

Abstract
Background/Aims
Intestinal fibrosis is a major complication of Crohn’s disease (CD). The profibrotic protein transforming growth factor-β (TGF-β) has been considered to be critical for the induction of the fibrotic program. TGF-β has the ability to induce not only the expression of extracellular matrix (ECM) including collagen, but also the production of plasminogen activator inhibitor-1 (PAI-1) that prevents enzymatic degradation of the ECM during the onset of fibrotic diseases. However, the significance of PAI-1 in the developing intestinal fibrosis has not been fully understood. In the present study, we examined the actual expression of PAI-1 in fibrotic legion of intestinal inflammation and its correlation with the abnormal ECM deposition.
Methods
Chronic intestinal inflammation was induced in BALB/c mice using 8 repeated intrarectal injections of 2,4,6-trinitrobenzene sulfonic acid (TNBS). TM5275, a PAI-1 inhibitor, was orally administered as a carboxymethyl cellulose suspension each day for 2 weeks after the sixth TNBS injection.
Results
Using a publicly available dataset (accession number, GSE75214) and TNBS-treated mice, we observed increases in PAI-1 transcripts at active fibrotic lesions in both patients with CD and mice with chronic intestinal inflammation. Oral administration of TM5275 immediately after the onset of intestinal fibrosis upregulated MMP-9 (matrix metalloproteinase 9) and decreased collagen accumulation, resulting in attenuation of the fibrogenesis in TNBS-treated mice.
Conclusions
PAI-1-mediated fibrinolytic system facilitates collagen degradation suppression. Hence, PAI-1 inhibitor could be applied as an anti-fibrotic drug in CD treatment.
Keywords: Plasminogen activator inhibitor-1; Matrix metalloproteinase 9; Intestinal fibrosis; Crohn disease; 2,4,6-Trinitrobenzene sulfonic acid
INTRODUCTION
Crohn’s disease (CD) is a chronic inflammatory condition of diverse etiology. Chronic inflammation lead to serious complications, especially extensive local intestinal fibrosis and digestive obstruction. Approximately 40% of CD patients develop clinically apparent strictures [1].
According to the different therapeutic approaches used in clinical practice, in addition to mixed types, the strictures can be subdivided into fibrotic and inflammatory types. Standard anti-inflammatory therapies, including treatment with anti-TNF-α antibodies, can improve inflammatory strictures by minimizing inflammation-mediated edema. Anti-inflammatory therapies currently used for IBD neither prevent nor reverse established fibrotic strictures, which may present years after the remission of active inflammation [2]. The immunological mechanisms related to acute phase of intestinal inflammation and repair have been extensively studied; in contrast, the pathophysiology of chronic mucosal wound healing and the late events of repair leading to fibrosis remain largely unexplored [3]. Therefore, an understanding of the cellular and molecular mechanisms of intestinal fibrosis and the development of a specific anti-fibrotic therapy for stricture complications in patients with CD are required because the existing therapeutic approaches are inadequate for addressing the problem [4].
In tissue fibrosis, accumulation of collagen occurs due to an imbalance between enhanced deposition and reduced degradation of the extracellular matrix (ECM) comprising mainly collagen [5]. Several reports have demonstrated that the exaggerated tissue deposition of collagen during the fibrotic process is largely due to an increase in the transcription of COL1A1 and COL1A2, which is governed by TGF-β and its intracellular mediators Smad proteins [6]. We previously reported that interferon-γ and its downstream effector Y-box binding protein-1 inhibit COL1A2 transcription [7] and that HSc025, a small compound, promotes nuclear Y-box binding protein-1 translocation, thereby resulting in reduced TGF-β production. To investigate the significance of transforming growth factor-β (TGF-β) and its signal transduction for the intestinal fibrosis, we took the chronic colitis model in mice induced by the frequent administrations of 2,4,6-trinitrobenzene sulfonic acid (TNBS), because the intestinal fibrosis has never been observed in the acute colitis model. Using this model, we showed that oral administration of HSc025 decreases TNBS-induced fibrosis in the gut, as well as in other organs, by reducing TGF-β production, suggesting the critical role of TGF-β for the intestinal fibrogenesis [7,8].
Of note, plasminogen activator inhibitor-1 (PAI-1) is another major downstream target of TGF-β signal transduction. The transcription of PAI-1 is directly regulated by Smad3 [9]. PAI-1 levels are reportedly elevated in fibrotic tissues; this disturbs the proteolytic activities of matrix metalloproteinases (MMPs) via tissue plasminogen activator (tPA) and plasmin, resulting in decreased rates of collagen degradation and tissue fibrogenesis [10]. Very recently, it has been reported that PAI-1 expression was highly enriched in active lesions in both patients with IBD and mice with experimental colitis, and confirmed that PAI-1 and its direct target, tPA, played an important role in regulating intestinal inflammation [11]. However, their functions have been only validated in the acute phase of experimetal colitis and its correlation with intestinal fibrogenesis observed in the chronic phase has remained obscure.
In the present study, we confirmed the increase in PAI-1 transcripts at the active fibrotic lesions in the terminal ileum of patients with CD, which was closely correlated with those of TGF-β1 and COL1A2. These correlations were also observed in TNBS-treated mice with intestinal fibrosis, and the oral administration of TM5275, originally developed PAI-1 inhibitor, ameliorates the pathogenesis in the chronic colitis model. Such observations provide novel insights into the potential treatment strategies for intestinal fibrosis in patients with CD.
METHODS
1. Mice and Induction of Chronic Colitis
Female (6-week-old) BALB/c mice were from CLEA Japan Inc. (Tokyo, Japan). Mice were maintained in a specific pathogen-free environment. All experiments were approved by the Animal Experimentation Committee of Tokai University, Kanagawa, Japan (approved Nos. #183031 and #194027). Fibrotic inflammation was produced via intrarectal injection by 3.5-F catheter instillation of a 2% solution of TNBS (Research Organics, Cleveland, OH, USA) in 50% ethanol under light anesthesia with isoflurane. TNBS was provided as 8 weekly injections, with the dose sequentially increasing in the 0.4–1.6 mg range (0.4, 0.4, 0.8, 0.8, 1.2, 1.2, 1.6, and 1.6 mg). The mice were picked up food for 24 hours before TNBS injection and were held vertically for 60 seconds after the intrarectal injection. The ethanol group mice injected a similar volume of 50% ethanol. Tissues were harvested 3 days after the completion of the 8th times injections. The colon was opened longitudinally and snap frozen for preparation of frozen sections or RNA extraction. The formalin-fixed paraffin-embedded sections were prepared for H&E or Masson trichrome staining as collagen detection.
2. Administration of PAI-1 Inhibitor
TM5275 is a specific inhibitor of PAI-1 molecules, and does not interfere with other serpin/serine protease systems [12].
TM5275 was orally administered as a carboxymethyl cellulose (CMC) suspension each day for 2 weeks after the 6th TNBS injection. The treatment had no effect on body weight or food intake throughout the experimental duration.
3. Experimental Design
The following groups of mice were included in the present chronic study: (1) ethanol-treated mice (n = 10); (2) TNBS and CMC-treated mice (n = 10); (3) TNBS and 10 mg/kg/day TM5275-treated mice (n = 9); or (4) TNBS and 50 mg/kg/day TM5275-treated mice (n = 10).
4. Histopathological Evaluation and Image Analysis of Intestinal Fibrosis
The evaluator blinded to the groups scored the macroscopic colonic lesions which is shown in Table 1 [7,13].
Table 1.
Score of Colonic Fibrosis
| Scoring | Score | |
|---|---|---|
| Macroscopic score | ||
| Adhesions | Absent | 0 |
| Mild/focal-zonal | 1 | |
| Severe/diffuse | 2 | |
| Thickness | Normal | 0 |
| Mild increase | 1 | |
| Severe increase | 2 | |
| Strictures | Absent | 0 |
| Mild | 1 | |
| Severe | 2 | |
| Dilation | Absent | 0 |
| Mild | 1 | |
| Severe | 2 | |
| Mucosal edema | Absent | 0 |
| Mild | 1 | |
| Severe | 2 | |
| Mucosal ulcers | Absent | 0 |
| 1 Or 2 ulcers at one site | 1 | |
| Ulcers at several sites | 2 | |
| Maximum possible score | 12 | |
| Microscopic score | ||
| Ulcerations | Absent | 0 |
| Small ulcers | 1 | |
| Large ulcers | 2 | |
| Degree of inflammation | Absent | 0 |
| Mild | 1 | |
| Moderate | 2 | |
| Severe | 3 | |
| Depth of lesions | Absent | 0 |
| Submucosa | 1 | |
| Muscularis propria | 2 | |
| Serosa | 3 | |
| Degree of fibrosis | Absent | 0 |
| Mild | 1 | |
| Severe | 2 | |
| Maximum possible score | 10 |
Specimens obtained from the colons of all groups were immersed in 10% buffered formalin in phosphate-buffered saline overnight at room temperature, and followed by the standard procedure for paraffin embedding. H&E stain assess the degree of inflammation and then with collagen staining to detect tissue fibrosis. The evaluator who was unaware of the group assignments, scored all the microscopic histological scores which is shown in Table 1 [14].
5. Collagen Assay Methods
Intestinal collagen content was evaluated using the Sircol Collagen Assay Kit (Biocolor, Carrickfergus, UK). The colonic specimens obtained from of all mice were homogenized in 0.5 M acetic acid containing 1% pepsin. The resulting mixture was stirred over night at 4°C. The total soluble collagen content of the mixture was determined. The acid-soluble type-I collagen supplied with the kit was used to generate a standard curve [15].
6. Quantitative PCR Assay
mRNA expressions of murine Pai-1 in the snap-frozen samples were assessed using quantitative PCR (qPCR). The amounts of target mRNA were quantified as described previously [16]. Total RNA (50 ng) was reverse transcribed using ImProm-II reverse transcriptase (Promega). The primers used for amplification of Pai-1 was 5′- GACACCCTCAGCATGTTCATC-3′ (forward primer) and 5′- AGGGTTGCACTAAACATGTCAG-3′ (reverse primer). The relative mRNA expressions of Pai-1 was normalized against that of the glyceraldehyde-3-phosphate dehydrogenase (Gapdh), gene in the same RNA preparation. qPCR was performed using SYBR Green qPCR Kits.
7. Detection of MMP-9 in the Colon
MMP-9 expression in the colon was evaluated using an ELISA kit (R&D Systems Inc., Minneapolis, MN, USA). Frozen colon tissues were grounded in phosphate-buffered saline with 1% protease inhibitor cocktail and 1% phosphatase inhibitor cocktail. After centrifugation using the Shake Master NEO system (15,000 rpm, 90 seconds, 4°C; Biomedical Science, Kobe, Japan), the supernatants of the homogenate were collected. The protein concentrations were determined using DC protein assays (Bio-Rad, Hercules, CA, USA).
8. Statistical Analysis
All analyses were performed using Graph Prism5.0 (GraphPad Software Inc. San Diego, CA, USA). Between-group differences were evaluated using the Mann-Whitney U-test (nonparametric). For the comparison of more than 3 groups, statistical analyses were performed using two-way ANOVA (parametric) or the Kruskal-Wallis test (nonparametric), followed by Bonferroni correction for parametric samples as a post-hoc test. P< 0.05 was considered statistically significant.
RESULTS
1. PAI-1 Is Overexpressed in the Terminal Ileum of Patients with CD
An increase in TGF-β production, which directly induces PAI-1 transcription, is correlated with the abnormal deposition of collagen during the onset of fibrotic disease [6]. Therefore, we validated the expression of PAI-1 in patients with CD. Intestinal fibrotic strictures are most commonly located in the terminal ileum [17], therefore, using a publicly available dataset (accession number GSE75214) [18], we discovered that PAI-1 transcript was significantly upregulated in the terminal ileal mucosa of active patients with CD than in that of normal controls or inactive patients with CD (Fig. 1A). In addition, we confirmed the correlation between the expressions of PAI-1 and TGF-β1 and demonstrated that PAI-1 transcript was positively regulated by TGF-β1 in patients with CD (Fig. 1B). Of note, the correlation coefficients between PAI-1 and COL1A1 and between PAI-1 and COL1A2 were 0.73 and 0.75, respectively (Fig. 1B). These results indicated that PAI-1 expression increased with fibrosis-related genes in the fibrotic ileal lesion of patients with CD.
Fig. 1.

The mRNA expression of PAI-1 is elevated in the terminal ileum of CD patients. The mRNA expression of PAI-1 (PAI-1), Col1A1 (COL1A1), Col1A2 (COL1A2) and TGF-β1 (TGF-β1) genes in the ileal tissue from control subjects (n=11), patients with inactive CD (n=16) and active CD (n=51). Data were derived from a Gene Expression Omnibus (GEO) dataset GSE75214. (A) The expression of PAI-1 (PAI-1) was significantly upregulated in the terminal ileal mucosa of active CD patients compared with that of normal controls or inactive CD patients. (B) COL1A1 and TGF-β1 are positively correlated. Correlation coefficient between PAI-1 and Col1A1 or Col1A2 are 0.73 or 0.75. P-value by Kruskal-Wallis test. Correlation of 2 values using Pearson correlation coefficient. a P<0.001. PAI-1, plasminogen activator inhibitor-1; TFG-β, transforming growth factor β.
2. Pai-1 Expression Is Also Detected in TNBS-Induced Murine Fibrotic Model
To examine whether Pai-1 expression facilitates intestinal fibrosis, we employed a TNBS-induced murine chronic colitis model. In the model, intestinal fibrosis gradually emerges with transition from the acute inflammatory phase to the chronic phase [7,15]. As demonstrated in our previous report, collagen deposition is completed following intrarectal injection of TNBS in the 8th week after the initial administration [7]. TNBStreated mice exhibited obvious collagen deposition, particularly in the submucosal layer (H&E and Masson trichrome staining) (Fig. 2A). A similar trend was observed in the collagen assay for quantifying actual deposition (Fig. 2B). We previously revealed that TGF-β1 production is increased in the colonic mucosa of TNBS-treated mice [7]. In addition, we detected an increase in local Pai-1 expression in fibrotic lesions when compared with the control mice (Fig. 2C). The results suggested that repeated intrarectal injection of TNBS-induced intestinal fibrosis along with local high expression of Pai-1, which was similar to the findings in patients with CD.
Fig. 2.
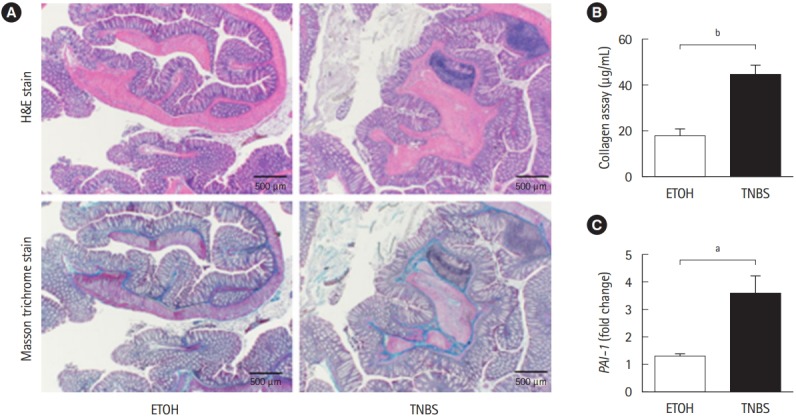
TNBS-induced fibrotic model demonstrated the enhancement of plasminogen activator inhibitor-1 (Pai-1) expression. BALB/c mice were intrarectal injected with 2% TNBS. (A) Representative histological examples of each group (H&E and Masson trichrome staining). (B) Collagen deposition was determined by the Sircol collagen assay. (C) The mRNA expression of murine Pai-1 in the colon samples was assessed by real-time PCR. Expression of the Pai-1 gene was normalized against that of the Gapdh gene in the same RNA preparation (5 ETOH, 10 TNBS). Bars represent the median. P-value by Mann-Whitney U-test. a P<0.05; b P<0.01. TNBS, 2,4,6-trinitrobenzene sulfonic acid; ETOH, ethanol.
3. PAI-1 Inhibitor Suppresses Intestinal Fibrosis in TNBS-Induced Murine Colitis
To assess the role of PAI-1 in TNBS-elicited intestinal fibrosis and to improve outcomes, we attempted to inhibit the signal by administering a PAI-1 inhibitor TM5275 after the collagen had accumulated (5 weeks later from the first treatment of TNBS) [7,12]. TM5275 is originally developed by ourselves and prevents PAI-1/tPA complex formation, thereby preserving active tPA [12,19]. Histological analysis revealed that TM5275 administration decreased TNBS-induced collagen deposition only in the 50 mg/kg group, compared with that in the CMCadministered control group (Fig. 3A). The effects of TM5275 on the macroscopic and microscopic score of colitis were clearly observed in the thickness, strictures and dilation of colon (Table 2) and the depth of lesion, the degree of fibrosis (Table 3), which is also shown as the score in Fig. 3B and C. This indicated that TM5275 attenuated the TNBS-induced colonic fibrosis. Consistently, tissue collagen content was significantly reduced in the 50 mg/kg TM5275 group but not in the 15 mg/kg group (Fig. 3D). These results suggested that the PAI-1 induction promoted collagen deposition and exacerbated colorectal fibrosis. In contrast, neither the loss of body weight nor the shortening of the gut was attenuated by the administration of TM5275 (Fig. 3E and F). These could be realized that the blockade of PAI-1 does not affect the chronic inflammation in the gut. PAI-1 reportedly promotes ECM deposition in fibrotic tissue by inhibiting MMP-9 production, and PAI-1 inhibition increases MMP-9 production [10,20,21]. Consistent with these reports, we detected the MMP-9 upregulation in the 50 mg/kg TM5275 group (Fig. 3G). The results indicated that targeting PAI-1 inhibition reduced collagen deposition via actual MMP-9 induction and that PAI-1 inhibitor could be useful in the treatment of intestinal fibrosis in patients with CD (Fig. 4).
Fig. 3.

PAI-1 inhibitor (TM5275) suppressed intestinal fibrosis in TNBS-induced murine colitis. (A) Representative histological examples of each group (H&E and Masson trichrome staining). (B, C) Analysis of macroscopic score and microscopic score. (D) Collagen deposition was determined by the Sircol collagen assay. (E) Body weight changes of each group. (F) Colon length of each group. (G) Local production of MMP-9 in the colonic mucosa of chronic colitis by ELISA. Bars represent the median. P-value by two-way ANOVA with Bonferroni post-hoc test. a P<0.05; b P<0.001. PAI-1, plasminogen activator inhibitor-1; TNBS, 2,4,6-trinitrobenzene sulfonic acid; MMP-9, matrix metalloproteinase 9; ETOH, ethanol; CMC, carboxymethyl cellulose.
Table 2.
Macroscopic Score of Colonic Fibrosis
| Group | Macroscopic score of colonic fibrosis |
|||||
|---|---|---|---|---|---|---|
| Adhesion | Thickness | Strictures | Dilation | Mucosal edema | Total macroscopic score | |
| Ethanol (n = 10) | 0.20 ± 0.42 | 0.10 ± 0.32 | 0 | 0.20 ± 0.42 | 0.50 ± 0.42 | 1.00 ± 0.67 |
| CMC (n = 10) | 0.90 ± 0.31 | 1.80 ± 0.42 | 1.80 ± 0.47 | 0.80 ± 0.42 | 0.80 ± 0.42 | 6.10 ± 0.87 |
| TM5275 10 mg/kg (n = 9) | 0.74 ± 0.44 | 1.38 ± 0.72 | 1.65 ± 0.44 | 0.74 ± 0.50 | 0.74 ± 0.44 | 5.40 ± 0.60 |
| TM5275 50 mg/kg (n = 10) | 0.80 ± 0.42 | 0.70 ± 0.67a,c | 0.80 ± 0.63a,c | 0.30 ± 0.48a,b | 0.50 ± 0.53a,b | 3.10 ± 0.73a,c |
Values are presented as mean±SD.
Statistical analyses were performed using two-way ANOVA with Bonferroni post-hoc test, which were compared with those of carboxymethyl cellulose (CMC) group.
P<0.05.
P<0.001.
Table 3.
Microscopic Score of Colonic Fibrosis
| Group | Microscopic score of colonic fibrosisa |
||||
|---|---|---|---|---|---|
| Ulcerations | Degree of inflammation | Depth of lesions | Degree of fibrosis | Total microscopic score | |
| Ethanol (n = 10) | 0.10 ± 0.32 | 0.20 ± 0.42 | 0.20 ± 0.42 | 0.30 ± 0.48 | 0.80 ± 0.63 |
| CMC (n = 10) | 0.90 ± 0.31 | 1.60 ± 0.51 | 1.80 ± 0.48 | 1.90 ± 0.31 | 6.20 ± 0.63 |
| TM5275 10 mg/kg (n = 9) | 0.83 ± 0.63 | 1.46 ± 0.52 | 1.56 ± 0.51 | 1.56 ± 0.50 | 5.30 ± 0.62 |
| TM5275 50 mg/kg (n = 10) | 0.70 ± 0.48 | 1.30 ± 0.67 | 0.90 ± 0.56b | 0.40 ± 0.51b | 3.30 ± 0.67b |
Values are presented as mean±SD.
Statistical analyses were performed using two-way ANOVA with Bonferroni post-hoc test, which were compared with those of carboxymethyl cellulose (CMC) group.
P<0.001.
Fig. 4.

The anti-fibrotic mechanism of plasminogen activator inhibitor-1 (PAI-1) inhibitor. (A) Induced transforming growth factor β (TGF-β) and its autocrine signaling (straight and semicircular arrows with +) promote the deposition of collagen during colonic fibrosis in mice as shown in previous report.7 Simultaneously, it induces PAI-1 (arrow with red +), which inactivates tissue plasminogen activator (tPA)/plasmin axis, and reduces the total amount and active matrix metalloproteinase 9 (MMP-9) (downward red arrows), resulting in the accumulation of collagen (upward red arrows). (B) Conversely, PAI-1 inhibitor activates tPA/plasmin axis, up-regulates MMP-9 (upward blue arrows) and causes the degradation of collagen (downward blue arrows).
DISCUSSION
Although the antifibrinolytic PAI-1 functions have received most of the attention in research on tissue fibrosis [22], more recent report has demonstrated a correlation between enhanced PAI-1 expression and inflammation in IBD patients and murine acute colitis model [11]. In the present study, we observed PAI-1 overexpression at active lesions in both patients with CD and mice with the chronic colitis; this overexpression seems to exacerbate intestinal fibrosis and PAI-1 inhibitor is a potential drug candidate for targeted therapy. As PAI-1 inhibition induced MMP-9 activation in the TNBS colitis model, PAI-1-mediated MMP-9 activity inhibition could be a key mechanism via which ECM is deposited in the lesions [20].
A large number of studies have focused on the molecular mechanisms responsible for the TGF-β-stimulated type-I collagen gene transcription and its pathological roles during the fibrotic process [23-25]. Because TGF-β is a potent anti-inflammatory cytokine, its signal blockade seems to exacerbate inflammation. TGF-β1-deficient mice develop severe multi-organ inflammation [26,27], and targeted deletions of Smad 2 and 4 are associated with early death in mice [28,29], highlighting the need for caution in applying anti-TGF-β strategies as anti-fibrotic therapies. In addition to the direct induction of collagen, we focused on TGF-β attenuating the fibrinolytic system via PAI-1 expression as its downstream target. Therefore, targeting PAI-1 inhibition could facilitate intestinal fibrosis treatment with minimal or no side effects.
MMP-9 has previously been identified as a key marker of inflammation in both UC and CD [30,31] and as effective in the development of murine colitis [32]. Similarly, plasmin inhibition reportedly protects against colitis in mice by suppressing MMP-9 [21]. However, a recent report argued against its significance because MMP-9 inhibition does not attenuate murine colitis [33]. In addition, recent clinical trials with MMP-9 antagonist have not demonstrated significant efficacy in patients with moderate-to-severe UC or CD [34]. Such results obscure the significance of MMP-9 for the regulation of inflammation. Contrary to their validation methods, which focus on the acute phase of colitis, in the present study, we focus on the significance of PAI-1 for intestinal fibrosis in the chronic phase of colitis, where MMP-9 seems to be effective in the degradation of fibrotic deposition. Because MMP-9 plays multiple roles at different stages of IBD development, anti-MMP-9 therapy should be developed with caution. Previous report indicated that tPA-mediated active plasmin increases the total amount of MMP-9 in the gut with acute inflammation [21]. Although its precise mechanism has not been fully understood yet, there may be another pathway to induce MMP-9 by active plasmin. This is quite consistent with our finding that the blockade of PAI-1 activity, probably activates tPA/plasmin axis, resulting in the upregulation of total MMP-9 (Fig. 4). Alternatively, PAI-1-mediated fibrinolytic system could regulate other protease(s) to degrade ECM deposition; however, this aspect requires further investigation.
PAI-1 inhibitors that we developed are currently under evaluation in phase II clinical trials for the treatment of chronic myeloid leukemia with Imatinib in Japan. If the drugs exhibit a potential to attenuate intestinal fibrosis in IBD, it could facilitate further indication and clinical research. Because only few anti-fibrotic drugs are available for the treatment of various fibrotic lesions in multiple organs, more novel approaches are required to facilitate processes that could lead to the development of effective therapies.
Acknowledgments
Authors would like to thank the members of the Support Center for Medical Research and Education, Tokai University, for their experimental support.
Footnotes
FINANCIAL SUPPORT
This study was supported by the Ministry of Education, Culture, Sports, Science and Technology (MEXT; 17H05802 for Hozumi K); Japan Society for the Promotion of Science (JSPS; 19K017413 for Imai J); a MEXT-Supported Program for the Strategic Research Foundation at Private University (Tokai University) for Imai Y.
CONFLICT OF INTEREST
No potential conflict of interest relevant to this article was reported.
AUTHOR CONTRIBUTION
Conceptualization: Hozumi K, Imai J, Yahata T. Funding acquisition: Hozumi K, Imai J. Methodology: Hozumi K, Imai J, Yahata T, Ichikawa H, Ibrahim AA, Yazawa M, Sumiyoshi H. Supervision: Ichikawa H, Inagaki Y, Matsushima M, Suzuki T, Mine T, Ando K, Miyata T. Writing original draft: Imai J, Hozumi K. All authors read and approved the final version of the manuscript.
REFERENCES
- 1.Cosnes J, Gower-Rousseau C, Seksik P, Cortot A. Epidemiology and natural history of inflammatory bowel diseases. Gastroenterology. 2011;140:1785–1794. doi: 10.1053/j.gastro.2011.01.055. [DOI] [PubMed] [Google Scholar]
- 2.Latella G, Sferra R, Speca S, Vetuschi A, Gaudio E. Can we prevent, reduce or reverse intestinal fibrosis in IBD? Eur Rev Med Pharmacol Sci. 2013;17:1283–1304. [PubMed] [Google Scholar]
- 3.Speca S, Giusti I, Rieder F, Latella G. Cellular and molecular mechanisms of intestinal fibrosis. World J Gastroenterol. 2012;18:3635–3661. doi: 10.3748/wjg.v18.i28.3635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rieder F, Fiocchi C, Rogler G. Mechanisms, management, and treatment of fibrosis in patients with inflammatory bowel diseases. Gastroenterology. 2017;152:340–350. doi: 10.1053/j.gastro.2016.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bettenworth D, Rieder F. Medical therapy of stricturing Crohn’s disease: what the gut can learn from other organs: a systematic review. Fibrogenesis Tissue Repair. 2014;7:5. doi: 10.1186/1755-1536-7-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Verrecchia F, Mauviel A. Transforming growth factor-beta and fibrosis. World J Gastroenterol. 2007;13:3056–3062. doi: 10.3748/wjg.v13.i22.3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Imai J, Hozumi K, Sumiyoshi H, et al. Anti-fibrotic effects of a novel small compound on the regulation of cytokine production in a mouse model of colorectal fibrosis. Biochem Biophys Res Commun. 2015;468:554–560. doi: 10.1016/j.bbrc.2015.10.123. [DOI] [PubMed] [Google Scholar]
- 8.Higashi K, Tomigahara Y, Shiraki H, et al. A novel small compound that promotes nuclear translocation of YB-1 ameliorates experimental hepatic fibrosis in mice. J Biol Chem. 2011;286:4485–4492. doi: 10.1074/jbc.M110.151936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Samarakoon R, Higgins PJ. Integration of non-SMAD and SMAD signaling in TGF-beta1-induced plasminogen activator inhibitor type-1 gene expression in vascular smooth muscle cells. Thromb Haemost. 2008;100:976–983. [PMC free article] [PubMed] [Google Scholar]
- 10.Ghosh AK, Vaughan DE. PAI-1 in tissue fibrosis. J Cell Physiol. 2012;227:493–507. doi: 10.1002/jcp.22783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaiko GE, Chen F, Lai CW, et al. PAI-1 augments mucosal damage in colitis. Sci Transl Med. 2019;11:e. doi: 10.1126/scitranslmed.aat0852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ibrahim AA, Yahata T, Onizuka M, et al. Inhibition of plasminogen activator inhibitor type-1 activity enhances rapid and sustainable hematopoietic regeneration. Stem Cells. 2014;32:946–958. doi: 10.1002/stem.1577. [DOI] [PubMed] [Google Scholar]
- 13.Latella G, Vetuschi A, Sferra R, et al. Smad3 loss confers resistance to the development of trinitrobenzene sulfonic acid-induced colorectal fibrosis. Eur J Clin Invest. 2009;39:145–156. doi: 10.1111/j.1365-2362.2008.02076.x. [DOI] [PubMed] [Google Scholar]
- 14.Lawrance IC, Wu F, Leite AZ, et al. A murine model of chronic inflammation-induced intestinal fibrosis down-regulated by antisense NF-kappa B. Gastroenterology. 2003;125:1750–1761. doi: 10.1053/j.gastro.2003.08.027. [DOI] [PubMed] [Google Scholar]
- 15.Fichtner-Feigl S, Fuss IJ, Young CA, et al. Induction of IL-13 triggers TGF-beta1-dependent tissue fibrosis in chronic 2,4,6-trinitrobenzene sulfonic acid colitis. J Immunol. 2007;178:5859–5870. doi: 10.4049/jimmunol.178.9.5859. [DOI] [PubMed] [Google Scholar]
- 16.Higashi K, Inagaki Y, Fujimori K, Nakao A, Kaneko H, Nakatsuka I. Interferon-gamma interferes with transforming growth factor-beta signaling through direct interaction of YB-1 with Smad3. J Biol Chem. 2003;278:43470–43479. doi: 10.1074/jbc.M302339200. [DOI] [PubMed] [Google Scholar]
- 17.Gasche C, Scholmerich J, Brynskov J, et al. A simple classification of Crohn’s disease: report of the Working Party for the World Congresses of Gastroenterology, Vienna 1998. Inflamm Bowel Dis. 2000;6:8–15. doi: 10.1097/00054725-200002000-00002. [DOI] [PubMed] [Google Scholar]
- 18.Vancamelbeke M, Vanuytsel T, Farré R, et al. Genetic and transcriptomic bases of intestinal epithelial barrier dysfunction in inflammatory bowel disease. Inflamm Bowel Dis. 2017;23:1718–1729. doi: 10.1097/MIB.0000000000001246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yahata T, Ibrahim AA, Muguruma Y, et al. TGF-beta-induced intracellular PAI-1 is responsible for retaining hematopoietic stem cells in the niche. Blood. 2017;130:2283–2294. doi: 10.1182/blood-2017-02-767384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oh CK, Ariue B, Alban RF, Shaw B, Cho SH. PAI-1 promotes extracellular matrix deposition in the airways of a murine asthma model. Biochem Biophys Res Commun. 2002;294:1155–1160. doi: 10.1016/S0006-291X(02)00577-6. [DOI] [PubMed] [Google Scholar]
- 21.Munakata S, Tashiro Y, Nishida C, et al. Inhibition of plasmin protects against colitis in mice by suppressing matrix metalloproteinase 9-mediated cytokine release from myeloid cells. Gastroenterology. 2015;148:565–578. doi: 10.1053/j.gastro.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 22.Pincha N, Hajam EY, Badarinath K, et al. PAI1 mediates fibroblast-mast cell interactions in skin fibrosis. J Clin Invest. 2018;128:1807–1819. doi: 10.1172/JCI99088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rieder F, Bettenworth D, Imai J, Inagaki Y. Intestinal fibrosis and liver fibrosis: consequences of chronic inflammation or independent pathophysiology? Inflamm Intest Dis. 2016;1:41–49. doi: 10.1159/000445135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Inagaki Y, Okazaki I. Emerging insights into transforming growth factor beta Smad signal in hepatic fibrogenesis. Gut. 2007;56:284–292. doi: 10.1136/gut.2005.088690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li C, Iness A, Yoon J, et al. Noncanonical STAT3 activation regulates excess TGF-beta1 and collagen I expression in muscle of stricturing Crohn’s disease. J Immunol. 2015;194:3422–3431. doi: 10.4049/jimmunol.1401779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Diebold RJ, Eis MJ, Yin M, et al. Early-onset multifocal inflammation in the transforming growth factor beta 1-null mouse is lymphocyte mediated. Proc Natl Acad Sci U S A. 1995;92:12215–12219. doi: 10.1073/pnas.92.26.12215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kulkarni AB, Ward JM, Yaswen L, et al. Transforming growth factor-beta 1 null mice: an animal model for inflammatory disorders. Am J Pathol. 1995;146:264–275. [PMC free article] [PubMed] [Google Scholar]
- 28.Nomura M, Li E. Smad2 role in mesoderm formation, leftright patterning and craniofacial development. Nature. 1998;393:786–790. doi: 10.1038/31693. [DOI] [PubMed] [Google Scholar]
- 29.Yang X, Li C, Xu X, Deng C. The tumor suppressor SMAD4/ DPC4 is essential for epiblast proliferation and mesoderm induction in mice. Proc Natl Acad Sci U S A. 1998;95:3667–3672. doi: 10.1073/pnas.95.7.3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.de Bruyn M, Arijs I, Wollants WJ, et al. Neutrophil gelatinase B-associated lipocalin and matrix metalloproteinase-9 complex as a surrogate serum marker of mucosal healing in ulcerative colitis. Inflamm Bowel Dis. 2014;20:1198–1207. doi: 10.1097/MIB.0000000000000068. [DOI] [PubMed] [Google Scholar]
- 31.de Bruyn M, Vandooren J, Ugarte-Berzal E, Arijs I, Vermeire S, Opdenakker G. The molecular biology of matrix metalloproteinases and tissue inhibitors of metalloproteinases in inflammatory bowel diseases. Crit Rev Biochem Mol Biol. 2016;51:295–358. doi: 10.1080/10409238.2016.1199535. [DOI] [PubMed] [Google Scholar]
- 32.Castaneda FE, Walia B, Vijay-Kumar M, et al. Targeted deletion of metalloproteinase 9 attenuates experimental colitis in mice: central role of epithelial-derived MMP. Gastroenterology. 2005;129:1991–2008. doi: 10.1053/j.gastro.2005.09.017. [DOI] [PubMed] [Google Scholar]
- 33.De Bruyn M, Breynaert C, Arijs I, et al. Inhibition of gelatinase B/MMP-9 does not attenuate colitis in murine models of inflammatory bowel disease. Nat Commun. 2017;8:15384. doi: 10.1038/ncomms15384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.De Bruyn M, Ferrante M. Failure of MMP-9 antagonists in IBD: demonstrating the importance of molecular biology and well-controlled preclinical studies. J Crohns Colitis. 2018;12:1011–1013. doi: 10.1093/ecco-jcc/jjy102. [DOI] [PubMed] [Google Scholar]