

Abstract
While the mechanistic understanding of proton-coupled electron transfer (PCET) has advanced significantly, few reports have sought to elucidate the factors that control chemoselectivity in these reactions. Here we present a kinetic study that provides a quantitative basis for understanding the chemoselectivity in competitive PCET activations of amides and thiols relevant to catalytic olefin hydroamidation reactions. These results demonstrate how the interplay between PCET rate constants, H-bonding equilibria, and rate-driving force relationships jointly determine PCET chemoselectivity under a given set of conditions. In turn, these findings predict reactivity trends in a model hydroamidation reaction, rationalize the selective activation of amide N−H bonds in the presence of much weaker thiol S−H bonds, and deliver strategies to improve the efficiencies of PCET reactions employing thiol co-catalysts.
Graphical Abstract
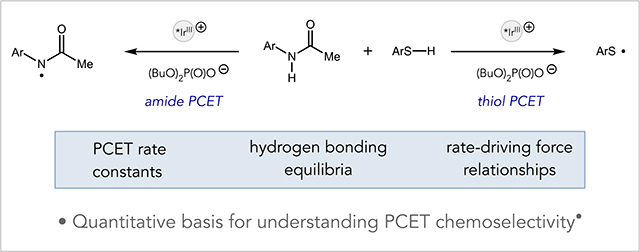
Useful synthetic methods exhibit reliable selectivities, enabling users to confidently predict reaction outcomes in complex settings. To this end, we have recently become interested in trying to understand the features governing chemoselectivity in multi-site proton-coupled electron transfer (MS-PCET) reactions. Oxidative MS-PCET reactions are redox processes wherein protons and electrons are exchanged between a substrate and two independent molecular acceptors – a Brønsted base and one electron oxidant – in a concerted elementary step. Similar to related hydrogen-atom transfer (HAT) reactions,1 prior kinetic studies have shown that MS-PCET processes generally exhibit linear rate-driving force relationships,2 suggesting that abstraction selectivity should track with bond strength differential with weaker bonds to hydrogen reacting preferentially. These quantitative insights reinforce a common intuition that homolytic activation of very strong bonds (BDFEs ≥ 100 kcal/mol) is generally not practical when much weaker bonds to hydrogen (BDFEs ≤ 80 kcal/mol) are also present.
We recently observed a curious exception to this principle in the development of a catalytic method for alkene hydroamidation.3a, 3b In these reactions, which were subsequently studied in detail by Nocera and coworkers,3c a substrate amide N–H bond engages in a concerted MS-PCET reaction with an excited-state Ir(III) oxidant and a dialkyl phosphate base to furnish a reactive amidyl radical. Subsequent addition of the amidyl to a pendant olefin creates a new C–N bond and a vicinal carbon-centered radical that is then reduced by HAT from a thiophenol co-catalyst to form the closed-shell product. A surprising aspect of this reaction relates to the selectivity of the initial MS-PCET step. Both amides3 and thiols4 were demonstrated to be competent substrates for MS-PCET activation under the reaction conditions, and the thiophenol S–H bond (BDFE ~ 79 kcal/mol)5 is significantly weaker than the substrate amide N–H bond (BDFE ~ 99 kcal/mol).6 Accordingly, one might expect that the thiophenol would inhibit the desired N–H oxidation reaction by sequestering the Ir/phosphate catalysts in a highly favorable, but unproductive, PCET process. However, competitive luminescence quenching studies in a model system revealed a rate law for deactivation of the Ir excited state that exhibited a first-order concentration dependence on the amide and phosphate and a zero-order dependence on the concentration of thiol (Scheme 1).3a Efficient and selective amide activation in opposition to such a large thermodynamic bias raises intriguing questions about the physical origins of MS-PCET selectivity in these systems.
Scheme 1.

MS-PCET chemoselectivity relevant to catalytic hydroamidation reactions
Here we present a kinetic study of both amide and thiol PCET activations that sheds light on this surprising selectivity. These experiments demonstrate that both pre-equilibrium hydrogen bonding and the sensitivity of the rate-driving force relationship for each substrate class plays a key role in determining PCET selectivity under a given set of conditions. In turn these findings provide insights into potential pitfalls associated with PCET-based catalysis with thiol H-atom donors, as well as actionable strategies to overcome them. The details of these investigations and their applications to predicting reactivity trends in a catalytic hydroamidation reaction are described herein.
Oxidative MS-PCET reactions typically occur through hydrogen-bonded complexes between the substrate E–H bond and the Brønsted base.7 As such, the free energy profiles of these reactions are determined by both the kinetic barrier for the PCET event and the favorability of forming the reactive hydrogen-bonding complex, as illustrated in Figure 1. Based on this understanding, we studied the PCET reactions of four N-aryl amides (A1 – A4) and four aryl thiols (T1 – T4), mediated by two distinct Ir(III)-based photooxidants (Ir-1 and Ir-2) and a dibutyl phosphate base (NBu4OP(O)(OBu)2) in 1,2-dichloroethane (DCE) at room temperature (Table 1). The hydrogen bonding equilibrium constant (KA) for association with the phosphate base and the PCET rate constant for oxidation of this H-bonded complex (kPCET) were determined simultaneously for both the amide and the thiol series via a simple luminescence quenching method we recently described in a study of ketone PCET activation.2c This method enabled us to evaluate the sensitivity of the reaction rates to changes in driving force associated with varying either the substrate or the potential of the excited state oxidant. The PCET rate constants and H-bonding equilibrium constants for both the amide and thiol series are presented in Table 1 and the corresponding rate-driving force correlations are shown in Figure 2.9
Figure 1.

Free energy surface for the MS-PCET activation of amides and thiols.
Table 1.
Kinetic and H-bonding equilibrium data
| entry | amide/thiol | *Ir(III) | BDFEN-H/S-H (kcal/mol)6,8 | KA(M−1) | ΔG°’PCET (kcal/mol) | kPCET (M−1s−1) |
|---|---|---|---|---|---|---|
| 1 | A1 | Ir-1 | 98.9 | 1050 | 8.4 | 8.4×109 |
| 2 | A1 | Ir-2 | 98.9 | 1050 | 10.9 | 1.1×109 |
| 3 | A2 | Ir-1 | 101.1 | 3550 | 11.3 | 9.3×108 |
| 4 | A2 | Ir-2 | 101.1 | 3550 | 13.8 | 6.8×107 |
| 5 | A3 | Ir-1 | 101.6 | 1390 | 11.2 | 9.3×108 |
| 6 | A3 | Ir-2 | 101.6 | 1390 | 13.7 | 7.5×107 |
| 7 | A4 | Ir-1 | 100.0 | 1500 | 9.7 | 3.0×109 |
| 8 | A4 | Ir-2 | 100.0 | 1500 | 12.2 | 1.8×108 |
| 9 | T1 | Ir-1 | 79.1 | 200 | −12.4 | 9.5×109 |
| 10 | T1 | Ir-2 | 79.1 | 200 | −9.8 | 3.6×109 |
| 11 | T2 | Ir-1 | 76.9 | 44 | −15.6 | 1.0×1010 |
| 12 | T2 | Ir-2 | 76.9 | 44 | −13.0 | 7.0×109 |
| 13 | T3 | Ir-1 | 84.0 | 5600 | −5.5 | 4.0×109 |
| 14 | T3 | Ir-2 | 84.0 | 5600 | −3.0 | 1.0×109 |
| 15 | T4 | Ir-1 | 81.3 | 2150 | −8.8 | 8.3×109 |
| 16 | T4 | Ir-2 | 81.3 | 2150 | −6.3 | 2.2×109 |
 | ||||||
Figure 2.

Rate-driving force relationships
These results provide a compelling explanation for selective amide activation in the competitive luminescence quenching studies discussed above, as seen through comparison of the data for PCET activation of (A1) and (T1) by Ir-1 and the phosphate base. Surprisingly, the rate constants for PCET with both A1 (8.4 × 109 M−1s−1) and T1 (9.5 × 109 M−1s−1) approached the diffusion limit in DCE (~1 × 1010 M−1s−1).10 However, the KA values indicated that the amide forms a more favorable hydrogen-bonded complex (KA = 1050 M−1) with the phosphate base than the thiol does (KA = 200 M−1), ensuring that the concentration of the reactive amide-phosphate H-bond complex in solution is significantly higher than that of the competing thiol-phosphate complex. As both elementary steps occur with similar rate constants, the thiol reaction is unable to take advantage of the additional 20 kcal/mol of driving force, and the resulting selectivity is gated by the relative concentration of the reactive H-bonded complexes in solution. Based on the KA and kPCET values obtained, the relative rates for amide and thiol activation at the concentrations of the synthetic hydroamidation reactions is ~ 50:1, consistent with the previously determined rate law for luminescence quenching.3a
Evaluating a series of amide and thiol substrates with less favorable driving forces enabled us quantify the rate-driving force relationship for each substrate class outside of the diffusion-limited regime. Linear rate-driving force correlations were observed for both reactant classes. The amide series exhibited rate constants comparable to those obtained previously by Nocera,3c and a Brønsted slope (α) of 0.53, similar to the value expected from Marcus theory.11 However, the thiol series exhibits a much shallower dependence (α = 0.10), indicating a difference in intrinsic barriers for the two sets of substrates.12,13 This value is similar in magnitude to the small Brønsted slope observed in our previous study of PCET activations of ketones that was attributed to non-perfect synchronization (NPS),2c wherein factors that serve to stabilize the product are only partially realized at the transition state.14 Mayer and coworkers recently proposed a similar NPS-based explanation for modest Bronsted slopes in the MS-PCET oxidations of C–H bonds, and we anticipate that similar explanations may be operative here. 15
Importantly, MS-PCET chemoselectivity between competing substrates is determined by differences in both KA and kPCET. The differing slopes of the plots in Figure 2 highlights that changes in driving force effect kPCET-amide and kPCET-thiol with different sensitivities. Therefore, the selectivity between N–H and S–H activation pathways can be modified by changing the overall driving force for the reaction, which is jointly determined by the identity of the oxidant and the base catalysts. Similarly, differences in KA values modulate the concentrations of the reactive H-bonded adducts, whose relative abundance also factors into the observed chemoselectivity.
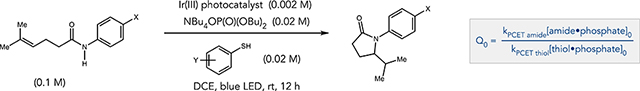
To demonstrate how the interplay of these factors can influence the chemoselectivity of the PCET step, we define a selectivity factor Q, which is taken as □—the ratio of the PCET rate constant for oxidation of the amide-phosphate H-bond adduct times its concentration, to that of the thiol adduct counterpart (equation 1).
| (1) |
Q reflects the competition between the two H-bonded adducts to serve as the electron donor for a limiting concentration of the excited-state oxidant. As such, we hypothesized that the PCET selectivity reflected in Q may correlate with efficiency in a catalytic hydroamidation reaction, and that variation of the factors comprising Q could provide useful insight into the ways in which reaction outcomes can be rationally modulated (and complex reactivity trends understood) through careful choice of reaction conditions. Therefore, we evaluated a series of hydroamidation reactions where the value of Q and its component factors were systematically varied (Table 2). Importantly, during these catalytic reactions the amide substrate is consumed while the concentration of the thiol catalyst remains relatively constant.3c This will cause the initial value of Q (Q0 in Table 2) to decrease as the reaction proceeds, and below a certain threshold of Q the thiol is expected to compete kinetically with the amide substrate in the PCET event, effectively halting reaction progress.
Table 2.
Correlation of Selectivity Factor Q0 with Catalytic Reaction Outcomes
 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| entry | thiol | amide X= | *Ir(lll) | kPCET amide(M−1s−1) | KA amide(M−1) | kPCET thiol(M−1s−1) | KA thiol(M−1) | Q0 | % yieldb | % recoveryb |
| 1 | PhSH | H | Ir-1 | 8.4×109 | 1050 | 9.5×109 | 200 | 96 : 4 | 100 | 0 |
| 2 | PhSH | COMe | Ir-1 | 3.0×109 | 1500 | 9.5×109 | 200 | 91 : 9 | 100 | 0 |
| 3 | PhSH | CN | Ir-1 | 9.3×108 | 3550 | 9.5×109 | 200 | 87 : 13 | 100 | 0 |
| 4 | PhSH | H | Ir-2 | 1.1×109 | 1050 | 3.6×109 | 200 | 86 : 12 | 100 | 0 |
| 5 | PhSH | COMe | Ir-2 | 1.8×108 | 1500 | 3.6×109 | 200 | 62 : 38 | 80 | 9 |
| 6 | PhSH | CN | Ir-2 | 6.8×107 | 3550 | 3.6×109 | 200 | 57 : 43 | 8 | 85 |
| 7 | 3,5-(CF3)2C6H3SH | H | Ir-1 | 8.4×109 | 1050 | 8.3×109 | 2100 | 78 : 22 | 95 | 0 |
| 8 | 3,5-(CF3)2C6H3SH | H | Ir-2 | 1.1×109 | 1050 | 2.2×109 | 2100 | 64 : 36 | 85 | 0 |
| 9 | 3,5-(CF3)2C6H3SH | COMe | Ir-1 | 3.0×109 | 1500 | 8.3×109 | 2100 | 56 : 44 | 60 | 30 |
| 10 | 3,5-(CF3)2C6H3SH | CN | Ir-1 | 9.3×108 | 3550 | 8.3×109 | 2100 | 47 : 53 | 13 | 62 |
| 11 | 3,5-(CF3)2C6H3SH | COMe | Ir-2 | 1.8×108 | 1500 | 2.2×109 | 2100 | 23 : 77 | 10 | 47 |
| 12 | 3,5-(CF3)zC6H3SH | CN | Ir-2 | 6.8×107 | 3550 | 2.2×109 | 2100 | 18 : 82 | 0 | 84 |
| 13 | C6F5SH | H | Ir-1 | 8.4×109 | 1050 | 4.0×109 | 5600 | 75 : 25 | 91 | 0 |
| 14 | C6F5SH | H | Ir-2 | 1.1×109 | 1050 | 1.0×109 | 5600 | 61 : 39 | 61 | 23 |
| 15 | C6F5SH | COMe | Ir-1 | 3.0×109 | 1500 | 4.0×109 | 5600 | 56 : 44 | 17 | 70 |
| 16 | C6F6SH | CN | Ir-1 | 9.3×108 | 3550 | 4.0×109 | 5600 | 44 : 56 | 3 | 54 |
| 17 | C6F5SH | COMe | Ir-2 | 1.8×108 | 1500 | 1.0×109 | 5600 | 24 : 76 | trace | 54 |
| 18 | C6F5SH | CN | Ir-2 | 6.8×107 | 3550 | 1.0×109 | 5600 | 18 : 82 | 0 | 88 |
| In the following two entries, [thiol] = 0.04 M, [base] = 0.005 M | ||||||||||
| 19 | PhSH | H | Ir-2 | 1.1×109 | 1050 | 3.6×109 | 200 | 81 : 19 | 100 | 0 |
| 20 | C6F6SH | H | Ir-2 | 1.1×109 | 1050 | 1.0×109 | 5600 | 35 : 65 | 15 | 50 |
Reactions performed on 0.05 mmol scale.
Yield and recovery assessed after 12h by 1H-NMR relative to an internal standard.
The reactions in the upper portion of Table 2 are divided into three groups. Within each group, a single thiol is employed, while the identities of the amide and oxidant are varied. Notably, in all cases the reaction yields were found to trend together with the Q0 values. The mass balance for reactions that did not reach full conversion was comprised predominantly of recovered starting material. Further evidence that PCET chemoselectivity had a direct impact on reaction outcomes could be found in entry 9, which reached a reaction endpoint of 60% yield and 30% recovery after 12 hours. However, adding a second equivalent of starting material after 12 hours (and thus re-raising the Q value above the critical threshold) resulted in restored hydroamidation reactivity and an additional 36% yield of product formation. This indicates that while the productive reaction had stalled, the catalyst system was still active and that the lack of reactivity was principally a function of a kinetically dominant but non-productive PCET activation of the thiol.
Evaluating the results in Table 2 more broadly, we can distinguish several general reactivity trends. First, amide substrates bearing electron-withdrawing groups (EWGs) are more favorable H-bonding partners for the anionic phosphate, resulting in an increased value of KA-amide and a higher equilibrium concentration of the amide phosphate adduct. However, kPCET-amide decreases for the same substrates as EWGs increase the strength of the N–H bond. Since the Brønsted α value for amides is comparatively large (~0.53), the change in driving force dominates and the overall rate of N–H PCET is suppressed. Accordingly, Q0 also decreases, and the hydroamidation reactions of these substrates are less efficient than the parent compound (e.g. comparing entries 4, 5, 6; 8, 11, 12).
Incorporating EWGs into the aryl thiol also increases KA-thiol and the equilibrium concentration of the thiol-phosphate complex, while simultaneously diminishing kPCET-thiol as the S–H BDFE increases. However, the Brønsted slope for the thiol is comparatively small (α =0.10), indicating that changes in the driving force have comparatively little impact on kPCET-thiol. As such, the increase in adduct concentration is the dominant factor, and serves to increase the overall rate of S–H PCET activation. This in turn decreases Q0 and makes the hydroamidation reaction less efficient (e.g. comparing entries 5, 11, 17; 19, 20).
Finally, as the reduction potential of the Ir photocatalyst becomes more positive the driving force for both PCET reactions becomes more favorable. This causes both kPCET-amide and kPCET-thiol to increase while both KA values remain constant. However, since kPCET-amide is more sensitive to changes in the driving force than kPCET-thiol, Q0 increases when a stronger oxidant is employed, resulting in a more efficient hydroamidation reaction (e.g. comparing entries 3, 6; 9, 11). These results demonstrate how chemoselectivity in MS-PCET reactions and their attendant effects on reaction efficiency can be modulated simply by varying the driving force associated with the one-electron oxidant. Considered altogether, we note that every oxidant and thiol can effectively facilitate the hydroamidation reaction of at least one amide substrate; moreover, every amide substrate can be effectively cyclized with at least one combination of oxidant and thiol. These studies provide quantitative framework for interpreting reactivity trends and suggest general strategies for increasing selectivity for amide activation in marginal reactions. We anticipate that the framework presented here will provides a road map for studying chemoselectivity in other PCET reactions, where the interplay of hydrogen bonding affinities, differential PCET kinetics, and the sensitivities of the rate driving force relationships can be rationally exploited to improve the selectivity of a given process.
Supplementary Material
ACKNOWLEDGMENT
Financial support was provided by the NIH (R01 GM113105).
Footnotes
No competing financial interests have been declared.
ASSOCIATED CONTENT
The Supporting Information is available free of charge on the ACS Publications website.
Experimental procedures, luminescence quenching data, and characterization data (PDF)
References
- (1).(a) Mayer JM Understanding hydrogen atom transfer: from bond strengths to Marcus theory. Acc. Chem. Res 2011, 44, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Xue X; Ji P; Zhou B; Cheng J-P The Essential Role of Bond Energetics in C −H Activation/ Functionalization. Chem. Rev 2017, 117, 8622. [DOI] [PubMed] [Google Scholar]
- (2).(a) Morris WD; Mayer JM Separating Proton and Electron Transfer Effects in Three-Component Concerted Proton-Coupled Electron Transfer Reactions. J. Am. Chem. Soc, 2017, 139, 10312. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Markle TF; Darcy JW; Mayer JM A new strategy to efficiently cleave and form C–H bonds using proton-coupled electron transfer. Sci. Adv 2018, 4, eaat5776. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Qiu G; Knowles RR Rate-Driving Force Relationships in the Multisite-PCET Activation of Ketones. J. Am. Chem. Soc 2019, 141, 2721. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Nomrowski J; Wenger OS Photoinduced PCET in ruthenium–phenol systems: thermodynamic equivalence of Uni-and bidirectional reactions. Inorg. Chem 2015, 54, 3680. [DOI] [PubMed] [Google Scholar]; (e) Rhile IJ; Mayer JM One-electron oxidation of a hydrogen-bonded phenol occurs by concerted proton-coupled electron transfer. J. Am. Chem. Soc 2004, 126, 12718. [DOI] [PubMed] [Google Scholar]; (f) Markle TF; Rhile IJ; DiPasquale AG; Mayer JM Probing concerted proton–electron transfer in phenol–imidazoles. Proc. Natl. Acad. Sci. U.S.A 2008, 105, 8185. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Markle TF; Tronic TA; DiPasquale AG; Kaminsky W; Mayer JM Effect of Basic Site Substituents on Concerted Proton–Electron Transfer in Hydrogen-Bonded Pyridyl–Phenols. J. Phys. Chem. A 2012, 116, 12249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).(a) Miller DC; Choi GJ; Orbe HS; Knowles RR Catalytic Olefin Hydroamidation Enabled by Proton-Coupled Electron Transfer. J. Am. Chem. Soc 2015, 137, 13492. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nguyen ST; Zhu Q; Knowles RR PCET-Enabled Olefin Hydroamidation Reactions with N-Alkyl Amides. ACS Catal 2019, 9, 4502. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ruccolo S; Qin Y; Schnedermann C; Nocera DG General Strategy for Improving the Quantum Efficiency of Photoredox Hydroamidation Catalysis. J. Am. Chem. Soc 2018, 140, 14926. [DOI] [PubMed] [Google Scholar]
- (4).(a) Kuss-Petermann M; Wenger OS Mechanistic Diversity in Proton-Coupled Electron Transfer between Thiophenols and Photoexcited [Ru (2, 2′-Bipyrazine) 3] 2+. J. Phys. Chem. Lett 2013, 4, 2535. [Google Scholar]; (b) Medina-Ramos J; Oyesanya O; Alvarez JC Buffer Effects in the Kinetics of Concerted Proton-Coupled Electron Transfer: The Electrochemical Oxidation of Glutathione Mediated by [IrCl6] 2–at Variable Buffer p K a and Concentration. J. Phys. Chem. C 2013, 117, 902. [Google Scholar]; (c) Gagliardi CJ; Murphy CF; Binstead RA; Thorp HH; Meyer TJ Concerted Electron–Proton Transfer (EPT) in the Oxidation of Cysteine. J. Phys. Chem. C 2015, 119, 7028. [DOI] [PubMed] [Google Scholar]
- (5).Bordwell FG; Cheng J-P; Ji G-Z; Satish AV; Zhang X Bond dissociation energies in DMSO related to the gas phase values. J. Am. Chem. Soc 1991, 113, 9790. [Google Scholar]
- (6).Cheng JP; Zhao YY Homolytic bond cleavage energies of the acidic N–H bonds in dimethyl sulfoxide solution and properties of the corresponding radicals and radical cations. Tetrahedron 1993, 49, 5267. [Google Scholar]
- (7).Mader EA; Mayer JM The Importance of Precursor and Successor Complex Formation in a Bimolecular Proton− Electron Transfer Reaction. Inorg. Chem 2010, 49, 3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).(a) Bordwell FG; Zhang XM; Satish AV; Cheng JP Assessment of the importance of changes in ground-state energies on the bond dissociation enthalpies of the OH bonds in phenols and the SH bonds in thiophenols. J. Am. Chem. Soc 1994, 116, 6605. [Google Scholar]; (b) Dené s F; Pichowicz M; Povie G; Renaud P Thiyl Radicals in Organic Synthesis. Chem. Rev 2014, 114, 2587. [DOI] [PubMed] [Google Scholar]; (c) dos Santos JVA; Newton AS; Bernardino R; Guedes RC Substituent effects on O–H and S–H bond dissociation enthalpies of disubstituted phenols and thiophenols. International Journal of Quantum Chemistry 2008, 108, 754 See SI for calibration of S–H bond energies among different studies. [Google Scholar]
- (9).As in previous studies, we are unable to quantify the favorability of any potential post-PCET H-bonding. Thus, the reported driving force, ΔG°’PCET, is taken to be the free energy change from the H-bonded reactant state to the unbound product state, which is expected to be more endergonic than the actual MS-PCET driving force
- (10).Romero N; Nicewicz DA. Mechanistic Insight into the Photoredox Catalysis of Anti-Markovnikov Alkene Hydrofunctionalization Reactions. J. Am. Chem. Soc 2014, 136, 17024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).(a) Dempsey JL; Winkler JR; Gray HB Proton-coupled electron flow in protein redox machines. Chem. Rev 2010, 110, 7024. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Huynh MHV; Meyer TJ “Proton-coupled electron transfer.” Chem. Rev 2007, 107, 5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Mayr H; Breugst M; Ofial AR Farewell to the HSAB Treatment of Ambident Reactivity. Angew. Chem., Int. Ed 2011, 50, 6470. [DOI] [PubMed] [Google Scholar]
- (13).The correlation in the thiol series also suggests that any impact of varying the position of the substituents on aryl ring is accurately captured by the variance in the BDFEs of the thiol S-H bonds and in their ability to serve as H-bond donors as reflected in the measured KA values.
- (14).(a) Bernasconi CF The principle of imperfect synchronization: I. Ionization of carbon acids. Tetrahedron 1985, 41, 3219. [Google Scholar]; (b) Bernasconi CF Intrinsic barriers of reactions and the principle of nonperfect synchronization. Acc. Chem. Res 1987, 20, 301. [Google Scholar]; (c) Bernasconi CF The principle of nonperfect synchronization: More than a qualitative concept? Acc. Chem. Res 1992, 25, 9. [Google Scholar]; (d) Bernasconi CF The principle of non-perfect synchronization. Adv. Phys. Org. Chem 1992, 27, 119. [Google Scholar]; (e) Bernasconi CF The principle of nonperfect synchronization: recent developments. Adv. Phys. Org. Chem 2010, 44, 223. [Google Scholar]
- (15).Darcy JW; Kolmar SS; Mayer JM Transition State Asymmetry in C–H Bond Cleavage by Proton-Coupled Electron Transfer. J. Am. Chem. Soc 2019, 141, 10777. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.