

Abstract
During mitosis, Kif11, a kinesin motor protein, promotes bipolar spindle formation and chromosome movement, and during interphase, Kif11 mediates diverse trafficking processes in the cytoplasm. In humans, inactivating mutations in KIF11 are associated with (1) retinal hypovascularization with or without microcephaly and (2) multi-organ syndromes characterized by variable combinations of lymphedema, chorioretinal dysplasia, microcephaly and/or mental retardation. To explore the pathogenic basis of KIF11-associated retinal vascular disease, we generated a Kif11 conditional knockout (CKO) mouse and investigated the consequences of early postnatal inactivation of Kif11 in vascular endothelial cells (ECs). The principal finding is that postnatal EC-specific loss of Kif11 leads to severely stunted growth of the retinal vasculature, mildly stunted growth of the cerebellar vasculature and little or no effect on the vasculature elsewhere in the central nervous system (CNS). Thus, in mice, Kif11 function in early postnatal CNS ECs is most significant in the two CNS regions—the retina and cerebellum—that exhibit the most rapid rate of postnatal growth, which may sensitize ECs to impaired mitotic spindle function. Several lines of evidence indicate that these phenotypes are not caused by reduced beta-catenin signaling in ECs, despite the close resemblance of the Kif11 CKO phenotype to that caused by EC-specific reductions in beta-catenin signaling. Based on prior work, defective beta-catenin signaling had been the only known mechanism responsible for monogenic human disorders of retinal hypovascularization. The present study implies that retinal hypovascularization can arise from a second and mechanistically distinct cause.
Introduction
Monogenic disorders in which the retina is insufficiently vascularized are inherited in X-linked, autosomal recessive or autosomal dominant patterns, and they range in severity from subclinical defects in the peripheral retina to congenital blindness secondary to severe retinal gliosis ((1,2); OMIM 13370, 305390, 310600, 605750, 601813 and 613310). Among individuals in which the disease phenotype is confined entirely or predominantly to the retina, 25–30% carry mutations in genes coding for the components of a signaling system in which the Muller glia-derived ligand Norrin binds to receptor Frizzled4 (Fz4), co-receptor Lrp5 and co-activator Tspan12 on the surface of vascular endothelial cells (ECs) to activate beta-catenin signaling ((3–6); beta-catenin signaling is also referred to as canonical Wnt signaling).
Hemizygous inactivating mutations in the gene coding for Norrin (NDP) cause Norrie disease (OMIM 310600), in which severe retinal hypovascularization leads to congenital blindness. Mutations in other beta-catenin pathway genes generally produce a milder degree of hypovascularization, and the resulting phenotypes are referred to as familial exudative vitreoretinopathy (FEVR). In mice, targeted mutations in each of these genes similarly lead to retinal vascularization defects (2,7). In the brain and spinal cord, ligands Wnt7a and Wnt7b, Fz4 and other Frizzled receptors, co-receptors Lrp5 and Lrp6 and co-activators Gpr124 and Reck function analogously to the Norrin/Fz4/Lrp5/Tspan12 system in the retina (8–14). Although the Norrin/Fz4/Lrp5/Tspan12 system is also active in the brain and spinal cord, mutations in genes coding for Norrin signaling components cause relatively mild vascular phenotypes in the brain and spinal cord because of redundancy with Wnt7a/Wnt7b signaling (10,15).
Retinal hypovascularization can also be part of more complex syndromes. For example, Norrie disease is frequently associated with progressive hearing loss and, in a subset of affected individuals, with neuropsychiatric features (16). Whereas heterozygous mutations in LRP5 cause FEVR (OMIM 601813), homozygous mutations in LRP5 cause Osteoporosis-Pseudoglioma Syndrome (OMIM 259770), which is characterized by both severe retinal hypovascularization and retarded bone development (17). Recently, heterozygous mutations in the gene coding for beta-catenin (CTNNB1) have been identified in individuals with isolated defects in retinal vascularization (FEVR) as well as FEVR accompanied by intellectual disability (18,19). CTNNB1 mutations have also been found in individuals who exhibit some or all of a distinctive constellation of defects, including intellectual disability, autism spectrum disorder, abnormal craniofacial features, childhood hypotonia, progressive spasticity of the lower limbs and sparse hair ((20–22); OMIM 615075). The diversity of these defects likely reflects the widespread role of beta-catenin signaling in mammalian development and the important role of beta-catenin-cadherin complexes in epithelial integrity (23).
The most recent addition to the set of syndromes that feature retinal hypovascularization is microcephaly, lymphedema and chorioretinal dysplasia (MLCRD) syndrome (OMIM 152950), which is caused by mutations in the gene coding for the kinesin motor protein Kif11 (also known as Kinesin-5, BLM-C or Eg5; (24–31)). As with CTNNB1 mutations, individuals with KIF11 mutations can exhibit retinal hypovascularization with or without the other features of MLCRD (4,32–35). During mitosis, Kif11, a plus-end directed motor, interacts with microtubules in the mitotic spindle to promote bipolar spindle formation and separation of duplicated centrosomes (36,37). Kif11 has also been implicated in multiple functions unrelated to the mitotic spindle. Kif11 interacts with ribosomes to enhance translation, possibly by serving as a link to microtubules (38); Kif11 enhances transport of vesicles from the trans-Golgi network to the plasma membrane (39); and in postmitotic neurons, Kif11 is involved in cell migration and in axonal and dendritic transport and morphology (40–42). Because of its role in cell division, Kif11 has been intensively studied as a potential target for cancer chemotherapy, and several Kif11 inhibitors are currently in clinical trials (43,44).
The present experiments were undertaken to establish a mouse model of retinal hypovascularization due to Kif11 deficiency. In previous work, a targeted null allele and a gene trap allele in Kif11 produced no apparent phenotype when present in the heterozygotes state with a wild-type (WT) allele but caused pre-implantation embryonic lethality in the homozygous state, precluding an analysis of Kif11 function in vascular development (45,46). To permit cell-type-specific analyses of Kif11 loss-of-function effects at later ages, we have generated a Kif11 conditional knockout (CKO) allele. We report here that postnatal EC-specific knockout of Kif11 leads to a profound hypoplasia of the retinal vasculature and a milder hypoplasia of the cerebellar vasculature. Importantly, EC-specific constitutive activation of beta-catenin signaling—induced by artificially stabilizing beta-catenin—does not rescue the Kif11 retinal vascular phenotype. These data imply that Kif11 causes retarded retinal vascular development by a mechanism that is independent of the beta-catenin pathway. The Kif11 conditional allele described here should be useful for genetic dissections of Kif11 function in the contexts of normal development, for modeling Kif11 deficiency syndromes and for investigating the effects of Kif11 inhibition in cancer chemotherapy.
Results
Construction and characterization of a Kif11 CKO mouse line
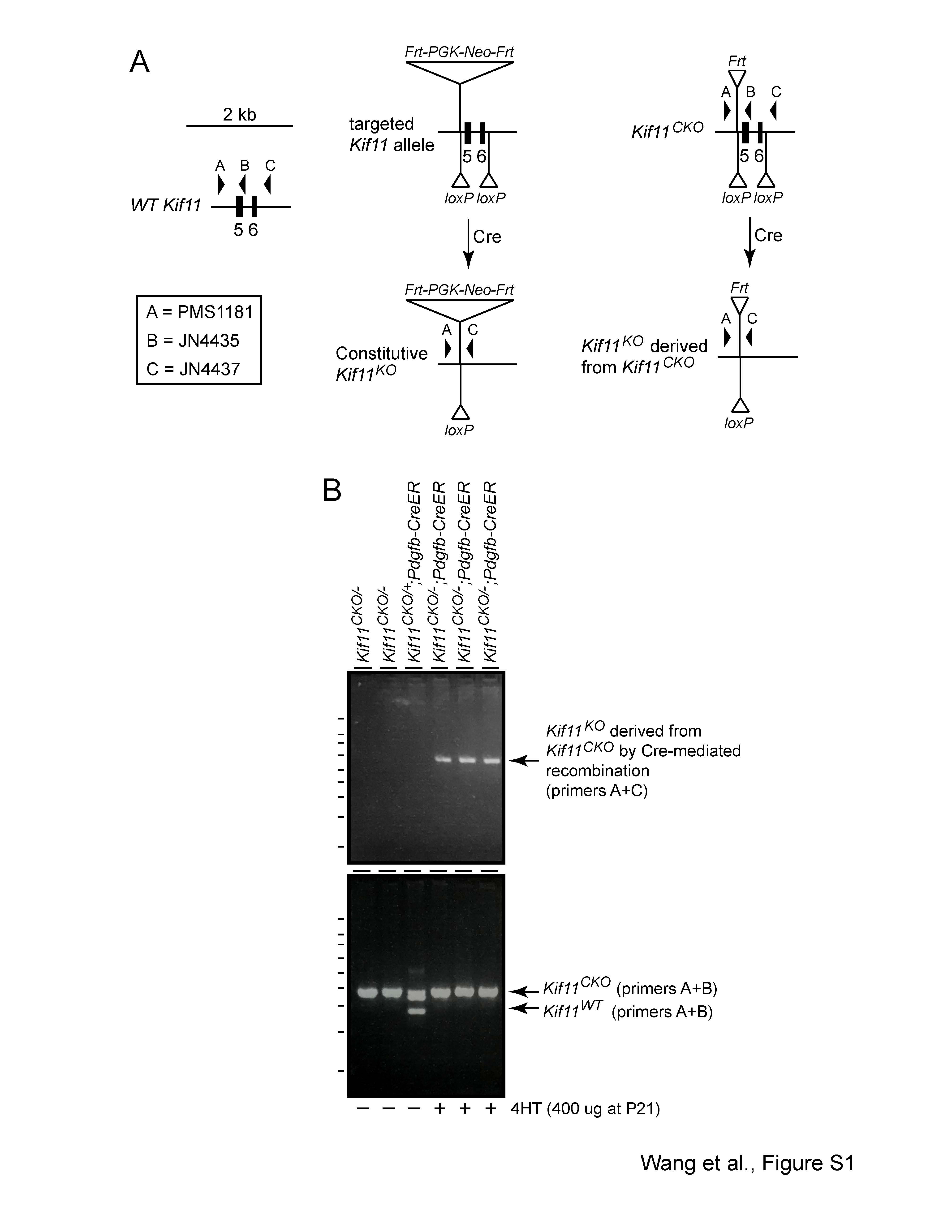
To construct a Kif11CKO allele, exons 5 and 6 were flanked by loxP sites (Fig. 1). Deletion of these two exons leads to a 311 base pair deletion within the Kif11 coding region, resulting in a frameshift starting at codon 129 of the 1052 codon open reading frame. As a technical point, we note that in the construction of the Kif11 null allele by Chauvière et al. (46), in which exons 2 and 3 were replaced by a Beta-Geo reporter/selectable marker, the efficiency of homologous recombination in embryonic stem (ES) cells was ~ 1/400. As the Kif11 introns contain multiple regions with dispersed repetitive sequences and some of these sequences are present in the homology arms of the targeting vector designed by Chauvière et al. (46), we designed the targeting vector for the Kif11CKO allele to minimize such sequences on the assumption that this would increase the efficiency of correctly targeted events. With the Kif11CKO targeting vector shown in Figure 1, the frequency of correctly targeted events was ~ 20% as determined by southern blotting of ES cell DNA with flanking probes.
Figure 1.
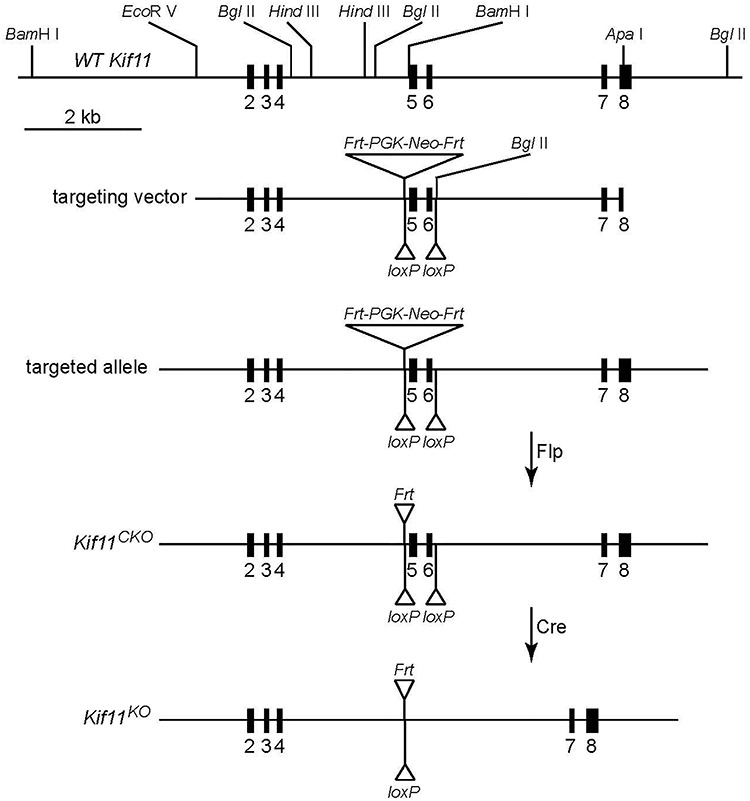
Structure of the Kif11 CKO allele. Shown from top to bottom are the region of the WT Kif11 allele encompassing exons 2–8, the ES cell targeting vector, the targeted allele, the CKO allele and the KO allele following the Cre-mediated recombination of the CKO allele.
Kif11CKO/CKO mice are healthy and fertile. We used Sox2-Cre-mediated germline deletion of Kif11 exons 5 and 6 to generate a recessive loss-of-function allele (Kif11−). We note that this Kif11− allele retains the Frt-Neo-Frt (FNF) selectable marker 5′ of exon 5 and, therefore, can be distinguished by PCR genotyping from the Kif11− allele generated by Cre-mediated recombination of Kif11CKO, which lacks the FNF insertion. This distinction was essential for assessing the efficiency of Cre-mediated recombination in Kif11CKO/− retinas, as described below. In a Kif11+/− intercross, no Kif11−/− mice were found among 61 progeny genotyped, consistent with the previously reported embryonic lethality of a conventional Kif11 null allele. As there is no apparent phenotype associated with a reduction in the number of WT Kif11 alleles from two to one, we have used both Kif11+/+ and Kif11+/− as WT controls in the experiments described below.
Retinal response to ablation of Kif11 in vascular ECs
To explore the role of Kif11 in retinal vascular development, which occurs in mice during the first 2 postnatal weeks, we used a single intraperitoneal injection of 4-hydroxytamoxifen (4HT) in Kif11CKO/−;Pdgfb-CreER mice to initiate Cre-mediated elimination of Kif11 function in vascular ECs. (Pdgfb-CreER is also expressed in some hematopoietic cells; effects of Kif11 recombination on hematopoietic cells were not studied.) Retinal flatmounts were prepared from postnatal day (P)8 and P12 Kif11CKO/−;Pdgfb-CreER mice and phenotypically WT littermate controls that had received 100 μg 4HT at P4. This comparison showed that EC-specific loss of Kif11 produces a severe retardation of retinal vascular development, with reduced radial growth of the vascular front and reduced vascular density within the developing vascular plexus (Fig. 2). This phenotype of retarded angiogenesis is strikingly similar to the retarded angiogenesis produced by the loss of beta-catenin signaling in ECs, as seen in mice lacking Ndp or Fz4 (47–49). Based on analyses of other retinal hypovascularization models (50–52), the retarded vascular development in the Kif11CKO/−;Pdgfb-CreER retina is predicted to cause severe retinal hypoxia.
Figure 2.
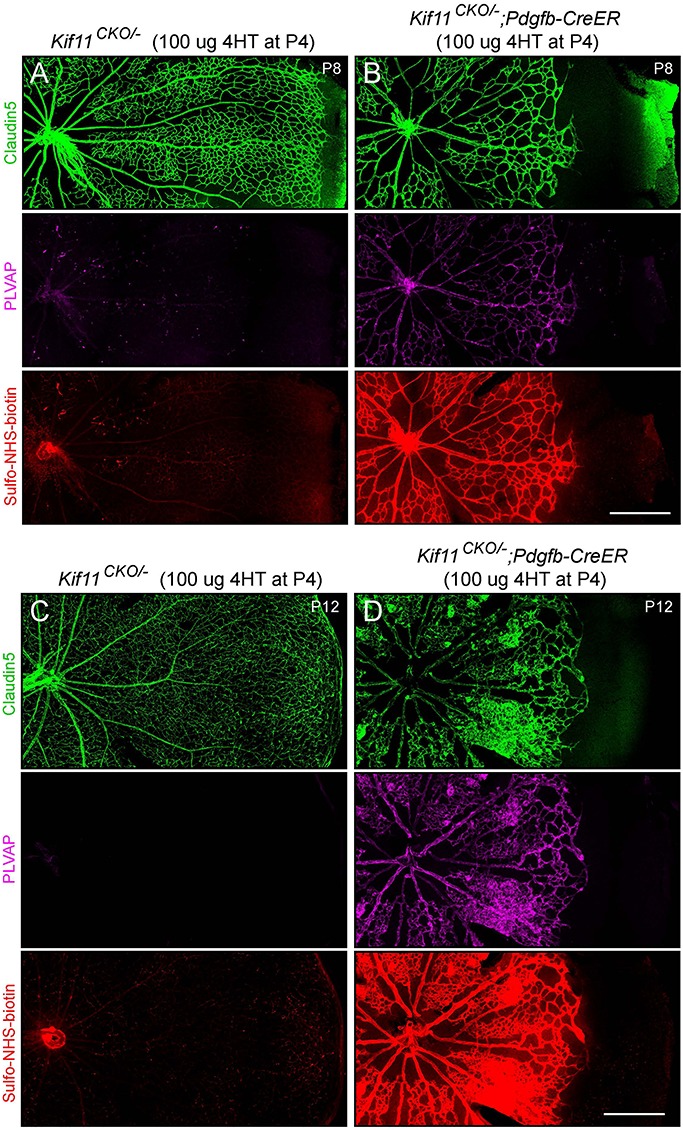
Severely retarded retinal vascular growth following the early postnatal EC-specific knockout of Kif11. Retinal flatmounts were prepared at P8 or P12 from littermate control Kif11CKO/− mice (A,C) and Kif11CKO/−;Pdgfb-CreER mice (B,D) following the treatment with 100 μg 4HT at P4. Each image shows one retinal quadrant, with the optic disc at the left and the retinal periphery at the right. Loss of Kif11 leads to retarded vascular development and reduced vascular coverage of the retinal surface. At both ages, Kif11CKO/−;Pdgfb-CreER retinas show increased expression of PLVAP and increased Sulfo-NHS-biotin leakage. Scale bars, 500 μm.
The severity of the angiogenesis phenotype seen in Figure 2 reflects the efficiency of Cre-mediated recombination. Since Kif11 is presumed to act cell autonomously, any unrecombined ECs should have continued to proliferate and follow the angiogenic program, as has been observed in mosaic retinas with a mixture of Frizzled4+ and Frizzled4 null ECs (48). Our failure to observe mosaic vascular phenotypes in Kif11CKO/−;Pdgfb-CreER retinas implies that, with the 4HT dosage used here, the vast majority of ECs were converted from a Kif11CKO/− state to a Kif11−/− state.
During the first 2 weeks of postnatal development, the WT murine retinal vasculature develops a blood-retina barrier (BRB), which includes the assembly of tight junctions between ECs and suppression of fenestrae formation and transcytosis (53). These cell biological changes are accompanied by accumulation of the tight junction protein Claudin5 and suppression of Plasmalemma Vesicle-Associated Protein (PLVAP), a structural component of EC fenestrations and caveolae (49,54). In Kif11CKO/−;Pdgfb-CreER retinal ECs at P8, there is an accumulation of PLVAP, especially near the vascular front, which correlates with leakage of the intravascular amine-reactive low molecular weight tracer Sulfo-N-hydroxysuccinimide (NHS)-biotin (Fig. 2A and B). At P12, PLVAP accumulation and Sulfo-NHS-biotin leakage are even more prominent (Fig. 2C and D). Accumulation of PLVAP and increased vascular permeability can occur as a direct effect of reduced beta-catenin signaling, but they can also occur in response to elevated levels of retina-derived vascular endothelial growth factor (VEGF; also known as vascular permeability factor), which is produced at a high level by the hypoxic retina (50,51,55).
Kif11 promotes cell proliferation but not beta-catenin signaling
The retarded angiogenesis phenotype produced by EC-specific loss of Kif11, as described above, could be consistent with either of two models: (1) loss of Kif11 impairs beta-catenin signaling in ECs or (2) loss of Kif11 impairs EC proliferation independent of beta-catenin signaling. As described in the following paragraphs, we have conducted three experimental tests to distinguish these two models.
In the first test, we analyzed the accumulation of LEF1, a transcription factor and beta-catenin partner, in retinal ECs. In addition to functioning as a mediator of beta-catenin signaling, LEF1 is also a target of beta-catenin regulation, and during retinal and brain vascular development in WT mice, beta-catenin signaling leads to the nuclear accumulation of LEF1 in vein and capillary ECs (Fig. 3A). Under conditions of reduced beta-catenin signaling—as seen in Ndp or Fz4 mutant mice—there is a corresponding reduction in nuclear LEF1 (56,57). In contrast, in Kif11CKO/−;Pdgfb-CreER retinal ECs at P8, LEF1 accumulates in all or nearly all EC nuclei and at levels that are higher than in WT ECs (Fig. 3B), perhaps reflecting feedback regulation produced by retinal hypoxia. These data imply that the beta-catenin pathway is active (and potentially elevated) in ECs in Kif11CKO/−;Pdgfb-CreER retinas. This conclusion is also consistent with the absence of Claudin5 suppression and the relatively modest PLVAP accumulation in Kif11CKO/−;Pdgfb-CreER retinas at P8. In contrast, loss of Ndp or Fz4 (i.e. loss of EC beta-catenin signaling) leads to Claudin5 suppression and high-level accumulation of PLVAP at all postnatal ages (49).
Figure 3.
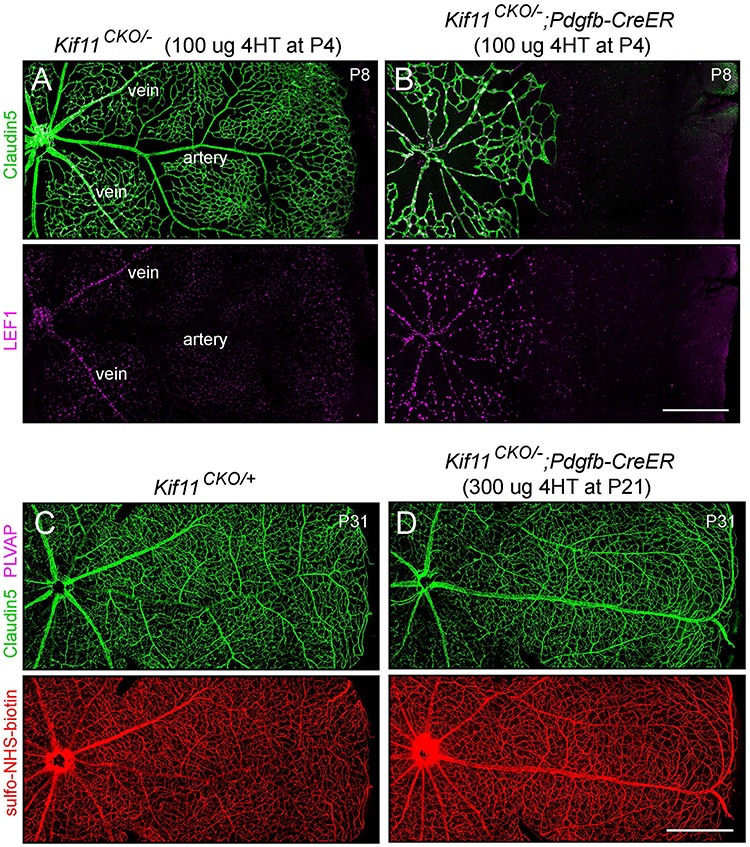
EC-specific knockout of Kif11 leads to continued EC accumulation of LEF1 during development and no effect of post-mitotic knockout on vascular permeability. (A,B) Retinal flatmounts were prepared at P8 from littermate control Kif11CKO/− mice (A) and Kif11CKO/−;Pdgfb-CreER mice (B) following the treatment with 100 μg 4HT at P4. Each image shows one retinal quadrant, with the optic disc at the left and the retinal periphery at the right. LEF1, a marker of beta-catenin signaling, accumulates in the nuclei of vein and capillary ECs but not in arterial ECs in control retinas and in all ECs in mutant retinas. (C,D) Retinal flatmounts were prepared at P31 from littermate control Kif11CKO/+ mice (C) and Kif11CKO/−;Pdgfb-CreER mice (D) following the treatment with 300 μg 4HT at P21. Each image shows one retinal quadrant as in (A) and (B). Loss of Kif11 has no effect on vascular architecture, expression of Claudin5, suppression of PLVAP or BRB integrity, as seen by the absence of Sulfo-NHS-biotin leakage from the intravascular space into the retinal parenchyma. See Supplementary Material Figure S1 for an analysis of Cre-mediated recombination at P21. Scale bars, 500 μm.
In the second test, we asked whether initiating EC-specific loss of Kif11 in the mature retinal vasculature converts ECs from a Claudin5+/PLVAP− state to a Claudin5−/PLVAP+ state and compromises the BRB, as is observed with acute loss of beta-catenin signaling in mature ECs (49,58). For this test, Kif11CKO/−;Pdgfb-CreER mice were treated with 4HT at P21, when the retinal vasculature was fully developed. When these retinas were examined 10 days later (at P31), the retinal vasculature was still Claudin5+/PLVAP−, and it showed no detectable leakage of intravascular Sulfo-NHS-biotin (Fig. 3C and D). Importantly, the retina flatmount analysis in Figure 3C and D is sufficiently sensitive to detected BBB loss at single-cell resolution because (1) expression of PLVAP in single ECs can be readily observed and (2) loss of barrier integrity by single ECs gives rise to a detectable accumulation of sulfo-NHS-biotin leakage around the affected cells, as demonstrated in Figure 6D of Zhou et al. (58).
Since the results described in the preceding paragraph are consistent with either (1) no effect of Kif11 loss in the mature retinal vasculature or (2) a failure of Cre-mediated recombination in the mature retinal vasculature, it was important to independently assess the presence of Cre-mediated recombination. Therefore, we PCR amplified total retina genomic DNA with primers that distinguish the relevant genotypes to assess Cre-mediated recombination (Supplementary Material, Fig. S1A). Specifically, we used primers that flank the two Kif11CKO loxP sites and amplified DNA from P31 Kif11CKO/−;Pdgfb-CreER retinas obtained from mice that were injected with 300 μg 4HT at P21, as shown in Figure 3C and D. This reaction generated readily detectable amounts of the ~ 550 bp PCR product derived from Cre-mediated recombination of the Kif11CKO allele in ECs (Supplementary Material, Fig. S1B, right three lanes), despite the relatively low abundance of EC DNA in the retinal DNA sample (ECs comprise < 5% of retinal cells). In contrast, PCR amplification of DNA from littermate Kif11CKO/+;Pdgfb-CreER or Kif11CKO/− retinas that were not exposed to 4HT did not show this product (Supplementary Material, Fig. S1B, left three lanes). Similar results were obtained with seven additional experimental mice and six additional control mice that were injected with 4HT at P4 or P5. We note that under the PCR amplification conditions used here, longer PCR products (e.g. ~ 1.2 kb from the unrecombined Kif11CKO allele and ~ 2.5 kb from the constitutive Kif11− allele) do not accumulate to detectable levels. These results imply that a substantial number of Kif11CKO/−;Pdgfb-CreER ECs were converted by Cre-mediated recombination to a Kif11−/− genotype at P21. Thus, our failure to observe the conversion of any retinal ECs from PLVAP− to PLVAP+ or any localized sulfo-NHS-biotin leakage implies that loss of Kif11 in the mature retina has little or no effect on BBB properties.
In the third test, we asked whether constitutive activation of beta-catenin signaling in ECs can rescue the retinal vascular phenotype caused by loss of Kif11. In this experiment, 4HT treatment induces Cre-mediated recombination of a Ctnnb1 allele to generate a deletion derivative that codes for a stabilized version of beta-catenin. More specifically, Cre-mediated deletion of Ctnnb1 exon 3 leads to a small in-frame deletion that encompasses the phosphorylation site that controls beta-catenin ubiquitination and its subsequent degradation by the proteasome. The Ctnnb1 allele that has loxP sites flanking exon 3 is referred to as Ctnnb1flex3. In earlier work that used the same timing and dose of 4HT, Pdgfb-CreER-mediated recombination of the Ctnnb1flex3 allele was observed to fully rescue the retinal vascular defects caused by loss of Norrin (58), which, as described above, closely resembles the defects exhibited by Kif11CKO/−;Pdgfb-CreER retinas. In a WT background, Pdgfb-CreER-mediated recombination of the Ctnnb1flex3 allele has little effect, leading only to a small increase in vascular density (58). For the present experiment, both Kif11 loss and beta-catenin stabilization were initiated by 4HT treatment at P3. In contrast to the rescue of NdpKO retinas, EC-specific stabilization of beta-catenin had no effect on the Kif11 loss-of-function phenotype (Fig. 4A–D). At P10, retinas with or without stabilized beta-catenin exhibited indistinguishable patterns of vascular growth retardation (as quantified in Fig. 4E), disorganized capillary structure, and induction of PLVAP. Thus, artificially activating beta-catenin signaling fails to rescue the Kif11 loss-of-function phenotype.
Figure 4.
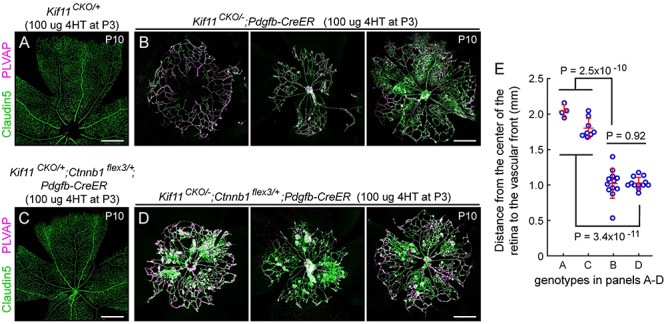
Postnatal EC-specific stabilization of beta-catenin does not rescue the Kif11 retinal vascular phenotype. (A,B) Retinal flatmounts were prepared at P10 from control Kif11CKO/+ mice (A) and Kif11CKO/−;Pdgfb-CreER mice (B; three examples shown) following the treatment with 100 μg 4HT at P3 to assess the Kif11 retinal vascular phenotype. (C,D) From the same set of crosses, retinal flatmounts were prepared at P10 from control Kif11CKO/+;Ctnnb1flex3/+;Pdgfb-CreER mice (C) and Kif11CKO/−;Ctnnb1flex3/+;Pdgfb-CreER mice (D; three examples shown) following the treatment with 100 μg 4HT at P3. Retinal vascular development in both sets of control mice is indistinguishable from WT. Beta-catenin stabilization following the Cre-mediated recombination of Ctnnb1flex3/+ fails to rescue the Kif11 retinal vascular phenotype. In (A) and (C), each image shows one retinal quadrant, with the optic disc at the bottom and the retinal periphery at the top; in (B) and (D), each image is centered on the optic disc. All images in (A–D) are at the same magnification. Scale bars, 500 μm. (E) Quantification of the radial distance from the optic disc to the vascular front at P10 for individual retinal quadrants for the genotypes shown in panels A–D. Plots show mean ± standard deviation.
In sum, these three tests imply that the Kif11 retinal vascular phenotype is not caused by reduced beta-catenin signaling. The most parsimonious explanation for the Kif11 phenotype is that it reflects Kif11’s role in cell proliferation and, more specifically, in mitotic spindle dynamics during mitosis. In the context of this model, we can explain PLVAP accumulation in ECs and Sulfo-NHS-biotin leakage in Kif11CKO/−;Pdgfb-CreER retinas as secondary consequences of retinal hypoxia rather than as a direct effect of decreased beta-catenin signaling. As noted above, in multiple models of retinal vascular insufficiency, retinal hypoxia results in increased expression of VEGF, which increases PLVAP expression and vascular permeability (50,51,55).
Effect of Kif11 loss on cerebellar vascular development
We next asked whether EC-specific loss of Kif11 has effects on vascular development beyond the retina. The ~ 100% survival of Kif11CKO/−;Pdgfb-CreER mice treated with 4HT at P3–P4 and followed for > 1 week implies that during this period of rapid postnatal growth the vascular supply to most tissues accommodates the increase in demand. As earlier studies had shown highly efficient recombination by Pdgfb-CreER throughout the early postnatal brain vasculature (10,15), we focused our comparative analysis on the Kif11CKO/−;Pdgfb-CreER phenotype in the brain. Specifically, we compared sagittal sections of brain from eight Kif11CKO/−;Pdgfb-CreER mice and nine control Kif11CKO/− and Kif11CKO/+ littermates that were treated with 4HT at P3 or P4 and sacrificed between P8 and P12. This analysis showed that the brain sizes between the two cohorts were closely matched, and the vascular densities in the cortex, thalamus, and brain stem were indistinguishable by visual inspection (Fig. 5A and B). However, the cerebella of Kif11CKO/−;Pdgfb-CreER mice showed a modestly reduced vascular density (Fig. 5A–C), an effect that was statistically significant (Fig. 5D). (The P-value of 3.9 × 10−4 for the experimental versus control comparison in Figure 5D was determined by a conservative calculation in which the comparison was between the mean values of vascular density for individual mice, determined by averaging the fraction of the length of the granule cell layer/molecular layer boundary that was vascularized in seven cerebellar folia per mouse.)
Figure 5.
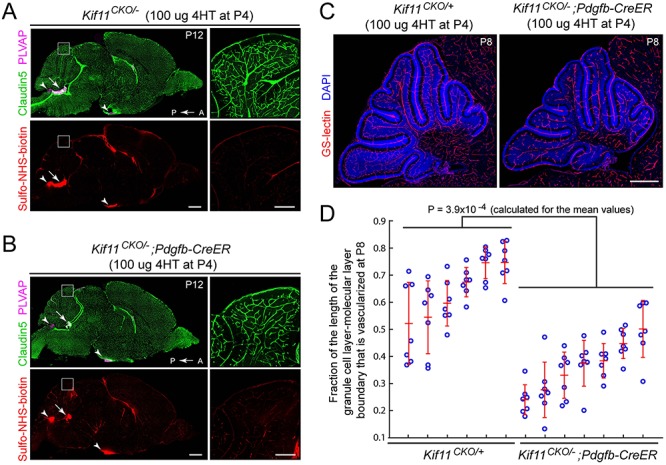
Retarded density of cerebellar vasculature following the early postnatal EC-specific knockout of Kif11. (A,B) Sagittal brain sections were prepared at P12 from control Kif11CKO/− mice (A) and Kif11CKO/−;Pdgfb-CreER mice (B) following the treatment with 100 μg 4HT at P4. The region of the dorsal cerebellum enclosed by the white square is enlarged at right. Scale bars, 1 mm (left images) and 200 μm (right images). Except for modestly reduced vascular density in the cerebellum (inset at right), brain size and vascular density are unaffected by EC-specific loss of Kif11. Sulfo-NHS-biotin leakage is seen in the choroid plexus (arrow) and in the circumventricular organs (area postrema and median eminence; arrowheads) as expected but is not observed elsewhere in the brain. (C) Sagittal brain sections were prepared at P8 from control Kif11CKO/+ mice and Kif11CKO/−;Pdgfb-CreER mice, following the treatment with 100 μg 4HT at P4. Scale bar, 500 μm. (D) With early postnatal loss of Kif11, vascular density in the P8 cerebellum is reduced, as determined by quantifying the fraction of the molecular layer/granule cell layer border that is occupied by blood vessels in six control Kif11CKO/+ and seven Kif11CKO/−;Pdgfb-CreER P8 mice. For each mouse, seven folia were quantified from a single confocal stack of the cerebellum [examples in (C)]. The plot in (D) shows mean ± standard deviation for each mouse.
Although the entire brain is growing during early postnatal life in mice, the cerebellum is distinctive in exhibiting extremely rapid growth during this period, with most granule cell divisions and much of the Purkinje cell and granule cell arbor growth occurring postnatally (59). Thus, the early postnatal requirement for Kif11 function in central nervous system (CNS) vascular cells is most significant in two CNS regions—the retina and the cerebellum—that exhibit rapid rates of postnatal growth.
Discussion
The experiments reported here establish a model for KIF11-based retinal vascular disease using EC-specific elimination of Kif11 in mice. The principal finding is that postnatal EC-specific loss of Kif11 leads to severely stunted growth of the retinal vasculature that is not caused by reduced EC beta-catenin signaling despite its close resemblance to the phenotype caused by mutations in beta-catenin signaling components. Interestingly, one other region of the CNS in which early postnatal EC-specific loss of Kif11 produces a prominent hypovascularization phenotype is the cerebellum, suggesting that the most significant requirements for Kif11 function occur in rapidly developing CNS regions that require correspondingly rapid vascular growth.
The phenotypic spectrum of KIF11 disorders
The present work complements a growing literature on the phenotypic consequences of KIF11 mutations in humans. One set of reports defined a syndrome in which autosomal dominant MLCRD is associated with KIF11 mutations (25,27). Other studies have noted the extensive overlap with a closely related syndrome characterized by chorioretinal dysplasia, microcephaly and mental retardation, with or without lymphedema that is also associated with KIF11 mutations (28,30). Finally, analyses that focused on large cohorts of patients with ocular disease have found KIF11 mutations to be associated with FEVR with or without microcephaly (4,32–35). Taken together, these studies imply a substantial degree of phenotypic variability, potentially related to the diverse functions of Kif11 in mitosis and in intracellular trafficking in non-dividing cells. The present work suggests that reduced Kif11 function in retinal and brain ECs may explain part of the pathophysiology of these human disorders.
The role of Kif11 in rapidly dividing cells: implications for retinal vascular disease
Kif11, Kif15 (also called Hklp2 or Kinesin-12) and dynein are essential force-generating proteins involved in forming the bipolar spindle during mitosis and directing its movement before and during anaphase (60). Interestingly, Kif15 exhibits partial redundancy with Kif11 in a manner that varies among cell lines (61) and that can account for partial escape from pharmacologic inhibition of Kif11 (62,63). These observations suggest that the rank order of sensitivity of ECs in the early postnatal CNS to loss of Kif11—retina > cerebellum > the rest of the brain—may reflect different degrees of dependence on Kif11 versus Kif15, either as intrinsic differences between the vasculatures in different CNS regions or as functional differences that reflect the rapidity of EC proliferation. In this regard, it is interesting that in experiments aimed at slowing tumor growth with a Kif11 inhibitor, Exertier et al (64) observed that Kif11 expression in ECs was increased by VEGF, and that pharmacologic inhibition of Kif11 had potent anti-angiogenic effects in cell culture, in chick and zebrafish embryos, and in a tumor model in mice. Thus, in each of the paradigms in which Kif11 loss or blockade has a large effect, the unifying feature may be the extreme rapidity of EC proliferation.
In sum, the present experiments implicate Kif11’s function in mitotic spindle assembly as the likely link to its role in diseases of retinal vascular development, and they suggest that genes coding for other components or regulators of the mitotic spindle could be plausible candidates for diseases or syndromes with the same or overlapping features. Importantly, these data imply that retinal hypovascularization disorders can arise from two mechanistically distinct causes: defects in beta-catenin signaling and defects in the mitotic machinery.
Materials and Methods
Mice
The following mouse alleles were used: Pdgfb-CreER (65), Sox2-Cre ((66); JAX 008454), R26-Flp ((67); JAX 009086), Ctnnb1flex3 (68), Kif11CKO and Kif11− (see below). Experiments were conducted, and mice were housed and handled according to the approved Institutional Animal Care and Use Committee protocol MO16M369 of the Johns Hopkins Medical Institutions.
Construction of the Kif11 CKO line
A BAC clone with one endpoint 3.5 kb 5′ of exon 2 and extending through the 3′ end of the Kif11 gene was used to construct the targeting vector, which consisted of (1) a ~ 3.5 kb 5′ homology arm starting at an EcoR V site ~ 1 kb 5′ of exon 2 and ending ~ 100 bp 5′ of exon 5; (2) loxP sites ~ 100 bp 5′ of exon 5 and 3′ of exon 6, with a Frt-PGK-Neo-Frt (FNF) cassette adjacent to the 5′ loxP site and (3) a ~ 3.5 kb 3′ homology arm starting ~ 100 bp 3′ of exon 6 and ending at an Apa I site in exon 8 (Fig. 1). The loxP sites are 640 bp apart. The targeting sequences were inserted into a vector with a PGK promoter and Herpes Simplex Virus Thymidine Kinase coding region for negative selection with ganciclovir. Following electroporation of R1 ES cells, positive selection with G418 and negative selection with ganciclovir correctly targeted ES clones were identified by southern blotting with probes distal to the 5′ and 3′ homology arms. Correctly targeted clones with a normal karyotype were injected into mouse blastocysts. To create the constitutive null allele (Kif11−), the original Kif11 targeted allele with FNF still present was subjected to germline Cre-mediated recombination by crossing to Sox2-Cre. To generate the Kif11CKO allele, the FNF cassette was excised by crossing to germline Flp-expressing mice (R26-Flp). Genotyping to distinguish WT and Kif11CKO alleles with a single PCR reaction was performed with a pair of primers flanking the 3′ loxP site [sense: ACTGCAGCAACCTTGATGAATGCTT (PMS1134) and antisense: CAATCTTCCAGTATTGCGAGCCTC (PMS1161)]. Three PCR primers were used in two separate PCR reactions to distinguish the WT Kif11 allele, the Kif11CKO allele and the Kif11 null allele resulting from Cre-mediated recombination of the Kif11CKO allele (Supplementary Material, Fig. S1): GATGGGAGGTGTAGCTGAGTAGTA (PMS1181), AGATTTCCAATAGGGACACTTTAACTG (JN4435) and ATGGTTGATTTTGAGACACAGTC (JN4437). PCR conditions were 1 × 94°C for 4 min, 35 × (94°C for 10 s; 63°C for 30 s; 72°C for 30 s) and 1 × 72°C for 7 min.
Antibodies and other reagents
The following antibodies were used for tissue immunohistochemistry and immunoblotting: rat anti-mouse PLVAP (MECA-32; BD Biosciences 553 849); mouse anti-Claudin5, Alexa Fluor 488 conjugate (Thermo Fisher Scientific 352 588); rabbit mAb anti-LEF1 (Cell Signaling Technologies C12A5); Texas Red Streptavidin (Vector Laboratories SA-5006). Alexa Fluor-labeled secondary antibodies and GS Lectin (Isolectin GS-IB4) were from Thermo Fisher Scientific. Sulfo-NHS-biotin was from Thermo Fisher Scientific (21217).
Tissue processing and immunohistochemistry
Tissues were prepared and processed for immunohistochemical analysis as described by Wang et al. (49) and Zhou et al. (58). Mice were injected intraperitoneally with Sulfo-NHS-biotin (100 μl of 20 mg/ml Sulfo-NHS-biotin in PBS for P8–P12 mice and 200 μl of 20 mg/ml Sulfo-NHS-biotin in PBS for P31 mice) ~ 30 min prior to sacrifice. Mice were deeply anesthetized with ketamine and xylazine and then perfused via the cardiac route with 1% PFA in PBS without calcium or magnesium, and the brains were dissected and dehydrated in 100% MeOH overnight at 4°C. Tissues were re-hydrated the following day in 1× PBS at 4°C for at least 3 h before embedding in 3% agarose. Tissue sections of 100–200 μm thickness were cut using a vibratome (Leica). For retina flatmounts, intact eyes were immersion fixed in 1% PFA in PBS at room temperature for 1 h, and then, the retinas were dissected and processed as described below.
Sections were incubated overnight with primary antibodies or Texas Red streptavidin diluted in 1× PBSTC (1× PBS + 1% Triton X-100 + 0.1 mM CaCl2) + 10% normal goat serum (NGS). Incubation and washing steps were performed at 4°C. Sections were washed at least three times with 1× PBSTC over the course of 6 h and subsequently incubated overnight with secondary antibodies diluted in 1× PBSTC +10% NGS. If a primary rat antibody was used, secondary antibodies were additionally incubated with 1% normal mouse serum as a blocking agent. The next day, sections were washed at least three times with 1× PBSTC over the course of 6 h and flat mounted using Fluoromount G (EM Sciences 17 984-25). Sections were imaged using a Zeiss LSM700 confocal microscope and processed with ImageJ, Adobe Photoshop and Adobe Illustrator software.
4HT preparation and administration
Solid 4HT (Sigma-Aldrich H7904) was dissolved in an ethanol:sunflower seed oil (Sigma-Aldrich S5007) mixture (1:10 volume:volume) to a final concentration of 2 mg/ml and stored in aliquots at −80°C. All injections were performed intraperitoneally.
Quantification of retinal vascular growth and cerebellar EC vascular density
For quantifying the distance from the optic disc to the vascular front, four distance measurements at 90-degree intervals were made from each image of a flat-mounted retina. For quantifying the density of blood vessels at the granule cell layer/molecular layer junction, 100 μm thick sagittal vibratome sections were stained with GS-lectin and DAPI. Confocal images were scanned at 10 μm intervals along the Z-axis, and four of these scans were Z-stacked. Starting with each Z-stacked image (as seen in Fig. 5), the Adobe Illustrator pencil tool was used to first trace the length of the granule cell layer/molecular layer junction for each of the seven cerebellar folia and then to trace those regions of the granule cell layer/molecular layer junction that included a blood vessel. The length the traces for each folium was quantified by calculating its pixel coverage as a fraction of a standard rectangular area, using ImageJ. For the granule cell layer/molecular layer junction of each folium, the vascularized length was divided by the total length to yield the fraction of the granule cell layer/molecular layer junction that was vascularized. MATLAB and Excel were used to generate plots and to perform statistical analysis. The mean ± standard deviations are shown. Statistical significance was determined by the unpaired t test, using the mean values for each mouse.
Supplementary Material
{kind=link}
Acknowledgements
The authors thank Chip Hawkins, Ann Lawler and Johnisha Witherspoon (Johns Hopkins Transgenic Core Facility) for ES cell injection, Dr Maketo Taketo (Kyoto University) for the Ctnnb1flex3 mice and Drs. Chris Cho, Amir Rattner and Jie Wang for assistance and helpful discussions.
Funding
This work was supported by the Howard Hughes Medical Institute.
Conflict of Interest: None declared.
References
- 1. Gilmour D.F. (2015) Familial exudative vitreoretinopathy and related retinopathies. Eye (Lond.), 29, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sun Y. and Smith L.E.H. (2018) Retinal vasculature in development and diseases. Annu. Rev. Vis. Sci., 4, 101–122. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 3. Nikopoulos K., Venselaar H., Collin R.W., Riveiro-Alvarez R., Boonstra F.N., Hooymans J.M., Mukhopadhyay A., Shears D., van Bers M., de Wijs I.J. et al. (2010) Overview of the mutation spectrum in familial exudative vitreoretinopathy and Norrie disease with identification of 21 novel variants in FZD4, LRP5, and NDP. Hum. Mutat., 31, 656–666. [DOI] [PubMed] [Google Scholar]
- 4. Rao F.Q., Cai X.B., Cheng F.F., Cheng W., Fang X.L., Li N., Huang X.F., Li L.H. and Jin Z.B. (2017) Mutations in LRP5,FZD4, TSPAN12, NDP, ZNF408, or KIF11 genes account for 38.7% of Chinese patients with familial exudative vitreoretinopathy. Invest. Ophthalmol. Vis. Sci., 58, 2623–2629. [DOI] [PubMed] [Google Scholar]
- 5. Tang M., Sun L., Hu A., Yuan M., Yang Y., Peng X. and Ding X. (2017) Mutation spectrum of the LRP5, NDP, and TSPAN12 genes in Chinese patients with familial exudative vitreoretinopathy. Invest. Ophthalmol. Vis. Sci., 58, 5949–5957. [DOI] [PubMed] [Google Scholar]
- 6. Li J.K., Li Y., Zhang X., Chen C.L., Rao Y.Q., Fei P., Zhang Q., Zhao P. and Li J. (2018) Spectrum of variants in 389 Chinese probands with familial exudative vitreoretinopathy. Invest. Ophthalmol. Vis. Sci., 59, 5368–5381. [DOI] [PubMed] [Google Scholar]
- 7. Ye X., Wang Y. and Nathans J. (2010) The Norrin/Frizzled4 signaling pathway in retinal vascular development and disease. Trends Mol. Med., 16, 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Stenman J.M., Rajagopal J., Carroll T.J., Ishibashi M., McMahon J. and McMahon A.P. (2008) Canonical Wnt signaling regulates organ-specific assembly and differentiation of CNS vasculature. Science, 322, 1247–1250. [DOI] [PubMed] [Google Scholar]
- 9. Daneman R., Agalliu D., Zhou L., Kuhnert F., Kuo C.J. and Barres B.A. (2009) Wnt/beta-catenin signaling is required for CNS, but not non-CNS, angiogenesis. Proc. Natl. Acad. Sci. U. S. A., 106, 6422–6422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhou Y. and Nathans J. (2014) Gpr124 controls CNS angiogenesis and blood-brain barrier integrity by promoting ligand-specific canonical Wnt signaling. Dev. Cell, 31, 248–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Posokhova E., Shukla A., Seaman S., Volate S., Hilton M.B., Wu B., Morris H., Swing D.A., Zhou M., Zudaire E. et al. (2015) GPR124 functions as a WNT7-specific coactivator of canonical β-catenin signaling. Cell Rep., 10, 123–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vanhollebeke B., Stone O.A., Bostaille N., Cho C., Zhou Y., Maquet E., Gauquier A., Cabochette P., Fukuhara S., Mochizuki N. et al. (2015) Tip cell-specific requirement for an atypical Gpr124- and Reck-dependent Wnt/β-catenin pathway during brain angiogenesis. elife, 4, e06489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cho C., Smallwood P.M. and Nathans J. (2017) Reck and Gpr124 are essential receptor cofactors for Wnt7a/Wnt7b-specific Signaling in mammalian CNS angiogenesis and blood-brain barrier regulation. Neuron, 95, 1056–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Eubelen M., Bostaille N., Cabochette P., Gauquier A., Tebabi P., Dumitru A.C., Koehler M., Gut P., Alsteens D., Stainier D.Y.R., Garcia-Pino A. and Vanhollebeke B. (2018) A molecular mechanism for Wnt ligand-specific signaling. Science, 361, 6403. [DOI] [PubMed] [Google Scholar]
- 15. Wang Y., Cho C., Williams J., Smallwood P.M., Zhang C., Junge H.J. and Nathans J. (2018) Interplay of the Norrin and Wnt7a/Wnt7b signaling systems in blood-brain barrier and blood-retina barrier development and maintenance. Proc. Natl. Acad. Sci. U. S. A., 115, E11827–E11836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Smith S.E., Mullen T.E., Graham D., Sims K.B. and Rehm H.L. (2012) Norrie disease: extraocular clinical manifestations in 56 patients. Am. J. Med. Genet. A, 158A, 1909–1917. [DOI] [PubMed] [Google Scholar]
- 17. Maupin K.A., Droscha C.J. and Williams B.O. (2013) A comprehensive overview of skeletal phenotypes associated with alterations in Wnt/β-catenin Signaling in humans and mice. Bone Res., 1, 27–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Panagiotou E.S., Sanjurjo Soriano C., Poulter J.A., Lord E.C., Dzulova D., Kondo H., Hiyoshi A., Chung B.H., Chu Y.W., Lai C.H.Y. et al. (2017) Defects in the cell signaling mediator β-catenin cause the retinal vascular condition FEVR. Am. J. Hum. Genet., 100, 960–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sun W., Xiao X., Li S., Jia X., Wang P. and Zhang Q. (2019) Germline mutations in CTNNB1 associated with syndromic FEVR or Norrie disease. Invest. Ophthalmol. Vis. Sci., 60, 93–97. [DOI] [PubMed] [Google Scholar]
- 20. Tucci V., Kleefstra T., Hardy A., Heise I., Maggi S., Willemsen M.H., Hilton H., Esapa C., Simon M., Buenavista M.T. et al. (2014) Dominant β-catenin mutations cause intellectual disability with recognizable syndromic features. J. Clin. Invest., 124, 1468–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kuechler A., Willemsen M.H., Albrecht B., Bacino C.A., Bartholomew D.W., van Bokhoven H., van den Boogaard M.J., Bramswig N., Büttner C., Cremer K. et al. (2015) De novo mutations in beta-catenin (CTNNB1) appear to be a frequent cause of intellectual disability: expanding the mutational and clinical spectrum. Hum. Genet., 134, 97–109. [DOI] [PubMed] [Google Scholar]
- 22. Kharbanda M., Pilz D.T., Tomkins S., Chandler K., Saggar A., Fryer A., McKay V., Louro P., Smith J.C., Burn J. et al. (2017) Clinical features associated with CTNNB1 de novo loss of function mutations in ten individuals. Eur. J. Med. Genet., 60, 130–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Valenta T., Hausmann G. and Basler K. (2012) The many faces and functions of β-catenin. EMBO J., 31, 2714–2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Limwongse C., Wyszynski R.E., Dickerman L.H. and Robin N.H. (1999) Microcephaly-lymphedema-chorioretinal dysplasia: a unique genetic syndrome with variable expression and possible characteristic facial appearance. Am. J. Med. Genet., 86, 215–218. [PubMed] [Google Scholar]
- 25. Hazan F., Ostergaard P., Ozturk T., Kantekin E., Atlihan F., Jeffery S. and Ozkinay F. (2012) A novel KIF11 mutation in a Turkish patient with microcephaly, lymphedema, and chorioretinal dysplasia from a consanguineous family. Am. J. Med. Genet. A, 158A, 1686–1689. [DOI] [PubMed] [Google Scholar]
- 26. Kelly M.N., Khuddus N., Motamarry S. and Tuli S. (2012) Microcephaly, lymphedema, chorioretinal dysplasia (MLCRD) syndrome. J. Pediatr. Health Care, 26, 306–311. [DOI] [PubMed] [Google Scholar]
- 27. Ostergaard P., Simpson M.A., Mendola A., Vasudevan P., Connell F.C., van Impel A., Moore A.T., Loeys B.L., Ghalamkarpour A., Onoufriadis A. et al. (2012) Mutations in KIF11 cause autosomal-dominant microcephaly variably associated with congenital lymphedema and chorioretinopathy. Am. J. Hum. Genet., 90, 356–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mirzaa G.M., Enyedi L., Parsons G., Collins S., Medne L., Adams C., Ward T., Davitt B., Bicknese A., Zackai E. et al. (2014) Congenital microcephaly and chorioretinopathy due to de novo heterozygous KIF11 mutations: five novel mutations and review of the literature. Am. J. Med. Genet. A, 164A, 2879–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mears K., Bakall B., Harney L.A., Penticoff J.A. and Stone E.M. (2015) Autosomal dominant microcephaly associated with congenital lymphedema and chorioretinopathy due to a novel mutation in KIF11. JAMA Ophthalmol., 133, 720–721. [DOI] [PubMed] [Google Scholar]
- 30. Balikova I., Robson A.G., Holder G.E., Ostergaard P., Mansour S. and Moore A.T. (2016) Ocular manifestations of microcephaly with or without chorioretinopathy, lymphedema or intellectual disability (MCLID) syndrome associated with mutations in KIF11. Acta Ophthalmol., 94, 92–98. [DOI] [PubMed] [Google Scholar]
- 31. Birtel J., Gliem M., Mangold E., Tebbe L., Spier I., Müller P.L., Holz F.G., Neuhaus C., Wolfrum U., Bolz H. and Charbel Issa P. (2017) Novel insights into the phenotypical spectrum of KIF11-associated retinopathy, including a new form of retinal ciliopathy. Invest. Ophthalmol. Vis. Sci., 58, 3950–3959. [DOI] [PubMed] [Google Scholar]
- 32. Robitaille J.M., Gillett R.M., LeBlanc M.A., Gaston D., Nightingale M., Mackley M.P., Parkash S., Hathaway J., Thomas A., Ells A. et al. (2014) Phenotypic overlap between familial exudative vitreoretinopathy and microcephaly, lymphedema, and chorioretinal dysplasia caused by KIF11 mutations. JAMA Ophthalmol., 132, 1393–1399. [DOI] [PubMed] [Google Scholar]
- 33. Hu H., Xiao X., Li S., Jia X., Guo X. and Zhang Q. (2016) KIF11 mutations are a common cause of autosomal dominant familial exudative vitreoretinopathy. Br. J. Ophthalmol., 100, 278–283. [DOI] [PubMed] [Google Scholar]
- 34. Li J.K., Fei P., Li Y., Huang Q.J., Zhang Q., Zhang X., Rao Y.Q., Li J. and Zhao P. (2016) Identification of novel KIF11 mutations in patients with familial exudative vitreoretinopathy and a phenotypic analysis. Sci. Rep., 6, 26564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Karjosukarso D.W., Cremers F.P.M., van Nouhuys C.E. and Collin R.W.J. (2018) Detection and quantification of a KIF11 mosaicism in a subject presenting familial exudative vitreoretinopathy with microcephaly. Eur. J. Hum. Genet., 26, 1819–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Blangy A., Lane H.A., d'Herin P., Harper M., Kress M. and Nigg E.A. (1995) Phosphorylation by p34(cdc2) regulates spindle association of human Eg5, a kinesin-related motor essential for bipolar spindle formation in vivo. Cell, 83, 1159–1169. [DOI] [PubMed] [Google Scholar]
- 37. Ferenz N.P., Gable A. and Wadsworth P. (2010) Mitotic functions of kinesin-5. Semin. Cell Dev. Biol., 21, 255–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bartoli K.M., Jakovljevic J., Woolford J.L. Jr. and Saunders W.S. (2011) Kinesin molecular motor Eg5 functions during polypeptide synthesis. Mol. Biol. Cell, 22, 3420–3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wakana Y., Villeneuve J., van Galen J., Cruz-Garcia D., Tagaya M. and Malhotra V. (2013) Kinesin-5/Eg5 is important for transport of CARTS from the trans-Golgi network to the cell surface. J. Cell Biol., 202, 241–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Falnikar A., Tole S. and Baas P.W. (2011) Kinesin-5, a mitotic microtubule-associated motor protein, modulates neuronal migration. Mol. Biol. Cell, 22, 1561–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kahn O.I., Sharma V., González-Billault C. and Baas P.W. (2015) Effects of kinesin-5 inhibition on dendritic architecture and microtubule organization. Mol. Biol. Cell, 26, 66–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Freixo F., Martinez Delgado P., Manso Y., Sánchez-Huertas C., Lacasa C., Soriano E., Roig J. and Lüders J. (2018) NEK7 regulates dendrite morphogenesis in neurons via Eg5-dependent microtubule stabilization. Nat. Commun., 9, 2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rath O. and Kozielski F. (2012) Kinesins and cancer. Nat. Rev. Cancer, 12, 527–539. [DOI] [PubMed] [Google Scholar]
- 44. Myers S.M. and Collins I. (2016) Recent findings and future directions for interpolar mitotic kinesin inhibitors in cancer therapy. Future Med. Chem., 8, 463–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Castillo A. and Justice M.J. (2007) The kinesin related motor protein, Eg5, is essential for maintenance of pre-implantation embryogenesis. Biochem. Biophys. Res. Commun., 357, 694–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chauvière M., Kress C. and Kress M. (2008) Disruption of the mitotic kinesin Eg5 gene (Knsl1) results in early embryonic lethality. Biochem. Biophys. Res. Commun., 372, 513–519. [DOI] [PubMed] [Google Scholar]
- 47. Luhmann U.F., Lin J., Acar N., Lammel S., Feil S., Grimm C., Seeliger M.W., Hammes H.P. and Berger W. (2005) Role of the Norrie disease pseudoglioma gene in sprouting angiogenesis during development of the retinal vasculature. Invest. Ophthalmol. Vis. Sci., 46, 3372–3382. [DOI] [PubMed] [Google Scholar]
- 48. Ye X., Wang Y., Cahill H., Yu M., Badea T.C., Smallwood P.M., Peachey N.S. and Nathans J. (2009) Norrin, frizzled-4, and Lrp5 signaling in endothelial cells controls a genetic program for retinal vascularization. Cell, 139, 285–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang Y., Rattner A., Zhou Y., Williams J., Smallwood P.M. and Nathans J. (2012) Norrin/Frizzled4 signaling in retinal vascular development and blood brain barrier plasticity. Cell, 151, 1332–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rattner A., Yu H., Williams J., Smallwood P.M. and Nathans J. (2013) Endothelin-2 signaling in the neural retina promotes the endothelial tip cell state and inhibits angiogenesis. Proc. Natl. Acad. Sci. U. S. A., 110, E3830–E3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rattner A., Wang Y., Zhou Y., Williams J. and Nathans J. (2014) The role of the hypoxia response in shaping retinal vascular development in the absence of Norrin/Frizzled4 signaling. Invest. Ophthalmol. Vis. Sci., 55, 8614–8625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Heng J.S., Rattner A., Stein-O'Brien G.L., Winer B.L., Jones B.W., Vernon H.J., Goff L.A. and Nathans J. (2019) Hypoxia tolerance in the Norrin-deficient retina and the chronically hypoxic brain studied at single-cell resolution. Proc. Natl. Acad. Sci. U. S. A., 116, 9103–9114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chow B.W. and Gu C. (2017) Gradual suppression of transcytosis governs functional blood-retinal barrier formation. Neuron, 93, 1325–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Stan R.V., Tkachenko E. and Niesman I.R. (2004) PV1 is a key structural component for the formation of the stomatal and fenestral diaphragms. Mol. Biol. Cell, 15, 3615–3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kaur C., Foulds W.S. and Ling E.A. (2008) Blood-retinal barrier in hypoxic ischaemic conditions: basic concepts, clinical features and management. Prog. Retin. Eye Res., 27, 622–647. [DOI] [PubMed] [Google Scholar]
- 56. Sabbagh M.F., Heng J.S., Luo C., Castanon R.G., Nery J.R., Rattner A., Goff L.A., Ecker J.R. and Nathans J. (2018) Transcriptional and epigenomic landscapes of CNS and non-CNS vascular endothelial cells. elife, 7, e36187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wang Y., Sabbagh M.F., Gu X., Rattner A., Williams J. and Nathans J. (2019) Beta-catenin signaling regulates barrier-specific gene expression in circumventricular organ and ocular vasculatures. elife, 8, e43257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zhou Y., Wang Y., Tischfield M., Williams J., Smallwood P.M., Rattner A., Taketo M.M. and Nathans J. (2014) Canonical WNT signaling components in vascular development and barrier formation. J. Clin. Invest., 124, 3825–3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chizhikov V. and Millen K.J. (2003) Development and malformations of the cerebellum in mice. Mol. Genet. Metab., 80, 54–65. [DOI] [PubMed] [Google Scholar]
- 60. van Heesbeen R.G., Tanenbaum M.E. and Medema R.H. (2014) Balanced activity of three mitotic motors is required for bipolar spindle assembly and chromosome segregation. Cell Rep., 8, 948–956. [DOI] [PubMed] [Google Scholar]
- 61. Gayek A.S. and Ohi R. (2014) Kinetochore-microtubule stability governs the metaphase requirement for Eg5. Mol. Biol. Cell, 25, 2051–2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tanenbaum M.E., Macůrek L., Janssen A., Geers E.F., Alvarez-Fernández M. and Medema R.H. (2009) Kif15 cooperates with eg5 to promote bipolar spindle assembly. Curr. Biol., 19, 1703–1711. [DOI] [PubMed] [Google Scholar]
- 63. Sturgill E.G., Norris S.R., Guo Y. and Ohi R. (2016) Kinesin-5 inhibitor resistance is driven by kinesin-12. J. Cell Biol., 213, 213–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Exertier P., Javerzat S., Wang B., Franco M., Herbert J., Platonova N., Winandy M., Pujol N., Nivelles O., Ormenese S. et al. (2013) Impaired angiogenesis and tumor development by inhibition of the mitotic kinesin Eg5. Oncotarget, 4, 2302–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Claxton S., Kostourou V., Jadeja S., Chambon P., Hodivala-Dilke K. and Fruttiger M. (2008) Efficient, inducible Cre-recombinase activation in vascular endothelium. Genesis, 46, 74–80. [DOI] [PubMed] [Google Scholar]
- 66. Hayashi S., Lewis P., Pevny L. and McMahon A.P. (2002) Efficient gene modulation in mouse epiblast using a Sox2Cre transgenic mouse strain. Gene Expr. Patterns, 2, 93–97. [DOI] [PubMed] [Google Scholar]
- 67. Farley F.W., Soriano P., Steffen L.S. and Dymecki S.M. (2000) Widespread recombinase expression using FLPeR (flipper) mice. Genesis, 28, 106–110. [PubMed] [Google Scholar]
- 68. Harada N., Tamai Y., Ishikawa T., Sauer B., Takaku K., Oshima M. and Taketo M.M. (1999) Intestinal polyposis in mice with a dominant stable mutation of the beta-catenin gene. EMBO J., 18, 5931–5942. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.