

Abstract
Pre-B cell leukemia factor 1 (PBX1) is an essential developmental transcription factor, mutations in which have recently been associated with CAKUTHED syndrome, characterized by multiple congenital defects including congenital heart disease (CHD). During analysis of a whole-exome-sequenced cohort of heterogeneous CHD patients, we identified a de novo missense variant, PBX1:c.551G>C p.R184P, in a patient with tetralogy of Fallot with absent pulmonary valve and extra-cardiac phenotypes. Functional analysis of this variant by creating a CRISPR-Cas9 gene-edited mouse model revealed multiple congenital anomalies. Congenital heart defects (persistent truncus arteriosus and ventricular septal defect), hypoplastic lungs, hypoplastic/ectopic kidneys, aplastic adrenal glands and spleen, as well as atretic trachea and palate defects were observed in the homozygous mutant embryos at multiple stages of development. We also observed developmental anomalies in a proportion of heterozygous embryos, suggestive of a dominant mode of inheritance. Analysis of gene expression and protein levels revealed that although Pbx1 transcripts are higher in homozygotes, amounts of PBX1 protein are significantly decreased. Here, we have presented the first functional model of a missense PBX1 variant and provided strong evidence that p.R184P is disease-causal. Our findings also expand the phenotypic spectrum associated with pathogenic PBX1 variants in both humans and mice.
Introduction
Congenital heart disease (CHD) affects approximately 1 in a 100 live births (1). These structural malformations of the heart that can affect the ventricles, atria, valves or the great vessels can vary in severity and require variable degrees of intervention. They may occur in isolation or as part of a broader syndrome comprising multiple extra-cardiac phenotypes. Sporadic incidence of CHD, in families with no history of heart defects, is higher than familial cases of CHD (2). Evidence that there is a genetic basis to CHD has been growing over decades of research; however, currently a clinically actionable genetic diagnosis is achieved for only 30% of cases (3). The clinical utility of a genetic diagnosis is highlighted by the vital role it plays in management of the patient’s phenotypic diagnosis and prognosis in cases of syndromic CHD (4), as well as calculating the recurrence risk. Currently, the number of genes implicated in CHD causality remains limited. Genes with the strongest support for causality are those that are recurrently mutated in CHD patients with robust functional evidence indicative of pathogenicity. The utilization of massively parallel sequencing techniques such as whole exome sequencing (WES) and whole genome sequencing has provided an opportunity for novel CHD gene discovery in an unbiased manner. Recent studies have put forward many genes with variants identified in CHD patients that have predicted-pathogenicity in silico but so far have not been verified functionally (3,5,6).
We recently analysed variants identified in a cohort of CHD patients who underwent WES (5). A variant of uncertain significance, PBX1:c.551G>C p.R184P, was identified in pre-B cell leukemia factor 1 (PBX1) in a patient with tetralogy of Fallot (TOF) and extra-cardiac phenotypes. PBX1 belongs to the TALE class of transcription factors of which there are three others (PBX2, PBX3 and PBX4). All four proteins share strong sequence homology within three main protein domains: PBC-A, PBC-B and the C-terminal homeodomain (7,8). They are highly expressed during embryonic development with Pbx1, Pbx2 and Pbx3 strongly expressed in the heart (9,10). Recently, pathogenic variants in PBX1 were implicated in the causality of a novel syndrome, congenital anomalies of kidney and urinary tract syndrome with or without hearing loss, abnormal ears or developmental delay (CAKUTHED, OMIM: 617641). This syndrome has high phenotypic variability; however, it is generally characterized by congenital anomalies of the urogenital system, craniofacial anomalies and developmental delay (11–15). Congenital heart defects have also been observed in some patients (11).
At the time of discovery, the contribution of PBX1-R184P towards disease causality was uncertain, and in vitro functional assessment was inconclusive. In order to elucidate the contribution of the PBX1 variant towards the phenotypes observed in our patient, we created a knock-in mouse model of the variant, PBX1-R184P, to functionally assess the impact of the variant during embryonic development. Homozygosity of the R184P variant was embryonic lethal, with embryos affected by multiple developmental anomalies including cardiac phenotypes similar to our patient. We also observed cardiac defects in a proportion of heterozygous embryos, indicating a dominant model of inheritance. Here, we describe the generation and comprehensive analysis of the first animal model of a PBX1 missense variant and provide functional evidence of the pathogenicity of the variant observed in our patient.
Results
Identification of a de novo missense PBX1 variant in a patient with CHD and extra-cardiac phenotypes
The proband is the youngest of six children of healthy parents (Fig. 1A). He has four healthy older brothers and one healthy older sister. He was born at a gestational age of 39 weeks. Echocardiography revealed that the proband had TOF with absent pulmonary valve. There was a single large ventricular septal defect (VSD) with anterior and rightward deviation of the infundibular septum and the aorta overriding the interventricular septum. There was moderate pulmonary annular hypoplasia measuring 12 mm and the pulmonary valve was noted to be absent with severe stenosis and regurgitation (peak gradient of 99 mmHg, median gradient of 67 mmHg). The pulmonary arteries were dilated with the right artery at 1.7 cm and the left pulmonary artery at 1.2 cm. A small patent foramen ovale (PFO) was observed during surgery. The right ventricle was mildly dilated and hypertrophied with reasonable systolic function. The proband underwent surgical correction 3 months after birth with RV-PA conduit insertion, PA plication, VSD repair and closure of PFO. Transthoracic echo 1 month after surgery revealed a small residual VSD with left-to-right blood flow, normal left ventricular systolic function and hypertrophied right ventricle. Electrocardiogram did not reveal any conduction defects.
Figure 1.
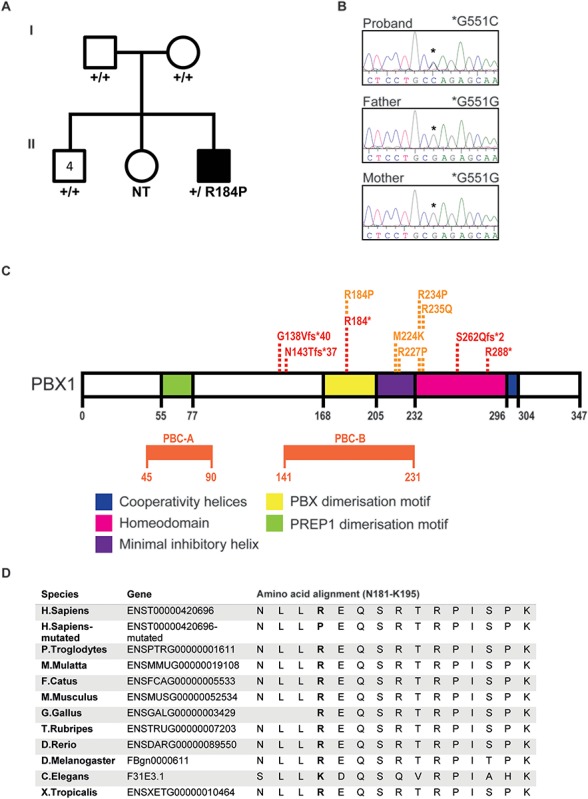
A novel de novo variant in PBX1 was identified in a patient with syndromic CHD by WES. (A) Pedigree of the family with the affected individual (black) carrying the de novo PBX1 variant PBX1:c.551G>C p.R184P. The patient presented with TOF with absent pulmonary valve and later also diagnosed with bronchiolitis, autism, hyperechogenic kidneys, skeletal (clinodactyly) and craniofacial defects (broad nasal tip, thick lips, prominent philtrum and anteverted ears) (B) De novo presence of the variant was confirmed by Sanger sequencing. The older sister’s DNA was not available for testing (NT). Asterisk represents the position of the nucleotide variation in the proband (c.551G>C) versus the reference allele in the parents (c.551G). (C) A schematic representation of the PBX1 protein indicating the functional domains; PREP1 dimerization domain (green), PBX dimerization domain (yellow), inhibitory helix (purple), homeodomain (pink) and cooperativity helices (Blue). The positions of the pathogenic loss-of-function (red) and missense (orange) variants that are currently reported in the literature are presented. (D) Amino acid sequence alignment of reference and mutated human PBX1 protein (residues 181–195) with selected eukaryotes shows that R184 is within a highly conserved region of the protein and that the arginine residue is highly conserved between species.
Assessment at 1 year of age placed the proband in the 25th percentile of body weight, 90th percentile of length and 50–90th percentile of head circumference. The proband was noted to suffer from recurrent bronchiolitis episodes during the first year of life. Renal ultrasound imaging of the proband revealed increased echogenicity of both kidneys. The proband was later diagnosed with autism and mild craniofacial (broad nasal tip, thick lips, prominent philtrum and anteverted ears), and skeletal phenotypes (clinodactyly) were also observed.
Preliminary cytogenetic tests (Multiplex Ligation-dependent Probe Amplification (MLPA) and whole genome Single Nucleotide Polymorphism (SNP) microarray) did not detect chromosomal or copy number abnormalities. WES was undertaken on the proband and the parents to potentially identify a monogenic cause of disease. No damaging variants in genes known to cause CHD were identified. All variants that segregated with disease were then considered (Supplementary Material, Table S1). A novel de novo missense variant (c.551G>C p.R184P) was identified in the proband within the PBX1 gene, which encodes a homeodomain-containing transcription factor (16). To verify the de novo status of the variant, the proband, parents and the four older brothers were Sanger sequenced (Fig. 1B). The variant was not detected in the parents or the siblings.
PBX1 contains three highly conserved regions; PBC-A, PBC-B and the homeodomain (Fig. 1C). The main functional domains of PBX1 are found in these conserved regions such as those motifs required for interaction with co-factors. R184P variant falls within the PBX1 dimerization motif where a pathogenic stop-gain variant was identified for CAKUTHED (Fig. 1C, Table 1 and (Supplementary Material, Table S2). PBX1:c. 551G>C, p.R184P is not present in healthy control individuals in the ExAC/gnomAD database. The arginine residue at position 184 is highly conserved in eukaryotes (Fig. 1D) and the substitution to a proline residue is predicted to be highly damaging towards protein function by multiple in silico predictors (Supplementary Material, Table S1).
Table 1.
Summary of reported patients with heterozygous, pathogenic, single gene, PBX1 mutations
| Missense | LOF | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Publication | Slavotinek, 2017 a | Eozenou, 2019 b | Riedhammer, 2017 c | Heidet, 2017 d | Slavotinek, 2017 a | Le Tanno, 2017 e | Heidet, 2017 d | |||||||||
| PBX1 amino acid position | R184P f | M224K | R227P | R234P | R235Q | R235Q | R235Q | G138Vfs*40 | N143Tfs*37 | R184* | S262Qfs*2 | R288* | 1.5 Mb deletion (only PBX1) | 0.276 MB deletion (only PBX1) | Deletion of one PBX1 allele | Deletion of one PBX1 allele |
| CHD | Y | Y | Y | Y | Y | |||||||||||
| Renal/Urinary | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | ||||
| Reproductive | Y | Y | Y | Y | Y | Y | Y | Y | Y | |||||||
| Skeletal | Y | Y | Y | Y | Y | Y | Y | |||||||||
| Respiratory | Y | Y | Y | Y | Y | |||||||||||
| Digestive | Y | Y | Y | Y | ||||||||||||
| Craniofacial | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | |||
| Nervous system | Y | Y | Y | Y | Y | |||||||||||
| Growth/size/body | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | |||||
| Otherg | Y | Y | Y | Y | Y | Y | ||||||||||
| Sex | Male | Male | Male | Female | Male | Male | Male | Male | Female | Female | Female | Female | Male | Male | Female | Male |
The variant positions refer to PBX1 transcript NM_002585.4. Presence of a defect in the patient is indicated by Y. Patients are separated by those that harbour missense versus loss-of-function variants. For more details, refer to Supplementary Material, Table S2.
aSlavotinek et al. (11).
bEozenou et al. (12).
cRiedhammer et al. (13).
dHeidet et al. (14).
eLe Tanno et al. (15).
fCurrent manuscript.
gOthers include phenotypes that do not fall within any other category.
Creation of a knock-in mouse model of the R184P variant
Despite the novelty, the high conservation and the in silico pathogenicity of the PBX1-R184P variant, we were unable to unequivocally assign pathogenicity as per the guidelines established by the American College of Medical Genetics and determine the variant as the cause of the phenotypes observed in our patient. Therefore, we pursued functional assessment of the variant to determine its role in disease causality. We engineered a mouse model with the R184P variant using the CRISPR-Cas9 system. The exact variant was produced in the mouse as the PBX1 protein shares identity between human and mouse (Supplementary Material, Fig. S1). Heterozygous mice were intercrossed to produce the three possible genotypes: wild-type (+/+), heterozygous (+/R184P) and homozygous (R184P/R184P). Mendelian ratios of the embryos were analysed at six stages of embryonic (E) development: E11.5, E13.5, E14.5, E15.5, E17.5 and at birth (Supplementary Material, Fig. S2). The expected number of embryos for each genotype was observed at each embryonic stage except for at birth where there was a complete loss of the homozygous genotype and a significant loss of the heterozygous genotype. Analysis of embryonic death during development revealed that a proportion of homozygous embryos began to die from E15.5 (15%), with 8% found to be dead at E17.5, and none were born alive (Fig. 2A). Heterozygous embryos began to die at E17.5 (3%), and 45% died at birth.
Figure 2.
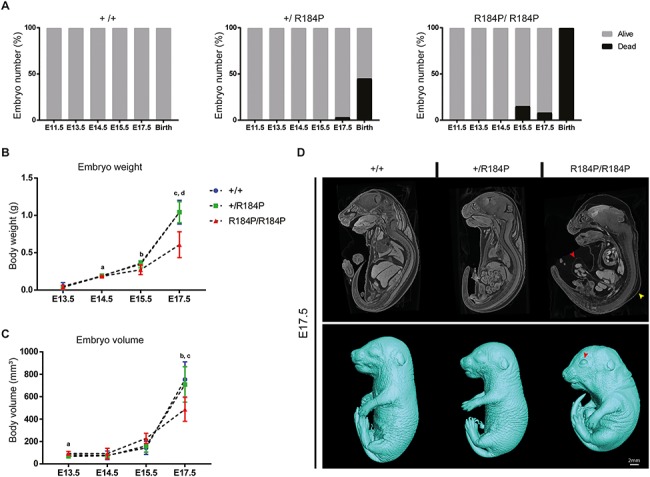
Functional assessment of the R184P knock-in mouse model. (A). Embryo survival was analysed at six stages during embryonic development, embryonic day (E) 11.5, E13.5, E14.5, E15.5, E17.5 and birth. Embryonic death was observed in both homozygous and heterozygous embryos. Death during development was not observed in wild-type littermates (n > 40 per stage). (B) Measurement of body weights of wild-type, heterozygous and homozygous embryos shows that homozygous embryos displayed lower body weight compared to wild-type and heterozygous embryos during development (n ≥ 12 per stage; a, P < 0.01; b, P < 0.05; c, P < 0.01; d, P < 0.001). (C) Whole body volume quantification of embryos during development shows that homozygous embryos have lower body volumes during late development (n ≥ 12 per stage; a, P < 0.05; b, P < 0.05; c, P < 0.05). (D) Whole embryo analysis by microCT shows the dysmorphic homozygous embryos at E17.5. Top panel: red arrow, umbilical hernia; yellow arrow, subcutaneous oedema. Bottom panel: red arrow, open eyes. Results are presented as mean ± SD.
Measurements revealed that from E14.5, the homozygous mutant embryos weighed significantly less than their wild-type counterparts (Fig. 2B). The weights of the homozygous embryos remained lower than wild-type embryos at E15.5, as well as at E17.5 when their weights were also significantly lower than heterozygous embryos. We utilized micro-CT imaging as a non-invasive method of producing high resolution 3D images of the embryos. This method permits detailed scrutiny of the internal organs of the embryos, and allowed detection of phenotypes that deviated from typical organ structures and organization. Collection of high resolution data also permits quantitative volumetric analysis of organs and the entire embryo. We observed that at E13.5, the homozygous embryos were larger than heterozygous embryos; however, by E17.5 the homozygous embryos were smaller than both wild-type and heterozygous embryos (Fig. 2C, Supplementary Material, Fig. S3). The homozygous embryos also displayed hunched posture, subcutaneous edema, open eyes, and umbilical hernia (Fig. 2D).
Phenotypic analysis of the R184P knock-in mouse model
Analysis of the micro-CT data revealed that the homozygous embryos had multiple organ system defects (Figs 3–6, Supplementary Material, Figs S4–S6). Heterozygous embryos also exhibited defects, although to a lesser extent than homozygous embryos.
Figure 3.
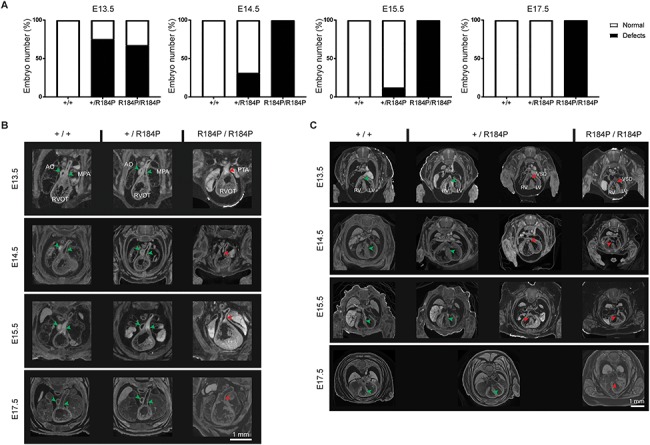
Analysis of heart defects observed in the R184P knock-in mouse model. (A) Heart defects were observed in homozygous embryos throughout development and in a proportion of heterozygous embryos until E15.5 (n ≥ 14 per stage). (B) Homozygous embryos exhibited PTA at all stages examined (E13.5–E17.5). (C) VSDs were observed in homozygous embryos examined at all stages. VSDs were also observed in a proportion of heterozygous embryos at E13.5, E14.5 and E15.5 (see A of this figure). AO, aorta; MPA, main pulmonary trunk; RVOT, right ventricular outflow tract; PTA, persistent truncus arteriosus; RV, right ventricle; LV, left ventricle; VSD, ventricular septal defect. Green arrows, typical heart morphology; red arrows, variation from the norm or presence of a defect.
Figure 6.
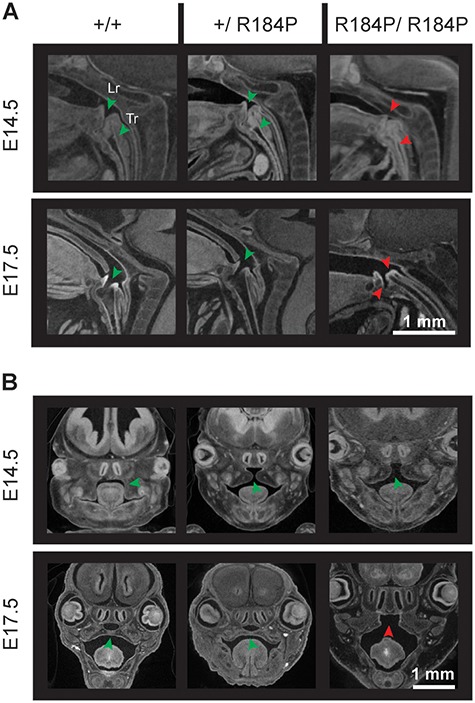
Defects in trachea, larynx and palate were observed in R184P homozygous mice. (A) Larynges of the homozygous embryos fail to open during development. In wild-type and heterozygous embryos, the larynx is open from E13.5. The trachea of the homozygous embryos remains atretic at all examined stages. The trachea of wild-type and heterozygous embryos are patent by E17.5 (n ≥ 14 per stage). (B) The secondary palate of the homozygous embryos does not fuse. Palatal defects are not observed in wild-type and heterozygous embryos (n ≥ 14 per stage). Data are presented at E14.5 and E17.5. Lr, larynx; Tr, trachea. Green arrows, typical organ development. Red arrows, deviation from the norm or presence of defect.
As our proband has CHD, we first focused on heart development of the mutant mice. Persistent truncus arteriosus (PTA), where the aorta and the main pulmonary artery do not septate, and VSDs where the septum between the right and left ventricles does not completely separate the two ventricles, were observed in the hearts of homozygous embryos at all developmental stages (Fig. 3A–C). PTA was not identified in the hearts of heterozygous or wild-type embryos. However, VSDs were observed in the hearts of heterozygous embryos from E13.5 to E15.5 (Fig. 3A and C). At E13.5, 75% of heterozygous embryos had VSDs, 31% of heterozygous embryos at E14.5, 12.5% heterozygous embryos had VSDs at E15.5, but none were observed in heterozygotes at E17.5. Heart defects were not observed in wild-type embryos at any stage.
Severe defects were also observed during lung development. We noted that the lungs were consistently dysmorphic in homozygous embryos at all stages examined from E13.5 to E17.5 (Fig. 4A). Homozygous lungs had the typical number of lung lobes (four lobes on the right and one lobe on the left); however, they were poorly separated and hypoplastic. In contrast, lungs of both heterozygous and wild-type embryos were indistinguishable. The quantification of this phenotype confirmed the visual inspection of the homozygous lungs. At E17.5 when the greatest difference in lung size was observed, lung volume of the R184P/R184P embryos (mean ± SD; 0.007 ± 0.001 mm3) remained consistently below the volumes of the heterozygous (0.019 ± 0.003 mm3) and wild-type embryos (0.020 ± 0.003 mm3) (Fig. 4B).
Figure 4.
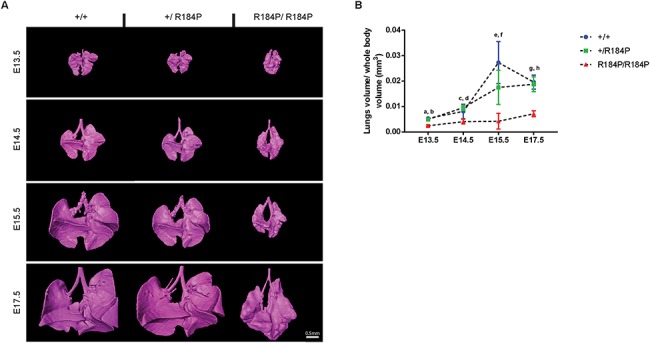
Analysis of lung development in R184P knock-in mouse model. (A) Lungs in the homozygous embryos were smaller and dysmorphic compared to heterozygous and wild-type lungs at all analysed stages. (B) Quantification of the 3D reconstructions of the lungs showed a significant decrease in lung volume of the homozygous embryos at all stages (n ≥ 12 per stage; a and b, P < 0.0001; c, P < 0.05; d, P < 0.0001; e and f, P < 0.01; g and h, P < 0.0001). Results are presented as mean ± SD.
We also noted a range of other phenotypes in the R184P mutant embryos. Homozygous embryos displayed spleen agenesis (failed spleen development), a phenotype not observed in the heterozygous or wild-type embryos at any stage of development (Supplementary Material, Fig. S4). Physiological umbilical hernia, whereby the intestines herniate outwards from the body to form a bulge on the abdomen, was observed in embryos of all genotypes up to E15.5 (Supplementary Material, Fig. S5). At E17.5, umbilical hernia was not observed in wild-type embryos, but was present in all homozygous embryos (Fig. 2D). A total of 12.5% of heterozygous embryos also had umbilical hernia at E17.5. We observed that embryos of all genotypes had open eyes until E17.5 when heterozygous and wild-type embryos exhibited closed eyes, by which time the eye-lids had formed (Fig. 2D, Supplementary Material, Fig. S6). However, at E17.5, the eyes of homozygous embryos remained open. We also observed that the homozygous embryos displayed defects in thymus development where the thymi were either ectopic or absent (Supplementary Material, Fig. S4). The homozygous embryos had abnormal development of the trachea and the larynx (Fig. 6A, Supplementary Material, Fig. S6). In the majority of heterozygous (88%) and wild-type embryos (100%), the larynx was patent from the earliest stage of our observations (at E13.5) and in all analysed embryos by E17.5. In contrast, in the homozygous embryos, the larynx failed to open until E17.5. Furthermore, in both heterozygous and wild-type embryos, the trachea is atretic at all stages prior to E17.5, at which time the trachea opens to the larynx. However, in the homozygous embryos, the trachea remained atretic at all stages analysed. Homozygous embryos also displayed palatal defects whereby the palatal shelves of the maxilla that fuses in heterozygous and wild-type embryos by E15.5 do not fuse in the homozygous embryos (Fig. 6B, Supplementary Material, Fig. S6). At E13.5, all embryos displayed lack of fusion of the palatal shelves, whereas at E14.5, 87% of wild-type, 88% of heterozygous and 100% of homozygous embryos exhibited lack of palate fusion. By E17.5, all wild-type and heterozygous embryos showed fusion of the palatal shelves, while all homozygous embryos continued to display cleft palates.
The urogenital system is consistently affected in CAKUTHED (Table 1, Supplementary Material, Table S2). Therefore, we examined the urogenital system of the mutant embryos for defects. Analysis of the homozygous embryonic kidneys revealed that they were hypoplastic and failed to ascend within the body cavity during embryonic development. Analysis of the reconstructed kidneys showed that the volume of kidneys of homozygous embryos (right kidney: 0.00072 ± 0.00032 mm3, left kidney: 0.00074 ± 0.00033 mm3) remained consistently lower than that of their heterozygous (right kidney: 0.0019 ± 0.0010 mm3, left kidney: 0.0020 ± 0.0010 mm3) and wild-type (right kidney: 0.0021 ± 0.0014 mm3, left kidney: 0.0021 ± 0.0013 mm3) counterparts during development (Fig. 5A and C). We also measured the position of the kidneys within the body cavities of the embryos and noted that in homozygous embryos (right kidney: 0.7864 ± 0.0138 mm, left kidney: 0.7955 ± 0.0153 mm), the kidneys remained mispositioned caudally within the body while the kidneys of the heterozygous (right kidney: 0.7494 ± 0.0453 mm, left kidney: 0.7584 ± 0.0371 mm) and wild-type embryos ascended up the body cavity (right kidney: 0.7363 ± 0.0400 mm, left kidney: 0.7361 ± 0.0453 mm) (Fig. 5B and C). In homozygous embryos, the adrenal glands did not develop while they were present in the heterozygous and wild-type embryos (Fig. 5C).
Figure 5.
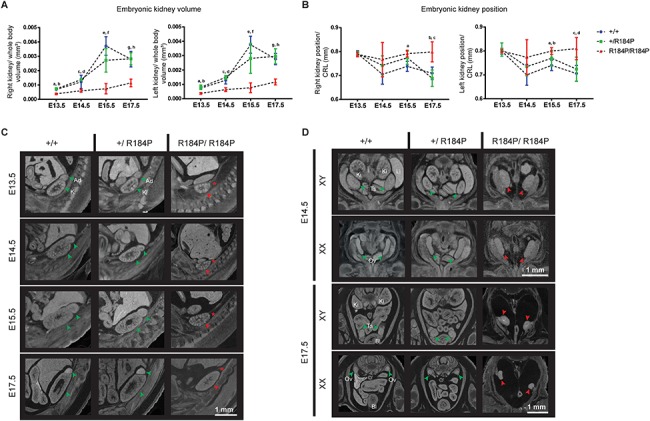
Analysis of urogenital phenotypes of the R184P knock-in mouse model. (A) Quantification of 3D reconstructions of the embryonic kidneys showed that both kidneys (right kidney and left kidney) of the homozygous embryos remained consistently smaller than the kidneys of the heterozygous and wild-type embryos at all stages examined. (Right kidney: n ≥ 12; a, P < 0.001; b, P < 0.0001; c, P < 0.05; d, P < 0.0001; e, P < 0.001; f, P < 0.01; g, P < 0.001; h, P < 0.0001. Left kidney: n ≥ 12; a and b, P < 0.001; c, P < 0.01; d, P < 0.0001; e, P < 0.0001; f, P < 0.01; g and h, P < 0.0001). (B) Measurement of the position of the kidneys within the body cavity relative to crown-to-rump length (CRL). In homozygous embryos, the position of the kidneys remains unchanging between E13.5 and E17.5. In both heterozygous and wild-type embryos, the kidneys ascend within the body cavity. (Right kidney: n ≥ 13; a, P < 0.01; b, P < 0.01; c, P < 0.001. Left kidney: n ≥ 13; a, P < 0.001; b, P < 0.05; c, P < 0.01; d, P < 0.001). (C) MicroCT analysis of the hypoplastic, pelvic kidneys of the homozygous embryos. Adrenal glands are absent in the homozygous embryos from the earliest stage examined at E13.5. (D) Gonads of homozygous R184P XY and XX embryos are dysmorphic and situated ectopically, presented at E14.5 and E17.5. Ad, adrenal glands; Bl, bladder; Ki, kidneys; Li, liver; Ov, ovary; Ts, testis. Green arrows, typical organ development. Red arrows, deviation from the norm or presence of defect. Red asterisk, absence of adrenal gland. Results are presented as mean ± SD.
In terms of gonadal development, we observed that in homozygous embryos until E15.5, gonads could not be distinguished by sex where the shape of the gonads were neither ovarian nor testicular but an intermediate shape (Fig. 5D). However, by E17.5, the testes could be identified morphologically although they appeared to be at an earlier developmental stage than E17.5, and their position within the body appeared ectopic. Similarly at this stage, the ovaries, although no longer morphologically similar to the testes, appeared to be at an intermediate stage of development, earlier than E17.5, as assessed by morphological examination. Gonadal development of heterozygous and wild-type embryos proceeded as typical per stage and sex.
Molecular analysis of the R184P defect
The observed spectrum of phenotypes of the R184P model is very similar to those previously reported in the Pbx1 knockout (KO) model (17). In order to examine whether the R184P variant affects PBX1 expression, we assessed protein levels and transcripts in whole embryos at E11.5. By western blot, we observed that levels of PBX1-R184P protein, as both PBX1a and PBX1b isoforms, were decreased when compared to heterozygous or wild-type embryos (Fig. 7A). This was confirmed by quantification of band intensity where protein levels in homozygous embryos were reduced relative to heterozygous and wild-type embryos by 2.26-fold and 2.71-fold, respectively (Fig. 7B). There was no difference in PBX1 protein levels between heterozygous and wild-type embryos. We assessed Pbx1 transcript to identify whether the missense variant was affecting Pbx1 mRNA possibly leading to lower protein levels in the homozygous embryos. Analysis of transcript levels by qPCR showed that Pbx1 expression was up-regulated in both homozygous (3.2-fold) and heterozygous embryos (2.4-fold) (Fig. 7C) relative to wild-type. There was no difference between transcript expression of homozygous and heterozygous embryos. We also assessed the expression of closely related Pbx genes, Pbx2 and Pbx3, as well as PBX1-interacting partner, Meis2, and genes regulated by PBX1 during development of the heart (Pax3), lungs (Fgf10), skeleton (Sox9) and kidneys (Pdgfrb) (Fig. 7C). We observed up-regulation of Pbx2 transcript in R184P homozygous embryos, relative to wild-type (3.8-fold) and heterozygous (3.1-fold) embryos. Expression of Fgf10, directly regulated by PBX1 during lung development (18), was down-regulated in heterozygous (3.2-fold) and homozygous (3.1-fold) embryos compared to wild-type embryos. Sox9, hypothesized to function in parallel with PBX1 during chondrogenesis (17), was up-regulated in both heterozygous (4.5-fold) and homozygous (6.8-fold) embryos relative to wild-type embryos. Pdgfrb is repressed by PBX1 during early stages of kidney development (19), and in the R184P homozygous embryos its expression was up-regulated relative to both wild-type (4.4-fold) and heterozygous (5.4-fold) embryos. In contrast, the expression levels of Pbx3, Meis2 and Pax3 were not altered between genotypes.
Figure 7.
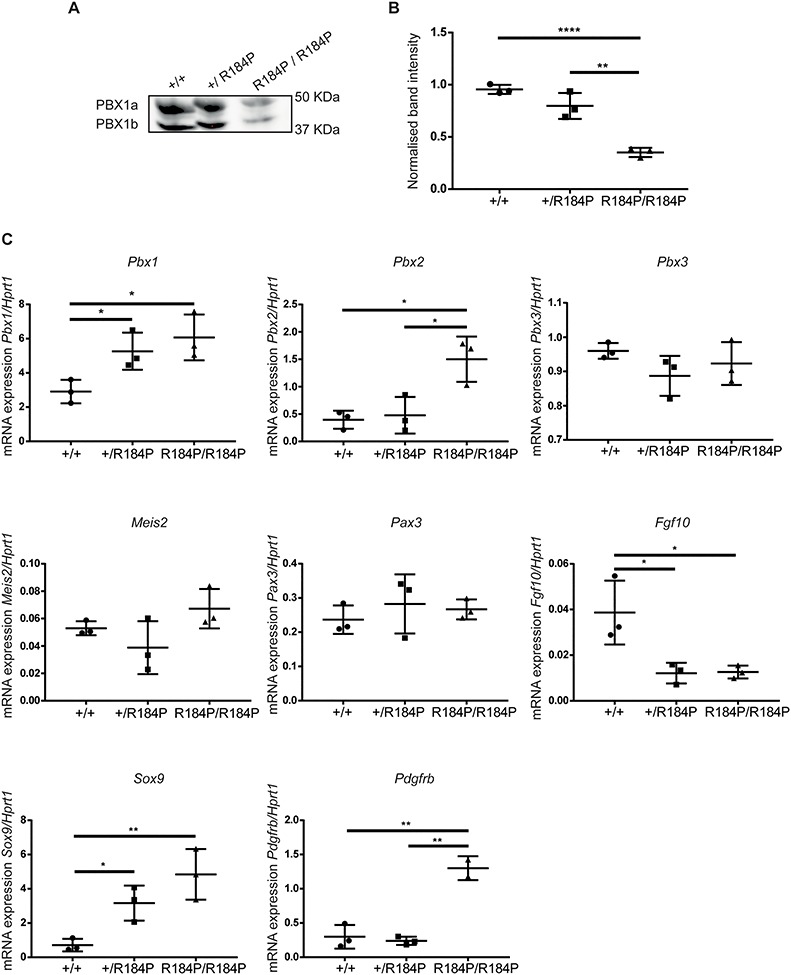
Assessment of PBX1 protein and mRNA expression in the R184P mouse model. (A) Western blot analysis of PBX1 protein expression in E11.5 embryos revealed that the protein is present at lower levels in the homozygous mutant embryos. (B) Quantification of PBX1 protein normalized to total protein loaded per lane (n = 3 embryos per genotype, **P < 0.01, ****P < 0.0001). (C) Analysis of the Pbx1, Pbx2, Pbx3, Meis2, Pax3, Fgf10, Sox9 and Pdgfrb transcripts in E11.5 embryos relative to Hprt1 transcript levels. The expression of Pbx1 and Sox9 are up-regulated in heterozygous and homozygous embryos. Expression of Pbx2 and Pdgfrb are up-regulated in homozygous embryos. The expression of Fgf10 is down-regulated in heterozygous and homozygous embryos. The expression of Pbx3, Meis2 and Pax3 are not altered (n = 3 embryos per genotype,*P < 0.05, **P < 0.01). Results are presented as mean ± SD.
Discussion
Here we have presented the first in vivo functional evidence that a missense variant in PBX1 is able to cause congenital defects. We characterized the R184P model in a comprehensive temporal manner, following the phenotypes throughout development and were able to observe the progression of defects from early in development until later stages.
One of the most startling findings was the observation that the R184P homozygous embryos displayed phenotypes nearly identical to the Pbx1 constitutive KO embryos (9,16,17,20–22). Comprehensive analysis of the constitutive Pbx1 KO, first reported by Selleri et al., examined the embryos at E14.5 (17). The constitutive KO displays multi-organ defects as summarized in Table 2. Analysis of the R184P embryos at the same developmental stage showed that the major organs were affected in a similar manner. For example, the heart defects reported in the constitutive KO were PTA and VSD (9,20), which were the same heart phenotypes that were present in the R184P model. Hypoplastic lungs, hypoplastic pelvic kidneys, absent adrenal glands and spleen were all phenotypes shared by the R184P model and the constitutive KO (17,19,21–23).
Table 2.
Phenotype comparison of the R184P and the original Pbx1 constitutive knock-out mouse models
| R184P model | Pbx1 constitutive KOa | ||
|---|---|---|---|
| R184P/R184P | +/R184P | −/− | |
| Cardiac | VSD; PTA | VSD | VSD; PTA |
| Hematopoietic | Aplastic spleen | Not observed | Aplastic spleen; Anemia |
| Renal/Urinary | Hypoplastic, underdifferentiated, pelvic kidneys | Not observed | Hypoplastic, underdifferentiated, ectopic kidneys |
| Reproductive | Delayed gonad differentiation, indifferent at E14.5, ectopic at E17.5 | Not observed | Sex differentiation arrest; loss of Müllerian duct in both sexes |
| Skeletal | Not observed | Not observed | Skeletal agenesis malformations |
| Respiratory | Atretic larynx (resolves by E17.5); Atretic trachea; hypoplastic lungs | Not observed | Hypoplastic lungs |
| Digestive | Narrow esophagus; hypoplastic, ectopic stomach; hypoplastic liver | Not observed | Hypoplastic liver; hypoplastic stomach; hypoplastic gut |
| Craniofacial | Open eyelids at E17.5; unfused secondary palate (cleft palate) | Not observed | Mandibular hypoplasia; hypoplastic pinna |
| Nervous system | Hypoplastic brain | Not observed | Not reported |
| Endocrine | Aplastic adrenal glands; hypoplastic, ectopic thymi | Not observed | Hypoplastic pancreas; aplastic adrenal glands; hypoplastic, ectopic, aplastic thymi; ectopic, aplastic parathyroid |
| Embryo (growth/size) | Umbilical hernia; subcutaneous edema; hunched posture | Umbilical hernia; subcutaneous edema | Subcutaneous edema; hunched posture |
Abbreviations: ACMG, American College of Medical Genetics; Ad, adrenal glands; AO, aorta; Bl, bladder; CAKUTHED, congenital anomalies of kidney and urinary tract syndrome with or without hearing loss, abnormal ears or developmental delay; CHD, congenital heart disease; CRL, crown to rump length; Ki, kidneys; KO, knockout; Li, liver; LOF, loss of function; Lr, larynx; LV, left ventricle; MPA, main pulmonary trunk; Ov, ovary; PA, pulmonary artery; PBX, pre B-cell leukemia; PFO, patent foramen ovale; PTA, persistent truncus arteriosus; RV, right ventricle; RVOT, right ventricular outflow tract; TALE, three amino acid loop extension; TOF, tetralogy of Fallot; Tr, trachea; Ts, testis; VMC, vascular mural cells; VSD, ventricular septal defect; WES, whole exome sequencing.
Molecular analysis of PBX1 protein in the R184P embryos revealed significant loss of the PBX1 protein in the R184P homozygous embryos at E11.5. This finding suggests that R184P may be a loss-of-function allele where protein levels have fallen below a critical functional threshold. It is possible that the substitution of an arginine to a proline, whose rigid, cyclic structure is predicted to introduce a turn and disrupt secondary structure of the protein, may also impact the tertiary structure of PBX1. Consequently, such misfolding may destabilize the mutant protein, thus leading to lower protein levels as observed.
We eliminated the possibility that the lower protein levels are due to decreased Pbx1 transcripts by assessing whole embryo mRNA by qPCR. We discovered that levels of Pbx1 transcript were in fact up-regulated in both homozygous and heterozygous embryos. This finding suggests the possibility of a compensatory mechanism whereby the mutant embryos react to decreased PBX1 protein levels by up-regulating gene transcription. Our transcriptional analysis also revealed that PBX family member, Pbx2, is transcriptionally upregulated in the homozygous embryos. Similar to the up-regulation of Pbx1 transcript in the homozygous embryos, this observation suggests an attempt at rescue of the R184P phenotypes by Pbx2, but not Pbx3, whose expression was not altered in the mutants. This indicates that PBX1 and PBX2 may act cooperatively during development and may share the regulation of developmental processes. This is consistent with previous findings that used compound mutant embryos of the Pbx genes (Pbx1, Pbx2 and Pbx3) to show that heart development is dependent on combined dosages of the PBX proteins (9).
Multiple examples of genetic compensation have been reported in species such as yeast, drosophila, zebrafish and mice (reviewed in 24). This mechanism that becomes activated in response to genetic variations functions to ensure the genetic robustness of an organism by maintaining viability and fitness. In a majority of embryos heterozygous for the R184P variant, this mechanism appears to have succeeded, and indeed PBX1 protein levels are similar to wild-type protein levels. However, in those heterozygous embryos with abnormal phenotypes, it is possible that PBX1 protein levels may not reach wild-type levels. Furthermore, as we have observed, the same mechanism fails to increase protein levels in the R184P homozygous embryos, suggesting that the mutant protein is actively degraded. The penetrance of phenotypes in these embryos indicates that loss of PBX1 functionality is not recovered and that PBX1-controlled functions cannot be mediated by other factors such as PBX2 during development. However, constitutive Pbx1 KO embryos die at E17.5, whereas in the R184P model only 8% of homozygous embryos succumb at E17.5. It is possible that presence of some remaining PBX1 protein, despite the structural deviance, may moderate the severity that is described when both alleles of the Pbx1 gene are lost.
This compensatory mechanism may underlie the normal development of the majority of heterozygous embryos in the R184P model. It is also likely to be the reason why developmental defects were not reported in any heterozygous embryos in the constitutive Pbx1 KO. Our findings also suggest that protein levels may not reach the required threshold during early development as we observe heart defects in 75% of heterozygous embryos. However, by E17.5 none of the heterozygous embryos display heart defects, suggesting that PBX1 protein levels have reached the crucial amount required for typical heart development. However, subcutaneous edema and umbilical hernia persist in a small proportion of heterozygous embryos (Supplementary Material, Fig. S5), indicating that compensation may not recover all PBX1 functions affected by the variant.
It appears that this compensatory mechanism may not be conserved in humans or that it might not be able to overcome the presence of a loss of function (LOF) allele. It is possible that either the necessary gene-gene or gene-environmental interactions that regulate this mechanism in mice are not preserved in humans. This is evident in patients with heterozygous LOF PBX1 mutations. The compensatory mechanism would otherwise upregulate protein to functional levels, meaning that heterozygous LOF variants would not lead to disease. But as has been noted, heterozygous LOF PBX1 variants cause disease and are not found in control populations (Table 1, Supplementary Material, Table S2).
The advantage of assessing a missense variant is that it allows us to elucidate possible mechanisms of disease causality. Where a LOF causes disease by absence of protein product, missense variants have the ability to disrupt protein folding, protein–protein interactions, and protein–DNA interactions. Our variant is missense; in fact, it is the only missense variant in the PBX dimerization domain associated with an abnormal human phenotype. As PBX1 is not a transcription factor that can bind DNA as a monomer, the association of binding partners plays an essential role in its functionality (16,25). The central PBX dimerization domain facilitates its interactions with itself as well as other cofactors (26). It is possible that the R184P variant interferes with homodimerization and/or heterodimerization. However, if the process being disturbed is homodimerization, only PBX1 functions would be affected and our model would be identical to the constitutive Pbx1 KO mouse. Aside from observing phenotypes in some heterozygous embryos, we also observed novel phenotypes in the homozygous embryos that have not been reported in the constitutive KO such as ocular defects, cleft palate and tracheal defects (Table 2). We expect that it is likely heterodimerization that is perturbed by the R184P variant. Previously, missense variants have permitted the identification of protein regions that are responsible for its tissue-specific functions (12). As our patient is affected by CHD, it is likely that a heart-specific interaction is being impaired by the variant. Other patients with pathogenic variants at p.M224, p.R234 and p.R235 also have congenital heart defects, suggesting that this region (p.R184—p.R235) is required for protein–protein interactions essential for heart development (see Table 1, Supplementary Material, Table S2) (11).
The most likely candidate is MEIS2, which interacts with PBX1 and most intriguingly shares overlapping KO phenotypes with the R184P model. Meis2 KO model displays such phenotypes as PTA, cleft palate, eye-lid defect and absent pulmonary valves (27), which when combined with TOF is an extremely rare phenotype (1/10 000 TOF cases (28)) as observed in our patient. Unlike its family member MEIS1, which interacts with PBX1 via the N-terminal PBC-A domain, the location of MEIS2 interaction with PBX1 is not yet firmly established. Interestingly, human patients have been reported with de novo missense variants and copy number variants encompassing MEIS2 who share certain common phenotypes; cleft palate, VSD and hearing loss (29–32). These phenotypes are also observed in human patients with pathogenic PBX1 mutations (see Table 1, Supplementary Material, Table S2). Craniofacial anomalies and cardiac defects are also observed in our patient with the R184P variant. Our transcriptional analysis revealed that Meis2 is not PBX1-dependent. Thus, given the commonality of the phenotypes shared between the R184P model and Meis2 loss-of-function model, as well as the similarity of phenotypes observed in human patients with PBX1 and MEIS2 mutations, we believe that it is likely that the interaction between PBX1 and MEIS2 is being perturbed by the R184P variant. We cannot discount that other interactions may also be perturbed; however, this would require the identification and functional assessment of tissue-specific PBX1 binding partners.
Interestingly, we did not observe changes in expression of the PBX1 cardiac target gene, Pax3, in the R184P model. As Pax3 is expressed in pre-migratory cardiac neural crest cells at E8.0 where it is under PBX1 regulation (20), it is possible that any change in Pax3 expression is masked by the generalized Pax3 expression levels observed in the whole embryo where other factors may regulate its expression. Perhaps, it is also possible that the PBX1-Pax3 regulation occurs in a very refined temporal manner and that at E11.5 where we assessed transcription, Pax3 is no longer under PBX1 control. However, we observed expression changes in genes regulated by PBX1 in other organ systems. PBX1 regulates the expression of Fgf10 in the lungs to direct surfactant production during development. Loss of Pbx1 in the lungs leads to reduced expression of Fgf10, reduced production of surfactants and perinatal lethality associated with failure of alveolar expansion (18). In the R184P model, we observed down-regulation of Fgf10 in both heterozygous and homozygous embryos. We did not observe significant morphological changes in lungs of heterozygous embryos, indicating that lung development is not impaired in heterozygous embryos. However, we cannot dismiss that production of surfactants by the lungs may be impaired in these embryos that may compromise lung function at a post-natal stage. We also assessed the expression of Sox9, which was hypothesized by Selleri et al. (17) to function in parallel with PBX1 during chondrogenesis to maintain a population of proliferating chondrocytes. Loss of Pbx1 led to loss of proliferating chondrocytes and multiple skeletal defects in the constitutive Pbx1 KO. The authors saw no overall impact on Sox9 expression from E13.5-E15.5 as assessed by in situ hybridization. We were unable to assess skeletal defects in the R184P model due to technical limitations imposed by micro-CT analysis. However, given the similarity between the R184P model and the constitutive KO, and the observed clinodactyly in the PBX1-R184P patient, it is highly likely that skeletal development is affected in the R184P mutant embryos. Sox9 expression is up-regulated in both heterozygous and homozygous embryos of the R184P model at E11.5. This suggests that SOX9 and PBX1 share overlapping functions during chondrogenesis and SOX9 may compensate for the loss of PBX1 to a certain extent. Furthermore, we observed an up-regulation in expression of Pdgfrb in the kidneys of the homozygous embryos. Previous assessment of kidney development has shown that PBX1 is crucial for maintaining a population of vascular mural cell (VMC) progenitors by repressing the expression of Pdgfrb (19). The loss of Pbx1 led to premature up-regulation of Pdgfrb in the VMC progenitors leading to premature differentiation of the VMC population as well as disruption of renal vasculature development. The up-regulation of Pdgfrb in the R184P homozygous embryos indicates that the VMC progenitors in these kidneys are becoming prematurely differentiated, and that this loss of progenitors may underlie the hypoplasticity of the kidneys that we observe in these embryos.
The spectrum of phenotypes reported in the R184P and the constitutive KO models are becoming increasingly recapitulated in humans with PBX1 mutations (Table 1, Supplementary Material, Table S2). Heart defects are reported in 5/16 patients who also have respiratory defects, and genitourinary defects are observed in 16/16 patients. Craniofacial defects are seen in 13/16 patients, and developmental defects (physical and neurological) are observed in 12/16 patients. Patients with previously unobserved phenotypes such as ectopic thymus, small spleen and small adrenal glands have been discovered (unpublished data, manuscript in preparation). These observations suggest that the full spectrum of Pbx1-related phenotypes should be expected in human patients with mutations in PBX1 and that the pre-natal identification of a pathogenic PBX1 variant may allow prognostication that would permit early intervention of some phenotypes.
What is also notable when comparing the two patient groups presented in Table 2 is the clear phenotypic variability prevalent between patients with pathogenic PBX1 variants. Interestingly, a patient has been reported with a loss-of-function variant at the exact amino acid position as the R184P patient, p.R184* (see Table 2, Supplementary Material, Table S1) (14). This patient was reported within a small cohort of CAKUTHED patients and was diagnosed with renal defects, dysmorphic features, developmental delay, but no heart defects, whereas the R184P has CHD associated with syndromic phenotypes. Aside from variable expressivity, incomplete penetrance of damaging variants has also been observed whereby a patient has been reported with a mutation in PBX1 at p.R288* with renal, craniofacial, cardiac and auditory defects (see Table 2, Supplementary Material, Table S1) (11). An individual with the same variant is reported in the gnomAD genome browser as a healthy reference individual without congenital defects. Although it is evident that there are several mutational hotspots within PBX1 that are susceptible to disease-causing mutations, the penetrance of PBX1 variants and the expressivity of phenotypes appear to be highly variable and possibly dependent on multiple factors such as interacting partners and influence of genetic modifiers.
Our findings show that the missense variant, R184P, leads to loss of protein, which results in a near-phenocopy of the Pbx1 constitutive KO mouse. However, the presence of novel phenotypes indicates a secondary mechanism of disease causality whereby the interaction with cofactors may be perturbed. This in vivo functional assessment of a disease-causal variant observed in a patient with CHD, asserts PBX1 missense variants as capable of causing syndromic CHD where complex, cyanotic CHD falls within the spectrum of PBX1-related phenotypes. With the evidence generated by our functional study, we are now able to update the ACMG classification of the R184P variant to pathogenic thus assigning significant clinical utility to the variant.
Materials and Methods
Patient details—recruitment and consent
Ethical approval for this study was obtained from the Sydney Children’s Hospital Network Human Research Ethics Committee (approval number HREC/16/SCHN/73). The patient and family were recruited at Princess Margaret Hospital for Children, Perth, Australia at presentation to a pre-admission clinic prior to cardiac surgery. Heart defect in the proband was confirmed by echocardiography.
Whole exome sequencing
The generation and analysis of WES data for this family have been reported elsewhere (5,11).
Creation of the CRISPR knock-in and mouse study
The PBX1-R184P knock-in mouse was created using the Mouse engineering Garvan/ABR (MEGA) service of Australian Bioresources (ABR) using CRISPR-Cas9 technology. The following CRISPR guide was used to engineer the variant: GCTGAACTTGCGGTGGATGATGCTCACCATC CGCTCGATCTCCTTCGGAGAGATGGGCCTGGTCCGGCTTTGCTCTGGAAGCAGGTTCATCACGTGGGTGGTGAATTCATTGCATGCCTGCAGTGGTAACACGG TAAACATGGGGTGAAC within exon 4 of the mouse Pbx1 gene. The guide was injected into C57BL/6 J embryos using electroporation and one male founder mouse with the variant was produced. Primers: 5′ TGAGAAAGACAGAGAAAGCTCGT 3′ and 5′ ATCTCCTGCAACCATGGATT 3′ were used to amplify the target region, and induction of the variant was confirmed by Sanger sequencing (Supplementary Material, Fig. S1).
Phenotypic analysis of the mouse line was performed following the guidelines and the approval of Garvan Institute of Medical Research/St. Vincent’s Animal Experimentation Ethics Committee (research approval 15/27, 18/27). Male and female mice heterozygous for the knock-in allele were used in timed-matings to produce wild-type, heterozygous and homozygous embryos at ages E11.5, E13.5, E14.5, E15.5 and E17.5 when the mothers were sacrificed and embryos harvested.
MicroCT
Embryo preparation
Embryos were fixed in 4% PFA/1% glutaraldehyde for 3 days at 4°C. Afterwards, the fixed embryos were incubated in hydrogel (4% acrylamide, 0.05% BIS, 4% PFA, 0.25% VA044, 0.05% Saponin) for 3 days at 4°C. The embryos were then polymerized in hydrogel using the X-Clarity Polymerization System (Logos Biosystems) (−90 kPa, 37°C, 3 h 10 min). Hydrogel was then removed from the embryos which were incubated in Lugol’s solution (Sigma Aldrich) for 5 days prior to microCT scanning.
Scanning
Skyscan1272 (Bruker) was used for embryo scanning. Embryos at all stages were scanned using the following settings: resolution 4904 × 3280, filter 0.5 μm aluminium, rotation 180 degrees and rotation step 0.3. Image pixel size varied between stages: E13.5: 2 μm, E14.5: 2.5–4 μm, E15.5: 2 μm, E17.5: 4 μm. Scans were reconstructed using NRecon (v1.7.1.0) and optimized for post-alignment, smoothing, ring artefacts and beam hardening.
Analysis
Reconstructed scans were visualized via CTvox (v3.3.0), CT Analyser (v1.17.7.2+) and resized using DataViewer (v1.5.4.0). Quantification of the volumes of embryos and organs was performed using AMIRA software (v2019.1). 3D models of organs were created by selecting the organ of interest and using the watershed function to remove all regions outside the organ of interest. Organs that could not be watershed were selected manually from the scanned data and interpolated. Label analysis function was used to quantify body/organ volumes.
Western blot analysis
Proteins were extracted from wild-type, heterozygous and homozygous whole embryos at E11.5 in Radioimmunoprecipitation Assay (RIPA) buffer (150 mM NaCl, 1% Octylphenoxypolyethoxyethanol (IGEPAL) CA-630, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris pH 8.0). Protein (30 μg) was loaded on a 4–15% GTX Stain-Free acrylamide gels (Biorad) in denaturing conditions with SeeBlue Plus2 prestained protein ladder (Biorad). The Stain-Free gel was activated using Chemidoc Stain-Free gel activation protocol (Biorad). The proteins were transferred to a nitrocellulose membrane (0.45 μM pore size, Amersham). Transference of protein was confirmed by imaging the membrane using the Stain-Free blot protocol of the Chemidoc. Rabbit-anti PBX1 antibody (#4342, Cell Signalling Technologies) was used to detect PBX1 protein using WesternBreeze (Thermofisher Scientific) according to the manufacturer’s instructions. Quantification of band intensity was performed using ImageJ.
Quantitative real-time PCR
E11.5 whole embryos were collected, snap frozen in liquid nitrogen and stored at −80°C. Total RNA was isolated using TriReagent (Life Technologies) following the manufacturer’s instructions. The tissues were homogenized using an insulin syringe. RNA purity (A260/A280 ratio ≥ 1.8) and concentration were measured using the NanoDrop ND-1000 Spectrophotometer. Total RNA 1 μg was converted into cDNA using the QuantiTect Reverse Transcription kit according to the manufacturer’s instructions. The qPCR reaction contained 4 ng of cDNA, LightCycler 480 SYBR Green 1 Master (Roche), forward and reverse primers (0.3 μM). All samples were run in triplicate on CFX384 (BioRad). The amplification protocol consisted of 3′ 95°C for one cycle and 10″ 95°C 30″ 60°C for 39 cycles and finished with a standard melting curve protocol. Melting curve analysis and quantification were performed using the CFX384 software (BioRad). Each primer used was validated to be specific for the gene by confirming a single peak on the melting curve. Raw values were exported to Microsoft Excel and analysed for absolute quantification using a standard curve. Primers used for qPCR: Pbx1 For 5′ GGGTGCAGGTTCAGACAACT 3′, Rev 5′ TGCATGCCTGCTCATACTTCT 3′; Pbx2 For 5′ AGACATCGGGGACATTCTGC 3′, Rev 5′ TCCGAATGCTAAGGCCAGTT 3′; Pbx3 For 5′ ATGTGCAGTCACAGGTGGAT 3′, Rev 5′ GCCAGCCTCCATTAGCGTTT 3′; Meis2 For 5′ GATCGCACAGGATGCGTTTC 3′, Rev 5′ GTCTAACCCATCGCCTCCAG 3′; Pax3 For 5′ CGCTTTTTCGTCTCGCCTTC 3′, Rev 5′ CAAGAGGGGTGGACACTTCC 3′; Fgf10 For 5′ AAGCCATCAACAGCAACTAT 3′, Rev 5′ ATTGTGCTGCCAGTTAAAAG 3′; Sox9 For 5′ AGTACCCGCATCTGCACAAC 3′, Rev 5′ TACTTGTAATCGGGGTGGTCT 3′; Pdgfrb For 5′ GTGGTCCTTACCGTCATCTCTC 3′, Rev 5′ GTGGAGTCGTAAGGCAACTGCA 3′; Hprt1 For 5′ GCTTGCTGGTGAAAAG GACCTCTCGAAG 3′, Rev 5′ CCCTGAAGTACTCATTATAGTCAAG.
GGCAT 3′.
Statistical analysis
All statistical analyses were performed with Prism 7.03 (GraphPad Software). All datasets were tested for statistical significance using Student’s t-test. Chi-squared test was used to analyse expected versus observed Mendelian ratios. In all figures, data represent mean ± standard deviation.
Funding
National Health and Medical Research Council (NHMRC) (Fellowships ID1135886, ID1042002 to S.L.D., and Program Grant ID1074386 to S.L.D.); Australian National Heart Foundation (Fellowship ID101204 to E.G.); Australian Postgraduate Award (UNSW) (J.O.S.); Office of Health and Medical Research NSW Government to S.L.D; Chain Reaction (The Ultimate Corporate Bike Challenge) to S.L.D.; Channel 7 Telethon to S.L.D.; The Key Foundation to S.L.D and The National Institutes of Health (R01 grant DE024745 to L.S.).
Conflict of Interest Statement
Authors declare no conflicts of interest exist.
Supplementary Material
Acknowledgements
The authors would like to thank the family for their participation in the study. The authors acknowledge the technical assistance of Ella M.M.A. Martin and Joelene A. Greasby.
References
- 1. van der Linde D., Konings E.E.M., Slager M.A., Witsenburg M., Helbing W.A., Takkenberg J.J.M. and Roos-Hesselink J.W. (2011) Birth prevalence of congenital heart disease worldwide. J Am. Coll. Cardiol., 58, 2241–2247. [DOI] [PubMed] [Google Scholar]
- 2. Blue G.M., Kirk E.P., Giannoulatou E., Sholler G.F., Dunwoodie S.L., Harvey R.P. and Winlaw D.S. (2017) Advances in the genetics of congenital heart disease a Clinician's guide. J. Am. Coll. Cardiol., 69, 860–870. [DOI] [PubMed] [Google Scholar]
- 3. Alankarage D., Ip E., Szot J.O., Munro J., Blue G.M., Harrison K., Cuny H., Enriquez A., Troup M., Humphreys D.T. et al. (2019) Identification of clinically actionable variants from genome sequencing of families with congenital heart disease. Genetics In Medicine., 21, 1111–1120. [DOI] [PubMed] [Google Scholar]
- 4. Simmons M.A. and Brueckner M. (2017) The genetics of congenital heart disease...Understanding and improving long-term outcomes in congenital heart disease: a review for the general cardiologist and primary care physician. Curr. Opin. Pediatr., 29, 520–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Szot J.O., Cuny H., Blue G.M., Humphreys D.T., Ip E., Harrison K., Sholler G.F., Giannoulatou E., Leo P., Duncan E.L. et al. (2018) A screening approach to identify clinically actionable variants causing congenital heart disease in exome data. Circ. Genom. Precis. Med., 11:e001978. [DOI] [PubMed] [Google Scholar]
- 6. Jin S.C., Homsy J., Zaidi S., Lu Q.S., Morton S., DePalma S.R., Zeng X., Qi H.J., Chang W.I., Sierant M.C. et al. (2017) Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat. Genet. 49, 1593−+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Burglin T.R. and Ruvkun G. (1992) New motif in Pbx genes. Nat. Genet., 1, 319–320. [DOI] [PubMed] [Google Scholar]
- 8. Burglin T.R. (1997) Analysis of TALE superclass homeobox genes (MEIS, PBC, KNOX, Iroquois, TGIF) reveals a novel domain conserved between plants and animals. Nucleic Acids Res., 25, 4173–4180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stankunas K., Shang C., Twu K.Y., Kao S.C., Jenkins N.A., Copeland N.G., Sanyal M., Selleri L., Cleary M.L. and Chang C.P. (2008) Pbx/Meis deficiencies demonstrate multigenetic origins of congenital heart disease. Circ. Res., 103, 702–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schnabel C.A., Selleri L., Jacobs Y., Warnke R. and Cleary M.L. (2001) Expression of Pbx1b during mammalian organogenesis. Mech. Dev., 100, 131–135. [DOI] [PubMed] [Google Scholar]
- 11. Slavotinek A., Risolino M., Losa M., Cho M.T., Monaghan K.G., Schneidman-Duhovny D., Parisotto S., Herkert J.C., Stegmann A.P.A., Miller K. et al. (2017) De novo, deleterious sequence variants that alter the transcriptional activity of the homeoprotein PBX1 are associated with intellectual disability and pleiotropic developmental defects. Hum. Mol. Genet., 26, 4849–4860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Eozenou C., Bashamboo A., Bignon-Topalovic J., Merel T., Zwermann O., Lourenco D., Lottmann H., Lichtenauer U., Rojo S., Beuschlein F. et al. (2019) The TALE homeodomain of PBX1 is involved in human primary testis-determination. Hum. Mutat., 40, 1071–1076. [DOI] [PubMed] [Google Scholar]
- 13. Riedhammer K.M., Siegel C., Alhaddad B., Montoya C., Kovacs-Nagy R., Wagner M., Meitinger T. and Hoefele J. (2017) Identification of a novel heterozygous De novo 7-bp Frameshift deletion in PBX1 by whole-exome sequencing causing a multi-organ syndrome including bilateral dysplastic kidneys and Hypoplastic clavicles. Front. Pediatr., 5, 251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Heidet L., Moriniere V., Henry C., De Tomasi L., Reilly M.L., Humbert C., Alibeu O., Fourrage C., Bole-Feysot C., Nitschke P. et al. (2017) Targeted exome sequencing identifies PBX1 as involved in monogenic congenital anomalies of the kidney and urinary tract. J. Am. Soc. Nephrol., 28, 2901–2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Le Tanno P., Breton J., Bidart M., Satre V., Harbuz R., Ray P.F., Bosson C., Dieterich K., Jaillard S., Odent S. et al. (2017) PBX1 haploinsufficiency leads to syndromic congenital anomalies of the kidney and urinary tract (CAKUT) in humans. J. Med. Genet., 54, 502–510. [DOI] [PubMed] [Google Scholar]
- 16. Selleri L., Zappavigna V. and Ferretti E. (2019) ‘Building a perfect body’: control of vertebrate organogenesis by PBX-dependent regulatory networks. Genes. Dev., 33, 258–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Selleri L., Depew M.J., Jacobs Y., Chanda S.K., Tsang K.Y., Cheah K.S.E., Rubenstein J.L.R., O'Gorman S. and Cleary M.L. (2001) Requirement for Pbx1 in skeletal patterning and programming chondrocyte proliferation and differentiation. Development., 128, 3543–3557. [DOI] [PubMed] [Google Scholar]
- 18. Li W., Lin C.Y., Shang C., Han P., Xiong Y., Lin C.J., Yang J., Selleri L. and Chang C.P. (2014) Pbx1 activates Fgf10 in the mesenchyme of developing lungs. Genesis., 52, 399–407. [DOI] [PubMed] [Google Scholar]
- 19. Hurtado R., Zewdu R., Mtui J., Liang C., Aho R., Kurylo C., Selleri L. and Herzlinger D. (2015) Pbx1-dependent control of VMC differentiation kinetics underlies gross renal vascular patterning. Development., 142, 2653–U2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chang C.P., Stankunas K., Shang C., Kao S.C., Twu K.Y. and Cleary M.L. (2008) Pbx1 functions in distinct regulatory networks to pattern the great arteries and cardiac outflow tract. Development., 135, 3577–3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schnabel C.A., Godin R.E. and Cleary M.L. (2003) Pbx1 regulates nephrogenesis and ureteric branching in the developing kidney. Dev. Biol., 254, 262–276. [DOI] [PubMed] [Google Scholar]
- 22. Brendolan A. (2005) A Pbx1-dependent genetic and transcriptional network regulates spleen ontogeny. Development., 132, 3113–3126. [DOI] [PubMed] [Google Scholar]
- 23. McCulley D.J., Wienhold M.D., Hines E.A., Hacker T.A., Rogers A., Pewowaruk R.J., Zewdu R., Chesler N.C., Selleri L. and Sun X. (2018) PBX transcription factors drive pulmonary vascular adaptation to birth. J. Clin. Invest., 128, 655–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. El-Brolosy M.A. and Stainier D.Y.R. (2017) Genetic compensation: a phenomenon in search of mechanisms. PLoS Genet., 13, e1006780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Neuteboom S.T. and Murre C. (1997) Pbx raises the DNA binding specificity but not the selectivity of antennapedia Hox proteins. Mol. Cell. Biol., 17, 4696–4706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Calvo K.R., Knoepfler P., McGrath S. and Kamps M.P. (1999) An inhibitory switch derepressed by Pbx, Hox, and Meis/Prep1 partners regulates DNA-binding by Pbx1 and E2a-Pbx1 and is dispensable for myeloid immortalization by E2a-Pbx1. Oncogene., 18, 8033–8043. [DOI] [PubMed] [Google Scholar]
- 27. Machon O., Masek J., Machonova O., Krauss S. and Kozmik Z. (2015) Meis2 is essential for cranial and cardiac neural crest development. BMC Dev. Biol., 15, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kirshbom P.M. and Kogon B.E. (2004) Tetralogy of Fallot with absent pulmonary valve syndrome. Semin. Thorac. Cardiovasc. Surg. Pediatr. Card. Surg. Annu., 7, 65–71. [DOI] [PubMed] [Google Scholar]
- 29. Louw J.J., Corveleyn A., Jia Y.J., Hens G., Gewillig M. and Devriendt K. (2015) MEIS2 involvement in cardiac development, cleft palate, and intellectual disability. Am. J. Med. Genet. A., 167, 1142–1146. [DOI] [PubMed] [Google Scholar]
- 30. Johansson S., Berland S., Gradek G.A., Bongers E., de Leeuw N., Pfundt R., Fannemel M., Rodningen O., Brendehaug A., Haukanes B.I. et al. (2014) Haploinsufficiency of MEIS2 is associated with orofacial clefting and learning disability. Am. J. Med. Genet. A., 164, 1622–1626. [DOI] [PubMed] [Google Scholar]
- 31. Douglas G., Cho M.T., Telegrafi A., Winter S., Carmichael J., Zackai E.H., Deardorff M.A., Harr M., Williams L., Psychogios A. et al. (2018) De novo missense variants in MEIS2 recapitulate the microdeletion phenotype of cardiac and palate abnormalities, developmental delay, intellectual disability and dysmorphic features. Am. J. Med. Genet. A., 176, 1845–1851. [DOI] [PubMed] [Google Scholar]
- 32. Verheije R., Kupchik G.S., Isidor B., Kroes H.Y., Lynch S.A., Hawkes L., Hempel M., Gelb B.D., Ghoumid J., D'Amours G. et al. (2019) Heterozygous loss-of-function variants of MEIS2 cause a triad of palatal defects, congenital heart defects, and intellectual disability. Eur. J. Hum. Genet., 27, 278–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. DiMartino J.F., Selleri L., Traver D., Firpo M.T., Rhee J., Warnke R., O'Gorman S., Weissman I.L. and Cleary M.L. (2001) The Hox cofactor and proto-oncogene Pbx1 is required for maintenance of definitive hematopoiesis in the fetal liver. Blood., 98, 618–626. [DOI] [PubMed] [Google Scholar]
- 34. Kim S.K., Selleri L., Lee J.S., Zhang A.Y., Gu X.Y., Jacobs Y. and Cleary M.L. (2002) Pbx1 inactivation disrupts pancreas development and in Ipf1-deficient mice promotes diabetes mellitus. Nat. Genet., 30, 430–435. [DOI] [PubMed] [Google Scholar]
- 35. Schnabel C.A., Selleri L. and Cleary M.L. (2003) Pbx1 is essential for adrenal development and urogenital differentiation. Genesis., 37, 123–130. [DOI] [PubMed] [Google Scholar]
- 36. Manley N.R., Selleri L., Brendolan A., Gordon J. and Cleary M.L. (2004) Abnormalities of caudal pharyngeal pouch development in Pbx1 knockout mice mimic loss of Hox3 paralogs. Dev. Biol., 276, 301–312. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.