

Abstract
Autophagy is a process in which intracellular components and dysfunctional organelles are delivered to the lysosome for degradation and recycling. Autophagy has various connections to a large number of human diseases, as its functions are essential for cell survival, bioenergetic homeostasis, organism development, and cell death regulation. In the past two decades, substantial effort has been made to identify the roles of autophagy in tumor suppression and promotion, neurodegenerative disorders, and other pathophysiologies. This review summarizes the current advances and discusses the unanswered questions in understanding the involvement of autophagy in pathogenic mechanisms of disease, primarily focusing on cancer and neurodegenerative diseases.
Subject terms: Chaperone-mediated autophagy, Macroautophagy

Facts
Autophagy plays bidirectional roles in cancer largely depending on the context.
Autophagy-related prognostic signature varies within cancers.
Chaperone-mediated autophagy and macroautophagy are involved in the pathogenesis of most neurodegenerative disorders.
Autophagy plays context-dependent roles at various stages of neurodegenerative diseases.
A reversible correlation between tumorigenesis and degeneration exists upon autophagy modulation.
Open questions
What are the mechanisms of disease-specific autophagy signatures?
How can we avoid the side effects of autophagy modulation in disease treatment?
How do core autophagy-related genes escape from genetic alterations in cancer?
What is the difference between neuronal autophagy and nonneuronal autophagy?
What is the crosstalk between cancer suppression and neuroprotection?
Introduction
Two of our “unanswered question” series of reviews, entitled “the molecular machinery of autophagy: unanswered questions” and “the regulation of autophagy: unanswered questions”, have discussed fundamental concepts of the autophagy mechanism and unanswered aspects of auto-phagy regulation [1, 2]. With the progressively advanced understanding of the molecular mechanism of autophagy, a considerable number of studies have highlighted the role of abnormal autophagy modulation in various human diseases, particularly neurodegenerative diseases and cancers. In this review, we focus on the involvement of autophagy in cancer and neurodegenerative disorders and comment on the major unanswered questions in the field.
Autophagy machinery
Depending on the mechanism of how cargos are delivered to lysosomes for degradation, there are three general types of autophagy, including macroautophagy, microautophagy and chaperone-mediated autophagy (CMA) [3].
Macroautophagy has the best-characterized and most universal mechanism among the three types of autophagy, and underscores the pathogenesis of a vast majority of diseases with autophagic dysfunction. The catabolic process of macroautophagy is evolutionarily conserved among all eukaryotic cells with five steps, including initiation, nucleation, elongation, fusion, and degradation (Fig. 1) [4]. The primary feature distinguishing macroautophagy from the other two forms of autophagy is the formation of double-membrane structures named autophagosomes, which deliver damaged organelles and protein aggregates to lysosomes.
Fig. 1. Schematic model for macroautophagy and PINK1-PRKN-mediated mitophagy.

Macroautophagy initiation occurs in response to cellular stresses. The phagophore nucleates and expands to sequester unwanted organelles and macromolecules and forms a double-membrane structure named the autophagosome. The outer membrane of the autophagosome fuses with a lysosome to deliver the cargo to the newly formed autolysosome for degradation. The breakdown products are further recycled following the release from permeases present in the lysosome/autolysosome membrane. The bottom scheme represents nonselective macroautophagy with the feature of random sequestration of cytoplasm. The top scheme depicts PINK1-PRKN-mediated mitophagy. Under normal conditions, PINK1 is cleaved within mitochondria in healthy mitochondria, and retrotranslocates to the cytosol for proteasomal degradation. When mitochondria damage occurs, mitophagy is induced. First, PINK1 is stabilized on the outer membrane and phosphorylates ubiquitin and PRKN separately. By this action, PRKN is recruited to the mitochondria surface and further ubiquitinates outer mitochondrial membrane (OMM) proteins. Ubiquitin is able to recruit more PRKN to form a feedback loop. Phosphorylated ubiquitin also recruits mitophagy receptors such as CALCOCO2/NDP52 and OPTN, which bind to phagophore-attached LC3, stimulating the engulfment of abnormal mitochondria in a phagophore.
Microautophagy transports cytosolic material into lysosomes through direct invagination or protrusion of the lysosomal membrane. In the context of disease association, less is known about microautophagy, and thus this process is not further discussed in this review [5].
CMA is a unique autophagy process that occurs without major changes in either lysosomal or autophagosomal membranes. Instead, substrate proteins are selectively delivered to the lysosome individually based on the recognition of a CMA-specific consensus motif, the pentapeptide KFERQ [6]. As an efficient cellular quality control pathway, CMA plays a vital role in protecting neurons from numerous forms of damage, including brain injury and toxic protein aggregation in the setting of neurodegenerative diseases.
Of note, macroautophagy can be divided into selective autophagy and nonselective (also known as bulk) auto-phagy [7]. Selective autophagy is characterized by high specificity in the choice and delivery of cargo for degradation, whereas nonselective autophagy is purported to randomly take up cytoplasm and lacks cargo specificity. Different forms of selective autophagy are named based on the type of digested cargo, such as mitophagy, reticulophagy, pexophagy, aggrephagy, etc. Mitophagy, which is the best-understood among all the selective autophagy types, plays an important role in protection against many diseases, particularly several neurodegenerative diseases, as it assists with the clearance of damaged mitochondria and regulates mitochondria quality control.
To provide a comprehensive background for the following discussion in diseases, here we briefly describe how nonselective autophagy, mitophagy, and CMA are regulated in human cells. The process of macroautophagy involves a set of autophagy-related (ATG) proteins in a hierarchical manner. Nonselective autophagy is controlled by MTOR and AMPK signaling pathways in response to changes in nutrient conditions such as the availability of glucose and amino acids, or other types of stress. The first group of ATG proteins involved in the process is the ULK1 kinase complex, composed of ULK1, RB1CC1/FIP200, ATG13, and ATG101, which is responsible for the phosphorylation of certain downstream factors. Subsequently, the class III phosphatidylinositol 3-kinase (PtdIns3K) complex, which is composed of various proteins depending on the stage of the pathway being regulated, including BECN1/Beclin 1, PIK3C3/VPS34, PIK3R4/VPS15, ATG14, NRBF2, and UVRAG, is activated to synthesize phosphatidylinositol-3-phosphate (PtdIns3P) for the purpose of nucleation of the membrane that will form the autophagosome [8]. The ATG9 trafficking system, which is composed of ATG2A or ATG2B, WDR45/WIPI4 and the transmembrane protein ATG9A, supplies membrane and functions in elongation of the initial sequestering compartment, termed the phagophore. In addition, two ubiquitin-like (Ubl) conjugation systems participate in membrane expansion, involving the Ubl proteins ATG12 and Atg8-family proteins (including the LC3 and GABARAP subfamilies). ATG12 is conjugated to ATG5 via the action of E1 and E2 enzymes ATG7 and ATG10, and subsequently binds ATG16L1. Atg8-family proteins are conjugated to the membrane-resident lipid phosphatidylethanolamine (PE). The attachment of PE requires the action of the protease ATG4, the E1 enzyme ATG7, the E2 enzyme ATG3, and the ATG12–ATG5-ATG16L1 complex acting as an E3 enzyme. After completion of expansion of the phagophore and sequestration of substrates, the autophagosome is formed. The outer autophagosomal membrane fuses with a lysosome or first with an endosome (to generate an intermediate compartment, termed an amphisome) to form an autolysosome. Subsequent to these fusion events, the inner autophagosome membrane will be degraded to expose the enclosed cargo to lysosomal hydrolases. Different from what is characterized in yeast, the ATG conjugation systems may not play a key role in autophagosome formation or closure, but instead they greatly affect degradation efficiency of the inner autophagosomal membrane [9]. The fusion step relies on various factors, including RAB GTPases, the HOPS complex, SNAREs and other accessory proteins [10]. Last, the lysosomal hydrolases degrade the cargo, and the resulting metabolites get released through permeases present on the autolysosomal membrane.
Mitophagy, the selective autophagy of mitochondria, involves the specific engulfment of damaged or superfluous mitochondria into phagophores through a process involving cargo recognition. There are several pathways that mediate mitophagy [11]. Among them, PINK1-PRKN/PARK2/parkin-mediated mitophagy has been the most fully characterized. Under mitochondrial stress conditions, the kinase PINK1 acts in two ways (Fig. 1), including (1) inducing mitophagy directly via generation of phospho-ubiquitin and subsequent recruitment of autophagy receptors such as CALCOCO2/NDP52 and OPTN (optineurin); [12] (2) activating the E3 ubiquitin ligase PRKN, which generates ubiquitin chains on mitochondrial proteins, thus recruiting other receptors [13].
In CMA (Fig. 2), the chaperone HSPA8/HSC70 recognizes the CMA target motif KFERQ of cytosolic proteins and transports the substrate to the lysosomal membrane protein receptor LAMP2A. Once bound to LAMP2A, the substrate unfolds and is translocated across the lysosomal membrane directly into the lumen where it is degraded [14].
Fig. 2. Schematic model of chaperone-mediated autophagy.
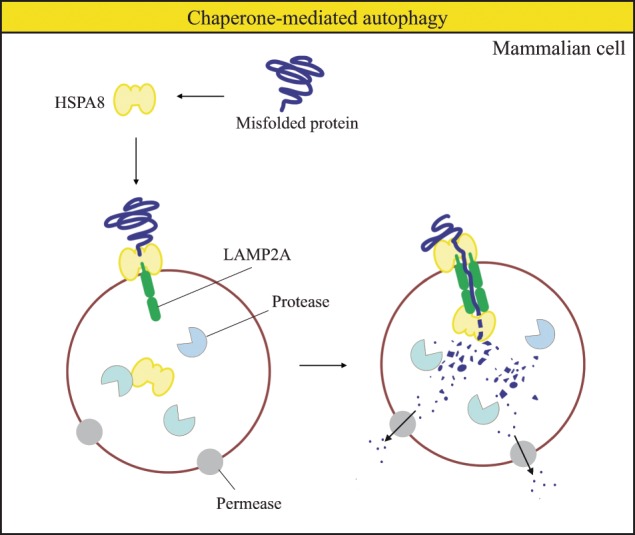
In chaperone-mediated autophagy, cytosolic proteins with a KFERQ motif are first recognized by HSPA8. The substrate-chaperone complex binds to a LAMP2A monomer. Subsequently, substrate translocation occurs via substrate unfolding and LAMP2A multimerization. Eventually, substrates are degraded in the lysosome and the resulting products are reused in the cell.
Autophagy and cancer
As mentioned in the Autophagy Machinery section, macroautophagy (hereafter autophagy in this section) works as a major survival mechanism by eliminating damaged organelles and protein aggregates, as well as promoting bioenergetic homeostasis, particularly under metabolic stress during nutrient deprivation. Thus, it is conceivable that autophagy is closely correlated with cancer pathology indicating that there are therapeutic implications. Intriguingly, autophagy plays context-dependent roles in cancer biology due to its complicated nature. In this section, we address the progress and remaining questions in understanding the dual roles of autophagy in cancer initiation and development, as well as efforts that have been made with regard to autophagy inhibition for clinical intervention.
Bidirectional roles of autophagy in cancer
Autophagy has been broadly recognized as a double-edged sword with distinct roles in tumor suppression and growth promotion [15]. Autophagy can be tumor-suppressive by preventing cancer initiation with its ability of eliminating oncogenic, unfolded or aggregated proteins and damaged organelles. Alternatively, autophagy can also be tumor-promotive through metabolite recycling to support tumor metabolism and cancer cell survival, and as a cytoprotective mechanism used by cancer cells in response to anticancer treatments.
Autophagy and tumor suppression
Autophagy serves as a tumor-suppressive mechanism through multiple established roles, including maintenance of genomic stability, eradication of endogenous sources of ROS, elimination of oncogenic proteins and induction of the immune response [16].
BECN1 is the first-identified ATG gene with recurrent genetic alterations in cancers. BECN1 is monoallelically deleted in 75% of the cases in ovarian cancers and 50% in breast cancers [17]. Heterozygous disruption of Becn1 in mice leads to increased incidence of spontaneous malignancies. By restoring BECN1 expression, breast cancer growth in the Becn1+/− mouse can be inhibited. Compelling evidence indicates that BECN1 is a haploinsufficient tumor suppressor gene [18]. Consistently, genetic modulations of several other ATG genes encoding components of the core machinery (i.e., proteins involved in autophagosome formation) are also identified in human cancers and lead to tumorigenesis in mice. Over 25% of gastric and colorectal cancers with microstatellite instability harbor frameshift mutations in ATG2B, ATG5, ATG9B, and ATG12 [19]. The identified ATG mutations are typically loss-of-function, as most of them lead to premature stops in ATG protein synthesis. A recent study demonstrates that in multiple tumor types, ATG5 somatic mutations prevent ATG12 conjugation by disrupting ATG5-ATG16L1 interactions, which leads to proteasomal degradation of ATG12 and ATG16L1, and thus inhibits autophagy. Furthermore, ATG16L2 is overexpressed in these tumor cells and functions as an endogenous dominant-negative inhibitor of autophagy by competing with ATG16L1 for ATG5 interaction [20]. Either mosaic knockout of Atg5 or liver-specific Atg7 deletion is able to cause benign liver tumor development in mice [21]. Beyond direct evidence from studies involving ATG genes, the antitumor effect of autophagy is supported by the fact that this process is stimulated by some tumor suppressors, including PTEN, TSC1-TSC2, STK11/LBK1, DAPK, and the most mutated tumor suppressor, TP53/p53 [22, 23].
Questions
Interestingly, biallelic BECN1 mutations have not been discovered in human cancers yet, and biallelic deletion of Becn1 is lethal in mice. This likely reflects the fact that autophagy is an essential process, and the complete elimination of autophagic function would result in embryonic lethality [24, 25], or, if occurring later in development, would cause severe physiological defects. Thus, the partial autophagy activity resulting from heteroallelic loss of BECN1/Becn1 is adequate to fulfill critical roles of auto-phagy that allow survival of the organism. Furthermore, this phenomenon might be explained by the type I interferon (IFN) signaling promotion and retinoic acid-inducible gene I (RIG-I)-like-receptors (RLR)-mediated anti-viral role of BECN1, which is not associated with autophagy [26]. From another perspective, the observation of monoallelic mutated BECN1/Becn1 suggests the requirement of functional autophagy for malignant transformation.
Although the overall mutation rates of ATG genes in gastric and colorectal cancers is above 25%, an interesting phenomenon is that loss of, or mutations in, single core ATG genes is uncommon in cancers. To systematically understand how autophagy genes may be involved in individual types of tumor initiation and progression, several groups have reported the global identification of mutations in ATG genes. In a large-scale human genomic analysis of somatic mutations in ATG genes across 11 cancer types, the core autophagy machinery was not targeted in almost all cancers [27], although many genes closely related to autophagy (for example, LRRK2 [leucine rich repeat kinase 2], the protein product of which is characterized controversially as both a positive and negative regulator in autophagy) have been identified with a relatively high mutation rate (frequency > 10%). Accordingly, the core autophagy machinery is very likely to escape from genomic alterations in cancers, in part because autophagy has an essential role in maintaining nuclear and mitochondria genomic stability [28]. The question of how these genes escape mutation should receive additional attention. Further investigation on this question may provide novel insights into targeting the genome in disease treatment.
Autophagy and tumor promotion
Once a tumor is developed, autophagy assists tumor survival and the metastatic cascade by providing beneficial metabolites under severe survival pressure in the tumor microenvironment (e.g., metabolic, hypoxic, genotoxic, and oxidative stresses) [29].
Many studies have demonstrated the vital role of autophagy in promoting tumorigenesis and growth. For example, Rb1cc1 deletion in mice suppresses breast cancer initiation and progression, suggesting the importance of autophagy in oncogene-induced tumors in vivo [30]. In addition, a few human studies have investigated how autophagy deficiency affects tumor growth from an in vitro perspective. BECN1 or ATG7 silencing eliminates the self-renewal ability and proliferation of cancer stem cells (CSCs) in human ALDH+ breast cancers in vitro [31, 32]. Knockout of ATG7, ATG12, or LC3 suppresses the maintenance of CD44+ CD24−/low breast CSCs in vitro [33, 34]. Nonetheless, it should be noted that ATG7 deficiency in several KRAS-driven tumor lines fails to inhibit cell proliferation in vitro as well as tumorigenesis in vivo [35]. The controversial observations in different tumor types emphasize the importance of further investigation into how autophagy affects the numbers of cancer cells differently depending on the type of cancer.
Questions
As aforementioned, the supportive role of autophagy in tumor progression and metastasis has been mostly clarified exploiting CSCs, which are known for self-renewal ability and potential for malignancy [36]. Intriguingly, even compared with other stem cells, CSCs harbor a higher basal autophagy level and often present different properties in response to autophagy modulation [37, 38]. For example, autophagy blockage in CSCs causes cell death in chronic myeloid leukemia CD34+ progenitor cells, but leads to the expansion of acute myeloid leukemia progenitor cells in hematopoietic stem cells (HSCs), suggesting autophagy may affect CSCs in a more complex manner with regard to origin, maintenance and differentiation [39]. It remains unclear as to the mechanism that endows CSCs with unique autophagy features. Currently, strategies for eradicating CSCs is a novel focus in cancer treatment. However, the question of how to specifically target CSCs in vivo has not been answered properly.
In the established model, tumor growth inhibition is prone to autophagy blockage. However, some opposite examples exist. Human ERBB2/HER2 (erb-b2 receptor tyrosine kinase 2), which is an oncogenic receptor tyrosine kinase, interacts with BECN1 and inhibits autophagy in breast cancer cells. Disrupting ERBB2-BECN1 binding and robustly inducing autophagy through either a genetic or pharmacological approach surprisingly inhibits the growth of ERBB2-positive tumors [40]. This observation is counter to the current general model that autophagy promotes tumor growth, and thus many questions are raised by this study. What accounts for the inhibition of both tumorigenesis and tumor progression by autophagy enhancement? Does it specifically occur in the receptor tyrosine kinase-driven breast cancer setting? Answers to these questions are critical to deepen our understanding of the bidirectional roles of autophagy in cancers and to make it possible to design corresponding therapeutic strategies.
Autophagy and cancer therapeutic applications
Proteins encoded by genes with altered expression levels are frequently considered as potential prognostic markers of cancers. It has been demonstrated that a high expression level of several ATG genes is significantly associated with a high patient survival rate in most cases, revealing the potential of ATG proteins as favorable prognostic markers [41, 42]. However, there are also some ATG proteins considered to be unfavorable prognostic markers in diagnosis [41]. The expression levels of ATG genes are variously regulated in distinct cancer types. Upregulation of ATG16L2, CAPN2, and TP63, and downregulation of ATG5 and five other genes related with autophagy (SIRT1, RPS6KB1, PEX3, UVRAG, and NAF1) are identified as markers with a high risk of disease recurrence in colon cancers, whereas in gastric cancer and pancreatic ductal adenocarcinoma, ULK1, BECN1, ATG3, and ATG10 are favorable prognostic factors [43, 44].
It is widely accepted that acute autophagy inhibition is effective in cancer treatment in two ways, including tumor-specific autophagy inhibition and systemic inhibition [45]. Tumor cell-specific inhibition of autophagy disrupts tumor metabolism, causing abnormal metabolic consequences, including redox imbalance, deficient mitochondrial metabolism, impaired energy homeostasis, and decreased nucleotide pools. All these effects together lead to growth impairment and death of tumor cells. Systemic inhibition of autophagy changes the microenvironment of tumors, impairs metabolic crosstalk circuits within tumor and stroma cells, and may alter host metabolism to achieve unexpected results. For example, systemic acute deletion of Atg7 in adult mice for five weeks is enough to shut off MTOR and MAP kinase signaling and lead to tumor cell death, while most normal tissues grow [46].
From a pharmacological perspective, the drug development targeting autophagy involves two primary directions, including (1) lysosomal inhibitors and derivatives such as chloroquine (CQ) and hyxdroxychloroquine (HCQ) [35, 47, 48], and (2) genomic alterations of ATG genes in the whole body or a specific organ [49]. However, although efficient in reducing tumor growth, most studies encounter the problem that autophagy disruption results in inflammation and histological tissue damage [46]. A recent study generated an inducible dominant-negative ATG4BC74A mutant mouse model, which is able to reversibly manipulate autophagy without a complete blockage. Interestingly, intermittent autophagy inhibition decreases fully-formed tumor growth with a tolerable toxicity in mice [50]. The inducible model can be regarded as a “genetic drug”, which mimics how a pharmacological inhibitor functions in a therapeutic scenario, rationally suggesting that intermittent dosing of autophagy inhibitors is a promising strategy in cancer treatment.
Questions
Although the recognition of ATG proteins as potential prognostic markers suggests the promising applicability of linking ATG proteins to clinicopathological parameters, a comprehensively assessed model is lacking. As noted above, only a small set of ATG genes are transcriptionally and translationally regulated in each cancer type, but the mechanism of the cancer-type specific autophagy signature is not clear. Furthermore, the correlation between the regulated ATG proteins and the type of cancer remains obscure in most cases. One possible reason is that many ATG proteins have multiple nonautophagic functions in several cellular pathways, including apoptosis, protein secretion, transcription, translation, etc. [51]. The complex nature of these proteins and pathways leads to different prognostic potential in cancer studies. Furthermore, the analysis of ATG gene expression as potential prognostic markers has been conducted within a small size of cohort. Thus, larger-scale explorations are required to guide the interpretation of various cancer-type-specific autophagy signatures. Besides, due to a lack of clinical data targeting autophagy, we have a poor understanding of whether ATG proteins can serve as predictive markers or not.
From the perspective of drug development, although CQ and HCQ are widely tested across cancers and have some relatively successful preliminary outcomes, a study exploiting KRAS-driven cancer cell lines demonstrates that ATG7 knockout cells in cancer are as sensitive as WT cells to CQ with regard to an antiproliferative effect [35]. Hence, it is in dispute as to whether the drug effect is from its role in inhibiting autophagy or other lysosomal processes with antitumor effects. Notably, a common occurrence of acquired resistance to autophagy inhibition in autophagy-dependent cancer cells was recently demonstrated [52]. The study shows that ATG7 knockout clones, following initial generation, undergo selection and adaptation to circumvent autophagy inactivation through NFE2L2 upregulation, providing an explanation accounting for the similar sensitivity to CQ in ATG7 knockout and WT cells. Moreover, this work emphasizes the need to address the question of how to reduce the incidence of adaptation to autophagy inactivation in autophagy-targeted therapies.
Another essential question about drug development under debate is which step we should inhibit to achieve viable therapeutic results, e.g., early steps of autophagosome formation, the fusion step between the autophagosome and lysosome, or the eventual degradation occurring in autolysosomes. Presumably, the BECN1-PIK3C3/VPS34 complex is not an ideal target, as these proteins also possess autophagy-independent functions in other cellular processes [53]. However, a Becn1F121A/F121A knock-in mouse model, which is associated with constitutively increased autophagy, demonstrates improved healthspan and longevity [54], pinpointing that disruption of the BECN1-BCL2 complex is an effective strategy to increase autophagy and benefit life span in mammals. The elegant study also reminds us that it is worthwhile generating autophagy-elevated models that are crossed with disease models to investigate disease targets. As autophagy may also serve as a scaffold to control the mechanism of cell death (apoptosis or necroptosis) with the requirement of ATG5 and SQSTM1/p62 [55], the development of intermittent autophagy-specific inhibitors may provide a finer therapeutic strategy. The aforementioned study exploiting the ATG4BC74A mouse model also suggests the potential of targeting ATG4 in cancers [50].
Although it is widely accepted that acute autophagy inhibition is effective in cancer treatment, an unsolved major issue of drug development is the side-effect of autophagy inhibition. Toxicity and histological tissue damage are often observed after autophagy-blocking treatments [46]. Thus, how to set an appropriate dose of autophagy inhibitors based on an individual patient’s requirements still remains a big challenge.
A large number of clinical trials testing autophagy inhibitors in cancers are undergoing, but there is still a lack of methods for tracking the response to autophagy in cancer patients. Currently, the most common way to monitor autophagy in cancer cells is through typical LC3 and SQSTM1/p62 markers, as well as tandem mRPF-GFP-LC3 fluorescence [56]. However, due to the participation of these autophagosomal markers in autophagy, the assays may not directly reveal the activity of the final autophagic degradation stage, and it is very hard to monitor the process over time. From this perspective, it is important to identify novel markers that are not involved as part of the autophagy machinery [41]. In addition, posttranslational modifications (phosphorylation, ubiquitination, and acetylation) are important in autophagy regulation. If novel methods that can precisely detect these modifications can be developed, it will provide important and useful information for cancer treatment.
Autophagy and neurodegenerative diseases
Abnormal protein accumulation in nerve cells is a hallmark of several neurodegenerative diseases, and harbors a direct link with impaired macroautophagy and CMA. Autophagy contributes to the protection against neurodegeneration due to its ability to remove damaged mitochondria and toxic proteins or micro-aggregates (Table 1) [57]. Compared with cancers, the roles of autophagy in neurodegenerative diseases seem to be even more mysterious, and relevant preclinical studies are lagging far behind. Here, we underline the relevance and unanswered questions between autophagy and some of the most common neurodegenerative diseases including Parkinson disease (PD), Alzheimer disease (AD), and Huntington disease (HD).
Table 1.
Neurodegenerative disorders (except PD, AD and HD) and correlated core ATG genes with genetic mutations.
| Disease | Gene(s) | Reference |
|---|---|---|
| ß-propeller protein-associated neurodegeneration (BPAN) | WDR45/WIPI4 | [115] |
| Amyotrophic lateral sclerosis (ALS) | PINK1, PRKN/PARK2, SQSTM1/p62, OPTN, and TBK1 | [116–118] |
| Ataxias | ATG4D, ATG5, VPS13D | [119–121] |
| Atypical apraxia of speech (AAS) | SQSTM1/p62 | [122] |
| Childhood-onset neurodegeneration | SQSTM1/p62 | [123] |
| Developmental and epileptic encephalopathy (DEE) | WDR45/WIPI4 | [124] |
| Early-onset epileptic encephalopathy (EOEE) | WDR45/WIPI4 | [125] |
| Frontotemporal dementia (FTD) | SQSTM1/p62 | [118] |
| Hereditary sensory and autonomic neuropathy II (HSANIIB) | RETREG1 | [126] |
| Neuromyelitis optica (NMO) | ATG5 | [127] |
| Normal-tension glaucoma (NTG) | OPTN, TBK1 | [128] |
| RETT-like syndrome | WDR45 | [129] |
Autophagy and Parkinson disease
PD is a progressive brain disorder with motor deficiency [58]. Aggregates of SNCA/alpha-synuclein, the main component of Lewy bodies, is the primary pathological hallmark of PD.
Notably, SNCA is a CMA substrate. Numerous studies exploiting animal models and cell cultures have demonstrated the involvement of CMA in the clearance of abnormal SNCA. Inhibition of CMA leads to aberrant SNCA accumulation and dopaminergic neurodegeneration in rats [59, 60]. Mutant SNCA that lacks the CMA recognition motif is degraded less efficiently in neuronal cell lines [61]. Overexpression of SNCA in mice is accompanied by a marked elevation of LAMP2A and HSPA8 [62]. Consistent with these observations, analysis of PD patients indicates that the expression of LAMP2A and HSPA8 is decreased by genetic alterations in the promoter region and microRNA upregulation, further confirming the involvement of compromised CMA in early PD development [63–66].
Accumulating evidence indicates that macroautophagy also plays a role in PD etiology. For example, extensive genetic analysis of PD patients confirms that abnormal expression levels of ATG genes of the conjugation machinery may contribute to PD. These genes include ATG5, ATG7, ATG12, and LC3B [67–70]. Consistently, macroautophagy dysregulation has been identified in the brain tissues from PD patients and animal models, suggesting the important involvement of macroautophagy in protein aggregate degradation in PD [71].
Overexpression and aggregation of SNCA are also associated with dysfunctional mitochondria, further implying impaired mitophagy in PD [72]. An interesting fact is that SNCA accumulation is able to cause delayed mitophagy. The ubiquitination of RHOT/Miro, an adaptor on the outer mitochondria membrane, on damaged mitochondria is required to promote recruitment of PRKN and mitochondrial clearance through mitophagy [73]. RHOT/Miro also plays a role in mitochondrial motility, and needs to be removed in order to allow efficient degradation of the organelle. A recent report indicates that SNCA accumulation leads to RHOT/Miro upregulation, and consequently retards mitophagy [74]. In addition, lowering RHOT/Miro levels is able to rescue the mitophagy phenotype and neurodegeneration in iPSC-derived neurons with SNCA mutations [74]. In addition to SNCA, gain-of-function or loss-of-function mutations in PD-associated genes in patients, including those in PRKN, PINK1, PARK7/DJ-1, LRRK2, and GBA, cause mitophagy and disturbed mitochondria homeostasis [75].
A recent study has indicated that inflammation after exhaustive exercise of mice with Prkn and Pink1 deletions can be fully rescued by the loss of STING, a pivotal regulator of the type I interferon response to cytosolic DNA, suggesting that dysfunctional mitophagy may lead to neurodegenerative diseases through abnormal inflammatory signaling [76].
Questions
As mentioned above, mutated PINK1, PRKN, and LRRK2 are frequently identified in autosomal recessive PD cases, suggesting the role of mitophagy as a PD causative factor. However, several pathways that mediate mitophagy, and mutations in other genes encoding mitophagy-related proteins, have not been yet been linked with PD. Whether PD etiology only involves PINK1-PRKN-mediated mitophagy or other mitophagy pathways is not clear. Of note, PRKN and PINK1 have been identified as suppressors of an immune-response-eliciting pathway, suggesting their role in autoimmunity [77]. Besides, the ubiquitination status of PRKN is essential for the selective escape of certain mitochondrial proteins to the endoplasmic reticulum to avoid the degradation that would otherwise occur in the mitophagy process [78]. The involvement of PRKN and PINK1 in all these biological processes other than autophagy makes it difficult to distinguish how much mitophagy contributes to neurodegenerative diseases.
Also, neither PRKN- nor PINK1-deficient mice present many PD-relevant phenotypes, including nigrostriatal, cognitive and noradrenergic deficits [79, 80]. To note, pink1 knockout and WT mice exhibit comparable levels of basal neural mitophagy, suggesting that the precise role of PINK1 in both basal and stress-induced mitophagy in vivo remains to be further defined [81]. Understanding the mechanism that prevents mutant mice from exhibiting robust signs of PD is essential to provide novel insights in PD treatment and advance our understanding of the role of PINK1-PRKN-mediated mitophagy in PD.
Whereas LRRK2 is considered to play an important role in macroautophagy regulation and greatly affects PD pathogenesis, there are many discordant reports, which lead to a debate as to whether autophagy is positively or negatively regulated by LRRK2 [82]. For example, LRRK2 deletion and LRRK2G2019S overexpression in human cells, which have opposite effects on adjusting LRRK2 kinase activity, lead to the same result of disrupting macroautophagy [83, 84]. This outcome may be associated with distinct roles of LRRK2 in the initiation and degradation process and its feedback loops. It requires further investigation to integrate these seemingly contradictory data and elucidate the dynamic nature of LRRK2 in autophagy. The answer of how LRRK2 regulates autophagy may shed light on understanding the molecular mechanism of PD pathogenesis.
Autophagy and Alzheimer disease
AD is a progressive neurodegenerative disease, with the major symptoms of memory impairment, cognitive defects, visual-spatial deficiency, and personality changes. AD patients present extracellular amyloid-beta (Aβ) accumulation and intracellular hyperphosphorylated MAPT (microtubule-associated protein tau) [85].
Similar to what happens in PD, the pathogenic mechanism of AD is, although not completely understood, closely related to mitophagy deficiency and CMA. The MAPT protein is identified as a substrate of both CMA and macroautophagy. Incomplete CMA of MAPT generates amyloidogenic fragments, and impaired macroautophagy fails to remove the MAPT aggregates [86]. In the AD mouse model, Aβ accumulation is accompanied by a cascade of upregulated mRNA levels of mitophagy-related genes, such as Sqstm1, Prkn, Dnm1l, Becn1, Bnip3, Pink1, and Map1lc3 [87, 88]. Similarly, patients with mild cognitive impairment display upregulation of several mitophagy transcripts [87].
Accumulation of autophagosomes in neurons is a prominent feature of AD [89]. Consistent with this hallmark, in a comprehensive study that utilized a customized microarray compromising neuropathology-associated genes to evaluate the entire autophagic process in CA1 neurons of hippocampus from early to late stages of AD, upregulation of autophagy and elevated lysosomal biogenesis were observed at the early stage. Autophagosome-lysosome fusion is not altered prominently in the whole process, whereas autophagic flux is impeded progressively probably due to failure of substrate clearance [90].
Notably, a recent study elucidated that mitophagy enhancement by urolithin A treatment is able to abolish AD-related MAPT hyperphosphorylation in human neuronal cells [91]. In addition, restoration of neuronal mitophagy also reverses cognitive decline and ameliorates p-MAPT pathology in both C. elegans and mouse AD models [91, 92]. This compelling evidence not only advances our knowledge of the important role of mitophagy defects in AD initiation and progression, but also broadly provides a promising therapeutic strategy for AD patients.
Questions
An interesting discovery is that APP (amyloid-beta precursor protein), which can be processed to Aβ, also contains a KFERQ motif at its C terminus, suggesting a role of CMA in Aβ clearance. By contrast, the deletion of the KFERQ motif fails to abolish its interaction with HSPA8, but increases the secretion of sAPP α/β, an intermediate product during APP processing [93]. Several questions can be considered: What is the role of KFERQ in autophagy other than as a CMA targeting motif in this disease context? How does APP processing develop this unique feature? As over 30% of cytosolic proteins possess a KFERQ motif to target them to CMA, these questions are critical to understand and further couple CMA and macroautophagy regulation in the AD context.
Autophagy and Huntington disease
HD is a progressive neurodegenerative disease with a primary feature of abnormal movement, following by cognitive dysfunction, motor deficits, and emotional problems. HD is caused by aberrant production and accumulation of mutant HTT (huntingtin; mHTT) protein due to an expansion of a polyglutamine (polyQ) tract [94].
HD is associated with abnormal CMA through failure to clear the HTT proteins [95, 96]. An interesting therapeutic strategy is to target mHTT with a fusion molecule of polyQ binding peptide 1 (QBP1) and the HSPA8-binding motif, as QBP1 specifically binds to an expanded polyQ tract instead of the polyQ motif in normal HTT. Delivery of the fusion molecule successfully ameliorates the disease phenotype in mice, suggesting a promising therapeutic solution [97]. Similarly, a recent study exploiting small-molecule-microarray-based screening, identified linker compounds for mHTT and LC3, which allows selective autophagic clearance of mHTT by delivering mHTT to phagophores, demonstrating a novel strategy to degrade disease-causative proteins through the use of phagophore-tethering compounds [98].
Not surprisingly, mounting evidence implicates the involvement of impaired macroautophagy in the HD pathogenic mechanism. mHTT accumulation is accompanied by a decreased level of basal autophagy [99]. Expanded polyQ tracts regulate BECN1 deubiquitination and decrease BECN1 protein levels, and thus are able to inhibit starvation-induced autophagy [100]. Polymorphisms in ATG7 are also associated with early age at onset in HD [101]. Moreover, enhanced autophagy by inhibition of either MTOR or the insulin signaling pathway facilitates the clearance of HTT aggregates and reduces toxicity in both fly and mouse models of HD [102, 103].
Questions
Previous evidence suggests autophagy suppresses HTT aggregate formation. Surprisingly, depletion of SQSTM1/p62, an autophagy receptor, significantly diminishes nuclear inclusions, partially abolishes disease phenotypes and extends life span in the HD mouse model [104]. The mechanism by which the controversial results occur between cargo receptor deletion and upstream signal alternation remains unclear (SQSTM1/p62 has additional roles in the cell, including regulation of MTOR). It has been proposed that the unexpected data might be caused by increased formation and accumulation of cytosolic mutant HTT due to deficient autophagy, and decreased HTT translocation into the nucleus after Sqstm1/p62 knockout [104]. It is important to perform further investigations affecting both core ATG machinery and cargo receptor proteins to fully understand the HD pathophysiology regarding its role in enhancing or diminishing autophagy.
As HTT is expressed ubiquitously and differently across the neuronal subtypes, more questions, such as how autophagy is utilized by the different cells of the CNS and how autophagy is regulated in distinct neuronal compartments, must be addressed to advance our understanding of HD pathogenesis.
As described above, the two proof-of-concept papers for drug discovery aim at harnessing autophagy for misfolded protein clearance in HD and other neurodegenerative diseases [97, 98]. However, it is possible that no linker compounds can be identified directly targeting LC3 and the relevant disease-causative proteins. How to tether LC3 and target proteins indirectly without affecting autophagy function will be a crucial issue.
Autophagy and other neurodegenerative diseases
Protein turnover and aggregation is involved in the cellular etiology of many other neurodegenerative diseases besides PD, AD, and HD [105]. Autophagy is able to, at least partially, account for the cellular pathology in these diseases (Table 1).
Similar to what occurs in cancers, autophagy may also play multiple roles in neurodegenerative diseases, largely depending on disease progression. For example, in amyotrophic lateral sclerosis (ALS), autophagy is necessary for neuromuscular innervation in the early state, and once the disease is developed, autophagy is responsible for promoting progression in a nonautonomous manner [106].
Questions
Although most of the neurodegenerative disorders are associated with CMA and mitophagy, the mutation frequency of some ATG genes still dramatically varies between diseases. For example, mutations in SQSTM1/p62, TBK1 and OPTN are often better correlated with ALS than PD, although the pathogenesis of both diseases are closely related to mitophagy. The precise mechanism behind the disease-specific mutations requires further investigation.
To date, most autophagy studies have been performed in nonneuronal cells. However, neurons may have a unique autophagy mechanism. For example, the MTORC1 inhibitor rapamycin is usually sufficient to induce autophagy in nonneuronal cells, but it has been reported that neither rapamycin nor starvation treatment is able to activate autophagy in neurons [107]. Instead, the combination of catalytic inhibitors of both MTORC1 and MTORC2 complexes robustly induces autophagy in this cell type [108]. These results suggest that the major physiological functions of autophagy in neurons are not sensitive to amino acid recycling in response to starvation as occurs in nonneuronal cells. Furthermore, neurons are highly postmitotic cells with a special morphology such as long dendrites and axons, and it has been reported that neuronal autophagosome biogenesis follows a spatially-ordered assembly pathway [109]. Briefly, autophagosomes form in the distal axon tip, and then traffic in a retrograde manner back to the soma [110]. The mechanism for how neuronal autophagy is precisely orchestrated remains unclear and requires comprehensive investigation. Understanding the spatiotemporal regulation of autophagy may contribute to our understanding of the unique vulnerability of neurons to defective mitophagy.
The exact mechanisms of how an autophagy deficit contributes to neurodegenerative disease remain unclear in most neurodegenerative disorders [111]. Although neurodegenerative diseases tend to develop in the aged brain, we should focus more on the resulting phenotypes of auto-phagy deficiency at an early stage. Solving this conundrum will help us shape a better understanding of neurodegenerative pathophysiology and develop corresponding therapies.
As mentioned previously in the Autophagy and Cancer section, it is worth noting that currently there is a lack of assays to validly detect autophagy defects in humans. Most commonly used assays are limited to intermediates in the pathway, which can lead to misinterpretation and largely constrain the performance of appropriate clinical studies. Thus, there is a great need to develop target-engaged biomarkers which can measure autophagy activity accurately and comprehensively to test putative autophagy inducers and inhibitors, and consequently guide the buildup of a dosing strategy [111].
Conclusions
Due to space limitations, we are not able to cover every corner of the enormous field of autophagy and disease in detail. To provide the readers with a more comprehensive understanding of how autophagy is involved in disease, we present a summarization of diseases with documented mutations in core ATG genes (Table 2) [112]. It is noteworthy that several core ATG genes are also involved in distinct membrane-trafficking processes other than canonical autophagy, including LC3-associated phagocytosis, ATG gene-dependent secretion, LC3-regulated exocytosis, LC3-associated endocytosis, etc. [113, 114]. The existence of these pathways requires awareness of whether pathological phenotypes of ATG gene-associated disease models is due to abnormal autophagy or defects in other pathways.
Table 2.
Diseases associated with abnormal core ATG machinery (except cancer and neurodegenerative diseases) and correlated genes.
| Gene(s) | Reference | |
|---|---|---|
| Inflammatory diseases | ||
| Behçet disease (BD) | ATG5 | [130] |
| Celiac disease (CeD) | ATG7, BECN1 | [131] |
| Crohn disease (CD) | ATG4A, ATG4D, ATG9A, ATG16L1, ULK1, CALCOCO2/NDP52 | [132–135] |
| Rheumatoid arthritis (RA) | ATG5 | [136] |
| Systemic lupus erythematosus (SLE) | ATG5, MAP1LC3B | [137, 138] |
| Systemic sclerosis | ATG5 | [139] |
| Vogt–Koyanagi–Harada syndrome (VKHS) | ATG10 | [130] |
| Respiratory diseases | ||
| Asthma | ATG5 | [140] |
| Chronic obstructive pulmonary disease (COPD) | ATG16L1, EGR1 | [141] |
| Myopathies | ||
| Distal myopathies with rimmed vacuoles (DMRV) | SQSTM1/p62 | [142] |
| Sporadic inclusion body myositis (SIBM) | SQSTM1/p62, FYCO1 | [143] |
| Others | ||
| Kashin–Beck disease (KB) | ATG4C | [144] |
| Paget disease of bone (PDB) | ATG5, ATG10, ATG16L1, SQSTM1/p62 | [145, 146] |
In this review, we focus on autophagy in cancer and neurodegenerative diseases, two of the most clinically problematic diseases. Although a plenitude of studies has emphasized the vital role of autophagy in diseases, current work only represents the first steps in understanding this dynamic and complex process in the disease context. A detailed integration of molecular understanding and clinical investigation is necessary for therapeutic applications.
It is notable that in many cases the blockage of cancer through autophagy modulation progressively leads to the development of neurodegeneration and vice versa, presumably suggesting their antagonistic relationships. Consequently, we should be cautious when considering whether simply elevating or inhibiting autophagy is or is not a feasible approach to treat diseases. The complexity of auto-phagy in the interplay between these diseases should be further untangled, and therefore provide a rational approach for modulating autophagy in an appropriate context-dependent manner.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Edited by F. Pentimalli
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Klionsky DJ. The molecular machinery of autophagy: unanswered questions. J Cell Sci. 2005;118:7–18. doi: 10.1242/jcs.01620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen Y, Klionsky DJ. The regulation of autophagy–unanswered questions. J Cell Sci. 2011;124:161–70. doi: 10.1242/jcs.064576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Parzych KR, Klionsky DJ. An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal. 2014;20:460–73. doi: 10.1089/ars.2013.5371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Feng Y, He D, Yao Z, Klionsky DJ. The machinery of macroautophagy. Cell Res. 2014;24:24. doi: 10.1038/cr.2013.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mijaljica D, Prescott M, Devenish RJ. Microautophagy in mammalian cells: revisiting a 40-year-old conundrum. Autophagy. 2011;7:673–82. doi: 10.4161/auto.7.7.14733. [DOI] [PubMed] [Google Scholar]
- 6.Wu H, Chen S, Ammar A-B, Xu J, Wu Q, Pan K, et al. Crosstalk between macroautophagy and chaperone-mediated autophagy: implications for the treatment of neurological diseases. Mol Neurobiol. 2015;52:1284–96. doi: 10.1007/s12035-014-8933-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gatica D, Lahiri V, Klionsky DJ. Cargo recognition and degradation by selective autophagy. Nat Cell Biol. 2018;20:233. doi: 10.1038/s41556-018-0037-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Russell RC, Tian Y, Yuan H, Park HW, Chang Y-Y, Kim J, et al. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol. 2013;15:741. doi: 10.1038/ncb2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tsuboyama K, Koyama-Honda I, Sakamaki Y, Koike M, Morishita H, Mizushima N. The ATG conjugation systems are important for degradation of the inner autophagosomal membrane. Science. 2016;354:1036–41. doi: 10.1126/science.aaf6136. [DOI] [PubMed] [Google Scholar]
- 10.Kriegenburg F, Ungermann C, Reggiori F. Coordination of autophagosome–lysosome fusion by ATG8 family members. Curr Biol. 2018;28:R512–R8. doi: 10.1016/j.cub.2018.02.034. [DOI] [PubMed] [Google Scholar]
- 11.Wei H, Liu L, Chen Q. Selective removal of mitochondria via mitophagy: distinct pathways for different mitochondrial stresses. Biochimica et Biophysica Acta (BBA)-Mol Cell Res. 2015;1853:2784–90. doi: 10.1016/j.bbamcr.2015.03.013. [DOI] [PubMed] [Google Scholar]
- 12.Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524:309. doi: 10.1038/nature14893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gladkova C, Maslen SL, Skehel JM, Komander D. Mechanism of parkin activation by PINK1. Nature. 2018;559:410. doi: 10.1038/s41586-018-0224-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li W, Yang Q, Mao Z. Chaperone-mediated autophagy: machinery, regulation and biological consequences. Cell Mol Life Sci. 2011;68:749–63. doi: 10.1007/s00018-010-0565-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.White E, DiPaola RS. The double-edged sword of autophagy modulation in cancer. Clin Cancer Res. 2009;15:5308–16. doi: 10.1158/1078-0432.CCR-07-5023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Galluzzi L, Pietrocola F, Bravo-San Pedro JM, Amaravadi RK, Baehrecke EH, Cecconi F, et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015;34:856–80. doi: 10.15252/embj.201490784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112:1809–20. doi: 10.1172/JCI20039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tang H, Sebti S, Titone R, Zhou Y, Isidoro C, Ross TS, et al. Decreased BECN1 mRNA expression in human breast cancer is associated with estrogen receptor-negative subtypes and poor prognosis. EBioMedicine. 2015;2:255–63. doi: 10.1016/j.ebiom.2015.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kang MR, Kim MS, Oh JE, Kim YR, Song SY, Kim SS, et al. Frameshift mutations of autophagy‐related genes ATG2B, ATG5, ATG9B and ATG12 in gastric and colorectal cancers with microsatellite instability. J Pathol. 2009;217:702–6. doi: 10.1002/path.2509. [DOI] [PubMed] [Google Scholar]
- 20.Wible DJ, Chao H-P, Tang DG, Bratton SB. ATG5 cancer mutations and alternative mRNA splicing reveal a conjugation switch that regulates ATG12–ATG5-ATG16L1 complex assembly and autophagy. Cell Disco. 2019;5:1–19. doi: 10.1038/s41421-019-0110-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, et al. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011;25:795–800. doi: 10.1101/gad.2016211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lorin Séverine, Hamaï Ahmed, Mehrpour Maryam, Codogno Patrice. Autophagy regulation and its role in cancer. Seminars in Cancer Biology. 2013;23(5):361–379. doi: 10.1016/j.semcancer.2013.06.007. [DOI] [PubMed] [Google Scholar]
- 23.Mrakovcic M, Fröhlich L. p53-mediated molecular control of autophagy in tumor cells. Biomolecules. 2018;8:14. doi: 10.3390/biom8020014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci. 2003;100:15077–82. doi: 10.1073/pnas.2436255100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tsukamoto S, Kuma A, Murakami M, Kishi C, Yamamoto A, Mizushima N. Autophagy is essential for preimplantation development of mouse embryos. Science. 2008;321:117–20. doi: 10.1126/science.1154822. [DOI] [PubMed] [Google Scholar]
- 26.Cui J, Jin S, Wang R-F. The BECN1-USP19 axis plays a role in the crosstalk between autophagy and antiviral immune responses. Autophagy. 2016;12:1210–1. doi: 10.1080/15548627.2016.1173801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lebovitz CB, Robertson AG, Goya R, Jones SJ, Morin RD, Marra MA, et al. Cross-cancer profiling of molecular alterations within the human autophagy interaction network. Autophagy. 2015;11:1668–87. doi: 10.1080/15548627.2015.1067362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vessoni A, Filippi-Chiela E, Menck CF, Lenz G. Autophagy and genomic integrity. Cell Death Differ. 2013;20:1444. doi: 10.1038/cdd.2013.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kocaturk NM, Akkoc Y, Kig C, Bayraktar O, Gozuacik D, Kutlu O. Autophagy as a molecular target for cancer treatment. Eur J Pharm Sci. 2019;134:116–37. doi: 10.1016/j.ejps.2019.04.011. [DOI] [PubMed] [Google Scholar]
- 30.Wei H, Wei S, Gan B, Peng X, Zou W, Guan J-L. Suppression of autophagy by FIP200 deletion inhibits mammary tumorigenesis. Genes Dev. 2011;25:1510–27. doi: 10.1101/gad.2051011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gong C, Bauvy C, Tonelli G, Yue W, Delomenie C, Nicolas V, et al. Beclin 1 and autophagy are required for the tumorigenicity of breast cancer stem-like/progenitor cells. Oncogene. 2013;32:2261. doi: 10.1038/onc.2012.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yue W, Hamaï A, Tonelli G, Bauvy C, Nicolas V, Tharinger H, et al. Inhibition of the autophagic flux by salinomycin in breast cancer stem-like/progenitor cells interferes with their maintenance. Autophagy. 2013;9:714–29. doi: 10.4161/auto.23997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maycotte P, Jones KL, Goodall ML, Thorburn J, Thorburn A. Autophagy supports breast cancer stem cell maintenance by regulating IL6 secretion. Mol Cancer Res. 2015;13:651–8. doi: 10.1158/1541-7786.MCR-14-0487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cufí S, Vazquez-Martin A, Oliveras-Ferraros C, Martin-Castillo B, Vellon L, Menendez JA. Autophagy positively regulates the CD44+ CD24-/low breast cancer stem-like phenotype. Cell Cycle. 2011;10:3871–85. doi: 10.4161/cc.10.22.17976. [DOI] [PubMed] [Google Scholar]
- 35.Eng CH, Wang Z, Tkach D, Toral-Barza L, Ugwonali S, Liu S, et al. Macroautophagy is dispensable for growth of KRAS mutant tumors and chloroquine efficacy. Proc Natl Acad Sci. 2016;113:182–7. doi: 10.1073/pnas.1515617113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hamaı A, Codogno P, Mehrpour M. Cancer stem cells and autophagy: facts and perspectives. J Cancer Stem Cell Res. 2014;2:e1005. [Google Scholar]
- 37.Boya P, Codogno P, Rodriguez-Muela N. Autophagy in stem cells: repair, remodelling and metabolic reprogramming. Development. 2018;145:dev146506. doi: 10.1242/dev.146506. [DOI] [PubMed] [Google Scholar]
- 38.Singh AK, Arya RK, Maheshwari S, Singh A, Meena S, Pandey P, et al. Tumor heterogeneity and cancer stem cell paradigm: updates in concept, controversies and clinical relevance. Int J Cancer. 2015;136:1991–2000. doi: 10.1002/ijc.28804. [DOI] [PubMed] [Google Scholar]
- 39.Auberger P, Puissant A. Autophagy, a key mechanism of oncogenesis and resistance in leukemia. Blood. 2017;129:547–52. doi: 10.1182/blood-2016-07-692707. [DOI] [PubMed] [Google Scholar]
- 40.Vega-Rubín-de-Celis S, Zou Z, Fernández ÁF, Ci B, Kim M, Xiao G, et al. Increased autophagy blocks HER2-mediated breast tumorigenesis. Proc Natl Acad Sci. 2018;115:4176–81. doi: 10.1073/pnas.1717800115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bortnik S, Gorski SM. Clinical applications of autophagy proteins in cancer: from potential targets to biomarkers. Int J Mol Sci. 2017;18:1496. doi: 10.3390/ijms18071496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ko YH, Cho Y-S, Won HS, Jeon EK, An HJ, Hong SU, et al. Prognostic significance of autophagy-related protein expression in resected pancreatic ductal adenocarcinoma. Pancreas. 2013;42:829–35. doi: 10.1097/MPA.0b013e318279d0dc. [DOI] [PubMed] [Google Scholar]
- 43.Mo Shaobo, Dai Weixing, Xiang Wenqiang, Li Yaqi, Feng Yang, Zhang Long, Li Qingguo, Cai Guoxiang. Prognostic and predictive value of an autophagy-related signature for early relapse in stages I–III colon cancer. Carcinogenesis. 2019;40(7):861–870. doi: 10.1093/carcin/bgz031. [DOI] [PubMed] [Google Scholar]
- 44.Cao Q-H, Liu F, Yang Z-L, Fu X-H, Yang Z-H, Liu Q, et al. Prognostic value of autophagy related proteins ULK1, Beclin 1, ATG3, ATG5, ATG7, ATG9, ATG10, ATG12, LC3B and p62/SQSTM1 in gastric cancer. Am J Transl Res. 2016;8:3831. [PMC free article] [PubMed] [Google Scholar]
- 45.Kimmelman AC, White E. Autophagy and tumor metabolism. Cell Metab. 2017;25:1037–43. doi: 10.1016/j.cmet.2017.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Karsli-Uzunbas G, Guo JY, Price S, Teng X, Laddha SV, Khor S, et al. Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov. 2014;4:914–27. doi: 10.1158/2159-8290.CD-14-0363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Junco JJ, Mancha-Ramirez A, Malik G, Wei S-J, Kim DJ, Liang H, et al. Ursolic acid and resveratrol synergize with chloroquine to reduce melanoma cell viability. Melanoma Res. 2015;25:103–12. doi: 10.1097/CMR.0000000000000137. [DOI] [PubMed] [Google Scholar]
- 48.Qin L, Xu T, Xia L, Wang X, Zhang X, Zhang X, et al. Chloroquine enhances the efficacy of cisplatin by suppressing autophagy in human adrenocortical carcinoma treatment. Drug Des Dev Ther. 2016;10:1035. doi: 10.2147/DDDT.S101701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gong C, Hu C, Gu F, Xia Q, Yao C, Zhang L, et al. Co-delivery of autophagy inhibitor ATG7 siRNA and docetaxel for breast cancer treatment. J Control Release. 2017;266:272–86. doi: 10.1016/j.jconrel.2017.09.042. [DOI] [PubMed] [Google Scholar]
- 50.Yang A, Herter-Sprie G, Zhang H, Lin EY, Biancur D, Wang X, et al. Autophagy sustains pancreatic cancer growth through both cell-autonomous and nonautonomous mechanisms. Cancer Discov. 2018;8:276–87. doi: 10.1158/2159-8290.CD-17-0952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Subramani S, Malhotra V. Non-autophagic roles of autophagy-related proteins. EMBO Rep. 2013;14:143–51. doi: 10.1038/embor.2012.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Towers CG, Fitzwalter BE, Regan D, Goodspeed A, Morgan MJ, Liu C-W, et al. Cancer cells upregulate Nrf2 signaling to adapt to autophagy inhibition. Dev Cell. 2019;50:690–703. doi: 10.1016/j.devcel.2019.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rohatgi RA, Shaw LM. An autophagy-independent function of Beclin 1 in cancer. Mol Cell Oncol. 2016;3:e1030539. doi: 10.1080/23723556.2015.1030539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fernández ÁF, Sebti S, Wei Y, Zou Z, Shi M, McMillan KL, et al. Disruption of the beclin 1–BCL2 autophagy regulatory complex promotes longevity in mice. Nature. 2018;558:136. doi: 10.1038/s41586-018-0162-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Goodall ML, Fitzwalter BE, Zahedi S, Wu M, Rodriguez D, Mulcahy-Levy JM, et al. The autophagy machinery controls cell death switching between apoptosis and necroptosis. Dev Cell. 2016;37:337–49. doi: 10.1016/j.devcel.2016.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Morgan MJ, Thorburn A. Measuring autophagy in the context of cancer. In: Tumor microenvironment. 2016;899:121–43. [DOI] [PubMed]
- 57.Yin Z, Pascual C, Klionsky DJ. Autophagy: machinery and regulation. Micro Cell. 2016;3:588. doi: 10.15698/mic2016.12.546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kalia LV, Lang AE. Parkinson disease in 2015: evolving basic, pathological and clinical concepts in PD. Nat Rev Neurol. 2016;12:65. doi: 10.1038/nrneurol.2015.249. [DOI] [PubMed] [Google Scholar]
- 59.Xilouri M, Brekk OR, Polissidis A, Chrysanthou-Piterou M, Kloukina I, Stefanis L. Impairment of chaperone-mediated autophagy induces dopaminergic neurodegeneration in rats. Autophagy. 2016;12:2230–47. doi: 10.1080/15548627.2016.1214777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xilouri M, Vogiatzi T, Vekrellis K, Park D, Stefanis L. Abberant α-synuclein confers toxicity to neurons in part through inhibition of chaperone-mediated autophagy. PLoS ONE. 2009;4:e5515. doi: 10.1371/journal.pone.0005515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vogiatzi T, Xilouri M, Vekrellis K, Stefanis L. Wild type α-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J Biol Chem. 2008;283:23542–56. doi: 10.1074/jbc.M801992200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mak SK, McCormack AL, Manning-Boğ AB, Cuervo AM, Di Monte DA. Lysosomal degradation of α-synuclein in vivo. J Biol Chem. 2010;285:13621–9. doi: 10.1074/jbc.M109.074617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sala G, Stefanoni G, Arosio A, Riva C, Melchionda L, Saracchi E, et al. Reduced expression of the chaperone-mediated autophagy carrier hsc70 protein in lymphomonocytes of patients with Parkinson's disease. Brain Res. 2014;1546:46–52. doi: 10.1016/j.brainres.2013.12.017. [DOI] [PubMed] [Google Scholar]
- 64.Murphy KE, Gysbers AM, Abbott SK, Spiro AS, Furuta A, Cooper A, et al. Lysosomal-associated membrane protein 2 isoforms are differentially affected in early Parkinson's disease. Mov Disord. 2015;30:1639–47. doi: 10.1002/mds.26141. [DOI] [PubMed] [Google Scholar]
- 65.Alvarez-Erviti L, Seow Y, Schapira AH, Rodriguez-Oroz MC, Obeso JA, Cooper J. Influence of microRNA deregulation on chaperone-mediated autophagy and α-synuclein pathology in Parkinson's disease. Cell death Dis. 2013;4:e545. doi: 10.1038/cddis.2013.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pang S, Chen D, Zhang A, Qin X, Yan B. Genetic analysis of the LAMP-2 gene promoter in patients with sporadic Parkinson's disease. Neurosci Lett. 2012;526:63–7. doi: 10.1016/j.neulet.2012.07.044. [DOI] [PubMed] [Google Scholar]
- 67.Chen D, Zhu C, Wang X, Feng X, Pang S, Huang W, et al. A novel and functional variant within the ATG5 gene promoter in sporadic Parkinson's disease. Neurosci Lett. 2013;538:49–53. doi: 10.1016/j.neulet.2013.01.044. [DOI] [PubMed] [Google Scholar]
- 68.Chen D, Pang S, Feng X, Huang W, Hawley RG, Yan B. Genetic analysis of the ATG7 gene promoter in sporadic Parkinson's disease. Neurosci Lett. 2013;534:193–8. doi: 10.1016/j.neulet.2012.12.039. [DOI] [PubMed] [Google Scholar]
- 69.Li Y, Huang J, Pang S, Wang H, Zhang A, Hawley RG, et al. Novel and functional ATG12 gene variants in sporadic Parkinson's disease. Neurosci Lett. 2017;643:22–6. doi: 10.1016/j.neulet.2017.02.028. [DOI] [PubMed] [Google Scholar]
- 70.Xu J, Yang Y, Pang S, Huang W, Qin X, Hawley RG, et al. Identification of a novel 21bp-insertion variant within the LC3B gene promoter in sporadic Parkinson’s disease. Transl Res. 2013;161:441–3. doi: 10.1016/j.trsl.2012.12.006. [DOI] [PubMed] [Google Scholar]
- 71.Friedman LG, Lachenmayer ML, Wang J, He L, Poulose SM, Komatsu M, et al. Disrupted autophagy leads to dopaminergic axon and dendrite degeneration and promotes presynaptic accumulation of α-synuclein and LRRK2 in the brain. J Neurosci. 2012;32:7585–93. doi: 10.1523/JNEUROSCI.5809-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu J, Liu W, Li R, Yang H. Mitophagy in Parkinson’s disease: from pathogenesis to treatment. Cells. 2019;8:712. doi: 10.3390/cells8070712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Safiulina D, Kuum M, Choubey V, Hickey MA, Kaasik A. Mitochondrial transport proteins RHOT1 and RHOT2 serve as docking sites for PRKN-mediated mitophagy. Autophagy. 2019;15:930–1. doi: 10.1080/15548627.2019.1586260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shaltouki A, Hsieh C-H, Kim MJ, Wang X. Alpha-synuclein delays mitophagy and targeting Miro rescues neuron loss in Parkinson’s models. Acta Neuropathol. 2018;136:607–20. doi: 10.1007/s00401-018-1873-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yonova-Doing E, Atadzhanov M, Quadri M, Kelly P, Shawa N, Musonda ST, et al. Analysis of LRRK2, SNCA, Parkin, PINK1, and DJ-1 in Zambian patients with Parkinson's disease. Parkinsonism Relat Disord. 2012;18:567–71. doi: 10.1016/j.parkreldis.2012.02.018. [DOI] [PubMed] [Google Scholar]
- 76.Sliter DA, Martinez J, Hao L, Chen X, Sun N, Fischer TD, et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature. 2018;561:258. doi: 10.1038/s41586-018-0448-9. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 77.Matheoud D, Sugiura A, Bellemare-Pelletier A, Laplante A, Rondeau C, Chemali M, et al. Parkinson’s disease-related proteins PINK1 and Parkin repress mitochondrial antigen presentation. Cell. 2016;166:314–27. doi: 10.1016/j.cell.2016.05.039. [DOI] [PubMed] [Google Scholar]
- 78.Saita S, Shirane M, Nakayama KI. Selective escape of proteins from the mitochondria during mitophagy. Nat Commun. 2013;4:1410. doi: 10.1038/ncomms2400. [DOI] [PubMed] [Google Scholar]
- 79.Kitada T, Pisani A, Porter DR, Yamaguchi H, Tscherter A, Martella G, et al. Impaired dopamine release and synaptic plasticity in the striatum of PINK1-deficient mice. Proc Natl Acad Sci. 2007;104:11441–6. doi: 10.1073/pnas.0702717104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Perez FA, Palmiter RD. Parkin-deficient mice are not a robust model of parkinsonism. Proc Natl Acad Sci. 2005;102:2174–9. doi: 10.1073/pnas.0409598102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.McWilliams TG, Prescott AR, Montava-Garriga L, Ball G, Singh F, Barini E, et al. Basal mitophagy occurs independently of PINK1 in mouse tissues of high metabolic demand. Cell Metab. 2018;27:439–49. doi: 10.1016/j.cmet.2017.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Manzoni C. The LRRK2–macroautophagy axis and its relevance to Parkinson's disease. Biochem Soc Trans. 2017;45:155–62. doi: 10.1042/BST20160265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Schapansky J, Nardozzi JD, Felizia F, LaVoie MJ. Membrane recruitment of endogenous LRRK2 precedes its potent regulation of autophagy. Hum Mol Genet. 2014;23:4201–14. doi: 10.1093/hmg/ddu138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bang Y, Kim K-S, Seol W, Choi HJ. LRRK2 interferes with aggresome formation for autophagic clearance. Mol Cell Neurosci. 2016;75:71–80. doi: 10.1016/j.mcn.2016.06.007. [DOI] [PubMed] [Google Scholar]
- 85.Huang J, Klionsky DJ. Autophagy and human disease. Cell Cycle. 2007;6:1837–49. doi: 10.4161/cc.6.15.4511. [DOI] [PubMed] [Google Scholar]
- 86.Wang Y, Martinez-Vicente M, Krüger U, Kaushik S, Wong E, Mandelkow E-M, et al. Synergy and antagonism of macroautophagy and chaperone-mediated autophagy in a cell model of pathological tau aggregation. Autophagy. 2010;6:182–3. doi: 10.4161/auto.6.1.10815. [DOI] [PubMed] [Google Scholar]
- 87.Sorrentino V, Romani M, Mouchiroud L, Beck JS, Zhang H, D’amico D, et al. Enhancing mitochondrial proteostasis reduces amyloid-β proteotoxicity. Nature. 2017;552:187. doi: 10.1038/nature25143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Martín-Maestro P, Gargini R, Perry G, Avila J, García-Escudero V. PARK2 enhancement is able to compensate mitophagy alterations found in sporadic Alzheimer's disease. Hum Mol Genet. 2015;25:792–806. doi: 10.1093/hmg/ddv616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kerr JS, Adriaanse BA, Greig NH, Mattson MP, Cader MZ, Bohr VA, et al. Mitophagy and Alzheimer’s disease: cellular and molecular mechanisms. Trends Neurosci. 2017;40:151–66. doi: 10.1016/j.tins.2017.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bordi M, Berg MJ, Mohan PS, Peterhoff CM, Alldred MJ, Che S, et al. Autophagy flux in CA1 neurons of Alzheimer hippocampus: increased induction overburdens failing lysosomes to propel neuritic dystrophy. Autophagy. 2016;12:2467–83. doi: 10.1080/15548627.2016.1239003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fang EF, Hou Y, Palikaras K, Adriaanse BA, Kerr JS, Yang B, et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat Neurosci. 2019;22:401. doi: 10.1038/s41593-018-0332-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ye X, Sun X, Starovoytov V, Cai Q. Parkin-mediated mitophagy in mutant hAPP neurons and Alzheimer's disease patient brains. Hum Mol Genet. 2015;24:2938–51. doi: 10.1093/hmg/ddv056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Park J-S, Kim D-H, Yoon S-Y. Regulation of amyloid precursor protein processing by its KFERQ motif. BMB Rep. 2016;49:337. doi: 10.5483/BMBRep.2016.49.6.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wild EJ, Tabrizi SJ. Targets for future clinical trials in Huntington's disease: what's in the pipeline? Mov Disord. 2014;29:1434–45. doi: 10.1002/mds.26007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Koga H, Martinez-Vicente M, Arias E, Kaushik S, Sulzer D, Cuervo AM. Constitutive upregulation of chaperone-mediated autophagy in Huntington's disease. J Neurosci. 2011;31:18492–505. doi: 10.1523/JNEUROSCI.3219-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Qi L, Zhang X-D, Wu J-C, Lin F, Wang J, DiFiglia M, et al. The role of chaperone-mediated autophagy in huntingtin degradation. PLoS ONE. 2012;7:e46834. doi: 10.1371/journal.pone.0046834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bauer PO, Goswami A, Wong HK, Okuno M, Kurosawa M, Yamada M, et al. Harnessing chaperone-mediated autophagy for the selective degradation of mutant huntingtin protein. Nat Biotechnol. 2010;28:256. doi: 10.1038/nbt.1608. [DOI] [PubMed] [Google Scholar]
- 98.Li Z, Wang C, Wang Z, Zhu C, Li J, Sha T, et al. Allele-selective lowering of mutant HTT protein by HTT–LC3 linker compounds. Nature. 2019;575:203–9. [DOI] [PubMed]
- 99.Wold MS, Lim J, Lachance V, Deng Z, Yue Z. ULK1-mediated phosphorylation of ATG14 promotes autophagy and is impaired in Huntington’s disease models. Mol Neurodegener. 2016;11:76. doi: 10.1186/s13024-016-0141-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ashkenazi A, Bento CF, Ricketts T, Vicinanza M, Siddiqi F, Pavel M, et al. Polyglutamine tracts regulate beclin 1-dependent autophagy. Nature. 2017;545:108. doi: 10.1038/nature22078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Metzger S, Saukko M, Van Che H, Tong L, Puder Y, Riess O, et al. Age at onset in Huntington’s disease is modified by the autophagy pathway: implication of the V471A polymorphism in Atg7. Hum Genet. 2010;128:453–9. doi: 10.1007/s00439-010-0873-9. [DOI] [PubMed] [Google Scholar]
- 102.Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- 103.Yamamoto A, Cremona ML, Rothman JE. Autophagy-mediated clearance of huntingtin aggregates triggered by the insulin-signaling pathway. J Cell Biol. 2006;172:719–31. doi: 10.1083/jcb.200510065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kurosawa M, Matsumoto G, Kino Y, Okuno M, Kurosawa-Yamada M, Washizu C, et al. Depletion of p62 reduces nuclear inclusions and paradoxically ameliorates disease phenotypes in Huntington's model mice. Hum Mol Genet. 2014;24:1092–105. doi: 10.1093/hmg/ddu522. [DOI] [PubMed] [Google Scholar]
- 105.Ariosa AR, Klionsky DJ. Autophagy core machinery: overcoming spatial barriers in neurons. J Mol Med. 2016;94:1217–27. doi: 10.1007/s00109-016-1461-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Rudnick ND, Griffey CJ, Guarnieri P, Gerbino V, Wang X, Piersaint JA, et al. Distinct roles for motor neuron autophagy early and late in the SOD1G93A mouse model of ALS. Proc Natl Acad Sci. 2017;114:E8294–E303. doi: 10.1073/pnas.1704294114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Krüger U, Wang Y, Kumar S, Mandelkow E-M. Autophagic degradation of tau in primary neurons and its enhancement by trehalose. Neurobiol Aging. 2012;33:2291–305. doi: 10.1016/j.neurobiolaging.2011.11.009. [DOI] [PubMed] [Google Scholar]
- 108.Roscic A, Baldo B, Crochemore C, Marcellin D, Paganetti P. Induction of autophagy with catalytic mTOR inhibitors reduces huntingtin aggregates in a neuronal cell model. J Neurochem. 2011;119:398–407. doi: 10.1111/j.1471-4159.2011.07435.x. [DOI] [PubMed] [Google Scholar]
- 109.Maday S, Holzbaur EL. Autophagosome biogenesis in primary neurons follows an ordered and spatially regulated pathway. Dev Cell. 2014;30:71–85. doi: 10.1016/j.devcel.2014.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Maday S, Holzbaur EL. Compartment-specific regulation of autophagy in primary neurons. J Neurosci. 2016;36:5933–45. doi: 10.1523/JNEUROSCI.4401-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Finkbeiner S. The autophagy lysosomal pathway and neurodegeneration. Cold Spring Harb Perspect Biol. 2019:a033993. 10.1101/cshperspect.a033993 [Epub ahead of print]. [DOI] [PMC free article] [PubMed]
- 112.van Beek N, Klionsky DJ, Reggiori F. Genetic aberrations in macroautophagy genes leading to diseases. Biochimica et Biophysica Acta (BBA)-Mol Cell Res. 2018;1865:803–16. doi: 10.1016/j.bbamcr.2018.03.002. [DOI] [PubMed] [Google Scholar]
- 113.Heckmann Bradlee L., Teubner Brett J.W., Tummers Bart, Boada-Romero Emilio, Harris Lacie, Yang Mao, Guy Clifford S., Zakharenko Stanislav S., Green Douglas R. LC3-Associated Endocytosis Facilitates β-Amyloid Clearance and Mitigates Neurodegeneration in Murine Alzheimer’s Disease. Cell. 2019;178(3):536-551.e14. doi: 10.1016/j.cell.2019.05.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Levine B, Kroemer G. Biological functions of autophagy genes: a disease perspective. Cell. 2019;176:11–42. doi: 10.1016/j.cell.2018.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Haack TB, Hogarth P, Kruer MC, Gregory A, Wieland T, Schwarzmayr T, et al. Exome sequencing reveals de novo WDR45 mutations causing a phenotypically distinct, X-linked dominant form of NBIA. Am J Hum Genet. 2012;91:1144–9. doi: 10.1016/j.ajhg.2012.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Oakes JA, Davies MC, Collins MO. TBK1: a new player in ALS linking autophagy and neuroinflammation. Mol Brain. 2017;10:1–10. doi: 10.1186/s13041-017-0287-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mizuno Y, Amari M, Takatama M, Aizawa H, Mihara B, Okamoto K. Immunoreactivities of p62, an ubiqutin-binding protein, in the spinal anterior horn cells of patients with amyotrophic lateral sclerosis. J Neurol Sci. 2006;249:13–8. doi: 10.1016/j.jns.2006.05.060. [DOI] [PubMed] [Google Scholar]
- 118.Du Y, Wooten MC, Wooten MW. Oxidative damage to the promoter region of SQSTM1/p62 is common to neurodegenerative disease. Neurobiol Dis. 2009;35:302–10. doi: 10.1016/j.nbd.2009.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kyöstilä K, Syrjä P, Jagannathan V, Chandrasekar G, Jokinen TS, Seppälä EH, et al. A missense change in the ATG4D gene links aberrant autophagy to a neurodegenerative vacuolar storage disease. PLoS Genet. 2015;11:e1005169. doi: 10.1371/journal.pgen.1005169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kim M, Sandford E, Gatica D, Qiu Y, Liu X, Zheng Y, et al. Mutation in ATG5 reduces autophagy and leads to ataxia with developmental delay. eLife. 2016;5:e12245. doi: 10.7554/eLife.12245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Seong E, Insolera R, Dulovic M, Kamsteeg EJ, Trinh J, Brüggemann N, et al. Mutations in VPS13D lead to a new recessive ataxia with spasticity and mitochondrial defects. Ann Neurol. 2018;83:1075–88. doi: 10.1002/ana.25220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Boutoleau-Bretonnière C, Camuzat A, Le Ber I, Bouya-Ahmed K, Guerreiro R, Deruet A-L, et al. A phenotype of atypical apraxia of speech in a family carrying SQSTM1 mutation. J Alzheimer's Dis. 2015;43:625–30. doi: 10.3233/JAD-141512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Haack TB, Ignatius E, Calvo-Garrido J, Iuso A, Isohanni P, Maffezzini C, et al. Absence of the autophagy adaptor SQSTM1/p62 causes childhood-onset neurodegeneration with ataxia, dystonia, and gaze palsy. Am J Hum Genet. 2016;99:735–43. doi: 10.1016/j.ajhg.2016.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Carvill GL, Liu A, Mandelstam S, Schneider A, Lacroix A, Zemel M, et al. Severe infantile onset developmental and epileptic encephalopathy caused by mutations in autophagy gene WDR 45. Epilepsia. 2018;59:e5–e13. doi: 10.1111/epi.13957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Abidi A, Mignon-Ravix C, Cacciagli P, Girard N, Milh M, Villard L. Early-onset epileptic encephalopathy as the initial clinical presentation of WDR45 deletion in a male patient. Eur J Hum Genet. 2016;24:615. doi: 10.1038/ejhg.2015.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kurth I, Pamminger T, Hennings JC, Soehendra D, Huebner AK, Rotthier A, et al. Mutations in FAM134B, encoding a newly identified Golgi protein, cause severe sensory and autonomic neuropathy. Nat Genet. 2009;41:1179. doi: 10.1038/ng.464. [DOI] [PubMed] [Google Scholar]
- 127.Cai P-P, Wang H-X, Zhuang J-C, Liu Q-B, Zhao G-X, Li Z-X, et al. Variants of autophagy-related gene 5 are associated with neuromyelitis optica in the Southern Han Chinese population. Autoimmunity. 2014;47:563–6. doi: 10.3109/08916934.2014.929668. [DOI] [PubMed] [Google Scholar]
- 128.Minegishi Y, Nakayama M, Iejima D, Kawase K, Iwata T. Significance of optineurin mutations in glaucoma and other diseases. Prog Retin Eye Res. 2016;55:149–81. doi: 10.1016/j.preteyeres.2016.08.002. [DOI] [PubMed] [Google Scholar]
- 129.Hoffjan S, Ibisler A, Tschentscher A, Dekomien G, Bidinost C, Rosa AL. WDR45 mutations in Rett (-like) syndrome and developmental delay: case report and an appraisal of the literature. Mol Cell Probes. 2016;30:44–9. doi: 10.1016/j.mcp.2016.01.003. [DOI] [PubMed] [Google Scholar]
- 130.Zheng M, Yu H, Zhang L, Li H, Liu Y, Kijlstra A, et al. Association of ATG5 gene polymorphisms with Behçet's disease and ATG10 gene polymorphisms with VKH syndrome in a Chinese Han population. Investig Ophthalmol Vis Sci. 2015;56:8280–7. doi: 10.1167/iovs.15-18035. [DOI] [PubMed] [Google Scholar]
- 131.Comincini S, Manai F, Meazza C, Pagani S, Martinelli C, Pasqua N, et al. Identification of autophagy-related genes and their regulatory miRNAs associated with celiac disease in children. Int J Mol Sci. 2017;18:391. doi: 10.3390/ijms18020391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Brinar M, Vermeire S, Cleynen I, Lemmens B, Sagaert X, Henckaerts L, et al. Genetic variants in autophagy-related genes and granuloma formation in a cohort of surgically treated Crohn's disease patients. J Crohn's Colitis. 2012;6:43–50. doi: 10.1016/j.crohns.2011.06.008. [DOI] [PubMed] [Google Scholar]
- 133.Meddens CA, Harakalova M, van den Dungen NA, Asl HF, Hijma HJ, Cuppen EP, et al. Systematic analysis of chromatin interactions at disease associated loci links novel candidate genes to inflammatory bowel disease. Genome Biol. 2016;17:247. doi: 10.1186/s13059-016-1100-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Hampe J, Franke A, Rosenstiel P, Till A, Teuber M, Huse K, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet. 2007;39:207. doi: 10.1038/ng1954. [DOI] [PubMed] [Google Scholar]
- 135.Henckaerts L, Cleynen I, Brinar M, John JM, Van Steen K, Rutgeerts P, et al. Genetic variation in the autophagy gene ULK1 and risk of Crohn's disease. Inflamm Bowel Dis. 2011;17:1392–7. doi: 10.1002/ibd.21486. [DOI] [PubMed] [Google Scholar]
- 136.LU X-l, Zhou X-j, Guo J-p, Jia R-l, Yi Z, Jiang Q, et al. Rs548234 polymorphism atPRDM1-ATG5 region susceptible to rheumatoid arthritis in Caucasians is not associated with rheumatoid arthritis in Chinese Han population. Chin Med J. 2011;124:2863–7. [PubMed] [Google Scholar]
- 137.Pierdominici M, Vomero M, Barbati C, Colasanti T, Maselli A, Vacirca D, et al. Role of autophagy in immunity and autoimmunity, with a special focus on systemic lupus erythematosus. FASEB J. 2012;26:1400–12. doi: 10.1096/fj.11-194175. [DOI] [PubMed] [Google Scholar]
- 138.Qi YY, Zhou XJ, Nath SK, Sun C, Wang YN, Hou P, et al. A rare variant (rs933717) at FBXO 31‐MAP 1 LC 3B in Chinese is associated with systemic lupus erythematosus. Arthritis Rheumatol. 2018;70:287–97. doi: 10.1002/art.40353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Mayes MD, Bossini-Castillo L, Gorlova O, Martin JE, Zhou X, Chen WV, et al. Immunochip analysis identifies multiple susceptibility loci for systemic sclerosis. Am J Hum Genet. 2014;94:47–61. doi: 10.1016/j.ajhg.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Le Pham D, Kim S-H, Losol P, Yang E-M, Shin YS, Ye Y-M, et al. Association of autophagy related gene polymorphisms with neutrophilic airway inflammation in adult asthma. Korean J Intern Med. 2016;31:375. doi: 10.3904/kjim.2014.390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Chen C-Z, Ou C-Y, Wang R-H, Lee C-H, Lin C-C, Chang H-Y, et al. Association of Egr-1 and autophagy-related gene polymorphism in men with chronic obstructive pulmonary disease. J Formos Med Assoc. 2015;114:750–5. doi: 10.1016/j.jfma.2013.07.015. [DOI] [PubMed] [Google Scholar]
- 142.Bucelli RC, Arhzaouy K, Pestronk A, Pittman SK, Rojas L, Sue CM, et al. SQSTM1 splice site mutation in distal myopathy with rimmed vacuoles. Neurology. 2015;85:665–74. doi: 10.1212/WNL.0000000000001864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Gang Q, Bettencourt C, Machado PM, Brady S, Holton JL, Pittman AM, et al. Rare variants in SQSTM1 and VCP genes and risk of sporadic inclusion body myositis. Neurobiol Aging. 2016;47:218. doi: 10.1016/j.neurobiolaging.2016.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Wu C, Wen Y, Guo X, Yang T, Shen H, Chen X, et al. Genetic association, mRNA and protein expression analysis identify ATG4C as a susceptibility gene for Kashin–Beck disease. Osteoarthr Cartil. 2017;25:281–6. doi: 10.1016/j.joca.2016.09.019. [DOI] [PubMed] [Google Scholar]
- 145.Usategui-Martín R, García-Aparicio J, Corral-Gudino L, Calero-Paniagua I, Del Pino-Montes J, Sarmiento RG. Polymorphisms in autophagy genes are associated with paget disease of bone. PLoS ONE. 2015;10:e0128984. doi: 10.1371/journal.pone.0128984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Hocking LJ, Lucas GJ, Daroszewska A, Mangion J, Olavesen M, Cundy T, et al. Domain-specific mutations in sequestosome 1 (SQSTM1) cause familial and sporadic Paget's disease. Hum Mol Genet. 2002;11:2735–9. doi: 10.1093/hmg/11.22.2735. [DOI] [PubMed] [Google Scholar]