

Abstract
Glutaric aciduria type 1 (GA1) is an inborn error of lysine degradation characterized by a specific encephalopathy that is caused by toxic accumulation of lysine degradation intermediates. Substrate reduction through inhibition of DHTKD1, an enzyme upstream of the defective glutaryl-CoA dehydrogenase, has been investigated as a potential therapy, but revealed the existence of an alternative enzymatic source of glutaryl-CoA. Here, we show that loss of DHTKD1 in glutaryl-CoA dehydrogenase-deficient HEK-293 cells leads to a 2-fold decrease in the established GA1 clinical biomarker glutarylcarnitine and demonstrate that oxoglutarate dehydrogenase (OGDH) is responsible for this remaining glutarylcarnitine production. We furthermore show that DHTKD1 interacts with OGDH, dihydrolipoyl succinyltransferase and dihydrolipoamide dehydrogenase to form a hybrid 2-oxoglutaric and 2-oxoadipic acid dehydrogenase complex. In summary, 2-oxoadipic acid is a substrate for DHTKD1, but also for OGDH in a cell model system. The classical 2-oxoglutaric dehydrogenase complex can exist as a previously undiscovered hybrid containing DHTKD1 displaying improved kinetics towards 2-oxoadipic acid.
Introduction
2-Oxoadipic acid (OA) is a metabolite in the lysine, hydroxylysine and tryptophan degradation pathways. It undergoes oxidative decarboxylation catalyzed by a recently discovered 2-oxoadipate dehydrogenase complex (OADHc) (Supplementary Material, Fig. S1) (1–4). The existence of this complex is based on the identification of mutations in DHTKD1 in individuals with 2-aminoadipic and 2-oxoadipic aciduria (AMOXAD, MIM #204750), an established inborn error of lysine metabolism (5–7). DHTKD1 encodes the dehydrogenase E1 and transketolase domain-containing protein 1 (DHTKD1), a close protein homolog of OGDH, the E1 component of the 2-oxoglutarate dehydrogenase complex (OGDHc) (8). The OADHc and OGDHc belong to the super family of 2-oxo acid dehydrogenase complexes, which consist of multiple subunits of the E1, E2 and E3 components. The E1 and E2 components of these complexes are unique and catalyze the oxidative decarboxylation and transacylation of the substrate to form a coenzyme A (CoA) ester. The E3 component (dihydrolipoyl dehydrogenase [DLD]) is shared among all complexes and regenerates the oxidized lipoamide cofactor of the E2 component. OADHc and OGDHc most likely share the E2 component (dihydrolipoyl succinyltransferase [DLST]) given the lack of novel candidate E2 components and the homology between OGDH and DHTKD1. Indeed, recombinant DHTKD1, DLST and DLD are able to assemble into an active OADHc in vitro (9). Thiamin has been shown to regulate OGDHc and OADHc function in rat brain (10), but the biochemical and enzymatic properties of the native OADHc in mammalian tissues have not been fully characterized.
Lysine degradation is a clinically relevant pathway, because of two severe inborn errors of metabolism. Pyridoxine-dependent epilepsy (MIM #266100) is a condition characterized by severe seizures that can be treated by pyridoxine. It is caused by mutations in ALDH7A1 (11,12). Glutaric aciduria type 1 (GA1, MIM #231670) is a cerebral organic aciduria caused by a defect in glutaryl-CoA dehydrogenase encoded by GCDH (Supplementary Material, Fig. S1). Patients can present with macrocephaly and may develop a complex movement disorder because of striatal injury with acute (acute encephalopathic crisis) or insidious onset (13–16). Remarkably, hyperlysinemia (MIM #238700) and AMOXAD, two other defects in lysine metabolism, are considered biochemical phenotypes of questionable clinical significance (7,17–19).
Substrate reduction therapies in inborn errors of metabolism aim to decrease the levels of toxic metabolite by inhibiting an enzyme upstream of the defective enzyme. The occurrence of seemingly non-harmful enzyme defects in the lysine degradation pathway suggests that substrate reduction therapy by inhibiting these enzymes is safe in humans. Accordingly, inhibition of DHTKD1 has been proposed as a therapeutic intervention in GA1 (20). This approach was tested in Dhtkd1/Gcdh double knockout (KO) mice. Unexpectedly, knocking out Dhtkd1 did not appear to mitigate the clinical and biochemical phenotype of the Gcdh KO mouse (20). This result indicates that DHTKD1 is not an exclusive source of glutaryl-CoA. Here, we reassessed the role of DHTKD1 in lysine degradation and identified the alternative source of glutaryl-CoA.
Results
Hypomorphic C57BL/6 Dhtkd1 allele is not protective in a GA1 mouse model
The identification of DHTKD1 mutations in individuals with AMOXAD in combination with the biochemical characterization of in vitro assembled OADHc suggests that DHTKD1 plays an important role in the conversion of OA into glutaryl-CoA (5–7,9,21). Additional evidence was provided by the characterization of the hypomorphic C57BL/6 Dhtkd1 allele. A mutation in the Dhtkd1B6 allele causes decreased Dhtkd1 mRNA and DHTKD1 protein, increased plasma 2-aminoadipic acid (AA) and increased urine OA (22–24). Previously, Sauer et al. reported that Gcdh KO mice on a C57BL/6J inbred background remained asymptomatic and survived lysine exposure (25). Based on these observations, we originally hypothesized that Dhtkd1B6 was a modifier of the phenotype in Gcdh KO mice. To test this hypothesis, we crossed the C57BL/6 congenic Gcdh KO mouse with DBA/2N to generate an experimental cohort of Gcdh−/− B6D2F2 mice. The genotype distribution in the B6D2F2 was Mendelian with no lethality of GA1 mice (Supplementary Material, Table S1).
We next evaluated the consequences of the different Gcdh and Dhtkd1 genotypes on plasma and urine biomarkers for GA1. The hypomorphic C57BL/6 Dhtkd1 allele did increase plasma AA and urine OA, but did not decrease the accumulation of plasma glutarylcarnitine (C5DC), and urine glutaric and 3-hydroxyglutaric acid (Fig. 1). These results confirm the work by Biagosch et al. by using a different DHTKD1 deficient model (20) and firmly establish that DHTKD1 is not an exclusive source of glutaryl-CoA. Interestingly, we noted that Gcdh KO mice had an increase in plasma AA and urine OA, which appears independent of the Dhtkd1 genotype (Fig. 1). This indicates that the OADHc is partially inhibited in the Gcdh KO likely because of inhibition by its accumulating product glutaryl-CoA.
Figure 1.
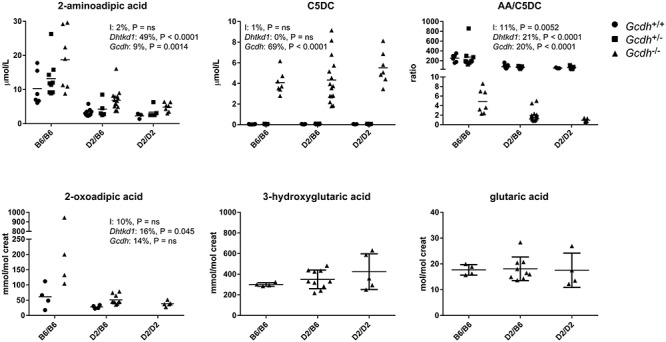
DHTKD1 deficiency does not limit the accumulation of GCDH substrates in Gcdh KO mice. Biochemical analyses of plasma C5DC and AA and urine OA, glutaric and 3-hydroxyglutaric acids in Gcdh KO mice generated with wild type (DBA/2N: D2) and hypomorphic Dhtkd1 (C57BL/6: B6) alleles. Plasma C5DC and AA are also shown for Gcdh+/+ and Gcdh+/− and urine OA is also shown for Gcdh+/+. The ratio of AA/C5DC is displayed. A two-way analysis of variance was performed and the results (% variation explained and P value) are displayed for each graph. I denotes the interaction term; Dhtkd1 denotes the effect of the Dhtkd1 allele (B6 or D2); Gcdh denotes the effect of the Gcdh genotype (Gcdh+/+, Gcdh+/− and Gcdh−/−); ns, not significant. Error bars indicate SD.
DHTKD1 KO decreases glutarylcarnitine in GCDH-deficient cell lines
To further investigate the enzymatic sources of glutaryl-CoA, we used CRISPR-Cas9 genome editing in HEK-293 cells. Single and double KO cell lines for GCDH and DHTKD1 were generated simultaneously (Supplementary Material, Fig. S2A). CRISPR-Cas9-induced mutations resulted in full KO cell lines as demonstrated by the absent GCDH and DHTKD1 protein in immunoblots (Supplementary Material, Fig. S2A). As predicted, single GCDH and DHTKD1 KO cell lines had increased C5DC and AA, respectively (Supplementary Material, Fig. S2B and C). AA was also elevated in GCDH/DHTKD1 double KO cell lines (Supplementary Material, Fig. S2C). The level of C5DC was reduced 2-fold in GCDH/DHTKD1 double KO cell lines when compared with single GCDH KO cell lines. C5DC levels, however, did not decrease to control levels (Supplementary Material, Fig. S2B).
The variation in C5DC between the different GCDH/DHTKD1 double KO cell lines was relatively large, which may be related to the clonal isolation. We therefore selected three different GCDH single KO cell lines, and then targeted DHTKD1 using CRISPR-Cas9 to generate GCDH/DHTKD1 double KO cell lines (Fig. 2A and Supplementary Material, Fig. S2D). These consecutively generated GCDH/DHTKD1 double KO cell lines reproduced the results obtained in the simultaneously generated GCDH/DHTKD1 double KOs and showed an overall decrease in C5DC, but not to control levels (Fig. 2A and Supplementary Material, Fig. S2E and F). Of the 11 isolated GCDH/DHTKD1 double KO cell lines eight had decreased (between 18 and 60% decrease), two had similar and one had increased C5DC levels (Fig. 2A). As expected the level of AA increased in all double KO cell lines. In conclusion, these data suggest that DHTKD1 inhibition can limit C5DC accumulation in GCDH deficient HEK-293 cell lines, but also confirm that there is an alternative source of glutaryl-CoA that may be able to compensate partially for the loss of DHTKD1.
Figure 2.
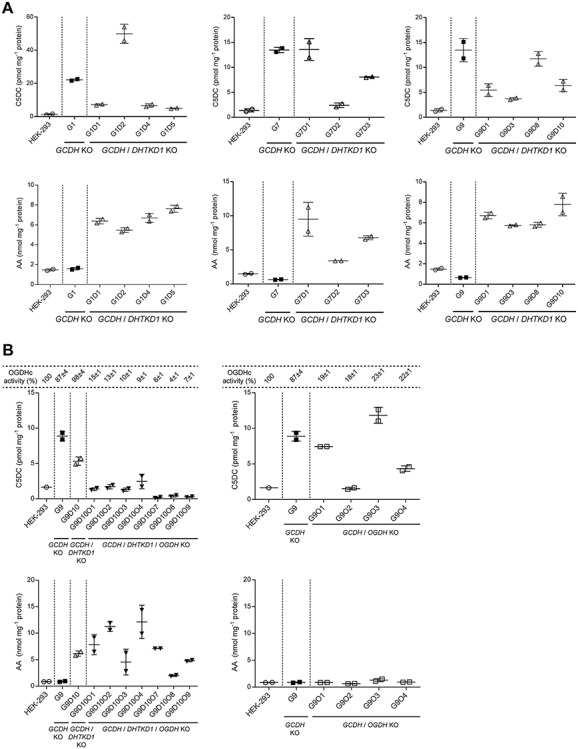
Genetic dissection of the source of glutaryl-CoA in GCDH KO cells. (A) Biochemical analyses of C5DC (upper panel) and AA (lower panel) levels in HEK-293 GCDH/DHTKD1 double KO (white triangle△) compared with HEK-293 GCDH single KO (black square■) and wild-type HEK-293 cells (white circle○). Single GCDH KO clones are indicated as G# and double GCDH/DHTKD1 KO clones as G#D#. Three HEK-293 GCDH KO clones (G1, G7 and G9) were selected for the generation of the DHTKD1 KO. Error bars indicate SD. (B) Biochemical analyses of C5DC (upper panel) and AA (lower panel) levels in HEK-293 GCDH/DHTKD1/OGDH triple KO (inverted black triangle▼) and HEK-293 GCDH/OGDH double KO (white square□) compared with HEK-293 GCDH/DHTKD1 double KO (white triangle△), HEK-293 GCDH single KO (black square■) and wild-type HEK-293 cells (white circle○). The oxidative decarboxylation of OG in cell lysates of HEK-293 KOs was measured to evaluate OGDH knockout (KO) efficiency and is represented as a percentage relative to wild-type activity in HEK-293 cell lysates. Single GCDH KO clones are indicated as G#, double GCDH/DHTKD1 KO clones as G#D#, double GCDH/OGDH KO clones as G#O# and triple GCDH/DHTKD1/OGDH KO clones as G#D#O#. The HEK-293 GCDH/DHTKD1 double KO clone G9D10 and HEK-293 GCDH single KO G9 were selected for the generation of the OGDH KO. Error bars indicate SD. See also Supplementary Material, Fig. S2.
OGDH partially compensates for the loss of DHTKD1
OGDHc from different sources including the reconstituted human complex displays activity towards OA albeit at much lower catalytic efficiency (9,26–30). To demonstrate that OGDHc (i.e. OGDH) is the alternative source of glutaryl-CoA in the GCDH/DHTKD1 double KO cell lines, we generated GCDH/DHTKD1/OGDH triple KO and GCDH/OGDH double KO cell lines. For this, we selected one GCDH/DHTKD1 double KO and one GCDH single KO cell line and targeted OGDH using CRISPR-Cas9 (Supplementary Material, Fig. S2G and H). OGDH protein was decreased in double and triple KO cell lines, but a variable amount of residual OGDH protein remained detectable. The apparent incomplete KO of OGDH may indicate that partial OGDH function is essential for the growth and proliferation in HEK-293 cells. In addition, the OGDH antibody may cross-react with the oxoglutarate dehydrogenase-like protein (OGDHL) homolog that has 79% sequence identity with OGDH. Residual OGDHc activity in HEK-293 KO cell lysates, however, was 4–15% in GCDH/DHTKD1/OGDH triple KO cell lines and 18–46% in GCDH/OGDH double KO cell lines illustrating that the targeting of OGDH led to a significant impairment of OGDHc function. The overall lower OGDHc activities in the GCDH/DHTKD1/OGDH triple KO cell lines may indicate that DHTKD1 contributes to the oxidative decarboxylation of 2-oxoglutarate (OG). Clones with the lowest OGDHc activities were selected for further evaluation.
When compared with the parental GCDH/DHTKD1 double KO, all GCDH/DHTKD1/OGDH triple KO cell lines showed a decrease in C5DC to control levels or lower (Fig. 2B and Supplementary Material, Fig. S2I). As expected, the level of AA remained increased in all triple KO cells, but with some variation between clones (Fig. 2B and Supplementary Material, Fig. S2J). Of the four GCDH/OGDH double KO cell lines, two cell lines had similar C5DC as the parental single GCDH KO, one had an intermediate C5DC and one had control C5DC levels (Fig. 2B and Supplementary Material, Fig. S2I). The level of AA remained at control level in all four GCDH/OGDH double KO cell lines (Fig. 2B and Supplementary Material, Fig. S2J). Combined, these cell line data demonstrate that OGDH is responsible for the residual C5DC formation in GCDH/DHTKD1 double KO cell lines. In the presence of DHTKD1 expression, OGDH does not have a major contribution to the production of C5DC.
We analyzed 24 h and 72 h organic acid accumulation in the cell culture media of one clonal cell line for control, GCDH KO, DHTKD1 KO, GCDH/DHTKD1 double KO and GCDH/DHTKD1/OGDH triple KO (Supplementary Material, Fig. S2K and L). Glutaric acid accumulation was increased in the GCDH KO cell line at both time points. Glutaric acid accumulation decreased in the GCDH/DHTKD1 double KO cell line at 24 h, but only slightly at 72 h. We observed an additional decrease of glutaric acid in GCDH/DHTKD1/OGDH triple KO cell line at 72 h. OA accumulation was only observed in DHTKD1 KO, GCDH/DHTKD1 double KO and GCDH/DHTKD1/OGDH triple KO cell lines with the latter displaying the highest accumulation. OG was observed in the media of all cell lines, but the accumulation was increased in GCDH/DHTKD1/OGDH triple KO cell lines most likely reflecting the OGDHc defect. We conclude that in the HEK-293 cells the accumulation of glutaric acid mimics intracellular C5DC levels. The observation that the levels of OA increase in the GCDH/DHTKD1/OGDH triple KO cell line when compared with the GCDH/DHTKD1 double KO cell line adds further prove to the described substrate overlap between the OGDH and OADH complexes in cells.
DHTKD1 and OGDH interact and share DLST and DLD
We next studied the impact of transient overexpression of the different E1, E2 and E3 components on the activity of OGDHc and OADHc. In HEK-293 cell lysates, the oxidative decarboxylation of OA is ~40 times lower than that of OG (Fig. 3A and Supplementary Material, Fig. S3A). The activity with OG increased only upon transfection of OGDH, while the activity with OA increased upon transfection of DHTKD1 and/or OGDH (Fig. 3A and Supplementary Material, Fig. S3B and C). Similar results were obtained when DLST and DLD were co-transfected with OGDH and DHTKD1. We also assessed enzyme activity after transfection of OGDHL (31) and MRPS36, which encodes a protein that has been implicated in the recruitment of DLD to the OGDHc complex (32). Transient transfection of OGDHL or MRPS36 did not increase 2-oxo acid dehydrogenase activity in HEK-293 cell lysates using either OA or OG substrates (Supplementary Material, Fig. S3C). These data further show that in cells DHTKD1 is highly specific for OA, whereas OGDH has activity with OA and OG. Because of this substrate overlap, OGDH is able to compensate for the loss of DHTKD1 function.
Figure 3.
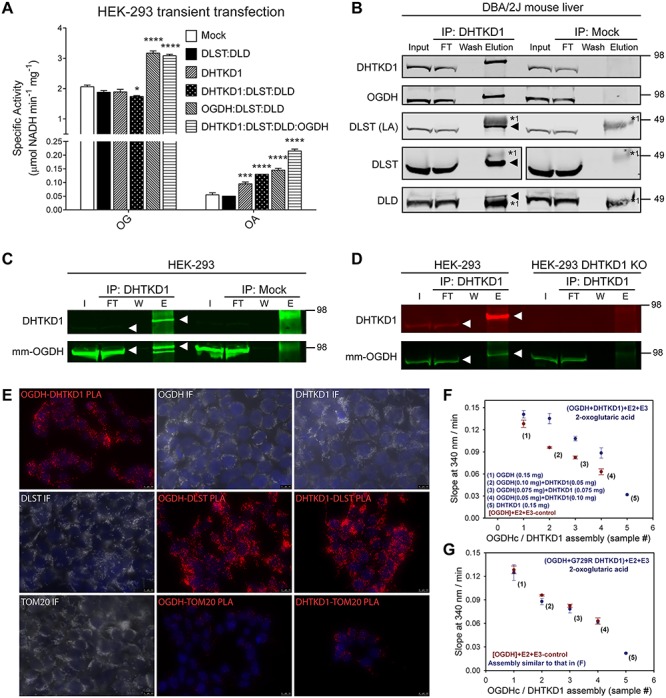
The OADHc complex contains DLST, DLD but also OGDH. (A) DHTKD1 and OGDH overexpression leads to increased oxidative decarboxylation of OA. OG and OA oxidative decarboxylation catalyzed by OGDHc and OADHc complexes in cell lysates of HEK-293 cells upon transient transfection of the indicated genes. HEK-293 cells were transfected with constructs encoding DHTKD1, DLST, DLD and OGDH. The ratio of DNA between the different gene combinations was maintained constant. *P < 0.05; ***P < 0.001 and ****P < 0.0001. (B) OGDH co-purifies with DHTKD1. DHTKD1 was immunoprecipitated from a liver homogenate of a DBA/2J mouse. DLST (using either an antibody against the lipoic moiety or the protein), DLD and OGDH were confirmed as interacting partners of DHTKD1. Aliquots of homogenate (I, input: 2.5% of total fraction volume), the unbound protein after incubation with IP antibody (FT, flow-through: 2.5% of total fraction volume), the last wash (W: 2% of total fraction volume) and eluted fraction (E: 100% of total fraction volume) were analyzed by WB by successively using anti-DHTKD1, anti-OGDH, anti-lipoic acid, anti-DLST and anti-DLD antibodies. Heavy (*1) chain of IP antibody is indicated. The triangle indicates the position of the co-IP protein when close to the heavy chain of the IP antibody. The position of molecular mass marker proteins (in kDa) is given. All proteins in the elution fraction migrate slightly higher than the proteins in the lanes containing input and FT likely because of lower protein amount in this lane. In DLST immunoblot a lane corresponding to the protein marker was omitted for clarity. (C and D) OGDH is co-immunoprecipitated from HEK-293 cell lysate by DHTKD1 (C) and the anti-DHTKD1 antibody is specific for DHTKD1 and does not immunoprecipitate OGDH (D). DHTKD1 was immunoprecipitated from HEK-293 cell lysate and from a DHTKD1 KO HEK-293 cell lysate. Aliquots of homogenate (I, FT, W and E, as in (B)) were analyzed by WB by successively using a mouse monoclonal anti-OGDH (mm-OGDH, 66285–1-lg) and anti-DHTKD1. The white triangle indicates the immunoblotted protein. The position of molecular mass marker proteins (in kDa) is given. (E) In situ PLA analysis and immunofluorescence of DHTKD1, OGDH and DLST in HEK-293 cells. TOM20 served as a negative control. Representative images of PLA (red). Scale bars: 10 μm. (F and G) Analysis of the rates of the NADH production by assembled hybrid complexes between OGDH and DHTKD1 (F), and OGDH and DHTKD1 G729R variant (G) with OG as a substrate. See also Supplementary Material, Figs S3 and S4 and Supplementary Material, Table S2.
The composition of the OADHc has not been characterized in mammalian tissues. To identify the other members of the complex, we performed immunoprecipitation (IP) experiments. DHTKD1 was successfully immunoprecipitated from DBA/2J mouse liver. Immunoblot analysis showed that DLST and DLD are the interacting E2 and E3 components (Fig. 3B). Surprisingly, the immunoprecipitate also contained a protein with a molecular weight slightly larger than DHTKD1. Given the molecular weight and the interaction with DLST and DLD, we hypothesized that this could be OGDH. Immunoblotting using OGDH-specific antibodies confirmed this hypothesis (Fig. 3B). Additional evidence came from proteomic analysis of gel slices of interest, which identified OGDH, DLST and DLD as interacting partners of DHTKD1 (Supplementary Material, Fig. S3D and Table S2). This result indicates that DHTKD1 and OGDH can form a hybrid OGDHc and OADHc.
Given the homology between DHTKD1 and OGDH (38% amino acid identity), we considered the possibility that the IP of OGDH with the anti-DHTKD1 antibody could be the result of direct binding of the anti-DHTKD1 antibody to OGDH. In an attempt to further confirm the DHTKD1–OGDH interaction, we tried IP of OGDH from DBA/2J mouse liver and kidney using two different commercially available OGDH antibodies (Supplementary Material, Fig. S3E–G). A rabbit polyclonal antibody was able to immunoprecipitate OGDH, but we could only demonstrate the interaction with DLST, and not with DLD and DHTKD1 (Supplementary Material, Fig. S3E and F). An IP of DLST from DBA/2J mouse liver did reveal the formation of stable complexes between DLST, DLD and OGDH, and DHTKD1 (Supplementary Material, Fig. S3H), but this result does not necessarily provide additional evidence for the existence of a hybrid complex. Therefore, to rule out the potential of unspecific pull-down of OGDH by the anti-DHTKD1 antibody, we performed IP experiments in control HEK-293 cells and DHTKD1 KO HEK-293 cells. Using the anti-DHTKD1 antibody, OGDH was co-immunoprecipitated in HEK-293 control lysates (Fig. 3C), but was not detected in a pull-down from HEK-293 DHTKD1 KO lysates (Fig. 3D). These data demonstrate the specificity of the anti-DHTKD1 antibody and support the existence of a hybrid OGDHc and OADHc containing DHTKD1, OGDH, DLST and DLD.
To further address the composition of OADHc, we first performed immunohistology on human kidney. We found that DHTKD1, OGDH, DLST and DLD are co-expressed in kidney cortex and medulla, most prominently in proximal tubular epithelium (Supplementary Material, Fig. S4A and data not shown). In situ protein–protein interactions were investigated by proximity ligation assays (PLAs) in HEK-293 cells and selected DHTKD1 and/or OGDH KO cell lines (Fig. 3E, Supplementary Material, Fig. S4B and C). PLA demonstrated the close proximity of OGDH and DHTKD1 to DLST (Fig. 3E and Supplementary Material, Fig. S4B) and DLD (data not shown), but also to each other (Supplementary Material, Fig. S4C).
Additional supporting evidence for the formation of a hybrid OGDHc and OADHc was obtained by in vitro reconstitution of the individual components. While keeping the total amount of E1 protein the same, OGDH was replaced by DHTKD1 (Fig. 3F, in blue). Controls contained the same amount of OGDH, but without DHTKD1 addition (Fig. 3F, in red). The higher rate of NADH production in the presence of DHTKD1 for each analyzed hybrid assembly suggests that both OGDH and DHTKD1 contribute to the oxidative decarboxylation of OG. The overall lower efficiency of the OADHc with OG is consistent with our previous work (21). As a control, OGDH was replaced by an inactive DHTKD1 variant (p.G729R) (6), which did not contribute to the oxidative decarboxylation of OG (Fig. 3G). Taken together, our data indicate that OGDH and DHTKD1 can be assembled into a hybrid dehydrogenase complex with activity towards OG.
Discussion
Mutations in DHTKD1 lead to a defect in OADHc activity and cause AMOXAD (5,6). This condition is currently regarded as a biochemical phenotype of questionable clinical significance, which means that it can be diagnosed through biochemical and genetic methods, but is not considered harmful (19,33,34). This observation led to the hypothesis that GA1 can be treated through substrate reduction by inhibiting DHTKD1, which was speculated to divert the accumulation of neurotoxic glutaryl-CoA into less harmful accumulation of AA and OA (5,20). Unexpectedly, Gcdh/Dhtkd1 double KO mice revealed similar biochemical and clinical outcomes as observed for the existing GA1 mouse model (20). We confirmed these results by making use of the naturally occurring hypomorphic Dhtkd1 allele in C57BL/6 and the wild type Dhtkd1 allele in DBA/2. Combined these animal studies show that DHTKD1 deficiency does not limit the accumulation of plasma C5DC and urine 3-hydroxyglutaric and glutaric acid in GA1 mice, and point to the existence of an alternative enzymatic source of glutaryl-CoA.
To further investigate the enzymatic origins of glutaryl-CoA in the lysine degradation pathway, we generated CRISPR-Cas9 gene KOs in HEK-293 cells. We show that in DHTKD1/GCDH double KO cell lines, the GA1 biomarker C5DC is reduced 2-fold compared with GCDH single KO cell lines. This decrease in C5DC suggests that DHTKD1 contributes to the production of GCDH substrates, but it also further establishes the existence of a significant alternative source of glutaryl-CoA, as C5DC is not completely reduced to control levels (Fig. 4). Biagosch et al. proposed two possible alternative sources of glutaryl-CoA, including the formation via an unknown enzymatic conversion in the pipecolic acid pathway, and production by OGHDc (with OGDH or OGDHL) (Supplementary Material, Fig. S1) (20). DHTKD1 is a paralog of OGDH, which is the E1 component of the OGDHc, a key enzyme from the tricarboxylic acid (TCA) cycle that catalyzes the conversion of OG into succinyl-CoA (35,36). In fact, OGDHc from many sources including in vitro reconstituted human OGDHc can use OA as substrate (9,26–30), albeit with lower affinity and catalytic efficiency. The OGDHc contains multiple copies of OGDH, DLST and DLD, as the E1, E2 and E3 components, respectively (37). OGDHL, a second paralog, shares high homology with OGDH (31) and is highly expressed in brain, kidney and liver, but its function remains unknown. We now show that overexpression of the individual DHTKD1 and OGDH components in HEK-293 cells resulted in increased oxidative decarboxylation of OA, which argues in favor of OGDHc as the source of C5DC in GCDH/DHTKD1 double KO cells. Indeed, knocking out OGDH in GCDH/DHTKD1 double KO cells led to a reduction in C5DC to control or even lower levels. These data are the first genetic proof that OGDH is the enzyme responsible for the partial compensation of deficient DHTKD1 activity (Fig. 4).
Figure 4.
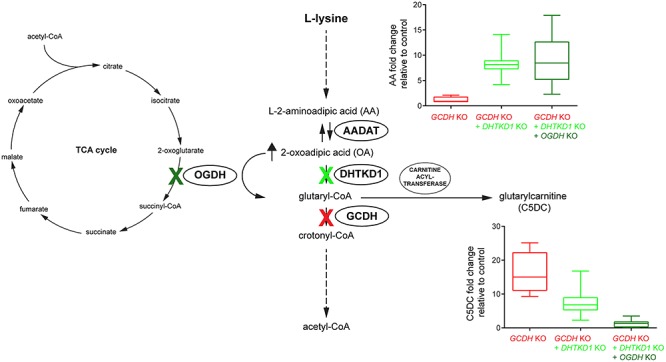
Schematic representation of the l-lysine degradation pathway with a summary of the results. Knockout of GCDH in HEK-293 cells leads to increased levels of glutaryl-CoA and its metabolite C5DC. This condition mimics the biochemical state occurring in GA1. A substrate reduction therapy for treatment of GA1 by inhibition of DHTKD1 was evaluated by knocking out DHTKD1 in GCDH KO cells. Knocking out DHTKD1 increased AA and decreased C5DC levels by ~2-fold. Knockout of OGDH in GCDH/DHTKD1 double KO cells further decreased C5DC to control levels. High concentrations of OA (and AA) such as those that occur after DHTKD1 inhibition or KO stimulate OGDHc activity leading to near normal pathway flux.
We also investigated the composition of the OADHc complex by IP of DHTKD1 from mouse liver and kidney, and human HEK-293 cells, as well as in situ PLA in HEK-293 cells. We show that DHTKD1 interacts with DLST and DLD, which corroborates the previously reported in vitro assembly of a functional OADHc with DHTKD1, DLST and DLD (9). Interestingly, we show that DHTKD1 also interacts with OGDH indicating that OGDH and DHTKD1 not only exist as isolated OGDHc and OADHc, but also as a hybrid complex. Further functional evidence for the existence of such a hybrid was obtained by in vitro reconstitution of a functional complex.
As predicted based on the human and mouse genetics data, knocking out DHTKD1 increased cellular AA levels. In contrast, knocking out OGDH did not change cellular AA levels confirming that DHTKD1 is relatively specific for OA, whereas OGDH is more promiscuous and can accept not only OG, but also OA. Although OGDH is active with OA, DHTKD1 deficiency leads to increased levels of AA and OA, which indicates that higher substrate levels are necessary for OGDHc to maintain adequate pathway flux. This nicely recapitulates the 49-fold preference in catalytic efficiency of reconstituted OADHc with the OA substrate over OG (9). High levels of pathway intermediates may be disadvantageous as they causes increased urinary loss of these metabolites and can limit overall flux by inhibiting upstream enzymes in the pathway.
We furthermore show that AA levels are higher in GCDH/DHTKD1 double KO cells when compared with DHTKD1 single KO cells (Supplementary Material, Fig. S2F). A similar phenomenon was observed in the Gcdh KO mouse. Gcdh KO mice had an increase in plasma AA and urine OA, which appeared independent of the Dhtkd1 genotype (Fig. 1). This increase in AA and OA can be attributed to product inhibition of OGDHc and OADHc by glutaryl-CoA (9), which is mediated via the shared E2 component (DLST) (38). Interestingly, affinity chromatography, co-precipitation and protein complementation assays have shown that GCDH interacts with DLST (39). The lipoyl group of DLST undergoes reductive glutarylation by E1, TPP and 2-oxoadipate; the glutaryl group is subsequently transferred to CoA in the DLST active center generating glutaryl-CoA. Close proximity of DLST and GCDH allows efficient channeling of glutaryl-CoA to GCDH for oxidative decarboxylation into crotonyl-CoA and as such limit the product inhibition of DLST. The presence of DHTKD1 and OGDH, two enzymes with overlapping and specific substrate specificities, in a single complex with the shared E2 and E3 components, and possibly GCDH increases the complexity of the enzyme complex, but could offer additional possibilities to coordinate metabolic flux in the TCA cycle and the lysine degradation pathway.
In conclusion, our study has identified both physical and functional interactions between DHTKD1 and OGDH. We show that OGDH and DHTKD1 can exist as a novel hybrid between OGDHc and OADHc in vivo and display substrate overlap in the HEK-293 cell system. The loss of DHTKD1 in GCDH-deficient cells does not prevent the accumulation of GCDH substrates because of the activity of OGDH driven by the elevated cellular levels of OA. This close functional relationship between DHTKD1 and OGDH limits the therapeutic potential of DHTKD1 inhibition for the treatment of GA1. It is, however, conceivable that DHTKD1 inhibition can limit the production of GCDH substrates under the conditions of increased lysine degradation, such as catabolic episodes. This option should be addressed in future studies.
Materials and Methods
Mouse strains
B6.129S4-Gcdhtm1Dmk/Mmnc mice (40) were provided by Dr Matthew Hirschey (Duke Molecular Physiology Institute, Duke University Medical Center, Durham, NC) after the approval of the Mutant Mouse Resource and Research Centers. All animal experiments were approved by the Institutional Animal Care and Use Committee of the Icahn School of Medicine at Mount Sinai (#2016–0490) and comply with the National Institutes of Health guide for the care and use of laboratory animals (NIH Publications No. 8023, revised 1978).
Cell lines
HEK-293 cells (ATCC, Cat#CRL-1573; RRID: CVCL_0045) were cultured in Dulbecco's modified Eagle medium (DMEM) with 4.5 g/L glucose, 584 mg/L l-glutamine and 110 mg/L sodium pyruvate, supplemented with 10% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin, in a humidified atmosphere of 5% CO2 at 37°C.
Animal experiments
B6.129S4-Gcdhtm1Dmk/Mmnc mice (40) were fully backcrossed to C57BL/6NJ (homozygous for WT Nnt). DBA/2NHsd females were crossed with C57BL/6NCrl. The resulting B6D2F1 was crossed again with C57BL/6NCrl. The Dhtkd1D2/B6 mice from these litters were crossed with Gcdh+/− mice in order to obtain double heterozygous (Gcdh+/−Dhtkd1D2/B6) mice. The Gcdh+/−Dhtkd1D2/B6 mice were then intercrossed to generate an experimental cohort of B6D2F2 mice (with 87.5% B6). Urine of individual mice was collected on multiple days and pooled in order to obtain sufficient sample volume. Mice were euthanized in a random fed state (afternoon) at 10 weeks of age using CO2. Blood from the inferior vena cava was collected for EDTA plasma isolation, and organs were rapidly excised and snap frozen in liquid nitrogen and subsequently stored at −80°C.
Generation of CRISPR-Cas9 KO cell lines
The generation of gene KO cell lines using the CRISPR-Cas9 genome editing technique was performed essentially as described (41), with minor modifications (42). For each gene, at least two different guides were chosen and cloned into the pSpCas9(BB)-2A-GFP vector. Following plasmid purification, HEK-293 cells were transfected using Lipofectamine 2000. Forty-eight hours after transfection, cells with GFP signal were sorted as single cells into 96-well plates by fluorescence-activated cell sorting analysis. After expansion, the clonal cell lines were characterized by DNA sequencing, immunoblotting and functional analysis essentially as described by Violante et al. (42). Guide and primer sequences are available upon request. We were not able to resolve the exact mutation for all used clonal cell lines, but all mutations were demonstrated to be deleterious through immunoblotting and functional assays.
Immunoblotting
Cells were lysed in radioimmunoprecipitation assay buffer supplemented with protease inhibitors cocktail (11836170001, Roche), centrifuged 10 min at 12 000 × g, 4°C and total protein determined by the bicinchoninic acid assay (BCA) method (23227, Thermo Fisher Scientific). Proteins were separated on a Bolt™ 4–12% Bis-Tris Plus Gel (Thermo Fisher Scientific), blotted onto a nitrocellulose membrane (926–31092, LI-COR) and detected using the following primary antibodies: DHTKD1 (GTX32561, Genetex), OGDH (66285–1-Ig, Proteintech), GCDH (gift of Michael Woontner, Children’s Hospital Colorado) and citrate synthase [GT1761] (GTX628143, Genetex) or [N2C3] (GTX110624; RRID:AB_1950045, Genetex). Proteins were visualized using IRDye 800CW or IRDye 680RD secondary antibodies (LI-COR, 926–32210; RRID:AB_621842, 926–68070; RRID:AB_10956588, 926–32211; RRID:AB_621843, and 926–68071; RRID:AB_10956166) in a Odyssey CLx Imager (LI-COR) with Image Studio Lite software (version 5.2, LI-COR). Equal loading was checked by Ponceau S (161470100, Acros Organics) staining and the citrate synthase signal.
Metabolite analyses
Plasma acylcarnitines, plasma amino acids and urine organic acids were measured by the Mount Sinai Biochemical Genetic Testing Laboratory (now Sema4). Urine organic acids were quantified using a standard curve and pentadecanoic acid as internal standard.
For quantification of AA and glutarylcarnitine (C5DC) in HEK-293 KO, the cell lines were incubated in supplemented DMEM with 0.4 mM l-carnitine (C0158, Sigma) for 24 h. Cells were collected, washed in PBS, flash-frozen in liquid nitrogen and stored at −80°C until further use. Samples were sonicated in ice-cold demineralized water and immediately thereafter deproteinized by adding ice-cold acetonitrile (34967-1 L, Honeywell) up to 80% (v/v). After 10 min centrifugation at maximum speed (4°C), the supernatant was dried under nitrogen and the residue was dissolved in 75 μl of demineralized water. Acylcarnitine internal standards were obtained from Cambridge Isotope Laboratories (Andover, MA) (NSK-B-1, containing 2H9-carnitine (d9-C0), 2H3-acetylcarnitine (d3-C2), 2H3-propionylcarnitine (d3-C3), 2H3-butyrylcarnitine (d3-C4), 2H9-isovalerylcarnitine (d9-C5), 2H3-octanoylcarnitine (d3-C8), 2H9-myristoylcarnitine (d9-C14) and 2H3-palmitoylcarnitine (d3-C16) and dissolved in 40 ml liquid chromatography–mass spectrometry (LC-MS)-grade methanol (1060352500, Millipore-Sigma). QCs and samples were prepared by mixing 20 μl of each with 100 μl of internal standard solution. Samples were centrifuged at 10 000 rpm for 10 min. Twenty microliters of the supernatant was transferred to a 96-well microplate and evaporated under a stream of nitrogen. Butylation was performed by adding 50 μl of 3 N n-butanol-HCl (1–2010007-200, REGIS Technologies) at 65°C for 15 min. After drying under nitrogen, the samples were reconstituted in 200 μl of methanol:water (8:2 v/v) and injected into the LC-MS/MS system. Amino acids were measured by LC-MS/MS as described (43). Protein pellets were dissolved in 25 mM KOH, 0.1% Triton X-100 and the protein amount determined by the BCA method for subsequent normalization of the amino acid and acylcarnitines levels.
Immunoprecipitation
Frozen mouse tissues (liver and kidney) were homogenized in ice-cold lysis buffer (50 mM MOPS, pH 7.4, 0.1% Triton X-100: ~20 mg per 1 ml lysis buffer) using a TissueLyser II apparatus (Qiagen). HEK-293 cell pellets were sonicated in ice-cold lysis buffer. Protein A or G magnetic Surebeads (161–4013 and 161–4021, Bio-Rad) were used for IPs depending on the affinity for the specific antibody. Antibodies were incubated with appropriate magnetic beads for 10 min at RT and beads washed to remove unbound antibody. Lysates were centrifuged at 1000 × g, 10 min at 4°C, and pre-cleared with an unrelated antibody of the same isotype as the antibody used for IP. Pre-cleared lysates were incubated with 5 μg of the specific IP antibody bound beads or a mock control antibody for 2 h at 4°C under gentle rotation. Immunoprecipitates were washed in PBS + 0.1% Tween 20, resuspended in 2× SDS-PAGE loading buffer (Bolt LDS sample buffer and sample reducing agent), boiled for 10 min at 70°C and analyzed by western blot. Anti-DHTKD1 (GTX32561, Genetex), anti-lipoic acid (437695; RRID:AB_212120, Calbiochem), anti-GCDH (gift of Michael Woontner, Children’s Hospital Colorado), anti-DLD (G-2) (sc-365 977; RRID:AB_10917587, Santa Cruz), anti-OGDH (GTX33374, Genetex), anti-OGDH (66285–1-lg, Proteintech), anti-DLST (ab110306; RRID:AB_10862702, Abcam) and mock anti-ABCD3 (TA310030, Origene) or anti-DCPS (ab57314; RRID:AB_941255, Abcam) antibodies were used for IP or western blot as indicated. Total protein staining was obtained using the REVERT total protein stain kit (926–11 010, LI-COR) and Quick Western Kit—IRDye® 680RD (926–68100, LI-COR) was used to avoid detection of the IP antibody, when appropriate.
Mass spectrometry analysis
Forty microliters of co-IP eluted samples were loaded on 4–12% Bolt gel, stained with Coomassie SimplyBlue SafeStain (LC6060, ThermoFisher) and submitted to the Microchemistry and Proteomics Core Facility at Memorial Sloan Kettering Cancer Center for in-gel tryptic digestion and protein identification by mass spectrometry. Scaffold 4.8.4 (Proteome Software Inc., Portland, OR) was used to validate MS/MS-based peptide and protein identifications. Peptides were identified from MS/MS spectra by searching against a library of known/likely interactors of DHTKD1.
Overexpression in HEK-293 cells
A human cDNA clone for DHTKD1 (NM_018706) was obtained from Origene (RC200967), encoding a C-terminal tagged DHTKD1-Myc-DDK in the pCMV6 plasmid. One rare variant (L308) was detected in the DHTKD1 cDNA and was therefore replaced by the common variant R308 by site directed mutagenesis using the following forward and reverse primers: 5′-GGC AGC AGT CTC GCC AAG ACG GCG-3′ and 5′-CGC CGT CTT GGC GAG ACT GCT GCC-3′ (mutagenic nucleotides in bold). In order to obtain the untagged protein, a STOP codon was introduced before the C-terminal tags by site directed mutagenesis using as template pCMV6-DHTKD1(R308)-Myc-DDK and the following forward and reverse primers: 5′-CCA AGA CCT TCG CTT AAC GTA CGC GGC CGC-3′ and 5′-GCG GCC GCG TAC GTT AAG CGA AGG TCT TGG-3′ (mutagenic nucleotides in bold). However, no impact on activity was detected for the presence of the variant or the C-terminal tag (Supplementary Material, Fig. S3A).
Human cDNA clones for OGDH (BC004964), DLST (BC001922), DLD (BC018696) and MRPS36 (KGD4) (BC015966) were provided by TransOMIC technologies (Huntsville, AL). The genes were amplified by using High Fidelity PrimeSTAR GXL Polymerase (R050A, Takara) and cloned into the AsiSI(SgfI)/MluI cloning sites of the pCMV6-Entry vector (PS100001, Origene). Each construct was analyzed by restriction digestion and sequencing. A human cDNA clone for OGDHL (NM_018245) was obtained from Origene (RC205225), encoding a C-terminal tagged OGDHL-Myc-DDK in the pCMV6 plasmid. Two rare variants (L511 and N573) were detected on OGDHL cDNA and were therefore replaced by the common variant P511 and D573 by site directed mutagenesis using the following forward and reverse primers: 5′-CCC ATG TTC ACC CAG CCG CTC ATG TAC AAG CAG-3′, 5′-CTG CTT GTA CAT GAG CGG CTG GGT GAA CAT GGG-3′ and 5′-ATA AAG CAC TGG TTG GAC TCC CCC TGG CCT G-3′, 5′-CAG GCC AGG GGG AGT CCA ACC AGT GCT TTA T-3′, respectively (mutagenic nucleotides in bold). Cell transfection grade plasmid DNA was obtained for every plasmid construct with NucleoBond Xtra Midi Plus EF DNA purification kit (740422.10, Macherey-Nagel).
HEK-293 cells were transfected with different combinations of vectors performing a total of 6.9 μg/T-25 flask using Lipofectamine 2000 as transfection agent and Opti-MEM I Reduced Serum (31985070, Thermo Fisher Scientific). After 24 h, cells were harvested, washed and flash-frozen in liquid nitrogen. Pellets were stored at −80°C until further use for immunoblot and/or enzyme activity analysis.
OGDHc and OADHc activity
HEK-293 cells were lysed in 50 mM MOPS, pH 7.4, 0.1% Triton X-100, sonicated and centrifuged for 10 min at 1000 × g, 4°C. The supernatant (100 μg total protein) was mixed with assay buffer (final concentration: 50 mM MOPS, pH 7.4, 0.2 mM MgCl2, 0.01 mM CaCl2, 0.3 mM TPP [C8754, Sigma], 0.12 mM Coenzyme-A [C3144, Sigma], 2 mM NAD [N6522, Sigma] and 2.6 mM β-mercaptoethanol). The reaction was started by the addition of 1 mM substrates: OG (K1128, Sigma) and OA (75447, Sigma). The activity of the OGDHc and OADHc was followed by measuring the NADH production at 340 nm at 30°C and steady-state velocities were calculated from the linear portion of the time curve. The activity of OGDHc from porcine heart (Sigma K1502) was measured as described above using 1 mM OG or OA as substrate.
Immunohistology and in situ PLA analysis
Archival human renal tissue specimens (n = 3) from normal portions of nephrectomy specimens of localized renal cell carcinoma were studied according to the national ethical guidelines of the ‘Code for Proper Secondary Use of Human Tissue’ developed by Federation of Medical Societies in the Netherlands (http://www.federa.org). The 4-μm-thick sections were cut from formalin-fixed, paraffin-embedded tissue and mounted on glass slides. After deparafinization and rehydration, heat-induced epitope retrieval (HIER) was performed using 10 mM Tris-HCl and 1 mM EDTA, pH 9.0, in a pressure-cooker for 20 min. After washing and blocking with Tris-buffered saline containing 0.01% Tween20 (TBST) and 5% normal goat serum (X0907 DAKO; Agilent, Santa Clara, CA), sections were incubated overnight at 4°C with either rabbit IgG anti-OGDH (GTX33374; 1:1000) or rabbit IgG anti-DHTKD1 (GTX32561; 1:1000) in TBST plus 0.1% BSA (TBSTBSA). After washing in TBST, sections were incubated for 30 min at RT with intestinal alkaline phosphatase-conjugated poly-AP goat anti-rabbit IgG (KLINHK331-5 K; BrightVision; Immunologic, Klinipath, Duiven, The Netherlands). AP activity was detected using PermaBlue Plus/AP chromogen (K058, Diagnostic Biosystems, Pleasonton, CA) in presence of 0.2 mM levamisole to inhibit endogenous non-intestinal alkaline phosphatase activity. After washing, a second HIER was applied to denature and remove all antibodies from the first staining sequence while leaving the precipitated chromogen unchanged (44). Subsequently, sections were incubated for 2 h at RT with mouse IgG2b anti-OGDH (pt66285–1-Ig; 1:500), mouse IgG1 anti-DLST (9F4BD5; 1:500), mouse IgG1 anti-DLD (G2; 1/500), or mouse IgG1 anti-TOM20 (MABT166; 1:1000), washed and incubated for 30 min at RT with poly-AP goat anti-mouse IgG (BrightVision). AP activity was detected using PermaRed-AP AutoPlus chromogen (K057, Diagnostic Biosystems) in presence of 0.2 mM levamisole. After washing, sections were mounted in VectaMount (H-5000, Burlingame, CA). In addition to sequential brightfield immunohistology, single and double indirect immunofluorescences were performed on renal tissue sections. Primary antibodies were applied as indicated above, secondary antibodies were Alexa Fluor 488-conjugated goat anti-rabbit IgG (A-11034 Invitrogen, ThermoFisher Scientific, Waltham, MA), Alexa Fluor 568-conjugated goat anti-mouse IgG2b (A-21144 Invitrogen) and Alexa Fluor 594-conjugated goat anti-mouse IgG1 (A-21125 Invitrogen), diluted 1:200 in TBSTBSA, containing 1 μg/ml Hoechst 33342 (H3570 Invitrogen). After washing, sections were mounted in ProLong Gold antifade reagent (P10144 Invitrogen). HEK-293 cells cultured on type I collagen-coated cover glass were fixed in 4% paraformaldehyde (in PBS), permeabilized for 20 min with 0.3% Triton X-100 (in PBS) and blocked for 60 min with 5% normal goat serum, 0.01% Tween20 (in PBS). Single and double immunofluorescences were performed as described above, however with secondary antibodies diluted 1:500.
In situ PLA (DUO92101; Sigma) was performed according to the manufacturer’s instructions. Primary antibodies were GTX33374 or GTX32561, in combination with 66285–1-Ig, ab110306, sc-365 977 or control MABT166; secondary antibodies conjugated with oligonucleotides were DuoLink anti-rabbit MINUS and DuoLink anti-mouse PLUS; the DuoLink Red detection kit was applied; for mounting either VectaShield with DAPI (H-1200; Vector Laboratories, Burlingame, CA) or ProLong Gold with DAPI (P36941) were used.
Analysis of multiple non-fluorescent chromogen stained slides was performed with spectral imaging using brightfield microscopy (DM5500B; Leica Microsystems, Wetzlar, Germany) with an HC PLAN APO 20×/0.70 objective. Multispectral data sets were acquired using a Nuance N-MSI-420-20 camera with Nuance 3.0.2 software (Perkin Elmer, Hopkinton, MA) from 420 to 720 nm at intervals of 10 nm. Spectral libraries for each chromogen were obtained from single-stained sections and were used to unmix the multiple staining patterns. Nuance software was used to display intensity heat maps for single channels and to construct composite images applying color universal design. Epifluorescence imaging was performed with PL FLUOTAR 40×/1.00–0.50 and HCX PL APO 63×/1.40–0.60 oil immersion objectives. Images were recorded using a Leica DFC365 FX camera and LASX imaging software (Leica Microsystems). For PLA imaging, z-stacks were recorded, and subsequently deconvolved and analyzed with Huygens Professional 19.03 software (Scientific Volume Imaging B.V., Hilversum, The Netherlands).
NADH production by in vitro assembled hybrid complexes
Expression and purification of recombinant human OGDH, DHTKD1, DLST and DLD were performed as described earlier (9,45). For hybrid complex assembly, the OGDH by itself or in the presence of different amounts of DHTKD1 (the total amount of OGDH+DHTKD1 = 0.15 mg) were incubated in 0.15 ml of the reaction mixture with DLST (0.30 mg) and DLD (0.75 mg) at E1/E2/E3 mass ratio (mg/mg/mg) of 1:2:5 (9). The total amount of the E1 proteins was kept constant (0.15 mg), while varying the mass ratios of OGDH:DHTKD1 in assembly with DLST and DLD: (1) OGDH by itself; (2) OGDH:DHTKD1 = 2:1; (3) OGDH:DHTKD1 = 1:1; (4) OGDH:DHTKD1 = 1:2; (5) DHTKD1 by itself. After 40 min of incubation at 25°C, a 0.01 ml aliquot of the reaction mixture containing 0.01 mg of total E1 and the corresponding amounts of E2 and E3 was withdrawn to start the reaction. The reaction medium contained the following in 1.0 ml: 0.1 M Tris.HCl (pH 7.5), 0.50 mM TPP, 2.0 mM MgCl2, 2.0 mM DTT and 2.5 mM NAD+. The reaction was initiated by addition of OG (2.0 mM) and CoA (0.30 mM) after 1 min of equilibration at 37°C. Steady-state velocities (slope at 340/min) were taken from the linear portion of the progress curve (y axis) and were plotted for each assembly analyzed (x axis).
Quantification and statistical analysis
Data are displayed as the mean ± standard deviation (SD). One-way analysis of variance with Bonferroni’s multiple comparison test and two-way analysis of variance as used for statistical analysis using GraphPad Prism 7 software (GraphPad, San Diego, CA). Significance is indicated as follows: *P < 0.05; **P < 0.01; ***P < 0.001 and ****P < 0.0001.
Author contributions
Conceptualization, S.M.H., R.J.D.; Methodology, F.J., J.A., R.C.H., R.S., C.Y., S.M.H.; Investigation, J.L., T.D., X.Z., N.S.N., S.M.H.; Writing—Original Draft, J.L.; Writing—Review and Editing, S.M.H.; Funding Acquisition, S.M.H., R.J.D.
Supplementary Material
Acknowledgements
We thank Dr Jonna Westover, University of Utah, Dr Frank Frerman, University of Colorado and Dr Michael Woontner from the Children’s Hospital Colorado for providing the anti-GCDH antibody; Dr Matthew Hirschey for providing the Gcdh KO mice. We thank Ethellyn Panta, Hongjie Chen, Jianqiang Zeng, Purvika Patel and Xing Ni for their excellent technical assistance with the clinical biochemical analyses, and Amelia Gabler for her excellent technical assistance with the proteomic analysis.
Conflict of Interest statement
None declared.
Funding
The Eunice Kennedy Shriver National Institute of Child Health and Human Development of the National Institutes of Health under award number R03HD092878 (to S.M.H.) and R21HD088775 (to S.M.H. and R.J.D.) and by grant UL1TR001433 from the National Center for Advancing Translational Sciences, National Institutes of Health, as well as National Institutes of Health, grant number R15GM116077 (to F.J.).
References
- 1. Pena I.A., Marques L.A., Laranjeira A.B., Yunes J.A., Eberlin M.N., MacKenzie A. and Arruda P. (2017) Mouse lysine catabolism to aminoadipate occurs primarily through the saccharopine pathway; implications for pyridoxine dependent epilepsy (PDE). Biochim. Biophys. Acta, 1863, 121–128. [DOI] [PubMed] [Google Scholar]
- 2. Struys E.A. and Jakobs C. (2010) Metabolism of lysine in alpha-aminoadipic semialdehyde dehydrogenase-deficient fibroblasts: evidence for an alternative pathway of pipecolic acid formation. FEBS Lett., 584, 181–186. [DOI] [PubMed] [Google Scholar]
- 3. Struys E.A., Jansen E.E. and Salomons G.S. (2014) Human pyrroline-5-carboxylate reductase (PYCR1) acts on delta(1)-piperideine-6-carboxylate generating L-pipecolic acid. J. Inherit. Metab. Dis., 37, 327–332. [DOI] [PubMed] [Google Scholar]
- 4. Crowther L.M., Mathis D., Poms M. and Plecko B. (2019) New insights into human lysine degradation pathways with relevance to pyridoxine-dependent epilepsy due to antiquitin deficiency. J. Inherit. Metab. Dis., 42, 620–628. [DOI] [PubMed] [Google Scholar]
- 5. Danhauser K., Sauer S.W., Haack T.B., Wieland T., Staufner C., Graf E., Zschocke J., Strom T.M., Traub T., Okun J.G. et al. (2012) DHTKD1 mutations cause 2-aminoadipic and 2-oxoadipic aciduria. Am. J. Hum. Genet., 91, 1082–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hagen J., Brinke H., Wanders R.J., Knegt A.C., Oussoren E., Hoogeboom A.J., Ruijter G.J., Becker D., Schwab K.O., Franke I. et al. (2015) Genetic basis of alpha-aminoadipic and alpha-ketoadipic aciduria. J. Inherit. Metab. Dis., 38, 873–879. [DOI] [PubMed] [Google Scholar]
- 7. Stiles A.R., Venturoni L., Mucci G., Elbalalesy N., Woontner M., Goodman S. and Abdenur J.E. (2016) New cases of DHTKD1 mutations in patients with 2-ketoadipic aciduria. JIMD Rep., 25, 15–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bunik V.I. and Degtyarev D. (2008) Structure-function relationships in the 2-oxo acid dehydrogenase family: substrate-specific signatures and functional predictions for the 2-oxoglutarate dehydrogenase-like proteins. Proteins, 71, 874–890. [DOI] [PubMed] [Google Scholar]
- 9. Nemeria N.S., Gerfen G., Nareddy P.R., Yang L., Zhang X., Szostak M. and Jordan F. (2018) The mitochondrial 2-oxoadipate and 2-oxoglutarate dehydrogenase complexes share their E2 and E3 components for their function and both generate reactive oxygen species. Free Radic. Biol. Med., 115, 136–145. [DOI] [PubMed] [Google Scholar]
- 10. Tsepkova P.M., Artiukhov A.V., Boyko A.I., Aleshin V.A., Mkrtchyan G.V., Zvyagintseva M.A., Ryabov S.I., Ksenofontov A.L., Baratova L.A., Graf A.V. et al. (2017) Thiamine induces long-term changes in amino acid profiles and activities of 2-oxoglutarate and 2-oxoadipate dehydrogenases in rat brain. Biochemistry (Mosc.), 82, 723–736. [DOI] [PubMed] [Google Scholar]
- 11. Mills P.B., Struys E., Jakobs C., Plecko B., Baxter P., Baumgartner M., Willemsen M.A., Omran H., Tacke U., Uhlenberg B. et al. (2006) Mutations in antiquitin in individuals with pyridoxine-dependent seizures. Nat. Med., 12, 307–309. [DOI] [PubMed] [Google Scholar]
- 12. Karnebeek C.D., Tiebout S.A., Niermeijer J., Poll-The B.T., Ghani A., Coughlin C.R. 2nd, Van Hove J.L., Richter J.W., Christen H.J., Gallagher R. et al. (2016) Pyridoxine-dependent epilepsy: an expanding clinical spectrum. Pediatr. Neurol., 59, 6–12. [DOI] [PubMed] [Google Scholar]
- 13. Boy N., Garbade S.F., Heringer J., Seitz A., Kolker S. and Harting I. (2019) Patterns, evolution, and severity of striatal injury in insidious- versus acute-onset glutaric aciduria type 1. J. Inherit. Metab. Dis., 42, 117–127. [DOI] [PubMed] [Google Scholar]
- 14. Goodman S.I., Stein D.E., Schlesinger S., Christensen E., Schwartz M., Greenberg C.R. and Elpeleg O.N. (1998) Glutaryl-CoA dehydrogenase mutations in glutaric acidemia (type I): review and report of thirty novel mutations. Hum. Mutat., 12, 141–144. [DOI] [PubMed] [Google Scholar]
- 15. Greenberg C.R., Reimer D., Singal R., Triggs-Raine B., Chudley A.E., Dilling L.A., Philipps S., Haworth J.C., Seargeant L.E. and Goodman S.I. (1995) A G-to-T transversion at the +5 position of intron 1 in the glutaryl CoA dehydrogenase gene is associated with the island lake variant of glutaric acidemia type I. Hum. Mol. Genet., 4, 493–495. [DOI] [PubMed] [Google Scholar]
- 16. Harting I., Neumaier-Probst E., Seitz A., Maier E.M., Assmann B., Baric I., Troncoso M., Muhlhausen C., Zschocke J., Boy N.P. et al. (2009) Dynamic changes of striatal and extrastriatal abnormalities in glutaric aciduria type I. Brain, 132, 1764–1782. [DOI] [PubMed] [Google Scholar]
- 17. Dancis J., Hutzler J., Ampola M.G., Shih V.E., Gelderen H.H., Kirby L.T. and Woody N.C. (1983) The prognosis of hyperlysinemia: an interim report. Am. J. Hum. Genet., 35, 438–442. [PMC free article] [PubMed] [Google Scholar]
- 18. Houten S.M., Te Brinke H., Denis S., Ruiter J.P., Knegt A.C., Klerk J.B., Augoustides-Savvopoulou P., Haberle J., Baumgartner M.R., Coskun T. et al. (2013) Genetic basis of hyperlysinemia. Orphanet J. Rare Dis., 8, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Goodman S.I. and Duran M. (2014) In Blau N., Duran M., Gibson K.M. and Dionisi-Vici C. (eds), Physician's Guide to the Diagnosis, Treatment, and Follow-Up of Inherited Metabolic Diseases. Springer, Berlin, pp. 691–705. [Google Scholar]
- 20. Biagosch C., Ediga R.D., Hensler S.V., Faerberboeck M., Kuehn R., Wurst W., Meitinger T., Kolker S., Sauer S. and Prokisch H. (2017) Elevated glutaric acid levels in Dhtkd1−/Gcdh- double knockout mice challenge our current understanding of lysine metabolism. Biochim. Biophys. Acta, 1863, 2220–2228. [DOI] [PubMed] [Google Scholar]
- 21. Nemeria N.S., Gerfen G., Yang L., Zhang X. and Jordan F. (2018) Evidence for functional and regulatory cross-talk between the tricarboxylic acid cycle 2-oxoglutarate dehydrogenase complex and 2-oxoadipate dehydrogenase on the l-lysine, l-hydroxylysine and l-tryptophan degradation pathways from studies in vitro. Biochim. Biophys. Acta Bioenerg., 1859, 932–939. [DOI] [PubMed] [Google Scholar]
- 22. Chick J.M., Munger S.C., Simecek P., Huttlin E.L., Choi K., Gatti D.M., Raghupathy N., Svenson K.L., Churchill G.A. and Gygi S.P. (2016) Defining the consequences of genetic variation on a proteome-wide scale. Nature, 534, 500–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wu Y., Williams E.G., Dubuis S., Mottis A., Jovaisaite V., Houten S.M., Argmann C.A., Faridi P., Wolski W., Kutalik Z. et al. (2014) Multilayered genetic and omics dissection of mitochondrial activity in a mouse reference population. Cell, 158, 1415–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Leandro J., Violante S., Argmann C.A., Hagen J., Dodatko T., Bender A., Zhang W., Williams E.G., Bachmann A.M., Auwerx J. et al. (2019) Mild inborn errors of metabolism in commonly used inbred mouse strains. Mol. Genet. Metab., 126, 388–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sauer S.W., Opp S., Komatsuzaki S., Blank A.E., Mittelbronn M., Burgard P., Koeller D.M., Okun J.G. and Kolker S. (2015) Multifactorial modulation of susceptibility to l-lysine in an animal model of glutaric aciduria type I. Biochim. Biophys. Acta, 1852, 768–777. [DOI] [PubMed] [Google Scholar]
- 26. Bunik V., Westphal A.H. and Kok A. (2000) Kinetic properties of the 2-oxoglutarate dehydrogenase complex from Azotobacter vinelandii evidence for the formation of a precatalytic complex with 2-oxoglutarate. Eur. J. Biochem., 267, 3583–3591. [DOI] [PubMed] [Google Scholar]
- 27. Bunik V.I. and Pavlova O.G. (1997) Inactivation of alpha-ketoglutarate dehydrogenase during its enzymatic reaction. Biochemistry (Mosc.), 62, 973–982. [PubMed] [Google Scholar]
- 28. Bunik V.I. and Pavlova O.G. (1997) Inhibition of pigeon breast muscle alpha-ketoglutarate dehydrogenase by structural analogs of alpha-ketoglutarate. Biochemistry (Mosc.), 62, 1012–1020. [PubMed] [Google Scholar]
- 29. Hirashima M., Hayakawa T. and Koike M. (1967) Mammalian alpha-keto acid dehydrogenase complexes. II. An improved procedure for the preparation of 2-oxoglutarate dehydrogenase complex from pig heart muscle. J. Biol. Chem., 242, 902–907. [PubMed] [Google Scholar]
- 30. Sauer S.W., Opp S., Hoffmann G.F., Koeller D.M., Okun J.G. and Kolker S. (2011) Therapeutic modulation of cerebral L-lysine metabolism in a mouse model for glutaric aciduria type I. Brain, 134, 157–170. [DOI] [PubMed] [Google Scholar]
- 31. Bunik V., Kaehne T., Degtyarev D., Shcherbakova T. and Reiser G. (2008) Novel isoenzyme of 2-oxoglutarate dehydrogenase is identified in brain, but not in heart. FEBS J., 275, 4990–5006. [DOI] [PubMed] [Google Scholar]
- 32. Heublein M., Burguillos M.A., Vogtle F.N., Teixeira P.F., Imhof A., Meisinger C. and Ott M. (2014) The novel component Kgd4 recruits the E3 subunit to the mitochondrial alpha-ketoglutarate dehydrogenase. Mol. Biol. Cell, 25, 3342–3349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fischer M.H. and Brown R.R. (1980) Tryptophan and lysine metabolism in alpha-aminoadipic aciduria. Am. J. Med. Genet., 5, 35–41. [DOI] [PubMed] [Google Scholar]
- 34. Fischer M.H., Gerritsen T. and Opitz J.M. (1974) Alpha-aminoadipic aciduria, a non-deleterious inborn metabolic defect. Humangenetik, 24, 265–270. [DOI] [PubMed] [Google Scholar]
- 35. Sheu K.F. and Blass J.P. (1999) The alpha-ketoglutarate dehydrogenase complex. Ann. NY Acad. Sci., 893, 61–78. [DOI] [PubMed] [Google Scholar]
- 36. Yeaman S.J. (1989) The 2-oxo acid dehydrogenase complexes: recent advances. Biochem. J., 257, 625–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Reed L.J. and Hackert M.L. (1990) Structure-function relationships in dihydrolipoamide acyltransferases. J. Biol. Chem., 265, 8971–8974. [PubMed] [Google Scholar]
- 38. Sauer S.W., Okun J.G., Schwab M.A., Crnic L.R., Hoffmann G.F., Goodman S.I., Koeller D.M. and Kolker S. (2005) Bioenergetics in glutaryl-coenzyme A dehydrogenase deficiency: a role for glutaryl-coenzyme a. J. Biol. Chem., 280, 21830–21836. [DOI] [PubMed] [Google Scholar]
- 39. Schmiesing J., Schluter H., Ullrich K., Braulke T. and Muhlhausen C. (2014) Interaction of glutaric aciduria type 1-related glutaryl-CoA dehydrogenase with mitochondrial matrix proteins. PLoS One, 9, e87715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Koeller D.M., Woontner M., Crnic L.S., Kleinschmidt-DeMasters B., Stephens J., Hunt E.L. and Goodman S.I. (2002) Biochemical, pathologic and behavioral analysis of a mouse model of glutaric acidemia type I. Hum. Mol. Genet., 11, 347–357. [DOI] [PubMed] [Google Scholar]
- 41. Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A. and Zhang F. (2013) Genome engineering using the CRISPR-Cas9 system. Nat. Protoc., 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Violante S., Achetib N., Roermund C.W.T., Hagen J., Dodatko T., Vaz F.M., Waterham H.R., Chen H., Baes M., Yu C. et al. (2019) Peroxisomes can oxidize medium- and long-chain fatty acids through a pathway involving ABCD3 and HSD17B4. FASEB J., 33, 4355–4364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Le A., Ng A., Kwan T., Cusmano-Ozog K. and Cowan T.M. (2014) A rapid, sensitive method for quantitative analysis of underivatized amino acids by liquid chromatography-tandem mass spectrometry (LC-MS/MS). J. Chromatogr. B Analyt. Technol. Biomed. Life Sci., 944, 166–174. [DOI] [PubMed] [Google Scholar]
- 44. Boer O.J., Meer J.J., Teeling P., Loos C.M., Idu M.M., Maldegem F., Aten J. and Wal A.C. (2010) Differential expression of interleukin-17 family cytokines in intact and complicated human atherosclerotic plaques. J. Pathol., 220, 499–508. [DOI] [PubMed] [Google Scholar]
- 45. Nemeria N.S., Ambrus A., Patel H., Gerfen G., Adam-Vizi V., Tretter L., Zhou J., Wang J. and Jordan F. (2014) Human 2-oxoglutarate dehydrogenase complex E1 component forms a thiamin-derived radical by aerobic oxidation of the enamine intermediate. J. Biol. Chem., 289, 29859–29873. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.