−/−-AngII mice compared with IgG controls (Fig. S5a). We next questioned whether anti-CD47 blocking antibodies given during established atherosclerosis would prevent atheroprogression. For these experiments, Ldlr−/− mice were fed the WD for 10 weeks, after which 100 μg/mouse of IgG or anti-CD47 were administered (3x/week i.p.) for 3 weeks. Cholesterol, body weight, and blood glucose levels were not statistically different between the groups (Table 1). Circulating leukocytes were not different between the groups, whereas RBCs were significantly decreased by the anti-CD47 treatment (Table 2). Anti-CD47 antibody treatment significantly decreased p-MLKL+ cells (Fig. 2a) and lesional necrosis (Fig. 2b). Representative H&E images are shown on the left of Fig. 2b. We next questioned the percentage of necroptotic macrophages. We co-stained p-MLKL and F4/80 (a macrophage marker) and found that there was significantly less F4/80+p-MLKL+ in the mice treated with anti-CD47 antibody compared with IgG controls (Fig. S5b). Representative images of plaques are shown on the left and F4/80 (green), p-MLKL (pink), and DAPI (blue) are shown (Fig. S5b). These results suggest that macrophages, as well as other lesional cells, undergo necroptosis in plaques. Moreover, these data also suggest that anti-CD47 treatment given during established atherosclerosis promoted the clearance of NCs in plaques.
Table 1.
CBC counts from IgG and anti-CD47 antibody treatment to WD-fed Ldlr−/− mice with established atherosclerosis
| Baseline | +IgG | +anti-CD47 | Significance | |
|---|---|---|---|---|
| % neutrophils | 37.3 ± 8.8 | 25.0.5 ± 1.1 | 31 ± 4.9 | NS |
| % lymphocytes | 48.7 ± 8.2 | 62.6 ± 3.2 | 54.7 ± 3.8 | NS |
| % monocytes | 5 ± 0.7 | 3.1 ± 0.6 | 4.4 ± 0.7 | NS |
| Red blood cells (×106/μL) | 8.4 ± 0.1 | 8.3 ± 0.2 | 7.2 ± 0.1** | |
| White Blood cells (×103/μL) | 6.8 ± 1.0 | 7.6 ± 1.6 | 6.4 ± 1.1 | NS |
| Hemoglobin (g/dL) | 13 ± 0.4 | 13.7 ± 0.5 | 12.8 ± 0.2 | NS |
| Platelets (×103/μL) | 569.2 ± 211.5 | 593.5 ± 140.1 | 619 ± 148.7 | NS |
Results are mean ± sem, One-way ANOVA with Tukey’s multiple comparison test
**p < 0.01 +IgG vs. +anti-CD47
Table 2.
Lesion area, body weight, blood glucose and cholesterol for IgG and anti-CD47 antibody treatment to WD-fed Ldlr−/− mice with established atherosclerosis
| Baseline | +IgG | +anti-CD47 | Significance | |
|---|---|---|---|---|
| Lesion area (μm2) | 22456.8 ± 2575.1 | 34228.0 ± 2960.8* | 28171.1 ± 2932.9 | |
| Body weight (g) | 35.6 ± 3.0 | 39.8 ± 1.6 | 36.5 ± 2.0 | NS |
| Blood glucose (mg/dL) | 169.5 ± 8.3 | 153.4 ± 10.7 | NS | |
| Cholesterol (mg/dL) | 1416.3 ± 153.9 | 1447.2 ± 153.0 | NS |
Results are mean ± sem, n = 5 for baseline, n = 6 for IgG, and n = 7 for anti-CD47 separate mice per group, One-way ANOVA with Tukey’s multiple comparison test
*p < 0.05 baseline vs. +IgG
Fig. 2.

Administration of IgG or anti-CD47 to WD-fed Ldlr−/− mice with established atherosclerosis decreases lesional necroptotic cells and increases SPM in plaques. a Aortic roots lesion sections from Ldlr−/− mice were immunostained with an anti-p-MLKL antibody and Hoechst and the percent of p-MLKL+ cells was quantified. b Aortic root lesions were stained with H&E and necrotic areas were quantified with Olympus DP2-BSW software. Necrotic core is abbreviated as NC. Baseline indicates mice that were sacrificed after 10 weeks of WD feeding. Results are mean ± sem, n = 5 for baseline, n = 6 for IgG, and n = 7 for anti-CD47 separate mice per group. *p < 0.05, One-way ANOVA, Tukey’s multiple comparisons test. c–e Aortas were collected and subjected to LC-MS/MS. c An SPM cluster that includes 15R-Lipoxin A4 (15R-LXA4), RvD1, RvD5, Maresin 1 (Mar1), 12-trans Mar1, and 10 S,17S-diHDHA was identified and the MRM chromatogram is shown. d Representative MS/MS spectra for 15R-LXA4, RvD1, Mar-1, and RvD5 are shown. e Quantification of the SPM cluster. Results are mean ± sem, n = 6 separate mice per group, *p < 0.05, student’s t-test
We next questioned whether decreased lesional necroptosis was associated with increased SPMs. Indeed, anti-CD47 treatment led to an increase in a lesional SPM cluster that included 15R-Lipoxin A4 (15R-LXA4), RvD1, RvD5, Maresin 1 (Mar1), 12-trans Mar1, and 10 S,17S-diHDHA (Fig. 2c–e). A representative MRM chromatogram of the SPM cluster is shown in Fig. 2c and representative MS/MS spectra are shown in Fig. 2d. Together, these results suggest that local clearance of NCs in atherosclerotic plaques is associated with increased SPM biosynthesis.
The SPM Resolvin D1 (RvD1) enhances the engulfment of NCs
Because RvD1 was identified in the above “cluster” and was previously shown to reduce plaque necrosis [8], we next questioned whether RvD1 further enhanced the clearance of NCs in a feed-forward manner. We observed that 10 nM RvD1 significantly increased NC uptake compared with controls (Fig. 3a). Three D-rendered images of RvD1-stimulated macrophages display more cyan, which is indicative of enhanced engulfment of NCs. Non-rendered images are also shown (Fig. S6a). Importantly, the size of ingested NCs in RvD1-stimulated macrophage was significantly larger than the vehicle (Veh) treatment (Fig. S6b), which suggests that RvD1 promoted whole-cell engulfment of NCs. Along these lines, the live-cell video revealed that RvD1-stimulated macrophages swiftly internalized NCs (Videos 5 and 6). RvD1 also enhanced the engulfment of apoptotic cells (Fig. S7a) and promoted the uptake of several ACs at once (Fig. S7b). These results suggest that RvD1 signaling stimulated macrophages to target and swiftly engulf whole NCs.
Fig. 3.

Resolvin D1 enhances the clearance of necroptotic cells through limiting RhoA signaling and activating CDC42. Macrophages were stimulated with Veh or 10 nM RvD1 for 20 min, prior to addition of NCs. Images were acquired and analyzed as in Fig. 1. a Representative images from live-cell imaging of macrophages (gray) and NCs (magenta), ingested NCs (light magenta-to-cyan) at 0, 15, and 30 min. Images were captured on a Zeiss LSM 880 microscope and rendered using Imaris software. Scale bar is 10 μm. Results are mean ± sem, of n = 8 separate NC and n = 7 separate NC + RvD1 cells, **p < 0.01 Two-way ANOVA. b p-MLC was quantified as in Fig. 1. Results are mean ± sem, of n = 6 separate cells per group, **p < 0.01 student’s t-test. c Macrophages were stimulated with Veh or 10 nM RvD1 or in combination with 10 μM of the RhoA inhibitor (Rhosin) for 20 min prior to addition of NCs. Imaging and quantification were carried out as in panel a. Results are mean ± sem, of n = 9–10 cells per, **p < 0.01, + NC vs. other groups, ^^p < 0.01 + NC + RhoAi vs. other groups, Two-way ANOVA, Tukey’s multiple comparison. d Representative 3D-rendered images of RvD1-stimulated macrophages (red) in the presence of NCs (green) are shown. e Representative phagocytic synapses are shown on the left and quantification is shown on the right. Results are mean ± sem, of n = 18 separate cells per group, **p < 0.01 student’s t-test. f Imaging and quantification were carried out as in panel a. Results are mean ± sem, of n = 8 cells per group,**p < 0.01 Two-way ANOVA. g Representative MIP images display macrophages in red and NCs in green. The white arrows denote the phagocytic synapses
Because we observed that aberrant CD47 on NCs increased a RhoA-p-MLC signaling pathway on macrophages during internalization, we first questioned whether RvD1 decreased p-MLC in macrophages. Indeed, RvD1 significantly reduced p-MLC compared with Veh controls (Fig. 3b). Moreover, RhoA inhibition significantly increased NC ingestion compared with Veh controls (Fig. 3c). RvD1 significantly increased engulfment compared with RhoA inhibition, which suggests that RvD1 was in part limiting RhoA signaling, but also activating an additional signaling pathway (Fig. 3c, Fig. S6c).
To probe mechanism we assessed 3D-rendered macrophages that were transfected with LifeAct-RFP and NCs that were labeled with PKH67 (Fig. 3d, Video 7). The images in Fig. 3d are snap-shots at different orientations and demonstrate that RvD1 promotes whole-cell NC engulfment. Representative 3D-rendered phagocytic synapses are shown in which the actin network is visibly branched in the RvD1-stimulated macrophages (Fig. 3e, left panel). Also, RvD1-treated macrophages had significantly larger synapses, compared with Veh controls (Fig. 3e). The phagocytic synapses from RvD1-stimulated macrophages were also larger than those from RhoA-inhibited macrophages (Fig. S4b), which further suggests that RvD1 initiates signaling that is distinct from simply limiting RhoA.
Importantly, RvD1 was able to enhance the uptake of different necroptotic cell types. We stimulated necroptosis in L929, aortic and airway smooth muscle cells. Macrophages in the presence of different necroptotic cell types exhibited similar arm-like projections. We found that RvD1-stimulated macrophages had increased phagocytic synapses in the presence of the above necroptotic cells types (Fig. S8a-c). These results suggest that RvD1 promotes whole-cell engulfment in these contexts as well.
We next questioned whether whole-cell engulfment by RvD1 limited inflammation. CXCL1 (a potent pro-inflammatory chemokine) was released from macrophages that nibble NCs (Fig. S9a). RvD1 significantly reduced CXCL1 during whole-cell engulfment of NCs. RvD1 did not regulate IL-10 levels from macrophages (Fig. S9b) at this time point. These results suggest that RvD1 dampened pro-inflammatory CXCL1.
To further assess mechanism, we questioned which GTPase is activated by RvD1 signaling. CDC42 is a GTPase that is known to promote branched actin and previous studies indicated that a structurally similar SPM (i.e., LXA4) regulates CDC42 in THP1 cells [20]. Therefore, we questioned whether RvD1 activates CDC42 for efficient engulfment of NCs. For these experiments, we treated macrophages with a CDC42 inhibitor in the presence of Veh or RvD1 and performed confocal z-stack live-cell imaging. First, inhibition of CDC42 did not affect the engulfment of NCs on its own, which suggests that CDC42 is not involved in NC internalization (Fig. 3g). However, CDC42 inhibition completely blocked RvD1’s ability to efficiently engulf NCs (Fig. 3f). Macrophages transfected with LifeAct-RFP also reveal that CDC42 inhibition did not alter how macrophages nibble NCs (Fig. 3g, left panels), but completely blocked RvD1’s ability to promote whole-cell engulfment and resulted in a small “arm-like” phagocytic synapse, similar to controls. These results suggest that when RvD1’s signaling is blocked, the CD47-RhoA-p-MLC signaling dominates. The next question is how RvD1 stimulates the macrophage to swiftly detect and target NCs.
RvD1 stimulates macrophages to secrete “eat me” signals for efficient engulfment
Macrophages release calreticulin (i.e., a known “eat me” signal [21]) as a means to “label” cancer or effete cells for their removal [22, 23]. Because RvD1-stimulated macrophages swiftly recognized NCs (even in the presence of elevated levels of CD47 on the NCs), we questioned whether RvD1 promoted the release of calreticulin onto NCs. Therefore, we co-transfected macrophages with LifeAct-RFP and Calreticulin-emerald (green) to label actin and the endoplasmic reticulum (ER), respectively. The NCs were stained with Alexa647-cell mask (pink). We first showed that Veh-treated macrophages extended “arms” that do not contain ER (Fig. 4a, top right panel, denoted by asterisks). In contrast, the Calreticulin-emerald green signal was prominent throughout the macrophage as well as in the phagocytic synapse of RvD1-stimulated macrophages (Fig. 4a bottom panels). The phagocytic synapse on the macrophage again resembles a “mouth-like” structure and is denoted by asterisks (Fig. 4a bottom panel). Importantly, the NCs had significantly more Calreticulin-emerald green signal on the NCs (Fig. 4a bottom left panel, 4B), which suggests that the ER from the macrophage was released onto NCs. Visualization of calreticulin on NCs is shown in Fig. 4a, right panel and highlighted with arrows. We next used an immunofluorescence-based approach to determine extruded ER components on NCs. For these experiments, we transfected macrophages with LifeAct-RFP and labeled NCs with PKH67 (green). NCs were co-cultured with macrophages as above and cells were fixed and stained with another ER marker called calnexin (cyan) (Fig. 4c). RvD1-stimulated macrophages had significantly more regions of externalized calnexin (Fig. 4c, d), as well as more calnexin on NCs (Fig. 4c, e), compared with Veh controls. Together, these results suggest that RvD1 stimulates macrophages to mobilize and release ER components onto NCs.
Fig. 4.

Resolvin D1 promotes the release of calreticulin in a CDC42 dependent manner. BMDMs co-transfected with LifeAct-RFP (red) and calreticulin-Emerald green were cultured with NCs (pink) as above and images were acquired on a Zeiss LSM 880 microscope. a Representative MIP images of n = 15 cells/ group are shown. Light blue boxes indicate regions that are expanded and are displayed on the right side of the panel. Asterisks denote the phagocytic synapse and arrows represent released calreticulin. b Quantification of calreticulin on NCs was carried out using Imaris software. Results are mean ± sem, n = 15 separate cells per group, **p < 0.01, student’s t-test. c BMDMs were transfected with LifeAct-RFP (red) and were co-cultured with NCs (green) as above and calnexin immunostaining is visualized in cyan. The images are representative of n = 13 cells/ group. d Quantification of externalized calnexin on the macrophages or e external calnexin on NCs was carried out with Imaris software. Results are mean ± sem, n = 13 separate cells per group, **p < 0.01 student’s t-test. f Experiments were carried out as in Fig. 3e. Representative images of n = 15 cells/group are shown
Actin is known to facilitate the movement and function of the ER [24] and because we know that CDC42 is required for RvD1’s efficient eating, we next questioned whether inhibition of CDC42 prevented the mobilization of the ER at the phagocytic synapse and limited the amount of released calreticulin. Indeed, inhibition of CDC42 prevented RvD1’s ability to mobilize the ER to the phagocytic synapse and to release calreticulin on NCs (Fig. 4f, bottom panels). Together these results suggest an entirely new mechanism by which RvD1 activates CDC42 within macrophages to mobilize the ER and release calreticulin to target NCs for swift engulfment.
RvD1 treatment of Ldlr−/− mice increases the clearance of lesional CD47+ cells and decreases necrosis
Because our in vitro results revealed that RvD1 increased the uptake of NCs, we next questioned whether RvD1 could decrease lesional NCs during advanced atherosclerosis. For these experiments, Ldlr−/− mice were fed the WD for 11 weeks, after which Veh or 100 ng/mouse of RvD1 were administered (3x/wk i.p.) for 3 weeks while still on the WD. Indeed, RvD1 significantly decreased lesional CD47+ cells (Fig. 5a) and lesional p-SHP-1, which is a marker of decreased CD47 signaling (Fig. 5b). Lastly, RvD1 significantly decreased necrotic areas compared with controls (Fig. 5c). Together these findings suggest that RvD1 enhances the clearance of CD47+ NCs in advanced atherosclerosis.
Fig. 5.

Administration of RvD1 to WD-fed Ldlr−/− mice with established atherosclerosis decreases lesional CD47 and necrosis. Ldlr−/− mice were fed a WD for 12 weeks, after which Veh or 100 ng/mouse of RvD1 (3x/week i.p.) was administered for 3 weeks. Aortic root lesions were immunostained with anti-CD47 (a) or anti-p-SHP1 (b) antibody and counterstained with Hoechst. The mean fluorescence intensity of CD47 was quantified. c Lesional necrosis was quantified with Olympus DP2-BSW software. All results are mean ± sem, n = 8 separate mice per group. *p < 0.05, Student’s t-test
Discussion
The findings of this study provide a new molecular clue as to why NCs are not efficiently cleared by macrophages and a previously unappreciated link between the accumulation of NCs and inflammation-resolution. Prior studies that investigated NC clearance revealed that there is a lethargic uptake of NCs by macrophages [12, 15, 25]. In fact, methods such as scanning and transmission electron microscopy were used to investigate NC internalization [26], which was critical to lay the foundation that NC uptake was a unique process. These studies revealed arm-like structures that encountered NCs. To our knowledge, our work presents the first visualization in real time as well as detailed cytoskeletal changes and signaling associated with NC clearance. We established live-cell z-stack confocal imaging methods with Airyscan resolution to determine temporal and spatial engulfment of NCs. Our methods for imaging and quantification are rapid, which opens the field to exploring and visualizing new mechanisms associated with dead cell recognition and internalization.
In this regard, we present evidence that NCs have increased and aberrant surface levels of CD47 that promotes inefficient clearance in which the macrophage nibbles instead of engulfs. NC nibbling by macrophages resembles a process called trogocytosis (from the ancient Greek trogo, meaning ‘gnaw’ or ‘nibble’). Recent work suggests that trogocytosis is a common process that is shared by amoeba [27], lymphocytes, neutrophils, macrophages [28–31], and microglial cells [32]. Why a macrophage commits to a trogocytosis program versus other internalization processes was not previously known. We postulate that in our context, macrophages nibble around clustered surface CD47. An interesting parallel to our work that microglial cells carry out trogocytosis to prune synapses [25]. These regions where microglia undergo trogocytosis may be due to the presence of CD47 on the surface of the neurons [33].
Additionally, our results also suggest that “eat me” signals may be conserved between ACs and NCs. Previous work suggests that there is at least one common “eat me” signal between NCs and ACs called phosphatidylserine (PS) [25]. Importantly, ACs release several pro-resolving “eat me” signals including Annexin A1 and SPMs [34, 35], to further enhance the clearance of ACs. Because NCs are not efficiently taken-up macrophages, NCs may not release pro-resolving “eat me signals” which would be an interesting topic to pursue in future studies.
Mechanistically, we observed that RhoA facilitated the nibbling of NCs by macrophages. We found that whole-cell NC clearance occurred when RhoA was limited. Inhibition of RhoA also enhances the uptake of ACs [17], which suggests that limiting RhoA may be a general mechanism for efficient engulfment of dead cells by macrophages. Because RvD1-stimulated macrophages decreased RhoA signaling and required CDC42 for efficient internalization, our results suggest that efficient engulfment of NCs is dependent on switching the balance away from RhoA and toward CDC42.
Our results also suggest that RvD1 promotes the release of calreticulin and other ER proteins near sites of ingestion to target NCs for swift clearance. Our work also suggests that RvD1 may regulate ER-mediated phagocytosis. ER-mediated phagocytosis has been observed previously in different contexts and is thought to be important for eating large cargo [36, 37]. Also, these general observations are in agreement with published studies [22, 23] but offer a distinct mechanism that adds important new links as to how RvD1 augments the removal of NCs.
We also found a previously unexplored link between NC clearance and inflammation-resolution. We observed that blockade of CD47 in advanced murine plaques locally stimulated the biosynthesis of SPM. Our work is the first to suggest a feed-forward circuit in which NC clearance by macrophages stimulates SPMs and SPMs like RvD1 further enhance the uptake of NCs (Fig. 6). Moreover, our results also suggest that the accumulation of NCs may derail inflammation-resolution. These findings are particularly important because they provide a new framework to approach treatment strategies for diseases in which NC accumulation promotes the pathology. SPMs and other pro-resolving ligands act to limit pro-inflammatory factors in a manner that does not cause immune suppression [2, 3] and therefore suggests a potentially safer way to treat long-term diseases like atherosclerosis. SPMs like RvD1, RvD2, and Mar1 and 15R-LXA4 were recently shown to reduce atheroprogression in mice [8, 38, 39]. Interestingly, these SPMs were part of the “cluster” we identified in advanced plaques where NCs were actively removed.
Fig. 6.
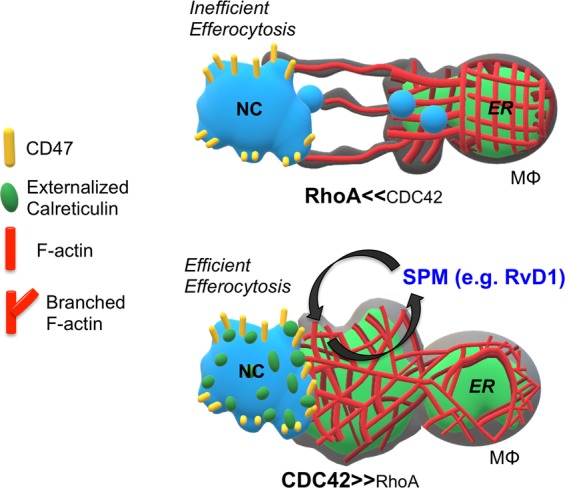
Overall scheme. NCs have high levels of CD47 that are clustered on the cell surface. Macrophages nibble the NCs in regions devoid of CD47 in a RhoA dependent manner. The uptake of NC enhances the biosynthesis of SPM. SPM, like RvD1 then enhances the engulfment of NCs in a CDC42 dependent manner. Additionally, RvD1 stimulates the release of ER proteins like calreticulin to target and label the NCs for swift engulfment in a CDC42 dependent manner
In summary, necroptosis evolved to amplify pro-inflammatory processes for efficient neutralization of pathogens [40]. Therefore, in certain contexts, inefficient clearance of NCs may be advantageous to the host. However, in the context of progressive age-related diseases, like atherosclerosis, the accumulation of NCs is maladaptive [10–12]. Our results suggest that RvD1 may be a new way to treat long-term diseases in which the accumulation of NCs exacerbates the pathology.
Methods
Generation of apoptotic or necroptotic cells
Bone marrow-derived macrophages (BMDMs) were stimulated with 1 µM staurosporine (Enzo Life Sciences, cat #ALX-380-014-C250), in 2% FBS media, for 48 h., 37 °C to initiate apoptosis. Flow cytometry was used to assess Annexin V and propidium iodide (PI) staining (BD Pharmingen cat# 556547). BMDMs were stimulated with 20 µM Z-VAD-FMK (ApexBio, cat #1902), in combination with 50 µg/mL of aggregated human oxidized LDL [12] (Kalen BioMed, cat # 770252-6) in 2% FBS media, for 24 h, 37 °C to obtain NCs. Necroptosis was assessed by PI positivity and increased p-MLKL (Abcam, cat#:ab196436) signal that was sensitive to Nec-1 inhibition (Fig. S10). For some experiments, CD47 (eBioscience, cat #17-0471-82) surface levels were assessed by flow cytometry and quantified with FlowJo software. For experiments carried out in Fig. S8, murine L929 cells [41] were stimulated with 50 ng/mL recombinant murine TNFα (R&D, cat # 410-MT-010) for 24 h. Human aortic or airway smooth muscle cells were stimulated with 20 µM Z-VAD-FMK, in combination with 50 µg/mL of aggregated human oxidized LDL twice over a 48 h period.
Identification and quantification of lipid mediators by LC-MS/MS
Murine lesions were harvested and immediately placed in Eppendorf tubes with 1 mL of ice-cold methanol, followed by mincing. Samples were then stored at −80 °C until lipid mediator extraction was performed. Sample preparation and metabololipidomic analysis was then conducted using methods and instrumentation as described recently in detail [42]. Briefly, deuterium-labeled internal standards (e.g., d5-RvD2 and d4-LXA4) were added to the samples to assess extraction recovery. Samples were then centrifuged and supernatants were collected and subjected to solid phase extraction using C18 cartridges. Lipid mediators were eluted from the column with methyl formate and the collected fraction was immediately taken to dryness using a steady stream of N2 gas. The samples were then resuspended in methanol:water (50:50) before injection and analysis by liquid chromatography-tandem mass spectrometry (LC-MS/MS). Lipid mediators were identified using criteria including retention time, specific multiple reaction monitoring (MRM) transitions and diagnostic MS/MS fragmentation spectra. Quantification of mediators was accomplished using standard curves determined for each mediator with authentic standards following normalization of extraction recovery based on internal deuterium-labeled standards.
Fixed and live-cell confocal microscopy
Imaging for fixed and live-cells was conducted on a Zeiss LSM 880 NLO confocal microscope with Airyscan module (Carl Zeiss Microscopy Jena, Germany) equipped with 63× oil 1.4 numerical aperture (NA) objective lens and collected through a 32-channel GaAsP detector as 0.2 Airy units per channel (Huff, 2015). The microscope has an incubation chamber set to 37 °C and has 5% CO2 for live-cell imaging. Cells were imaged using Argon405 (488 nm) laser and BP 420-480/BP 495–620 filter for GFP; DPSS 561-10 (561 nm) laser and BP 495–550/LP 570 filter for RFP. Z-stack collecting was under Nyquist sampling and with the SR settings. Z-stack images were acquired at an average of 30 slices, with 0.17 μm distance between each slice. Live-cell images were collected every 3 min for the duration of 33-min using the multi-position module through the Zen Black 2 software (Carl Zeiss Microscopy Jena, Germany), which allowed for the collection of up to three separate ROI’s per imaging sequence in each well. Macrophages were pre-incubated with 10 nM RvD1 for 20 min. Live-cell imaging for the Macrophage Engulfment Assay began within 10 min post-addition of apoptotic or necroptotic cells.
For fixed confocal microscopy macrophages were electroporated (details below) with LifeAct-RFP and/or Calreticulin-Emerald and plated onto an eight-well glass bottom coverslip and allowed to adhere for 18–24 h, 37 °C, 5% CO2. Macrophages were then pre-incubated with treatments such as vehicle control, 10 nM of RvD1, 10 μM of RhoA inhibitor (Rhosin, cat # 555460, EMD Millipore), or 800 nM of CDC42 inhibitor (ML 141, cat # SML0407, Sigma–Aldrich). In some experiments, NCs were pre-incubated with 10 μg/mL of IgG or anti-CD47 (BioXCell, cat# MOPC-21 or MIAP410, respectively) for 20 min. ACs or NCs were then added at a 3:1 ratio of dead cells: macrophages for an additional 40 min, after which cells were fixed with 4% PFA for 15 min in room temperature. For some experiments, cells were then blocked in 2% BSA in PBS for 30 min at room temperature. and then stained with either, anti-phospho-MLC (cat# 3675 S, Cell Signaling Technologies), anti-Mouse CD47 APC (clone miap301, 17–0471–82, eBioscience), or anti-Calnexin (sc-11397 Santa Cruz Biotechnology) antibodies for 1 h, at room temperature. Phospho-MLC antibody was suspended in perm wash buffer (BD, cat#51-2091KZ), whereas anti-CD47 or anti-calnexin primary antibody incubations were carried out in PBS. Excess antibodies were gently washed and an Alexafluor-647 secondary antibody was added and incubated for 1 h. Lastly, cells were fixed again with 4% PFA and gently washed with PBS. Imaging of fixed cells was conducted on the Zeiss LSM 880 with Fast Airyscan module as described above. The Zen Black 2 software was utilized for Airyscan processing post-imaging. Image analysis was carried out using the Imaris 9.2.0 software as described below.
Macrophage transfection
BMDMs were suspended in 500,000 cells in 1 ml of PBS and centrifuged for 5 min (1000 rpms, room temperature). Supernatants were removed and 1.5 µg of DNA plasmid (LifeAct-RFP and/or Calreticulin-Emerald) were then added directly to the cell pellet. mTagRFP-T-Lifeact-7 (Addgene plasmid #54586; http://n2t.net/addgene:54586; RRID:Addgene_54586) and mEmerald-Calreticulin-N-16 (Addgene plasmid #54023; http://n2t.net/addgene:54023; RRID:Addgene_54023) were gifts from Michael Davidson’s laboratory. Cells and DNA were resuspended in 40 µl of R-buffer (included in the Neon Invitrogen Electroporation kit). BMDMs and DNA were taken up in a Neon 10 µL pipette. The pipette tips containing the cells and plasmid were then placed in electroporation wells and subjected to 1650mV, 10 millisecond intervals, for 2 pulses. Once the electroporation process was complete the cells were added directly to an eight-well glass bottom coverslip with pre-warmed 20% FBS/DMEM/F12 imaging medium with no phenol red (Gibco) and incubated for 18–24 h, 5% CO2, 37 °C. The following day, macrophages were stimulated as above.
Murine atherosclerosis
Eight to ten-week-old male Ldlr−/− mice on the C57BL/6 J background were purchased from Jackson Laboratory (stock # 002207) and placed on a WD (TD.88137, Harlan Talked). IgG and anti-CD47: Ldlr−/− mice were fed a WD for 10 weeks, after which mice were randomly assigned into three groups, baseline, IgG, or anti-CD47. The baseline group was immediately sacrificed after 10 weeks of WD feeding. The remaining mice were injected with 100 μg/mouse of IgG or anti-CD47 (BioXCell, cat# MOPC-21 or MIAP410, respectively) for 3 weeks (3×/week, i.p.) while still on the WD. For aortic LC-MS/MS, the experiments were carried out as above, with exception to the treatment time in which mice were administered IgG or anti-CD47 (3×/week, i.p.) for only 1 week while still on the WD.
Vehicle and RvD1 treatment: Ldlr−/− mice were fed a WD for 12 weeks, after which mice were randomly assigned into two groups, namely Vehicle or RvD1. Mice in each group were injected were injected i.p. three times per week with 500 μl of sterile PBS (vehicle control) or RvD1 (100 ng/mouse) for an additional 3 weeks, while still on the WD [8]. The Albany Medical College IACUC provided ethical approval for these studies and procedures were conducted in accordance with approved animal protocols.
Aortic roots were harvested for histological analysis and the aorta and BCA were subjected to LC-MS/MS (above). Necrotic core analysis was carried out on H&E-stained lesional cross sections as previously described [8]. Cross sections of murine aortic root lesion were stained at 4 °C with anti-CD47 antibody (BioXCell as above), p-MLKL (Abcam, as above) p-SHP1 (Abcam, cat#: ab131500), or anti-rat F4/80 (BioRad, CI-A3-1). For p-MLKL and F4/80 lesion staining, frozen sections were placed in ice-cold acetone for 5 min. Slides were washed with PBS and lesions were permeabilized for 10 min in room temperature, followed by a PBS wash. Slides were then blocked with 1% BSA and 10% goat serum for 1 h, 4 °C. Blocking reagent was removed and primary antibodies were added in 1% BSA overnight at 4 °C. After 24 h, slides were washed with PBS and secondary antibodies were added in 1% BSA and 5% goat serum for 2 h at room temperature. The sections were washed again and counterstained with Hoechst or DAPI to identify nuclei, viewed on a Leica SPE confocal microscope, and analyzed using ImageJ software.
apoE-Angiotensin model: apoE−/− mice were purchased from Jackson Laboratory (stock #002052) and bred in a pathogen-free environment. Alzet osmotic pumps (Model 2004, Durect Corporation, CA) were implanted into 8-week-old male apoE−/− mice to deliver Angiotensin II (Sigma–Aldrich, 1000 ng/kg/min) subcutaneously and the mice were maintained on a Western diet (101511, Dyets Inc., PA) for 4 weeks. The day before pump implantation, mice were randomly assigned into two groups, IgG or anti-CD47, and received intraperitoneal injections of either 200 μg of mouse IgG isotype control or anti-CD47 antibody (BioXcell, as above) every other day for 4 weeks as previously described in ref. 16.
Image stream flow cytometry
BMDMs were labeled with PKH67, a green fluorescent cell membrane stain (Sigma–Aldrich). After induction of apoptosis or necroptosis, cells were co-cultured in a 1:1 ratio with live, adherent BMDMs for 3 h. at 37 °C, 5% CO2. The BMDMs were then washed with PBS to ensure removal of excess dead cells. BMDMs were then harvested using Cellstripper® (Corning, cat#23-25-056-CI-PK), and suspended in 50 uL of a 5% (vol/vol) FBS in PBS solution, for analysis via ImageStream flow cytometry. ImageStream data were acquired using the Amnis ImageStream Analyzer instrument equipped with the Amnis INSPIRE software. Analysis was performed with the Amnis IDEAS software [43]. Briefly, uptake of NCs and ACs was quantified through a two-step analysis protocol. First, all cells that were out of focus were gated on a histogram of the bright field gradient RMS feature and thus removed from further analysis. Second, single cells were identified based on a dot plot of bright field area versus aspect ratio and events with aspect ratios larger than ~0.6 (AU). Single cells were then plotted on a dot plot of bright field area versus intensity of the green fluorescence. Positive (fluorescently labeled NCs or ACs) and negative (live, unlabeled macrophages) controls were utilized to determine the appropriate gating strategies and ultimately the percentage uptake.
Cytokine and chemokine analysis
Macrophages (0.5 × 106 cells/well in a 12-well plate) were stimulated with 10 nM RvD1 or Veh for 20 mins, 37 °C, 5% CO2. NCs were then added at a 3:1 ratio of dead cells: macrophages for an additional 60 min. After 60 min, free NCs were removed and 400 μL of serum-free media was added for an additional 2 h, Supernatants were then collected and subjected to murine CXCL1 or murine IL-10 ELISA analysis (R&D).
Image analysis
Live-cell macrophage engulfment assay
Imaris 9.2.0 software was used to quantify the volume of apoptotic or necroptotic engulfment for individual macrophages in the presence or absence of RvD1. Using the surface module, macrophage cells were 3D-reconstructed based upon fluorescence intensity, quality of rendering, and area to ensure separation of macrophage and apoptotic/necroptotic populations. The same surface module was used to 3D-reconstruct both apoptotic and necroptotic populations. Following the rendering step, a mask was applied to the macrophage surface, which creates a separate channel based on fluorescence intensity. Next, a filter was applied to the apoptotic or necroptotic 3D-rendered surfaces, which is based on the mean fluorescence intensity of the newly masked macrophage channel. This allowed for the separation of apoptotic and/or necroptotic objects colocalized or not colocalized with the masked macrophage channel. We then created a new surface object from this filter set, which we designated as “Engulfed volume” indicated by the color cyan. Next, we quantified the macrophage to dead cell volume ratio as it changed over time using the Vantage plot module, which is plotted as “Engulfment (AU)”. We also quantified the ingested dead cell volume to obtain the size of the ingested material at 33-min. All vantage plots were exported as Excel spreadsheets and graphs were made using GraphPad Prism version 6.00 software (Windows, GraphPad Software, La Jolla, CA). For visualization purposes, the macrophage volume is colored as gray (at 70% transparency) and the AC or NC volume is represented as magenta. The overlap between the surface of the dead magenta cells and the transparent gray macrophages is depicted as a gradient from light magenta-to-cyan.
Analysis of phagocytic synapses on macrophages
Imaris 9.2.0 software was used to quantify the LifeAct-RFP volume of the phagocytic synapse, which has an arm- or mouth-like appearance. Macrophage synapses were 3D-rendered based on fluorescence intensity of LifeAct-RFP. Once all structures were analyzed, a collection vantage plot was utilized to batch and compare all individual structures. The vantage plot setting was graphed in a gallery view for visualization purposes of each structure type organized by volume and sphericity. The volumes for each structure are color coordinated as indicated by the spectral scale whereby the color purple = ~5 µm3 and red = ~650 µm3. The volumes for each individual structure were exported as an Excel spreadsheet and statistics and graphing were applied using GraphPad to compare the change in volume between treatments.
Fixed-cell macrophage phospho-MLC analysis
Macrophages were electroporated with LifeAct-RFP and incubated with NCs as above. Phospho-MLC on macrophages was 3D-rendered as a surface based on its fluorescence intensity. The mean fluorescence intensity of each surface on the macrophage with or without treatment was then exported into an Excel spreadsheet. Graphing and statistical analysis were completed using GraphPad.
Fixed-cell macrophage calnexin release analysis
Macrophages were electroporated with LifeAct-RFP and incubated with NCs as above. Images were analyzed with Imaris 9.2.0 software to compare the fluorescence intensity of Calnexin staining on both the macrophage and necroptotic cells with or without RvD1 stimulation. Calnexin on the macrophage was rendered as a surface based on its fluorescence intensity. The mean fluorescence intensity of each surface on the macrophage was exported into an Excel spreadsheet. Graphing and statistical analysis were completed using GraphPad. In order to quantify the released Calnexin that was stained on the necroptotic cells, necroptotic cells were 3D-rendered as a surface based on the PKH67 (green) staining. Next, Calnexin was rendered as a surface with the same algorithm used to quantify the Calnexin on the macrophage. To separate the Calnexin on the NC from that of the macrophage, a masked channel was created from the 3D-rendered NCs. A filter was applied on the Calnexin that was overlapping the fluorescence intensity of the masked channel of the NCs, thus separating the Calnexin on the necroptotic cells from the Calnexin on the macrophage. Then a new surface was created for the Calnexin on the necroptotic cells and the mean fluorescence intensity of released Calnexin was exported to an Excel spreadsheet and graphing and statistics were completed using GraphPad.
Fixed-cell macrophage calreticulin release analysis
Macrophages were electroporated with LifeAct-RFP and Calreticulin-Emerald and incubated with NCs as above. Images were analyzed with Imaris 9.2.0 software to compare the fluorescence intensity of Calreticulin staining on the necroptotic cells with or without RvD1 treatment. 3D-rendering and quantification methods followed the same protocol as calnexin analysis above.
Statistical analysis
All experiments were repeated at least three times and the statistical significance was evaluated. Results are shown as mean ± sem, and statistical differences were determined using the two-tailed Student’s t-test, one-way ANOVA or two-way ANOVA depending on the appropriate contexts. Prism (GraphPad Inc., La Jolla, CA) software was used and a p < 0.05 was considered statistically significant. Details can be found in the figure legends.
Supplementary information
Acknowledgements
We thank the AMC imaging core facility for the use of the Zeiss LSM880 confocal microscope. IMARIS software by Bitplane Inc was purchased using funding from NSF #1725984.
Funding
This work was supported by NIH grants HL119587 (G.F.), HL141127 (G.F.), HL119047 (K.J.R), HL106173 (M.S.), GM095467 (M.S.; Core B), and BRG-CA207725 (M.B.), NIH R35 HL144475 (N.J.L.), HL110951 (D.D.T), HL113208 (D.D.T), and HL130304 (D.D.T). BES was supported by a National Research Service Award from the NIH (HL136044), and KJR by the Canadian Institutes for Health Research.
Author contributions
GF and KJR designed research. GF, BDG, MM, and MB conducted and analyzed in vitro imaging experiments. GF, JH, NR, SS, ZH, YK, and NJL conducted and analyzed atherosclerosis experiments. BES, COR, and MS performed LC-MS/MS analysis. GF, BDG, and MM wrote the paper and all authors including DDT reviewed/edited the manuscript.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Edited by S. Nagata
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Brennan D. Gerlach, Michael Marinello
Supplementary information
The online version of this article (10.1038/s41418-019-0370-1) contains supplementary material, which is available to authorized users.
References
- 1.Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature. 2014;510:92–101. doi: 10.1038/nature13479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Spite M, Norling LV, Summers L, Yang R, Cooper D, Petasis NA, et al. Resolvin D2 is a potent regulator of leukocytes and controls microbial sepsis. Nature. 2009;461:1287–91. doi: 10.1038/nature08541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chiang N, Fredman G, Backhed F, Oh SF, Vickery T, Schmidt BA, et al. Infection regulates pro-resolving mediators that lower antibiotic requirements. Nature. 2012;484:524–8. doi: 10.1038/nature11042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Godson C, Mitchell S, Harvey K, Petasis NA, Hogg N, Brady HR. Cutting edge: lipoxins rapidly stimulate nonphlogistic phagocytosis of apoptotic neutrophils by monocyte-derived macrophages. J Immunol. 2000;164:1663–7. doi: 10.4049/jimmunol.164.4.1663. [DOI] [PubMed] [Google Scholar]
- 5.Schwab JM, Chiang N, Arita M, Serhan CN. Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature. 2007;447:869–74. doi: 10.1038/nature05877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cai B, Thorp EB, Doran AC, Subramanian M, Sansbury BE, Lin CS, et al. MerTK cleavage limits proresolving mediator biosynthesis and exacerbates tissue inflammation. Proc Natl Acad Sci USA. 2016;113:6526–31. doi: 10.1073/pnas.1524292113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tabas I, Bornfeldt KE. Macrophage phenotype and function in different stages of atherosclerosis. Circ Res. 2016;118:653–67. doi: 10.1161/CIRCRESAHA.115.306256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fredman G, Hellmann J, Proto JD, Kuriakose G, Colas RA, Dorweiler B, et al. An imbalance between specialized pro-resolving lipid mediators and pro-inflammatory leukotrienes promotes instability of atherosclerotic plaques. Nat Commun. 2016;7:12859. doi: 10.1038/ncomms12859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tabas I. Macrophage death and defective inflammation resolution in atherosclerosis. Nat Rev Immunol. 2010;10:36–46. doi: 10.1038/nri2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin J, Li H, Yang M, Ren J, Huang Z, Han F, et al. A role of RIP3-mediated macrophage necrosis in atherosclerosis development. Cell Rep. 2013;3:200–10. doi: 10.1016/j.celrep.2012.12.012. [DOI] [PubMed] [Google Scholar]
- 11.Meng L, Jin W, Wang X. RIP3-mediated necrotic cell death accelerates systematic inflammation and mortality. Proc Natl Acad Sci USA. 2015;112:11007–12. doi: 10.1073/pnas.1514730112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karunakaran D, Geoffrion M, Wei L, Gan W, Richards L, Shangari P, et al. Targeting macrophage necroptosis for therapeutic and diagnostic interventions in atherosclerosis. Sci Adv. 2016;2:e1600224. doi: 10.1126/sciadv.1600224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wallach D, Kang TB, Dillon CP, Green DR. Programmed necrosis in inflammation: Toward identification of the effector molecules. Science. 2016;352:aaf2154. doi: 10.1126/science.aaf2154. [DOI] [PubMed] [Google Scholar]
- 14.Enyedi B, Jelcic M, Niethammer P. The Cell nucleus serves as a mechanotransducer of tissue damage-induced inflammation. Cell. 2016;165:1160–70. doi: 10.1016/j.cell.2016.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Krysko DV, Denecker G, Festjens N, Gabriels S, Parthoens E, D'Herde K, et al. Macrophages use different internalization mechanisms to clear apoptotic and necrotic cells. Cell Death Differ. 2006;13:2011–22. doi: 10.1038/sj.cdd.4401900. [DOI] [PubMed] [Google Scholar]
- 16.Kojima Y, Volkmer JP, McKenna K, Civelek M, Lusis AJ, Miller CL, et al. CD47-blocking antibodies restore phagocytosis and prevent atherosclerosis. Nature. 2016;536:86–90. doi: 10.1038/nature18935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nakaya M, Tanaka M, Okabe Y, Hanayama R, Nagata S. Opposite effects of rho family GTPases on engulfment of apoptotic cells by macrophages. J Biol Chem. 2006;281:8836–42. doi: 10.1074/jbc.M510972200. [DOI] [PubMed] [Google Scholar]
- 18.Zhang S, Yeap XY, DeBerge M, Naresh NK, Wang K, Jiang Z, et al. Acute CD47 blockade during ischemic myocardial reperfusion enhances phagocytosis-associated cardiac repair. JACC Basic Transl Sci. 2017;2:386–97. doi: 10.1016/j.jacbts.2017.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chao MP, Weissman IL, Majeti R. The CD47-SIRPalpha pathway in cancer immune evasion and potential therapeutic implications. Curr Opin Immunol. 2012;24:225–32. doi: 10.1016/j.coi.2012.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reville K, Crean JK, Vivers S, Dransfield I, Godson C. Lipoxin A4 redistributes myosin IIA and Cdc42 in macrophages: implications for phagocytosis of apoptotic leukocytes. J Immunol. 2006;176:1878–88. doi: 10.4049/jimmunol.176.3.1878. [DOI] [PubMed] [Google Scholar]
- 21.Gardai SJ, McPhillips KA, Frasch SC, Janssen WJ, Starefeldt A, Murphy-Ullrich JE, et al. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell. 2005;123:321–34. doi: 10.1016/j.cell.2005.08.032. [DOI] [PubMed] [Google Scholar]
- 22.Feng M, Chen JY, Weissman-Tsukamoto R, Volkmer JP, Ho PY, McKenna KM, et al. Macrophages eat cancer cells using their own calreticulin as a guide: roles of TLR and Btk. Proc Natl Acad Sci USA. 2015;112:2145–50. doi: 10.1073/pnas.1424907112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Feng M, Marjon KD, Zhu F, Weissman-Tsukamoto R, Levett A, Sullivan K, et al. Programmed cell removal by calreticulin in tissue homeostasis and cancer. Nat Commun. 2018;9:3194. doi: 10.1038/s41467-018-05211-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gurel PS, Hatch AL, Higgs HN. Connecting the cytoskeleton to the endoplasmic reticulum and Golgi. Curr Biol. 2014;24:R660–R72. doi: 10.1016/j.cub.2014.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brouckaert G, Kalai M, Krysko DV, Saelens X, Vercammen D, Ndlovu MN, et al. Phagocytosis of necrotic cells by macrophages is phosphatidylserine dependent and does not induce inflammatory cytokine production. Mol Biol Cell. 2004;15:1089–100. doi: 10.1091/mbc.E03-09-0668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krysko DV, Vanden Berghe T, Parthoens E, D'Herde K, Vandenabeele P. Methods for distinguishing apoptotic from necrotic cells and measuring their clearance. Methods Enzymol. 2008;442:307–41. doi: 10.1016/S0076-6879(08)01416-X. [DOI] [PubMed] [Google Scholar]
- 27.Ralston KS, Solga MD, Mackey-Lawrence NM, Somlata, Bhattacharya A, Petri WA., Jr. Trogocytosis by Entamoeba histolytica contributes to cell killing and tissue invasion. Nature. 2014;508:526–30. doi: 10.1038/nature13242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Valgardsdottir R, Cattaneo I, Klein C, Introna M, Figliuzzi M, Golay J. Human neutrophils mediate trogocytosis rather than phagocytosis of CLL B cells opsonized with anti-CD20 antibodies. Blood. 2017;129:2636–44. doi: 10.1182/blood-2016-08-735605. [DOI] [PubMed] [Google Scholar]
- 29.Velmurugan R, Challa DK, Ram S, Ober RJ, Ward ES. Macrophage-mediated trogocytosis leads to death of antibody-opsonized tumor cells. Mol Cancer Ther. 2016;15:1879–89. doi: 10.1158/1535-7163.MCT-15-0335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pham T, Mero P, Booth JW. Dynamics of macrophage trogocytosis of rituximab-coated B cells. PLoS One. 2011;6:e14498. doi: 10.1371/journal.pone.0014498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sarvari AK, Doan-Xuan QM, Bacso Z, Csomos I, Balajthy Z, Fesus L. Interaction of differentiated human adipocytes with macrophages leads to trogocytosis and selective IL-6 secretion. Cell Death Dis. 2015;6:e1613. doi: 10.1038/cddis.2014.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weinhard L, di Bartolomei G, Bolasco G, Machado P, Schieber NL, Neniskyte U, et al. Microglia remodel synapses by presynaptic trogocytosis and spine head filopodia induction. Nat Commun. 2018;9:1228. doi: 10.1038/s41467-018-03566-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lehrman EK, Wilton DK, Litvina EY, Welsh CA, Chang ST, Frouin A, et al. CD47 protects synapses from excess microglia-mediated pruning during development. Neuron. 2018;100:120–34 e6. doi: 10.1016/j.neuron.2018.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scannell M, Flanagan MB, deStefani A, Wynne KJ, Cagney G, Godson C, et al. Annexin-1 and peptide derivatives are released by apoptotic cells and stimulate phagocytosis of apoptotic neutrophils by macrophages. J Immunol. 2007;178:4595–605. doi: 10.4049/jimmunol.178.7.4595. [DOI] [PubMed] [Google Scholar]
- 35.Dalli J, Serhan CN. Specific lipid mediator signatures of human phagocytes: microparticles stimulate macrophage efferocytosis and pro-resolving mediators. Blood. 2012;120:e60–72. doi: 10.1182/blood-2012-04-423525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gagnon E, Duclos S, Rondeau C, Chevet E, Cameron PH, Steele-Mortimer O, et al. Endoplasmic reticulum-mediated phagocytosis is a mechanism of entry into macrophages. Cell. 2002;110:119–31. doi: 10.1016/s0092-8674(02)00797-3. [DOI] [PubMed] [Google Scholar]
- 37.Becker T, Volchuk A, Rothman JE. Differential use of endoplasmic reticulum membrane for phagocytosis in J774 macrophages. Proc Natl Acad Sci USA. 2005;102:4022–6. doi: 10.1073/pnas.0409219102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Viola JR, Lemnitzer P, Jansen Y, Csaba G, Winter C, Neideck C, et al. Resolving lipid mediators maresin 1 and resolvin D2 prevent atheroprogression in mice. Circ Res. 2016;119:1030–8. doi: 10.1161/CIRCRESAHA.116.309492. [DOI] [PubMed] [Google Scholar]
- 39.Petri MH, Laguna-Fernandez A, Arnardottir H, Wheelock CE, Perretti M, Hansson GK, et al. Aspirin-triggered lipoxin inhibits atherosclerosis progression in ApoE−/− mice. Br J Pharmacol. 2017;174:4043–54. [DOI] [PMC free article] [PubMed]
- 40.Kaczmarek A, Vandenabeele P, Krysko DV. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity. 2013;38:209–23. doi: 10.1016/j.immuni.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 41.Robinson N, McComb S, Mulligan R, Dudani R, Krishnan L, Sad S. Type I interferon induces necroptosis in macrophages during infection with Salmonella enterica serovar Typhimurium. Nat Immunol. 2012;13:954–62. doi: 10.1038/ni.2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dalli J, Colas RA, Walker ME, Serhan CN. Lipid mediator metabolomics Via LC-MS/MS profiling and analysis. Methods Mol Biol. 2018;1730:59–72. doi: 10.1007/978-1-4939-7592-1_4. [DOI] [PubMed] [Google Scholar]
- 43.Fei C, Lillico DME, Hall B, Rieger AM, Stafford JL. Connected component masking accurately identifies the ratio of phagocytosed and cell-bound particles in individual cells by imaging flow cytometry. Cytom A. 2017;91:372–81. doi: 10.1002/cyto.a.23050. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.