

Abstract
Sonic hedgehog (SHH) signaling is crucial for the maintenance of the physiological self-renewal of granule neuron progenitor cells (GNPs) during cerebellar development, and its dysregulation leads to oncogenesis. However, how SHH signaling is controlled during cerebellar development is poorly understood. Here, we show that Trim32, a cell fate determinant, is distributed asymmetrically in the cytoplasm of mitotic GNPs, and that genetic knockout of Trim32 keeps GNPs at a proliferating and undifferentiated state. In addition, Trim32 knockout enhances the incidence of medulloblastoma (MB) formation in the Ptch1 mutant mice. Mechanistically, Trim32 binds to Gli1, an effector of SHH signaling, via its NHL domain and degrades the latter through its RING domain to antagonize the SHH pathway. These findings provide a novel mechanism that Trim32 may be a vital cell fate regulator by antagonizing the SHH signaling to promote GNPs differentiation and a tumor suppressor in MB formation.
Subject terms: Cancer models, Tumour-suppressor proteins, Development, Gene regulation
Introduction
Dysregulation occurring in developmental processes is often a key contribution to the pathogenesis of cancer [1, 2]. The postnatal cerebellum is one of the supreme models to study these events happening from neurogenesis to tumorigenesis. At early stages of postnatal neurogenesis, cerebellar granule neuron progenitor cells (GNPs) undergo massive proliferative, symmetric cell divisions to expand the pool of GNPs at the surface of the developmental cerebellum to form the external granular layer (EGL) [3–5]. Math1, also called Atoh1, is a basic helix–loop–helix transcription factor required for GNP production, and can serve as a marker for the GNPs [6, 7]. After expansion, GNPs then switch to undergo asymmetric cell divisions from symmetric cell divisions to exit out of cell cycle and differentiate into postmitotic granule neurons that will migrate inwards through the Purkinje cell layer to become mature granule neurons in the internal granule layer (IGL) at 3 weeks after birth [2, 8]. By the end of this period, the EGL will be disappeared [2, 8].
During this early postnatal period, sonic hedgehog (SHH), secreted by Purkinje cells, binds to its receptor Ptch1 expressed on the GNP surface [9]. This binding leads to activation of the SHH target genes, such as MycN, cyclin-D1, and Gli1, driving GNP proliferation while maintaining them at an undifferentiated state [10–12]. Our previous work has demonstrated that SHH enhances proliferation by regulating the balance between asymmetric and symmetric cell divisions of GNPs [4]. Aberrant activation of the SHH signaling can lead to an increase in progenitor SCDs that delay GNP differentiation, resulting in an overproduction of GNPs susceptible to malignant transformation, and inducing a distinct subgroup of medulloblastoma (MB), in which Math1 is found to be upregulated [1, 11, 13–16]. However, how the SHH signaling is controlled at the early stages of cerebellar development and during MB formation is still poorly understood.
Trim32 is a protein of the tripartite motif (TRIM) protein family, which is defined as a subfamily of the RING type E3 ubiquitin ligase family [17–19]. Trim32 mutations or knock-out are reported to be involved in the pathogenesis of LGMD2H, a disease with a myopathic phenotype accompanied by neurogenic features [20]. Trim32 has been shown to play an important role in regulation of muscle stem cell regeneration and induction of muscle autophagy [21, 22]. In addition, its function has also been studied in neural differentiation and cancer [19]. For example, Brat, the ortholog of Trim32 in the Drosophila, controls the neuroblast stem cell fate by degrading dMyc and that Brat mutant can lead to the formation of larval brain tumors [23–25]. Consistent with these findings, with highest expression in the brain [26, 27], Trim32 has been reported to act as a cell fate determinant to control the cell fate in mouse neural stem cell lineages by regulating the transcription factor c-Myc degradation and microRNA activities [28–31]. Furthermore, a recent study showed that Trim32 is a positive regulator of asymmetric cell division and exerts an effect against MycN and should be considered as a tumor suppressor candidate [30]. However, the role of Trim32 in cerebellar development and tumorigenesis is poorly defined.
In this study, we discovered that Trim32 knockout disrupts the balance between differentiation and proliferation of cerebellar GNPs and increased the incidence of MB in Ptch1+/− mice. Importantly, Trim32 negatively regulated the SHH pathway activity by interacting with the transcription factor Gli1, and degrading it. These results demonstrate a novel function of Trim32 as a determinant of cerebellar GNP differentiation and a tumor suppressor for MB, by regulating Gli1 and Gli1-dependent SHH signaling activity.
Results
Trim32 is expressed in GNPs of inner EGL, and displays an asymmetrical distribution in GNPs in the postnatal cerebellum
To investigate the expression and distribution patterns of Trim32 protein in the developing postnatal cerebellum, we first assessed the expression pattern of Trim32 at different stages of cerebellar development. Cerebella from postnatal day P0, P7, P14, and P60 (adult) were harvested. Expression of Trim32 became progressively stronger in the first postnatal 2 weeks (P7–P14) and reached the peak at postnatal 60 days, compared with the low expression levels at P0 (Fig. 1a, b). To further determine the cellar distribution of Trim32 in EGL, we utilized the Math1-GFP transgenic mouse model, in which Math1 is used as the promoter driving expression of the GFP reporter gene [6, 7]. In this way, GFP can be used as a marker of GNPs. As shown in Fig. 1c (left panel), at P7, Trim32-positive cells were found increased gradually from the EGL to IGL. In the IGL, postmitotic and differentiated granule neurons were almost all Trim32-positive. As shown in Fig. 1c (right panel), expression of Trim32 and Math1-GFP seemed to be mutually exclusive in the EGL. Trim32 was found predominantly in the inner EGL, where those GNPs expressing lower levels of Math1-GFP appeared to stop proliferation and to begin differentiation. In contrast, Math1-GFP-positive GNPs were mainly restricted in the outer EGL, where GNPs were kept in a proliferating and undifferentiated state. Thus, Trim32 is highly expressed in differentiated granule neurons at a lower level in GNPs of inner EGL, implying that expression of Trim32 increases gradually with cerebellar development and GNPs differentiation.
Fig. 1.

Trim32 is selectively expressed in inner EGL and displays an uneven cytoplasmic distribution during GNP differentiation. a RT-qPCR analysis of Trim32 in P0, P7, P14, and adult mouse cerebella. Data are expressed as means ± SD (n = 3). An asterisk indicates P < 0.05 and triple asterisks indicate P < 0.001. b Immunoblotting analysis of Trim32, Gli1, and MycN in P0, P7, P14, and adult mouse cerebellums. c Immunofluorescence staining of Trim32 (red) in the EGL of P7 Math1-GFP transgenic mouse cerebellum. Nuclei were counterstained with DAPI (blue). EGL external granule layer, ML molecular layer, PCL Purkinje cells layer, and IGL internal granule layer. The scale bars represent 100 µm in the first panel and 25 µm in the second panel. d Confocal analysis (left) and quantification of immunofluorescence intensities (right) of distribution of Trim32 (green) in the dividing GNPs in different phases of the cell cycle. The dashed line highlights the cells that are in the indicated phase of cell cycle. The cell-cycle phases were identified by PH3 staining (red) for DNA. Nuclei were counterstained with DAPI (blue). The scale bars represent 10 µm. Data are expressed as means ± SD (n = 3). ns indicates P > 0.05 and triple asterisks indicate P < 0.001
Since Trim32 has been shown to disperse asymmetrically in neocortex development as a cell fate determinant [28], we followed the subcellular distribution of Trim32 in dividing GNPs. Quantification of immunofluorescence intensities in the two emerging daughter cells revealed that Trim32 fluorescence is two times stronger in one daughter cell than the other, so Trim32 was distributed asymmetrically (Fig. 1d). By performing co-staining of Trim32 and PH3 (a uniformly mitotic nuclear protein), we found that at P7–P9, Trim32 was preferentially inherited by one of the two daughter cells in ~7.8% of the dividing GNPs (n = 8/103) (Fig. 1d). Thus, Trim32 segregated asymmetrically in dividing GNPs in the postnatal cerebellum, which is consistent with the findings in neural stem/progenitor cells in the mouse neocortex [28].
Trim32 knockout enhances cerebellar GNP proliferation
To examine the biological function of Trim32 in postnatal cerebellar neurogenesis, we constructed the Trim32 knockout mice. We sought to define whether knockout of Trim32 affected the proliferation and differentiation of GNPs of the EGL at an early postnatal stage. We observed that GNPs from Trim32KO/Math1-GFP mice cerebella expressed higher level of Math1, compared with those in Trim32WT/Math1-GFP mice (Fig. 2a). Immunostaining of NeuN that was used as a marker of the postmitotic differentiated granule neurons revealed that Trim32 knockout induced a decrease in the number of the differentiated NeuN-positive cells at P7, in Trim32KO mouse, compared with those in the control group (Fig. 2b). Quantitative analysis of Ki67+ cells, representing proliferative cells, revealed that in the Trim32KO mouse cerebellum, more Ki67-positive cells were found in the EGL. In addition, unlike what was observed in the control mice in which Ki67-positive cells were restricted to the outer EGL, these Ki67-positive cells were seen to be located beyond the outer EGL in the Trim32KO mouse cerebellum (Fig. 2c). Consistent with these results, Trim32 knockout led to an overgrowth of cerebellar GNPs and enlargement of the IGL in a subset of cerebellar lobes during late development (Fig. 2d). These findings indicate that the ablation of Trim32 can lead to an overproliferation and an interference with the differentiation of GNPs.
Fig. 2.

Trim32 knockout enhances GNP proliferation in the postnatal developing cerebellum. a Expression of the Math1-GFP protein (GFP, green) in P7 mouse cerebellar sections from the Math1-GFP/Trim32wt mice and Math1-GFP/Trim32KO mice. Nuclei were counterstained with DAPI (blue). Graph in a representing Math1 positive cells normalized to the length of the EGL edge. The scale bar represents 25 µm. Immunofluorescence staining of NeuN (red, b) and Ki67 (red, c) in P7 mouse cerebellar sections from the Math1-GFP/Trim32wt mice and Math1-GFP/Trim32KO mice. Nuclei were counterstained with DAPI (blue). Graph in b and c respectively representing NeuN or Ki67-positive cells normalized to the length of the EGL edge. The scale bar in b represents 50 µm. The scale bar in c represents 25 µm. oEGL outer external granule layer, iEGL inner external granule layer, ML molecular layer, and IGL internal granule layer. d P7 and P18 cerebellar midsagittal sections were stained for DAPI to show the overall morphology of cerebellum in Trim32wt mice and Trim32ko mice. The scale bar represents 500 µm
Trim32 overexpression antagonizes SHH signaling
Considering that SHH signaling is the driver for GNP proliferation and that Trim32 appears to inhibit GNP proliferation, we wondered if Trim32 might be a suppressor of the SHH pathway. To test this hypothesis, we first collected the P7 cerebellar GNPs from Trim32WT mice and Trim32KO mice, and then examined their expression levels of SHH target genes involved in the proliferation of GNPs, including Ccnd1, Ccnd2, McyN, and Gli1. As shown in Fig. 3a, b, these target genes were significantly upregulated at both mRNA and protein levels due to Trim32 knockout, indicating that loss of endogenous Trim32 enhances the SHH pathway activation. We next investigated whether Trim32 overexpression could inhibit the SHH pathway activity. We thus transduced HEK293T cells with lentiviruses encoding either Trim32-GFP or GFP (as a control). HEK293T cells have been previously shown to be responsive to SHH [32]. As expected, in HEK293T cells, SHH treatment transcriptionally activated expression of the target genes such as Gli1 and MycN (Fig. 3c, d), while Trim32 overexpression significantly inhibited the stimulatory effect induced by SHH (Fig. 3c, d). Furthermore, Trim32 overexpression significantly decreased Gli-RE-luciferase reporter activity induced by Gli1 in both GNP-derived D283 human MB cells and HEK293T cells (Fig. 3e, f). Hence, these findings suggest that functionally Trim32 may antagonize SHH signaling via inhibiting Gli1.
Fig. 3.

Trim32 antagonizes the SHH signaling activity. a RT-qPCR analysis of Trim32, the granule neuronal progenitor marker Maht1, and SHH target genes in P7 cerebellar granule neuronal progenitors (cGNPs) from Trim32wt mice and Trim32KO mice. Data are expressed as means ± SD (n = 3). Double asterisks indicate P < 0.01 and triple asterisks indicate P < 0.001. b Immunoblotting analysis of Trim32, Gli1, Ccnd1, Ccnd2, and MycN in P7 mouse cerebellum from Trim32wt mice and Trim32KO mice. c, d RT-PCR mRNA expression of the SHH target genes Gli1 and MycN in HEK293T cells overexpressing Trim32-GFP or GFP control vector. The cells were cultured with or without SHH for 24 and 48 h. Data are expressed as means ± SD (n = 3). Double asterisks indicate P < 0.01 and triple asterisks indicate P < 0.001. Gli-RE-luciferase activity in human medulloblastoma cell line D283 (e) and HEK293T cells (f), following transfected the indicated vectors. The luciferase activity was evaluated relative to Renilla activity. The means ± SD from three experiments are shown
Trim32 interacts with Gli1 through the NHL domain
The transcription factor Gli1 is a popular biomarker for activated SHH pathway [33]. To investigate whether Trim32 directly targets this component of the SHH pathway to inhibit its activation, we first examined whether Trim32 could bind to Gli1. We exogenously expressed Flag-tagged Gli1 and Trim32-GFP in HEK293T cells, and found that Trim32 and Gli1 could be reciprocally co-immunoprecipitated (Fig. 4a). Moreover, we also found that the endogenous Gli1 protein could be co-immunoprecipitated by exogenous Trim32 in HEK293T cells (Fig. 4b).
Fig. 4.

Trim32 interacts with Gli1 though the NHL domain. a In vivo assay for binding between Trim32 and Gli1. Expression vectors encoding TRIM32-GFP and Flag-tagged Gli1 were transfected into HEK293T cells. Whole cell lysates were immunoprecipitated with anti-Trim32 or anti-FLAG antibody and immunoblotted with anti-Trim32 and anti-Gli1 antibodies. b Co-immunoprecipitation of exogenously expressed Trim32-HA with the endogenous Gli1 in HEK293T cells. c Co-immunoprecipitation of exogenously expressed Trim32-GFP with the Gli1 truncated mutants. d Schematic drawing of full-length Trim32 (Trim32-wt) and deletion mutants. Trim32 contains a RING finger (R), two B-boxes (B), a coiled-coil region (Coil), and an NHL domain (NHL). e Co-immunoprecipitation of exogenously expressed Flag-tagged Gli1 with the Trim32 truncated mutants shown as in d
To further determine whether the precise binding region(s) of Gli1 is responsible for the interaction between Gli1 and Trim32, we constructed multiple truncated mutants, and found that all of them could be bound by Trim32 (Fig. 4c). Reciprocally, we also examined which domain(s) of Trim32 facilitated the interaction with Gli1. Co-immunoprecipitation experiments revealed that among four truncated mutants, including Trim32/ΔRING lacking the RING domain, Trim32/ΔB-box lacking the B-box domain, Trim32/ΔCC lacking the coil-coil domain, and Trim32/ΔNHL lacking the NHL domain, only Trim32/ΔNHL lost the ability to associate with Gli1, while all others did not (Fig. 4d, e). These results demonstrate that Trim32 binds to Gli1 through its NHL domain.
Trim32 promotes Gli1 ubiquitination and degradation
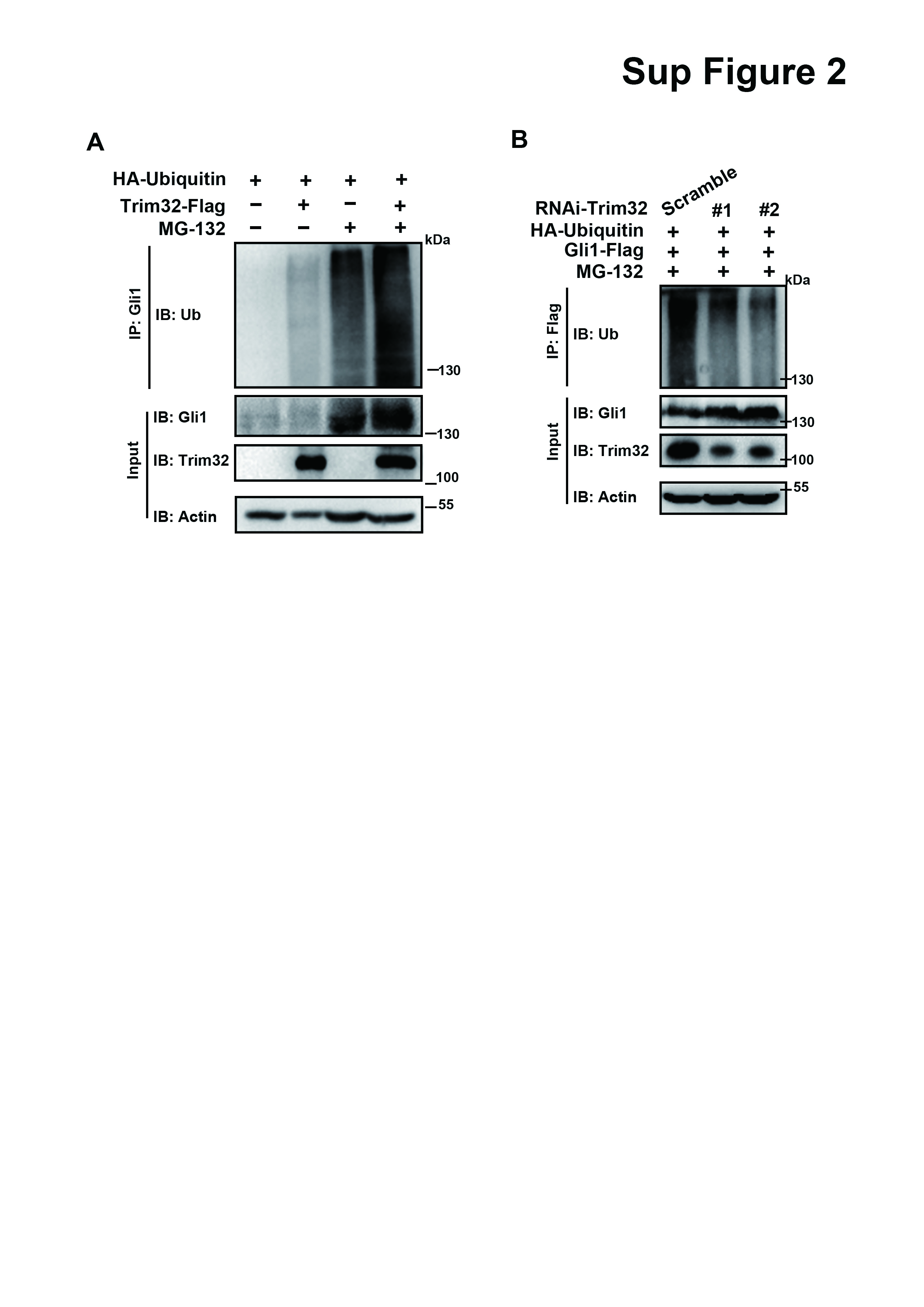
As Trim32 functions as an E3 ubiquitin ligase through its Ring domain [29, 34], we first determined whether Gli1 stability could be regulated by Trim32. We found that Trim32 could reduce Gli1 expression at the protein level in a dosage-dependent manner (Fig. 5a, b and Supplementary Fig. 1A, B). In contrast, Trim32 knockdown resulted in an accumulation of Gli1 protein, not Gli1 mRNA (Fig. 5c and Supplementary Fig. 1C–F), implying that Trim32 negatively regulates expression of Gli1 at protein level. Furthermore, we performed a cycloheximide (CHX) chase assay, and observed a shortened half-life of exogenous Gli1 in the presence of Trim32, but not that of Trim32/ΔRING (Fig. 5d, e), suggesting that the RING domain is required for the regulation of Trim32 on the degradation of Gli1 protein. Consistently, Trim32 knockdown prolonged the half-life of Gli1 protein (Fig. 5f, g), indicating that Trim32 negatively affected Gli1 stability. In addition, we found that the presence of MG132, a proteasome inhibitor, significantly reversed Gli1 reduction induced by Trim32 (Fig. 5h), suggesting that Trim32 triggers Gli1 degradation through the proteasome pathway.
Fig. 5.

Trim32 promotes Gli1 ubiquitination and degradation. a HEK293T cells transfected with Gli1 and increasing doses of Trim32 were collected for immunoblotting with the indicated antibodies. b Quantification of Gli1 and Trim32 protein levels (normalized to actin) from a. The means ± SEM from three experiments are shown. c Cells stably expressing scramble RNA or shRNA targeting Trim32 (#1, #2) were harvested for immunoblotting with the indicated antibodies. d HEK293T cells were transfected with plasmids encoding Flag-tagged Gli1 and control GFP vector or Trim32 or Trim32-ΔR, treated with 20 µg/ml cycloheximide (CHX) and harvested at the indicated times points. The levels of Gli1 and Trim32 in the lysates were investigated by immunoblotting. e Quantification of Gli1 remaining protein levels (normalized to actin) from d. The means ± SEM from three experiments are shown. f Cells were transfected with scramble RNA or shRNA targeting Trim32 (#1), treated with 20 µg/ml cycloheximide (CHX), collected at the indicated time points, and then immunoblotted with the indicated antibodies. g Quantification of Gli1 protein levels (normalized to actin) from f. The means ± SEM from three experiments are shown. h Cells transfected with Gli1 or Trim32 were treated with or without 10 µM MG132 for 8 h, collected, and then immunoblotted with the indicated antibodies. Graph representing quantification of Gli1 protein levels (normalized to actin). i Anti-ubiquitin immunoblotting of immunoprecipitated exogenous Gli1 in HEK293T cells transfected with the indicated plasmids. j The effect of Trim32-wt or Trim32 deletion mutant (Trim32-ΔR) to the activation of Gli-RE-luciferase induced by Gli1 in HEK293T cells. The means ± SD from three experiments are shown. Double asterisks indicate P < 0.01
Next, we investigated whether degradation of Gli1 was mediated via ubiquitination. We treated cells expressing Gli1 and HA-Ubiquitin with Trim32 or Trim32/ΔRING losing its E3 ubiquitin ligase activity in the presence or absence of MG132. The ubiquitination of Gli1 was augmented by Trim32, but not by Trim32/ΔRING, regardless of whether or not MG132 treatment was applied (Fig. 5i and Supplementary Fig. 2A). We also found that the levels of degradative ubiquitination of Gli1 were decreased clearly in Trim32 knockdown cells (Supplementary Fig. 2B). Moreover, Trim32 mutant lacking the RING domain weakened evidently the suppression of Trim32 on Gli-RE-luciferase reporter activity (Fig. 5j). Together, these results indicate that Trim32 promotes Gli1 degradation following its RING domain-mediated ubiquitination.
Trim32 knockout increases the incidence of MB in Ptch1+/− mice
MB is believed to arise from cerebellar GNPs containing highly proliferating ability in the EGL due to an aberrant activation of the SHH pathway at the early developing stage [1]. Given that we had demonstrated above that Trim32 antagonizes SHH signaling activity in development, we then examined whether it was involved in cerebellar tumorigenesis. As shown in Fig. 6a, b, we found that compared with adjacent normal cerebellar tissues, mouse SHH MB tissues presented reduced Trim32 expression at both mRNA and protein levels. These data demonstrated that expression level of Trim32 was significantly downregulated in SHH MBs. We next interrogated whether low Trim32 levels and high Gli1 levels were correlated negatively in human SHH MBs. By analyzing clinical SHH MB samples from Northcott brain samples 3 [35], we found a strong inverse correlation between Trim32 and Gli1 levels (P = 0.036, r = −0.3) (Fig. 6c). These findings together highlight the importance of Trim32 to antagonize SHH signaling in MB.
Fig. 6.

Trim32 knockout increases the incidence of MB in Ptch1+/− mice. Quantitative PCR mRNA (a; mean ± SD; n = 9) and protein levels (b; n = 3) of Trim32 in mouse medulloblastomas (MB) from Ptch1+/− mice and normal cerebella. c RNA expression level of Trim32 negatively correlated with Gli1 (n = 51) in human SHH MB samples. Data are analyzed from Oncomine database. d Kaplan–Meier analysis of MB incidence in 63 Ptch1+/−/Trim32wt mice (gray line) versus 17 Ptch1+/−/Trim32KO mice (black line). Ptch1+/−/Trim32wt mice and Ptch1+/−/Trim32KO mice, obtained by interbreeding for at least three generations the progeny of Ptch1 heterozygous and Trim32 knock-out mice, were then monitored for the onset of medulloblastoma; (triple asterisks indicate P < 0.001, Logrank test). e Expression of the Math1-GFP protein (GFP, green) in the Math1-GFP mouse medulloblastomas and adjacent cerebellar cortices sections from Ptch1+/−/Trim32wt mice and Ptch1+/−/Trim32KO mice. Nuclei were counterstained with DAPI (blue). The scale bar represents 200 µm. MB medulloblastoma, IGL internal granule layer. RT-qPCR analysis of SHH target genes (f), Trim32 (g), the granule neuronal progenitor marker Maht1 (h), and the differentiated granule cell markers including Tuj1 and NeuN (i) in adult mouse cerebellums and medulloblastomas from Ptch1+/−/Trim32wt mice and Ptch1+/−/Trim32KO mice. Data are expressed as means ± SD (n = 3). An asterisk indicates P < 0.05, double asterisks indicate P < 0.01, and triple asterisks indicate P < 0.001. j Immunoblotting analysis of Trim32, Gli1, Ccnd2, MycN, and NeuN in MB from Ptch1+/−/Trim32wt mice and Ptch1+/−/Trim32KO mice
To further study the role of Trim32 in MB oncogenesis, we analyzed the incidence of MB developed at 40 weeks in Ptch1+/− mice that were reported to give low incidence of MB and that in the offsprings that were derived from the crossing between Ptch1+/− and Trim32 knockout mice. We observed that 25.4% of the Ptch1+/−/Trim32WT mice (16/63) and 52.9% of the Ptch1+/−/Trim32KO mice (9/17) developed cerebellar MBs, and that none of the 50 wild-type mice or the 30 Ptch1+/+/Trim32KO mice developed MB, showing that Trim32 knockout resulted in a much higher incidence of MB in Ptch1+/− mice (p < 0.001, Fig. 6d). For further assessment of the molecular characteristics of these MBs, we first crossed the Math1-GFP mice with the Ptch1+/−/Trim32WT mice or with the Ptch1+/−/Trim32KO mice. As shown in Fig. 6e, MBs of the two genotypes all contained lots of Math1-GFP positive cells, indicating these MBs from the two genotypes are both originated from the malignant transformation of GNPs. Then we measured expression levels of the SHH pathway components in normal cerebellar tissues and MBs from two genotypes. At mRNA level, a significant upregulation of the SHH pathway target genes including Gli1, Gli2, Ccnd1, Ccnd2, and MycN were observed in the MB tissues than normal cerebellar tissues (Fig. 6f). Interestingly, higher expression of these genes was generally observed in Ptch1+/−/Trim32KO MBs than in Ptch1+/−/Trim32WT MBs. Furthermore, Trim32 knockout resulted in lower expression of the granule neuron cell differentiation markers including NeuN and Tuj1 in Ptch1+/−/Trim32KO MB compared with that in Ptch1+/−/Trim32WT MB, though no significant difference was seen for expression of the GNP marker Math1 (Fig. 6g–i). These data were further confirmed by immunoblotting experiments (Fig. 6j). Thus, Trim32 knockout increases the malignance of MB in the Ptch1+/− mice model by enhancing the SHH pathway activity and suppressing GNP differentiation.
Discussion
In the current study, we demonstrated histopathologically and genetically that cell fate determinant Trim32 is distributed asymmetrically in the cytoplasm of mitotic GNPs and that genetic deletion of Trim32 in the mouse cerebellum results in an imbalance between proliferation and differentiation of GNPs. Importantly, Trim32 knockout holds the GNPs in a proliferating state, prevents them from differentiation, and leads to an increased incidence of MB in Ptch1+/− mice. Mechanistically, at a biochemical level, Trim32 suppresses the activity of the SHH signaling so to promote GNP differentiation. These findings add Trim32 as a new molecule critical for the regulation of proliferation and differentiation of GNPs and implicate that Trim32 gene mutation or loss might be another cause for the formation of MBs.
It is interesting to point out that mounting evidence show that asymmetric distribution of Trim32 during cell divisions induces differentiation of neural stem/progenitor cells in mouse neocortex [28, 29, 36]. Consistent with this fact, we first show an asymmetric distribution of Trim32 in mitotic GNPs and then demonstrate a biologic function of Trim32 in cerebellar development and tumorigenesis. As we have reported previously, SHH signaling determines GNP fate by maintaining the balance between symmetric and asymmetric cell division in cerebellar neurogenesis [4]. In the current experiments, we further revealed that Trim32 negatively regulates the SHH signaling to act as a vital cell fate determinant to regulate the balance between differentiation and proliferation of GNPs in the cerebellum.
An important finding of the present study is to identify a posttranslational mechanism by which Trim32 suppresses the SHH signaling pathway, a major regulator of cerebellar development and MB formation. We demonstrated that Trim32 binds to Gli1, the sensitive readout of SHH output, and degrades it, to suppress SHH signaling pathway activity. Consistent with the fact that Trim32 possess the E3 ubiquitin ligase activity [29, 37], our present study provides clear evidence for protein degradation of Gli1 by Trim32. First, Trim32 and Gli1, not Gli2, can be co-immunoprecipitated through the NHL domain, indicating that two proteins are present in a complex. Second, Gli1 level is significantly reduced upon Trim32 co-expression, but is increased upon Trim32 knockdown in 293T cells, which is in accordance with the findings in the Trim32 knockout mice. Third, using a protein synthesis inhibitor cycloheximide, we further demonstrate that Trim32 decreases the Gli1 protein half-life, and that the degradation of Gli1 by Trim32 is prevented in the presence of MG132, a proteasome inhibitor. In addition, deletion of the RING domain rescues the ubiquitination and degradation of Gli1 by Trim32, weakening the suppression of Trim32 on the SHH pathway activity. Therefore, our current biochemical study provides a new insight into the mechanism as to how the SHH activity is controlled, that is, Trim32 interacts with Gli1, by promoting ubiquitination and degradation of Gli1 to suppress the SHH pathway activity. In this regard, it is noteworthy that Trim32 knockout or mutations are shown to be causative in muscular dystrophy, such as LGMD2H [20], via regulation of muscle stem cell regeneration and induction of muscle autophagy [21, 22]. As the SHH pathway is also functionally important for skeletal muscle regeneration [38], our findings may provide a new potential direction for exploring the pathogenesis of LGMD2H myopathy, which needs further investigation in the future.
In addition, our present experiments shed a new light on the causes for the formation of MB besides reinforcing the notion that SHH signaling is the main driving force for the formation of MB. In particular, Ptch1 has been identified as a frequently mutated gene or a genomically lost gene in sporadic MBs that belong to the molecular subtype hallmarked by an aberrant activity of hedgehog signaling [13, 39–42]. The monoallelic inactivation of the Ptch1 gene in mice and thus downstream activation of the SHH pathway leads to MB development at a frequency of about 15–25% [43–46]. In this study, we showed that Trim32 knockout increases the incidence of MB from 25.4 to 52.9% in the background of Ptch1+/− mice. These data support the notion that Trim32 might be a tumor suppressor. Gene expression analysis reveals that higher expression of SHH target genes are found in Ptch1+/−/Trim32KO MB tissues, compared with Ptch1+/−/Trim32WT MB tissues, as well as in Trim32KO GNPs than in Trim32WT GNPs. Hence, we propose that deletion of Trim32, in coordination with Ptch1 mutation, probably further enhances the aberrant activation of the SHH pathway, leading to MB formation. Meanwhile, in clinical SHH MB samples, we find a negative correlation between Trim32 and Gli1 levels (P = 0.036, r = −0.3). All of these data emphasize the significance of Trim32 in mouse or human SHH MB formation.
In conclusion, we identified a novel molecular mechanism by which Trim32 interacts with Gli1 and degrades it to suppress the SHH pathway in cerebellar development and tumorigenesis. A better understanding of formation of this SHH signaling-mediated MBs will facilitate the identification of new potential targets for therapeutic interventions of MBs.
Materials and methods
Mouse husbandry
Trim32 knock-out (Trim32KO) mice have been generated previously as described previously [47]. Ptch1+/− mice were obtained from the Jackson Laboratory (#003081). Ptch1+/− males crossed with Trim32KO females to generate Ptch1/Trim32 double heterozygous mice that were backcrossed with Trim32ko mice to obtain Ptch1+/−/Trim32KO mice; Math1-GFP transgenic mice were a gift from Dr Jane Johnson at the University of Texas Southwestern Medical Center at Dallas, Dallas, TX, USA. Math1-GFP mice were crossed with Ptch1/Trim32 double mutant mice to obtain the different genotypes under study, which were interbred at least three times before further analysis. All animal experiments were approved by the Animal Research Ethics Committee of Renji Hospital.
Cell culture
HEK293T cells were cultured in DMEM (Gibco) with 10% fetal bovine serum (Gibco) and 1% l-glutamine (Sigma). D283 cells were obtained from the American Type Culture Collection and maintained in MEM medium (Gibco) supplemented with 20% fetal bovine serum (Gibco) and 1% l-glutamine (Sigma). GNPs were isolated from postnatal day 7 mouse cerebellum according to the procedures described before and maintained in Neurobasal medium (Gibco) with B27 supplement (Gibco), 1% l-glutamine (Sigma), and 100 U/ml penicillin/streptomycin.
Plasmids construction
The human Trim32 cDNA and GLi1 cDNA plasmid were ordered from OriGene (#RC201289L2, #RC201110). The full-length or truncated Trim32 cDNA were amplified by PCR from the Trim32 cDNA plasmid, and then cloned to an expression vector pCMV6-AC-GFP (#PS100010, OriGene) respectively. The full-length or truncated Gli1 cDNA were amplified by PCR from the Gli1 cDNA plasmid, and then cloned to an expression vector pCMV-Flag, respectively. The primers for Trim32 mutations cloning and Gli1 mutations cloning were provide in the following table. The shRNA of Trim32 was ordered from OriGene (#TL308646), and the sequences of shRNA were as followed: #1: 5′-GTCCAATAGTCAAGTGGTAGAGGAGCAGA-3′;
#2: 5′-AGGCAGATGTAGCACTACTGGAGGAGACA-3′.
| Prim. | Sequence | Prim. | Sequence |
|---|---|---|---|
| TF1 | CGGGATCCCGCCACCATGGCTGCAGCAGCAGCT | TR5 | CCGCTCGAGTGGGGTGGAATATCT |
| TF2 | CGGGATCCCGCCACCATGAAGATTACCCGCATAACCA | TR6 | CGACGCGTGGTTATGCGGGTAATCTTGCT |
| TF3 | CGACGCGTCTGCAGCGGCGGAAGGCA | TR7 | CGACGCGTTGCCTTCCGCCGCTGCAGCTC |
| TF4 | CGACGCGTGTCGCTGACCGTGGTAACT | TR8 | CGACGCGTCATGGCCCAGGAATCTTC |
| GF1 | CGGATATCCGCCACCATGTTCAACTCGATGACC | GR4 | CCAAGCTTGGCTCCACTGTAGAAATGGA |
| GF2 | CGGATATCCGCCACCATGAGGGAGGAAAGCAGACTGA | GR5 | CCAAGCTTGGAGGCCCAAGAGGCAGAGA |
| GR3 | CCAAGCTTGGTTCATACACAGATTCAGGC | GR6 | CCAAGCTTGGGGCACTAGAGTTGAGGAAT |
mRNA expression analysis
Total RNA was extracted from GNPs, or from cultured cells with Trizol reagent (Invitrogen) according to the manufacturer’s instructions. cDNA was generated with the PrimeScript RT reagent kit (TakaRa) and subjected to real time qPCR using 2 × SYBR Premix Ex Taq (TakaRa). Relative abundance of mRNA was calculated by normalization to ACTB expression. The following primer pairs were used:
| Gene | Species | Forward | Reverse |
|---|---|---|---|
| ACTB | M.mus | GGCTGTATTCCCCTCCATCG | CCAGTTGGTAACAATGCCATGT |
| Trim32 | M.mus | CCTCCGGGAAGTGCTAGAATG | CACTGGCGGCAGATGGTATG |
| Math1 | M.mus | GAGTGGGCTGAGGTAAAAGAGT | GGTCGGTGCTATCCAGGAG |
| Gli1 | M.mus | CCAAGCCAACTTTATGTCAGGG | AGCCCGCTTCTTTGTTAATTTGA |
| Gli2 | M.mus | CAACGCCTACTCTCCCAGAC | GAGCCTTGATGTACTGTACCAC |
| Ccnd1 | M.mus | GCGTACCCTGACACCAATCTC | CTCCTCTTCGCACTTCTGCTC |
| Ccnd2 | M.mus | GAGTGGGAACTGGTAGTGTTG | CGCACAGAGCGATGAAGGT |
| MycN | M.mus | ACCATGCCGGGGATGATCT | ATCTCCGTAGCCCAATTCGAG |
| Tuj1 | M.mus | TAGACCCCAGCGGCAACTAT | GTTCCAGGTTCCAAGTCCACC |
| NeuN | M.mus | ATCGTAGAGGGACGGAAAATTGA | GTTCCCAGGCTTCTTATTGGTC |
| ACTB | Homo | CATGTACGTTGCTATCCAGGC | CTCCTTAATGTCACGCACGAT |
| Trim32 | Homo | CCGGGAAGTGCTAGAATGCC | CAGCGGACACCATTGATGCT |
| Gli1 | Homo | AGCGTGAGCCTGAATCTGTG | CAGCATGTACTGGGCTTTGAA |
| MycN | Homo | ACCCGGACGAAGATGACTTCT | CAGCTCGTTCTCAAGCAGCAT |
Immunostaining
The postnatal cerebella or mouse MB tissues were fixed with 4% paraformaldehyde for 2 h or overnight at 4 °C, dehydrated in 30% sucrose for overnight at 4 °C, and then sectioned for 7 µm. The slices were fixed with 15 min at room temperature, and then permeabilized with 0.3% Triton X-100. After blocking in donkey serum for 1 h at room temperature, slices were incubated with primary antibody for overnight at 44 °C. Then washed three times with PBS, the slices were incubated with fluorescence-conjugated secondary antibodies (1:1000, Invitrogen) for 1 h at room temperature in dark. The following antibodies were used for immunofluorescence: antibodies against Trim32 (1:200, Genetex, #GTX113936), NeuN (1:200, CST, #12,943), and Ki67 (1:500, Abcam, #ab15580). The slices were then washed three times with PBS and mounted with Prolong Gold antifade reagent with DAPI (Invitrogen). Cell images were captured with a confocal microscope (Nikon). Quantification was performed using ImageJ software.
Luciferase assays
Gli-RE-Luciferase reporter vector was obtained from Genomeditech (Shanghai, China). Luciferase assay was performed using the Dual-Luciferase® Reporter Assay System (Promega) according to the manufacturer’s protocol (Promega).
Immunoprecipitation and immunoblot analysis
For immunoprecipitation, cells were lysed 48 h after transfection with the indicated plasmids in lysis buffer (50 mM Tris, 150 mM NaCl, 1 mM EDTA, 0.25% Triton X-100 and protease inhibitors, pH 7.4). The lysates were centrifuged at 12,000 × g for 15 min and the resulting supernatants were bisected: one incubated with anti-Flag (Sigma) or anti-Trim32 (Genetex) antibodies, and the other incubated with IgG as a control for 2 h at 4 °C. After incubating with protein G-Agarose (Roche) for 1 h with rotation, the immunoprecipitated products were washed four times with the lysis buffer, and separated by SDS-polyacrylamide gel electrophoresis. For the CHX chase assay, cells were treated with 20 µg/ml CHX (Sigma) and harvested with RIPA at the indicated times. The lysates were separated by SDS-PAGE. For protein detection, the following antibodies were used: Trim32 (1:1000, GeneTex, #GTX113,937), Gli1 (1:1000, CST, #2643), Ccnd2 (1:1000, CST, #3741), Math1(1:1000, GeneTex, #GTX111,898), NeuN (1:1000, CST, #12,943), Ubiquitin (1:1000, CST, #3936), and actin (1:10,000, CST, #4970). The ImageJ program was used for quantitative analyses of western blots.
Statistical analysis
All experiments were performed independently and at least in triplicates. Statistical analysis was carried out using the GraphPad Prism software (version 5.0). Data are shown as mean ± SD. If the p value was less than 0.05, differences were considered significant.
Supplementary information
{kind=link}
{kind=link}
Acknowledgements
We are grateful to Dr Quan-Hong Ma and Dr Jens C. Schwamborn for the generous sharing of Trim32 knockout mice.
Funding
This study was supported by funds from the Ministry of Science and Technology of the People’s Republic of China (2017YFA0102900 to W-QG), the National Natural Science Foundation of China (81672485 to RY, 81872406 and 81630073 to W-QG), the Science and Technology Commission of Shanghai Municipality (16140904100 to RY and 16JC1405700 to W-QG), High Peak IV fund from the Education Commission of Shanghai Municipality on Stem Cell Research (to W-QG), KC Wong foundation (to W-QG), and Shanghai Municipal Commission of Health and Family Planning (201540193 to RY).
Author contributions
MW, RY, and W-QG designed the experiments. MW, WL, YZ, RY, and XL performed the experiments. YG and CZ performed the computational analysis. MW, RY, and W-QG wrote the manuscript. All authors approved the manuscript.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no competing interest.
Footnotes
Edited by M. Piacentini
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Ru Yang, Phone: +86 21 68383917, Email: yangru@yahoo.com.
Wei-Qiang Gao, Email: gao.weiqiang@sjtu.edu.cn.
Supplementary information
The online version of this article (10.1038/s41418-019-0415-5) contains supplementary material, which is available to authorized users.
References
- 1.Roussel MF, Hatten ME. Cerebellum development and medulloblastoma. Curr Top Dev Biol. 2011;94:235–82. doi: 10.1016/B978-0-12-380916-2.00008-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hatten ME, Roussel MF. Development and cancer of the cerebellum. Trends Neurosci. 2011;34:134–42. doi: 10.1016/j.tins.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Espinosa JS, Luo L. Timing neurogenesis and differentiation: insights from quantitative clonal analyses of cerebellar granule cells. J Neurosci. 2008;28:2301–12. doi: 10.1523/JNEUROSCI.5157-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang R, Wang M, Wang J, Huang X, Yang R, Gao WQ. Cell division mode change mediates the regulation of cerebellar granule neurogenesis controlled by the sonic hedgehog signaling. Stem Cell Rep. 2015;5:816–28. doi: 10.1016/j.stemcr.2015.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dahmane N, Ruiz i Altaba A. Sonic hedgehog regulates the growth and patterning of the cerebellum. Development. 1999;126:3089–100. doi: 10.1242/dev.126.14.3089. [DOI] [PubMed] [Google Scholar]
- 6.Ben-Arie N, Bellen HJ, Armstrong DL, McCall AE, Gordadze PR, Guo Q, et al. Math1 is essential for genesis of cerebellar granule neurons. Nature. 1997;390:169–72. doi: 10.1038/36579. [DOI] [PubMed] [Google Scholar]
- 7.Ayrault O, Zhao H, Zindy F, Qu C, Sherr CJ, Roussel MF. Atoh1 inhibits neuronal differentiation and collaborates with Gli1 to generate medulloblastoma-initiating cells. Cancer Res. 2010;70:5618–27. doi: 10.1158/0008-5472.CAN-09-3740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.White JJ, Sillitoe RV. Development of the cerebellum: from gene expression patterns to circuit maps. Wiley Interdiscip Rev Dev Biol. 2013;2:149–64. doi: 10.1002/wdev.65. [DOI] [PubMed] [Google Scholar]
- 9.Wallace VA. Purkinje-cell-derived sonic hedgehog regulates granule neuron precursor cell proliferation in the developing mouse cerebellum. Curr Biol. 1999;9:445–8. doi: 10.1016/s0960-9822(99)80195-x. [DOI] [PubMed] [Google Scholar]
- 10.Wechsler-Reya RJ, Scott MP. Control of neuronal precursor proliferation in the cerebellum by Sonic hedgehog. Neuron. 1999;22:103–14. doi: 10.1016/s0896-6273(00)80682-0. [DOI] [PubMed] [Google Scholar]
- 11.Ruiz i Altaba A, Sanchez P, Dahmane N. Gli and hedgehog in cancer: tumours, embryos and stem cells. Nat Rev Cancer. 2002;2:361–72. doi: 10.1038/nrc796. [DOI] [PubMed] [Google Scholar]
- 12.Zhang C, Ji Z, Wang M, Zhang W, Yang R, An H, et al. Stox1 as a novel transcriptional suppressor of Math1 during cerebellar granule neurogenesis and medulloblastoma formation. Cell Death Differ. 2016;23:2042–53. doi: 10.1038/cdd.2016.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goodrich LV. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science. 1997;277:1109–13. doi: 10.1126/science.277.5329.1109. [DOI] [PubMed] [Google Scholar]
- 14.Taylor MD, Liu L, Raffel C, Hui CC, Mainprize TG, Zhang X, et al. Mutations in SUFU predispose to medulloblastoma. Nat Genet. 2002;31:306–10. doi: 10.1038/ng916. [DOI] [PubMed] [Google Scholar]
- 15.Svard J, Heby-Henricson K, Persson-Lek M, Rozell B, Lauth M, Bergstrom A, et al. Genetic elimination of suppressor of fused reveals an essential repressor function in the mammalian hedgehog signaling pathway. Dev Cell. 2006;10:187–97. doi: 10.1016/j.devcel.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 16.Hallahan AR, Pritchard JI, Hansen S, Benson M, Stoeck J, Hatton BA, et al. The SmoA1 mouse model reveals that notch signaling is critical for the growth and survival of sonic hedgehog-induced medulloblastomas. Cancer Res. 2004;64:7794–800. doi: 10.1158/0008-5472.CAN-04-1813. [DOI] [PubMed] [Google Scholar]
- 17.Hatakeyama S. TRIM family proteins: roles in autophagy, immunity, and carcinogenesis. Trends Biochemical Sci. 2017;42:297–311. doi: 10.1016/j.tibs.2017.01.002. [DOI] [PubMed] [Google Scholar]
- 18.Zhou Z, Ji Z, Wang Y, Li J, Cao H, Zhu HH, et al. TRIM59 Is up-regulated in gastric tumors, promoting ubiquitination and degradation of p53. Gastroenterology. 2014;147:1043–54. doi: 10.1053/j.gastro.2014.07.021. [DOI] [PubMed] [Google Scholar]
- 19.Lazzari E, Meroni G. TRIM32 ubiquitin E3 ligase, one enzyme for several pathologies: from muscular dystrophy to tumours. Int J Biochem Cell Biol. 2016;79:469–77. doi: 10.1016/j.biocel.2016.07.023. [DOI] [PubMed] [Google Scholar]
- 20.Kudryashova E, Wu J, Havton LA, Spencer MJ. Deficiency of the E3 ubiquitin ligase TRIM32 in mice leads to a myopathy with a neurogenic component. Hum Mol Genet. 2009;18:1353–67. doi: 10.1093/hmg/ddp036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kudryashova E, Kramerova I, Spencer MJ. Satellite cell senescence underlies myopathy in a mouse model of limb-girdle muscular dystrophy 2H. J Clin Investig. 2012;122:1764–76. doi: 10.1172/JCI59581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Di Rienzo M, Antonioli M, Fusco C, Liu Y, Mari M, Orhon I, et al. Autophagy induction in atrophic muscle cells requires ULK1 activation by TRIM32 through unanchored K63-linked polyubiquitin chains. Sci Adv. 2019;5:eaau8857. doi: 10.1126/sciadv.aau8857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Betschinger J, Mechtler K, Knoblich JA. Asymmetric segregation of the tumor suppressor brat regulates self-renewal in Drosophila neural stem cells. Cell. 2006;124:1241–53. doi: 10.1016/j.cell.2006.01.038. [DOI] [PubMed] [Google Scholar]
- 24.Lee CY, Wilkinson BD, Siegrist SE, Wharton RP, Doe CQ. Brat is a Miranda cargo protein that promotes neuronal differentiation and inhibits neuroblast self-renewal. Dev Cell. 2006;10:441–9. doi: 10.1016/j.devcel.2006.01.017. [DOI] [PubMed] [Google Scholar]
- 25.Bello B, Reichert H, Hirth F. The brain tumor gene negatively regulates neural progenitor cell proliferation in the larval central brain of Drosophila. Development. 2006;133:2639–48. doi: 10.1242/dev.02429. [DOI] [PubMed] [Google Scholar]
- 26.Shieh PB, Kudryashova E, Spencer MJ. Limb-girdle muscular dystrophy 2H and the role of TRIM32. Handb Clin Neurol. 2011;101:125–33. doi: 10.1016/B978-0-08-045031-5.00009-8. [DOI] [PubMed] [Google Scholar]
- 27.Horn EJ, Albor A, Liu Y, El-Hizawi S, Vanderbeek GE, Babcock M, et al. RING protein Trim32 associated with skin carcinogenesis has anti-apoptotic and E3-ubiquitin ligase properties. Carcinogenesis. 2004;25:157–67. doi: 10.1093/carcin/bgh003. [DOI] [PubMed] [Google Scholar]
- 28.Schwamborn JC, Berezikov E, Knoblich JA. The TRIM-NHL protein TRIM32 activates microRNAs and prevents self-renewal in mouse neural progenitors. Cell. 2009;136:913–25. doi: 10.1016/j.cell.2008.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nicklas S, Hillje AL, Okawa S, Rudolph IM, Collmann FM, van Wuellen T, et al. A complex of the ubiquitin ligase TRIM32 and the deubiquitinase USP7 balances the level of c-Myc ubiquitination and thereby determines neural stem cell fate specification. Cell Death Differ. 2018;26:728–40. doi: 10.1038/s41418-018-0144-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Izumi H, Kaneko Y. Trim32 facilitates degradation of MYCN on spindle poles and induces asymmetric cell division in human neuroblastoma cells. Cancer Res. 2014;74:5620–30. doi: 10.1158/0008-5472.CAN-14-0169. [DOI] [PubMed] [Google Scholar]
- 31.Hillje AL, Worlitzer MM, Palm T, Schwamborn JC. Neural stem cells maintain their stemness through protein kinase C zeta-mediated inhibition of TRIM32. Stem Cells. 2011;29:1437–47. doi: 10.1002/stem.687. [DOI] [PubMed] [Google Scholar]
- 32.Di Marcotullio L, Ferretti E, Greco A, De Smaele E, Po A, Sico MA, et al. Numb is a suppressor of hedgehog signalling and targets Gli1 for Itch-dependent ubiquitination. Nat Cell Biol. 2006;8:1415–23. doi: 10.1038/ncb1510. [DOI] [PubMed] [Google Scholar]
- 33.De Luca A, Cerrato V, Fuca E, Parmigiani E, Buffo A, Leto K. Sonic hedgehog patterning during cerebellar development. Cell Mol life Sci. 2015;73:291–303. doi: 10.1007/s00018-015-2065-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Locke M, Tinsley CL, Benson MA, Blake DJ. TRIM32 is an E3 ubiquitin ligase for dysbindin. Hum Mol Genet. 2009;18:2344–58. doi: 10.1093/hmg/ddp167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Northcott PA, Shih DJ, Peacock J, Garzia L, Morrissy AS, Zichner T, et al. Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature. 2012;488:49–56. doi: 10.1038/nature11327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sato T, Okumura F, Kano S, Kondo T, Ariga T, Hatakeyama S. TRIM32 promotes neural differentiation through retinoic acid receptor-mediated transcription. J Cell Sci. 2011;124( Pt 20):3492–502. doi: 10.1242/jcs.088799. [DOI] [PubMed] [Google Scholar]
- 37.Liu J, Zhang C, Wang XL, Ly P, Belyi V, Xu-Monette ZY, et al. E3 ubiquitin ligase TRIM32 negatively regulates tumor suppressor p53 to promote tumorigenesis. Cell Death Differ. 2014;21:1792–804. doi: 10.1038/cdd.2014.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Straface G, Aprahamian T, Flex A, Gaetani E, Biscetti F, Smith RC, et al. Sonic hedgehog regulates angiogenesis and myogenesis during post-natal skeletal muscle regeneration. J Cell Mol Med. 2009;13(8B):2424–35. doi: 10.1111/j.1582-4934.2008.00440.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tukachinsky H, Petrov K, Watanabe M, Salic A. Mechanism of inhibition of the tumor suppressor patched by sonic hedgehog. Proc Natl Acad Sci USA. 2016;113:E5866–75. doi: 10.1073/pnas.1606719113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kool M, Koster J, Bunt J, Hasselt NE, Lakeman A, van Sluis P, et al. Integrated genomics identifies five medulloblastoma subtypes with distinct genetic profiles, pathway signatures and clinicopathological features. PloS One. 2008;3:e3088. doi: 10.1371/journal.pone.0003088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Northcott PA, Korshunov A, Witt H, Hielscher T, Eberhart CG, Mack S, et al. Medulloblastoma comprises four distinct molecular variants. J Clin Oncol. 2011;29:1408–14. doi: 10.1200/JCO.2009.27.4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wolter M, Reifenberger J, Sommer C, Ruzicka T, Reifenberger G. Mutations in the human homologue of the Drosophila segment polarity gene patched (PTCH) in sporadic basal cell carcinomas of the skin and primitive neuroectodermal tumors of the central nervous system. Cancer Res. 1997;57:2581–5. [PubMed] [Google Scholar]
- 43.Kessler JD, Hasegawa H, Brun SN, Emmenegger BA, Yang ZJ, Dutton JW, et al. N-myc alters the fate of preneoplastic cells in a mouse model of medulloblastoma. Genes Dev. 2009;23:157–70. doi: 10.1101/gad.1759909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Farioli-Vecchioli S, Cina I, Ceccarelli M, Micheli L, Leonardi L, Ciotti MT, et al. Tis21 knock-out enhances the frequency of medulloblastoma in Patched1 heterozygous mice by inhibiting the Cxcl3-dependent migration of cerebellar neurons. J Neurosci. 2012;32:15547–64. doi: 10.1523/JNEUROSCI.0412-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim JY, Nelson AL, Algon SA, Graves O, Sturla LM, Goumnerova LC, et al. Medulloblastoma tumorigenesis diverges from cerebellar granule cell differentiation in patched heterozygous mice. Dev Biol. 2003;263:50–66. doi: 10.1016/s0012-1606(03)00434-2. [DOI] [PubMed] [Google Scholar]
- 46.Corcoran RB, Bachar Raveh T, Barakat MT, Lee EY, Scott MP. Insulin-like growth factor 2 is required for progression to advanced medulloblastoma in patched1 heterozygous mice. Cancer Res. 2008;68:8788–95. doi: 10.1158/0008-5472.CAN-08-2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nicklas S, Otto A, Wu X, Miller P, Stelzer S, Wen Y, et al. TRIM32 regulates skeletal muscle stem cell differentiation and is necessary for normal adult muscle regeneration. PloS One. 2012;7:e30445. doi: 10.1371/journal.pone.0030445. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.