

Abstract
Objective:
To understand role of barrier molecules in melanomas.
Background:
We have reported poor patient survival and low immune infiltration of melanomas that overexpress a set of genes that include filaggrin (FLG), dystonin (DST), junction plakoglobin (JUP), and plakophilin-3 (PKP3), and are involved in cell-cell adhesions. We hypothesized that these associations are causal, either by interfering with immune cell infiltration or by enhancing melanoma cell growth.
Methods:
FLG and DSTwere knocked out by CRISPR/Cas9 in human DM93 and murine B16-F1 melanoma cells. PKP3 and JUP were overexpressed in murine B16-AAD and human VMM39 melanoma cells by lentiviral transduction. These cell lines were evaluated in vitro for cell proliferation and in vivo for tumor burden, immune composition, cytokine expression, and vascularity.
Results:
Immune infiltrates were not altered by these genes. FLG/DST knockout reduced proliferation of human DM93 melanoma in vitro, and decreased B16-F1 tumor burden in vivo. Overexpression of JUP, but not PKP3, in B16-AAD significantly increased tumor burden, increased VEGF-A, reduced IL-33, and enhanced vascularity.
Conclusions:
FLG and DST support melanoma cell growth in vitro and in vivo. Growth effects of JUP were only evident in vivo, and may be mediated, in part, by enhancing angiogenesis. In addition, growth-promoting effects of FLG and DST in vitro suggest that these genes may also support melanoma cell proliferation through angiogenesis-independent pathways. These findings identify FLG, DST, and JUP as novel therapeutic targets whose down-regulation may provide clinical benefit to patients with melanoma.
Keywords: angiogenesis, B16 melanoma, barrier molecules, cell-cell adhesions, immune cell infiltrate, vascularity
Immune infiltrates in tumors are associated with patient survival and responses to immune therapy in melanoma and other cancers.1–5 Consequently, patients with sparsely infiltrated tumors tend to have worse outcomes and respond poorly to immune therapy. The extent of immune infiltrates is determined by several processes: extravasation of immune cells from vasculature, intratumoral immune cell proliferation, immune cell survival, and strength of the immune response. One possible reason for low numbers of immune cells in tumors is the presence of barriers that prevent their extravasation from the vasculature. Therefore, understanding barriers to immune infiltrates can identify better targets to improve efficacy of therapy.
In prior work, we identified a set of 8 genes that were significantly up-regulated in melanomas lacking immune gene signatures and associated with significantly shortened patient survival.6 These genes [filaggrin (FLG), dystonin (DST), junction plakoglobin (JUP), plakophilin-3 (PKP3), desmoplakin (DSP), desmocollin-3 (DSC3), periplakin (PPL), and trop-2 (TACSTD2)] encode proteins that are key components of desmosomes and hemidesmosomes or other cell junctions and mediate mechanical barrier function in normal skin.6,7 Thus, we refer to them collectively as barrier molecules (BMs). Filaggrin is unique in that it forms a cornified cell envelope that provides barrier function in the skin,8,9 and it is involved in keratinocyte differentiation.10 Its function is key for epidermal homeostasis, and filaggrin deficiency in atopic dermatitis leads to impaired skin barrier function and enhanced immune responses.11–13 Interestingly, FLG, DST, PKP3, and JUP are localized intracellularly, and also at the cell surface.6,14–17 Thus, these differentially expressed genes may participate in intracellular processes that support melanoma cell growth and survival directly or indirectly.
To evaluate the potential roles of BMs in melanoma, we tested 3 hypotheses. First, we hypothesized that BMs reduce immune cell infiltrates by mechanical barrier function. Alternatively, we hypothesized that BM expression may be down-regulated by immune cell infiltrates or by cytokines and chemokines that recruit immune cells. Finally, based on the intracellular localization of some BMs, we hypothesized that BM expression directly supports melanoma cell growth. To test these hypotheses, we focused on 4 BM genes, FLG, DST, PKP3, and JUP, which we stably overexpressed or knocked out in human and murine melanoma cell lines. We studied those lines to assess the impact of BMs on melanoma cell proliferation in vitro and tumor growth in vivo, and also immune cell infiltration, cytokine production, and angiogenesis in vivo.
METHODS
Cell Culture
DM93 and VMM39 human melanoma cell lines were established at Duke University and University of Virginia, respectively, and were grown in RPMI media supplemented with 5% fetal bovine serum (FBS) and 1% penicillin/streptomycin. The C57BL/6-derived melanoma cell line B16-F1 (CRL-6323) was obtained from the American Type Culture Collection. The B16-F1 parental cell line was transfected to express the chimeric class I MHC molecule AAD (B16-AAD), as previously described.18,19 B16-F1 transfectants were cultured in RPMI-1640 containing 5% FBS. B16-AAD transfectants were cultured in RPMI-1640 containing 5% FBS supplemented with 15 mM HEPES and 300 μg/mL G418 (Life Technologies). All lines were Mycoplasma-negative, used within 10 passages, and cultured at 378C in 5% CO2 and 21% O2. To establish murine tumors, 4 × 105 tumor cells were injected subcutaneously (s.c) or intraperitoneally (i.p.), and were allowed to establish for 14 days before harvest.
Mice
C57BL/6 mice (NCI-Frederick Animal Production Program) were maintained in pathogen-free facilities at the University of Virginia. Protocols were approved by the University of Virginia Institutional Animal Care and Use Committee. Animals were used at ~8 to 12 weeks of age.
Plasmid Transfection and Lentiviral Transduction
Filaggrin and DST are large genes that presented challenges for cloning. Thus, alternate BMs, JUP and PKP3, were selected for overexpression studies. cDNA encoding human JUP and PKP3 were amplified from The CCSB Human ORFEOME Collection (V5.1) and subcloned with an N-terminal flag tag into a modified version of the pCW-Cas9 plasmid (Addgene #50661) after removal of the Cas9 gene and substitution with JUP or PKP3 using standard restriction digestion (AgeI and BamHI) and ligation (Fig. 1). An inducible Tet promoter was swapped with EF-1α promoter, and puromycin selection marker was swapped with blasticidin selection marker by standard restriction-enzyme cloning methods. 293T cells were transfected with the lentiviral vector, along with packaging plasmids, to generate PKP3 and JUP expressing lentiviruses. Melanoma cell lines (B16-AAD and VMM39) overexpressing human PKP3 or human JUP were generated by lentiviral transduction. Blasticidin was used to select for blasticidin-resistant clones containing the transfected gene. Human PKP3 is 97% aligned with murine PKP3 mRNA, whereas human JUP is 99% aligned with murine JUP mRNA. Confirmation of overexpression of PKP3 and JUP was performed via RT-qPCR using the following forward and reverse primer sets: human JUP: 5’-AGGTGACTTCCTGCTTCCTGAC-3’ and 5’-GCAGGCCTCATCCTCCTCCATG-3’; human PKP3: 5’-TCGAGGGACAGGACGTGAAGATAG-3’ and 5’-CCACCGCCTACCTCTGGCAGTC-3’. Protein expression of JUP and PKP3 was confirmed by immunoblotting cell extracts using antiflag antibodies.
FIGURE 1.
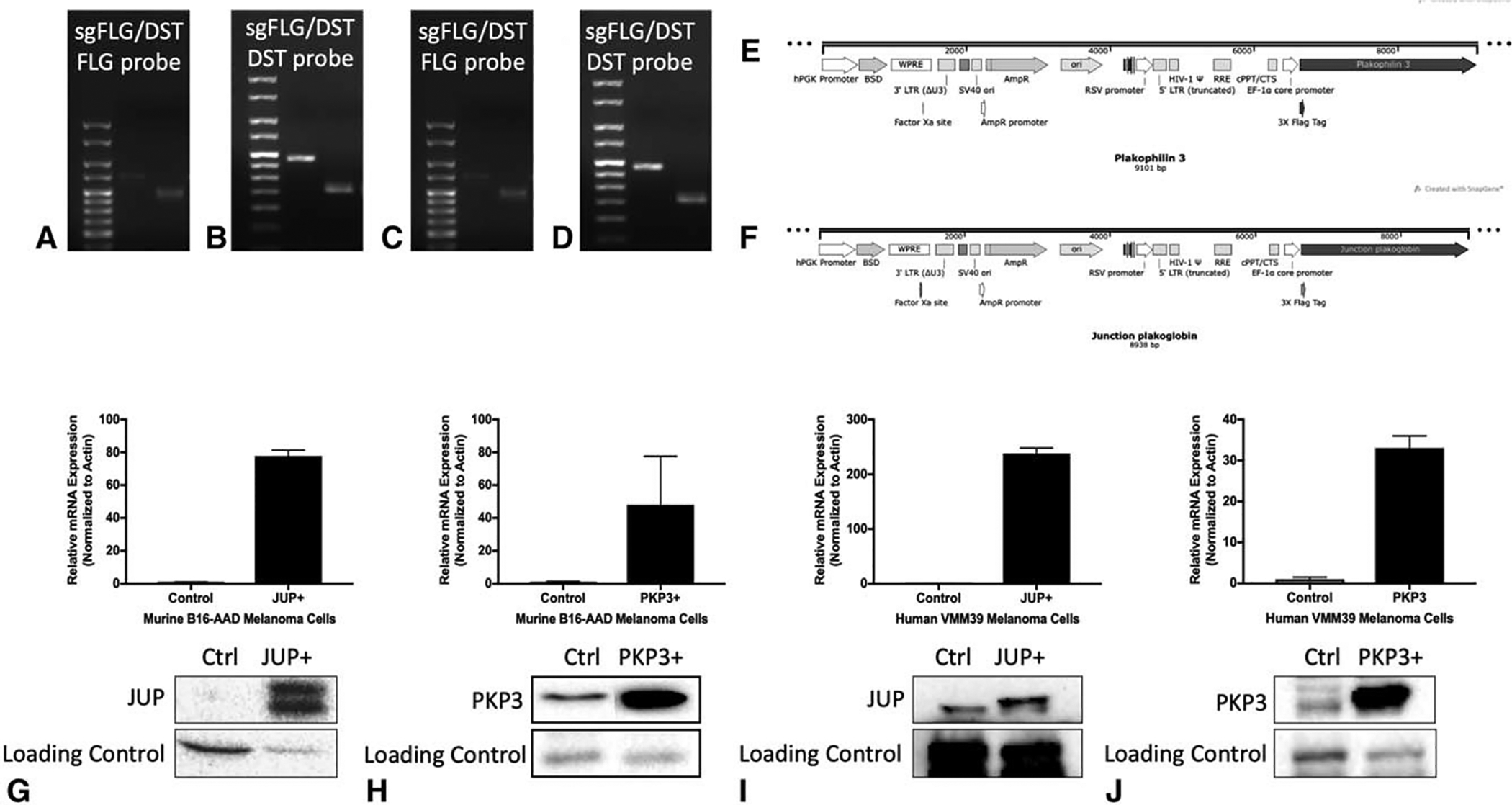
Confirmation of desired deletion or overexpression of barrier molecule genes in human and murine cell lines. Genotyping of murine B16-F1 cells transfected with sgRNA for FLG and DST using DNA gel demonstrates DNA ladder in lane 1 (left lane), Px330 empty vector in lane 2 (middle lane), and B16-F1 sgFLGDST in lane 3 (right lane) probed for FLG (A) and DST (B), and demonstrating lower mass compared to empty vector control. This is similarly demonstrated in human DM93 cells in (C) and (D). PKP3 (E) and JUP (F) plasmid design for cloning. Expression of JUP mRNA and protein (G) and PKP3 mRNA and protein (H) in murine B16-AAD control, JUP+, and PKP3+ melanoma cell lines with respective loading controls. Expression of JUP (I) and PKP3 (J) in human VMM39 control, JUP+, and PKP3+ melanoma cell lines.
Gene Targeting by CRISPR/Cas9
Single guide RNAs (sgRNAs) targeting FLG and DST were cloned into pX330 vector containing a human codon-optimized SpCas9 endonuclease (Addgene #42230) and transfected in the various cell lines. Control empty vector was pX330 vector. Targets of the sgRNAs are as follows: human FLG: 5’-caccgTCAACCATATCTGGGTCATC-3’ and 5’-caccgAAAGAGAATTTACCGATATC-3’; murine FLG: 5’-caccgTCAGCGATGTCTTGGTCATC-3’ and 5’-caccgCGAAATCTAGTTTGTCATCG-3’; human DST: 5’-caccgATATCTGATATCCATGTTAC-3’ and 5’-caccGTGAAAATTTCACTACCTGC-3’; murine DST: 5’-caccgTTCACGTGCTTCCGAACCTA-3’, 5’-caccGCACAATTTGATCTCCCTCC-3’. Plasmids containing each sgRNA were isolated by QIAprep Spin Miniprep Kit (Qiagen) and confirmed by sanger sequencing (Eurofins Scientific). Using CRISPR/Cas9 and sgRNA, co-knockout of both FLG and DST or control Empty Vector (EV) was performed in human DM93 melanoma cells and in murine B16-F1 melanoma cells. After puromycin selection, single cells were selected and expanded. Genomic DNA was extracted, and genotyping was performed using PCR amplification of genomic DNAwith the following forward and reverse primer sets: human FLG: 5’-TCTGTCTGATGCAGTCTCCCTC-3’ and 5’-CTTCTTTCCAGACTTGAGGGTC-3’; murine FLG: 5’-GACATCATAAATGTATATTGCA-3’ and 5’-CTTCTCCCATTTGATCCATGTG-3’; human DST: 5’-GTTAGCTTCCTTCCTTCCTAAAG-3’ and 5’-ATGTGTCTTACAGTTGTATAAGG-3’; murine DST: 5’-CGTGAAGCATGTGTCTGTTGTTA-3’ and 5’-ATCAGGAAAGCGCACACTTACC-3’.
Cell Lysis and Immunoblotting
Melanoma cells were lyzed using RIPA lysis buffer (50 mM Tris, pH 8.0; 150 mM NaCl, 1% NP-40; 0.5% sodium deoxycolate; 0.1% SDS; 1 mM Benzamidin-HCl; 0.5 μg/mL Leupeptin; 0.5 mg/mL Aprotinin; 1 mg/mL pepstatin; 20 mM NaF; 20 mM Na3VO4) with protease inhibitors. Equal amounts of protein were electropho-retically separated in 12% polyacrylamide (BioRad), trans-blotted to a nitrocellulose membrane, and incubated overnight with primary antibodies at 4°C. Immunoblot signals were detected by enhanced chemiluminescence.
Meso Scale Discovery Multiplex Assay
Tumors were homogenized and lyzed using lysis buffer (150 mM NaCl, 20 mM Tris, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, pH 7.5), sonicated, and centrifuged. Supernatants were collected and protein concentrations were quantified by BCA assay (Pierce, Thermo Fisher Scientific). Supernatants were analyzed for concentrations of 27 cytokines and chemokines (IL-12/IL-23p40, IL-21, IL-22, IL-23, GM-CSF, IFNγ, IL-10, IL-12p70, IL-13, IL-1b, IL-2, IL-4, IL-6, TNF-α, IL-15, IL-27p28/IL-30, IL-31, IL-33, IP-10, KC/GROa, MCP-1, MIP-1α, MIP-1β, VEGF-A, TGF-β2, TGF-β3, TGF-β1) with the Meso Scale Discovery assay (Meso Scale Diagnostics, Rockville, MD) following the manufacturer protocol. Plates were read with MSD Sector Imager, and data were analyzed by MSD Discovery Workbench software. Data were normalized against protein concentration and tumor size and compared to control.
RNA Extraction and Quantitative Real-time PCR
Human melanoma DM93 cells cultured in RPMI 1640 with 5% FBS were plated onto 60 mm dishes and cultured 24 hours with synthetic chemokines CCL5, CXCL9, CXCL10, CXCL11, and CXCL12, and cytokines IFNγ, IL-2, IL-4, and TGFβ1 (Peprotech). Cells were subsequently harvested, and total RNA was isolated with TRIZOL (Invitrogen). Complementary DNA (cDNA) was generated from 1 μg of total RNA using SuperScript III Reverse Transcriptase (Invitrogen). The quantitative real-time PCR (qRT-PCR) analysis was performed using Power SYBR Green PCR Master Mix (Invitrogen) following manufacturer instructions. Forward and reverse primers for FLG, DST, PKP3, and JUP were: FLG: 5’-GTTGGCTCAAGCATATTATGAG-3’ and 5’-caccgAAAGAGAATTTACCGATATC-3’; DST: 5’-GTTATGCTGGAATTCGGTGTG-3’ and 5’-caccGTGAAAATTTCACTACCTGC-3’; PKP3: 5’-GCTGCCCTCTGACCTGCAGCTG-3’ and 5’-CCACCGCCTACCTCTGGCAGTC-3’; JUP: 5’-GCCTATCAAGGTGACTGAGTGG-3’ and 5’-GCAGGCCTCATCCTCCTCCATG-3’. Primers for beta-actin (5’-CTCCTGAGCGCAAGTACTCC-3’ and 5’-GACTCGTCATACTCCTGCTTGCT-3’) were used to amplify beta-actin mRNA for normalization. Results were expressed relative to the basal expression of these genes in parental untreated cells.
Cell Proliferation and Viability Assays
CellTiter96 Non-radioactive cell proliferation assay (Promega) was used to measure proliferation/viability of cultured cells. Various genetically modified melanoma cell lines were seeded in 96-well plates next to control parental melanoma cell lines. Cells were stained with dye solution according to the manufacturer’s protocol 120 hours after seeding. Absorbance was recorded at 570 nm. Data were normalized to control.
Tissue Preparation and Immune Cell Isolation
Tissue was incubated in medium containing 0.42U/mL Liberase (Roche) for 15 minutes at 37°C and homogenized. CD45+ cells were purified using anti-CD45 magnetic beads (Miltenyi) and the Possel AutoMACS protocol.
Flow Cytometry Analysis
Cells were Fc blocked (BioXCell) and stained with fluores-cent antibodies to CD45, CD3, CD8, IFNγ, CD44, CD4, and FoxP3. Transcription factor Cytofix/Cytoperm Kit was used for fixation/permeabilization (BD Pharmingen). Live/Dead Aqua (Invitrogen) was used to exclude dead cells from analysis. Cells were run on FACS Cytoflex (Beckman Coulter) flow cytometers. FlowJo software was used for analysis.
Immunofluorescence Microscopy
Images were collected on an AxioImager with Apotome (Zeiss). The percent CD31+ area of each tumor section was evaluated for 10 areas of interest for each tumor using ImageJ Software (NIH) on acquired images from AxioImager. A single section per tumor and 10 random fields of view were used for analysis. Brightness and contrast were linearly adjusted and color-merged images were generated using Photoshop CS6 Software (Adobe).
Antibodies
Fluorophore- or biotin-conjugated antibodies specific for murine cell surface antigens and intracellular proteins are as follows: CD45 (30-F11), CD3 (145–2C11), CD4 (GK1.5), CD8 (53–6.7), CD44 (IM7), IFNγ (XMG1.2), FoxP3 (FJK-16 s), and CD31 (390) were from Biolegend. PKP3 (E-10) was from Santa Cruz. JUP (#2309) was from Cell Signaling Technology.
Statistical Analysis
Analyses were performed with GraphPad Prism 7 software. Student t test was used to compare 2 groups. One-way analysis of variance (ANOVA) with Tukey post-test was used to compare more than 2 groups. Multiple ANOVA tests for cytokine multiplex were corrected using Holm-Sidak method.20,21 A P value of less than 0.05 was considered to be significant.
Ethical Approval and Ethical Standards
All procedures performed in studies involving human participants/tissues were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. The animal study was approved by the University of Virginia Animal Care and Use Committee (IACUC Protocol 1068). All protocols and procedures used in this study were approved by and performed in accordance with the ethical standards of the University of Virginia Animal Care and Use Committee and with the National Institute of Health’s Guidelines for the Care and Use of Laboratory Animals.
The C57BL/6 mice were purchased from NCI-Frederick Animal Production Program. All mice were maintained in pathogen-free facilities. The C57BL/6-derived melanoma cell line B16-F1 (CRL-6323) was obtained from the American Type Culture Collection (Manassas, VA).
RESULTS
T-cell Attracting Chemokines/Cytokines do not Alter BM Expression
We first tested the hypothesis that T-cell-derived proinflammatory cytokines decrease BM gene expression.22,23 FLG was more highly overexpressed in human melanomas than the other BMs, and the other BMs, except DST, are overexpressed concordantly with FLG.6 Thus, we tested the impact of the cytokines and chemokines on FLG and DST expression in human DM93 melanoma cells. qRT-PCR demonstrated that neither FLG nor DST mRNA expression was significantly decreased by IFNγ, IL-2, or IL-4 in DM93 human melanoma cells (Fig. 2A). Chemokines CCL5, CXCL9, CXCL10, CXCL11, and CXCL12 can recruit activated CD8+ and Th1 CD4+ T cells to tissues,24,25 but none of them reduced FLG or DST mRNA expression (Fig. 2B). TGFβ has been linked to immune cell exclusion by stromal activation and creation of a physical barrier to immune infiltration.26–28 However, TGFβ1 failed to increase FLG or DST expression (Fig. 2C). Collectively, these results suggest that the inverse correlation of BM genes with Th1 immune genes is not explained by an inhibitory effect of cytokines or chemokines associated with Th1 immunity in the tumor microenvironment.
FIGURE 2.
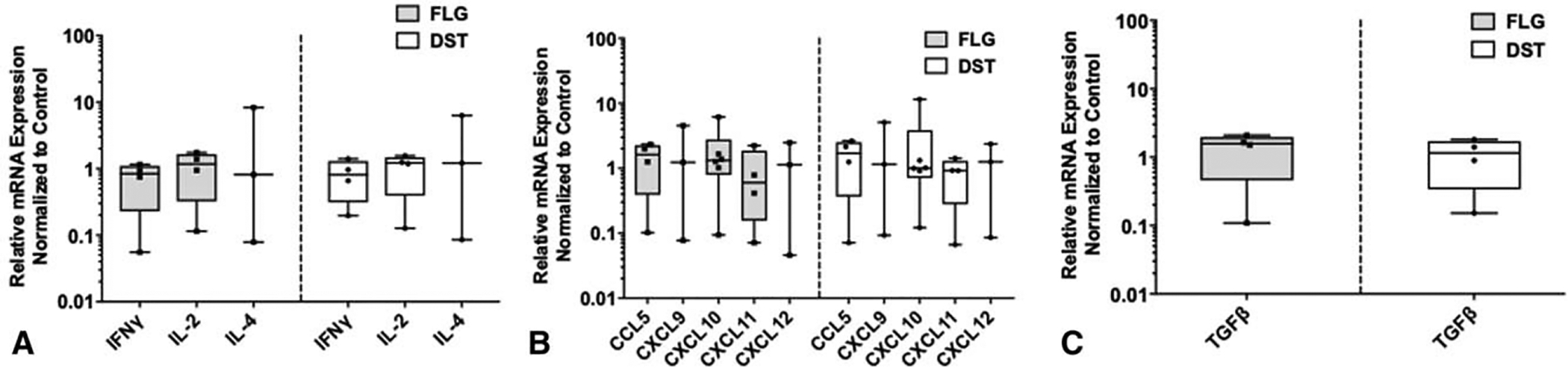
Proinflammatory and immunosuppressive cytokines/chemokines do not affect FLG or DST expression in melanoma. Normalized expression of FLG and DST mRNA to untreated control by quantitative RT-PCR on human DM93 melanoma cells following 24 hours of treatment with IFNγ, IL-2, and IL-4 (A); CCL5, CXCL9, CXCL10, CXCL11, and CXCL12 (Bb); and TGF-β1 (C). Experiments were performed in triplicates.
BM Expression Does Not Limit T-cell Infiltration Into Melanomas
Next, we tested the hypothesis that BM overexpression limits T-cell infiltration into tumor. The B16-F1 cell line and subcutaneous location were selected because these features result in poorly infiltrated tumors,29,30 which makes this model a good candidate to evaluate whether deletion of both FLG and DST in B16-F1 would increase immune infiltration in vivo. FLG and DST were targeted with sgRNA to delete both genes (sgFLGDST). After Cas9-sgRNA transfection, cells were selected with puromycin, and single clone selection and expansion was performed. The PCR-amplified DNA from a single clone was run on an agarose gel (Fig. 1A and B) and detected a smaller product, corresponding to the predicted size of the edited gene compared with wild-type. Deletion of the desired segment was confirmed via sanger sequencing and indicated a frameshift mutation in both FLG and DST (data not shown). B16-F1 melanoma cells were implanted subcutaneously to determine whether FLG and DST co-knockout would result in more extensively infiltrated tumors. Interestingly, FLG and DST co-knockout significantly decreased tumor burden compared with control (Fig. 3A), and there was a significant increase in CD45+ immune cells per gram of tumor (Fig. 3B). However, there was no change in intratumoral CD8+ or CD4+ T-cells (Fig. 3C and D), nor was there a change in the fraction of these cells that were antigen experienced or IFNγ-producing (Fig. 3E and F) or in the proportion of Tregs (FoxP3+ CD4+ T cells; Fig. 3G). Without controlling for tumor weight, there were no differences in total numbers of CD45+ cells, CD8+, and CD4+ T cells per tumor (Supplemental Fig. 1A–C, http://links.lww.com/SLA/B737). These results suggest that FLG and DST do not limit T-cell infiltration or alter the balance of effector and regulatory subsets. Nonetheless, and interestingly, deletion of these genes has a negative impact on tumor growth.
FIGURE 3.
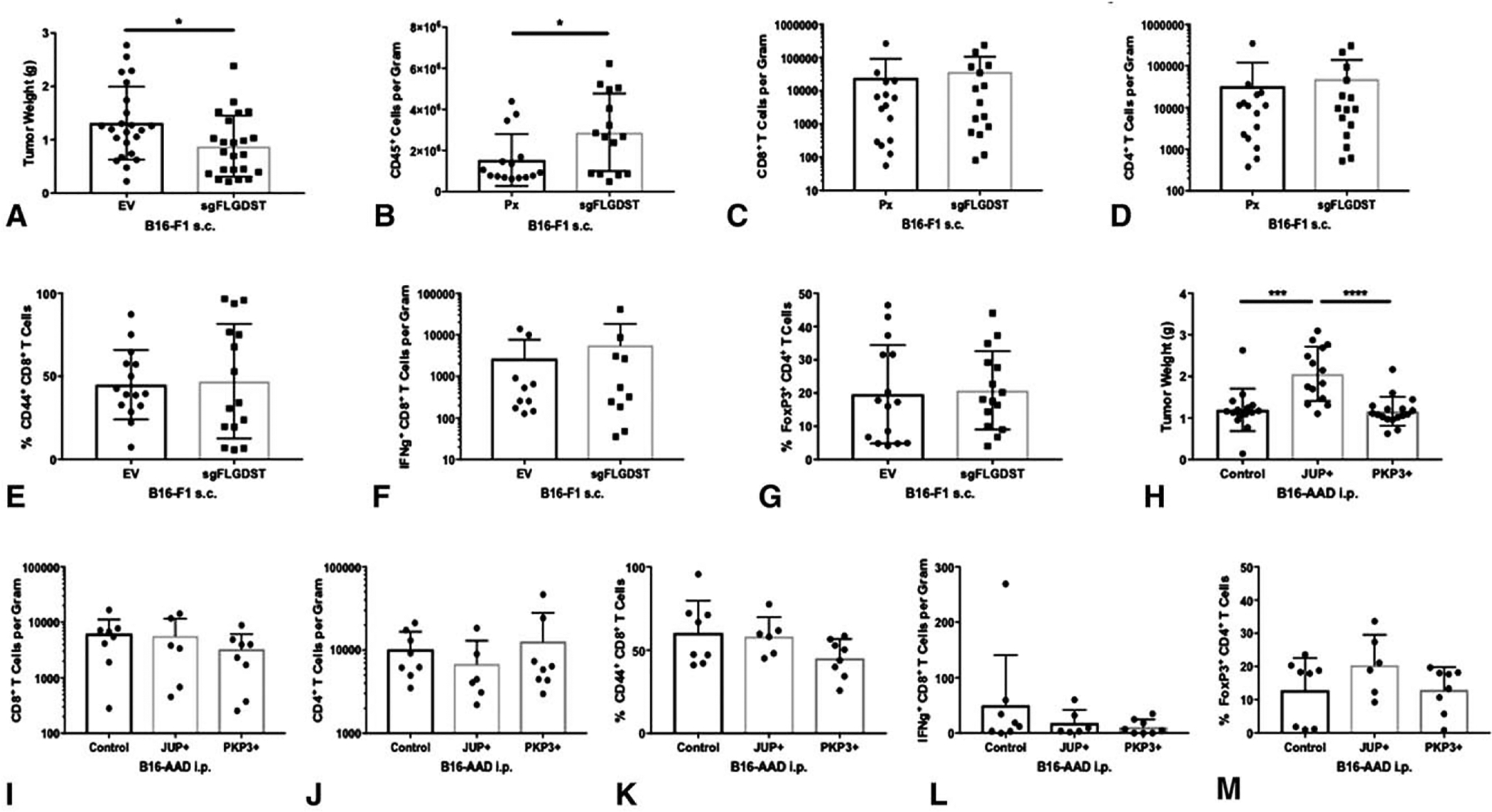
Barrier molecules drive B16 melanoma tumor growth, but do not affect intratumoral T-cell infiltration. (A) Tumor burden was determined with tumor weights obtained in day 14 B16-F1 Px and sgFLGDST2 s.c. tumors. CD45+ immune cells (B), CD8+ T cells (C), and CD4+ T cells (D) per gram of tumor tissue were determined by flow cytometry. Percentage of CD44+ CD8+ T cells (E), IFNg+ CD8+ T cells per gram (F), and percentage of FoxP3+ CD4+ T cells (G) were determined by flow cytometry. (H) Tumor weights were used for tumor burden in day 14 B16-AAD control, JUPþ, and PKP3 i.p. tumors. CD8+ T cells (I), CD4+ T cells (J), IFNγ+ CD8+ T cells (L) per gram of tumor tissue, and percentage of CD44+ CD8+ T cells (K) and FoxP3+ CD4+ T cells (M) were quantitated by flow cytometry. Differences were assessed by 1-way ANOVA, with significance noted by asterisks: *<0.05, **<0.01, ***<0.001, ****<0.0001. Experiments were performed in triplicates.
To further assess the impact of BMs on melanoma growth and immune infiltration, we evaluated the consequences of overexpression of other BMs, JUP, and PKP3. We cloned PKP3 and JUP into a lentiviral vector and stably transduced B16-AAD to overexpress these genes (Fig. 1E and F). The B16-AAD cell line and intraperitoneal location were selected because of their ability to produce heavily infiltrated tumors,30 making this a promising model to test whether overexpression of JUP or PKP3 will decrease immune infiltration.JUP mRNA expression was increased 78-fold and PKP3 mRNA expression 48-fold, and overexpression was confirmed by immunoblotting (Fig. 1G and H). PKP3 overexpression did not impact tumor growth; however, JUP overexpression significantly increased tumor burden compared with control (Fig. 3H). Neither JUP nor PKP3 overexpression impacted intratumoral CD8+ or CD4+ T cells (Fig. 3I and J), nor was there a change in the fraction of CD8+ T cells that were antigen experienced or IFNγ-secreting (Fig. 3K and L) or of CD4+ T cells that were FoxP3+ (Fig. 3M). Also, there were no significant differences in numbers of CD8+ and CD4+ T cells, or in IFNg+ CD8+ T cells per tumor (Supplemental Fig. 1D and F, http://links.lww.com/SLA/B737). These results support the conclusions reached above using knockout of FLG and DST, and suggest that these 4 BMs do not control T-cell infiltration or effector function in tumors. However, the enhanced growth of JUP-overexpressing tumors, coupled with growth reduction in FLG/DST co-knockout tumors, supports our third hypothesis, that BMs directly promote melanoma growth.
FLG and DST Have Cell-intrinsic Effects on Melanoma Cell Proliferation
To evaluate whether the impact of FLG and DSTon melanoma growth is cell-intrinsic, we tested whether the BMs alter growth properties of melanoma cells themselves. First, additional FLG and DST co-knockout transfectants of the human DM93 melanoma cell line were generated using CRISPR/Cas9 gene editing. DM93 was selected for FLG and DST deletion due to high mRNA expression of FLG and DST compared with other melanoma cell lines (data not shown). Deletion of these genes was confirmed with genotyping (Fig. 1C and D). Interestingly, FLG/DST co-knockout significantly decreased proliferation of human DM93 melanoma cells (Fig. 4A), but not of murine B16-F1 cells (Fig. 4B) in vitro.
FIGURE 4.
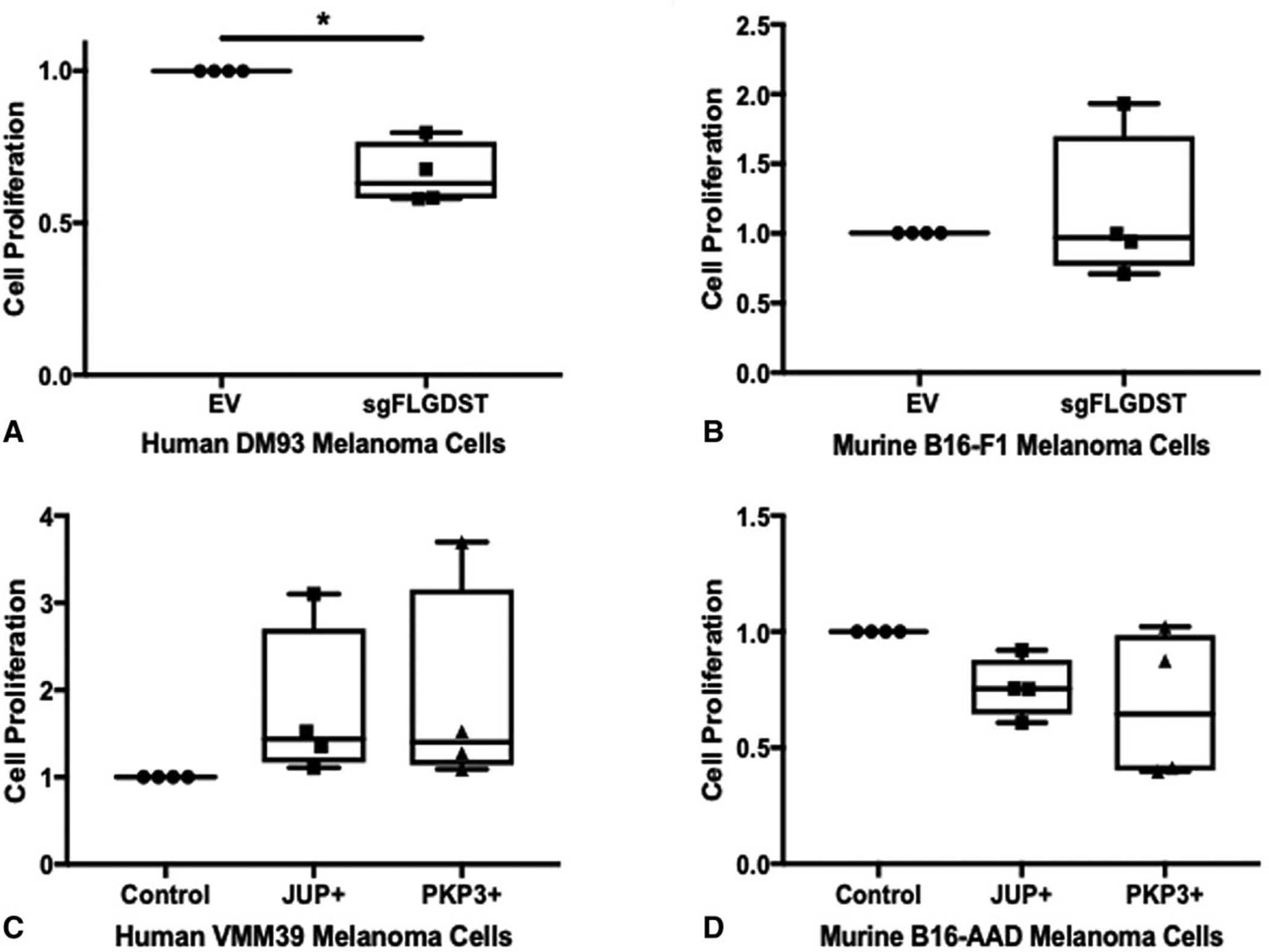
FLG and DST knockout decreases melanoma cell proliferation in DM93 human melanoma. MTT viability/cell proliferation assays for human DM93 melanoma cells (A) and murine B16-F1 cells (B) with Empty Vector (EV) and sgFLGDST. MTT cell proliferation/viability assays of human VMM39 melanoma cells (C) or murine B16-AAD melanoma cells (D) with control, JUP+, and PKP3+. Results are normalized to Control/EV. Data represent the average of 3 independent experiments SD.± Differences were assessed by 1-way ANOVA, with significance noted by asterisks: *<0.05.
Growth-promoting Effects of JUP on Melanoma Proliferation are Extrinsic
We also overexpressed JUP and PKP3 in the human melanoma cell line, VMM39, which had low mRNA expression of those genes compared with other human melanoma cell lines (data not shown). This increased mRNA expression of JUP 238-fold and of PKP3 33-fold and was confirmed by immunoblotting (Fig. 1I and J). In vitro, there was no impact on proliferation of B16-AAD or VMM39 cells by overexpression of JUP or PKP3 (Fig. 4C and D). Therefore, the growth enhancement with JUP overexpression may be tumor cell extrinsic, whereas FLG and DST knockout decreases melanoma growth through impaired proliferation, consistent with intrinsic effects that may be mediated through proliferation or apoptosis pathways.
JUP Supports Melanoma Growth Through Angiogenesis
We hypothesized that the tumor-promoting effect of JUP in vivo could be due to cytokines or chemokines with growth or angiogenic functions. Thus, we screened lysates of JUP-overexpressing and untransfected B16-AAD i.p. tumors for 27 cytokines and chemokines, of which, values were within range of detection for 17 analytes (Fig. 5). Comparing JUP overexpressing tumors to control, there were nonsignificant trends for decreases in IL-6, KC/GROα, MIP1a, and MIP1b (Fig. 5). There was also a nonsignificant trend for an increase in CXCL10 in JUP-overexpressing tumors. There were trends towards selective increases in TNFα, IL-27p28/IL-30, and selective decreases in TGFβ1 with PKP3 expression compared with control. Other cytokines associated with IL-33 (TGFβ2, TGFb3, IL-15, IL-31, MCP-1, and IL-1B) were not altered or were not evaluable. Significant changes were observed for 2 analytes: JUP-overexpressing lysates contained significantly lower levels of IL-33 (Fig. 6) and higher concentrations of VEGF-A (Fig. 7A), compared with both control and PKP3-overexpressing tumors. The increased VEGF-A in JUP-overexpressing tumors led us to evaluate vascular density, measured by the proportion of tumor section area occupied by CD31+ vasculature. Tumors with JUP overexpression had significantly greater intratumoral CD31+ area (Fig. 7B–D). This suggests that JUP may support tumor growth in an angiogenesis-dependent manner mediated by VEGF-A.
FIGURE 5.
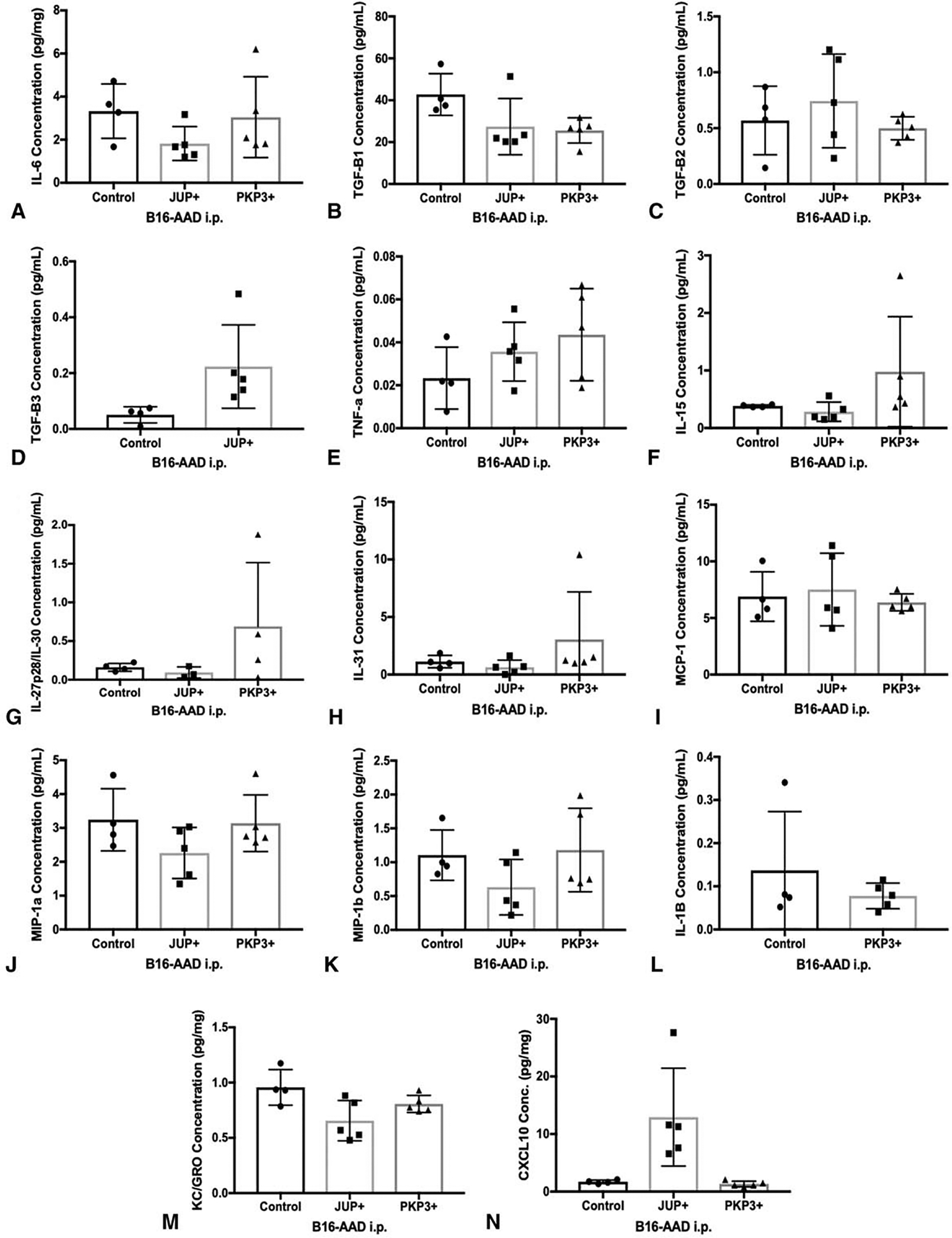
Intratumoral concentrations of various growth-promoting and pro-angiogenic chemokines and cytokines in B16-AAD transfectants. (A) IL-6, (B) TGF-β1, (C) TGF-β2, (D) TGF-β3, (E) TNF-a, (F) IL-15, (G) IL-27/p28/IL-30, (H) IL-31, (I) MCP-1, (J) MIP-1a, (K) MIP-1b, (L) IL-1b, (M) CXCL10, and (N) KC/GROα (CXCL1) concentrations per milligram of tumor tissue were quantitated using multiplex assay in day 14 B16-AAD control, JUP+, and PKP3+ i.p. tumors. Data were not within range of detection for IL-2, IL-4, IL-10, IL-12/IL-23p40, IL-12p70, IL-13, IL-21, IL-22, IL-23, GM-CSF, and IFNγ. ND indicates nondetectable. Values were plotted at 0 for TGF-β3 and IL-1b below detection limits for JUP- and PKP3-overexpressing tumors, respectively. Values are shown as 0 on the representative graphs.
FIGURE 6.
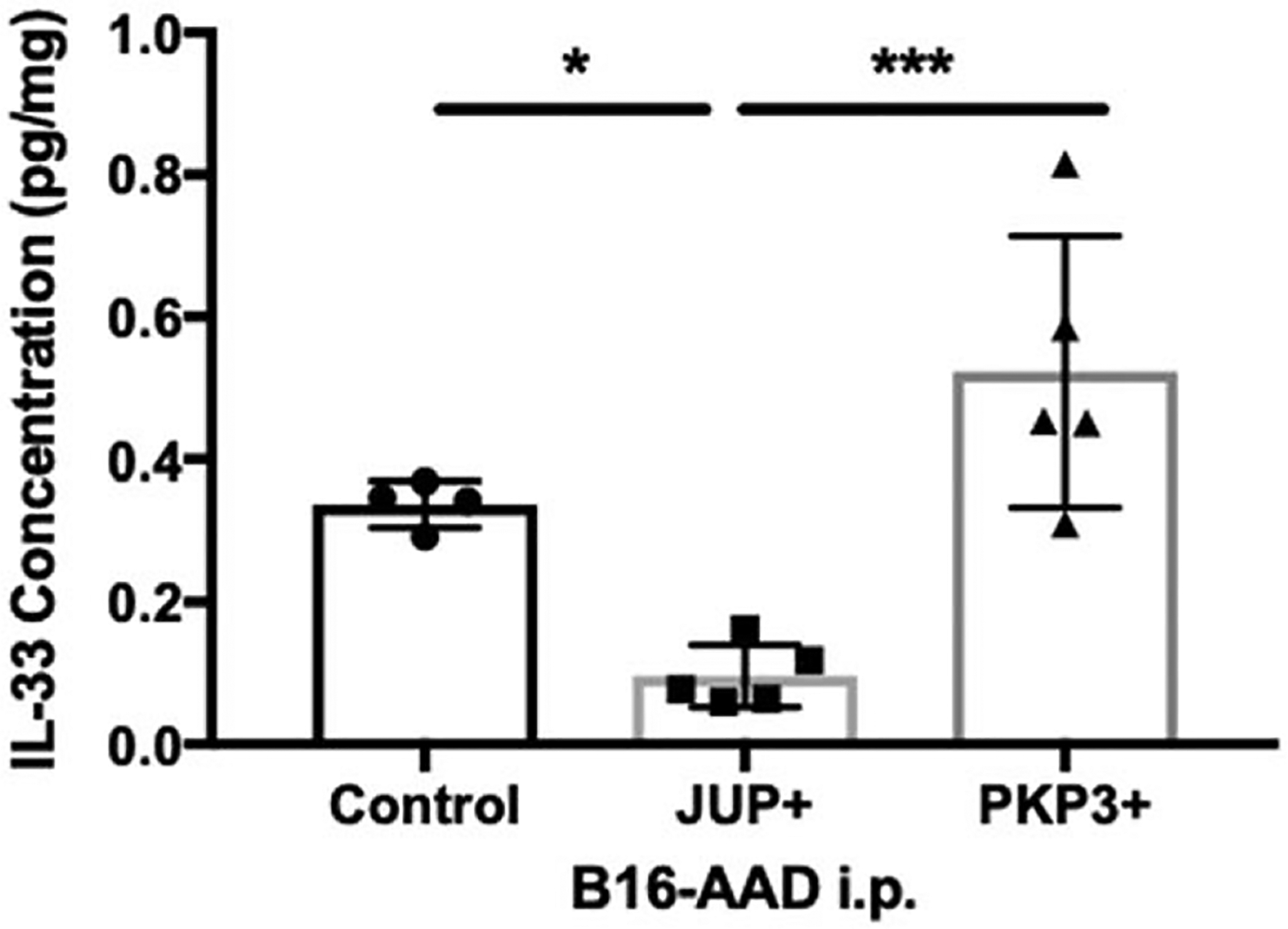
JUP overexpression down-regulates IL-33. IL-33 per milligram of tumor tissue was quantitated using multiplex assay in day 14 B16-AAD control (n = 4 tumors), JUP+ (n = 5 tumors), and PKP3+ (n = 5 tumors) i.p. tumors. Differences were assessed by 1-way ANOVA using Holm-Sidak correction method with significance noted by asterisks: *<0.05, **<0.01, ***<0.001.
FIGURE 7.
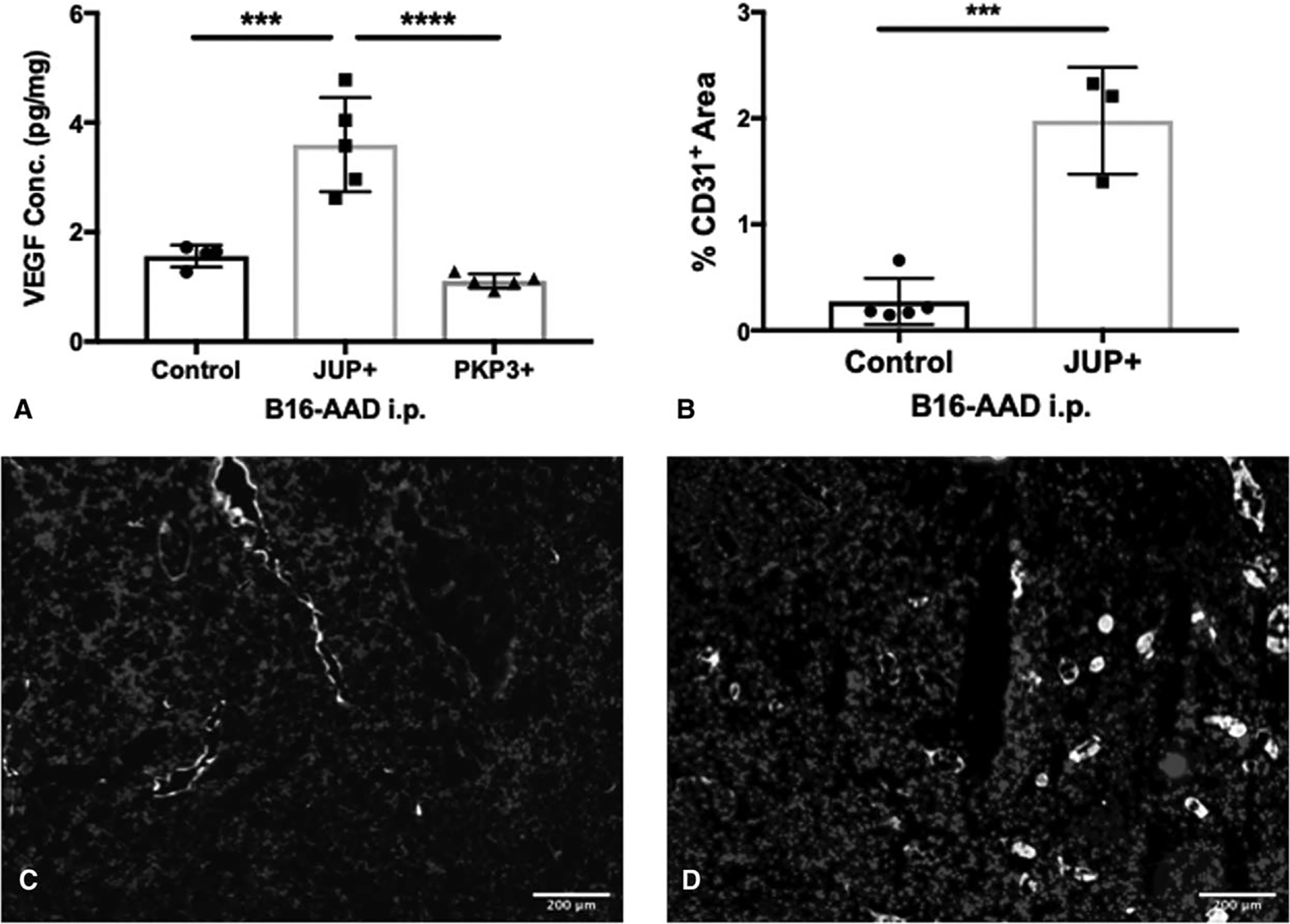
JUP overexpression drives increased angiogenesis in B16 murine melanomas. (A) VEGF-A concentration per milligram of tumor tissue was quantitated using multiplex assay in day 14 B16-AAD control, JUP +, and PKP3 + i.p. tumors. (B) Percentage of CD31+ pixel area was quantitated in B16-AAD control (n = 5 tumors) and JUP + (n = 4 tumors) i.p. tumors harvested at day 14 using ImageJ software. Representative immunofluorescence images stained with antibodies to CD31 (white), CD45 (green), and DAPI and analyzed of frozen sections of day 14 i.p. B16-AAD Control (C) and JUP + (D) tumors. Scale bar, 200 μm. Differences were assessed by 1-way ANOVA and unpaired t tests, with significance noted by asterisks: *<0.05, **<0.01, ***<0.001, ****<0.0001.
DISCUSSION
In the present study, we evaluated 3 hypotheses concerning the function of 4 barrier molecules, which have been associated with shortened patient survival in melanoma. Despite their known involvement in physical barriers that prevent cell movement, we found that FLG, DST, JUP, and PKP3 did not limit T-cell infiltration into tumors. Thus, our hypothesis that these 4 BMs impact immune infiltration was not supported. In addition, neither FLG nor DST expression was modulated by the cytokine milieu characteristic of well-infiltrated tumors. Instead, our work supported our hypothesis that FLG/DST and JUP, but not PKP3, promote melanoma growth, both in mice and in humans. In particular, we found that growth-promoting effects of FLG and DST are tumor cell intrinsic through direct control of proliferation. On the contrary, those mediated by JUP were extrinsic and may be mediated by up-regulating angiogenesis.
These 4 BMs did not impact immune cell infiltrate, challenging our hypothesis that BMs could provide a mechanical barrier to prevent immune cells from entering into tumor from vasculature. They also were not modulated by cytokines and chemokines associated with Th1 immune signatures, challenging our second hypothesis. This raises questions regarding other mechanisms to explain the strong inverse association of BM overexpression and immune cell infiltration. Most of the BMs are interconnected components within desmosomes, tight junctions, or adherens junctions.31 Thus, it is possible that all or most BMs must be co-expressed together to mediate an immune exclusion effect and that an individual barrier molecule alone is unable to create a sum effect within an intercellular junction that then limits immune infiltrate. Interestingly, we did not detect an effect on immune infiltration or tumor growth with PKP3 overexpression, which may be dependent on JUP or other BMs to initiate desmosome formation.32,33 Thus, overexpression of PKP3 in combination with JUP or other BMs may yield positive findings. One limitation of these studies is that infiltrating immune cells in murine melanomas were enumerated by flow cytometry, so effects on intratumoral distribution of immune cells cannot be ruled out. Another limitation is that murine studies of JUP and PKP3 were performed with human versions of those genes. There is very high homology between species, but it is possible that some differential effects could be mediated by murine versions of the genes. However, the significant effect of JUP overexpression suggests biologic relevance of the construct used.
Our data show that FLG, DST, and JUP support melanoma cell growth; however, their mechanisms of action remain unknown. Some BMs can translocate to the nucleus and bind transcription factors, whereby they are involved in signaling pathways.15–17,34 JUP inhibits the ubiquitin-proteasome pathway in fibrosarcoma35 and anti-apoptotic protein BCL-2 in squamous cell carcinoma.36 Additionally, FLG knockdown in keratinocytes is associated with increased apoptosis.9,37,38 However, the role of these genes in melanoma is not known. FLG, PKP3, and JUP also have been implicated in MAPK signaling pathways in keratinocytes and ovarian cancer.39–41 Never-theless, the identification and targeting of BMs that play a key role in promoting melanoma proliferation may provide novel opportunities for improving the survival of melanoma patients.
Our data identify JUP as supporting tumor growth through extrinsic effects, which is associated with increased VEGF-A. VEGF-A is a primary driver of angiogenesis, but also has immune-suppressive roles in cancer.42 VEGF inhibits effector T-cell function and increases Tregs, myeloid-derived suppressor cells, and tumor-associated macrophages.43,44 JUP is important for maintenance of vascular endothelial cell junctions and is linked to VEGF-mediated modification of vascular endothelial cadherin.45 Our data suggest JUP may be a driver of angiogenesis in melanoma in a VEGF-dependent manner.
Junction plakoglobin overexpression also reduced IL-33, which is an alarmin released by tissue damage. Mechanisms underlying the actions of IL-33 are not well understood; however, IL-33 supports antitumor immunity and inhibits tumor growth primarily by stimulating Th1 immune responses in B16 melanoma.46,47 Down-regulation of IL-33 has been linked to the onset of angiogenesis in wound healing in endothelial cells,48 which suggests that IL-33 could be part of an axis that enhances tumor growth and angiogenesis while reducing Th1 immune function. Interestingly, IL-6 is reported to be induced by IL-33.49 Weak trends towards decreases in IL-6 were observed in JUP overexpressing tumors, which supports suppression of the IL-33 pathway.
This study provides insights into the biology that underlies the aggressive phenotype of melanomas overexpressing FLG, DST, and JUP. Additional questions remain about the signaling pathways downstream of these genes and of PKP3. Our study identifies JUP as a driver of angiogenesis in a VEGF-dependent manner. Thus, VEGF is a potential target for the angiogenic-related effects of JUP on tumor growth for which VEGF inhibitors or VEGFR blocking antibodies may be considered. FLG, DST, and JUP warrant further study to identify upstream regulatory targets of these genes to intervene on their expression to confer clinical benefit by reducing mechanical barrier function and supporting antitumor immunity.
DISCUSSANTS
Dr Sandra Wong (Lebanon, NH):
Dr Leick, congratulations to you and your team for this nice work, which gets us closer to understanding barriers to immune infiltration with an end goal of better understanding poorly infiltrated melanomas so that we can better select patients for immunotherapy and define future potential therapeutic targets. I thank you for providing me a draft of your manuscript in advance of your presentation.
Your work focuses on genes that are involved in barrier functions in normal skin because of their involvement in desmosome formation and cell-cell mediation. JUP and PKP3 are two barrier genes that appear to be upregulated in these poor prognosis melanomas. As such, it’s reasonable to hypothesize that JUP and PKP3 enhance melanoma cell growth, either by directly or indirectly enhancing melanoma cell growth or, alternatively, by reducing immune cell infiltrates by a mechanical barrier function.
In your in vitro assessments, you show that JUP and PKP3 over-expression do not have an intrinsic effect on cell proliferation. In your in vivo experiments, you do show that JUP overexpression was associated with significantly increased tumor burden (though PKP3 was not). Further, your in vivo studies demonstrated no impact of JUP or PKP3 overexpression on CD8 or CD4 cells compared to control and, therefore, reasonably conclude that the genes actually have no involvement in T cell infiltration into tumors. So, digging deeper after these negative results, you found that there were increased concentrations of VEGF (at least with JUP overexpression), suggesting a role for angiogenesis in this process.
I have a few questions for you:
How do you know that JUP is actually regulating angiogenesis as opposed to having angiogenesis occur as a consequence of tumor growth itself?
Did you consider that there could be something else modulating cell behavior? You have shown that JUP and PKP3 have a variable impact on cell growth, with PKP3 overexpression not having an impact on increased tumor burden in your mouse in vivo studies. Is there a possible need to evaluate how these barrier molecular genes interact or need to be co-expressed in order to mediate immune cell infiltration?
Finally, I have an overarching question in terms of future directions for this work, especially in the current context of melanoma treatments. Could it be that there are some novel combinations of anti-angiogenic agents and immunotherapy to consider? Do you think there could be improved immune status after angiogenic therapy? And, what is a possible role for hypoxia in the relationship between angiogenesis and immune suppressive mechanisms? Thank you.
Response From Dr Katie M. Leick:
Thank you. With regards to your first question, regarding whether JUP is directly required for angiogenesis or if it mediates this effect via an indirect effect of the tumor microenvironment, we currently have work underway studying VEGF production in JUP over-expression models in states of normoxia and hypoxia in order to serve as a surrogate for the growth effects of JUP. Our findings suggest that the angiogenesis-promoting effects of JUP may be directly related to JUP overexpression and are independent of its growth effects; however, the data is preliminary and underpowered to say for certain at this time.
Also, JUP has been linked to VEGF-mediated modification of vascular endothelial cell cadherin in the literature, which may further support its potential role in directly promoting angiogenesis. Future experiments will explore this further to confirm whether or not JUP directly supports angiogenesis, but currently, we do have some evidence that may support this early on.
Then with regards to your second question, which I believe was regarding modulation of cell behavior.
Dr Sandra L. Wong (Lebanon, NH):
I was trying to get you to think about how the barrier molecular genes work together. You identified a suite of them, and it looked like JUP and PKP3 actually don’t have the same effect.
Response From Dr Katie M. Leick:
That is correct. We have studied four of the eight barrier molecule genes that were initially identified and later expanded that list to almost 50 barrier molecule genes. Most of these genes are involved in desmosome cell components so they may have some function that requires them to work together in order to elicit their effects, such as blocking out immune cell infiltration.
Initially, we set out to try to over-express all eight barrier molecules that were initially identified, as well as delete them, in melanoma cell lines. However, JUP and PKP3 were two of the initial barrier molecule genes that we had success in over-expressing. Once we found interesting effects of their overexpression, such as the phenotype changes in the JUP over-expressing model, we decided to pursue and focus on that further. But there remains a question as to whether some of the other barrier molecule genes that are unexplored might have an effect on immune cell infiltration, individually or independent of the other genes. However, another hypothesis would be that all 8 genes may need to be coexpressed together, which is technically challenging to test. Currently, we plan to continue to study the individual effects of the individual barrier molecule genes.
We have also identified a potential transcription factor in other work presented at SSO that may modulate all of these genes together. It would be interesting to identify a way to target that transcription factor in order to modulate the expression of all the genes together and then study whether or not they have effects on immune cell infiltration through global modulation.
Then, with regard to your last question about novel angiogenic/anti-angiogenic targets or targeted therapy in patients, there are a number of ongoing clinical trials using siRNA-directed therapy. Other groups at University of Virginia are involved in designing nanoparticle-directed drug delivery systems for siRNA using focused ultrasound and noninvasive approaches. It would be useful to consider using siRNA to knock down JUP expression, which could be done by interlesional injection or systemically.
Consideration could be extended to targeting other barrier molecule genes in this manner as well, especially if they have an interesting phenotypic effect, or to use multiple siRNAs to knock them all down together, which would be very useful. It could also be worthwhile to target a transcription factor that is capable of regulating all of these barrier molecule genes together to then knock it down and downregulate the expression of the other barrier molecule genes together.
Dr Sandra L. Wong (Lebanon, NH):
What’s really exciting, especially in the era of immunotherapy for melanoma, is the potential to actually combine it with novel angiogenic agents.
Response From Dr Katie M. Leick:
Yes. We also think it would be useful to consider use of Avastin in patients. Patients with high expression of JUP in their tumors could be identified and targeted for Avastin therapy, which may enable improved tumor control and potentially reduce blood loss for an operation or even the amount of tissue coverage needed in terms of a flap, if tumor control could be improved to such an extent. It may also serve as a great adjuvant for other therapies.
Dr Ronald J. Weigel (Iowa City, IA):
No conflict of interest.
I had two questions. First, you showed some data initially that demonstrated changes in patterns of expression associated with immune infiltration, and JUP was one example of a gene that was regulated, but it was over expressed when there was a decrease with immune infiltration. The question is, are there genes in that pattern of expression that are upregulated in association with an increase in immune infiltration, potentially identifying an antigen that’s inducing that increase in immune infiltration?
My second question relates to some of the discussion that we just heard. Do you have examples of JUP over expressing melanoma tumors where you can show that knockdown of JUP actually decreases angiogenesis and affects tumor growth? I don’t recall those data in what you presented.
Response From Dr Katie M. Leick:
With regards to your first question as to whether we have seen an increase in barrier molecule gene expression or any of the individual genes that are associated with an increased T-cell infiltration or immune signature at the very least, we have not. There are a few patients that might have that profile, but for the most part, they are very concordantly or discordantly expressed. The barrier molecule genes are discordantly expressed from immune genes.
We also included the barrier molecule genes in a large data set with almost 400 other genes in the wnt/beta catenin pathway. We found that the barrier molecule genes were much more concordantly expressed together and upregulated than the wnt/beta catenin genes. It seems as though there may be a strong association of those genes to be upregulated together and associated with a lack of immune signature.
Then with regards to your second question, we have plans underway to knock out JUP using CRISPR/Cas9. That work is in progress, so we do not have a CRISPR knockout of JUP for the mouse model at this time, but plan to do that in the future.
Dr Ronald J. Weigel (Iowa City, IA):
You could use an siRNA or shRNA rather than use CRISPR. For example, a simple approach would be to use an shRNA to JUP in one of the melanoma lines that over expresses JUP, and see if you can decrease angiogenesis in a xenograft model. Have you tried that?
Response From Dr Katie M. Leick:
Yes, that is something we are working on and plan to do in the future, but we are not quite there yet. It is a work in progress.
Dr Jeffrey Drebin (New York, NY):
Junction plakoglobin is also called gamma catenin, and it’s known that gamma catenin and beta catenin can compete for presence at the membrane. Beta catenin, of course, is the critical element in Wnt signaling downstream. One of the Wnt signaling elements downstream of beta catenin is VEGF.
So is it possible that when you over-express JUP or gamma catenin, you then free beta catenin, which can then do its thing in the nucleus, and you’re driving gene expression that way? If that’s the case, can you really link all of your very nice work thus far to Wnt signaling pathway and then take it to the next level?
Response From Dr Katie M. Leick:
That is a great question. I think that is a possibility, which we have not yet explored. With gamma catenin as the alternative name for JUP, it was interesting to find that gamma catenin was associated with beta catenin and the Wnt/beta catenin pathway. That connection would certainly be worth exploring in the future. Thank you.
Supplementary Material
ACKNOWLEDGMENTS
We thank the University of Virginia Histology Core for tissue sectioning and the Flow Cytometry Core for cell sorting experiments, both of which are supported by United States Public Health Services Cancer Center Support Grant P30 CA44579.
Funding: Support was provided by United States Public Health Services Research grants R01 CA78400 (VH Engelhard), R00CA140774 (T Abbas), and R01 CA057653 (CL Slingluff), United States Public Health Services Training grants in Surgical Oncology T32 CA163177 (KM Leick), Immunology T32 AI007496 (AB Rodriguez, RS Lindsay), Cancer Biology T32 CA009109 (R Eki), Cancer Center Support Grant P30 CA44579, and Cancer Research Institute Clinical Laboratory Integration Project (CL Slingluff, KM Leick). AB Rodriguez was the recipient of a Wagner Fellowship, MM Melssen was the recipient of the Rebecca Clary Harris Fellowship, and M Benamar was the recipient of the Farrow Fellowship, all from the University of Virginia.
Footnotes
Disclosures: The following disclosures apply to Dr Slingluff, but are not related to this work: Research support to the University of Virginia from Celldex (funding, drug), Glaxo-Smith Kline (funding), Merck (funding, drug), 3 M (drug), Theraclion (device staff support), Polynoma (PI of clinical trial of melanoma vaccine); Funding to the University of Virginia for Scientific Advisory Boards for Immatics, CureVac, Celldex. Also Dr. Slingluff receives license fee payments through the UVA Licensing and Ventures Group for patents for peptides used in cancer vaccines.
The following disclosures apply to Dr Engelhard, but are not related to this work: Research support to the University of Virginia from Agenus, Inc. Dr Engelhard is also a scientific consultant and shareholder for Agenus.
The authors report no conflicts of interest.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal’s Web site (www.annalsofsurgery.com).
REFERENCES
- 1.Erdag G, Schaefer JT, Smolkin ME, et al. Immunotype and immunohistologic characteristics of tumor-infiltrating immune cells are associated with clinical outcome in metastatic melanoma. Cancer Res. 2012;72:1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bogunovic D, O’Neill DW, Belitskaya-Levy I, et al. Immune profile and mitotic index of metastatic melanoma lesions enhance clinical staging in predicting patient survival. Proc Natl Acad Sci U S A. 2009;106:20429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mihm MC Jr, Clemente CG, Cascinelli N. Tumor infiltrating lymphocytes in lymph node melanoma metastases: a histopathologic prognostic indicator and an expression of local immune response. Lab Invest. 1996;74:43. [PubMed] [Google Scholar]
- 4.Wu X, Li J, Connolly EM, et al. Combined anti-VEGF and anti-CTLA-4 therapy elicits humoral immunity to galectin-1 which is associated with favorable clinical outcomes. Cancer Immunol Res. 2017;5:446–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hamid O, Schmidt H, Nissan A, et al. A prospective phase II trial exploring the association between tumor microenvironment biomarkers and clinical activity of ipilimumab in advanced melanoma. J Transl Med. 2011;9:204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Salerno EP, Bedognetti D, Mauldin IS, et al. Human melanomas and ovarian cancers overexpressing mechanical barrier molecule genes lack immune signatures and have increased patient mortality risk. Oncoimmunology. 2016;5:e1240857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nakatsukasa M, Kawasaki S, Yamasaki K, et al. Tumor-associated calcium signal transducer 2 is required for the proper subcellular localization of claudin 1 and 7: implications in the pathogenesis of gelatinous drop-like corneal dystrophy. Am J Pathol. 2010;177:1344–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Presland RB, Kuechle MK, Lewis SP, et al. Regulated expression of human filaggrin in keratinocytes results in cytoskeletal disruption, loss of cell-cell adhesion, and cell cycle arrest. Exp Cell Res. 2001;270:199–213. [DOI] [PubMed] [Google Scholar]
- 9.Pendaries V, Malaisse J, Pellerin L, et al. Knockdown of filaggrin in a three-dimensional reconstructed human epidermis impairs keratinocyte differentiation. J Invest Dermatol. 2014;134:2938–2946. [DOI] [PubMed] [Google Scholar]
- 10.Kypriotou M, Huber M, Hohl D. The human epidermal differentiation complex: cornified envelope precursors, S100 proteins and the ‘fused genes’ family. Exp Dermatol. 2012;21:643–649. [DOI] [PubMed] [Google Scholar]
- 11.Kawasaki H, Nagao K, Kubo A, et al. Altered stratum corneum barrier and enhanced percutaneous immune responses in filaggrin-null mice. J Allergy Clin Immunol. 2012;129 1538–46e6. [DOI] [PubMed] [Google Scholar]
- 12.Mildner M, Jin J, Eckhart L, et al. Knockdown of filaggrin impairs diffusion barrier function and increases UV sensitivity in a human skin model. J Invest Dermatol. 2010;130:2286–2294. [DOI] [PubMed] [Google Scholar]
- 13.Wallmeyer L, Dietert K, Sochorova M, et al. TSLP is a direct trigger for T cell migration in filaggrin-deficient skin equivalents. Sci Rep. 2017;7:774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pearton DJ, Dale BA, Presland RB. Functional analysis of the profilaggrin N-terminal peptide: identification of domains that regulate nuclear and cytoplasmic distribution. J Invest Dermatol. 2002;119:661–669. [DOI] [PubMed] [Google Scholar]
- 15.Bonne S, van Hengel J, Nollet F, et al. Plakophilin-3, a novel armadillo-like protein present in nuclei and desmosomes of epithelial cells. J Cell Sci. 1999;112(Pt 14):2265–2276. [DOI] [PubMed] [Google Scholar]
- 16.Simcha I, Shtutman M, Salomon D, et al. Differential nuclear translocation and transactivation potential of beta-catenin and plakoglobin. J Cell Biol. 1998;141:1433–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Young KG, Pool M, Kothary R. Bpag1 localization to actin filaments and to the nucleus is regulated by its N-terminus. J Cell Sci. 2003;116(Pt 22):4543–4555. [DOI] [PubMed] [Google Scholar]
- 18.Mullins DW, Bullock TN, Colella TA, et al. Immune responses to the HLA-A 0201-restricted epitopes of tyrosinase and glycoprotein 100 enable control of melanoma outgrowth in HLA-A 0201-transgenic mice,. J Immunol. 2001;167:4853. [DOI] [PubMed] [Google Scholar]
- 19.Newberg MH, Smith DH, Haertel SB, et al. Importance of MHC class 1 alpha2 and alpha3 domains in the recognition of self and non-self MHC molecules. J Immunol. 1996;156:2473. [PubMed] [Google Scholar]
- 20.Holm S. A simple sequentially rejective multiple test procedure. Scand J Stat. 1979;6:65–70. [Google Scholar]
- 21.Šidák Z Rectangular confidence regions for the means of multivariate normal distributions. J Am Stat Assoc. 1967;62:626–633. [Google Scholar]
- 22.Kaneko T, Tamai K, Matsuzaki Y, et al. Interferon-gamma down-regulates expression of the 230-kDa bullous pemphigoid antigen gene (BPAG1) in epidermal keratinocytes via novel chimeric sequences of ISRE and GAS. Exp Dermatol. 2006;15:308–314. [DOI] [PubMed] [Google Scholar]
- 23.Noh M, Yeo H, Ko J, et al. MAP17 is associated with the T-helper cell cytokine-induced down-regulation of filaggrin transcription in human keratinocytes. Exp Dermatol. 2010;19:355–362. [DOI] [PubMed] [Google Scholar]
- 24.Mauldin IS, Wages NA, Stowman AM, et al. Intratumoral interferon-gamma increases chemokine production but fails to increase T cell infiltration of human melanoma metastases. Cancer Immunol Immunother. 2016;65:1189–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harlin H, Meng Y, Peterson AC, et al. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. 2009;69:3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cremasco V, Astarita JL, Grauel AL, et al. FAP delineates heterogeneous and functionally divergent stromal cells in immune-excluded breast tumors. Cancer Immunol Res. 2018;6:1472–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mariathasan S, Turley SJ, Nickles D, et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018;554:544–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tauriello DVF, Palomo-Ponce S, Stork D, et al. TGFbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature. 2018;554:538–543. [DOI] [PubMed] [Google Scholar]
- 29.Peske JD, Thompson ED, Gemta L, et al. Effector lymphocyte-induced lymph node-like vasculature enables naive T-cell entry into tumours and enhanced anti-tumour immunity. Nat Commun. 2015;6:7114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leick KM, Pinczewski J, Mauldin IS, et al. Patterns of immune-cell infiltration in murine models of melanoma: roles of antigen and tissue site in creating inflamed tumors. Cancer Immunol Immunother. 2019;68:1121–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bonne S, Gilbert B, Hatzfeld M, et al. Defining desmosomal plakophilin-3 interactions. J Cell Biol. 2003;161:403–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gosavi P, Kundu ST, Khapare N, et al. E-cadherin and plakoglobin recruit plakophilin3 to the cell border to initiate desmosome assembly. Cell Mol Life Sci. 2011;68:1439–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gurjar M, Raychaudhuri K, Mahadik S, et al. Plakophilin3 increases desmosome assembly, size and stability by increasing expression of desmocollin2. Biochem Biophys Res Commun. 2018;495:768–774. [DOI] [PubMed] [Google Scholar]
- 34.Hu P, Berkowitz P, O’Keefe EJ, et al. Keratinocyte adherens junctions initiate nuclear signaling by translocation of plakoglobin from the membrane to the nucleus. J Invest Dermatol. 2003;121:242–251. [DOI] [PubMed] [Google Scholar]
- 35.Sadot E, Simcha I, Iwai K, et al. Differential interaction of plakoglobin and beta-catenin with the ubiquitin-proteasome system. Oncogene. 2000; 19:1992–2001. [DOI] [PubMed] [Google Scholar]
- 36.Hakimelahi S, Parker HR, Gilchrist AJ, et al. Plakoglobin regulates the expression of the anti-apoptotic protein BCL-2. J Biol Chem. 2000; 275:10905–10911. [DOI] [PubMed] [Google Scholar]
- 37.Dang N, Ma X, Meng X, et al. Dysregulated function of normal human epidermal keratinocytes in the absence of filaggrin. Mol Med Rep. 2016;14:2566–2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kuechle MK, Presland RB, Lewis SP, et al. Inducible expression of filaggrin increases keratinocyte susceptibility to apoptotic cell death. Cell Death Differ. 2000;7:566–573. [DOI] [PubMed] [Google Scholar]
- 39.Wang S, Qiu L, Meng X, et al. Knock-down of filaggrin influences the mitogen-activated protein kinases signaling pathway in normal human epidermal keratinocytes. Med Sci (Paris). 2018;34:94–98. [DOI] [PubMed] [Google Scholar]
- 40.Spindler V, Dehner C, Hubner S, et al. Plakoglobin but not desmoplakin regulates keratinocyte cohesion via modulation of p38MAPK signaling. J Invest Dermatol. 2014;134:1655–1664. [DOI] [PubMed] [Google Scholar]
- 41.Lim V, Zhu H, Diao S, et al. PKP3 interactions with MAPK-JNK-ERK1/2-mTOR pathway regulates autophagy and invasion in ovarian cancer. Biochem Biophys Res Commun. 2019;508:646–653. [DOI] [PubMed] [Google Scholar]
- 42.Li YL, Zhao H, Ren XB. Relationship of VEGF/VEGFR with immune and cancer cells: staggering or forward? Cancer Biol Med. 2016;13:206–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang H, Langenkamp E, Georganaki M, et al. VEGF suppresses T-lympho-cyte infiltration in the tumor microenvironment through inhibition of NF-kappaB-induced endothelial activation. FASEB J. 2015;29:227–238. [DOI] [PubMed] [Google Scholar]
- 44.Lapeyre-Prost A, Terme M, Pernot S, et al. Immunomodulatory activity of VEGF in cancer. Int Rev Cell Mol Biol. 2017;330:295–342. [DOI] [PubMed] [Google Scholar]
- 45.Muramatsu F, Kidoya H, Naito H, et al. Plakoglobin maintains the integrity of vascular endothelial cell junctions and regulates VEGF-induced phosphorylation of VE-cadherin. J Biochem. 2017;162:55–62. [DOI] [PubMed] [Google Scholar]
- 46.Lucarini V, Ziccheddu G, Macchia I, et al. IL-33 restricts tumor growth and inhibits pulmonary metastasis in melanoma-bearing mice through eosinophils. Oncoimmunology. 2017;6:e1317420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gao X, Wang X, Yang Q, et al. Tumoral expression of IL-33 inhibits tumor growth and modifies the tumor microenvironment through CD8 T and NK cells. J Immunol. 2015;194:438–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kuchler AM, Pollheimer J, Balogh J, et al. Nuclear interleukin-33 is generally expressed in resting endothelium but rapidly lost upon angiogenic or proinflammatory activation. Am J Pathol. 2008;173:1229–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pinto SM, Subbannayya Y, Rex DAB, et al. A network map of IL-33 signaling pathway. J Cell Commun Signal. 2018;12:615–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.