

Abstract
INTRODUCTION:
While some members of families with autosomal dominant Alzheimer disease mutations learn their mutation status, most do not. How knowledge of mutation status affects clinical disease progression is unknown. This study quantifies the influence of mutation awareness on clinical symptoms, cognition and biomarkers.
METHODS:
Mutation carriers and noncarriers from the Dominantly Inherited Alzheimer Network (DIAN) were stratified based on knowledge of mutation status. Rates of change on standard clinical, cognitive and neuroimaging outcomes were examined.
RESULTS:
Mutation knowledge had no associations with cognitive decline, clinical progression, amyloid deposition, hippocampal volume, or depression in either carriers or noncarriers. Carriers who learned their status mid-study had slightly higher levels of depression and lower cognitive scores.
DISCUSSION:
Knowledge of mutation status does not impact rates of change on any measured outcome. Learning of status mid-study may confer short-term changes in cognitive functioning, or changes in cognition may influence the determination of mutation status.
Introduction
Autosomal dominant Alzheimer disease (ADAD) is a rare form of Alzheimer disease (AD; < 1% of all cases) that arises from mutations in one of three genes, the amyloid precursor protein (APP), presenilin 1 (PSEN1) and 2 (PSEN2). These mutations are near 100% penetrant and onset of dementia symptoms typically occurs at a relatively young and predictable age across generations1. The Dominantly Inherited Alzheimer Network (DIAN) is a multinational, longitudinal study of ADAD families. Comparison of the rates of clinical, cognitive, and biological changes between mutation carriers and noncarriers in this population has provided critical insight into the order and timing of the AD pathological cascade2.
Members of ADAD families have a 50% risk of inheriting a causal mutation from an affected parent and some individuals choose to undergo genetic testing to determine if they are mutation carriers. However, whether knowledge of mutation status impacts clinical and cognitive outcomes in ADAD is currently unknown. In sporadic AD, disclosure of the most common genetic risk factor, apolipoprotein ε4 (APOE ε4), can produce differences on subjective and objective tests of memory3. However, such knowledge informs APOE ε4 carriers of only a higher risk of developing dementia as opposed to disclosure of a ~100% penetrant mutation in ADAD, which represents unavoidable dementia onset and a dramatically shortened lifespan. Without any available treatments, many ADAD family members, understandably, choose to avoid confirmation of mutation status, while others may choose testing in order to plan for what is to come.
Similar to knowledge of APOE, mutation carriers who are aware of their status may be expected to perform more poorly on clinical and cognitive outcomes than their unaware peers, independent of actual disease progression. Such a pattern reflects the concept of stereotype threat in which reinforcement of a negative stereotype (e.g., older adults typically perform poorly on cognitive tests) causes individuals to perform worse than they might otherwise4,5. Similarly, mutation carriers aware of their genetic status might be overly sensitive to normal “slips” of memory (e.g., “Where are my keys?”) and misinterpret that as an early sign of dementia. This sensitivity could bias clinical measures that rely on subjective reports from the participant and collateral sources. Assuming that knowledge of mutation status simply confers a performance bias, as opposed to altering the pathophysiology of the ADAD disease process, groups may differ on measures of clinical and cognitive function yet remain similar on biological indicators of disease progression such as the accumulation of amyloid or accelerated rates of brain atrophy. Conversely, noncarriers who learn their status would be expected to perform better than those who remain unaware.
Disproportionate rates of knowledge of mutation status may bias estimates of clinical and cognitive decline in ADAD and limit the use of ADAD as a model for sporadic AD. Moreover, such bias might affect the outcome of clinical trials that enroll a disproportionate number of “mutation aware” vs. “unaware” participants or if a large percentage of the study population becomes aware of their mutation status during the course of the study. To determine if prior mutation knowledge negatively impacts performance at baseline and longitudinally among ADAD family members, we examined clinical, cognitive and biological outcomes between participants who were aware or unaware of their mutation status. Second, to determine if learning one’s status mid-study also impairs performance, we compared outcomes between participants who became aware of their status at a post-baseline visit to those who have never learned their status.
Methods
Participants:
Data were selected from DIAN data freeze 13 (June 30th, 2018). The frequency of assessments in DIAN depends on the individual’s clinical status and overall estimated time to symptom onset (described below). Asymptomatic participants are evaluated in-person every three years (recently changed to every other year). Asymptomatic participants more than 5 years past their expected age of onset are no longer seen in-person, unless they have reported being aware of their mutation status, in which case they continue to be evaluated every other year. Symptomatic participants are seen yearly for as long as the site PI deems valuable data are being obtained and participant burden is still minimal. A full clinical, cognitive and biomarker panel is administered at every in-person visit. The consistency with which clinical symptoms appear within a specific mutation (i.e., all carriers of a given mutation tend to develop symptoms at the same age) allows for the calculation of “estimated years to symptom onset (EYO). For example, a person with an EYO of −5 is five years away from when they are expected to develop symptoms and hence EYO serves as an independent marker of disease stage. For all participants, estimated years to symptom onset (EYO) was calculated as the participant’s age minus the average age at onset of symptoms for all DIAN participants with that specific mutation. This average value has the highest correlation with actual age at onset1,2.
In the current project, our primary interest was the influence of awareness on rates of change. Therefore, we formed a subsample of the full cohort who had at least two assessments in order to be able to estimate a rate of change. We performed two primary analyses, each of which required a slightly different subset of the available cohort (see Figure 1 for a flowchart of the sample selection). Analysis 1 compared 100 participants (57 mutation carriers; 43 noncarriers) who had never learned their mutation status with 84 participants (66 mutation carriers; 18 noncarriers) who were aware of their mutation status at baseline. Participants aware of their mutation status were substantially higher in EYO compared to unaware participants. Given the non-overlap in EYO at the high and low ends across awareness groups, statistical control of EYO as a covariate would not adequately account for EYO differences. This issue was addressed by restricting the EYO range of the longitudinal cohort to include only −15 to + 10 years at baseline. This restriction resulted in a reasonable overlap in EYO across awareness groups allowing the use of EYO as a covariate. In addition, this EYO range is consistent with disease stages used in AD secondary prevention trials6. Analysis 2 examined the effects of learning mutation status mid-study (as opposed to having prior awareness) in 56 participants (31 carriers; 25 noncarriers). Furthermore, as the learner and unaware groups did not differ in terms of EYO at baseline, and in order to maintain as large of a sample size as possible, we did not apply any EYO restrictions to this sample.
Figure 1.
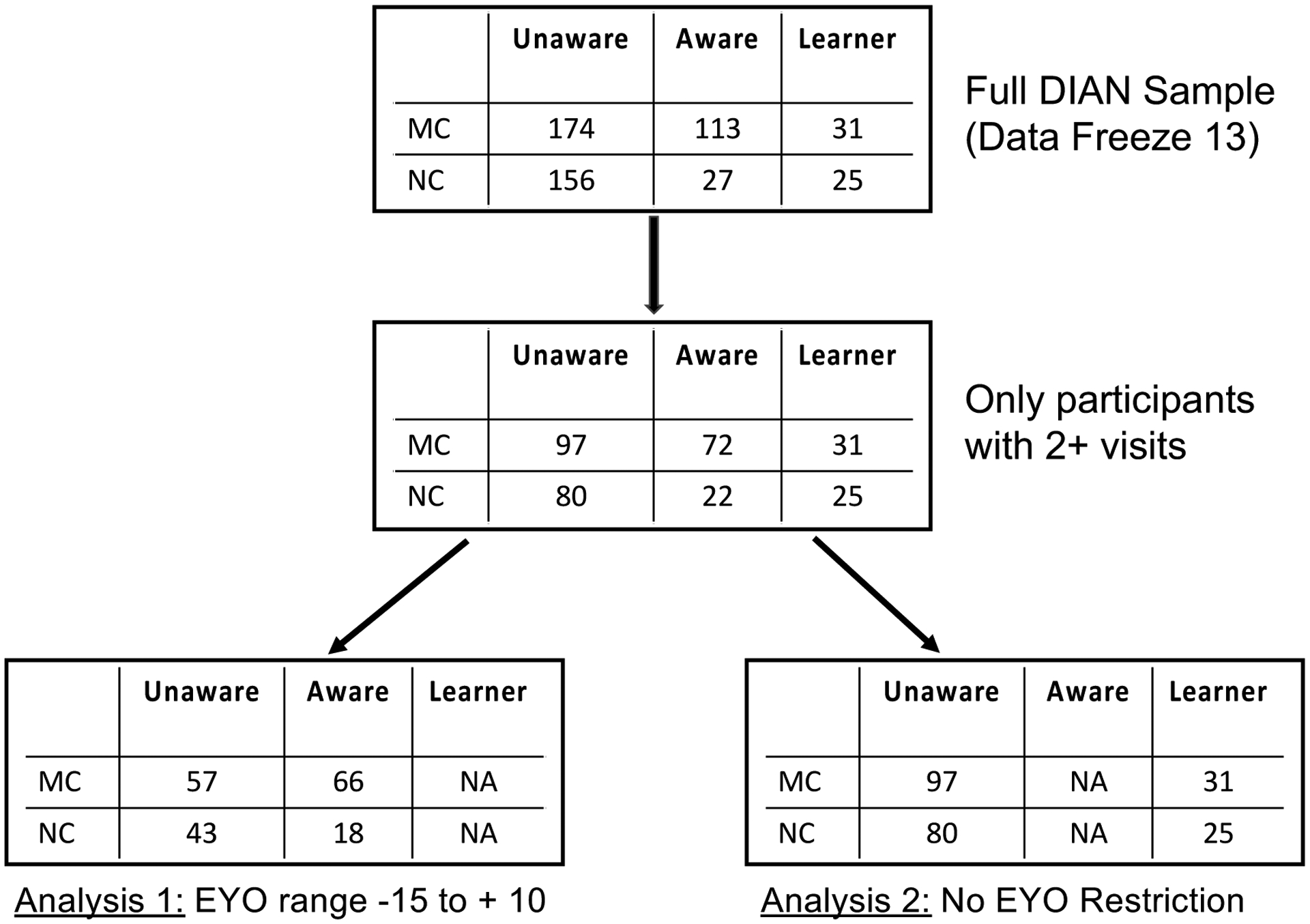
STROBE flowchart for participant selection. Unaware = unaware of mutation status, Aware = aware of mutation status, Learner = Became aware of mutation status following baseline visit. MC = mutation carrier, NC = Non-carrier, EYO = estimated years to symptom onset.
At each assessment, participants were verbally asked if they were aware of their mutation status. Based on this self-report, individuals were classified into one of three groups for analysis: “Mutation Unaware” participants reported not knowing their mutation status at the baseline visit and at every subsequent visit whereas “Mutation Aware” participants reported already knowing their status at baseline. Finally, the “Learner” group reported being unaware of their status at the initial visit but stated being aware after baseline. These classifications were formed regardless of an individual’s clinical status (i.e., global CDR rating).
Clinical Evaluation:
All participants were examined by an experienced clinician and rated for presence and severity of dementia using the Clinical Dementia Rating (CDR) scale7. A score of 0 on the CDR indicates absence of dementia symptoms and 0.5, 1, 2, and 3 reflect very mild, mild, moderate and severe dementia, respectively. In addition to the global CDR, the Sum of Boxes (CDR-SB) score provides a more detailed measure of symptom severity. Additional clinical and behavioral measures were also obtained including the Mini-Mental State Exam (MMSE)8 and the short-form (15 items) Geriatric Depression Scale (GDS)9. The DIAN Study protocol requires that all study staff performing clinical and cognitive evaluations are blinded to mutation status and mutation knowledge.
Cognitive Evaluation:
Each participant completed a comprehensive neuropsychological test battery described elsewhere10. We formulated a cognitive composite score that closely mirrors the primary cognitive endpoint for the DIAN Trials Unit (DIAN-TU)6. This composite consists of the MMSE total score, Logical Memory delayed recall11 the total score from the Digit Symbol Substitution test12, and delayed recall of a 16-item word list. Scores on each test were standardized to the mean and standard deviation of mutation carriers with EYO < −15. Due to substantial ceiling effects on the MMSE, the standard deviation is extremely small which effectively increases the weight of this measure in a composite score. Thus, an adjusted standard deviation for the MMSE was estimated from a smoothing spline model13. The resultant z-scores were then averaged to form a global cognitive composite score. Higher scores indicate better performance.
PET PiB Collection:
ß-Amyloid positron emission tomography (PET) neuroimaging was performed with a bolus injection of approximately 15 mCi of [11C] Pittsburgh Compound B (PiB). Data were used from 40 to 70 minutes post-injection and were motion corrected and partial volume corrected using methods described elsewhere14. For each region of interest, the standardized uptake value ratio (SUVR) was calculated using the cerebellum as a reference. A summary score was formed from the average SUVR across the following regions: precuneus, superior frontal, rostral middle frontal, lateral orbitofrontal, medial orbitofrontal, superior and medial temporal.
MRI Collection:
Structural MRI acquisition was performed using the Alzheimer Disease Neuroimaging Initiative (ADNI) protocol14. Participating sites were required to pass initial and regular follow-up quality control assessments to insure acquisition conformity. Each participant received an accelerated 3D sagittal T1-weighted MPRAGE on a 3T scanner. A high quality, whole-brain image with 1.1×1.1×1.2 mm voxels was acquired in approximately 5–6 minutes. Before analysis, images were screened for artifacts and protocol compliance by the ADNI imaging core. We used the average of the left and right hippocampal volumes, adjusted for total intracranial volume, as our outcome measure.
Data Analysis
Analysis 1:
Baseline demographics were first compared between the groups using independent samples t-tests for continuous variables and chi-square tests for categorical variables. Mutation aware and mutation unaware participants were then compared on each primary outcome measure (GDS, CDR-SB, Cognitive Composite, PiB SUVR, hippocampal volume) using separate linear mixed effects models with the lme4 package15 in the R statistical environment. Models were run separately for carriers and noncarriers. Each model included main effects, and the two-way interaction between years since entry into the study (hereafter referred to as “time”) and mutation awareness (aware of status vs. unaware, dummy coded with the unaware group as the reference). Baseline EYO and baseline CDR were included as covariates and allowed to interact with the time variable. As the focus of this study is on the influence of mutation knowledge, only effects involving this term are interpreted. Analysis 1 answers the critical question of whether individuals who enter a study aware of their genetic risk show differential rates of progression on key biomarkers of AD relative to those individuals who enter the study unaware. For example, mutation aware carriers might be expected to decline more quickly on cognition due to the stereotype threat phenomenon. If this is true, then a clinical trial would want to include “mutation awareness” as a factor in their randomization algorithms.
Analysis 2:
After comparing the groups’ baseline demographics using t-tests or chi-square tests, simple linear regressions compared performance between participants who learned their mutation status post-baseline and participants who were unaware of their status throughout the study. There was very little longitudinal data available following the visit at which participants learned their mutation status, so cross-sectional outcomes were examined at the visit when knowledge of mutation status was reported. For participants who remained unaware of their status, the first available post-baseline visit was selected so that comparisons on clinical and cognitive variables were less biased by familiarization with study procedures and practice effects. The models for these analyses included terms for mutation knowledge (dummy coded with the unaware group as the reference group), EYO as well as baseline performance on the outcome of interest in order to control for initial ability. The intent of Analysis 2 was to determine the extent to which learning mutation status after the study began might introduce differences in clinical, cognitive, and biomarker outcomes compared to persons who remain unaware of their status.
Results
Analysis 1: Mutation Unaware vs. Aware Participants
Baseline demographics on the sample included in this analysis are presented in Table 1. Compared to unaware mutation carriers, mutation aware carriers were higher in EYO based on an independent samples t-test (means: unaware = − 6.16y, aware = −0.11y; p < 0.001), were slightly older at baseline (p = 0.08), were more likely to have a CDR rating greater than zero (χ2 = 42.3, p < .001) but did not differ in years of education, sex or APOE status. Noncarriers who were aware of their status at baseline were no different from unaware noncarriers in EYO, age at baseline, education, sex, APOE status, or probability of CDR greater than zero.
Table 1:
Baseline demographics of the cohort used in Analysis 1
| Carriers | Noncarriers | |||
|---|---|---|---|---|
| Aware | Unaware | Aware | Unaware | |
| Number | 66 | 57 | 18 | 43 |
| Age | 43.95 (9.41) | 41.25 (7.86) | 43 (7.33) | 42.19 (8.2) |
| Education | 13.8 (2.75) | 13.88 (2.57) | 15.22 (3.59) | 15.02 (2.62) |
| Gender (% Female) | 52% | 65% | 61% | 53% |
| CDR greater than zero (%) | 71% | 25% | 0% | 5% |
| APOE Genotype | ||||
| 23 | 8 | 3 | 0 | 3 |
| 24 | 1 | 2 | 0 | 2 |
| 33 | 35 | 37 | 14 | 25 |
| 34 | 18 | 13 | 4 | 11 |
| 44 | 4 | 2 | 0 | 2 |
| EYO | −0.11 (5.69) | −6.16 (5.59) | −3.82 (6.36) | −4.69 (6.5) |
| Number of Visits | 3.08 (1.33) | 2.81 (1.01) | 2.72 (1.13) | 2.6 (0.93) |
| Time in Study (years) | 2.74 (1.80) | 3.33 (1.68) | 4.15 (1.88) | 3.42 (1.92) |
| GDS | 3.47 (3.33) | 2.44 (2.67) | 0.83 (1.1) | 1.19 (1.5) |
| CDR Sum | 2.82 (3.54) | 0.61 (1.51) | 0 (0) | 0.02 (0.11) |
| PIB SUVR | 2.53 (0.95) | 1.82 (0.92) | 1.03 (0.07) | 1.08 (0.29) |
| Cognition | −1.42 (1.08) | −0.58 (0.91) | 0.13 (0.52) | −0.04 (0.57) |
| Hippocampal Volume (Adjusted) | 0.26 (0.05) | 0.3 (0.04) | 0.31 (0.03) | 0.29 (0.03) |
Statistics are reported as mean (SD). Hippocampal volume is reported as a percentage of total intracranial volume.
In terms of our primary analysis, for ease of discussion, only the influence of mutation awareness on the outcomes of interest is described. Full regression output for mutation carriers is provided in Table 3. Change over time for each outcome was estimated from linear mixed effects models and plotted in Figure 2 as a function of mutation awareness. These analyses did not include participants who learned their status mid-study.
Table 3:
Estimated regression beta weights (SE) from Analysis 1 which compares mutation unaware (n=57) vs. mutation aware carriers (n = 66) in the restricted EYO sample. Beta weights for the noncarriers are provided in the supplementary material.
| GDS | CDR SB | Cognition | Amyloid | Hippocampal Volume | |
|---|---|---|---|---|---|
| Time | −0.04 | 1.39*** | −0.25*** | 0.09*** | −0.01*** |
| (0.18) | (0.22) | (0.05) | (0.03) | (0.002) | |
| Mutation Awareness | 0.34 | 0.21 | −0.01 | 0.23 | −0.01 |
| (0.59) | (0.53) | (0.16) | (0.20) | (0.01) | |
| Baseline EYO | −0.08 | 0.06 | −0.03** | 0.03 | −0.001 |
| (0.05) | (0.05) | (0.01) | (0.02) | (0.001) | |
| Baseline CDR | −2.85*** | −2.72*** | 1.30*** | −0.60*** | 0.04*** |
| (0.66) | (0.58) | (0.17) | (0.23) | (0.01) | |
| Baseline EYO*Time | 0.01 | 0.06*** | −0.01** | −0.002 | −0.0004*** |
| (0.01) | (0.02) | (0.004) | (0.002) | (0.0001) | |
| Baseline CDR*Time | 0.05 | −0.88*** | 0.15*** | −0.04 | 0.01*** |
| (0.19) | (0.25) | (0.06) | (0.03) | (0.002) | |
| Awareness*Time | 0.02 | 0.30 | −0.05 | −0.02 | 0.001 |
| (0.16) | (0.21) | (0.05) | (0.03) | (0.002) |
Note:
p<0.1;
p<0.05;
p<0.01
Figure 2.
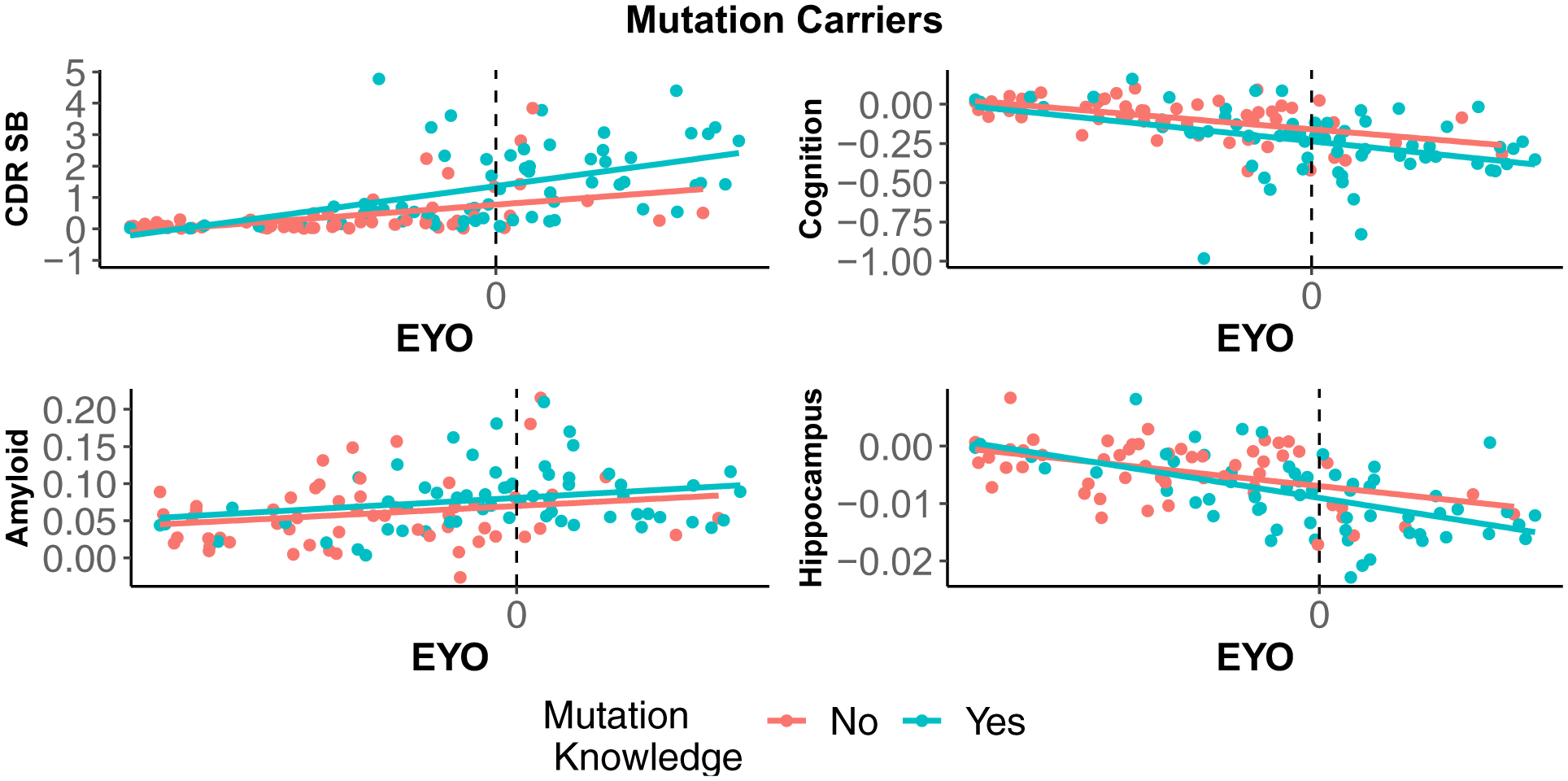
Estimated change over time for mutation carriers as a function of awareness (Ns = 57 unaware and 66 aware) and baseline EYO. Slopes were not different as a function of mutation knowledge of any of the outcomes. EYO ranged from −15 to + 10. Figures for the noncarriers are provided in the supplementary material.
Mutation Carriers:
Depressive symptoms, as measured by the GDS did not vary as a function of awareness at baseline and the interaction between awareness and time was not significant. Mutation aware carriers were not different from unaware carriers at baseline on the CDR-SB. As expected, CDR-SB scores increased over time significantly in the unaware carriers, and rates of change were similar between aware and unaware carriers. Awareness status was not associated with cognition at baseline. Cognition declined significantly over time in unaware carriers, and there was no time by awareness interaction on cognition. In terms of ß-amyloid burden (SUVR), mutation aware carriers did not differ from the unaware carriers at baseline. ß-Amyloid significantly increased over time, and this increase was similar for both aware and unaware participants. Finally, hippocampal volumes did not vary between aware and unaware carriers at baseline. Hippocampal volumes significantly decreased over time, and this change was similar in both awareness groups.
Noncarriers:
Among mutation non-carriers, awareness of mutation status had no relationships with baseline level or change in depressive symptoms, clinical status, cognition, ß-amyloid burden, or hippocampal volumes.
Analysis 2: Mutation unaware vs. Learner, no EYO restriction
As the unaware and those who learned their status mid-study did not differ in terms of EYO, we did not restrict the EYO range for this analysis. Furthermore, the groups did not differ in the length of time they have been enrolled in the study (Learners = 2.48 years, Unaware = 2.47 years, p = 0.97), EYO, age at baseline, probability of being CDR greater than zero, sex, APOE status, or education (all ps > 0.20).
Mutation Carriers:
Controlling for baseline performance on each variable, mutation carriers who learned their status exhibited higher levels of depressive symptoms, as measured by the GDS (β = 0.80, p = 0.02, Cohen’s d = 0.21) and lower scores on the cognitive composite (β = −0.24, p = 0.005, Cohen’s d = 0.25) at the post-disclosure visit relative to carriers who never learned their status. There were no differences in CDR-SB, ß-amyloid burden, hippocampal volume or suicidal ideation as a function of learning mutation status. Figure 3 presents the adjusted mean performance of each awareness group on each measure.
Figure 3.
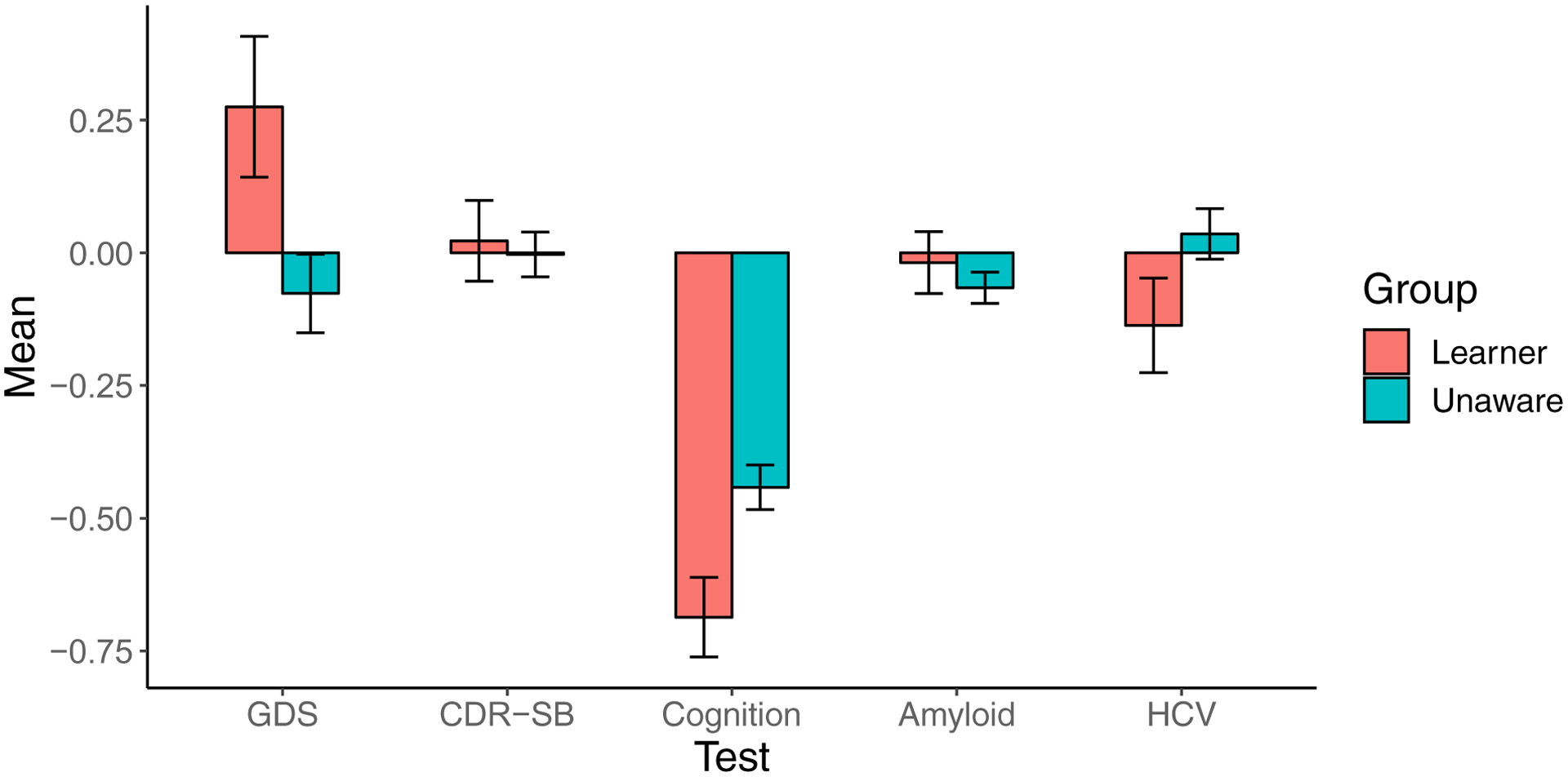
Associations of learning mutation status post-baseline assessment among ADAD mutation carriers. The first visit at which carriers reported having knowledge of mutation status (Learner, n = 31) was compared with the first post-baseline visit for all carriers who remained unaware throughout the study (Unaware n = 97). All measures are shown as adjusted z-scores (error bars represent the standard error of the mean) for ease of comparison.
Non-Carriers:
Controlling for baseline performance on each variable, no differences emerged on any variable between noncarriers who learned their status from those who remained unaware.
Discussion
The primary focus of the study was to determine whether knowledge of mutation status would have an impact on standard clinical, cognitive and biomarker outcomes at baseline and over time. We compared outcomes on participants who were better matched across the EYO range by reducing the sample to the EYO range eligible for entry into DIAN-TU. These analyses revealed that there was no evidence of baseline differences or accelerated declines on any of the measures (GDS, CDR Sum of Boxes, cognition, amyloid burden or hippocampal volume) in mutation aware participants compared to unaware individuals, either in carriers or noncarriers. These findings suggest that mutation knowledge does not alter the course of disease progression or accelerate clinical / cognitive trajectories in autosomal dominant AD.
There are a number of reasons that mutation aware carriers might have been expected to exhibit accelerated rates of change in clinical presentation and cognition. First, aware carriers may be more motivated to seek out genetic testing once they begin to notice changes in their day-to-day functioning. For example, the mutation aware carriers who are more advanced in the disease process may notice more overt changes in cognition and function (as reflected in the CDR-SB), prompting them to inquire about their mutation status. Therefore, carriers may be more likely to become aware of their mutation status because they are further along in the disease process. Indeed, mutation aware participants (both carriers and noncarriers) were significantly older and had a correspondingly higher EYO. This fact made it necessary to do an analysis on a restricted EYO sample to ensure this was not driving any observed differences.
Another interpretation draws from studies of stereotype threat4,5. Many individuals from ADAD families are understandably concerned with their cognitive abilities and are thus highly anxious during clinical and cognitive assessments. As a result, mutation aware participants, fearing that the assessment will reveal a significant increase of symptoms, may perform worse on cognitive tests and appear more impaired on their overall clinical evaluation due to stress or anxiety. Critically, our results show that awareness of mutation status does not appear to impart any effects on cognition, which is important for ADAD clinical trials that may require knowledge of mutation status for enrollment or for open-label study periods.
Although there were no differences on any outcomes as a function of awareness, it is not known precisely when the mutation aware participants learned of their status. Specifically, if negative consequences of mutation knowledge are relatively short-term, and participants were aware of their status for many years, any observed effects of awareness on subjective or performance-based outcomes should be attenuated. We attempted to address this by examining the subset of participants who became aware of their status during the course of the study. Despite exhibiting no differences in EYO or age, participants who learned their status after baseline exhibited slightly lower cognitive performance relative to carriers who never learned their status, although the effect size of this difference was small. No differences emerged in the noncarriers as a function of awareness. Thus, learning that one is a mutation carrier may have some immediate implications for cognitive performance whereas learning that one does not carry the mutation did not impart any protective benefits or other differences. Again, this pattern is consistent with stereotype threat whereby participants aware of their status may “expect” to perform poorly, which then biases their cognitive performance. These results could have implications for clinical trials if participants become aware of their status post-enrollment. Again, the effect size for this finding was small and cross-sectional, as we did not have enough assessments following the visit at which mutation status was learned to evaluate between-group differences in rates of change on any outcomes.
While these results are important in determining whether clinical and cognitive progression in the observational DIAN cohort is biased by individuals who are aware of their status, it is also important to consider the ramifications of mutation knowledge on mood. One may be cautioned against such testing if large and sustained changes in depression or anxiety were found. Although mutation aware carriers did exhibit higher GDS scores at the visit immediately following learning of their status, relative to unaware participants, the effect size was small (d = 0.21). Furthermore, were no differences in depression as a function of mutation knowledge in those who were aware of their status at baseline suggesting the slight increase in depression is likely a relatively short-lived phenomenon. Thus, knowledge of mutation status appears to confer limited effects on depressive symptoms, a finding which is similar to data from sporadic AD where disclosure of ß-amyloid imaging results16 and APOE genotype17 appears to have minimal impact on depression, anxiety or test-related distress.
Our results are otherwise similar to studies of APOE disclosure. Specifically, APOE ε4 carriers who were aware of their status rated themselves lower on a subjective measure of memory capacity in addition to performing worse on objective measures of immediate and delayed verbal recall3. At least in the short-term, the negative consequences of learning genetic status appear similar in both ADAD and sporadic AD and a direct comparison between the populations is warranted. Interestingly, accounting for APOE genotype in the current analyses did not alter the reported pattern of results. The duration, causality and generalizability of these negative consequences needs to be explicitly examined in a longitudinal analysis in both ADAD and sporadic AD.
We have focused largely on the influence of knowledge of risk on key outcomes in observational and clinical trials. However, there is ongoing debate on the practical and ethical considerations regarding risk disclosure in so-called “trial-ready” cohorts18,19. Such platforms aim to maintain a database of participants who meet certain criteria (e.g., are high risk) for rapid recruitment into clinical trials. The debate concerns whether these individuals should be informed of their risk as a requirement of being in the cohort. Our results suggest that minimal negative impact would occur from such disclosure. However, it is important to point out that participants aware of their status in DIAN have chosen to learn their status and it is possible that mandatory disclosure (e.g., as part of the requirements of a research study) may produce a different pattern of results. A recent meta-analysis of studies examining APOE disclosure as a part of study protocols yielded qualitatively similar results20, namely relatively little influence on depression or anxiety. Again, it is important to point out that APOE disclosure only informs of increased risk of AD, whereas mutation status in DIAN is a virtual guarantee. As individuals are very much aware of the implications of possessing the mutation, it is surprising that the results between the cohorts are so similar.
A few limitations of this study should be noted. First, the sample size in some of the mutation knowledge groups were quite small. It is possible that more differences would have emerged in a larger sample. The average number of follow-up assessments was also relatively small and significant differences may emerge with longer duration of follow-up. Because of this, Analysis 2, which discusses the impact of learning mutation status, was necessarily a cross-sectional analysis and duration of the negative consequences as a result of disclosure cannot be determined. Furthermore, the majority of participants who knew their status were relatively close to their age of onset and thus the influence of mutation knowledge very early in the disease cannot be determined from the current sample. Knowledge of status is based solely on self-report and we have no confirmation that genetic testing took place. Furthermore, we have limited information on when the participant became aware of their status and consequently how long they had to come to terms with the information. Finally, causality of knowledge of mutation status and any potential change in cognition and mood cannot be determined in this study, as worsening cognition and associated mood may lead to genetic testing and/or self-report of knowing mutation status. Future work should address whether knowledge of “risk” (genetic or otherwise) manifests similar on critical outcomes in other disorders (e.g., Huntington’s)21.
Conclusions
Among both mutation carriers and noncarriers, knowledge of mutation status at baseline was not associated with accelerated change on any clinical, cognitive or biological outcome. Self-report of learning one’s genetic status after baseline was associated with slight increases in depressive symptoms and lower cognitive scores but had no impact on clinical status or biomarkers. These findings suggest that mutation knowledge is an important variable for further study to determine causality when interpreting cognitive outcomes in ADAD, but do not appear to impact clinical or biological indicators of disease progression.
Supplementary Material
Table 2:
Baseline demographics of the cohort used in Analysis 2
| Carriers | Noncarriers | |||
|---|---|---|---|---|
| Learners | Unaware | Learners | Unaware | |
| Number | 31 | 97 | 25 | 80 |
| Age | 35.97 (12.04) | 35.76 (10.36) | 36.2 (8.46) | 36.98 (11.11) |
| Education | 13.9 (3.48) | 14.4 (2.76) | 14.88 (2.79) | 14.84 (2.48) |
| Gender (% Female) | 61% | 62% | 68% | 55% |
| CDR greater than zero (%) | 29% | 19% | 4% | 6% |
| EYO | −10.76 (10.84) | −12.11 (9.37) | −13.38 (9.29) | −10.43 (11.59) |
| APOE Genotype (N) | ||||
| 23 | 3 | 9 | 3 | 10 |
| 24 | 0 | 4 | 3 | 2 |
| 33 | 18 | 60 | 15 | 45 |
| 34 | 10 | 22 | 4 | 21 |
| 44 | 0 | 2 | 0 | 2 |
| Number of Visits | 2.97 (1.05) | 2.71 (0.89) | 3.16 (1.03) | 2.5 (0.8) |
| Time in Study (Years) | 3.68 (1.58) | 3.79 (1.88) | 4.55 (1.91) | 3.57 (1.80) |
| GDS | 2.42 (2.85) | 2.05 (2.36) | 2.04 (2.13) | 1.5 (2.02) |
| CDR Sum | 0.5 (0.94) | 0.39 (1.19) | 0.02 (0.1) | 0.07 (0.34) |
| PIB SUVR | 1.97 (1.13) | 1.56 (0.77) | 1.04 (0.08) | 1.07 (0.22) |
| Cognition | −0.43 (0.93) | −0.34 (0.86) | 0.25 (0.7) | −0.03 (0.58) |
| Hippocampal Volume (Adjusted) | 0.3 (0.03) | 0.3 (0.04) | 0.3 (0.03) | 0.3 (0.03) |
Statistics are reported as mean (SD). Hippocampal volume is reported as a percentage of total intracranial volume.
Acknowledgments
Data collection and sharing for this project was supported by The Dominantly Inherited Alzheimer’s Network (DIAN, U19AG032438) funded by the National Institute on Aging (NIA), the German Center for Neurodegenerative Diseases (DZNE), Raul Carrea Institute for Neurological Research (FLENI), Partial support by the Research and Development Grants for Dementia from Japan Agency for Medical Research and Development, AMED, and the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI).This manuscript has been reviewed by DIAN Study investigators for scientific content and consistency of data interpretation with previous DIAN Study publications. We acknowledge the altruism of the participants and their families and contributions of the DIAN research and support staff at each of the participating sites for their contributions to this study.
References
- 1.Ryman DC, Acosta-Baena N, Aisen PS, et al. Symptom onset in autosomal dominant Alzheimer disease: A systematic review and meta-analysis. Neurology. 2014;83(3):253–260. doi: 10.1212/WNL.0000000000000596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bateman RJ, Xiong C, Benzinger TL, et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. New England Journal of Medicine. 2012;367(9):795–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lineweaver TT, Bondi MW, Galasko D, Salmon DP. Effect of Knowledge of APOE Genotype on Subjective and Objective Memory Performance in Healthy Older Adults. American Journal of Psychiatry. 2014;171(2):201–208. doi: 10.1176/appi.ajp.2013.12121590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lamont RA, Swift HJ, Abrams D. A review and meta-analysis of age-based stereotype threat: Negative stereotypes, not facts, do the damage. Psychology and aging. 2015;30(1):180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pennington CR, Heim D, Levy AR, Larkin DT. Twenty years of stereotype threat research: A review of psychological mediators. PLOS one. 2016;11(1):e0146487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bateman RJ, Benzinger TL, Berry S, et al. The DIAN-TU Next Generation Alzheimer’s prevention trial: adaptive design and disease progression model. Alzheimer’s & dementia: the journal of the Alzheimer’s Association. 2017;13(1):8–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology. 1993. [DOI] [PubMed] [Google Scholar]
- 8.Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”: a practical method for grading the cognitive state of patients for the clinician. Journal of psychiatric research. 1975;12(3):189–198. [DOI] [PubMed] [Google Scholar]
- 9.Sheikh JI, Yesavage JA. Geriatric Depression Scale (GDS): recent evidence and development of a shorter version. Clinical Gerontologist: The Journal of Aging and Mental Health. 1986. [Google Scholar]
- 10.Storandt M, Balota DA, Aschenbrenner AJ, Morris JC. Clinical and psychological characteristics of the initial cohort of the Dominantly Inherited Alzheimer Network (DIAN). Neuropsychology. 2014;28(1):19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wechsler D Manual: Wechsler Memory Scale- Revised. San Antonio, TX: Psychological Corporation; 1987. [Google Scholar]
- 12.Wechsler D Manual: Wechsler Adult Intelligence Scale- Revised. New York, NY: Psychological Corporation; 1981. [Google Scholar]
- 13.Wang G, Berry S, Xiong C, et al. A novel cognitive disease progression model for clinical trials in autosomal-dominant Alzheimer’s disease. Statistics in Medicine. 2018;37(21):3047–3055. doi: 10.1002/sim.7811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Benzinger TLS, Blazey T, Jack CR, et al. Regional variability of imaging biomarkers in autosomal dominant Alzheimer’s disease. Proceedings of the National Academy of Sciences. 2013;110(47):E4502–E4509. doi: 10.1073/pnas.1317918110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bates D, Mächler M, Bolker B, Walker S. Fitting linear mixed-effects models using lme4. arXiv preprint arXiv:14065823. 2014. [Google Scholar]
- 16.Burns JM, Johnson DK, Liebmann EP, Bothwell RJ, Morris JK, Vidoni ED. Safety of disclosing amyloid status in cognitively normal older adults. Alzheimer’s & Dementia. 2017;13(9):1024–1030. doi: 10.1016/j.jalz.2017.01.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Green RC, Roberts JS, Cupples LA, et al. Disclosure of APOE Genotype for Risk of Alzheimer’s Disease. New England Journal of Medicine. 2009;361(3):245–254. doi: 10.1056/NEJMoa0809578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Milne R, Bunnik E, Tromp K, et al. Ethical Issues in the Development of Readiness Cohorts in Alzheimer’s Disease Research. 2017. doi: 10.14283/jpad.2017.5 [DOI] [PubMed] [Google Scholar]
- 19.Molinuevo JL, Cami J, Carné X, et al. Ethical challenges in preclinical Alzheimer’s disease observational studies and trials: Results of the Barcelona summit. Alzheimer’s & Dementia. 2016;12(5):614–622. doi: 10.1016/j.jalz.2016.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bemelmans SASA, Tromp K, Bunnik EM, et al. Psychological, behavioral and social effects of disclosing Alzheimer’s disease biomarkers to research participants: a systematic review. Alzheimer’s Research & Therapy. 2016;8(1). doi: 10.1186/s13195-016-0212-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kringlen G, Kinsley L, Aufox S, Rouleau G, Bega D. The Impact of Family History on the Clinical Features of Huntington’s Disease. JHD. 2017;6(4):327–335. doi: 10.3233/JHD-170256 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.