

Short abstract
http://aasldpubs.onlinelibrary.wiley.com/hub/journal/10.1002/(ISSN)2046-2484/video/15-4-reading-shroff-maddur a video presentation of this article
http://aasldpubs.onlinelibrary.wiley.com/hub/journal/10.1002/(ISSN)2046-2484/video/15-4-interview-shroff-maddur an interview with the author
https://www.wileyhealthlearning.com/Activity/7088613/disclaimerspopup.aspx questions and earn CME
Abbreviations
- CN
Crigler‐Najjar syndrome
- DJ
Dubin‐Johnson syndrome
- GS
Gilbert’s syndrome
- LDH
lactate dehydrogenase
- MRP
multidrug resistance–associated protein
- OATP
organic anion transporting polypeptide
- UGT1A1
uridine‐5′‐disphosphate gluconyltransferase 1A1
Clinic Consult
A 30‐year‐old man who is otherwise healthy presents to your clinic for evaluation of an elevated total bilirubin level of 3.5 mg/dL. The remainder of his liver enzymes, including aspartate aminotransferase, alanine aminotransferase, and alkaline phosphatase, are all normal. What is the approach to evaluating an isolated elevated bilirubin level?
Initial Approach
Although hyperbilirubinemia is frequently due to hepatic dysfunction, it can also occur in the absence of underlying liver disease. In the case of this patient, with an isolated bilirubin elevation and otherwise normal liver enzymes, the first step is to fractionate the bilirubin into direct and indirect components, because this will help frame the differential diagnosis. Table 1 lists the differential of an elevated bilirubin level, categorized by mechanism of action. Figure 1 summarizes the following approach to the patient in a concise algorithm.
Table 1.
Causative Factors of an Isolated Elevated Bilirubin Level by Mechanism
| Overproduction of Bilirubin |
|---|
|
Hemolytic anemias
|
| Dyserythropoiesis (e.g., vitamin/mineral deficiencies) |
| Hematoma resorption |
| Massive blood transfusion |
| Impaired hepatocellular uptake |
| Medications (e.g., rifampin, cyclosporine) |
| Rotor syndrome |
| Impaired bilirubin conjugation |
| Medications (e.g., protease inhibitors) |
| Gilbert’s syndrome |
| Crigler‐Najjar syndrome (types I and II) |
| Impaired hepatic excretion |
| DJ |
Fig 1.
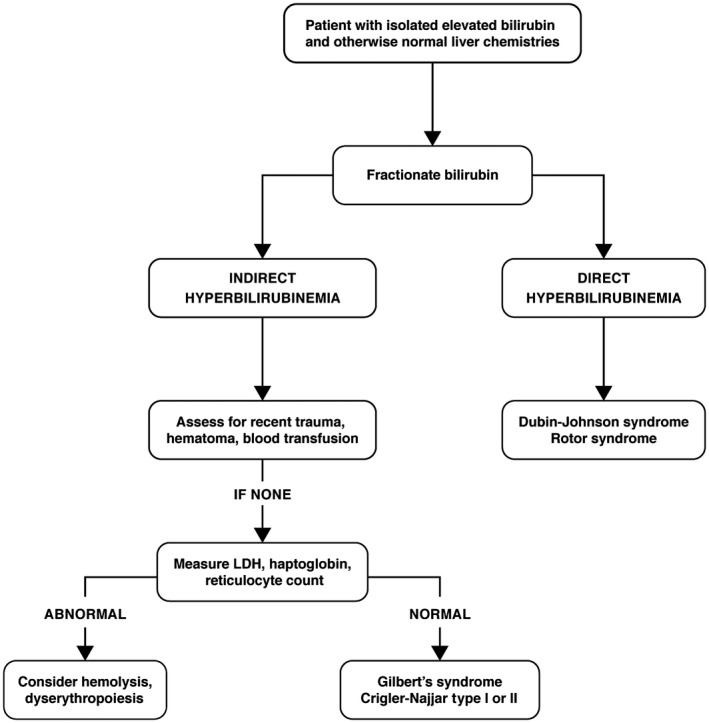
Algorithm in the evaluation of an isolated hyperbilirubinemia.
Indirect Hyperbilirubinemia
A predominantly indirect hyperbilirubinemia, which is usually classified as <20% direct fraction, reflects one of two mechanistic processes: (1) overproduction of bilirubin, or (2) impaired conjugation.
Bilirubin is formed as a product in the metabolism of heme. 1 The majority (80%) derives from the breakdown of hemoglobin from senescent erythrocytes, and the remaining 20% comes from nonhemoglobin proteins (e.g., myoglobin, cytochromes). Once produced, bilirubin is unconjugated and poorly water soluble, requiring albumin to circulate in plasma. The bilirubin‐albumin complex is able to pass through fenestrated hepatic sinusoidal endothelial cells into hepatocytes, where it dissociates from albumin. Once in the hepatocyte, the enzyme uridine‐5′‐disphosphate gluconyltransferase 1A1 (UGT1A1) conjugates bilirubin to glucuronic acid.
If this patient were to have an indirect hyperbilirubinemia, the clinician should first consider conditions that lead to increased erythrocyte turnover, and thus overproduction of bilirubin. 2 Hemolytic anemias can be ruled out by measuring serum levels of lactate dehydrogenase (LDH), haptoglobin, hemoglobin, and reticulocyte count. Massive blood transfusions, resorption of large hematomas, and hemoglobinopathies also lead to increased erythrocyte turnover and can easily be ruled out based on history. Ineffective erythropoiesis, as occurs in vitamin and micronutrient deficiencies, should also be considered. Otherwise, impaired conjugation can occur through inhibition of UGT1A1 by certain medications, including the protease inhibitors indinavir and atazanavir and the kinase inhibitor sorafenib. 3 , 4
In the absence of this, the likely etiology is a hereditary disorder of bilirubin metabolism (Table 2), either Gilbert’s syndrome (GS) or Crigler‐Najjar syndrome (CN).
Table 2.
Hereditary Disorders of Bilirubin Metabolism
| Disorder | Mechanism | Total Serum Bilirubin Level (mg/dL) | Treatment |
|---|---|---|---|
| Gilbert’s syndrome | Mutation in UGT1A1 causing activity <33% normal | Up to 4 | None |
| Crigler‐Najjar syndrome type 1 | UGT1A1 activity absent | Usually 20‐40 | Phototherapy |
| Exchange transfusions | |||
| Liver transplant | |||
| Crigler‐Najjar syndrome type 2 | UGT1A1 activity <10% normal | Usually <20 | Phenobarbital |
| DJ | MRP2 receptor mutation impairing transport across canalicular membrane | Usually 2‐5 | None |
| Rotor syndrome | OATP1B1 and OATP1B3 mutations affecting reuptake of conjugated bilirubin by hepatocytes | Usually 2‐5 | None |
GS is the most common hereditary disorder of bilirubin metabolism. Its prevalence rate in white populations, where it has been studied most, is estimated at 10%. 5 GS occurs as a result of one of several identified mutations in the UGT1A1 gene, 6 leading to a reduction in enzyme activity to approximately one‐third normal. The result is an impairment in bilirubin conjugation, which manifests as an unconjugated hyperbilirubinemia that is generally <4 mg/dL. The disease follows a benign course and requires no treatment.
CN, a much rarer condition with an estimated prevalence of 0.6 per million, 7 also results from mutations in the UGT1A1 gene. In type I CN, there is complete absence of UGT1A1, resulting in severe neonatal jaundice and the risk for kernicterus. Treatment generally requires exchange transfusion and phototherapy as a bridge to liver transplantation. In type II CN, UGT1A1 activity is reduced to <10% normal but not abolished. Patients may not be diagnosed until early childhood or teenage years, and bilirubin levels are usually <20 mg/dL. Treatment consists of lifelong phenobarbital therapy, which increases expression of UGT1A1 and enhances bilirubin conjugation.
Direct Hyperbilirubinemia
In patients with an isolated, direct hyperbilirubinemia (defined as >70%‐80% direct fraction), the mechanism is an impairment of bilirubin transport after conjugation.
After bilirubin is conjugated in the hepatocyte, it becomes water‐soluble and is excreted across the hepatocyte canalicular membrane into the biliary system via the multidrug resistance–associated protein 2 (MRP2). A small amount of conjugated bilirubin takes an alternate route and is secreted back across the sinusoidal pole of the hepatocyte into plasma via an MRP3 transporter. Once in plasma, the conjugated bilirubin can be secreted into urine or can undergo reuptake into the hepatocyte by the organic anion transporting polypeptides (OATP1B1 and OATP1B3). Conjugated bilirubin that enters the biliary system is excreted via the fecal route, with a small amount reabsorbed via the terminal ileum and secreted in urine. 1
Acquired causes of a direct hyperbilirubinemia are primarily the result of medications that affect the earlier membrane transporters. Rifampin and cyclosporine competitively inhibit the OATP1B1 transport protein, causing elevations in serum levels of conjugated bilirubin. 8 , 9
Otherwise, the likely cause is one of the remaining two hereditary disorders in bilirubin metabolism (Table 2): Dubin‐Johnson syndrome (DJ) or Rotor syndrome. 2
In DJ, a defect exists in the gene encoding MRP2, resulting in an impairment in bilirubin transport across the canalicular membrane. 10 In Rotor syndrome, the proposed mechanism is via gene mutations that affect OATP1B1 and OATP1B3. 11 In both, serum levels of conjugated bilirubin are increased, but bilirubin conjugation remains unaffected. Patients are typically asymptomatic, and pruritis is not observed (because serum bile acid levels are unchanged). The degree of hyperbilirubinemia in both is usually mild (<5 mg/dL), but in DJ it can be increased by intercurrent illness, oral contraceptives, or pregnancy. Neither DJ nor Rotor syndrome require treatment because both follow a benign disease course without progressive hepatocyte damage.
Summary
The approach to an isolated elevated bilirubin requires an understanding of bilirubin metabolism. Acquired causative factors can be ruled out easily by history, review of medications, and basic laboratory analysis. Hereditary disorders, especially when discovered in adulthood, are almost always benign and require no specific treatment.
Potential conflict of interest: Nothing to report.
References
- 1. Lidofsky SD. Jaundice In: Feldman M, Friedman LS, Brandt LJ, eds. Sleisenger and Fordtran’s Gastrointestinal and Liver Disease, 10th ed Philadelphia, PA: Saunders; 2016:336‐339. [Google Scholar]
- 2. Wolkoff AW, Berk PD. Bilirubin metabolism and jaundice In: Schiff ER, Sorrell MF, Maddrey WC, eds. Schiff’s Diseases of the Liver, 10th ed Philadelphia, PA: Lippincott William & Wilkins; 2007:226‐228. [Google Scholar]
- 3. Rotger M, Taffe P, Bleiber G, et al. Gilbert syndrome and the development of antiretroviral therapy‐associated hyperbilirubinemia. J Infect Dis 2005;192:1381‐1386. [DOI] [PubMed] [Google Scholar]
- 4. Peer CJ, Sissung TM, Kim A, et al. Sorafenib is an inhibitor of UGT1A1 but is metabolized by UGT1A9: Implications of genetic variants on pharmacokinetics and hyperbilirubinemia. Clin Cancer Res 2012;18:2099‐2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bosma PJ. Inherited disorders of bilirubin metabolism. J Hepatol 2003;38:107‐117. [DOI] [PubMed] [Google Scholar]
- 6. Canu G, Minucci A, Zuppi C, et al. Gilbert and Crigler Najjar syndromes: An update of the UDP‐glucuronosyltransferase 1A1 (UGT1A1) gene mutation database. Blood Cells Mol Dis 2013;50:273‐280. [DOI] [PubMed] [Google Scholar]
- 7. van der Veere CN, Sinaasappel M, McDonagh AF, et al. Current therapy for Crigler‐Najjar syndrome type 1: Report of a world registry. Hepatology 1996;24:311‐315. [DOI] [PubMed] [Google Scholar]
- 8. Vavrick SR, Van Montfoort J, Ha HR, et al. Interactions of rifamycin SV and rifampicin with organic anion uptake systems of human liver. Hepatology 2002;36:164‐172. [DOI] [PubMed] [Google Scholar]
- 9. Campbell SD, de Morais SM, Xu JJ. Inhibition of human organic anion transporting polypeptide OATP 1B1 as a mechanism of drug‐induced hyperbilirubinemia. Chem Biol Interact 2004;150:179‐187. [DOI] [PubMed] [Google Scholar]
- 10. Paulusma CC, Kool M, Bosma PJ, et al. A mutation in the human canalicular multispecific organic anion transporter gene causes the Dubin‐Johnson syndrome. Hepatology 1997;25:1539‐1542. [DOI] [PubMed] [Google Scholar]
- 11. van de Steeg E, Stranecky V, Hartmannova H, et al. Complete OATP1B1 and OATP1B3 deficiency causes human Rotor syndrome by interrupting conjugated bilirubin reuptake into the liver. J Clin Invest 2012;122:519‐528. [DOI] [PMC free article] [PubMed] [Google Scholar]