

Abstract
Epstein–Barr virus (EBV), which is ubiquitous in the adult population, is causally associated with human malignancies. Like many infectious agents, EBV has evolved strategies to block host cell death, including through expression of viral homologues of cellular BCL-2 pro-survival proteins (vBCL-2s), such as BHRF1. Small molecule inhibitors of the cellular pro-survival BCL-2 family proteins, termed ‘BH3-mimetics’, have entered clinical trials for blood cancers with the BCL-2 inhibitor venetoclax already approved for treatment of therapy refractory chronic lymphocytic leukaemia and acute myeloid leukaemia in the elderly. The generation of BH3-mimetics that could specifically target vBCL-2 proteins may be an attractive therapeutic option for virus-associated cancers, since these drugs would be expected to only kill virally infected cells with only minimal side effects on normal healthy tissues. To achieve this, a better understanding of the contribution of vBCL-2 proteins to tumorigenesis and insights into their biochemical functions is needed. In the context of Burkitt lymphoma (BL), BHRF1 expression conferred strong resistance to diverse apoptotic stimuli. Furthermore, BHRF1 expression in mouse haematopoietic stem and progenitor cells accelerated MYC-induced lymphoma development in a model of BL. BHRF1 interacts with the cellular pro-apoptotic BCL-2 proteins, BIM, BID, PUMA and BAK, but its capability to inhibit apoptosis could not be mapped solely to one of these interactions, suggesting plasticity is a key feature of BHRF1. Site-directed mutagenesis revealed a site in BHRF1 that was critical for its interaction with PUMA and blocking DNA-damage-induced apoptosis, identifying a potentially therapeutically targetable vulnerability in BHRF1.
Subject terms: Cancer models, Experimental models of disease
Introduction
The intrinsic/mitochondrial apoptotic pathway is governed by interactions between pro-apoptotic and pro-survival members of the BCL-2 family of proteins. This pathway culminates in permeabilisation of the mitochondrial outer membrane leading to caspase activation and subsequent cellular demolition. Pro-apoptotic BCL-2 family members are subdivided into the BH3-only proteins (BIM, PUMA, NOXA, BID, BMF, BIK, BAD, HRK) and the multi-BH domain apoptosis effectors (BAX, BAK and BOK). The pro-survival BCL-2 proteins include BCL-2, BCL-XL, BCL-W, MCL-1 and A1/BFL1. The relative expression and interactions of pro-survival with pro-apoptotic BCL-2 family proteins determines the fate of the cell [1].
Viruses have evolved mechanisms to manipulate cell death to alter the fate of infected host cells, which is particularly relevant for oncogenic viruses, including the Epstein–Barr virus (EBV) [2]. EBV is a human gamma herpesvirus that establishes a life-long asymptomatic infection in B lymphocytes of >90% of adult humans [3], but it is also associated with >200,000 cancers annually. EBV-associated tumours are frequently of B lymphoid origin, reflecting the natural reservoir for EBV, but may also be of epithelial or T/NK cell origin [4].
EBV-associated Burkitt lymphoma (BL) is a frequent childhood B lymphoid cancer in sub-Saharan Africa [4]. Its defining characteristic is deregulated expression of c-MYC due to a reciprocal chromosomal translocation between the c-MYC gene and an immunoglobulin (Ig) locus [5]. The association of EBV with BL varies geographically; in areas of high incidence (i.e. endemic disease) almost all tumours are EBV-positive, whereas in sporadic cases of BL the presence of EBV varies geographically (~15–85% of tumours) [3]. Viral gene expression in BL is restricted and the role of viral genes in BL is not fully resolved, although the consensus is that they contribute to overcoming the pro-apoptotic effects of deregulated c-MYC expression [6].
EBV can growth-transform resting B cells in vitro. Such EBV-transformed B cell lines express nine viral proteins (EBNAs1, 2, LP, 3A, 3B, 3C and LMPs 1, 2A, 2B) and various non-coding RNAs (EBERs, BARTs); a program termed Latency III. Initiation of transformation appears to additionally require transient expression of two vBCL-2 proteins [7], termed BHRF1 and BALF1, that share sequence and structural homology with cellular BCL-2 proteins [8–10]. BHRF1 is widely reported to inhibit apoptosis [11–14] but the role of BALF1 is unclear, with contradictory reports of pro-survival or, conversely, pro-death functions [9, 10, 15].
In most EBV-associated BLs, the virus assumes a Latency I pattern of infection that involves expression of EBNA1, the EBERs and BARTs only [16]. Latency I infection provides low level protection from apoptosis by blocking upregulation of BIM and PUMA [6, 17, 18]. In up to 15% of BLs, EBV exhibits a Wp-restricted latency characterised by selective expression of EBNA1, EBNA-LP, EBNA3A, 3B, 3C, together with BHRF1 and the non-coding EBERs and BARTs [19, 20]. Wp-restricted latency confers stronger protection from apoptosis, likely through modulation of pro-apoptotic BIM expression by the EBNA3 proteins [21] and also potently through expression of BHRF1 [20]. Whilst the chemoresistance conferred by BHRF1 is likely mediated through engagement with cellular BCL-2 family proteins, the key specific interactions are not established [20]. Furthermore, the implications of these interactions for the early pathogenesis of EBV-associated MYC-driven lymphomas are unknown.
BH3-mimetic drugs that bind to cellular pro-survival BCL-2 proteins and trigger tumour cells to undergo apoptosis have recently been developed as cancer therapeutics [22–25]. The most clinically advanced BH3-mimetic, venetoclax that binds specifically to BCL-2, has shown significant efficacy and tolerability in treatment refractory chronic lymphocytic leukaemia patients [26]. BH3-mimetics targeting vBCL-2 proteins would be anticipated to be well-tolerated since only virus-infected cells would be killed. To develop a BH3-mimetic targeting BHRF1, a clearer understanding is needed of the impact of BHRF1 expression in early pre-malignant cells and of the interactions of BHRF1 with the cellular apoptotic machinery that confer chemoresistance. We have addressed these progress-limiting issues in the context of MYC-driven lymphomas.
Methods and materials
Animal work
All work involving mice was conducted according to the guidelines of The Walter and Eliza Hall Institute of Medical Research Animal Ethics Committee. Mice were housed in a pathogen-free facility, cared for by trained animal technicians. The Eµ-Myc, PumaKO, BimKO and BakKO mice are maintained on a C57BL/6-Ly5.2+ background [27–32]. The impact of BHRF1 (mutated and wild-type (WT)) expression on MYC-induced tumorigenesis was examined using a haematopoietic stem and progenitor cells (HSPCs) reconstitution model as described before [33] and in the Supplementary Information. A sample size of seven (three male and four female) 7.5–8.5-week-old C57BL/6-Ly5.1+ mice for each condition/mutant was used and mice were randomised to the groups. The animal technicians assessing the welfare of the mice were blinded to the expected outcomes.
Cell lines
Human BL and mouse Eµ-Myc lymphoma-derived cell lines were cultured as described [6, 20, 29, 34]. HEK293T and 293T cells were cultured in DMEM with 10% HI FCS in a humidified incubator at 37 °C, 5% CO2. All cell lines were routinely tested for mycoplasma.
Retroviruses, lentiviruses and infection protocols
Schematics of pMIG retroviral and dox-inducible F-UTG lentiviral vectors [35] encoding WT or mutant BHRF1 are depicted in Supplementary Fig. 1. Virus-containing supernatants were produced by transient transfection of HEK293T or 293T cells with the expression constructs of interest alongside packaging plasmids; VSV-G, PAX2 (for lentiviral constructs) and ENV, GAG or VSV-G (for the retroviral constructs), using Lipofectamine 2000 or the standard CaPO4 method [35]. Lymphoma-derived cell lines were spin infected [33].
TP53 gene sequencing
DNA was extracted from suspensions of lymphoma cells using the DNEasy Tissue kit (Qiagen) according to the manufacturer’s instructions. Exons 4–11 encoding the DNA-binding domain of the mouse TP53 gene were amplified using GoTaq (Promega) as described in Supplementary Information with the primers detailed in Supplementary Table 1 and were sequenced on a MiSEQ analyser.
Co-immunoprecipitation (Co-IP)
Cells were lysed in 1% Triton X-100 ONYX buffer (20 mM Tris-HCl pH 7.4, 135 mM NaCl, 1.5 mM MgCl2, 1 mM EDTA, 10% glycerol) or 1% CHAPS ONYX buffer with complete protease inhibitor cocktail (Roche) on ice and anti-FLAG immunoprecipitation performed as described [36] and detailed in Supplementary Information.
Western blotting
RIPA buffer containing complete protease inhibitor cocktail (Roche) was used to extract proteins from lymphoma cells. Proteins were separated by SDS-PAGE using MES buffer and transferred to nitrocellulose membranes. Size markers were Precision Plus Protein Standards (Bio-Rad). Membranes were blocked in phosphate buffered saline containing 0.1% Tween 20 and 5% skimmed milk, and protein detected using the enhanced chemiluminescence reagent (Luminata Forte Western blot HRP substrate, Millipore WBLUF0500) with the Bio-Rad Chemidoc XRS+ using ImageLab v5.2 software (Bio-Rad, Hercules, CA).
Cell death assays
Lymphoma cells were plated at 3 × 104 cells per well of flat-bottomed 96-well plates. Etoposide (Sigma), ionomycin (Sigma), anti-IgM Fab2 antibody fragments (MP Biomedicals), Roscovitine and Staurosporine (both Cell Signalling Technologies) were added at the concentrations indicated in the figures. Cells were harvested and dead cells stained with 2.5 µg/mL propidium iodide (PI) (Sigma). GFP and PI were detected using a FACS Calibur. Relative cell viability was determined by comparison of drug treated to DMSO treated cells. Assays were performed in technical triplicates and the experiments carried out three to five times. The results shown are the mean and standard deviations of at least three independent experiments. Experiments with mouse cells were deemed suitable for inclusion if >70% death was induced in control pMIG cells.
Cell surface phenotyping
Lymphoma cells were stained with fluorochrome-conjugated antibodies as detailed in the Supplementary information. Cells were analysed in a LSRIIW FACS machine using PI, Ly5.2 staining and forward/side scatter to identify live lymphoma cells.
Statistical analysis
GraphPad Prism 7.0 software was used for statistical analyses. An unpaired two-tailed Student’s t test was carried out to compare the death induced in Wp-restricted BLs to Latency I BLs in Fig. 1. Unpaired two-tailed Student’s t tests were carried out to compare the viability of cells expressing mutant BHRF1 proteins or empty control virus to cells expressing WT BHRF1 (Figs. 3–5). For these data sets, we applied the Bonferroni adjustment to account for the multiple comparisons being performed where each of the cell lines expressing mutant BHRF1 or carrying the empty control virus were compared with cells expressing WT BHRF1 (this equated to a factor of 6 for each of the bars presented in the graphs in Figs. 3–5). Survival comparisons of mouse cohorts were calculated using log-rank (Mantel–Cox) analysis. Differences were considered significant where the p value fell below 0.05 and were classified as follows: *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001, p > 0.05 ns (not significant).
Fig. 1.

A subset of human BLs harbour EBV in a form of viral latency that involves expression of BHRF1 and these BLs are protected from diverse apoptotic stimuli. a Western blotting for EBV latent proteins EBNA1, EBNA2, LMP1, BHRF1, lytic protein BZLF1 and the cellular protein ACTIN (loading control) in a panel of representative BL derived cell lines that have different associations with EBV. Included are EBV-negative BLs (BL41 and BL2), Latency I BLs (Eze-BL and Sav-BL) that express EBNA1 protein only, and Wp-restricted BLs (Oku-BL and Sal-BL) that express EBNA1 and BHRF1 (and EBNA3A, 3B, 3C and truncated EBNA-LP, data not shown). As a control for western blotting, an LCL (X50-7) was included that expresses a Latency III pattern of infection, involving expression of EBNA1, EBNA2, low BHRF1 and LMP1 (plus additionally EBNAs 3A, 3B, 3C and -LP and LMP2s, data not shown). Lytic cycle-induced Akata-BL (lytic Akata-BL) cells were included as a control for cells undergoing viral replication and expressing high levels of the immediate early lytic antigen, BZLF1, and the early antigen BHRF1. b BL cell lines, Oku-BL and Sal-BL cells, that carry EBV in a Wp-restricted latency, involving expression of BHRF1, were significantly more protected from death induced by ionomycin (1 µg/mL) and anti-IgM Fab2 antibody fragments (5 µg/mL), both analysed after 48 h of treatment, compared with the Latency I BL cells, Eze-BL and Sav-BL. Cell death was determined by flow cytometry after staining with Annexin V and propidium iodide (PI) and cell death induction was calculated by comparing to control, untreated cells. Data are shown as the mean and standard deviation of three independent experiments. Unpaired two-tailed t-tests were performed to assess the significance of the differences between cells carrying EBV in Wp-restricted latency and Latency I patterns of infection (****p ≤ 0.0001)
Fig. 3.
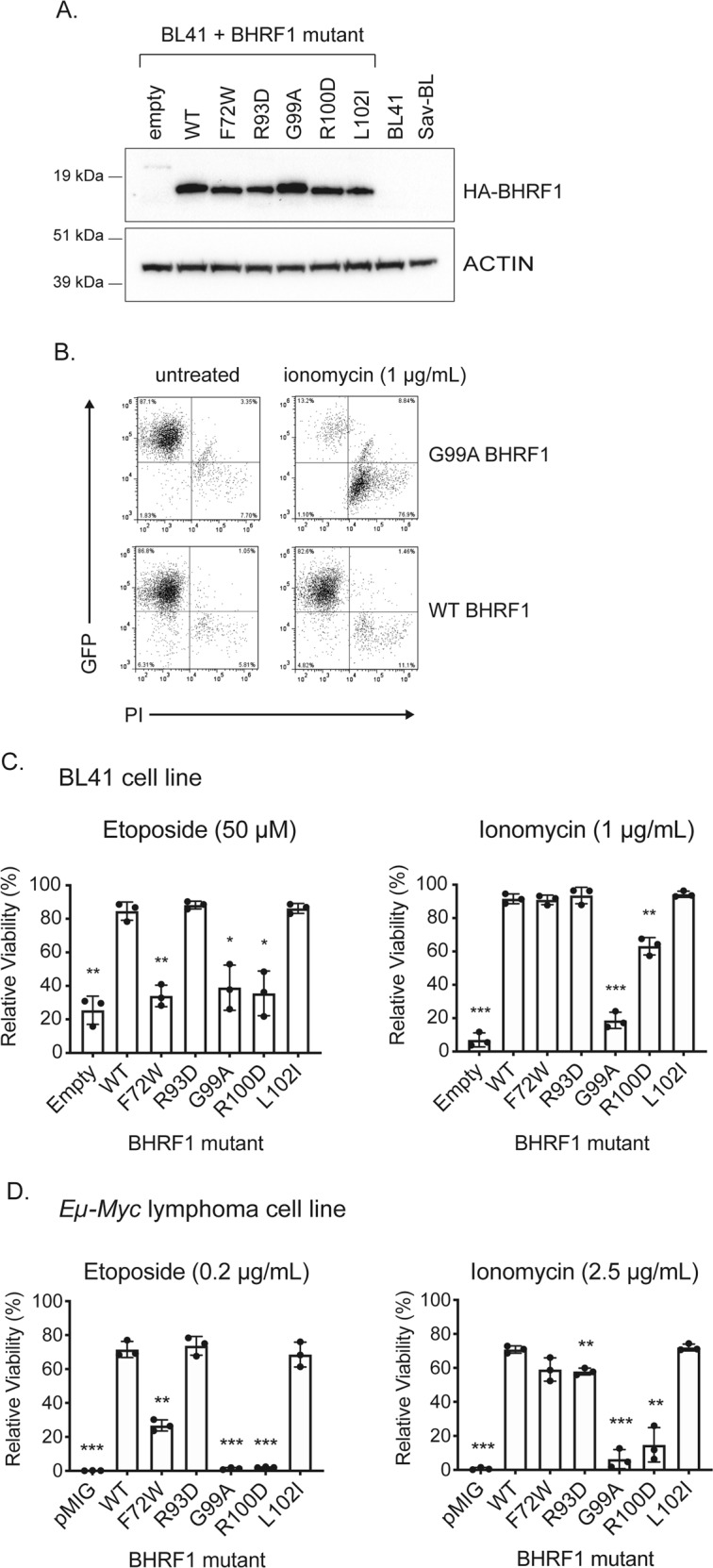
Analysis of mutant BHRF1 proteins in human BL cells and mouse Eµ-Myc lymphoma cell lines. a Western blots showing expression of HA-tagged WT or mutant BHRF1 proteins expressed from a dox-inducible lentiviral vector in EBV-negative BL41 cells. Probing for ACTIN was used as a loading control. b Representative FACS plots showing staining for GFP (expressed from the lentiviral vector that also encodes the WT or mutant BHRF1 proteins) and PI in BL41 cells expressing either HA-tagged WT BHRF1 or the loss-of-function mutant G99A BHRF1 protein, before and after treatment with ionomycin (1 µg/mL, analysed at 48 h). c Viability of BL41 cells expressing HA-tagged WT or mutant BHRF1 proteins following treatment with etoposide (50 µM) for 24 h or ionomycin (1 µg/mL) for 48 h. Results are expressed relative to untreated cells and represent the mean and standard deviation of triplicate experiments. d Viability of murine Eµ-Myc lymphoma cell lines expressing FLAG-tagged WT or mutant BHRF1 proteins constitutively from a retrovirus following 24 h treatment with etoposide (0.2 µg/mL) or ionomycin (2.5 µg/mL). Results are expressed relative to untreated cells and represent the mean and standard deviation of triplicate experiments. For c and d, unpaired two-tailed t-tests were performed and a Bonferroni adjustment of 6 applied to calculate the significance of the observed differences between cells expressing mutant forms of BHRF1 or negative controls (empty vector transduced cells) compared with those expressing WT BHRF1 protein; *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001
Fig. 5.

Analysis of WT and mutant BHRF1 proteins in mouse Eµ-Myc lymphoma cell lines deficient in key interacting pro-apoptotic BCL-2 family proteins. a Viability of mouse Eµ-Myc;BakKO lymphoma cells expressing FLAG-tagged WT or mutant BHRF1 proteins from a constitutively active retrovirus after treatment with etoposide (0.2 µg/mL) or ionomycin (2.5 µg/mL) for 24 h. Results are expressed relative to untreated cells and represent the mean and standard deviation of triplicate experiments or four experiments for ionomycin treatment of Eµ-Myc;BakKO variants. b Viability of mouse Eµ-Myc;PumaKO lymphoma cells expressing FLAG-tagged WT or mutant BHRF1 proteins from a constitutively active retrovirus after treatment with etoposide (0.2 µg/mL) or ionomycin (2.5 µg/mL) for 24 h. Results are expressed relative to untreated cells and represent the mean and standard deviation of triplicate experiments. Unpaired two-tailed t-tests were performed and a Bonferroni adjustment of 6 applied to calculate the significance of the observed differences in a and b data sets between cells expressing mutant BHRF1 proteins or empty vector (pMIG) compared with cells expressing WT BHRF1; *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001. c Western blots showing expression of the cellular pro-apoptotic BH3-only proteins BIM, BID and PUMA in co-immunoprecipitations with FLAG-tagged WT or mutant BHRF1 proteins (left panels) and whole cell inputs (right panels). Probing for FLAG-tagged BHRF1 served as a control for the co-immunoprecipitation samples (left) and probing for FLAG-tagged BHRF1 and HSP70 served as loading controls for the input samples (right)
Recombinant protein expression and isometric thermal calorimetry
Mutants of BHRF1∆C31 were generated by strand-overlap PCR and cloned into pET DUET using BamHI and EcoRI. WT and mutant BHRF1∆C31 proteins were expressed, purified and examined using isothermal titration calorimetry with human BH3 domain peptides as previously described [37, 38] and detailed further in the Supplementary information.
Results
BLs expressing BHRF1 are resistant to cytotoxic stimuli that induce apoptosis
Different associations with EBV are observed in the setting of BL. The majority of EBV-associated BLs display a Latency I gene expression pattern, characterised by EBNA1 protein expression (Fig. 1a) and the EBER and BART transcripts [16]. Some BLs, called Wp-restricted BLs, express a broader panel of latent proteins, including EBNA1, EBNA3A, 3B, 3C and BHRF1 in the absence of the oncoproteins EBNA2 and LMP1 because they carry an EBNA2-deleted virus. Latent BHRF1 is expressed at comparatively low levels in Wp-restricted BLs compared with the levels observed in cells undergoing lytic replication, which additionally co-express the lytic antigen BZLF1 (demonstrated using induced Akata-BL in Fig. 1a) [19, 20, 39]. The low levels of BHRF1 found in Wp-restricted BLs are sufficient to confer significant protection from apoptotic stimuli [20]. As shown in Fig. 1b, less cell death was induced in two representative Wp-restricted BL cell lines compared with two Latency I BL cell lines following exposure to the calcium ionophore, ionomycin and surface Ig cross linking.
Structural insights into BHRF1
Previously, the binding affinity of C-terminally truncated BHRF1 for BH3 domains of human cellular pro-apoptotic BCL-2 family proteins was examined by ITC [37]. BIM-BH3 was the strongest BHRF1 interactor (18 nM), followed by the BH3 domains from PUMA, BID and BAK (70, 109 and 150 nM, respectively) and a weak interaction with the BAX-BH3 domain (1400 nM) was also observed. Using the crystal structure of the BHRF1:BAK-BH3 peptide complex [37], key amino acid (aa) residues located in the hydrophobic binding groove of BHRF1 that mediate binding to BH3 domains were mutated to generate a panel of 5 mutants (F72W, R93D, G99A, R100D and L102I, Fig. 2) (denoted as WT aa-position-MUT aa). The aim was to generate BHRF1 mutants with more restricted interaction profiles than WT BHRF1 for functional analyses in whole cells. The affinities of the mutant BHRF1 proteins to BH3 domains from human BIM, PUMA, BID, BAK and BAX were quantitated by ITC (Supplementary Fig. 2). Compared with WT BHRF1, the G99A, R100D and F72W BHRF1 mutants had reduced binding, whereas others had increased binding to specific cellular pro-apoptotic proteins (e.g. R93D to BIM, L102I to BIM, PUMA and BID). For some BHRF1 mutants, no binding (denoted NB in Supplementary Fig. 2) to certain cellular BH3 domains was detectable.
Fig. 2.

Structural insights into BHRF1. Structure of BHRF1 protein bound to the BH3 peptide of human BIM as determined by X-ray diffraction (Protein Data Bank Accession number 2WH6). Amino acid residues selected for mutation are highlighted in colour on the grey protein surface and labelled in black
Identifying loss-of-function BHRF1 mutants
To determine the impact of these mutations on BHRF1 function, epitope-tagged WT and mutant forms of BHRF1 were cloned into dox-inducible lentiviral vectors (FTrex-UTG or FT-UTG) (HA-tagged) and constitutively active retroviral vectors (pMIG) (FLAG-tagged) (Supplementary Fig. 1), both of which constitutively co-express GFP. EBV-negative human BL41 cells were infected with the lentiviral expression constructs or a control empty vector and cell lines generated in which the WT or mutant BHRF1 proteins were expressed at comparable levels (see Fig. 3a for HA-BHRF1 protein and Supplementary Fig. 3 for GFP levels).
Virus-transduced BL41 cells were treated with diverse apoptosis-inducing agents and their viability measured. Representative FACS plots are shown in Fig. 3b. WT BHRF1-expressing BL41 cells remained highly viable (>90% GFP-positive/PI-negative), whereas the viability of cells expressing the G99A mutant BHRF1 protein dropped to <20% following exposure to ionomycin for 48 h (Fig. 3b). Results from BL41 cells with all of the mutant BHRF1 proteins following treatment with etoposide and ionomycin are summarised in Fig. 3c. Similar results were obtained for treatment with other cytotoxic stimuli, including roscovitine and staurosporine (Supplementary Fig. 4). These experiments revealed that the G99A and R100D BHRF1 mutants had significantly reduced ability to protect cells from diverse apoptotic stimuli, whereas the R93D and L102I BHRF1 mutants protected BL41 cells from all apoptotic stimuli to levels comparable to or more than WT BHRF1. Interestingly, the F72W BHRF1 mutant protected BL41 cells from ionomycin, which acts primarily through BIM [30], but not from etoposide which kills through activation of TP53 and concomitant upregulation of PUMA and NOXA [28].
To independently verify and extend these results, we utilised cell lines derived from lymphomas of Eµ-Myc transgenic mice, a model of MYC-driven aggressive B-cell lymphomas, like BL [27, 40]. Eµ-Myc mice carry a c-MYC transgene under the control of the Ig heavy chain enhancer, Eµ, leading to the development of pre-B or sIg+ B-cell lymphomas. This transgene recapitulates the c-MYC;Ig chromosomal translocation found in human BL patients that present with later stage germinal centre B-cell-derived lymphomas. FLAG-tagged WT and mutant BHRF1 proteins were expressed from a retrovirus in an Eµ-Myc lymphoma-derived cell line (Supplementary Fig. 3). The statistically significant reductions in viability observed with certain BHRF1 mutants in the BL41 cells were also apparent when these BHRF1 mutants were expressed in the mouse Eµ-Myc lymphoma-derived cell lines (Fig. 3d).
Binding to cellular BIM is not the sole interaction critical for BHRF1 function
It was reported that the ability of BHRF1 to protect cells from apoptosis is due solely to its capacity to bind cellular BIM [41]. To investigate this, we expressed HA-tagged WT and mutant BHRF1 proteins in the EBV-negative human BL cell line, BL2, that cannot express BIM due to bi-allelic deletion of the BIM locus [42] (Fig. 4a). BL2 cell viability was assessed following treatment with etoposide (50 µM for 24 h) or ionomycin (1 µg/mL for 48 h) (Fig. 4b). Due to the absence of pro-apoptotic BIM, the overall amount of killing was reduced in BL2 cells when treated with etoposide or ionomycin. Nevertheless, BL2 cells expressing WT BHRF1 were significantly more protected from the cytotoxic insults applied than BL2 cells carrying the control lentivirus (Fig. 4b). These results were independently verified in mouse Eµ-Myc;BimKO lymphoma-derived cell lines expressing FLAG-tagged WT BHRF1 (Fig. 4c). Here, the protection conferred by WT BHRF1 to etoposide and ionomycin was highly statistically significant compared to cells carrying the control retrovirus.
Fig. 4.

Analysis of mutant BHRF1 proteins in BIM-deficient human BL cells and mouse Eµ-Myc lymphoma cell lines. a Western blots showing expression of HA-tagged WT or mutant BHRF1 proteins expressed from a dox-inducible lentiviral vector in the EBV-negative BL2 cells that naturally do not express the BH3-only protein BIM. BL41 and Sav-BL cells act as positive controls for BIM expression. Probing for ACTIN was used as a loading control. b Viability of BL2 cells expressing WT or mutant BHRF1 proteins after treatment with etoposide (50 µM) for 24 h or ionomycin (1 µg/mL) for 48 h. Results are expressed relative to untreated cells and represent the mean and standard deviation of triplicate experiments c Viability of murine Eµ-Myc;BimKO lymphoma cells expressing FLAG-tagged WT or mutant BHRF1 proteins from a constitutively active retrovirus after treatment with etoposide (0.2 µg/mL) or ionomycin (2.5 µg/mL) for 24 h. Results are expressed relative to untreated cells and represent the mean and standard deviation of triplicate experiments, or five experiments for ionomycin treatment of Eµ-Myc;BimKO variants. For b and c, unpaired two-tailed t-tests were performed and a Bonferroni adjustment of 6 applied to calculate the significance of the observed differences between cells expressing mutant BHRF1 proteins or negative control (empty vector transduced cells) compared with cells expressing WT BHRF1 protein; *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001
In these BIM-deficient human and mouse cell lines, the BHRF1 mutants gave a trend of protection that was similar to that observed in the BL41 and wt Eµ-Myc lymphoma-derived cell lines but which just missed statistical significance for some BHRF1 mutants (Fig. 4b, c). This is likely because of the reduced amount of induced cell death in BIM-deficient cells and also the stringency of the Bonferroni adjustment that was used to correct for multiple comparisons. These results using multiple drugs in two independent cell systems indicate that BHRF1 does not function solely through binding and inhibiting pro-apoptotic BIM and that the loss-of-function observed for some BHRF1 mutants is not solely a reflection of their inability to bind and inhibit BIM.
BHRF1 protects from apoptotic stimuli in the absence of key cellular pro-apoptotic proteins
To determine whether interaction with a different cellular pro-apoptotic protein was essential for BHRF1 pro-survival function, the WT and mutant BHRF1 proteins were expressed in Eµ-Myc;BakKO and Eµ-Myc;PumaKO mouse lymphoma-derived cell lines. WT BHRF1 significantly protected cells from both drugs in the absence of either BAK or PUMA (Fig. 5a, b).
The overall pattern of protection afforded by the BHRF1 mutants was the same in the Eµ-Myc;BakKO lymphoma cells as in the BL41 cells and the WT Eµ-Myc lymphoma cells (Fig. 5a). In the Eµ-Myc;PumaKO lymphoma cells the R93D, L102I, G99A and R100D mutant BHRF1 proteins behaved in a similar manner to previously, although the ionomycin treatment results for G99A and R100D BHRF1 proteins were trends that just missed statistical significance, p = 0.07 for both mutants (Fig. 5b). Interestingly, in the absence of PUMA the F72W BHRF1 mutant protected from both ionomycin- and etoposide-induced apoptosis. This suggests this mutant cannot bind sufficient PUMA, and so can only protect from DNA-damaging drugs when this major TP53 driven initiator of apoptosis is absent and instead apoptosis must be mediated by NOXA and/or, in a TP53-independent manner, through BIM [30]. This finding identifies aa72 in BHRF1 as essential to protect from DNA-damage induced, PUMA-mediated apoptosis.
To directly examine the ability of the BHRF1 mutants to interact with cellular pro-apoptotic BCL-2 family proteins, we performed Co-IP experiments in the mouse Eµ-Myc lymphoma cells under unstressed conditions using two different lysis buffers containing either Triton X-100 (Fig. 5 and Supplementary Fig. 5) or CHAPS (Supplementary Fig. 6), to account for detergent-induced conformational changes that may affect the results [43]. Consistent with the ITC data, interactions were observed between WT BHRF1 and murine BIM, tBID and PUMA (Fig. 5c and in an independent cell line in Supplementary Fig. 5) when the cells were lysed with buffer containing Triton X-100. With this buffer, we were unable to detect an interaction between BHRF1 and BAK, even when the cells were treated with apoptotic stimuli in the presence of the pan-caspase inhibitor, QVD-OPh (data not shown). However, when the Co-IPs were performed with a buffer containing 1% CHAPS, WT and all mutant BHRF1 proteins showed similar binding to BAK (Supplementary Fig. 6).
In terms of differential binding to cellular BCL-2 family proteins by the BHRF1 mutants, the potent apoptosis protector, R93D mutant BHRF1, displayed equivalent or greater binding to murine BIM, PUMA and tBID than WT BHRF1. For the loss-of-function BHRF1 mutants, G99A and R100D, very little binding to BIM (although slightly more BIM binding was seen with the R100D than the G99A BHRF1 mutant; Supplementary Fig. 5), and no binding to PUMA or tBID was detected. For the L102I BHRF1 mutant, binding to BIM, PUMA and tBID was evident, but there was evidence that the binding to BIM was reduced compared with WT BHRF1 when buffers containing Triton X-100 were used (Fig. 5 and Supplementary Fig. 5). Interestingly, for the F72W BHRF1 mutant, binding to BIM was observed (relatively more binding of F72W mutant BHRF1 to BIM was observed with CHAPS buffer than with Triton X-100 buffer (Supplementary Fig. 6)), but no binding to PUMA or tBID was detected (Fig. 5). The lack of binding to PUMA is consistent with the finding that the F72W BHRF1 mutant cannot protect from DNA-damage-induced apoptosis.
Discrepancies were observed between the interactions predicted by ITC (Supplementary Fig. 2) and the results seen by Co-IP (Fig. 5 and Supplementary Figs. 5 and 6). This is likely due to the fundamental differences in the approaches. ITC relies on using exogenously titrated BH3 peptides (rather than proteins), whereas Co-IP studies examine interactions with endogenous BCL-2 family members in whole cell lysates, where at least some of these proteins may be localised on the mitochondrial outer membrane. In addition, some proteins, e.g. BAK, must undergo significant structural changes to bind to pro-survival BCL-2 proteins [44]. Finally, ITC measurements examine particular interactions in isolation, whereas interactions interrogated by Co-IP in a cellular context are subject to competition, with multiple BH3-only proteins as well as primed BAX and BAK available for binding.
Impact of BHRF1 mutations on MYC-induced lymphoma development
To investigate the impact of WT and mutant forms of BHRF1 on MYC-driven lymphoma development, Eµ-Myc transgenic HSPCs were infected in vitro with the retroviruses described above and injected into lethally irradiated congenic mice (Fig. 6a). These mice were monitored for signs of lymphoma by visual examination and elevated white blood cell counts in the peripheral blood (Supplementary Fig. 7A), and Kaplan–Meier survival curves plotted (Fig. 6b). As for cellular pro-survival BCL-2 proteins [45], enforced expression of WT BHRF1 in Eµ-Myc HSPCs substantially accelerated lymphoma development compared with control mice injected with Eµ-Myc HSPCs infected with a control retrovirus (median latency 52 days (WT BHRF1) compared with 206 days (pMIG), **p = 0.006). Interestingly, the R93D BHRF1 mutant accelerated lymphoma development to an even greater extent than WT BHRF1 (median latency of 31 days, R93D versus WT ***p = 0.0003). Only the G99A BHRF1 mutant did not significantly accelerate MYC-driven lymphoma development. Unexpectedly, the R100D BHRF1 mutant did accelerate MYC-driven lymphoma development (median survival 67 days) compared with pMIG controls, despite its inability to protect cells from apoptotic stimuli in vitro. In addition, the F72W BHRF1 mutant also accelerated lymphoma development (median survival 49 days, pMIG versus F72W BHRF1 **p = 0.007) to a similar extent as WT BHRF1, despite being unable to protect cells from DNA-damage-induced apoptosis in vitro and having only poor binding to PUMA.
Fig. 6.

Comparing the ability of WT BHRF1 versus mutant forms of BHRF1 to accelerate Eµ-Myc-driven lymphoma development and analysis of the lymphomas that arise in the mice. a Schematic of the experimental procedure. Foetal liver cells, a rich source of HSPCs, from embryonic day 14.5 (E14.5) Eµ-Myc transgenic C57BL/6-Ly5.2+ mice were infected with a control pMIG retrovirus or retroviruses expressing FLAG-tagged WT or mutant forms of BHRF1. These infected HSPCs were cultured in medium supplemented with cytokines and then injected into the tail vein of lethally irradiated (2 × 5.5 Gy, 3 h apart) C57BL/6-Ly5.1+ recipient mice. b Kaplan–Meier survival curves of C57BL/6-Ly5.1+ mice injected with the Eµ-Myc HSPCs infected with retroviruses expressing pMIG, WT BHRF1 or mutant forms of BHRF1; n = 7 mice in each cohort. The median latencies of lymphoma development for the different BHRF1 expression vectors are listed in the table. The survival curves of the mice transplanted with Eµ-Myc HSPCs expressing mutant BHRF1 proteins were statistically compared with the control pMIG and WT BHRF1 cohort survival curves using a Mantel–Cox test. WT BHRF1 and all BHRF1 mutants, with the exception of G99A mutant BHRF1, statistically significantly accelerated Eµ-Myc-driven lymphoma development compared with mice injected with Eµ-Myc HSPCs expressing pMIG alone. The mice injected with the Eµ-Myc HSPCs expressing the BHRF1 mutants R93D and L102I displayed significantly accelerated lymphoma development compared with mice injected with the Eµ-Myc HSPCs expressing WT BHRF1 (R93D BHRF1*** and L102I BHRF1*). c Immunophenotyping of the Eµ-Myc lymphomas arising in the different recipient mice, as described in a and b. d Western blotting to measure expression of FLAG-tagged BHRF1, TRP53 and P19ARF in lymphomas from the spleens or lymph nodes of the recipient mice. Probing for ACTIN was used as a loading control. e Summary of TP53 status in Wp-restricted human BL cell lines that express latent BHRF1
The lymphoma immunophenotypes were determined by staining with antibodies to the B-cell markers CD19, B220, IgM and IgD and flow cytometric analysis (Fig. 6c). Lymphomas from mice injected with Eµ-Myc HSPCs infected with control pMIG retrovirus or retroviruses expressing the functionally inactive G99A and R100D BHRF1 mutants were mostly of B-cell origin (B220+, CD19+and either IgM+ or IgM−) (Fig. 6c). Interestingly, lymphomas that arose from the Eµ-Myc HSPCs expressing WT BHRF1 or the BHRF1 mutants that could fully protect against ionomycin (R93D, F72W, L102I) instead exhibited a progenitor cell phenotype (B220+, CD19−). This shift to immature progenitor cell lymphomas was previously observed when Eµ-Myc-induced lymphomagenesis was accelerated by the expression of cellular pro-survival proteins (e.g. BCL-2, MCL-1) [45–47]. Western blotting revealed that the expression levels of the mutant BHRF1 proteins in the lymphoma cells inversely correlated with their ability to protect from DNA-damage-induced cell death in vitro; BHRF1 R93D was expressed at the lowest levels, and BHRF1 R100D, G99A and F72W at the highest levels (Fig. 6d).
The results generated with the R100D BHRF1 mutant were intriguing. This mutant could not significantly protect lymphoma cells from apoptosis in vitro, but could significantly accelerate MYC-driven lymphoma development in vivo, and yet the lymphomas retained the B-cell phenotype characteristic of control lymphomas carrying pMIG. Some mice transplanted with Eµ-Myc HSPCs expressing R100D mutant BHRF1 presented with unusually large lymph nodes (Supplementary Fig. 7B), however, the significance of this remains unclear.
Since Trp53 mutations are potently selected for in Eµ-Myc lymphomas (~20–30%) and loss of WT TRP53 function dramatically accelerates MYC-driven lymphoma development [48, 49], we investigated the incidence of Trp53 mutations in the lymphomas by western blotting for expression of a stabilised mutant TRP53 protein and/or high levels of p19ARF, indicative of Tp53 mutation/loss [50, 51] (Fig. 6d and Supplementary Fig. 7C). Two from four pMIG control lymphomas showed evidence of Trp53 mutation, whereas lymphomas expressing functional BHRF1 protein (WT, R93D, L102I and F72W BHRF1) showed no evidence of Trp53 mutations. Interestingly, many of the lymphomas expressing R100D BHRF1 showed relatively high expression of TRP53 and/or p19ARF proteins, but sequencing of exons 4-11 of Trp53 did not reveal mutations. Human BL cell lines have high rates (up to 70%) of TP53 mutations [52, 53] but, strikingly we found that TP53 was not mutated in the BL cell lines we examined, Sal-, Oku- and Ava-BL, that naturally express BHRF1 (Fig. 6e). Collectively, these findings indicate that expression of BHRF1 relieves the pressure to mutate TP53 during MYC-driven lymphomagenesis.
Discussion
Although BL is regarded as highly curable, survival for patients in resource-poor settings, those of older age or with higher stage or refractory disease can be extremely poor [54–56], prompting efforts to discover novel vulnerabilities in these tumours to exploit therapeutically. In this regard, a subset of aggressive BLs encode the EBV protein BHRF1, a vBCL-2 protein, which in in vitro experiments confers broad spectrum chemoresistance [20]. That BHRF1 is a bone fide therapeutic target for this subset of aggressive BLs was indicated by a previous study reporting a peptide, called BINDI, which specifically targeted BHRF1 and inhibited lymphoma growth in a xenograft model of BL [57]. Therefore, a BH3-mimetic targeting viral BHRF1 should be therapeutically beneficial for some BLs and is predicted to be well tolerated since it should only kill virally infected cells, thereby circumventing the on-target toxicities associated with the BH3-mimetic drugs that target cellular pro-survival BCL-2 proteins, such as navitoclax (ABT-263) [24, 25, 58].
Whilst there is compelling evidence supporting the need for a vBCL-2-specific BH3-mimetic drug, progress has been limited by a lack of clear understanding of how BHRF1 interacts with the cellular apoptotic machinery to achieve potent protection from cell killing. It has been reported that BHRF1 protects from apoptosis solely through inhibition of the cellular pro-apoptotic BH3-only protein BIM [41]. However, our data show that BHRF1 protects BIM-deficient human and murine lymphoma-derived cell lines and mouse lymphoma-derived cell lines deficient for PUMA, BID or BAK, from diverse cytotoxic insults. These findings indicate that a key feature of how BHRF1 functions to protect from apoptosis is its ability to bind multiple pro-apoptotic proteins, and that all of these interactions play a role. The observation that the R100D BHRF1 mutant can accelerate MYC-driven lymphomagenesis despite conferring minimal protection from apoptosis in vitro may indicate that only a low threshold level of interaction with cellular BH3-only proteins is required for BHRF1 to counter cell death in vivo and thereby promote lymphomagenesis. In BLs that express BHRF1, the virally encoded EBNA3 proteins are reported to repress BIM expression [19], and therefore BHRF1 could further contribute to chemoresistance by inhibiting other cellular initiators and mediators of apoptosis. This likely indicates that all of these interactions need to be inhibited to suppress the function of BHRF1 in a therapeutic setting. This should be feasible given that BH3-mimetic drugs targeting BCL-2, BCL-XL or MCL-1 inhibit all their interactions with pro-apoptotic BH3-only proteins, as well as the apoptosis effectors BAX and BAK [59–61]. However, a different interpretation of the results observed with the R100D BHRF1 mutant is that BHRF1 may interact with other, yet to be identified, cellular proteins involved in cell fates and that these interactions are more important in an in vivo setting. This could impact the potential effectiveness of a BH3-mimetic drug that specifically targets the BH3-binding groove of BHRF1.
We identified a mutant form of BHRF1 with enhanced capability to accelerate MYC-driven lymphoma development. In addition, we identified two amino acids (G99 and R100) in BHRF1 that are critical for its anti-apoptotic function following diverse cytotoxic stimuli. These residues are highly conserved in cellular pro-survival BCL-2 family proteins and are therefore likely to be poor areas of the protein to target when designing a BHRF1-selective drug [62]. More attractive from a drug design perspective is the phenylalanine (F) residue at aa72 of BHRF1, which is not conserved in other pro-survival BCL-2 proteins [63, 64]. Mutating this residue to tryptophan abolished the ability of BHRF1 to protect lymphoma cells from DNA-damaging agents that kill through TP53-mediated transcriptional upregulation of PUMA and NOXA [65]. Importantly, protection from ionomycin, which primarily kills through induction of BIM [30], was retained, proving that the F72W mutant BHRF1 protein is folded and active. Experiments in PUMA-deficient cells confirmed that the F72 residue in BHRF1 is critical for its interaction with this cellular pro-apoptotic BH3-only protein. Consequently, the region surrounding F72 within BHRF1 could be targeted specifically with novel BH3-mimetics to sensitise lymphoma cells to DNA-damaging chemotherapeutics.
Having characterised the key cellular interactions important for BHRF1 to confer protection from chemotherapeutic drugs in established lymphomas, we investigated if BHRF1 expression could contribute to the early stages of malignant transformation. Using a HSPC reconstitution model of MYC-driven lymphomagenesis we found that BHRF1, like the cellular anti-apoptotic BCL-2 proteins, potently synergises with MYC to drive lymphoma development. Interestingly, the resulting lymphomas had a progenitor cell phenotype (B220pos, CD19/IgM/IgDneg) rather than the classical pre-B or B-cell phenotype [45–47], suggesting that BHRF1 can potently block cell death associated with aberrant c-MYC expression, permitting uncontrolled proliferation of nascent neoplastic cells driving rapid lymphoma development.
These in vivo experiments using BHRF1 mutants provided interesting insights into the complementing cellular drivers of MYC-driven lymphoma development. It is reported that two pathways, one regulated by BIM and the other by TP53-mediated induction of PUMA and NOXA, can suppress MYC-induced lymphomagenesis [48, 66, 67]. The data generated using the different BHRF1 mutants support this notion. BHRF1 mutants that could potently bind BIM (e.g. R93D BHRF1) led to the most accelerated lymphoma development. MYC-driven lymphomas frequently harbour mutations in TP53; ~20% of mouse Eµ-Myc lymphomas [48], ~70% of human BL cell lines and ~20% of BL biopsies [52, 53]. Interestingly, BLs that naturally express BHRF1 retain a WT TP53 gene and all Eµ-Myc lymphomas that arose from the HSPCs expressing WT or fully functional BHRF1 mutants expressed WT TRP53. This suggests that BHRF1 expression relieves the selection pressure for MYC-driven lymphoma cells to acquire mutations in Tp53 (and potentially other genes) possibly through binding and inhibiting the pro-apoptotic proteins PUMA and BIM. This supports the proposition that therapeutic targeting of BHRF1 using novel BH3-mimetics alongside conventional chemotherapeutic drugs that act (in part) through TP53 by inducing the expression of PUMA, NOXA and indirectly also BIM, may be a promising therapeutic approach for these lymphomas.
Supplementary information
Acknowledgements
We thank G. Siciliano, H. Johnson, C. Alessandro, K. Hughes and D. Fayle for mouse husbandry, Dr S. Iyer for advice to establish experimental techniques and Prof. T. Speed for advice with statistical testing. This work was supported by funding from the Kay Kendall Leukaemia Fund Fellowship KKL331 awarded to GLK; Victorian Cancer Agency Fellowship (MCRF 17028) awarded to GLK, Cancer Research UK Programme Grant C5575/A15032 awarded to MR; Cancer Council Victoria, grants-in-aid #1086157 and #1147328 awarded to GLK; the National Health and Medical Research Council, Project Grant #1086291 awarded to GLK and #637336 to MK, Program Grant #101671 awarded to PMC, DCSH and AS, Fellowship #1020363 awarded to AS, Fellowship #1116934 awarded to PMC, Fellowship #1043149 awarded to DCSH, Fellowship #637372 awarded to MK; the Leukaemia Foundation Australia grant awarded to GLK and AS, the Leukaemia and Lymphoma Society Grant #7001-13, awarded to PMC, DCSH, AS and MJH; the estate of Anthony (Toni) Redstone OAM and The Craig Perkins Cancer Research Foundation; Australian Research Council Future Fellowship awarded to MK (FT130101349) and operational infrastructure grants through the Australian Government NHMRCS IRIISS and the Victorian State Government Operational Infrastructure Support.
Author contributions
LF, RC, CC, NS, LCAG, CDS, MK and GLK performed the experiments. GD, MJH, PMC, DCSH, AS, MK, MR and GLK provided resources. LF, MJH, RJT, CS, AIB, DCC, GD, MK, AS, PMC, DCSH, MR and GLK contributed to the conception and design of the research. LF, CC and GLK analysed the data and LF, MK, MR, AS and GLK interpreted the data. GLK wrote the manuscript and it was edited and critically revised by LF, AS, MK and MR.
Compliance with ethical standards
Conflict of interest
The Walter and Eliza Hall Institute receives milestone and royalty payments related to venetoclax, and employees (CC, LCAG, GD, MJH, PMC, DCSH, AS and GLK) may be eligible for benefits related to these payments. The laboratory of AS receives research funding from Servier. The other authors declare that they have no conflict of interest.
Footnotes
Edited by C. Borner
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: M. Kvansakul, M. Rowe, G. L. Kelly
Contributor Information
Leah Fitzsimmons, Email: l.fitzsimmons@bham.ac.uk.
Gemma L. Kelly, Email: gkelly@wehi.edu.au
Supplementary information
The online version of this article (10.1038/s41418-019-0435-1) contains supplementary material, which is available to authorized users.
References
- 1.Strasser A, Cory S, Adams JM. Deciphering the rules of programmed cell death to improve therapy of cancer and other diseases. EMBO J. 2011;30:3667–83. doi: 10.1038/emboj.2011.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fitzsimmons L, Kelly GL. EBV and apoptosis: the viral master regulator of cell fate? Viruses. 2017;9:e339. [DOI] [PMC free article] [PubMed]
- 3.Magrath I. The pathogenesis of Burkitt’s lymphoma. Adv Cancer Res. 1990;55:133–270. doi: 10.1016/s0065-230x(08)60470-4. [DOI] [PubMed] [Google Scholar]
- 4.Rickinson AB. Co-infections, inflammation and oncogenesis: future directions for EBV research. Semin Cancer Biol. 2014;26:99–115. doi: 10.1016/j.semcancer.2014.04.004. [DOI] [PubMed] [Google Scholar]
- 5.Dalla-Favera R, Bregni M, Erikson J, Patterson D, Gallo RC, Croce CM. Human c-myc onc gene is located on the region of chromosome 8 that is translocated in Burkitt lymphoma cells. Proc Natl Acad Sci USA. 1982;79:7824–7. doi: 10.1073/pnas.79.24.7824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fitzsimmons L, Boyce AJ, Wei W, Chang C, Croom-Carter D, Tierney RJ, et al. Coordinated repression of BIM and PUMA by Epstein-Barr virus latent genes maintains the survival of Burkitt lymphoma cells. Cell Death Differ. 2018;25:241–54. doi: 10.1038/cdd.2017.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Altmann M, Hammerschmidt W. Epstein-Barr virus provides a new paradigm: a requirement for the immediate inhibition of apoptosis. PLoS Biol. 2005;3:e404. doi: 10.1371/journal.pbio.0030404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cleary ML, Smith SD, Sklar J. Cloning and structural analysis of cDNAs for bcl-2 and a hybrid bcl-2/immunoglobulin transcript resulting from the t(14;18) translocation. Cell. 1986;47:19–28. doi: 10.1016/0092-8674(86)90362-4. [DOI] [PubMed] [Google Scholar]
- 9.Marshall WL, Yim C, Gustafson E, Graf T, Sage DR, Hanify K, et al. Epstein-Barr virus encodes a novel homolog of the bcl-2 oncogene that inhibits apoptosis and associates with Bax and Bak. J Virol. 1999;73:5181–5. doi: 10.1128/jvi.73.6.5181-5185.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bellows DS, Howell M, Pearson C, Hazlewood SA, Hardwick JM. Epstein-Barr virus BALF1 is a BCL-2-like antagonist of the herpesvirus antiapoptotic BCL-2 proteins. J Virol. 2002;76:2469–79. doi: 10.1128/jvi.76.5.2469-2479.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Henderson S, Huen D, Rowe M, Dawson C, Johnson G, Rickinson A. Epstein virus-coded BHRF 1 protein, a viral homologue of Bcl-2 protects human B cells from programmed cell death. Proc Natl Acad Sci USA. 1993;90:8479–83. doi: 10.1073/pnas.90.18.8479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McCarthy NJ, Hazlewood SA, Huen DS, Rickinson AB, Williams GT. The Epstein-Barr virus gene BHRF1, a homologue of the cellular oncogene Bcl-2, inhibits apoptosis induced by gamma radiation and chemotherapeutic drugs. Adv Exp Med Biol. 1996;406:83–97. doi: 10.1007/978-1-4899-0274-0_9. [DOI] [PubMed] [Google Scholar]
- 13.Fanidi A, Hancock DC, Littlewood TD. Suppression of c-Myc-induced apoptosis by the Epstein-Barr virus gene product BHRF1. J Virol. 1998;72:8392–5. doi: 10.1128/jvi.72.10.8392-8395.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kawanishi M. Epstein-Barr virus BHRF1 protein protects intestine 407 epithelial cells from apoptosis induced by tumor necrosis factor alpha and anti-Fas antibody. J Virol. 1997;71:3319–22. doi: 10.1128/jvi.71.4.3319-3322.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hsu WL, Chung PJ, Tsai MH, Chang CL, Liang CL. A role for Epstein-Barr viral BALF1 in facilitating tumor formation and metastasis potential. Virus Res. 2012;163:617–27. doi: 10.1016/j.virusres.2011.12.017. [DOI] [PubMed] [Google Scholar]
- 16.Rowe DT, Rowe M, Evan GI, Wallace LE, Farrell PJ, Rickinson AB. Restricted expression of EBV latent genes and T-lymphocyte-detected membrane antigen in Burkitt’s lymphoma cells. EMBO J. 1986;5:2599–607. doi: 10.1002/j.1460-2075.1986.tb04540.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Komano J, Sugiura M, Takada K. Epstein-Barr virus contributes to the malignant phenotype and to apoptosis resistance in Burkitt’s lymphoma cell line Akata. J Virol. 1998;72:9150–6. doi: 10.1128/jvi.72.11.9150-9156.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kennedy G, Komano J, Sugden B. Epstein-Barr virus provides a survival factor to Burkitt’s lymphomas. Proc Natl Acad Sci USA. 2003;100:14269–74. doi: 10.1073/pnas.2336099100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kelly G, Bell A, Rickinson A. Epstein-Barr virus-associated Burkitt lymphomagenesis selects for downregulation of the nuclear antigen EBNA2. Nat Med. 2002;8:1098–104. doi: 10.1038/nm758. [DOI] [PubMed] [Google Scholar]
- 20.Kelly GL, Long HM, Stylianou J, Thomas WA, Leese A, Bell AI, et al. An Epstein-Barr virus anti-apoptotic protein constitutively expressed in transformed cells and implicated in burkitt lymphomagenesis: the Wp/BHRF1 link. PLoS Pathog. 2009;5:e1000341. doi: 10.1371/journal.ppat.1000341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Anderton E, Yee J, Smith P, Crook T, White RE, Allday MJ. Two Epstein-Barr virus (EBV) oncoproteins cooperate to repress expression of the proapoptotic tumour-suppressor Bim: clues to the pathogenesis of Burkitt’s lymphoma. Oncogene. 2008;27:421–33. doi: 10.1038/sj.onc.1210668. [DOI] [PubMed] [Google Scholar]
- 22.Merino D, Kelly GL, Lessene G, Wei AH, Roberts AW, Strasser A. BH3-mimetic drugs: blazing the trail for new cancer medicines. Cancer Cell. 2018;34:879–91. doi: 10.1016/j.ccell.2018.11.004. [DOI] [PubMed] [Google Scholar]
- 23.Delbridge AR, Strasser A. The BCL-2 protein family, BH3-mimetics and cancer therapy. Cell Death Differ. 2015;22:1071–80. doi: 10.1038/cdd.2015.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wilson WH, O’Connor OA, Czuczman MS, Lacasce AS, Gerecitano JF, Leonard JP, et al. Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: a phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. lancet Oncol. 2010;11:1149–59. doi: 10.1016/S1470-2045(10)70261-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roberts AW, Seymour JF, Brown JR, Wierda WG, Kipps TJ, Khaw SL, et al. Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: results of a phase I study of navitoclax in patients with relapsed or refractory disease. J Clin Oncol. 2012;30:488–96. doi: 10.1200/JCO.2011.34.7898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roberts AW, Davids MS, Pagel JM, Kahl BS, Puvvada SD, Gerecitano JF, et al. Targeting BCL2 with venetoclax in relapsed chronic lymphocytic leukemia. N Engl J Med. 2016;374:311–22. doi: 10.1056/NEJMoa1513257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adams JM, Harris AW, Pinkert CA, Corcoran LM, Alexander WS, Cory S, et al. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature. 1985;318:533–8. doi: 10.1038/318533a0. [DOI] [PubMed] [Google Scholar]
- 28.Villunger A, Michalak EM, Coultas L, Mullauer F, Bock G, Ausserlechner MJ, et al. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science. 2003;302:1036–8. doi: 10.1126/science.1090072. [DOI] [PubMed] [Google Scholar]
- 29.Happo L, Cragg MS, Phipson B, Haga JM, Jansen ES, Herold MJ, et al. Maximal killing of lymphoma cells by DNA-damage inducing therapy requires not only the p53 targets Puma and Noxa but also Bim. Blood. 2010;116:5256–67. doi: 10.1182/blood-2010-04-280818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bouillet P, Metcalf D, Huang DC, Tarlinton DM, Kay TW, Kontgen F, et al. Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science. 1999;286:1735–8. doi: 10.1126/science.286.5445.1735. [DOI] [PubMed] [Google Scholar]
- 31.Kaufmann T, Tai L, Ekert PG, Huang DC, Norris F, Lindemann RK, et al. The BH3-only protein bid is dispensable for DNA damage- and replicative stress-induced apoptosis or cell-cycle arrest. Cell. 2007;129:423–33. doi: 10.1016/j.cell.2007.03.017. [DOI] [PubMed] [Google Scholar]
- 32.Lindsten T, Ross AJ, King A, Zong W, Rathmell JC, Shiels HA, et al. The combined functions of proapoptotic Bcl-2 family members Bak and Bax are essential for normal development of multiple tissues. Mol Cell. 2000;6:1389–99. doi: 10.1016/s1097-2765(00)00136-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aubrey BJ, Kelly GL, Kueh AJ, Brennan MS, O’Connor L, Milla L, et al. An inducible lentiviral guide RNA platform enables the identification of tumor-essential genes and tumor-promoting mutations in vivo. Cell Rep. 2015;10:1422–32. doi: 10.1016/j.celrep.2015.02.002. [DOI] [PubMed] [Google Scholar]
- 34.Kelly GL, Grabow S, Glaser SP, Fitzsimmons L, Aubrey BJ, Okamoto T, et al. Targeting of MCL-1 kills MYC-driven mouse and human lymphomas even when they bear mutations in p53. Genes Dev. 2014;28:58–70. doi: 10.1101/gad.232009.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Herold MJ, van den Brandt J, Seibler J, Reichardt HM. Inducible and reversible gene silencing by stable integration of an shRNA-encoding lentivirus in transgenic rats. Proc Natl Acad Sci USA. 2008;105:18507–12. doi: 10.1073/pnas.0806213105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vogel S, Raulf N, Bregenhorn S, Biniossek ML, Maurer U, Czabotar P, et al. Cytosolic Bax: does it require binding proteins to keep its pro-apoptotic activity in check? J Biol Chem. 2012;287:9112–27. doi: 10.1074/jbc.M111.248906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kvansakul M, Wei AH, Fletcher JI, Willis SN, Chen L, Roberts AW, et al. Structural basis for apoptosis inhibition by Epstein-Barr virus BHRF1. PLoS Pathog. 2010;6:e1001236. doi: 10.1371/journal.ppat.1001236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Burton DR, Caria S, Marshall B, Barry M, Kvansakul M. Structural basis of deerpox virus-mediated inhibition of apoptosis. Acta Crystallogr D Biol Crystallogr. 2015;71(Pt 8):1593–603. doi: 10.1107/S1399004715009402. [DOI] [PubMed] [Google Scholar]
- 39.Tierney RJ, Shannon-Lowe CD, Fitzsimmons L, Bell AI, Rowe M. Unexpected patterns of Epstein-Barr virus transcription revealed by a high throughput PCR array for absolute quantification of viral mRNA. Virology. 2015;474:117–30. doi: 10.1016/j.virol.2014.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Harris AW, Pinkert CA, Crawford M, Langdon WY, Brinster RL, Adams JM. The Eµ-myc transgenic mouse: a model for high-incidence spontaneous lymphoma and leukemia of early B cells. J Exp Med. 1988;167:353–71. doi: 10.1084/jem.167.2.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Desbien AL, Kappler JW, Marrack P. The Epstein-Barr virus Bcl-2 homolog, BHRF1, blocks apoptosis by binding to a limited amount of Bim. Proc Natl Acad Sci USA. 2009;106:5663–8. doi: 10.1073/pnas.0901036106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Richter-Larrea JA, Robles EF, Fresquet V, Beltran E, Rullan AJ, Agirre X, et al. Reversion of epigenetically mediated BIM silencing overcomes chemoresistance in Burkitt lymphoma. Blood. 2010;116:2531–42. doi: 10.1182/blood-2010-02-268003. [DOI] [PubMed] [Google Scholar]
- 43.Hsu Y-T, Youle RJ. Bax in murine thymus is a soluble monomeric protein that displays differential detergent-induced conformations. J Biol Chem. 1998;273:10777–83. doi: 10.1074/jbc.273.17.10777. [DOI] [PubMed] [Google Scholar]
- 44.Kvansakul M, Hinds MG. The structural biology of BH3-only proteins. Methods Enzymol. 2014;544:49–74. doi: 10.1016/B978-0-12-417158-9.00003-0. [DOI] [PubMed] [Google Scholar]
- 45.Strasser A, Harris AW, Bath ML, Cory S. Novel primitive lymphoid tumours induced in transgenic mice by cooperation between myc and bcl-2. Nature. 1990;348:331–3. doi: 10.1038/348331a0. [DOI] [PubMed] [Google Scholar]
- 46.Swanson PJ, Kuslak SL, Fang W, Tze L, Gaffney P, Selby S, et al. Fatal acute lymphoblastic leukemia in mice transgenic for B cell-restricted bcl-xL and c-myc. J Immunol. 2004;172:6684–91. doi: 10.4049/jimmunol.172.11.6684. [DOI] [PubMed] [Google Scholar]
- 47.Campbell KJ, Bath ML, Turner ML, Vandenberg CJ, Bouillet P, Metcalf D, et al. Elevated Mcl-1 perturbs lymphopoiesis, promotes transformation of hematopoietic stem/progenitor cells, and enhances drug resistance. Blood. 2010;116:3197–207. doi: 10.1182/blood-2010-04-281071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eischen CM, Weber JD, Roussel MF, Sherr CJ, Cleveland JL. Disruption of the ARF-Mdm2-p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes Dev. 1999;13:2658–69. doi: 10.1101/gad.13.20.2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Michalak EM, Jansen ES, Happo L, Cragg MS, Tai L, Smyth GK, et al. Puma and to a lesser extent Noxa are suppressors of Myc-induced lymphomagenesis. Cell Death Differ. 2009;16:684–96. doi: 10.1038/cdd.2008.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Quelle DE, Zindy F, Ashmun RA, Sherr CJ. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell. 1995;83:993–1000. doi: 10.1016/0092-8674(95)90214-7. [DOI] [PubMed] [Google Scholar]
- 51.Dittmer D, Pati S, Zambetti G, Chu S, Teresky AK, Moore M, et al. Gain of function mutations in p53. Nat Genet. 1993;4:42–46. doi: 10.1038/ng0593-42. [DOI] [PubMed] [Google Scholar]
- 52.Farrell PJ, Allan GJ, Shanahan F, Vousden KH, Crook T. p53 is frequently mutated in Burkitt’s lymphoma cell lines. EMBO J. 1991;10:2879–87. doi: 10.1002/j.1460-2075.1991.tb07837.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bhatia KG, Gutierrez MI, Huppi K, Siwarski D, Magrath IT. The pattern of p53 mutations in Burkitt’s lymphoma differs from that of solid tumors. Cancer Res. 1992;52:4273–6. [PubMed] [Google Scholar]
- 54.Kim H, Park ES, Lee SH, Koo HH, Kim HS, Lyu CJ, et al. Clinical outcome of relapsed or refractory burkitt lymphoma and mature B-cell lymphoblastic leukemia in children and adolescents. Cancer Res Treat. 2014;46:358–65. doi: 10.4143/crt.2013.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Costa LJ, Xavier AC, Wahlquist AE, Hill EG. Trends in survival of patients with Burkitt lymphoma/leukemia in the USA: an analysis of 3691 cases. Blood. 2013;121:4861–6. doi: 10.1182/blood-2012-12-475558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McGoldrick SM, Mutyaba I, Adams SV, Larsen A, Krantz EM, Namirembe C, et al. Survival of children with endemic Burkitt lymphoma in a prospective clinical care project in Uganda. Pediatric blood &. cancer. 2019;66:e27813. doi: 10.1002/pbc.27813. [DOI] [PubMed] [Google Scholar]
- 57.Procko E, Berguig GY, Shen BW, Song Y, Frayo S, Convertine AJ, et al. A computationally designed inhibitor of an Epstein-Barr viral Bcl-2 protein induces apoptosis in infected cells. Cell. 2014;157:1644–56. doi: 10.1016/j.cell.2014.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mason KD, Khaw SL, Rayeroux KC, Chew E, Lee EF, Fairlie DW, et al. The BH3 mimetic compound, ABT-737, synergizes with a range of cytotoxic chemotherapy agents in chronic lymphocytic leukemia. Leukemia. 2009;23:2034–41. doi: 10.1038/leu.2009.151. [DOI] [PubMed] [Google Scholar]
- 59.Kotschy A, Szlavik Z, Murray J, Davidson J, Maragno AL, Le Toumelin-Braizat G, et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature. 2016;538:477–82. doi: 10.1038/nature19830. [DOI] [PubMed] [Google Scholar]
- 60.Lessene G, Czabotar PE, Sleebs BE, Zobel K, Lowes KN, Adams JM, et al. Structure-guided design of a selective BCL-X(L) inhibitor. Nat Chem Biol. 2013;9:390–7. doi: 10.1038/nchembio.1246. [DOI] [PubMed] [Google Scholar]
- 61.Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013;19:202–8. doi: 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]
- 62.Kvansakul M, Hinds MG. The Bcl-2 family: structures, interactions and targets for drug discovery. Apoptosis. 2015;20:136–50. doi: 10.1007/s10495-014-1051-7. [DOI] [PubMed] [Google Scholar]
- 63.Kvansakul M, Hinds MG. Structural biology of the Bcl-2 family and its mimicry by viral proteins. Cell Death Dis. 2013;4:e909. doi: 10.1038/cddis.2013.436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Petros AM, Olejniczak ET, Fesik SW. Structural biology of the Bcl-2 family of proteins. Biochim Biophys Acta. 2004;1644:83–94. doi: 10.1016/j.bbamcr.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 65.Michalak EM, Villunger A, Adams JM, Strasser A. In several cell types the tumour suppressor p53 induces apoptosis largely via Puma but Noxa can contribute. Cell Death Differ. 2008;15:1019–29. doi: 10.1038/cdd.2008.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hemann MT, Bric A, Teruya-Feldstein J, Herbst A, Nilsson JA, Cordon-Cardo C, et al. Evasion of the p53 tumour surveillance network by tumour-derived MYC mutants. Nature. 2005;436:807–11. doi: 10.1038/nature03845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Egle A, Harris AW, Bouillet P, Cory S. Bim is a suppressor of Myc-induced mouse B cell leukemia. Proc Natl Acad Sci USA. 2004;101:6164–9. doi: 10.1073/pnas.0401471101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.