

Abstract
Regulatory variation plays a major role in complex disease and that cell type-specific binding of transcription factors (TF) is critical to gene regulation. However, assessing the contribution of genetic variation in TF-binding sites to disease heritability is challenging, as binding is often cell type-specific and annotations from directly measured TF binding are not currently available for most cell type-TF pairs. We investigate approaches to annotate TF binding, including directly measured chromatin data and sequence-based predictions. We find that TF-binding annotations constructed by intersecting sequence-based TF-binding predictions with cell type-specific chromatin data explain a large fraction of heritability across a broad set of diseases and corresponding cell types; this strategy of constructing annotations addresses both the limitation that identical sequences may be bound or unbound depending on surrounding chromatin context and the limitation that sequence-based predictions are generally not cell type-specific. We partitioned the heritability of 49 diseases and complex traits using stratified linkage disequilibrium (LD) score regression with the baseline-LD model (which is not cell type-specific) plus the new annotations. We determined that 100 bp windows around MotifMap sequenced-based TF-binding predictions intersected with a union of six cell type-specific chromatin marks (imputed using ChromImpute) performed best, with an 58% increase in heritability enrichment compared to the chromatin marks alone (11.6× vs. 7.3×, P = 9 × 10−14 for difference) and a 20% increase in cell type-specific signal conditional on annotations from the baseline-LD model (P = 8 × 10−11 for difference). Our results show that TF-binding annotations explain substantial disease heritability and can help refine genome-wide association signals.
Introduction
Genome-wide association studies have revealed that non-coding genetic variation plays a central role in complex diseases and traits (1–3). Partitioning disease heritability has further aided our understanding of the contribution of specific genomic features, shining a particular spotlight on cell type-specific regulation (4–7). Transcription factors (TFs) are key elements of transcriptional regulation (8–10), and changes in their binding are known to affect human disease (11–17). However, TFs are numerous and their binding is often cell type-specific; directly measuring TF binding is possible using ChIP-seq (18), but ChIP-seq data have been generated for only a limited number of TFs and cell types (19,20); a complete atlas of all TF-binding sites would require tens of thousands of experiments, requiring immense resources. Because of these complexities, the contribution of genetic variation in TF-binding sites to disease heritability has not been assessed for a broad set of diseases and corresponding cell types.
Many TFs bind specifically to unique motifs in the DNA sequence (21–23), and their binding preferences can be inferred using sequence alone (24–32). However, these sequence-based predictions often lack specificity as chromatin context has profound effects on TF binding. The vast majority of matches to a TF consensus sequence fall in regions of heterochromatin, which are inaccessible and therefore not actually bound (19). It has been shown that incorporating chromatin information and footprints from DNase-seq, ATAC-seq, or histone modification ChIP-seq in addition to sequence can greatly improve prediction of TF binding (33–35). However, methods that include footprinting depend on signal unique to ATAC-seq and DNase-seq, which is lost when the data are imputed, and are thus limited to cell types where directly measured ATAC-seq or DNase-seq data are available. Since ATAC-seq and DNase-seq are not currently available in most of the Roadmap cell types (20), we sought an alternate strategy for annotating cell type-specific TF-binding sites.
Here, we consider measurements of cell type-specific regulatory activity, which are available for more than 100 cell types (20,36). We intersect various sequence-based TF annotations with cell type-specific chromatin annotations [including those imputed using ChromImpute (37)], creating cell type-specific TF-binding annotations for many tissues and cell types. This strategy addresses both the limitation that identical sequences may be bound or unbound depending on surrounding chromatin context and the limitation that sequence-based predictions are often not cell type-specific and can easily be applied to a broad set of tissues and cell types. We use stratified LD score regression (S-LDSC) (4) with the baseline-LD model (5) plus the new annotations to partition the heritability of 49 diseases and complex traits (average N = 320 K) in order to evaluate the contribution of these cell type-specific TF binding annotations to disease.
Results
Cell type-specific TF-binding annotations predict direct measurements of TF binding
To create more accurate annotations of cell type-specific TF binding, we intersected sequence-based predictions with cell type-specific chromatin annotations (Fig. 1; see Materials and Methods). We constructed cell type-specific chromatin annotations by taking the union of ChIP-seq peaks from five histone modifications that have previously been associated with active enhancers and promoters (H3K4me1, H3K4me2, H3K4me3, H3K9ac and H3K27ac) as well as DNase1 hypersensitive sites (38), available in 127 tissues and cell types as part of the Roadmap Epigenomics project (20). As experimental data are not available for every chromatin mark in every cell type, we constructed two sets of annotations: one from all available directly measured peaks and one from imputed peaks computed for each chromatin mark and cell type using ChromImpute (37). We call these combined cell type-specific chromatin annotations ‘Chromatin.measured’ and ‘Chromatin.imputed’, respectively. We intersected the cell type-specific chromatin annotations with three sets of sequence-based TF-binding predictions: MotifMap (24), Kheradpour et al. (25) and CisBP (39). MotifMap uses sequence preferences from TRANSFAC (21) and JASPAR (22,23) as well as conservation to predict binding. Kheradpour et al. train many motif-finding methods on ENCODE TF ChIP-seq data and choose those that perform best to apply genome-wide. CisBP is a large database of TF-binding preferences from many sources. For each TF prediction set, we also tested annotations that include 20, 50, and 100 bp windows. These windows may capture effects of sequence outside of the core motif (40) and may capture cooperative binding sites for TFs that were not included in the datasets. We did not include sequence-based TF-binding predictions produced by deep learning methods (27–31) in our main analyses (see Discussion). We have made our annotations and partitioned LD scores freely available (see Web Resources).
Figure 1.
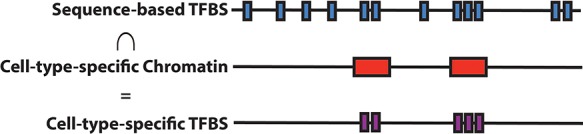
Strategy for constructing cell type-specific TF-binding annotations. We intersect sequence-based TF-binding annotations such as MotifMap±100 bp (blue bars; mean segment length 240 bp) with cell type-specific chromatin annotations (red bars; mean segment length 1200 bp) to create cell type-specific TF-binding annotations such as Chromatin∩MotifMap100 (purple bars; mean segment length 220 bp).
We assessed whether our new cell type-specific TF-binding annotations predict direct measurements of TF binding. We compared ChIP-seq peaks from 91 experiments for 76 factors in lymphoblastoid cell lines (LCLs) from ENCODE (19) with the corresponding LCL-specific TF-binding annotations and computed fold excess overlap (Fig. 2 and Supplementary Material, Table S1; see Materials and Methods). As expected, there was relatively small excess overlap for the sequenced-based predictions: mean 1.69× [standard error (SE) 0.02] across the three sequence-based predictions. However, the excess overlap was much larger when using either measured or imputed cell type-specific chromatin annotations: 12.9× (SE 0.4) or 9.6× (SE 0.3), respectively. [In addition to the Roadmap chromatin annotations, we also considered the recently published fitCons2 (36) annotations, because they also represent cell type-specific annotations of regions likely to be regulatory; these annotations are based on evolutionary constraint. However, they exhibited only 2.0× excess overlap with TF ChIP-seq (SE 0.02); see Supplementary Material, Table S1.] When the chromatin annotations were intersected with sequence-based predictions, the excess overlap increased, with the highest overlap in Chromatin.measured∩CisBP: 17.6× (SE 0.7). Analysis of five other cell types for which ChIP-seq TF-binding data were available for at least 20 TFs produced similar conclusions (Supplementary Material, Table S1). This confirms that the new annotations are more accurately capturing cell type-specific TF binding. However, ChIP-seq peak may not provide a true gold-standard metric for capture of TF-binding sites, as sequencing data peaks will also include regions surrounding the sites that are actually bound (41). Indeed, ChIP-seq peaks for TF binding ranged from 200 to 500 base pairs, while TF-binding sites are generally 6–20 base pairs (9). This may imply that enrichments for true TF binding are underestimated in this analysis. Moreover, enrichment for TF binding does not guarantee that the annotations will be informative for human disease. We therefore turn to analysis of disease heritability to evaluate our annotations and investigate their potential applications.
Figure 2.
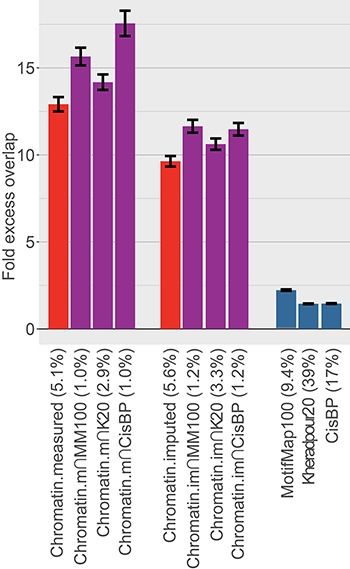
Comparison of excess overlap with TF ChIP-seq. We report the fold excess overlap with TF ChIP-seq peaks from ENCODE cell line GM12878 (LCL; data available from 91 experiments for 76 TFs) for sequence-based TF-binding annotations (blue bars), cell type-specific chromatin annotations (red bars) and cell type-specific TF-binding annotations (purple bars). Error bars denote one SE. The percentage under each bar indicates the proportion of SNPs in each annotation. Numerical results, including results for five other tissues for which ChIP-seq TF-binding data were available for at least 20 TFs, are reported in Supplementary Material, Table S1.
Predicted cell type-specific TF-binding annotations are enriched for disease heritability
We assessed whether our new cell type-specific TF-binding annotations are enriched for disease heritability. We used two metrics to quantify the contribution of an annotation to disease heritability: enrichment and standardized effect size (τ*; see Materials and Methods). Enrichment is defined as the proportion of heritability explained by single nucleotide polymorphisms (SNPs) in an annotation divided by the proportion of SNPs in the annotation (4). τ* is defined as the proportionate change in per-SNP heritability associated with an increase in the value of the annotation by one standard deviation, conditional on other annotations included in the model (5). Unlike enrichment, τ* quantifies effects that are unique to the focal annotation.
We analyzed 49 diseases and complex traits for which summary association statistics are publicly available (Table 1; average N = 320 k) and analyzed 127 Roadmap tissues and cell types (20). For each (trait, cell type) pair, we ran S-LDSC (4) using the baseline-LD model v2.0 (see Web Resources) (5) and the corresponding cell type-specific chromatin annotation. For each trait, we chose the best cell type based on statistical significance of τ* for the cell type-specific chromatin annotation, consistent with the previous work (Table 1) (4). We used this cell type for all cell type-specific annotations for that trait; this is a conservative choice when comparing our new cell type-specific TF-binding annotations to cell type-specific chromatin annotations.
Table 1.
Choice of best cell type for each trait
| Trait | Electronic identification (EID) | Tissue type | Z-score |
|---|---|---|---|
| Red blood cell distribution (UKBB) | E123 | Blood (K562) | 6.81 |
| High-density lipoprotein (HDL) | E066 | Liver | 4.43 |
| Coronary artery disease | E109 | Small intestine | 4.11 |
| Autoimmune traits | E116 | Blood (LCL) | 7.30 |
| Depressive symptoms | E082 | Fetal brain | 3.59 |
| Hypothyroidism (UKBB) | E116 | Blood (LCL) | 6.33 |
| Red blood count (UKBB) | E035 | Blood (CD34) | 4.58 |
| Dermatologic diseases (UKBB) | E044 | Blood (CD4) | 5.33 |
| White blood cell count (UKBB) | E031 | Blood (CD19) | 7.65 |
| Respiratory and ear–nose–throat diseases (UKBB) | E042 | Blood (CD4) | 5.45 |
| Age at menopause (UKBB) | E022 | IPSC | 3.28 |
| Platelet count (UKBB) | E036 | Blood (CD34) | 7.34 |
| Ulcerative colitis | E116 | Blood (LCL) | 4.45 |
| Age first birth | E082 | Fetal brain | 3.96 |
| Schizophrenia | E082 | Fetal brain | 5.97 |
| Low-density lipoprotein (LDL) | E066 | Liver | 3.57 |
| Eosinophil count (UKBB) | E046 | Blood (CD56) | 8.14 |
| Waist-hip ratio (UKBB) | E063 | Adipose | 9.19 |
| Systolic blood pressure (UKBB) | E097 | Ovary | 6.59 |
| Crohn’s disease | E041 | Blood (CD4) | 5.08 |
| Eczema | E042 | Blood (CD4) | 6.54 |
| FEV1-FVC ratio (UKBB) | E088 | Lung | 9.37 |
| Type 2 diabetes | E118 | Liver (HepG2) | 2.85 |
| Rheumatoid arthritis | E116 | Blood (LCL) | 5.86 |
| High cholesterol (UKBB) | E066 | Liver | 3.96 |
| Forced vital capacity (UKBB) | E088 | Lung | 6.95 |
| BMI | E072 | Brain | 2.60 |
| BMI (UKBB) | E070 | Brain | 6.99 |
| Type 2 diabetes (UKBB) | E118 | Liver (HepG2) | 3.12 |
| Number of children ever born | E015 | ESC | 3.19 |
| Height (UKBB) | E023 | Adipose | 8.35 |
| Years of education | E082 | Fetal brain | 7.50 |
| College education (UKBB) | E082 | Fetal brain | 8.55 |
| Age at menarche (UKBB) | E087 | Pancreas | 5.74 |
| Height | E049 | Mesenchyme | 6.94 |
| Neuroticism (UKBB) | E082 | Fetal brain | 7.11 |
| Balding type 1 (UKBB) | E086 | Fetal kidney | 4.99 |
| Anorexia | E082 | Fetal brain | 2.12 |
| Morning person (UKBB) | E082 | Fetal brain | 7.82 |
| Smoking status (UKBB) | E082 | Fetal brain | 7.30 |
| Heel T score (UKBB) | E086 | Fetal kidney | 8.32 |
| Tanning (UKBB) | E061 | Skin | 2.87 |
| Sunburn occasion (UKBB) | E061 | Skin | 2.63 |
| Skin color (UKBB) | E059 | Skin | 3.17 |
| Hair color (UKBB) | E061 | Skin | 4.55 |
| Ever smoked | E069 | Brain | 2.28 |
| Autism spectrum | E003 | ESC | 2.49 |
| Lupus | E116 | Blood (LCL) | 5.03 |
| Celiac | E042 | Blood (CD4) | 5.61 |
We report the fold excess overlap with TF ChIP-seq peaks from six tissues and cell lines from ENCODE. We test sequence-based TF-binding annotations, cell type-specific chromatin annotations and cell type-specific TF-binding annotations.
We sought to identify the most disease-informative way to combine sequence-based TF-binding predictions and cell type-specific chromatin annotations. For each combination of 24 cell type-specific TF-binding annotations [3 sequence-based TF predictions × 4 window sizes (0, 20, 50 and 100 bp) × 2 chromatin types (measured, imputed)], we ran S-LDSC conditional on the baseline-LD model and the cell type-specific chromatin annotation. We meta-analyzed results across the 49 traits and calculated three metrics for each annotation: heritability enrichment, τ* and combined τ*; combined τ* is a generalization of τ* that quantifies the combined information in the cell type-specific chromatin and cell type-specific TF-binding annotations, conditional on the baseline-LD model (see Materials and Methods). Statistical significance for combined τ* was assessed by jackknifing the difference between combined τ* and corresponding chromatin τ*. Statistical significance for enrichment was assessed by comparing the enrichment of TF-binding annotations and corresponding chromatin annotations [see Eq. (2) of (42)].
Results of the meta-analysis across 49 traits are reported in Figure 3 (6 cell type-specific TF-binding annotations; 3 sequence-based TF predictions × 2 chromatin types, with best window size for each) and Table 1. The sequence-based TF-binding annotations alone attained relatively low enrichment and τ*, corresponding to the relatively low overlap with TF ChIP-seq. We therefore compare to chromatin annotations, which performed much better, for the remainder of our analyses. We note that imputed chromatin consistently attained slightly smaller heritability enrichment but slightly higher τ* than measured chromatin. (The fitCons2 annotations exhibited smaller enrichment and slightly smaller τ* than the chromatin annotations; see Supplementary Material, Table S3. This, combined with our earlier results, indicates that they are indeed capturing unique disease-relevant information, but they are not ideal for capturing TF binding.) Since the imputed chromatin data performed well and are complete for all 127 Roadmap cell types, we focused on imputed chromatin for subsequent analyses. The Chromatin∩MotifMap100 annotations performed best, with a 59% higher heritability enrichment than Chromatin (11.6× vs. 7.3×, P = 9 × 10−14 for difference) and a 20% higher combined τ* (2.01 vs. 1.67 τ*; P = 8 × 10−11 for difference); these improvements are statistically significant after correcting for 24 hypotheses tested. The τ* values, reflecting information unique to these annotations, were very large relative to analogous values (τ* up to 0.52) that we recently estimated for non-cell-type-specific LD-related annotations (5) and molecular QTL annotations (43); as such, the Δτ* of 0.20 is a substantial improvement. Chromatin∩Kheradpour20 and Chromatin∩CisBP attained slightly worse results. Using smaller window sizes increased the disease heritability enrichment (Supplementary Material, Table S2), indicating that there is a concentration of activity in the predicted TF-binding sites. However, Chromatin∩MotifMap100 and Chromatin∩Kheradpour20 attained larger τ* values than the corresponding annotations without windows. Since the median feature size of annotations without windows was quite small (7–15 base pairs), our results indicate that the regions surrounding core TF-binding sites also contain important sequences. Finally, we tested an annotation of Chromatin intersected with the union of the three best TFBS annotations, which attained a lower enrichment and τ* than Chromatin∩MotifMap100 (8.8× enrichment and 1.89 τ*).
Figure 3.
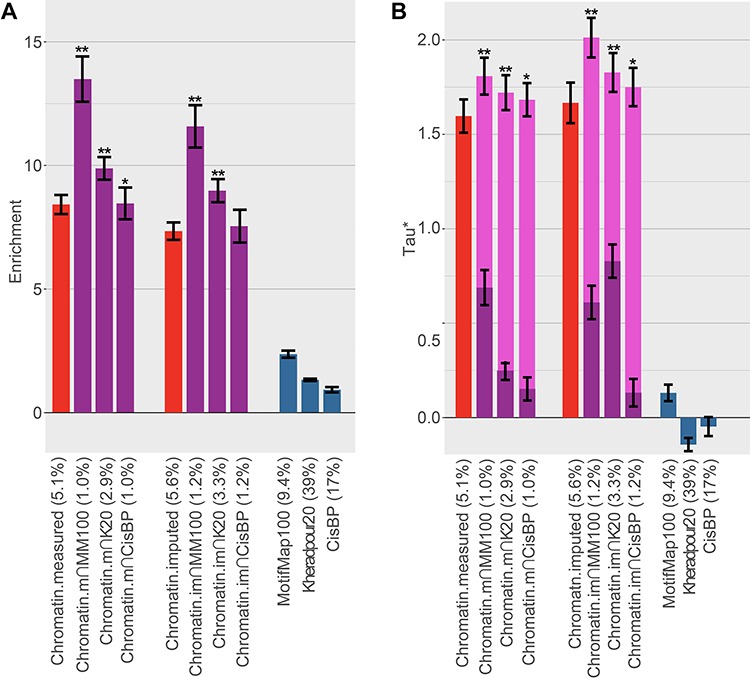
Comparison of heritability enrichment and τ* across 49 diseases and complex traits. We report (A) heritability enrichment and (B) τ* for sequence-based TF-binding annotations (blue bars), tissue-specific chromatin annotations (red bars) and tissue-specific TF-binding annotations (purple bars, including dark purple bars for τ* in a joint model and light purple bars for combined τ*). Error bars denote one SE. Single asterisk in (A) indicates > 2 SE difference in enrichment vs. corresponding chromatin annotation (we note that differences have much smaller SEs) and in (B) P-value < 0.05 for combined τ* vs. τ* of corresponding chromatin annotation. Double asterisks show > 4 SE difference and P-value <1e − 5. The percentage under each bar indicates the proportion of SNPs in each annotation. Numerical results are reported in Supplementary Material, Table S2A and S2B.
Results of targeted meta-analyses across 6 autoimmune, 5 blood, and 11 brain-related traits (see Materials and Methods) are reported in Figure 4 and Supplementary Material, Table S3. For the six autoimmune traits, Chromatin∩MotifMap100 attained a much higher heritability enrichment than Chromatin (23.6× vs. 11.1×; P = 0.001 for difference) and a substantially higher combined τ* (3.11 vs. 2.61; P = 0.004 for difference). Chromatin∩MotifMap100 also outperformed TF-binding annotations from ENCODE ChIP-seq (heritability enrichment = 23.6× vs. 5.32×; τ* = 3.11 vs. 1.75). Results were similar for the five blood traits, though enrichments were slightly smaller and differences less significant. On the other hand, the 11 brain-related traits attained substantially smaller enrichments, consistent with the previous work (4,5). However, Chromatin∩MotifMap100 still attained substantial improvements in heritability enrichment (7.04 vs. 4.98; P = 0.002 for difference) and τ* (1.17 vs. 1.07; P = 0.003 for difference). We also considered an annotation constructed from the union of all ENCODE ChIP-seq TF-binding experiments. Notably, this annotation underperformed Chromatin∩MotifMap100 for all trait classes and performed particularly poorly for the brain-related traits (heritability enrichment = 1.31, τ* = 0.07). This is likely because very few of the ENCODE ChIP-seq experiments were conducted in brain tissues, highlighting the importance of methods to create cell type-specific TF annotations when ChIP-seq data are unavailable.
Figure 4.
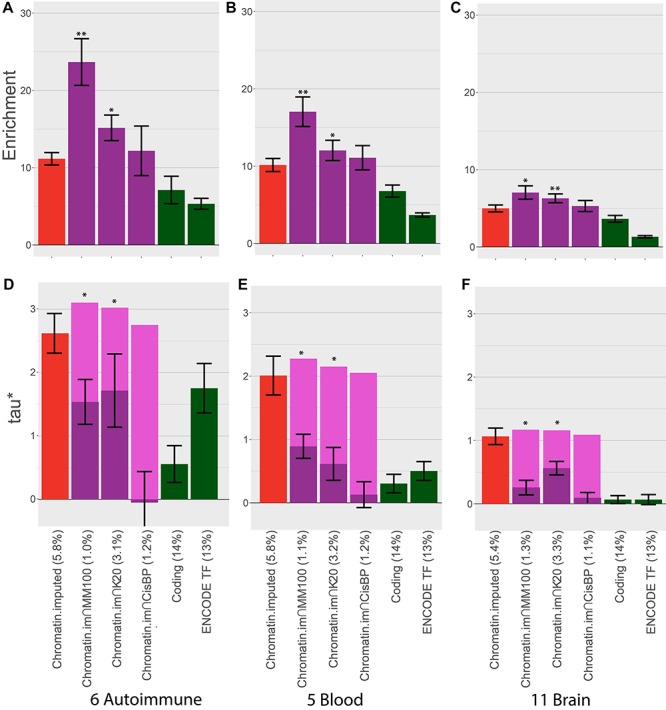
Comparison of heritability enrichment and τ* for autoimmune, blood and brain-related traits. We report (A–C) heritability enrichment for each trait class and (D–F) τ* for each trait class for cell type-specific chromatin annotations (red bars) and cell type-specific TF-binding annotations (purple bars, including dark purple bars for τ* in a joint model and light purple bars for combined τ*). We include coding regions (green bars) and an annotation constructed from the union of all ENCODE ChIP-seq TF-binding experiments (green bars) for comparison purposes. Error bars denote one SE. Single asterisk indicates P < 0.05 for (A) enrichment vs. corresponding chromatin annotation and (B) combined τ* vs. τ* of corresponding chromatin annotation. Double asterisks represent P-value <1e − 5. The percentage under each bar indicates the proportion of SNPs in each annotation. Numerical results are reported in Supplementary Material, Table S3A and S3B.
Finally, we compared the heritability enrichments of Chromatin and Chromatin∩MotifMap100 for each individual trait (Fig. 5 and Supplementary Material, Table S4). We determined that 43 of 44 traits with significant enrichment for at least one of these two annotations had higher heritability enrichment for Chromatin∩MotifMap100. However, some traits show only modest improvements in heritability enrichment, perhaps because binding preferences for the relevant TFs are not well captured by sequence-based predictions; alternatively, it is possible that TF-binding sites play smaller roles for these traits.
Figure 5.
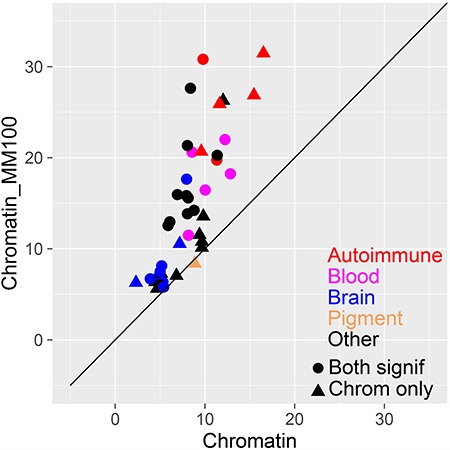
Comparison of heritability enrichment for each trait. We report the heritability enrichment of cell type-specific chromatin annotations (x-axis) and cell type-specific TF-binding annotations (y-axis). Results are displayed for 44 traits that have significant enrichment for at least one of these two annotations, assessed using P = 0.05/127 (correcting for 127 cell types analyzed). Numerical results are reported in Supplementary Material, Table S4.
Choice of baseline vs. baseline-LD model in cell type-specific analyses
Our main analyses (Figs 3–5) used the recently updated baseline-LD model (v2.0), which includes 6 LD-related annotations (5); using a more complete model is appropriate when the goal is to estimate heritability enrichment while minimizing bias due to model misspecification (4,5). On the other hand, our previous work (4,6) identified critical cell types for disease by computing the statistical significance of τ* conditioned on the baseline model, which does not include the LD-related annotations. The LD-related annotations reflect the action of negative selection (5); some of the LD-related annotations are correlated with cell type-specific annotations—particularly brain annotations, which show stronger signals of negative selection (44). Thus, we hypothesized that cell type-specific signals might be stronger when conditioning on the baseline model instead of the baseline-LD model. To assess this, we compared the statistical significance of the combined τ* for (Chromatin + Chromatin∩MotifMap100) using the baseline (v1.1) vs. baseline-LD (v2.0) models across 49 traits; in each case, we chose the most significant of the 127 Roadmap cell types. We determined that the baseline model generally produces more significant combined τ* values than the baseline-LD model, particularly for brain traits and cell types (Fig. 6 and Supplementary Material, Table S5). Thus, we recommend that the baseline model should be used when the goal is to identify critical cell types; however, the baseline-LD model should still be used when the goal is to obtain unbiased estimates of heritability enrichment.
Figure 6.
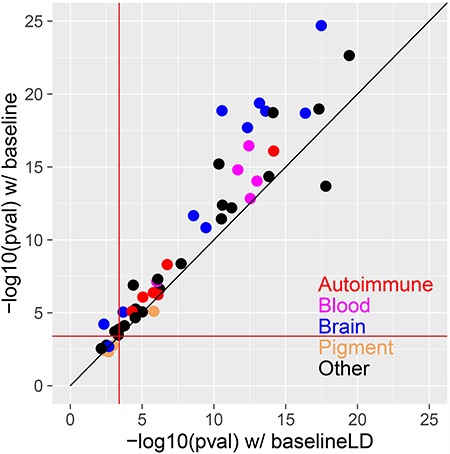
Comparison of combined cell type-specific annotations (Chromatin + Chromatin∩MotifMap100) conditioned on the baseline vs. baseline-LD models. We report the statistical significance (–log10P-value of combined τ*) of the combined cell type-specific annotations (Chromatin + Chromatin∩MotifMap100) for the baseline (y-axis) vs. baseline-LD (x-axis) models, for each of 49 traits. In each case, we report results for the most significant tissue/cell type. The red lines indicate the P = 0.05/127 significance threshold, correcting for testing of 127 cell types. Numerical results are reported in Supplementary Material, Table S5.
Discussion
We explored a new strategy for constructing cell type-specific TF-binding annotations by intersecting sequence-based TF predictions with cell type-specific chromatin annotations. We determined that the resulting cell type-specific TF-binding annotations significantly outperformed cell type-specific chromatin annotations across 49 diseases and complex traits, with highly significant improvements in both heritability enrichment and τ*; this strategy increased heritability enrichment for 43 of 44 traits with significant conditional signal for cell type-specific chromatin and greatly outperformed non-cell-type-specific sequence-based TF-binding annotations. These findings are consistent with the higher overlap of our cell type-specific TF-binding annotations with ENCODE TF ChIP-seq peaks. We also determined that annotations constructed using imputed chromatin (37) attained slightly higher τ* than annotations constructed using measured chromatin; we recommend the use of imputed chromatin annotations, since they are complete for all 127 Roadmap cell types. We note that there exist many other strategies for prioritizing disease-relevant cell types (3,4,6,45) and that strategies based on specifically expressed genes tend to identify similar cell types to the chromatin-based strategies that we consider here (6).
Our results confirm that TF binding is important for diseases and complex traits and provide a quantification of their contribution to heritability. In particular, a large proportion of the heritability explained by active chromatin regions comes from predicted TF-binding sites, particularly for autoimmune diseases. The proportion of disease heritability explained by TF-binding sites will only increase as our TF predictions improve. We note the variation in performance of different TF-binding prediction methods in our analyses. We hypothesize that this is because combining annotations across TFs adds an additional layer of complexity, as TF-binding predictions for different TFs are not on the same scale; in particular, TF consensus sequences vary in size and the number of sites bound by a TF varies greatly. MotifMap, which attained the highest enrichment and τ* in our comparisons, combines traditional position weight matrix scoring systems with measures of evolutionary conservation and Monte Carlo methods to estimate false discovery rate. Kheradpour annotations, which attained moderate enrichment and τ*, were created by curating a diverse set of motifs based on experimental validation with ChIP-seq. On the other hand, CisBP, which has the most complete set of TF-binding motifs, attained the lowest enrichment and τ* in our comparisons. This is likely due to naïve combining across all TFs using P-values for the match to the sequence at a specific location above the background.
We recommend that our cell type-specific TF-binding annotations should be incorporated into efforts to interpret GWAS signals using functionally informed fine mapping (3,26,46,47). For example, Chen et al. (47) reported an association between the TT>A polymorphism at chr6: 138231039 and lupus. The polymorphism was annotated as active chromatin in 14 of the 127 Roadmap cell types, including LCL and 12 other blood cell types. Furthermore, this polymorphism lies inside our Chromatin∩MotifMap100 annotation for these 14 cell types, whereas only 2 of the 37 common variants within a 10 kb window lie in the Chromatin∩MotifMap100 annotation for LCL. This example also demonstrates the potential for dissecting generally acting vs. cell type-specific GWAS loci and identifying active cell types for each locus. The cell type-specific TF-binding annotations may also be useful in efforts to use functional information to increase association power (48–50) and improve polygenic risk prediction (51–53).
We note four limitations of our work. First, our cell type-specific TF-binding annotations attain higher heritability enrichment than cell type-specific chromatin annotations, but explain less heritability in total due to their smaller size. We evaluated this tradeoff using the τ* metric (5), which demonstrated that our cell type-specific TF-binding annotations attain a highly significant increase in cell type-specific signal conditional on the baseline-LD model, compared to cell type-specific chromatin annotations alone. Second, we did not include sequence-based TF-binding predictions produced by deep learning methods in our main analyses (28–31). We investigated several strategies for combining TF-binding predictions produced by DeepBind (29) across TFs, but we were unable to devise a strategy that attained performance close to the strategies that we report here (see Materials and Methods). In particular, combining DeepBind predictions across TFs is a major challenge, as DeepBind was not designed for performing a genome-wide scan for all TFs jointly. Third, the sequence-based predictions that we incorporate are limited to TFs that have sufficient data available to learn the underlying consensus sequence. It is possible that TFs active in some cell types (e.g. skin) are underrepresented, potentially explaining why some traits (e.g. pigmentation traits) perform less well in our analyses. Fourth, inferences about components of heritability can potentially be biased by failure to account for LD-dependent architectures (5,54–56). All of our main analyses used the baseline-LD model, which includes six LD-related annotations (5). The baseline-LD model is supported by formal model comparisons using likelihood and polygenic prediction methods, as well as analyses using a combined model incorporating alternative approaches (7); however, there can be no guarantee that the baseline-LD model perfectly captures LD-dependent architectures. Despite these limitations, our tissue-specific TF-binding annotations significantly improve our understanding of disease and complex trait heritability. All annotations have been made publicly available (see Web Resources).
Materials and Methods
Constructing sequence-based TF-binding annotations
MotifMap: Predicted TF-binding sites for build hg19 were downloaded from the MotifMap website (http://motifmap.igb.uci.edu).
Kheradpour et al.: Predicted TF-binding sites for build hg19 were downloaded from http://compbio.mit.edu/encode-motifs/matches.txt.gz
CisBP: Position weight matrixes for all human TFs were downloaded from the CisBP website (http://cisbp.ccbr.utoronto.ca/). Genome-wide matches were created using MEME FIMO software (http://meme-suite.org/doc/fimo.html), which provides P-values for the match of a given sequence to a motif above the background genomic sequence. Matches with P-value <1e − 5 for each TF were kept.
DeepBind: We downloaded DeepBind and the human TF models from the DeepBind website (http://tools.genes.toronto.edu/deepbind). We then constructed fasta files spanning the entire genome with overlapping 101 base pair lines of sequence as input for a genome-wide DeepBind scan. We ran DeepBind genome-wide for each TF as well as on a gold-standard set of sequence from ChIP-seq data (also downloaded from the DeepBind site). We then assigned each 101 base pair line a z-score for binding based on (1) the mean and standard deviation of the gold-standard sequences or (2) the mean and standard deviation of the genome-wide scores. We constructed binding annotations using various thresholds for both, but no combination yielded positive results.
Assessing overlap with direct measurements of ChIP-seq
We downloaded TF ChIP-seq data from ENCODE (http://hgdownload.cse.ucsc.edu/goldenpath/hg19/encodeDCC/wgEncodeAwgTfbsUniform/). For each cell type with at least 30 experiments, we created a bed file with the union of ChIP-seq peaks from all TFs assayed. We then calculated the excess overlap between an annotation (A) and the ChIP-seq peaks (B) as
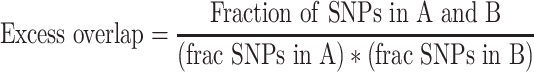 |
(1) |
We calculated SEs using a block-jackknife, dividing the genome into 200 blocks of equal genomic size.
Choosing best cell type for each disease
In order to identify the most relevant cell type for each disease, we applied S-LDSC conditional on the baseline-LD model (v2.0) with ‘Chromatin.imputed’ annotations for each pair of 127 Roadmap cell types and 49 traits. For each disease, we chose the most disease-relevant cell type based on significance of τ*.
Calculating combined τ*
In order to calculate combined τ*, we applied S-LDSC conditional on the baseline-LD model and including both Chromatin and one Chromatin∩TFBS annotation at a time. We then calculated
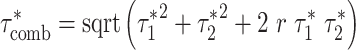 |
(2) |
where 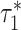 and
and 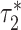 are the τ* for Chromatin and Chromatin∩TFBS, respectively, and
are the τ* for Chromatin and Chromatin∩TFBS, respectively, and 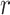 is the correlation between the Chromatin and Chromatin∩TFBS annotations. We calculated SEs for
is the correlation between the Chromatin and Chromatin∩TFBS annotations. We calculated SEs for 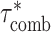 using a block-jackknife with 200 blocks. We also calculated P-values for the difference between
using a block-jackknife with 200 blocks. We also calculated P-values for the difference between 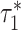 and
and 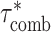 by jackknifing on the value (
by jackknifing on the value (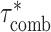 −
− 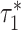 ). With this metric, we measure the combined information being captured by a set of cell type-specific annotations.
). With this metric, we measure the combined information being captured by a set of cell type-specific annotations.
Meta-analyzing across autoimmune, blood, and brain-related traits
In order to understand how heritability is localized across classes of traits, we created three groups: autoimmune [autoimmune traits (UKBB), ulcerative colitis, Crohn’s disease, rheumatoid arthritis, lupus and celiac], blood-related [red blood cell distribution (UKBB), red blood count (UKBB), white blood count (UKBB), platelet count (UKBB) and eosinophil count (UKBB)] and brain-related (depressive symptoms, schizophrenia, waist-hip ratio (UKBB), BMI, BMI (UKBB), years of education, college years (UKBB), age at menarche (UKBB), neuroticism (UKBB), smoking status (UKBB) and ever smoked]. We used the previously chosen most relevant cell type and meta-analyzed τ* using a random effects meta-analysis implemented in R by the rmeta package.
Web Resources
CisBP, http://cisbp.ccbr.utoronto.ca/
DeepBind, http://tools.genes.toronto.edu/deepbind
ENCODE, http://hgdownload.cse.ucsc.edu/goldenpath/hg19/encodeDCC
Kheradpour et al., http://compbio.mit.edu/encode-motifs
LDSC software, https://github.com/bulik/ldsc/wiki
LDSC annotations, https://data.broadinstitute.org/alkesgroup/LDSCORE/
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to Manolis Kellis, Yue Li and Babak Alipanahi for helpful discussions. This research was conducted using the UK Biobank Resource under Application 16549.
Funding
National Institutes of Health (U01 HG009379, R01 MH101244, R01 MH109978, R01 MH107649, F32 HG009615); McLennan Family Fund award.
Author Summary: Identifying regions of the genome that contribute the most to heritability is crucial to our understanding of mechanisms of human diseases. Here, we investigate the contribution of genetic variants in transcription factor-binding sites to disease heritability. These sites are known to be important for gene regulation and often carry specific sequence motifs. However, annotation of transcription factor binding is difficult, as it depends on cell type-specific factors as well. Since directly measured binding data are not always available, we investigate strategies to accurately infer binding for a wide range of cell types and factors by intersecting sequence-based binding predictions with cell type-specific chromatin data. The annotations we create have larger enrichment for disease heritability than other cell type-specific annotations and contain information that is not explained by previous models. This shows that annotating transcription factor-binding sites is important for understanding the cell types and regions that contribute to human disease.
Conflict of Interest Statement
The authors report no conflicts of interest that pertain to this research.
References
- 1. Hindorff L.A., Sethupathy P., Junkins H.A., Ramos E.M., Mehta J.P., Collins F.S. and Manolio T.A. (2009) Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl. Acad. Sci., 106, 9362–9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Maurano M.T., Humbert R., Rynes E., Thurman R.E., Haugen E., Wang H., Reynolds A.P., Sandstrom R., Qu H. and Brody J. (2012) Systematic localization of common disease-associated variation in regulatory DNA. Science, 337, 1190–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Trynka G., Sandor C., Han B., Xu H., Stranger B.E., Liu X.S. and Raychaudhuri S. (2013) Chromatin marks identify critical cell types for fine mapping complex trait variants. Nat. Genet., 45, 124–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Finucane H.K., Bulik-Sullivan B., Gusev A., Trynka G., Reshef Y., Loh P.-R., Anttila V., Xu H., Zang C. and Farh K. (2015) Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet., 47, 1228–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gazal S., Finucane H.K., Furlotte N.A., Loh P.-R., Palamara P.F., Liu X., Schoech A., Bulik-Sullivan B., Neale B.M., Gusev A. et al. (2017) Linkage disequilibrium–dependent architecture of human complex traits shows action of negative selection. Nat. Genet., 49, 1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Finucane H.K., Reshef Y.A., Anttila V., Slowikowski K., Gusev A., Byrnes A., Gazal S., Loh P.-R., Lareau C., Shoresh N. et al. (2018) Heritability enrichment of specifically expressed genes identifies disease-relevant tissues and cell types. Nat. Genet., 50, 621–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gazal S., Marquez-Luna C., Finucane H.K. and Price A.L. (2018) Reconciling S-LDSC and LDAK models and functional enrichment estimates. bioRxiv, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Voss T.C. and Hager G.L. (2013) Dynamic regulation of transcriptional states by chromatin and transcription factors. Nat. Rev. Genet., 15, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lambert S.A., Jolma A., Campitelli L.F., Das P.K., Yin Y., Albu M., Chen X., Taipale J., Hughes T.R. and Weirauch M.T. (2018) The human transcription factors. Cell, 172, 650–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Klemm S.L., Shipony Z. and Greenleaf W.J. (2019) Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet., in press. [DOI] [PubMed] [Google Scholar]
- 11. Cowper-Sal·lari R., Zhang X., Wright J.B., Bailey S.D., Cole M.D., Eeckhoute J., Moore J.H. and Lupien M. (2012) Breast cancer risk–associated SNPs modulate the affinity of chromatin for FOXA1 and alter gene expression. Nat. Genet., 44, 1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Karczewski K.J., Dudley J.T., Kukurba K.R., Chen R., Butte A.J., Montgomery S.B. and Snyder M. (2013) Systematic functional regulatory assessment of disease-associated variants. Proc. Natl. Acad. Sci., 110, 9607–9612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. McVicker G., van de Geijn B., Degner J.F., Cain C.E., Banovich N.E., Raj A., Lewellen N., Myrthil M., Gilad Y. and Pritchard J.K. (2013) Identification of genetic variants that affect histone modifications in human cells. Science, 342, 747–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Price A.L., Spencer C.C.A. and Donnelly P. (2015) Progress and promise in understanding the genetic basis of common diseases. Proc. R. Soc. B, 282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mathelier A., Shi W. and Wasserman W.W. (2015) Identification of altered cis-regulatory elements in human disease. Trends Genet., 31, 67–76. [DOI] [PubMed] [Google Scholar]
- 16. Whitington T., Gao P., Song W., Ross-Adams H., Lamb A.D., Yang Y., Svezia I., Klevebring D., Mills I.G., Karlsson R. et al. (2016) Gene regulatory mechanisms underpinning prostate cancer susceptibility. Nat. Genet., 48, 387. [DOI] [PubMed] [Google Scholar]
- 17. Liu Y., Walavalkar N.M., Dozmorov M.G., Rich S.S., Civelek M. and Guertin M.J. (2017) Identification of breast cancer associated variants that modulate transcription factor binding. PLoS Genet., 13, e1006761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zentner G.E. and Henikoff S. (2014) High-resolution digital profiling of the epigenome. Nat. Rev. Genet., 15, 814. [DOI] [PubMed] [Google Scholar]
- 19. Consortium E.P. (2012) An integrated encyclopedia of DNA elements in the human genome. Nature, 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Romanoski C.E., Glass C.K., Stunnenberg H.G., Wilson L. and Almouzni G. (2015) Epigenomics: Roadmap for regulation. Nature, 518, 314–316. [DOI] [PubMed] [Google Scholar]
- 21. Wingender E., Dietze P., Karas H. and Knüppel R. (1996) TRANSFAC: a database on transcription factors and their DNA binding sites. Nucleic Acids Res., 24, 238–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sandelin A., Alkema W., Engström P., Wasserman W.W. and Lenhard B. (2004) JASPAR: an open-access database for eukaryotic transcription factor binding profiles. Nucleic Acids Res., 32, D91–D94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mathelier A., Fornes O., Arenillas D.J., Chen C.-y., Denay G., Lee J., Shi W., Shyr C., Tan G., Worsley-Hunt R. et al. (2016) JASPAR 2016: a major expansion and update of the open-access database of transcription factor binding profiles. Nucleic Acids Res., 44, D110–D115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Daily K., Patel V.R., Rigor P., Xie X. and Baldi P.J.B.B. (2011) MotifMap: integrative genome-wide maps of regulatory motif sites for model species. BMC Bioinformatics, 12, 495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kheradpour P. and Kellis M. (2014) Systematic discovery and characterization of regulatory motifs in ENCODE TF binding experiments. Nucleic Acids Res., 42, 2976–2987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Weirauch M.T., Yang A., Albu M., Cote A.G., Montenegro-Montero A., Drewe P., Najafabadi H.S., Lambert S.A., Mann I., Cook K. et al. (2014) Determination and inference of eukaryotic transcription factor sequence specificity. Cell, 158, 1431–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lee D., Gorkin D.U., Baker M., Strober B.J., Asoni A.L., McCallion A.S. and Beer M.A. (2015) A method to predict the impact of regulatory variants from DNA sequence. Nat. Genet., 47, 955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhou J. and Troyanskaya O.G. (2015) Predicting effects of noncoding variants with deep learning–based sequence model. Nat. Methods, 12, 931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Alipanahi B., Delong A., Weirauch M.T. and Frey B.J. (2015) Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat. Biotechnol., 33, 831. [DOI] [PubMed] [Google Scholar]
- 30. Zeng H., Edwards M.D., Liu G. and Gifford D.K. (2016) Convolutional neural network architectures for predicting DNA–protein binding. Bioinformatics, 32, i121–i127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kelley D.R., Snoek J. and Rinn J.L. (2016) Basset: learning the regulatory code of the accessible genome with deep convolutional neural networks. Genome Res., 26, 990–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Reshef Y.A., Finucane H.K., Kelley D.R., Gusev A., Kotliar D., Ulirsch J.C., Hormozdiari F., Nasser J., O’Connor L., van de B. et al. (2018) Detecting genome-wide directional effects of transcription factor binding on polygenic disease risk. Nat. Genet., 50, 1483–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pique-Regi R., Degner J.F., Pai A.A., Gaffney D.J., Gilad Y. and Pritchard J.K. (2011) Accurate inference of transcription factor binding from DNA sequence and chromatin accessibility data. Genome Res., 21, 447–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Moyerbrailean G.A., Kalita C.A., Harvey C.T., Wen X., Luca F. and Pique-Regi R. (2016) Which genetics variants in DNase-Seq footprints are more likely to Alter binding? PLoS Genet., 12, e1005875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gusmao E.G., Dieterich C., Zenke M. and Costa I.G. (2014) Detection of active transcription factor binding sites with the combination of DNase hypersensitivity and histone modifications. Bioinformatics, 30, 3143–3151. [DOI] [PubMed] [Google Scholar]
- 36. Gulko B. and Siepel A. (2019) An evolutionary framework for measuring epigenomic information and estimating cell-type-specific fitness consequences. Nat. Genet., 51, 335–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ernst J. and Kellis M. (2015) Large-scale imputation of epigenomic datasets for systematic annotation of diverse human tissues. Nat. Biotechnol., 33, 364–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ernst J. and Kellis M. (2012) ChromHMM: automating chromatin-state discovery and characterization. Nat. Methods, 9, 215–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Weirauch M.T., Cote A., Norel R., Annala M., Zhao Y., Riley T.R., Saez-Rodriguez J., Cokelaer T., Vedenko A., Talukder S. et al. (2013) Evaluation of methods for modeling transcription factor sequence specificity. Nat. Biotechnol., 31, 126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rogers J.M., Barrera L.A., Reyon D., Sander J.D., Kellis M., Keith Joung J. and Bulyk M.L. (2015) Context influences on TALE–DNA binding revealed by quantitative profiling. Nat. Commun., 6, 7440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rhee H.S. and Pugh B.F. (2012) ChIP-exo: a method to identify genomic location of DNA-binding proteins at near single nucleotide accuracy. Curr. Protoc. Mol. Biol., 21. doi: 10.1002/0471142727.mb0471142124s0471142100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hujoel M.L.A., Gazal S., Hormozdiari F., van de Geijn B. and Price A.L. (2019) Disease heritability enrichment of regulatory elements is concentrated in elements with ancient sequence age and conserved function across species. Am. J. Hum. Genet., 104, 611–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hormozdiari F., Gazal S., van de Geijn B., Finucane H.K., Ju C.J.T., Loh P.-R., Schoech A., Reshef Y., Liu X., O’Connor L. et al. (2018) Leveraging molecular quantitative trait loci to understand the genetic architecture of diseases and complex traits. Nat. Genet., 50, 1041–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gazal S., Loh P.-R., Finucane H., Ganna A., Schoech A., Sunyaev S. and Price A. (2018) Functional architecture of low-frequency variants highlights strength of negative selection across coding and non-coding annotations. Nat. Genet., 50, 1600–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Backenroth D., He Z., Kiryluk K., Boeva V., Pethukova L., Khurana E., Christiano A., Buxbaum J.D. and Ionita-Laza I. (2018) FUN-LDA: a latent Dirichlet allocation model for predicting tissue-specific functional effects of noncoding variation: methods and applications. Am. J. Hum. Genet., 102, 920–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kichaev G., Yang W.-Y., Lindstrom S., Hormozdiari F., Eskin E., Price A.L., Kraft P. and Pasaniuc B. (2014) Integrating functional data to prioritize causal variants in statistical fine-mapping studies. PLoS Genet., 10, e1004722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chen W., McDonnell S.K., Thibodeau S.N., Tillmans L.S. and Schaid D.J. (2016) Incorporating functional annotations for fine-mapping causal variants in a Bayesian framework using summary statistics. Genetics, 204, 933–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pickrell J.K. (2014) Joint analysis of functional genomic data and genome-wide association studies of 18 human traits. Am. J. Hum. Genet., 94, 559–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sveinbjornsson G., Albrechtsen A., Zink F., Gudjonsson S.A., Oddson A., Másson G., Holm H., Kong A., Thorsteinsdottir U., Sulem P. et al. (2016) Weighting sequence variants based on their annotation increases power of whole-genome association studies. Nat. Genet., 48, 314. [DOI] [PubMed] [Google Scholar]
- 50. Kichaev G., Bhatia G., Loh P.-R., Gazal S., Burch K., Freund M., Scoech A., Pasaniuc B. and Price A. (2017) Leveraging polygenic functional enrichment to improve GWAS power. Am. J. Hum. Genet., in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Shi J., Park J.-H., Duan J., Berndt S.T., Moy W., Yu K., Song L., Wheeler W., Hua X., Silverman D. et al. (2016) Winner's curse correction and variable thresholding improve performance of polygenic risk modeling based on genome-wide association study summary-level data. PLoS Genet., 12, e1006493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hu Y., Lu Q., Powles R., Yao X., Yang C., Fang F., Xu X. and Zhao H. (2017) Leveraging functional annotations in genetic risk prediction for human complex diseases. PLoS Comput. Biol., 13, e1005589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Marquez-Luna C., Gazal S., Loh P.-R., Furlotte N., Auton A. and Price A.L. (2018) Modeling functional enrichment improves polygenic prediction accuracy in UK Biobank and 23andMe data sets. bioRxiv, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Speed D., Cai N., Johnson M.R., Nejentsev S. and Balding D.J. (2017) Reevaluation of SNP heritability in complex human traits. Nat. Genet., 49, 986–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Speed D., Hemani G., Johnson M.R. and Balding D.J. (2012) Improved heritability estimation from genome-wide SNPs. Am. J. Hum. Genet., 91, 1011–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yang J., Bakshi A., Zhu Z., Hemani G., Vinkhuyzen A.A.E., Lee S.H., Robinson M.R., Perry J.R.B., Nolte I.M., van Vliet-Ostaptchouk J.V. et al. (2015) Genetic variance estimation with imputed variants finds negligible missing heritability for human height and body mass index. Nat. Genet., 47, 1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.