

Abstract
Human longevity is a complex trait influenced by both genetic and environmental factors, whose interaction is mediated by epigenetic mechanisms like DNA methylation. Here, we generated genome-wide whole-blood methylome data from 267 individuals, of which 71 were long-lived (90–104 years), by applying reduced representation bisulfite sequencing. We followed a stringent two-stage analysis procedure using discovery and replication samples to detect differentially methylated sites (DMSs) between young and long-lived study participants. Additionally, we performed a DNA methylation quantitative trait loci analysis to identify DMSs that underlie the longevity phenotype. We combined the DMSs results with gene expression data as an indicator of functional relevance. This approach yielded 21 new candidate genes, the majority of which are involved in neurophysiological processes or cancer. Notably, two candidates (PVRL2, ERCC1) are located on chromosome 19q, in close proximity to the well-known longevity- and Alzheimer’s disease-associated loci APOE and TOMM40. We propose this region as a longevity hub, operating on both a genetic (APOE, TOMM40) and an epigenetic (PVRL2, ERCC1) level. We hypothesize that the heritable methylation and associated gene expression changes reported here are overall advantageous for the LLI and may prevent/postpone age-related diseases and facilitate survival into very old age.
Introduction
Human longevity is a rare phenomenon with approximately one centenarian in 5000 people (1). Many long-lived individuals (LLI) do not only reach an extreme age, but also exhibit a relatively good health status, which is achieved by postponing the onset of major age-related diseases and the beginning of functional decline (2). Although LLI epitomize the longevity phenotype, they obviously also show signs of physiological aging. Longevity has a clear genetic contribution of up to 40% in elderly who survive beyond 85 years (3), but environmental and lifestyle parameters are more relevant determinants for becoming long-lived (4). The interaction between the different factors can be mediated by epigenetic mechanisms such as DNA methylation, regulatory RNAs and/or histone modifications (5, 6). Up to now, only few genome-wide DNA methylation profiling studies in LLI have been performed, typically using array or immunoprecipitation sequencing technologies on blood-derived cells (reviewed in 7). The results show that nonagenarians and centenarians have a more youthful DNA methylation profile than expected from their chronological age (8, 9). Generally, both aging and longevity are associated with changes in DNA methylation; however, some changes can be attributed specifically to aging, others to longevity. Aging is characterized by a decrease in global DNA methylation (5, 10), which in turn is associated with higher frailty and mortality (11, 12). Interestingly, offspring of centenarians were shown to exhibit a less pronounced loss of DNA methylation (5). Thus, a better preservation of the methylation status may be one avenue of how LLI counteract or suppress age-related morbidities (13). Increased methylation of certain sites in genes influencing DNA/RNA synthesis, metabolism, and cellular signaling appears to be another longevity-promoting mechanism, since these sites are different from those that are age-dependently hypermethylated (genes involved in transcriptional regulation and development of anatomical structures) (5).
When transient biomarkers like methylation changes are investigated in pure cross-sectional studies (i.e. long-lived versus young), it is difficult to disentangle if the observed differences simply mark the longevity phenotype or actually drive it (6, 14, 15). This limitation can be partly overcome by applying a design in which the offspring of LLI are compared to offspring of parents that did not attain an exceptional old age (5). Another option, which is pursued here, is to link age-related methylation changes to genetic variation by methylation quantitative trait loci (mQTL) analysis. As the investigated variants are constitutional, they cannot be a consequence of longevity, but rather represent one of its regulators. When assessing causal effects in complex traits like longevity, it needs to be considered that allelic predisposition alone is not sufficient. In addition to specific genotypes, the manifestation of a phenotype also requires a non-genetic trigger (e.g. diet, lifestyle) with a molecular effect (e.g. change in methylation, expression).
In the present study, we generated whole-blood genome-wide methylation data from 267 individuals, of which 71 were long-lived (90–104 years). We decided against a cell type-specific analysis as Yuan et al. (16) had shown that most age-dependent DNA methylation changes observed in blood are robust and independent of the granulocyte/lymphocyte ratio, which is known to change with aging (16). We applied reduced representation bisulfite sequencing (RRBS), a technology that combines the accuracy and resolution of bisulfite sequencing with cost effectiveness and high sample throughput (17, 18). Compared to Infinium microarrays, RRBS provides higher genome-wide coverage, single-site and single-allele resolution, and insights into DNA methylation heterogeneity (19–21). Differentially methylated sites (DMSs) were identified in a stringent two-stage analysis using a discovery and a replication cohort. We subsequently correlated the obtained DMS information with previously published gene expression and single-nucleotide polymorphism (SNP) data from the same individuals (14). Through combination of several ‘omic’ techniques (epigenomics, transcriptomics, genomics) we narrowed down the DMSs to a list of 21 new longevity candidate genes, with the majority of them being linked with either neurophysiology or cancer.
Results
Description of DNA methylation data obtained by RRBS
Sequencing statistics were very similar for both study cohorts, i.e. the discovery (125 individuals) and the replication sample (142 individuals), with a median total number of sequenced reads of around 25 million, which were filtered to around 20 million reads and provided coverage for 3.6 to 3.7 million CpG sites in each sample (Supplementary Table 1). After filtering, 2 142 543 CpG sites remained in the discovery cohort and 2 007 932 CpG sites in the replication cohort that were covered by at least five reads in at least 50% of the samples. The median coverage per site was 10 reads and the median methylation ratio was 0.88 and 0.89 for the two sets of samples, respectively (Supplementary Fig. 1). About 1 944 778 sites were available in both cohorts covering 19 403 protein-coding genes, 16 669 promoters and 23 124 CGIs.
Differential methylation analysis
We first performed site-specific differential methylation analysis in the discovery sample and, applying an adjusted P-value < 0.05, we found 6381 CpG sites to be significantly differentially methylated between LLI (n = 53, age range: 91–104 years) and younger controls (n = 72, age range: 20–53 years) (either hypermethylated or hypomethylated; Fig. 1, Supplementary Table 2). Of these, 6265 DMSs were also available in the replication cohort. In that group, we did not perform an extreme group comparison (LLI versus younger controls), but recruited the individuals over a broad age range and used the age of the participants as a continuous covariate. In the replication cohort, 1851 of the 6265 DMSs showed a significant differential methylation and, strikingly, for 1797 DMSs (97.1% of the 1851 DMSs) we found the same direction of effect in both cohorts (Fig. 1, Supplementary Table 3). This overlap is significantly larger than the expected number of 995 replicated CpG sites (permutation test P-value < 10-4). About 70% (1256 CpG sites) were hypomethylated, i.e. showed lower methylation in LLI compared with younger subjects. However, the observed effect sizes were small. The median difference in mean DNA methylation levels between LLI and younger controls in the DMSs measured in the discovery sample was −0.14 and 0.116 for hypo- and hypermethylated sites, respectively (Supplementary Fig. 2). Hypomethylated sites were enriched for sites in promoter flanking regions (adjusted P-value = 1.57 × 10−98) and CTCF binding sites (adjusted P-value = 4.03 × 10−40), but depleted for sites in CpG islands (CGIs) (adjusted P-value = 7.43 × 10−126) (Supplementary Fig. 3). In contrast, hypermethylated sites were enriched for CGIs (adjusted P-value = 7.81 × 10−109), transcription factor binding sites (adjusted P-value = 4.43 × 10−61), exons (adjusted P-value = 1.09 × 10−22) and coding sequences (adjusted P-value = 2.52 × 10−22). To check for potential confounding by, e.g. differences in cell type composition, we performed an additional analysis in the discovery sample for all 6381 DMSs, adjusting for the first principal component estimated on the complete data set. After adjusting, all DMSs remained significant (adjusted P-values < 0.05) and showed the same direction of effect.
Figure 1.
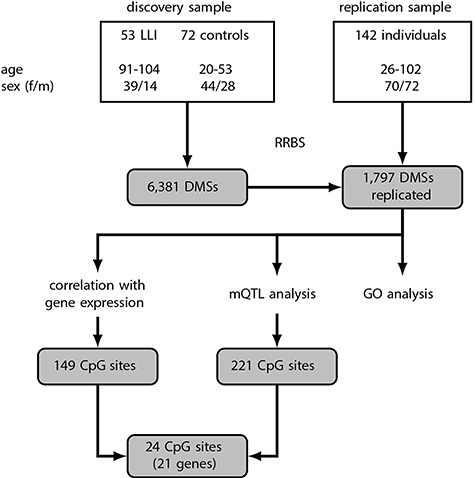
Flowchart summarizing the study design and statistical approach. DMS, differential methylated site; GO, gene ontology; LLI, long-lived individuals; mQTL, methylation quantitative trait locus.
Consecutive hypo- and hypermethylated DMSs were separately merged into 87 differentially methylated regions (DMRs) (Supplementary Table 4), of which more than 75% (65) were hypomethylated. The regions were short with a median length of 56 bp containing four CpG sites and 56 of the DMRs covered a transcript or a promoter.
Gene ontology and locus overlap analysis
A pathway analysis with the 1797 overlapping DMSs (listed in Supplementary Table 3) based on KEGG (22) and GO (23) revealed no significant GO terms or KEGG pathways for the hypomethylated DMSs. While also no significant KEGG pathways were found for the hypermethylated DMSs, 21 GO terms reached statistical significance (Supplementary Table 5); of these, 14 GO terms were over-represented and 7 GO terms were under-represented for the hypermethylated DMSs. The two most overrepresented groups were ‘homophilic cell adhesion via plasma membrane adhesion molecules’ and ‘cell-cell adhesion via plasma-membrane adhesion molecules’, while ‘protein binding’ was the most underrepresented one (Supplementary Table 5). Our observation that only for hypermethylated DMSs significant GO terms were detected is in concordance with Kananen et al. (24) and previous reports suggesting that hypomethylated DMSs form a less homogeneous group than hypermethylated DMSs (24, 25).
Enrichment analysis for genomic region sets (LOLA; http://databio.org/lola/; (26)) on the hypo- and hypermethylated DMSs yielded significant results for the hypermethylated DMSs only. Interestingly, regions that had previously been identified as binding regions of the chromatin-associated protein sirtuin 6 (SIRT6; (27)) were enriched in the subset of hypermethylated DMSs (adjusted P-value = 1.24 × 10-8, Supplementary Table 6). Increased methylation at SIRT6 binding sites could prevent protein binding and eventually lead to silencing of SIRT6-dependent gene expression (e.g. decreased expression of tumor suppressor or DNA repair genes).
Correlation analysis between CpG methylation sites and gene expression in the discovery sample
We performed correlation analysis for the 1797 DMSs. We generated 890 pairs of genes and CpG sites (CpG sites within the respective gene body or promoter) and identified 149 significant correlations (adjusted P-value < 0.05) that consisted of 149 DMSs in 102 genes (Fig. 1, Supplementary Table 7). Of these 149 correlated CpG-gene pairs, 78 exhibited a negative correlation (i.e. lower methylation associated with higher gene expression and vice versa) and 71 showed a positive correlation. The majority of correlated DMSs were observed in intronic regions. Sites with a negative correlation were more often located in exonic regions (27 versus 10%), while sites with a positive correlation were more frequent in promoters (38 versus 22%). Generally, differential methylation of genes was only to a limited extent reflected in changes of gene expression (16.7% of the 890 pairs tested). The correlation analysis between DMSs and gene expression yielded only little effects of age, i.e. aging-induced differential methylation seems to occur predominantly without concordant gene expression changes. This observation is in agreement with previous findings (16, 28, 29). For comparison, we repeated the correlation analysis for all 14 672 genes in the transcriptome data set which had at least one CpG site in their body or promoter region resulting in 1 216 157 pairs of CpG sites and genes. Only 553 pairs were significant after adjustment for multiple testing (0.045 versus 16.74% for the DMSs-based correlation analysis) covering 230 genes (1.57 versus 17.62%), but a negative correlation was observed for 73% of the significant pairs. Results were very similar when CpG sites in the gene body or promoter were considered separately (gene body: 0.045% out of 1 215 054 pairs were significant and 73% with negative correlation; promoter: 0.045% out of 1 155 976 pairs were significant and 70% with negative correlation).
Correlation between CpG sites and gene expression was stronger in the global analysis with median absolute correlation of 0.44 compared to 0.31 in the DMS-based analysis. However, 35 of the 149 significant DMS correlations were still significant after adjustment for the global analysis.
Methylation quantitative trait loci analysis
Furthermore, using genotyping data from the discovery sample (14), we tested if DNA methylation levels at the 1797 DMSs were associated with nearby common genetic variants. We tested 39 274 combinations of CpG sites and SNPs, of which 776 pairs containing 221 sites and 670 SNPs were significant (adjusted P-value < 0.05) (Fig. 1, Supplementary Table 8).
We also tested if known longevity-associated SNPs in the APOE and FOXO3 genes (30, 31) might act as mQTLs. However, neither the two tested SNPs in FOXO3 nor the APOE status (tested as dichotomous variant; ε4 versus ε2 or ε3) showed significant association with nearby CpG sites.
Identification of new longevity candidates through a combined ‘omics’ approach
To compile a list of candidate genes, we combined the results of the methylation, RNA-seq and SNP genotyping analyses. We found 24 DMSs to be significantly correlated with gene expression and to have at least one significant mQTL within 50 kb (Table 1). Hypomethylation with age was observed for all but one (LOC100128398) of these CpG sites. The effects on gene expression were diverse. Thirteen CpG sites were positively correlated with the expression of the annotated gene (i.e. the lower methylation was correlated with lower gene expression), while 11 sites showed a negative correlation (lower methylation correlated with higher gene expression). The 24 DMSs mapped to 21 genes (Table 1). A literature and database search (28, 32–41; https://gemex.eurac.edu/bioinf/age/; (42)) has shown significant age-related gene expression changes for 13 of these genes (marked with an asterisk in Table 1). In our replication cohort, the 24 DMSs showed a linear change in methylation with age that often tapered off in the elderly (Supplementary Fig. 4), confirming previous reports ((12), and references therein). As an example, the age-related methylation and concomitant gene expression changes for the CpG site chr6.150040098, annotated to the large tumor suppressor kinase 1 (LATS1), are shown in more detail (Fig. 2). Compared with younger controls, LLI showed lower levels of both methylation at the CpG site and CpG site-associated gene expression, consistent with the positive correlation between methylation and gene expression (Table 1, Fig. 2). However, when considering the methylation changes in more detail across a broad age range (in the replication sample) it becomes evident that the methylation levels decreased nearly linearly only between the ages 26 to ~ 70 years, but appear to stabilize thereafter (Fig. 2).
Table 1.
Information about differentially methylated sites (DMSs) that showed significant correlation with gene expression and association with nearby SNPs
| CpG site | Gene | Methylation changes | Gene expression changes | mQTL analysis | ||
|---|---|---|---|---|---|---|
| methylation difference (adjusted P-value) | correlation coefficient (adjusted P-value) | mQTL-SNP | maximum methylation difference (adjusted P-value) | |||
| discovery | replication | |||||
| chr14.24801071 | ADCY4* | −0.17 (6.60e−03) | −0.17 (1.44e−02) | −0.40 (2.15e−04) | rs11158632 | 0.18 (2.75e-02) |
| chr14.24801073 | ADCY4* | −0.17 (4.00e−02) | −0.19 (2.50e−02) | −0.29 (1.28e−02) | rs12891732 | 0.15 (1.29e-02) |
| chr10.49673509 | ARHGAP22* | −0.11 (6.52e−03) | −0.09 (3.74e−03) | −0.32 (4.25e−03) | rs9663790 | 0.12 (1.14e-02) |
| chr6.91007306 | BACH2* | −0.13 (8.67e−03) | −0.10 (6.24e−03) | 0.32 (4.60e−03) | rs2174281 | 0.14 (4.39e-02) |
| chr1.95393371 | CNN3* | −0.18 (1.47e−03) | −0.11 (3.80e−03) | 0.33 (2.72e−03) | rs859057 | 0.02 (1.91e-02) |
| chr17.56004243 | CUEDC1* | −0.09 (9.65e−05) | −0.05 (1.58e−03) | −0.38 (4.15e−04) | rs1017528 | 0.11 (4.15e-02) |
| chr19.45930589 | ERCC1* | −0.14 (1.84e−10) | −0.05 (1.79e−02) | −0.32 (3.29e−03) | rs11615 | 0.14 (3.07e-02) |
| chr21.40176597 | ETS2* | −0.17 (1.02e−03) | −0.10 (9.91e−04) | 0.24 (4.10e−02) | rs2283639 | 0.39 (3.23e-11) |
| chr11.61596996 | FADS2 | −0.17 (1.61e−02) | −0.14 (2.44e−02) | −0.32 (6.24e−03) | rs174535 | 0.23 (7.59e-03) |
| chr6.150040098 | LATS1 | −0.22 (8.28e−09) | −0.11 (2.27e−04) | 0.44 (4.23e−05) | rs10872646 | 0.24 (3.79e-02) |
| chr5.169694956 | LCP2* | −0.12 (2.58e−04) | −0.05 (4.01e−02) | 0.30 (6.89e−03) | rs2271146 | 0.18 (3.87e-03) |
| chr19.58513473 | LOC100128398 | 0.08 (4.95e−02) | 0.06 (1.26e−02) | −0.33 (2.72e−03) | rs11881126 | 0.14 (1.04e-03) |
| chr10.134166587 | LRRC27* | −0.10 (7.72e−05) | −0.07 (7.98e−06) | −0.31 (4.96e−03) | rs2474339 | 0.08 (2.65e-02) |
| chr13.113656361 | MCF2L* | −0.23 (3.74e−05) | −0.21 (3.72e−02) | 0.39 (3.26e−04) | rs2993282 | 0.25 (3.55e-02) |
| chr19.45351770 | PVRL2 | −0.18 (1.10e−05) | −0.06 (1.32e−02) | −0.42 (9.53e−05) | rs2075642 | 0.20 (8.22e-03) |
| chr19.45351779 | PVRL2 | −0.21 (1.81e−07) | −0.12 (3.44e−03) | −0.39 (2.15e−04) | rs395908 | 0.12 (3.79e-02) |
| chr19.45352168 | PVRL2 | −0.14 (2.62e−02) | −0.15 (1.46e−05) | −0.26 (2.39e−02) | rs4803760 | 0.36 (1.59e-03) |
| chr15.67430536 | SMAD3* | −0.07 (8.35e−04) | −0.09 (8.13e−04) | 0.39 (2.15e−04) | rs1065080 | 0.04 (5.34e-03) |
| chr22.39147165 | SUN2 | −0.11 (3.47e−02) | −0.10 (1.71e−05) | 0.39 (2.15e−04) | rs4821822 | 0.07 (1.54e-02) |
| chr12.50136491 | TMBIM6 | −0.13 (8.59e−03) | −0.10 (4.02e−02) | 0.26 (2.95e−02) | rs1993398 | 0.04 (1.29e-02) |
| chr17.10634582 | TMEM220* | −0.15 (1.11e−03) | −0.13 (1.51e−03) | 0.33 (2.87e−03) | rs406446 | 0.19 (8.05e-03) |
| chr10.73062374 | UNC5B | −0.12 (1.66e−03) | −0.04 (3.79e−03) | −0.29 (1.19e−02) | rs1538894 | 0.13 (5.45e-03) |
| chr6.30883001 | VARS2* | −0.14 (4.36e−03) | −0.21 (3.96e−02) | −0.26 (2.73e−02) | rs2249459 | 0.23 (1.99e-02) |
| chr3.22412739 | ZNF385D | −0.19 (2.19e−03) | −0.23 (9.34e−04) | −0.28 (1.86e−02) | rs9854991 | 0.22 (2.96e-02) |
Genes are listed alphabetically, sites are sorted by genes; columns show adjusted P-values as well as the difference in mean methylation levels between old and young individuals in the discovery and replication cohort; Spearman’s correlation coefficient and maximum difference in median methylation levels between genotypes. * indicates genes for which a literature and database search ((28, 32–41); https://gemex.eurac.edu/bioinf/age/ (42)) yielded significant age-related expression changes.
Figure 2.
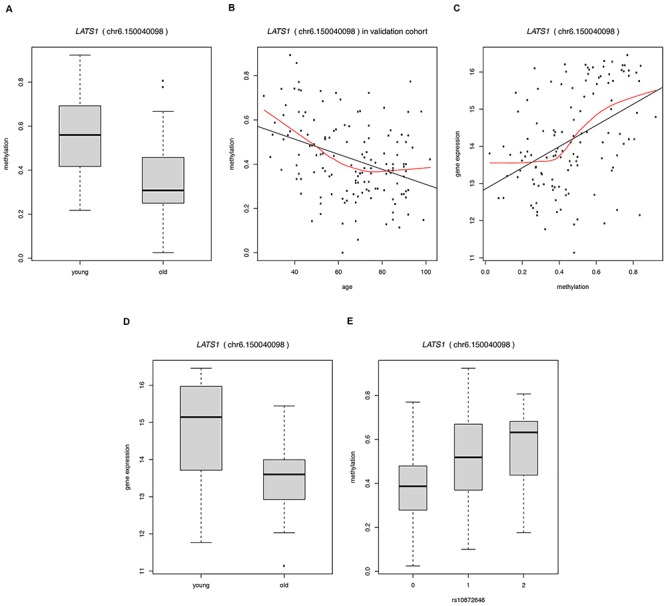
Depiction of age-related methylation at the CpG site chr6.150040098 annotated to large tumor suppressor kinase 1 (LATS1) and associated gene expression changes in the discovery and replication sample, respectively. (A) methylation difference between young and old (i.e. long-lived, ≥91 years) individuals in the discovery sample (extreme group comparison); (B) methylation changes over the age range 26–102 years in the replication sample; (C) positive correlation between methylation and gene expression; (D) gene expression difference between young and old (i.e. long-lived, ≥91 years) individuals in the discovery sample (extreme group comparison); (E) allele-specific methylation at rs10872646 with additive coding (0, homozygous for the major allele; 1, heterozygous; 2, homozygous for the minor allele).
In silico functional assessment of the longevity candidates
The functions of the 21 candidate genes and their potential involvement in age-related diseases were investigated by manual literature research in PubMed (Supplementary Table 9). Look-up of our mQTL-SNPs in the GWAS catalog showed entries for the two SNPs rs174535 and rs4803760 (Supplementary Table 10). An interaction analysis using the continuously updated InterMine database (43), which integrates data from a huge number of sources, revealed that various proteins may physically interact with the protein products of our candidate genes (Supplementary Table 11). Thirteen of these proteins showed interactions with three or more candidates from our query list (Supplementary Tables 11 and 12). Detailed information on the 13 proteins including relevant references is provided in Supplementary Table 12. Strikingly, almost all of them were previously reported in the context of neurodegeneration and neurophysiology, respectively, and all of them have also been described to play a role in human malignancies (Supplementary Table 12). The 13 proteins showed interactions with 16 of our 21 candidates (Supplementary Tables 11 and 12). Additionally, for a graphical depiction, an interaction network (Fig. 3) was generated using the STRING database (44) and the 21 candidate genes and their 13 top interaction partners (according to the InterMine database; (43)) as input variables. The protein–protein interaction P-value for the STRING network was P = 0.006, confirming more interactions than expected by chance. According to the EMBL-EBI Expression Atlas (www.ebi.ac.uk/gxa), apart from NTRK1 and EGFR (both not expressed in blood), all interaction partners and our gene candidates are expressed in blood and brain.
Figure 3.
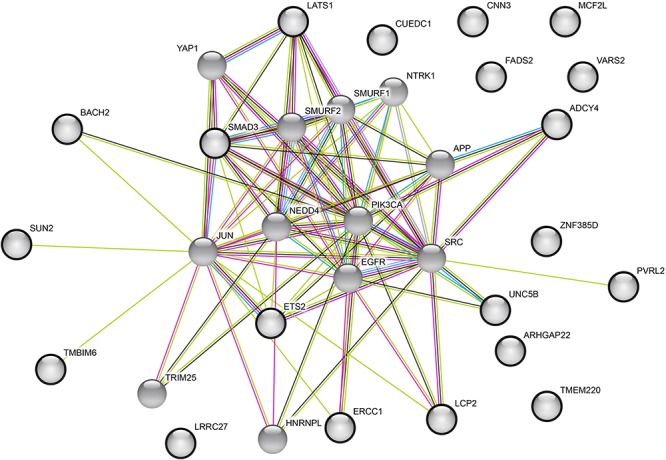
The protein–protein interaction network with the 21 candidate genes and their top 13 interaction partners as input variables analyzed using the STRING database. The top 13 interaction partners were selected based on the InterMine database. The colored edges represent the types of evidence used in the interaction predictions by the STRING software: experimental evidence (dark pink), text mining evidence (light green), database evidence (light blue), protein homology evidence (purple), and co-expression evidence (black). The 21 candidate genes are indicated by bold circles.
Investigation of allelic longevity associations
Since eight of the 24 mQTL-SNPs were present in our published German case-control data sets (45, 46), we looked up if they were associated with longevity. None of them showed an allelic association (Supplementary Table 13).
One of the 21 genes mapping to the 24 DMSs was poliovirus receptor-related 2 (PVRL2) with three significant mQTLs (Table 1, Supplementary Fig. 5). Variation in PVRL2 has previously been reported to be associated with longevity in Han Chinese (47). When we tested all 10 SNPs in the PVRL2 region that were available in our data sets (45, 46), the minor allele of rs6859 (A) was found to show an association with longevity (nominal P = 0.000792; OR = 0.87, 95% CI = 0.80–0.94; Table 2). This SNP, however, did not represent an mQTL. Since PVRL2 lies in close proximity to the APOE locus on chromosome 19q13.32, we analyzed whether the PVRL2 signal was independent of the APOE association. Conditioning for the TOMM40-SNP rs2075650 caused loss of the signal (Table 2). We observed the same finding in a Danish longevity sample (Table 2). SNP rs6859 was not among the variants investigated in the Han Chinese and also showed no high LD (R2 < 0.8) with any of the SNPs tested in that population (based on LDlink; https://ldlink.nci.nih.gov/; (48); accessed August 21, 2018).
Table 2.
Association results for SNPs in the PVRL2 gene in the German chip data sets (A) and replication of the finding for rs6859 in the Danish longevity sample (B)
| Without conditioning | Conditionede | |||||||
|---|---|---|---|---|---|---|---|---|
| SNP | MAFa LLI | MAFa Controls | P CCA b | ORc | 95% C.I.d | P CCA b | ORc | 95% C.I.d |
| A Germans | ||||||||
| 1. Illumina Immunochip data set (46) | ||||||||
| rs6859 | 0.400 | 0.432 | 0.000792 | 0.8669 | 0.798–0.942 | 0.205 | 0.944 | 0.864–1.032 |
| 2. Affymetrix® Genome-Wide Human SNP Array 6.0 data set (45) | ||||||||
| rs4081918 | 0.094 | 0.079 | 0.116 | 1.208 | 0.955–1.528 | 0.089 | 1.231 | 0.969–1.564 |
| rs11879589 | 0.093 | 0.079 | 0.137 | 1.197 | 0.945–1.516 | 0.106 | 1.220 | 0.959–1.552 |
| rs11672399 | 0.070 | 0.058 | 0.167 | 1.211 | 0.923–1.589 | 0.088 | 1.275 | 0.964–1.686 |
| rs519113 | 0.237 | 0.218 | 0.188 | 1.109 | 0.951–1.293 | 0.124 | 1.131 | 0.967–1.324 |
| rs7255063 | 0.388 | 0.394 | 0.662 | 0.970 | 0.848–1.110 | 0.465 | 0.948 | 0.820–1.095 |
| rs8104483 | 0.267 | 0.271 | 0.755 | 0.977 | 0.842–1.133 | 0.469 | 0.945 | 0.811–1.101 |
| rs17561351 | 0.055 | 0.053 | 0.762 | 1.047 | 0.779–1.407 | 0.485 | 1.114 | 0.822–1.510 |
| rs2927466 | 0.238 | 0.233 | 0.802 | 1.020 | 0.874–1.191 | 0.701 | 1.031 | 0.881–1.206 |
| rs12610605 | 0.169 | 0.166 | 0.820 | 1.021 | 0.855–1.218 | 0.833 | 0.979 | 0.807–1.188 |
| B Danes | ||||||||
| rs6859 | 0.389 | 0.436 | 0.013 | 0.8193 | 0.700–0.959 | 0.239 | 0.903 | 0.762–1.070 |
Listed are allele frequencies, allelic P-values, odds ratios and the 95% confidence intervals.
aMinor allele frequency, MAF; the definition of the minor allele is based on controls.
bAllelic P-values, PCCA; calculated with chi-squared (χ2)-test with one degree of freedom.
cOdds ratio for longevity, OR; based on the minor allele in controls.
d95% confidence interval, 95% C.I.; C.I. for the odds ratio.
Discussion
Here, we used a stringent two-stage analysis with a discovery and a replication cohort to monitor age-dependent methylation changes. DMSs observed in both study cohorts and with the same direction of effect were further combined with gene expression and SNP data from the discovery sample to identify DMSs that likely drive the longevity phenotype in humans. This approach led to the detection of 21 new candidates, the majority of which are involved in neurophysiological processes or cancer.
LLI represent a unique study population as they avoid, survive, and/or postpone age-related diseases and disability (49). An increasing number of reports indicate that LLI, and centenarians in particular, represent a paragon of successful healthy aging ((49) and references therein). Since LLI are characterized by an extraordinarily advanced age, they also show signs of the aging process. Thus, we expected the LLI in our study to exhibit molecular methylation profiles associated with both longevity and aging. This turned out to be the case.
First, we confirmed specific methylation changes that had been reported in previous studies, thus validating our approach. Various genes annotated to the DMSs identified in our study were earlier described as methylation-associated longevity genes such as KCNA3, DHX40, and TP53TG3 (all hypermethylated, Supplementary Table 3) (5). We also replicated aging-associated DMSs in e.g. LAG3 and ZEB2 (both hypomethylated) (50), FHL2 (hypermethylated) (50) or OTUD7A (hypermethylated) (51, 52) (Supplementary Table 3). One prominent marker of chronological age, ELOVL2 (53), was not covered in our RRBS data set.
In general, we found that with advanced age far more regions were hypo- than hypermethylated, which is in concordance with the literature (5, 10). Especially CTCF binding sites were affected by aging-dependent hypomethylation. CTCF is a conserved regulatory protein influencing, e.g. transcriptional activation and repression, chromatin organization, insulation, and imprinting (54). CTCF binding sites generally map to methylation-free sites genome-wide (55). In addition, Yuan et al. (16) showed that in differentiated cells versus human embryonic stem cells, CTCF binding sites were enriched in age-hypomethylated DMRs. Together with our results, this observation strengthens the hypothesis of a CTCF-dependent chromatin redistribution during aging that could contribute to altered gene expression (16).
The large extent of global hypomethylation during aging may, among other factors, be due to age-related changes in the one-carbon metabolism (crucial for methylation reactions) and to an age-related decrease in the activity of DNA methyltransferase 1 that plays an important role in maintaining genomic methylation (56). Hypomethylation likely contributes to a deregulation of gene expression and to the functional decline in aging (57).
Our strategy of combining various ‘omic’ data sets yielded 21 new candidates involved in human aging/longevity (Table 1). We defined a candidate gene as such if, in our analyses, we (i) observed DMSs in the gene consistently in both of our two study cohorts, (ii) if the differential methylation was reflected in a change in gene expression as an indicator of functional relevance, and (iii) if there was at least one mQTL-SNP within 50 kb of the CpG site. It is especially the combination of all three parameters in conjunction with non-genetic factors that provides an influence on longevity, which otherwise would not have been detectable when considering the effect of the SNPs alone (as seen in the negative allelic associations of the eight tested SNPs). Unfortunately, the currently available methods (e.g. (58)) to determine the direction of causality (e.g. SNP—> methylation—> expression changes) did not allow a robust and valid analysis of our data because of the limited sample size.
All the candidate genes, except LOC100128398, were hypomethylated with advanced age. However, the effects on gene expression were rather diverse: hypomethylation was associated with higher gene expression levels in the LLI for 10 of the 21 loci, and with lower gene expression for the remaining 10 genes. LOC100128398, the only hypermethylated locus with aging, showed a negative correlation between methylation and gene expression and therefore, the LLI exhibited lower expression levels. Interestingly, in previous aging studies, hypermethylation was generally, though not exclusively, linked to lower gene expression. Hypomethylation, in contrast, had more diverse effects on gene expression, in line with our results (29).
Most of the candidates meeting our criteria have previously been reported in humans or model organisms in the context of longevity/lifespan (CNN3, PVRL2), neurophysiology (ADCY4, CNN3, MCF2L, PVRL2, SMAD3, ZNF385D), metabolism (ADCY4, FADS2, SMAD3), cancer (e.g. ARHGAP22, BACH2, CUEDC1, ERCC1, ETS2, LATS1, LCP2, SMAD3, SUN2, TMBIM6, TMEM220, UNC5B), immune system/immunity (BACH2, LCP2, PVRL2) or lifestyle factors (smoking, ZNF385D). The remaining genes have either very diverse functions or are not well characterized yet (Table 1, Fig. 4, for more information incl. references, please refer to Supplementary Table 9).
Figure 4.
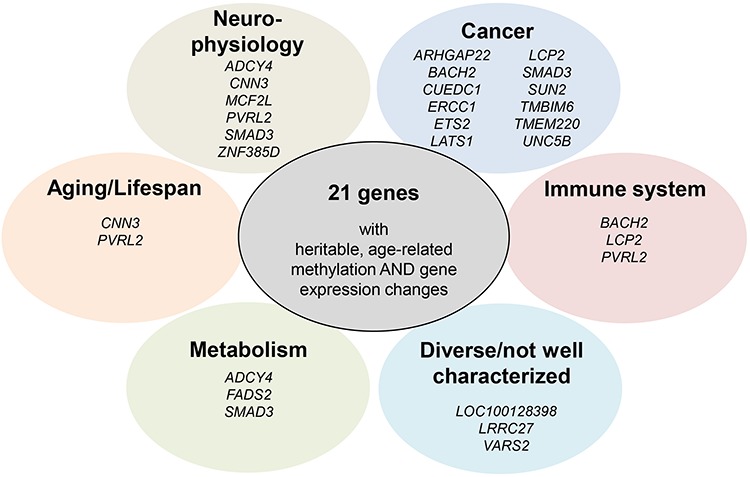
The 21 candidate genes clustered according to their functions and/or associations with diseases or other phenotypes.
Our in-depth analysis taking into account potential protein–protein interactions indicated that all gene products of our candidates may further interact with proteins involved in neuronal health (interaction partners like, e.g. APP, NTRK1, and NEDD4) or cancer (e.g. SRC, JUN, and YAP1; Fig. 3, Supplementary Tables 11 and 12). Interestingly, all our candidates are expressed in both blood and brain (EMBL-EBI Expression Atlas; www.ebi.ac.uk/gxa). This supports the potential involvement of our candidate genes in age-related conditions. It is tempting to speculate that our findings reflect the phenotype of the LLI, who, in this study, did not show overt signs of cognitive decline or cancer at the time of recruitment.
Notably, PVRL2 mapped to three mQTLs in our study and lies in very close proximity to APOE and TOMM40 on chromosome 19q (Fig. 5), both of which are negatively associated with longevity (46) and positively with Alzheimer’s disease (AD) (59, 60). SNP alleles in PVRL2 have also been previously reported in the context of longevity (47) and AD (59, 60); however, both the results from our Immunochip and from the Danish longevity sample (Table 2) indicate that the reported associations may mainly be driven by APOE and TOMM40, respectively. It needs to be clarified in future studies, if PVRL2 represents a gene that influences longevity via allelic variation and/or methylation. What remains apparent is the accumulation of aging and/or longevity loci on chromosome 19q. In addition to APOE, TOMM40, and PVRL2, also the candidate gene ERCC1 is located nearby. We observed correlations on both the methylation and the gene expression levels (Supplementary Fig. 6, Supplementary Table 14) between some of these genes, which supports their functional connection. Overall, the region on chromosome 19 might represent a longevity hub (similar to what has been proposed recently for chromosome 6; (61)) that is influenced by both genetic and epigenetic factors.
Figure 5.
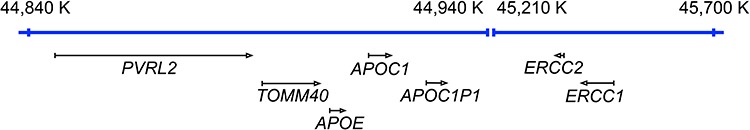
Genomic locations of PVRL2, TOMM40, APOE and ERCC1 with some adjacent genes on chromosome 19q.
Furthermore, we investigated in our study if genetic variants that have already been associated with human longevity are also correlated with methylation changes. For this purpose, we selected the APOE status (defined by the two SNPs rs429358 and rs7412 (30)) and the two FOXO3 SNPs rs4946935 and rs12206094 (31); however, we could not detect any association with nearby CpG sites. Therefore, in whole blood, the selected genetic variants do not function as mQTLs, at least not within our self-set boundary of a 50 kb distance to any CpG site. This result was not entirely surprising since it had been shown before that variation in APOE influences DNA methylation in some brain tissues (frontal cortex, temporal cortex, pons), but not in peripheral blood (62).
Our approach of linking differentially methylated sites with SNP data and gene expression yielded 21 new candidate genes for human aging/longevity. mQTLs represent heritable regions of DNA methylation, with a potential vulnerability to environmental factors. These regions are characterized by a higher stability over time and have already been proposed to be involved in the regulation of longevity (63). Indeed, a recent longitudinal study on Swedish twins showed that age-related methylation changes are fairly persistent and genetically controlled (64). However, further longitudinal studies are now needed to elucidate when during an individual’s lifespan the methylation changes occur and what the functional implications are.
The DMSs observed in the LLI mapped to a large extent to genes implicated in cancer and neurodegeneration. We hypothesize that the methylation and associated gene expression changes observed in this study are overall advantageous for the LLI, considering their good physical and cognitive health status at the time of recruitment. The pattern observed for LATS1 (Fig. 2; see also Results) is a good example. LATS1 is a tumor suppressor gene (Supplementary Table 9) and low expression levels have previously been associated with faster cancer progression (65). Our methylation data indicated a linear loss of DNA methylation with aging at the respective CpG site in LATS1 (especially in the age range from 26 to ~ 70 years); however, with a clear plateau in the elderly. Since the correlation between LATS1 methylation and gene expression was positive (higher methylation associated with higher gene expression), the LLI might benefit from a better preservation of gene expression levels. Accordingly, the methylation changes may help prevent or postpone diseases like cancer and facilitate overall survival into very old age.
Materials and Methods
Study cohorts
This study comprised two independent cohorts from Germany, one for a discovery and one for a replication stage, with a total of 267 whole blood samples that were collected with the support of the biobank PopGen (30). In the discovery sample (n = 125 individuals), we screened for DMSs using ‘extreme’ groups, i.e. LLIs who exceeded 90 years of age (n = 53, age range: 91–104 years) and younger control individuals (n = 72, age range: 20–53 years). For this set of samples, RNA-seq-based transcriptome and SNP genotyping data were available (14). For the replication, we recruited 142 individuals who represented a broad age range (26–102 years). This approach allowed us to trace the changes in methylation levels across different age groups. LLI were only included in the study if they did not show any overt signs of cognitive decline at the time of recruitment. All 267 participants signed a written informed consent. Approval for the study was received from the Ethics Committee of Kiel University. The demographics of the two study cohorts are shown in Figure 1. All German samples and information on their corresponding phenotypes were obtained from the PopGen Biobank (Schleswig-Holstein, Germany). They can be accessed through a Material Data Access Form at http://www.uksh.de/p2n/ Information+for+Researchers.html.
The Danish cases (n = 678, age range: 90.0–102.5 years) used for the replication of the rs6859-longevity-association consisted of participants drawn from the following five nation-wide surveys collected at the University of Southern Denmark: the Study of Danish Old Sibs (DOS), the 1905 Birth Cohort Study, the 1911–12 Birth Cohort Study, the Longitudinal Study of Danish Centenarians, and the Longitudinal Study of Ageing Danish Twins (LSADT) (66–68). From DOS and LSADT, one individual from each sib-ship or twin pair was randomly selected among participants that had reached an age of at least 91 years for DOS, and 90 years for LSADT. From the 1905 Birth Cohort Study, participants with a minimum age of 96 years were considered. The Danish controls (n = 738, age range: 55.9–79.9 years) consisted of individuals recruited by the Danish Twin Registry (68). Surviving participants were revisited from 2008 to 2011, when the blood samples for DNA extraction were collected. To ensure a control sample of unrelated individuals, only one twin from each twin pair was included. Collection and use of biological material and survey information were approved by the Regional Scientific Ethical Committees for Southern Denmark, and the study was approved by the Danish Data Protection Agency.
Reduced representation bisulfite sequencing
We generated genome-wide single CpG resolution methylation data for all 267 study participants using an optimized RRBS protocol (69, 70). Genomic DNA was extracted from EDTA whole blood using the AutoPure LS instrument and accessories (Qiagen GmbH, Hilden, Germany) according to the manufacturer’s instructions. For RRBS, 100 ng of extracted DNA were digested for 12 h at 37°C with 20 units of MspI (R0106L; New England Biolabs, Frankfurt am Main, Germany) in 30 μL of 1 × NEB buffer 2. Fill-in and A-tailing were performed by adding Klenow fragment (M0212L; New England Biolabs, Frankfurt am Main, Germany) and dNTP mix (10 mM dATP, 1 mM dCTP, 1 mM dGTP). After ligation to methylated Illumina TruSeq LT v2 adaptors using Quick Ligase (M2200L; New England Biolabs, Frankfurt am Main, Germany), the libraries were size-selected by performing a 0.75× clean-up with AMPure XP beads (A63881; Beckman Coulter, Brea, USA). On average six libraries were pooled in equal amounts based on qPCR data and bisulfite converted using the EZ DNA Methylation Direct Kit (D5020; Zymo Research, Irvine, CA, USA) with the following changes to the manufacturer’s protocol: conversion reagent was used at 0.9 × concentration, incubation was performed for 20 cycles of 1 min at 95°C, 10 min at 60°C and desulphonation time was extended to 30 min. Bisulfite-converted libraries were enriched for 17 cycles using PfuTurbo Cx Hotstart DNA Polymerase (600 412; Agilent Technologies, Inc., Santa Clara, CA, USA). After a 2 × AMPure XP clean-up, quality control was performed by a Qubit dsDNA HS (Q32854; Thermo Fisher Scientific Inc., Waltham, MA, USA) and Experion DNA 1 k assay (700–7107; Bio-Rad Laboratories, Inc., Hercules, CA, USA). All RRBS libraries were sequenced on the Illumina HiSeq 2000 platform using the 50-bp single-read setup.
RRBS data processing and analysis
Base calls provided by the Illumina Realtime Analysis software were converted into BAM files using Illumina2bam (https://github.com/wtsi-npg/illumina2bam) and subsequently demultiplexed using BamIndexDecoder from the same package. Initial quality control was performed with the FastQC software (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). All bioinformatic analyses were done relative to the hg19/GRCh37 assembly of the human genome. RRBS reads were aligned with BSMAP/RRBSMAP (71). Preprocessing of the sequenced reads (i.e. quality trimming, adapter removal) was done with Trimmomatic (72), and DNA methylation calling was conducted using a custom script, following a previously described method (73, 74).
Sample identity was verified by comparing predicted sex based on the DNA methylation data to annotated sex for each individual in the two cohorts. Sex was predicted using the proportion of gonosomal reads that map to the Y chromosome. CpG sites were removed when they had a common SNP (MAF > 0.05 in the European samples of the 1000 Genomes Project) (75) or tandem repeats (based on UCSC repeats from Tandem Repeats Finder) (76), or were located on the X or Y chromosome. Furthermore, CpG sites with a total coverage >105 or <5 for more than 50% of the samples were excluded. Methylation ratios at the remaining sites were set to missing if coverage was smaller than five reads.
CpG sites were annotated with regard to the location of CGIs, genes and regulatory information. CGI definitions were downloaded from UCSC (ftp://hgdownload.cse.ucsc.edu/goldenPath/hg19/database/cpgIslandExt.txt.gz). As in the Bioconductor package methylSig (77), CGI shores were defined as regions outside CGIs but within 2000 bp of any CGI. CGI shelves were defined as regions within 2000 bp from a CGI shore. Information about genes including transcript, introns, exons, 3'-UTR and 5'-UTR was extracted from Bioconductor packages Homo.sapiens and TxDb.Hsapiens.UCSC.hg19.knownGene. Promoters were defined as regions 1000 bp upstream of a transcription starting site according to the definition in methylSig. Regulatory information including enhancers and transcription factor binding sites was downloaded from Ensembl build 78 (ftp://ftp.ensembl.org/pub/release-78/regulation/homo_sapiens/RegulatoryFeatures_MultiCell.gff.gz). Chromosomal locations were mapped from genome build hg38 to hg19 using the liftOver function in the Bioconductor package rtracklayer (78).
For both study cohorts, site-based differential methylation analysis was performed using the beta regression model implemented in BiSeq (79) with sex as covariate. We selected this statistical approach since the beta distribution is an appropriate distribution for modeling methylation values that are restricted between 0 and 1. In the discovery sample, methylation values were compared between LLI and control individuals, whereas age was used as a continuous covariate in the replication sample. P-values were adjusted for multiple testing using the Benjamini–Hochberg procedure (80). To check for potential confounding, a sensitivity analysis was performed using beta regression adjusting for both sex and the first principal component.
We tested whether the observed number of replicated CpG sites (adjusted P-value < 0.05 in the validation cohort and same direction of effect) was different from the null hypothesis using a permutation test with 10 000 repetitions. To preserve correlation between neighboring CpG sites, we used a circular permutation scheme as described (81). CpG sites were placed on a ‘circular genome’ according to their genomic location and the P-values and corresponding effect sizes of the differential methylation analysis in the validation cohort were permuted by rotation with respect to the locations of the sites.
Differentially methylated regions (DMRs) were defined separately for hypo- and hypermethylated DMSs in a two-stage process. First, regions with at least three CpS sites were identified of which at least 50% were required to be DMSs. Second, overlapping regions were combined using the reduce function in the Bioconductor package GenomicRanges (82).
Generation of SNP genotype and transcriptome data in the discovery sample
For the discovery sample, we had already published genotyping data for about 700 000 SNPs (HumanOmniExpress BeadChip; Illumina, Inc., San Diego, CA, USA) as well as RNAseq-based transcriptome data (14). We removed SNPs with MAF < 5%, a call rate < 95%, and Hardy–Weinberg P-value < 0.0001. Finally, we had methylation and genotype (587 716 SNPs) data for 121 individuals, and methylation and transcriptome data for 125 individuals, respectively.
Methylation quantitative trait loci analysis in the discovery sample
We analyzed associations between common genetic variants and methylation levels in the discovery cohort. If fewer than five individuals were homozygous for the minor allele, we combined the homozygous minor allele and heterozygous genotype into one group. We tested mQTL associations between CpG sites that were differently methylated in both study cohorts and SNPs with a maximal distance of 50 kb to the CpG sites using beta regression with an additive coding of the genotypes. P-values were adjusted for multiple testing using the Benjamini-Hochberg procedure.
Correlation analysis between CpG methylation sites and gene expression in the discovery sample
Only those CpG sites that were differentially methylated in both study cohorts were considered for correlation analysis with gene expression data from the discovery sample. We further removed genes on alternate representations of chromosomes and with more than 50% zero counts. For gene names with several genomic locations, we used the location with the largest total count. We correlated cis-CpG sites and gene expression pairs, where cis refers to the CpG sites that were located within the gene body or promoter. Spearman’s rank correlation was used to assess the correlation between methylation levels and gene expression. P-values were corrected for multiple testing using the Benjamini–Hochberg method.
APOE and FOXO3 genotyping and mQTL association testing
Known longevity-associated SNPs in the APOE locus (rs429358, rs7412; together defining the 3 APOE alleles (30)) and the FOXO3 gene (rs12206094, rs4946935 (31)) were genotyped in both study cohorts using TaqMan technology (Thermo Fisher Scientific Germany BV & Co. KG, Braunschweig, Germany). For our analysis, we used a binary coding with ε4 as one allele and any other allele as the second allele. We tested mQTL associations with CpG sites within 50 kb around each SNP using the same approach as for the general mQTL analysis (see previous subsection).
In silico data exploration
By combining our methylation results with the genotyping and transcriptome data, we defined a list of 21 potential candidate longevity genes (Table 1). Hereof, a candidate gene must (i) exhibit age-associated DMSs (consistent in our two study cohorts), (ii) show methylation-associated gene expression, and (iii) contain at least one mQTL-SNP within 50 kb of the CpG site. Interaction analysis of the candidate genes was then performed with InterMine (www.humanmine.org; (43); accessed August 20, 2018), and an interaction network was generated with the STRING software (44) using the 21 candidate genes and their top 13 potential interaction partners suggested by InterMine (43) as input variables. mQTL-SNPs were looked up in the NHGRI-EBI GWAS Catalog (www.ebi.ac.uk/gwas/; accessed July 10, 2018). Expression patterns were evaluated using the EMBL-EBI Expression Atlas (www.ebi.ac.uk/gxa; accessed August 20, 2018).
For eight of the 24 mQTL-SNPs, genotypes were available in our published German case-control data sets (45, 46). These SNPs were investigated for a longevity-association using logistic regression with sex as covariate.
Since variants in the PVRL2 gene have previously been associated with longevity (47), all SNPs in the PVRL2 region were extracted from the Ensembl database (83). The 10 SNPs present in our data sets (45, 46) were subjected to association testing as described above.
For the replication of the rs6859-longevity-association in the Danish, for the cases, data on rs6859 and rs2075650 was extracted from quality controlled Illumina HumanOmniExpress Array (Illumina San Diego, CA, USA) genotype data. Quality control included filtering of SNPs on genotype call rate < 95%, HWE P < 10−4, and MAF < 1%, and individuals on sample call rate < 95%, relatedness and gender mismatch. For controls, data on rs6859 and rs2075650 was extracted from quality controlled genotype data created using the Illumina Infinium PsychArray (Illumina San Diego, CA, USA). Quality control included filtering SNPs on genotype call rate < 98%, HWE P < 10−6, and MAF = 0, and individuals on sample call rate < 99%, relatedness and gender mismatch. After merging of data from cases and controls, fulfillment of the Hardy-Weinberg equilibrium (P > 0.05) was confirmed for both SNPs. Association analysis was performed in Plink, where logistic regression was used to assess the association between case/control status and the rs6859 genotype. Sex was included as a covariate, and the condition-option in Plink was included to perform the analysis conditioned on rs2075650.
Supplementary Material
Acknowledgements
We thank the Biomedical Sequencing Facility at CeMM for assistance with next-generation sequencing, and Johanna Hadler for her help in the RRBS profiling. Genotyping of the Danish controls was conducted by the SNP&SEQ Technology Platform, Science for Life Laboratory, Uppsala, Sweden (http://snpseq.medsci.uu.se/genotyping/snp-services/).
Funding
Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy (EXC 306); EpiHealth (EU FP7, HEALTH-2012-F2-278418); INTERREG 4A Syddanmark-Schleswig-K.E.R.N; Bundesministerium für Bildung und Forschung (BMBF, Federal Ministry of Education and Research; grant no. 01EY1103); National Program for Research Infrastructure 2007 (grant no. 09-063256); Danish Agency for Science Technology and Innovation; Velux Foundation; US National Institute of Health (P01 AG08761); National Institutes of Health (R01 AG037985) (Pedersen).
Conflict of Interest Statement
The authors have no conflict of interest to declare.
Author Contributions
A.N. and A.F. conceived the project; S.Sc., F.F. and W.L. organized recruitment and collection of the samples. Genotyping data of the German individuals (apart from the published chip data) were collected and processed by S.Sz., J.D., G.V., R.H. and W.K.; M.N., J.M.-F. and K.C. were responsible for the recruitment, genotyping and analysis of the Danish longevity sample; RRBS data were generated by P.D. and C.B.; DNA methylation profiles were analyzed by S.Sz., F.-A.H. and G.V. Functional implications of the findings were assessed by J.D., G.G.T., A.N. and S.Sz. Statistical analyses were performed by S.Sz. A.N., J.D., G.G.T. and R.H. interpreted the results. A.N., J.D. and S.Sz. wrote the manuscript, with input from all other authors.
References
- 1. Sebastiani P. and Perls T.T. (2012) The genetics of extreme longevity: lessons from the New England centenarian study. Front. Genet., 3, 277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Andersen S.L., Sebastiani P., Dworkis D.A., Feldman L. and Perls T.T. (2012) Health span approximates life span among many supercentenarians: compression of morbidity at the approximate limit of life span. J. Gerontol. A Biol. Sci. Med. Sci., 67A, 395–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Murabito J.M., Yuan R. and Lunetta K.L. (2012) The search for longevity and healthy aging genes: insights from epidemiological studies and samples of long-lived individuals. J. Gerontol. A Biol. Sci. Med. Sci., 67A, 470–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. vB Hjelmborg, J., Iachine, I., Skytthe, A., Vaupel, J.W., McGue, M., Koskenvuo, M., Kaprio, J., Pedersen, N.L. and Christensen, K. (2006) Genetic influence on human lifespan and longevity. Hum. Genet., 119, 312–321. [DOI] [PubMed] [Google Scholar]
- 5. Gentilini D., Mari D., Castaldi D., Remondini D., Ogliari G., Ostan R., Bucci L., Sirchia S.M., Tabano S., Cavagnini F. et al. (2013) Role of epigenetics in human aging and longevity: genome-wide DNA methylation profile in centenarians and centenarians’ offspring. Age (Dordr.), 35, 1961–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Elsharawy A., Keller A., Flachsbart F., Wendschlag A., Jacobs G., Kefer N., Brefort T., Leidinger P., Backes C., Meese E. et al. (2012) Genome-wide miRNA signatures of human longevity. Aging Cell, 11, 607–616. [DOI] [PubMed] [Google Scholar]
- 7. Bacalini M.G., D’Aquila P., Marasco E., Nardini C., Montesanto A., Franceschi C., Passarino G., Garagnani P. and Bellizzi D. (2017) The methylation of nuclear and mitochondrial DNA in ageing phenotypes and longevity. Mech. Ageing Dev., 165, 156–161. [DOI] [PubMed] [Google Scholar]
- 8. Armstrong N.J., Mather K.A., Thalamuthu A., Wright M.J., Trollor J.N., Ames D., Brodaty H., Schofield P.R., Sachdev P.S. and Kwok J.B. (2017) Aging, exceptional longevity and comparisons of the Hannum and Horvath epigenetic clocks. Epigenomics, 9, 689–700. [DOI] [PubMed] [Google Scholar]
- 9. Horvath S., Pirazzini C., Bacalini M.G., Gentilini D., Di Blasio A.M., Delledonne M., Mari D., Arosio B., Monti D., Passarino G. et al. (2015) Decreased epigenetic age of PBMCs from Italian semi-supercentenarians and their offspring. Aging (Albany NY), 7, 1159–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Heyn H., Li N., Ferreira H.J., Moran S., Pisano D.G., Gomez A., Diez J., Sanchez-Mut J.V., Setien F., Carmona F.J. et al. (2012) Distinct DNA methylomes of newborns and centenarians. Proc. Natl. Acad. Sci. U. S. A., 109, 10522–10527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bellizzi D., D’Aquila P., Montesanto A., Corsonello A., Mari V., Mazzei B., Lattanzio F. and Passarino G. (2012) Global DNA methylation in old subjects is correlated with frailty. Age (Dordr.), 34, 169–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Christiansen L., Lenart A., Tan Q., Vaupel J.W., Aviv A., McGue M. and Christensen K. (2015) DNA methylation age is associated with mortality in a longitudinal Danish twin study. Aging Cell, 15, 149–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Xiao F.H., He Y.H., Li Q.G., Wu H., Luo L.H. and Kong Q.P. (2015) A genome-wide scan reveals important roles of DNA methylation in human longevity by regulating age-related disease genes. PLoS One, 10, e0120388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Häsler R., Venkatesh G., Tan Q., Flachsbart F., Sinha A., Rosenstiel P., Lieb W., Schreiber S., Christensen K., Christiansen L., Nebel A. (2017) Genetic interplay between human longevity and metabolic pathways — a large-scale eQTL study. Aging Cell, 16, 716–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wurmb-Schwark N., Schwark T., Caliebe A., Drenske C., Nikolaus S., Schreiber S. and Nebel A. (2010) Low level of the mtDNA(4977) deletion in blood of exceptionally old individuals. Mech. Ageing Dev., 131, 179–184. [DOI] [PubMed] [Google Scholar]
- 16. Yuan T., Jiao Y., Jong S., Ophoff R.A., Beck S. and Teschendorff A.E. (2015) An integrative multi-scale analysis of the dynamic DNA methylation landscape in aging. PLoS Genet., 11, e1004996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gu H., Bock C., Mikkelsen T.S., Jäger N., Smith Z.D., Tomazou E., Gnirke A., Lander E.S. and Meissner A. (2010) Genome-scale DNA methylation mapping of clinical samples at single-nucleotide resolution. Nat. Methods, 7, 133–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Meissner A., Gnirke A., Bell G.W., Ramsahoye B., Lander E.S. and Jaenisch R. (2005) Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res., 33, 5868–5877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bock C., Tomazou E.M., Brinkman A.B., Müller F., Simmer F., Gu H., Jäger N., Gnirke A., Stunnenberg H.G. and Meissner A. (2010) Quantitative comparison of genome-wide DNA methylation mapping technologies. Nat. Biotechnol., 28, 1106–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Klughammer J., Kiesel B., Roetzer T., Fortelny N., Nemc A., Nenning K.H., Furtner J., Sheffield N.C., Datlinger P., Peter N. et al. (2018) The DNA methylation landscape of glioblastoma disease progression shows extensive heterogeneity in time and space. Nat. Med., 24, 1611–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sheffield N.C., Pierron G., Klughammer J., Datlinger P., Schönegger A., Schuster M., Hadler J., Surdez D., Guillemot D., Lapouble E. et al. (2017) DNA methylation heterogeneity defines a disease spectrum in Ewing sarcoma. Nat. Med., 23, 386–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ogata H., Goto S., Sato K., Fujibuchi W., Bono H. and Kanehisa M. (1999) KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res., 27, 29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T. et al. (2000) Gene ontology: tool for the unification of biology. Nat. Genet., 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kananen L., Marttila S., Nevalainen T., Jylhävä J., Mononen N., Kähönen M., Raitakari O.T., Lehtimäki T. and Hurme M. (2016) Aging-associated DNA methylation changes in middle-aged individuals: the Young Finns study. BMC Genomics, 17, 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Reynolds L.M., Taylor J.R., Ding J., Lohman K., Johnson C., Siscovick D., Burke G., Post W., Shea S., Jacobs D.R. Jr. et al. (2014) Age-related variations in the methylome associated with gene expression in human monocytes and T cells. Nat. Commun., 5, 5366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sheffield N.C. and Bock C. (2016) LOLA: enrichment analysis for genomic region sets and regulatory elements in R and bioconductor. Bioinformatics, 32, 587–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ram O., Goren A., Amit I., Shoresh N., Yosef N., Ernst J., Kellis M., Gymrek M., Issner R., Coyne M. et al. (2011) Combinatorial patterning of chromatin regulators uncovered by genome-wide location analysis in human cells. Cell, 147, 1628–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hannum G., Guinney J., Zhao L., Zhang L., Hughes G., Sadda S., Klotzle B., Bibikova M., Fan J.B., Gao Y. et al. (2013) Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell, 49, 359–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Steegenga W.T., Boekschoten M.V., Lute C., Hooiveld G.J., Groot P.J., Morris T.J., Teschendorff A.E., Butcher L.M., Beck S. and Müller M. (2014) Genome-wide age-related changes in DNA methylation and gene expression in human PBMCs. Age (Dordr.), 36, 9648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nebel A., Croucher P.J., Stiegeler R., Nikolaus S., Krawczak M. and Schreiber S. (2005) No association between microsomal triglyceride transfer protein (MTP) haplotype and longevity in humans. Proc. Natl. Acad. Sci. U. S. A., 102, 7906–7909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Flachsbart F., Dose J., Gentschew L., Geismann C., Caliebe A., Knecht C., Nygaard M., Badarinarayan N., Elsharawy A., May S. et al. (2017) Identification and characterization of two functional variants in the human longevity gene FOXO3. Nat. Commun., 8, 2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Harries L.W., Hernandez D., Henley W., Wood A.R., Holly A.C., Bradley-Smith R.M., Yaghootkar H., Dutta A., Murray A., Frayling T.M. et al. (2011) Human aging is characterized by focused changes in gene expression and deregulation of alternative splicing. Aging Cell, 10, 868–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Passtoors W.M., Boer J.M., Goeman J.J., Akker E.B., Deelen J., Zwaan B.J., Scarborough A., Breggen Rv., Vossen R.H., Houwing-Duistermaat J.J. et al. (2012) Transcriptional profiling of human familial longevity indicates a role for ASF1A and IL7R. PLoS One, 7, e27759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Akker E.B., Passtoors W.M., Jansen R., Zwet E.W., Goeman J.J., Hulsman M., Emilsson V., Perola M., Willemsen G., Penninx B.W. et al. (2014) Meta-analysis on blood transcriptomic studies identifies consistently coexpressed protein–protein interaction modules as robust markers of human aging. Aging Cell, 13, 216–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Peters M.J., Joehanes R., Pilling L.C., Schurmann C., Conneely K.N., Powell J., Reinmaa E., Sutphin G.L., Zhernakova A., Schramm K. et al. (2015) The transcriptional landscape of age in human peripheral blood. Nat. Commun., 6, 8570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Reynolds L.M., Ding J., Taylor J.R., Lohman K., Soranzo N., Fuente A., Liu T.F., Johnson C., Barr R.G., Register T.C. et al. (2015) Transcriptomic profiles of aging in purified human immune cells. BMC Genomics, 16, 333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Irizar H., Goñi J., Alzualde A., Castillo-Triviño T., Olascoaga J., Lopez de Munain A. and Otaegui D. (2015) Age gene expression and coexpression progressive signatures in peripheral blood leukocytes. Exp. Gerontol., 72, 50–56. [DOI] [PubMed] [Google Scholar]
- 38. Yang J., Huang T., Petralia F., Long Q., Zhang B., Argmann C., Zhao Y., Mobbs C.V., Schadt E.E., Zhu J. et al. (2015) Synchronized age-related gene expression changes across multiple tissues in human and the link to complex diseases. Sci. Rep., 5, 15145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bryois J., Buil A., Ferreira P.G., Panousis N.I., Brown A.A., Viñuela A., Planchon A., Bielser D., Small K., Spector T. et al. (2017) Time-dependent genetic effects on gene expression implicate aging processes. Genome Res., 27, 545–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li C., Mo D., Li M., Zheng Y., Li Q., Ou S. and Zhang Z. (2019) Age-related but not longevity-related genes are found by weighted gene co-expression network analysis in the peripheral blood cells of humans. Genes Genet. Syst., 93, 221–228. [DOI] [PubMed] [Google Scholar]
- 41. Jia K., Cui C., Gao Y., Zhou Y. and Cui Q. (2018) An analysis of aging-related genes derived from the genotype-tissue expression project (GTEx). Cell Death Discov., 4, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Blankenburg H., Pramstaller P.P. and Domingues F.S. (2018) A network-based meta-analysis for characterizing the genetic landscape of human aging. Biogerontology, 19, 81–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Smith R.N., Aleksic J., Butano D., Carr A., Contrino S., Hu F., Lyne M., Lyne R., Kalderimis A., Rutherford K. et al. (2012) InterMine: a flexible data warehouse system for the integration and analysis of heterogeneous biological data. Bioinformatics, 28, 3163–3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Szklarczyk D., Morris J.H., Cook H., Kuhn M., Wyder S., Simonovic M., Santos A., Doncheva N.T., Roth A., Bork P. et al. (2017) The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res., 45, D362–D368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nebel A., Kleindorp R., Caliebe A., Nothnagel M., Blanché H., Junge O., Wittig M., Ellinghaus D., Flachsbart F., Wichmann H.E. et al. (2011) A genome-wide association study confirms APOE as the major gene influencing survival in long-lived individuals. Mech. Ageing Dev., 132, 324–330. [DOI] [PubMed] [Google Scholar]
- 46. Flachsbart F., Ellinghaus D., Gentschew L., Heinsen F.A., Caliebe A., Christiansen L., Nygaard M., Christensen K., Blanché H., Deleuze J.F. et al. (2016) Immunochip analysis identifies association of the RAD50/IL13 region with human longevity. Aging Cell, 15, 585–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lu F., Guan H., Gong B., Liu X., Zhu R., Wang Y., Qian J., Zhou T., Lan X., Wang P. et al. (2014) Genetic variants in PVRL2-TOMM40-APOE region are associated with human longevity in a Han Chinese population. PLoS One, 9, e99580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Machiela M.J. and Chanock S.J. (2015) LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics, 31, 3555–3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Engberg H., Oksuzyan A., Jeune B., Vaupel J.W. and Christensen K. (2009) Centenarians–a useful model for healthy aging? A 29-year follow-up of hospitalizations among 40,000 Danes born in 1905. Aging Cell, 8, 270–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bacalini M.G., Boattini A., Gentilini D., Giampieri E., Pirazzini C., Giuliani C., Fontanesi E., Remondini D., Capri M., Del Rio A. et al. (2015) A meta-analysis on age-associated changes in blood DNA methylation: results from an original analysis pipeline for Infinium 450k data. Aging (Albany NY), 7, 97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Marttila S., Kananen L., Häyrynen S., Jylhävä J., Nevalainen T., Hervonen A., Jylhä M., Nykter M. and Hurme M. (2015) Ageing-associated changes in the human DNA methylome: genomic locations and effects on gene expression. BMC Genomics, 16, 179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Florath I., Butterbach K., Müller H., Bewerunge-Hudler M. and Brenner H. (2014) Cross-sectional and longitudinal changes in DNA methylation with age: an epigenome-wide analysis revealing over 60 novel age-associated CpG sites. Hum. Mol. Genet., 23, 1186–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Garagnani P., Bacalini M.G., Pirazzini C., Gori D., Giuliani C., Mari D., Di Blasio A.M., Gentilini D., Vitale G., Rezzi S. et al. (2012) Methylation of ELOVL2 gene as a new epigenetic marker of age. Aging Cell, 11, 1132–1134. [DOI] [PubMed] [Google Scholar]
- 54. Phillips J.E. and Corces V.G. (2009) CTCF: master weaver of the genome. Cell, 137, 1194–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mukhopadhyay R., Yu W., Whitehead J., Xu J., Lezcano M., Pack S., Kanduri C., Kanduri M., Ginjala V., Vostrov A. et al. (2004) The binding sites for the chromatin insulator protein CTCF map to DNA methylation-free domains genome-wide. Genome Res., 14, 1594–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Jung M. and Pfeifer G.P. (2015) Aging and DNA methylation. BMC Biol., 13, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Richardson B. (2003) Impact of aging on DNA methylation. Ageing Res. Rev., 2, 245–261. [DOI] [PubMed] [Google Scholar]
- 58. Hemani G., Tilling K. and Davey Smith G. (2017) Orienting the causal relationship between imprecisely measured traits using GWAS summary data. PLoS Genet., 13, e1007081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Abraham R., Moskvina V., Sims R., Hollingworth P., Morgan A., Georgieva L., Dowzell K., Cichon S., Hillmer A.M., O’Donovan M.C. et al. (2008) A genome-wide association study for late-onset Alzheimer’s disease using DNA pooling. BMC Med. Genomics, 1, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Marioni R.E., Harris S.E., Zhang Q., McRae A.F., Hagenaars S.P., Hill W.D., Davies G., Ritchie C.W., Gale C.R., Starr J.M. et al. (2018) GWAS on family history of Alzheimer’s disease. Transl. Psychiatry, 8, 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Donlon T.A., Morris B.J., Chen R., Masaki K.H., Allsopp R.C., Willcox D.C., Elliott A. and Willcox B.J. (2017) FOXO3 longevity interactome on chromosome 6. Aging Cell, 16, 1016–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Smith A.K., Kilaru V., Kocak M., Almli L.M., Mercer K.B., Ressler K.J., Tylavsky F.A. and Conneely K.N. (2014) Methylation quantitative trait loci (meQTLs) are consistently detected across ancestry, developmental stage, and tissue type. BMC Genomics, 15, 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Bell J.T., Tsai P.C., Yang T.P., Pidsley R., Nisbet J., Glass D., Mangino M., Zhai G., Zhang F., Valdes A. et al. (2012) Epigenome-wide scans identify differentially methylated regions for age and age-related phenotypes in a healthy ageing population. PLoS Genet., 8, e1002629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wang Y., Karlsson R., Lampa E., Zhang Q., Hedman Å.K., Almgren M., Almqvist C., McRae A.F., Marioni R.E., Ingelsson E. et al. (2018) Epigenetic influences on aging: a longitudinal genome-wide methylation study in old Swedish twins. Epigenetics, 13, 975–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ji T., Liu D., Shao W., Yang W., Wu H. and Bian X. (2012) Decreased expression of LATS1 is correlated with the progression and prognosis of glioma. J. Exp. Clin. Cancer Res., 31, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Rasmussen S.H., Andersen-Ranberg K., Thinggaard M., Jeune B., Skytthe A., Christiansen L., Vaupel J.W., McGue M. and Christensen K. (2017) Cohort profile: the 1895, 1905, 1910, and 1915 Danish birth cohort studies - secular trends in the health and functioning of the very old. Int. J. Epidemiol., 46, 1746–1746j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Robine J.-M., Cheung S.L., Saito Y., Jeune B., Parker M.G. and Herrmann F.R. (2010) Centenarians today: new insights on selection from the 5-COOP study. Curr. Gerontol. Geriatr. Res., 2010, 120354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Pedersen D.A., Larsen L.A., Nygaard M., Mengel-From J., McGue M., Dalgård C., Hvidberg L., Hjelmborg J., Skytthe A., Holm N.V. et al. (2019) The Danish twin registry: an updated overview. Twin Res. Hum. Genet., 22, 499–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Klughammer J., Datlinger P., Printz D., Sheffield N.C., Farlik M., Hadler J., Fritsch G. and Bock C. (2015) Differential DNA methylation analysis without a reference genome. Cell Rep., 13, 2621–2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Veillard A.C., Datlinger P., Laczik M., Squazzo S. and Bock C. (2016) Diagenode® premium RRBS technology: cost-effective DNA methylation mapping with superior coverage. Nat. Methods, 13, 184. [Google Scholar]
- 71. Xi Y., Bock C., Müller F., Sun D., Meissner A. and Li W. (2012) RRBSMAP: a fast, accurate and user-friendly alignment tool for reduced representation bisulfite sequencing. Bioinformatics, 28, 430–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Bolger A.M., Lohse M. and Usadel B. (2014) Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics, 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Assenov Y., Müller F., Lutsik P., Walter J., Lengauer T. and Bock C. (2014) Comprehensive analysis of DNA methylation data with RnBeads. Nat. Methods, 11, 1138–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Bock C., Beerman I., Lien W.H., Smith Z.D., Gu H., Boyle P., Gnirke A., Fuchs E., Rossi D.J. and Meissner A. (2012) DNA methylation dynamics during in vivo differentiation of blood and skin stem cells. Mol. Cell, 47, 633–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. The 1000 Genomes Projekt Consortium, Auton A., Brooks L.D., Durbin R.M., Garrison E.P., Kang H.M., Korbel J.O., Marchini J.L., McCarthy S., McVean G.A. et al. (2015) A global reference for human genetic variation. Nature, 526, 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Benson G. (1999) Tandem repeats finder:a program to analyze DNA sequences. Nucleic Acids Res., 27, 573–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Park Y., Figueroa M.E., Rozek L.S. and Sartor M.A. (2014) MethylSig: a whole genome DNA methylation analysis pipeline. Bioinformatics, 30, 2414–2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Lawrence M., Gentleman R. and Carey V. (2009) Rtracklayer: an R package for interfacing with genome browsers. Bioinformatics, 25, 1841–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Hebestreit K., Dugas M. and Klein H.U. (2013) Detection of significantly differentially methylated regions in targeted bisulfite sequencing data. Bioinformatics, 29, 1647–1653. [DOI] [PubMed] [Google Scholar]
- 80. Hochberg Y. and Benjamini Y. (1990) More powerful procedures for multiple significance testing. Stat. Med., 9, 811–818. [DOI] [PubMed] [Google Scholar]
- 81. Cabrera C.P., Navarro P., Huffman J.E., Wright A.F., Hayward C., Campbell H., Wilson J.F., Rudan I., Hastie N.D., Vitart V. et al. (2012) Uncovering networks from genome-wide association studies via circular genomic permutation. G3 (Bethesda), 2, 1067–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Lawrence M., Huber W., Pagès H., Aboyoun P., Carlson M., Gentleman R., Morgan M.T. and Carey V.J. (2013) Software for computing and annotating genomic ranges. PLoS Comput. Biol., 9, e1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Aken B.L., Achuthan P., Akanni W., Amode M.R., Bernsdorff F., Bhai J., Billis K., Carvalho-Silva D., Cummins C., Clapham P. et al. (2017) Ensembl 2017. Nucleic Acids Res., 45, D635–D642. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.