

Abstract
The current gold standard for unambiguous molecular identification in metabolomics analysis is comparing two or more orthogonal properties from the analysis of authentic reference materials (standards) to experimental data acquired in the same laboratory with the same analytical methods. This represents a significant limitation for comprehensive chemical identification of small molecules in complex samples. The process is time consuming and costly, and the majority of molecules are not yet represented by standards. Thus, there is a need to assemble evidence for the presence of small molecules in complex samples through the use of libraries containing calculated chemical properties. To address this need, we developed a Multi-Attribute Matching Engine (MAME) and a library derived in part from our in silico chemical library engine (ISiCLE). Here, we describe an initial evaluation of these methods in a blinded analysis of synthetic chemical mixtures as part of the U.S. Environmental Protection Agency’s (EPA) Non-Targeted Analysis Collaborative Trial (ENTACT, Phase 1). For molecules in all mixtures, the initial blinded false negative rate (FNR), false discovery rate (FDR), and accuracy were 57%, 77%, and 91%, respectively. For high evidence scores, the FDR was 35%. After unblinding of the sample compositions, we optimized the scoring parameters to better exploit the available evidence and increased the accuracy for molecules suspected as present. The final FNR, FDR, and accuracy were 67%, 53%, and 96%, respectively. For high evidence scores, the FDR was 10%. This study demonstrates that multiattribute matching methods in conjunction with in silico libraries may one day enable reduced reliance on experimentally derived libraries for building evidence for the presence of molecules in complex samples.
Graphical Abstract
■ INTRODUCTION
Conventional metabolomics and small molecule identification approaches have demonstrated immense value for disease diagnosis, evaluation of environmental exposures, and discovery of novel molecules. This success is reflected in the large number of recent biomedical, environmental exposure, and soil and ecology studies employing metabolomics approaches.2–10 In contrast to genetic and proteomic information available from rapid genome and proteome sequencing, far less is understood about the totality of human exposure and small molecules found in the environment.12–14 Furthermore, driven by a broader interest in understanding biological impacts of chemical exposures, biomonitoring is undergoing a significant evolution.15,16 Traditional biomonitoring approaches, either targeted (seeking to identify specific compounds) or nontargeted (seeking to identify as many compounds as possible),17 and using either low- or high-resolution mass spectrometry18,19 rely on authentic, pure reference materials (standards) for unambiguous chemical identification and are therefore limited to the subset of molecules for which these standards exist.20 A wealth of information about human exposure continues to emerge from these methods for a subset of chemical space confined to the list of molecules represented by standards. The Centers for Disease Control and Prevention (CDC) National Health and Nutrition Examination Survey (NHANES) program and the National Institutes of Health (NIH) Children’s Health Exposure Analysis Resource (CHEAR) centers have provided such data, leading to examples of successful applications of these methods.21–23
Recent proposals to characterize the whole metabolome and exposome-the aggregate of all exposures-are driving a shift from traditional quantitative analytical chemistry and the typical strictures for chemical identification to new methods applicable to the discovery of molecules for which there are no standards.24 The vast chemical space of the metabolome and exposome together includes endogenous (e.g., metabolites) and exogenous (e.g., xenobiotics, industrial chemicals, consumer products, and transformation products of these) chemicals.16 There are no authentic reference materials for the preponderance of these molecules. For example, using an automated script we found only 17% of compounds found in the Human Metabolome Database, HMDB,25 and less than 2% of compounds found in exposure chemical databases like the EPA DSSTox (comptox.epa.gov) can be readily purchased in pure form. Without chemical standards, unambiguous chemical identification is limited to the small number of molecules amenable to nuclear magnetic resonance spectroscopy- or crystallography-based structural elucidation, while the vast majority is left as chemical “dark matter”.26 The need for more comprehensive and unambiguous chemical identification in these studies is driving innovations in analytical chemistry, computational chemistry, and cheminformatics.27,28 For example, new targeted and nontargeted methods have emerged as adaptions to traditional analytical chemistry.29
We are beginning to test methods for building evidence for the presence of small molecules in complex samples through the use of libraries of calculated chemical properties and associated matching to experimental data using multiple molecular attributes (i.e., multiattribute matching). Reliance on traditional, experimentally derived libraries for chemical identification will remain the gold standard for many common applications in the field. However, we also recognize that unless we look to computational methods for establishing libraries the majority of chemical space will remain unidentifiable. We hypothesize that libraries of chemical properties derived computationally can replace libraries derived from authentic standards for compound identification under specific conditions, for example, when the evidence of presence is strong enough and confidence high enough to support the intended application. Indeed, the entire field of proteomics has been built on this same premise, i.e., the in silico generation of reference proteomes based on corresponding genomes, and associated in silico digestion of proteins to their constituent peptides, followed then by in silico prediction of their tandem mass spectra.
The multiattribute matching approach described here utilizes multiple experimental data types, including accurate mass, isotopic distribution, and collisional cross section (CCS), and comparison of these values to entries in in silico libraries, leveraging instrumental and computational innovations developed at Pacific Northwest National Laboratory (PNNL).20,30–39 As an initial evaluation of this methodology, we participated in the U.S. Environmental Protection Agency’s (EPA) Non-Targeted Analysis Collaborative Trial (ENTACT, Phase 1), an interlaboratory challenge established to provide a consistent set of verified, blinded synthetic mixtures for the objective testing of nontargeted analytical chemistry methods.40–42 We performed blinded analysis of 10 synthetic mixtures each containing an, at the time, unknown number of substances as part of the multilaboratory challenge. Accurate mass, isotopic signature, and CCS measurements were collected using ion-mobility spectrometry-mass spectrometry (IMS-MS) and ultrahigh resolution 21-T Fourier transform ion cyclotron resonance-mass spectrometry (FTICR-MS) (Figure 1). These properties were also calculated for each molecule in the processed form of the EPA Toxicity Forecaster (ToxCast)43 library, allowing us to match observed features (e.g., peaks characterized by a measured mass and intensity, or a measured mass, CCS and intensity) to library entries. After unblinding, we performed statistical analysis on the results in order to assess the method’s performance. Scoring parameters were then optimized to improve the method and to better understand the importance of each parameter in the scoring algorithm. Our findings demonstrate the potential of building evidence for the presence of small molecules in complex samples using in silico libraries and multiattribute matching algorithms.
Figure 1.
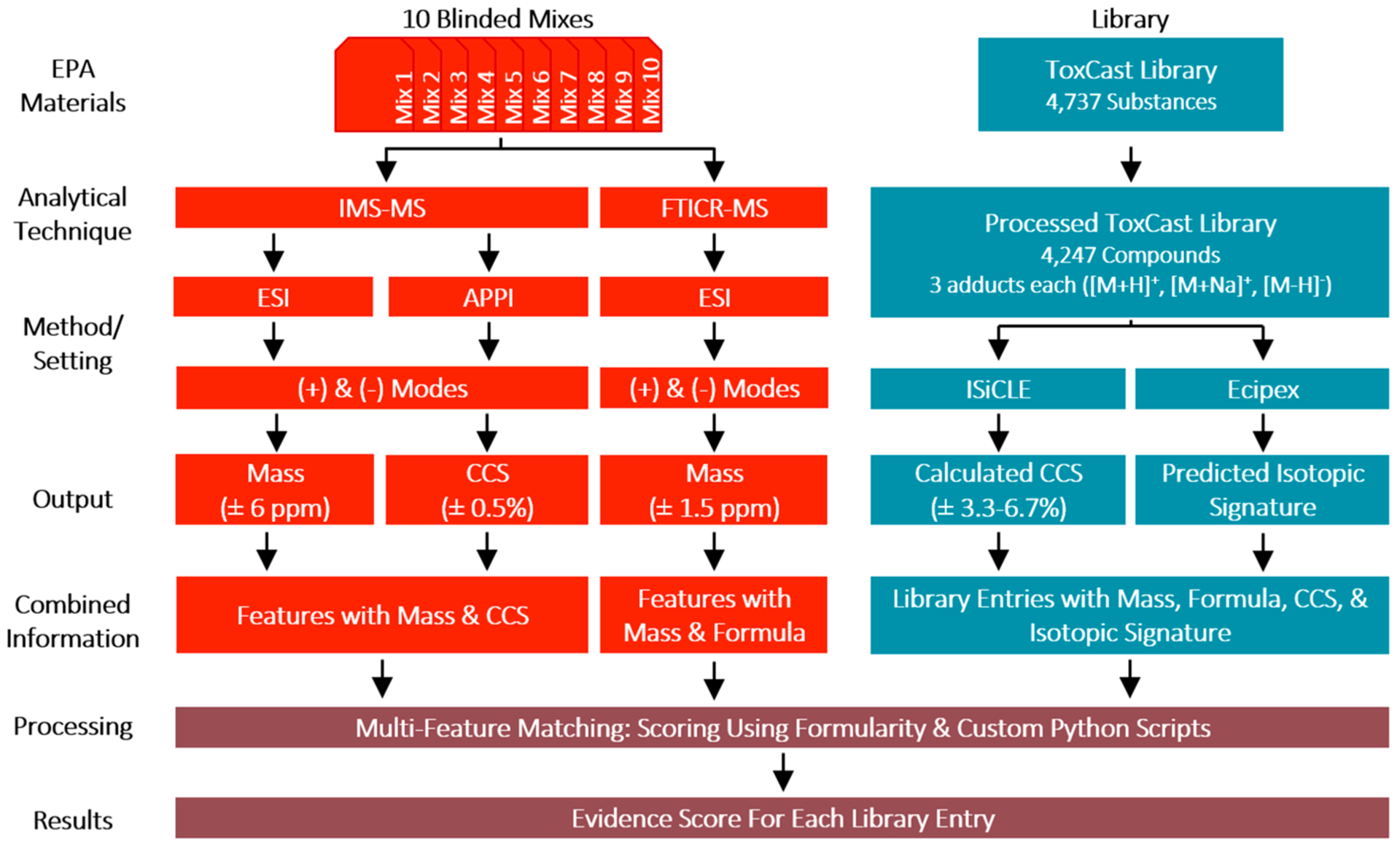
Project overview. Detailed project flow, starting from the blinded mixtures and ToxCast Library (the given suspect screening library). After instrumental analysis of the mixtures and computational property calculations for library entries, our multiattribute scoring algorithm was used for assigning evidence and determining compounds suspected to be present in each mixture. Note substances can be composed of one or more molecules that separate upon solvation in liquid. Molecules are single molecular structures.
■ EXPERIMENTAL SECTION
Mass Spectrometry of Blinded Samples.
The 10 synthetic mixtures and blanks, provided by the EPA, were analyzed using a drift tube ion mobility spectrometry-mass spectrometer (IMS-MS)30,44 and a 21-T Fourier transform-ion cyclotron resonance spectrometer-mass spectrometer (FTICR-MS)31,32,45 in both positive (+) and negative (−) ionization modes (Figure 1). This resulted in 14 disparate experimental data sets per sample. Details regarding the experimental protocol for sample preparation and mass spectrometry methods are provided in Supporting Information (SI) 1.0 –3.0.
Chemical Property Calculations.
CCS and isotopic signatures were calculated for the [M + H]+, [M + Na]+, and [M − H]− adducts of each entry in the suspect library using the in silico chemical library engine (ISiCLE)20,33,39 and Ecipex46 (based on chemical formula assignments from Formularity34), respectively. Please see SI 3.2 and 4 for additional details.
Multiattribute Downselection, Matching, and Scoring.
We developed a comprehensive package, written in Python, for assessing experimental evidence for the presence of molecules in complex samples, the Multi-Attribute Matching Engine (MAME), which includes feature downselection and a scoring system (broken into low, medium, and high levels of evidence). MAME is modular so that it can be customized in future studies for different types of data and different sets of experimental- and computational-based matching libraries. MAME scores evidence for the presence of a molecule, with the level of evidence increasing by the number and quality of experimental features that match to properties in the in silico library for a given entry. We scored the evidence of suspect library entries being in each mixture using our weighting method.
Note that the method described here does not label specific features as belonging to a specific compound (i.e., directly linking a feature arising due to instrument response to a specific compound), which is common in the literature. Instead, our scoring system considers all evidence indicating the presence of a compound, where multiple features consistent with possible instrument responses of a compound increase the probability of that compound’s presence. The focus is to connect the experimental evidence to the presence of specific compounds, rather than attempt to prove that specific features resulted from specific compounds. This is an important distinction as it is not always possible to label a feature as belonging to a particular compound, especially in the case of complex samples. Instead, we use multiple experimental features to lend evidence to the presence of a compound within a sample without attempting to label individual features.
Downselection of candidate features and molecular library entries and evidence scoring were performed using MAME, which processed all 14 raw data sets per sample to achieve multiattribute, aggregate evidence-based lists of molecules that are suspected to be present in each sample. A set of parameter cutoffs was used for data preprocessing (Table S1). For example, for an IMS-MS feature to be counted toward the evidence score of a molecule it needed to (i) be observed in all three technical replicates, (ii) have a signal intensity ≥ 1000 (arbitrary units), (iii) have a mass measurement error ≤ ±6 ppm, and (iv) not have been observed in more than one blank (which also had three technical replicates). For an FTICR-MS feature to be counted toward the evidence score of a molecule it needed to (i) have a mass measurement error ≤ ±1.5 ppm and (ii) not have been observed in the blank run.
Once all analytical features were processed and matched to corresponding entries in the suspect library, we scored the evidence of each library entry being present in each mixture using MAME, which uses a total of 11 independent scoring parameters (Table 1). These parameters were initially selected based on expert domain knowledge in our group, since this type of study had not been pursued previously. On the basis of our previous experience with these types of data sets collected individually, we subjectively attempted to add higher weight to criteria that were less susceptible to noise or was thought to provide better quality evidence for the presence of a molecule. For example, due to the high mass resolution of the FTICR-MS compared to the QTOF connected to our IMS, FTICR-MS features initially received a higher score. Additionally, features with an intensity higher than the 30th percentile of all passing feature intensities were assigned to provide more evidence than their low-intensity counterparts since a higher intensity across all samples was thought to be more likely to point to something real in the sample, rather than noise or low-level contamination. A library entry was labeled as “suspected present” in the mixture if its evidence score reached a threshold of 6.0 or more. Evidence scores of 6.0–11.0, 11.0–19.0, and 19.0+ were labeled as low, medium, and high evidence, respectively. A more detailed description of MAME is included in SI 5.2–5.3, and the full software package is available upon request. As an example, Figure 2 shows how pioglitazone was scored and correctly labeled as suspected present in one of the mixtures.
Table 1.
Initial Scoring Criteria and Their Associated Weights
| category | index | criteria | weight |
|---|---|---|---|
| IMS-MS | 1 | high intensity | 2.0 |
| 2 | low intensity | 1.0 | |
| 3 | low CCS error | 3.0 | |
| FTICR-MS | 4 | high intensity | 4.0 |
| 5 | low intensity | 2.0 | |
| 6 | isotopic signature | 3.0a | |
| IMS-MS and FTICR-MS | 7 | additional adducts | 1.0 |
| 8 | additional features | 0.5 | |
| 9 | detected by both MS | 2.0 | |
| library | 10 | unique mass | 4.0 |
| 11 | large mass | 1.0 |
Earned a maximum of one time per library entry.
Figure 2.
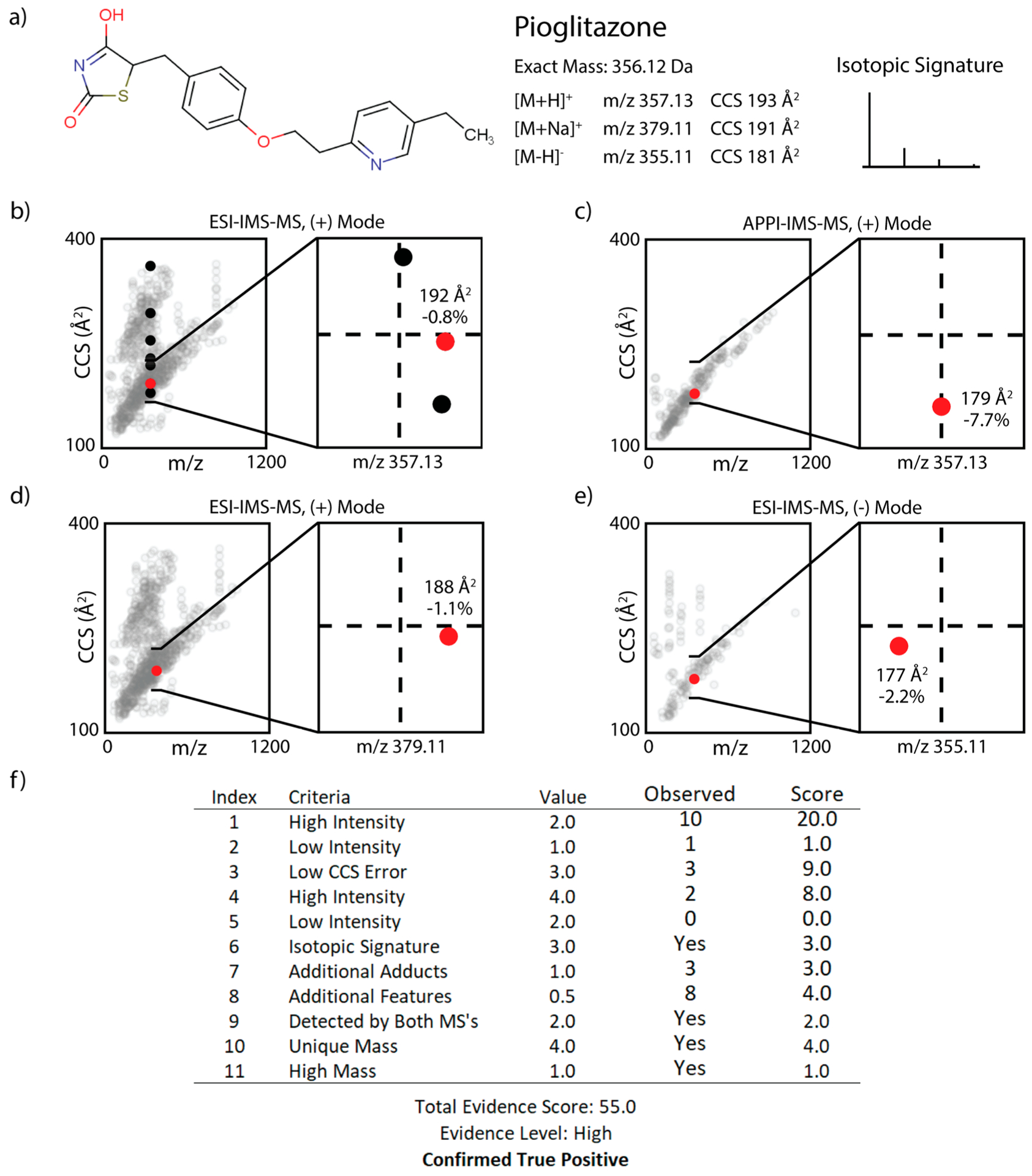
Example scoring of pioglitazone, a true positive evaluated using our multiattribute scoring system. Note, pioglitazone hydrochloride was in the ToxCast library and then changed to pioglitazone (the structure suspected present in solution) in our processed library. (a) Library entry for pioglitazone, a phenol ether1 drug (sold as Actos) used to control high blood sugar in patients with type 2 diabetes,11 with calculated CCS (using standard ISiCLE) for the three adduct types and its calculated isotopic signature. (b-e) IMS-MS features observed within a ±6 ppm mass error window of a given adduct mass. A magnified view is provided, centered around the calculated mass and CCS, with the mass and CCS ranges extending 6 ppm and 20 Å, respectively, on either side of this average. Percentages are with respect to the calculated CCS. Red points indicate the experimental feature closest to our prediction. (f) Combined scoring of all features. Number of features matching a specified criterion, or whether the criterion was met, is provided in the “Observed” column.
Analysis and Optimization of the Scoring Algorithm.
As metrics to quantify success, we used false discovery rate (FDR), false negative rate (FNR), and accuracy. Equations and more details for each of these metrics are provided in SI 5.4. Also, please see SI 5.5 for details on how new weights for each scoring criteria were optimized after unblinding.
■ RESULTS AND DISCUSSION
The foundation of our approach is the in silico construction of a library of chemical properties used to characterize experimental data collected for each sample. The method operates by considering the consistency between the library of predicted properties and the observed analytical features and subsequently quantifying and weighting their similarity. Calculated scores based on the evaluation of experimental features matched to library entries allow us to determine a single-evidence score for each library entry and, ultimately, whether there is enough evidence to indicate a given compound is suspected present in a sample. For the purpose of this study, any compounds labeled as suspected present that, after unblinding, were found to be intentionally spiked in are considered a true positive.
Construction of the in silico Library.
The EPA provided the ToxCast library as the suspect library (mixtures were only spiked with ToxCast substances). We processed all substances within this library as described in SI 5.1, which lead to a suspect library of 4348 total compounds that are theoretically observable by mass spectrometry. Approximately one-half of this library did not have a unique mass (assuming a mass error of ±6 ppm, Figure S1a). Further, 47% of library entries have at least one other formula conflict within the ToxCast, and over 13% had five or more conflicts. Even perfect mass accuracy can only result in determining a molecular formula, for which each map to numerous molecular structures in nearly all chemical libraries, and thus, high-resolution mass instruments alone are inadequate for high-accuracy identification without complementary, orthogonal data.47
CCS is a chemical property that provides additional information on which to increase the uniqueness of each library entry (Figure S1b). This is especially powerful when considering the CCS of each adduct as independent information, effectively adding corroborating dimensions of data for each adduct with a known CCS. The addition of CCS increased the evidence score of 79% of true positive compounds (average addition of 5.0 points, σ = 2.6 points). Broken down by evidence levels, addition of CCS increased the evidence score of 72%, 85%, and 95% of compounds for low, medium, and high evidence levels, respectively.
Multiattribute Matching for Building Evidence for the Presence of Small Molecules in Blinded Samples.
Before unblinding the true compositions of the mixtures, we performed multiattribute matching by comparing the measured properties of downselected experimental features to the in silico library of calculated properties and scoring each putative match using values given in Table 1. Note, for IMS-MS, the high-intensity cutoff (i.e., the 30th percentile value of downselected features) was 2123 and 2174 for positive and negative mode, respectively. For FTICR-MS, the high-intensity cutoff was 3358 and 110 for positive and negative mode, respectively. Please see SI 6 for details on the results of the analysis of samples and feature extraction. An example of the multi-attribute scoring method is demonstrated in Figure 2.
This same analysis was performed for all library entries, taking into consideration all 14 data sets (and blanks), using MAME, resulting in an evidence score for each library entry for each mix. We submitted the list of compounds labeled as suspected present in each mixture (and their associated evidence scores and evidence levels) to the EPA, who then unblinded the samples by returning the sample key to enable the assessment of our approach. An overview of the results is shown in Figure 3.
Figure 3.
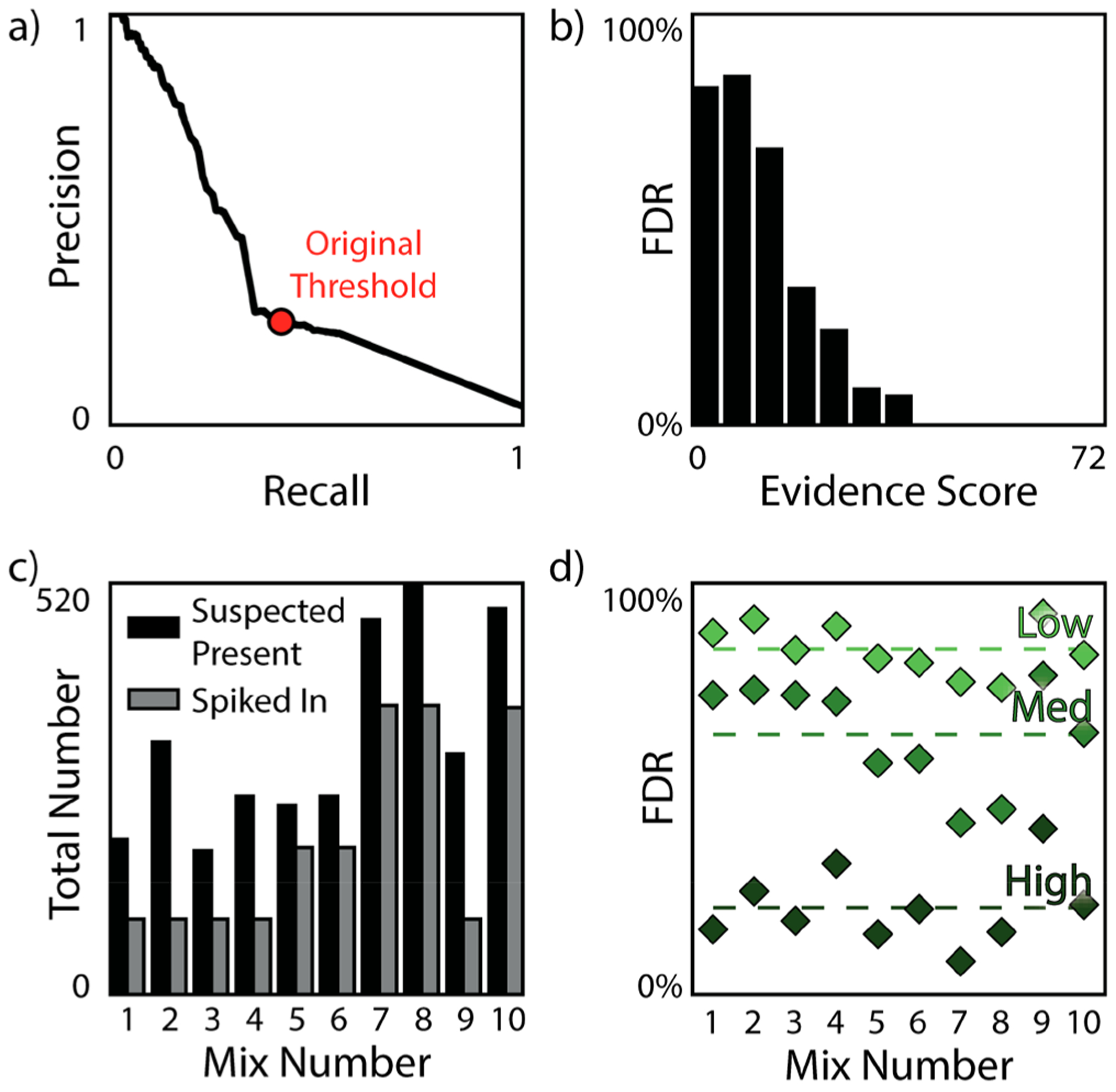
Blinded results using our multiattribute matching methods. (a) AUPR curve, with red dot showing our threshold (a total evidence score of 6.0). Please refer to the SI for details on the highest F1 score. (b) FDR as a function of evidence score. (c) Comparison between the number of molecules suspected present compared to the number of molecules spiked into each mixture. (d) FDR for each of the mixtures individually, split by evidence levels.
Overall FDR, FNR, and accuracy were 77%, 57%, and 91%, respectively. For molecules suspected to be present with high evidence scores (19.0 or more) provided by both mass and CCS, FDR was 35%. Additionally, FDR had a smooth inverse trend with increasing evidence score (Figure 3b). The capability to distinguish between compounds with the same mass (including isomers) was also demonstrated (Figure S10).
One issue, which caused a high FDR, was that 300–500 molecules were routinely suspected present in sample mixtures designed to contain 95–365 substances (Figure 3c). We hypothesized the high occurrence of false positives was attributable to one or more of the following factors: (i) noise present in raw data; (ii) low evidence score threshold; (iii) detection of molecules that were in the suspect library but unintentionally present in the samples due to reactions occurring in the highly concentrated mixtures; and/or (iv) multimer formation during the ionization process due to high sample concentrations. In the case of multimers, we hypothesized these formed during ionization, remained as multimers upon entry and flight through the IMS drift tube, and then dissociated to the constituent monomer prior to arriving at the MS detector.37 Support for this hypothesis was provided by much higher observed CCS values than expected in respect to their corresponding m/z values. Because the criteria for labeling a compound as suspected present required associated experimental features to be observed across all three technical replicates (in the case of IMS-MS features) and minimal presence (observed once at most) in blanks, it seems unlikely that low levels of equipment contamination were the cause of the high FDR. In a recently published initial report by the EPA on Phase 1 of the ENTACT study,41 it was revealed that nearly all participants that had reported their results to date also detected more compounds than were intentionally added (see Table 2 in Ulrich et al., 201941). Briefly, the other reporting laboratories predicted an average of 163 additional molecules to be present beyond what the EPA intentionally spiked in. This represents an average 116% increase over intentional spiked molecules. These are most likely not artifacts but represent real molecules that were not part of the intentionally spiked list. Ulrich et al. state that the additional compounds could originate from impurities, reaction or breakdown products, or laboratory contaminants added during the handling of the samples. Additionally, it was reported that some reference standards in mixture 10 had <90% purity.
As a clear example of a potential impurity or reaction product, Figure 4 shows tamoxifen and 4-hydroxytamoxifen (a hydroxylated form of tamoxifen) molecules both found in the suspect library and both receiving high evidence scores (45.5 and 25, respectively) in the same sample. However, only tamoxifen was classified as a true positive since it was intentionally added to the mixture, whereas 4-hydroxytamoxifen was not. It is possible that 4-hydroxytamoxifen may not be a genuine false positive and instead could have been formed in situ, due to reactions within the mixture, or been a trace contaminant from an impure standard.
Figure 4.
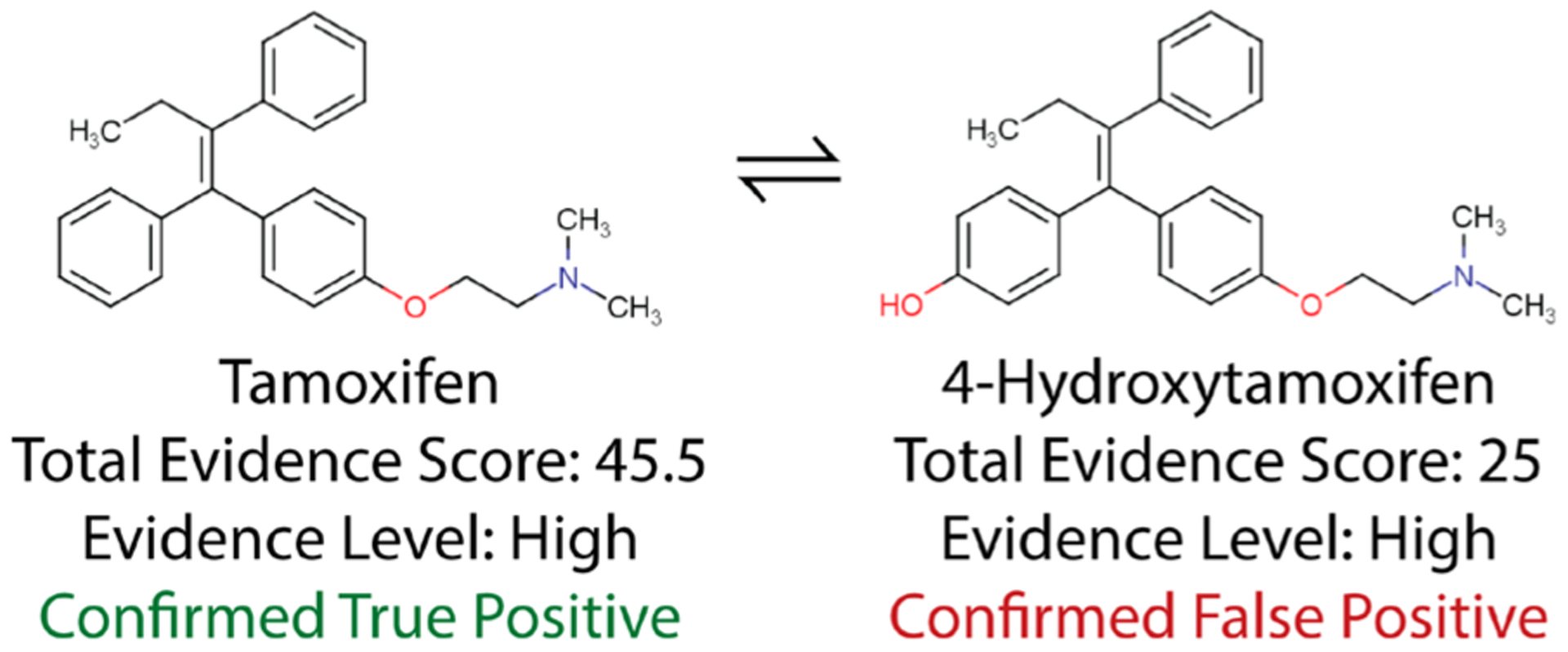
Tamoxifen and 4-hydroxytamoxifen. Both were suspected present with high evidence levels in the same mixture, but only tamoxifen was intentionally added to the mixture by the EPA.
Additionally, it is important to note the importance of choosing a threshold (i.e., minimum evidence score to warrant the label “suspected present”) that best reflects the desired balance of true positives to true negatives. For example, during a forensics study it may be desirable to decrease the number of false positives, and therefore, a higher threshold would be needed. This would decrease FDR but also increase FNR. For example, in our case, increasing the threshold from 6 to 19 leads to an FDR of 35%, FNR of 81%, and accuracy of 96%.
We optimized the threshold by finding the one that yielded the highest F1 score (a function of FNR and FDR, equation provided in SI 5.5). We found a threshold of 9.5 (and using the same set of weights as the blinded approach) decreased FDR by 14% (to 63%), increased FNR by 9% (to 66%), and increased accuracy by 4% (to 95%) (Figure S11).
On the basis of these initial results, we concluded the approach worked well but would likely be improved by optimizing the scoring parameters and threshold ranges for each evidence level. Beyond finding which false positives were present due to the reasons stated earlier, this was the most powerful way to improve the overall results and learn more about the algorithm before broader application.
Optimization of Multiattribute Matching Approach.
To determine the importance of each scoring parameter and to increase the accuracy of the approach (within the confines of these specific sample types), we set out to optimize the scoring method and subsequent evidence level cutoffs. The results of the Monte Carlo and particle swarm optimization methods are provided in the SI (SI 7, Figures S12–16). Optimization results were used to better understand the effect of each parameter and to update weights (Table S2), ultimately decreasing the combined FNR and FDR (Figure S14). Briefly, of the 11 adjustable scoring weights criteria, four were consistently awarded the highest weight by both optimization methods (in order from the highest): (i) multiple adducts being observed by a single instrument (index 7), (ii) high-intensity IMS-MS features (index 1), (iii) low-intensity IMS-MS features (index 2), and (iv) detection on both MS instruments (index 9) (Figure S13). These were deemed to be of very high importance for determining evidence. The lowest scoring criteria were (in order from lowest) (i) high-intensity FTICR-MS features (index 4), (ii) unique mass (index 10), and (iii) large mass (index 11). See SI 7 for the full discussion.
■ CONCLUSIONS
The capability to routinely measure and identify even a modest fraction of biologically, environmentally, or medically important chemicals within all of chemical space remains one of the grand challenges in science. The vast majority of molecules are not represented by standards. Furthermore, data for even fewer molecules have been added to reference libraries for use in identification (libraries currently cover much less than 1% of chemical space). This limit has remained a major constraint for decades in the global search for chemical biomarkers of disease, toxin exposure, and affliated efforts in the search for new drug candidates and attempts to sequence the complete metabolome. It is clear that relying on a single instrument and slow, costly establishment of reference libraries in the laboratory, restricted to standards available for purchase, is not a viable approach for identifying the tens to hundreds of thousands of small molecules in complex biological or environmental samples. Through advances in instrumentation, computation, and data integration, there has been a push for a shift in metabolomics and exposomics toward using multi-attribute matching with in silico libraries, in which the use of multiple molecular properties, accurately predicted computationally and consistently measured experimentally, are used for building evidence for the presence of molecules in complex samples with reduced reliance on standards.
This study was an initial demonstration of using in silico multifeature libraries for providing evidence for the presence of small molecules in blinded samples within in the context of an interlaboratory comparison. Our findings, both pre- and postoptimization, show the potential value in using multi-attribute-based methods with calculated chemical properties. Furthermore, evidence from CCS provided increased evidence for most true positives and was able to distinguish between isomers. Because these approaches are nascent and this study only represents a single set of synthetic samples, which covered only a small region of chemical space and only a single solvent matrix, there is still substantial work to be done to establish these methods as an accepted approach by the community for making actual molecular identifications with a specified level of confidence.
This work represents a small step toward a future in molecular identification that may have a reduced reliance on the use of authentic reference material for building reference libraries. The use of only m/z and calculated CCS, as demonstrated in this study, does not currently enable unambiguous molecular identification. What these libraries currently allow is downselection of candidate molecules from a list and evidence for their potential presence in samples. Thus, throughout the study we avoided labeling high evidence scores as actual identifications because, to date, there is currently (1) no community-wide accepted scheme for assigning levels of confidence (e.g., Schymanski48 or Metabolomics Standards Initiative levels49) for the matching of in silico data and (2) no established relationship between either claimed levels of confidence or evidence and true global false discovery rates in nonsynthetic samples. Therefore, for this study, discussion was limited to the evidence for the presence of molecules in samples and a subsequent reporting of how evidence scores based on multiple features related to the actual spiked-in compounds revealed after unblinding.
Importantly, the essential tool for molecular identification in complex samples is an accurate library of chemical properties for matching against experimental data, not the authentic reference material itself from which libraries may be built experimentally. Authentic standards have been the preferred approach for obtaining libraries because the error is limited to relatively small experimental errors. For computational tools that can produce chemical property libraries with known errors, those libraries can be of value for providing evidence for the presence of a molecule in a sample, similar to a library made from experimental analysis of authentic compounds with known experimental error. It will be the work of efforts such as the Metabolomics Standards Initiative and metabolomics societies to establish frameworks and criteria for assessing confidence when using in silico libraries. It is critical that researchers are transparent about the methods used to create libraries and their associated errors (through rigorous validation) and transparently include the scoring methods used to assess the evidence for the presence of a molecule when applying such libraries in their molecular identification pipelines.
To improve the results in the future we will need to add additional capabilities that can be predicted or calculated. The results indicated the value for future use of additional chemical attribute “dimensions”, such as MS/MS fragmentation patterns, chromatographic retention time, more accurate prediction of adduct formation (e.g., additional metal ion adducts not considered here), and infrared or Raman spectra. MAME was written so it can be modified in the future to handle different experimental data and library types. Through the generation of methods for quantification of false discovery rates, thorough validation, and agreement in the community on identification confidence criteria, it may eventually be possible for complete molecular identification, for even large library sizes, and potentially the complete molecular universe, through use of multiple accurately measured and calculated chemical properties. The value in increasing the accuracy of analytical and computational methods is important; however, adding orthogonal chemical properties for all researchers in the field to use will aid in building evidence for the presence of small molecules and will be essential for addressing major challenges within metabolomics. As additional chemical properties are added to this pipeline, the “distance” between the features of each library entry will become dramatically larger, thereby requiring a lower resolution for each property. The so-called “curse of dimensionality”50,51 can be used for our benefit to turn each library entry into a unique or nearly unique set of chemical properties. As metabolomics evolves and computational libraries are used more frequently, associated methods could eventually challenge the field’s current definition of and requirements for identification.
While methods have not yet been developed for measuring values such as accuracy and false discover rates in real (i.e., nonsynthetic) complex mixtures, the approach described here was developed using blinded results. In future studies we plan to again validate this approach using the optimized scoring parameters on other synthetic mixtures and real samples where molecules have already been confidently identified with standards. It remains to be determined how well this approach will work in natural samples, which typically have compounds spanning a large number of concentration ranges and chemical classes. In Phase 2 of the ENTACT study, our approach will be assessed against samples of standardized extracts (unaltered and fortified house dust, human serum, and silicone bands) with the main goal of determining the limitations and abilities of the method on natural samples and to determine how a matrix affects the ability to detect the presence of compounds. Our groups’ future work will include assessing how MAME and in silico libraries perform in multiple types of natural samples with validation through gold standard identification methods using authentic reference material. Consistent low FNR, FDR, and accuracy with the same scoring system will show the use and reliability of the method in building evidence for the presence of molecules in complex samples.
Supplementary Material
■ ACKNOWLEDGMENTS
This research was partially supported by the Genomic Science Program (GSP), Office of Biological and Environmental Research (OBER), the U.S. Department of Energy (DOE), and is a contribution of the Pacific Northwest National Laboratory (PNNL) Metabolic and Spatial Interactions in Communities (MOSAIC) Scientific Focus Area (SFA). The Multi-Attribute Matching Engine (MAME) was fully developed under MOSAIC funding. Portions of this research were also supported by the National Institutes of Health, National Institute of Environmental Health Sciences grant U2CES030170, the United States Environmental Protection Agency (Interagency Agreement DW-089–92452001-0 in support of DOE Project No. 68955A), the National Cancer Institute (grant R03CA222443), and a PNNL Laboratory Directed Research and Development program, the Microbiomes in Transition (MinT) Initiative. This work was performed in the W. R. Wiley Environmental Molecular Sciences Laboratory (EMSL), a DOE national scientific user facility at the PNNL. The NWChem calculations were performed using the Cascade supercomputer at the EMSL. PNNL is operated by Battelle for the DOE under contract DE-AC05–76RL0 1830.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at 10.1021/acs.jcim.9b00444.
Further detailed methods and additional figures (PDF) Suspect library, property predictions, and results broken down for each mixture in this challenge (XLSX)
Notes
The authors declare no competing financial interest.
■ REFERENCES
- (1).Djoumbou Feunang Y; Eisner R; Knox C; Chepelev L; Hastings J; Owen G; Fahy E; Steinbeck C; Subramanian S; Bolton E; Greiner R; Wishart DS ClassyFire: automated chemical classification with a comprehensive, computable taxonomy. J. Cheminform 2016, 8, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Vinayavekhin N; Homan EA; Saghatelian A Exploring Disease through Metabolomics. ACS Chem. Biol 2010, 5, 91–103. [DOI] [PubMed] [Google Scholar]
- (3).Gebregiworgis T; Powers R Application of NMR Metabolomics to Search for Human Disease Biomarkers. Combinatorial Chem. High Throughput Screening 2012, 15, 595–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Use of Metabolomics to Advance Research on Environmental Exposures and the Human Exposome: Workshop in Brief; The National Academies Press: Washington, DC, 2016; p 12. [PubMed] [Google Scholar]
- (5).Lu K; Abo RP; Schlieper KA; Graffam ME; Levine S; Wishnok JS; Swenberg JA; Tannenbaum SR; Fox JG Arsenic Exposure Perturbs the Gut Microbiome and Its Metabolic Profile in Mice: An Integrated Metagenomics and Metabolomics Analysis. Environ. Health Perspect 2014, 122, 284–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Glauser G; Boccard J; Rudaz S; Wolfender JL Mass spectrometry-based metabolomics oriented by correlation analysis for wound-induced molecule discovery: identification of a novel jasmonate glucoside. Phytochem. Anal 2010, 21, 95–101. [DOI] [PubMed] [Google Scholar]
- (7).Wu C; Zacchetti B; Ram AFJ; van Wezel GP; Claessen D; Hae Choi Y Expanding the chemical space for natural products by Aspergillus-Streptomyces co-cultivation and biotransformation. Sci. Rep 2015, 5, 10868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Pirhaji L; Milani P; Leidl M; Curran T; Avila-Pacheco J; Clish CB; White FM; Saghatelian A; Fraenkel E Revealing disease-associated pathways by network integration of untargeted metabolomics. Nat. Methods 2016, 13, 770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Griffin JL; Wang X; Stanley E Does Our Gut Microbiome Predict Cardiovascular Risk? Circ.: Cardiovasc. Genet. 2015, 8, 187–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Sampaio BL; Edrada-Ebel R; Da Costa FB Effect of the environment on the secondary metabolic profile of Tithonia diversifolia: a model for environmental metabolomics of plants. Sci. Rep 2016, 6, 29265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Goldberg RB; Kendall DM; Deeg MA; Buse JB; Zagar AJ; Pinaire JA; Tan MH; Khan MA; Perez AT; Jacober SJ A Comparison of Lipid and Glycemic Effects of Pioglitazone and Rosiglitazone in Patients With Type 2 Diabetes and Dyslipidemia. Diabetes Care 2005, 28, 1547–1554. [DOI] [PubMed] [Google Scholar]
- (12).Pearson H Meet the human metabolome. Nature 2007, 446, 8. [DOI] [PubMed] [Google Scholar]
- (13).Dettmer K; Aronov PA; Hammock BD Mass spectrometry-based metabolomics. Mass Spectrom. Rev 2007, 26, 51–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Worley B; Powers R Multivariate Analysis in Metabolomics. Curr. Metabolomics 2012, 1, 92–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Bohan DA; Vacher C; Tamaddoni-Nezhad A; Raybould A; Dumbrell AJ; Woodward G Next-Generation Global Biomonitoring: Large-scale, Automated Reconstruction of Ecological Networks. Trends Ecol. Evol 2017, 32, 477–487. [DOI] [PubMed] [Google Scholar]
- (16).Dennis KK; Marder E; Balshaw DM; Cui Y; Lynes MA; Patti GJ; Rappaport SM; Shaughnessy DT; Vrijheid M; Barr DB Biomonitoring in the Era of the Exposome. Environ. Health Perspect 2017, 125, 502–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Patti GJ; Yanes O; Siuzdak G Metabolomics: the apogee of the omic triology. Nat. Rev. Mol. Cell Biol 2012, 13, 263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Onghena M; Van Hoeck E; Van Loco J; Ibanez M; Cherta L; Portoles T; Pitarch E; Hernandez F; Lemiere F; Covaci A Identification of substances migrating from plastic baby bottles using a combination of low-resolution and high-resolution mass spectrometric analysers coupled to gas and liquid chromatography. J. Mass Spectrom 2015, 50, 1234–1244. [DOI] [PubMed] [Google Scholar]
- (19).Guo J; Yun BH; Upadhyaya P; Yao L; Krishnamachari S; Rosenquist TA; Grollman AP; Turesky RJ Multi-Class Carcinogenic DNA Adduct Quantification in Formalin-Fixed Paraffin-Embedded Tissues by Ultra-Performance Liquid Chromatography-Tandem Mass Spectrometry. Anal. Chem 2016, 88, 4780–4787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Metz TO; Baker ES; Schymanski EL; Renslow RS; Thomas DG; Causon TJ; Webb IK; Hann S; Smith RD; Teeguarden JG Integrating ion mobility spectrometry into mass spectrometry-based exposome measurements: what can it add and how far can it go? Bioanalysis 2017, 9, 81–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Patel CJ; Bhattacharya J; Butte AJ An Environment-Wide Association Study (EWAS) on type 2 diabetes mellitus. PLoS One 2010, 5, No. e10746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Eke PI; Dye BA; Wei L; Slade GD; Thornton-Evans GO; Borgnakke WS; Taylor GW; Page RC; Beck JD; Genco RJ Update on Prevalence of Periodontitis in Adults in the United States: NHANES 2009 to 2012. J. Periodontol 2015, 86, 611–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Cathey A; Ferguson KK; McElrath TF; Cantonwine DE; Pace G; Alshawabkeh A; Cordero JF; Meeker JD Distribution and predictors of urinary polycyclic aromatic hydrocarbon metabolites in two pregnancy cohort studies. Environ. Pollut 2018, 232, 556–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Bloszies CS; Fiehn O Using untargeted metabolomics for detecting exposome compounds. Curr. Opin. Toxicol 2018, 8, 87–92. [Google Scholar]
- (25).Wishart DS; Tzur D; Knox C; Eisner R; Guo AC; Young N; Cheng D; Jewell K; Arndt D; Sawhney S; Fung C; Nikolai L; Lewis M; Coutouly MA; Forsythe I; Tang P; Shrivastava S; Jeroncic K; Stothard P; Amegbey G; Block D; Hau DD; Wagner J; Miniaci J; Clements M; Gebremedhin M; Guo N; Zhang Y; Duggan GE; Macinnis GD; Weljie AM; Dowlatabadi R; Bamforth F; Clive D; Greiner R; Li L; Marrie T; Sykes BD; Vogel HJ; Querengesser L HMDB: the Human Metabolome Database. Nucleic Acids Res. 2007, 35, D521–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).da Silva RR; Dorrestein PC; Quinn RA Illuminating the dark matter in metabolomics. Proc. Natl. Acad. Sci. U. S. A 2015, 112, 12549–12550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Kurita KL; Glassey E; Linington RG Integration of high-content screening and untargeted metabolomics for comprehensive functional annotation of natural product libraries. Proc. Natl. Acad. Sci. U. S. A 2015, 112, 11999–12004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Barupal DK; Fan S; Fiehn O Integrating bioinformatics approaches for a comprehensive interpretation of metabolomics datasets. Curr. Opin. Biotechnol 2018, 54, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Newton SR; McMahen RL; Sobus JR; Mansouri K; Williams AJ; McEachran AD; Strynar MJ Suspect screening and non-targeted analysis of drinking water using point-of-use filters. Environ. Pollut 2018, 234, 297–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Ibrahim YM; Baker ES; Danielson WF III; Norheim RV; Prior DC; Anderson GA; Belov ME; Smith RD Development of a new ion mobility time-of-flight mass spectrometer. Int. J. Mass spectrom 2015, 377, 655–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Tfaily MM; Chu RK; Toyoda J; Tolić N; Robinson EW; Paša-Tolić L; Hess NJ Sequential extraction protocol for organic matter from soils and sediments using high resolution mass spectrometry. Anal. Chim. Acta 2017, 972, 54–61. [DOI] [PubMed] [Google Scholar]
- (32).Tfaily MM; Chu RK; Tolić N; Roscioli KM; Anderton CR; Paša-Tolić L; Robinson EW; Hess NJ Advanced Solvent Based Methods for Molecular Characterization of Soil Organic Matter by High-Resolution Mass Spectrometry. Anal. Chem 2015, 87, 5206–5215. [DOI] [PubMed] [Google Scholar]
- (33).Graham TR; Renslow R; Govind N; Saunders SR Precursor Ion-Ion Aggregation in the Brust-Schiffrin Synthesis of Alkanethiol Nanoparticles. J. Phys. Chem. C 2016, 120, 19837–19847. [Google Scholar]
- (34).Tolic N; Liu Y; Liyu A; Shen Y; Tfaily MM; Kujawinski EB; Longnecker K; Kuo LJ; Robinson EW; Pasa-Tolic L; Hess NJ Formularity: Software for Automated Formula Assignment of Natural and Other Organic Matter from Ultrahigh-Resolution Mass Spectra. Anal. Chem 2017, 89, 12659–12665. [DOI] [PubMed] [Google Scholar]
- (35).Zheng X; Renslow RS; Makola MM; Webb IK; Deng L; Thomas DG; Govind N; Ibrahim YM; Kabanda MM; Dubery IA; Heyman HM; Smith RD; Madala NE; Baker ES Structural Elucidation of cis/trans Dicaffeoylquinic Acid Photo-isomerization Using Ion Mobility Spectrometry-Mass Spectrometry. J. Phys. Chem. Lett 2017, 8, 1381–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Valiev M; Bylaska EJ; Govind N; Kowalski K; Straatsma TP; Van Dam HJJ; Wang D; Nieplocha J; Apra E; Windus TL; de Jong WA NWChem: A comprehensive and scalable open-source solution for large scale molecular simulations. Comput. Phys. Commun 2010, 181, 1477–1489. [Google Scholar]
- (37).Ma J; Casey CP; Zheng X; Ibrahim YM; Wilkins CS; Renslow RS; Thomas DG; Payne SH; Monroe ME; Smith RD; Teeguarden JG; Baker ES; Metz TO PIXiE: an algorithm for automated ion mobility arrival time extraction and collision cross section calculation using global data association. Bioinformatics 2017, 33, 2715–2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Zhang X; Romm M; Zheng X; Zink EM; Kim YM; Burnum-Johnson KE; Orton DJ; Apffel A; Ibrahim YM; Monroe ME; Moore RJ; Smith JN; Ma J; Renslow RS; Thomas DG; Blackwell AE; Swinford G; Sausen J; Kurulugama RT; Eno N; Darland E; Stafford G; Fjeldsted J; Metz TO; Teeguarden JG; Smith RD; Baker ES SPE-IMS-MS: An automated platform for sub-sixty second surveillance of endogenous metabolites and xenobiotics in biofluids. Clin. Mass. Spectrom 2016, 2, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Zheng X; Zhang X; Schocker NS; Renslow RS; Orton DJ; Khamsi J; Ashmus RA; Almeida IC; Tang K; Costello CE; Smith RD; Michael K; Baker ES Enhancing glycan isomer separations with metal ions and positive and negative polarity ion mobility spectrometry-mass spectrometry analyses. Anal. Bioanal. Chem 2017, 409, 467–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Sobus JR; Grossman JN; Chao A; Singh R; Williams AJ; Grulke CM; Richard AM; Newton SR; McEachran AD; Ulrich EM Using prepared mixtures of ToxCast chemicals to evaluate non-targeted analysis (NTA) method performance. Anal. Bioanal. Chem 2019, 411, 835–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Ulrich EM; Sobus JR; Grulke CM; Richard AM; Newton SR; Strynar MJ; Mansouri K; Williams AJ EPA’s non-targeted analysis collaborative trial (ENTACT): genesis, design, and initial findings. Anal. Bioanal. Chem 2019, 411, 853–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Sobus JR; Wambaugh JF; Isaacs KK; Williams AJ; McEachran AD; Richard AM; Grulke CM; Ulrich EM; Rager JE; Strynar MJ; Newton SR Integrating tools for non-targeted analysis research and chemical safety evaluations at the US EPA. J. Exposure Sci. Environ. Epidemiol 2018, 28, 411–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Richard AM; Judson RS; Houck KA; Grulke CM; Volarath P; Thillainadarajah I; Yang C; Rathman J; Martin MT; Wambaugh JF; Knudsen TB; Kancherla J; Mansouri K; Patlewicz G; Williams AJ; Little SB; Crofton KM; Thomas RS ToxCast Chemical Landscape: Paving the Road to 21st Century Toxicology. Chem. Res. Toxicol 2016, 29, 1225–1251. [DOI] [PubMed] [Google Scholar]
- (44).May JC; Goodwin CR; Lareau NM; Leaptrot KL; Morris CB; Kurulugama RT; Mordehai A; Klein C; Barry W; Darland E; Overney G; Imatani K; Stafford GC; Fjeldsted JC; McLean JA Conformational Ordering of Biomolecules in the Gas Phase: Nitrogen Collision Cross Sections Measured on a Prototype High Resolution Drift Tube Ion Mobility-Mass Spectrometer. Anal. Chem 2014, 86, 2107–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Shaw JB; Lin T-Y; Leach FE; Tolmachev AV; Tolić N; Robinson EW; Koppenaal DW; Paša-Tolić L 21 T Fourier Transform Ion Cyclotron Resonance Mass Spectrometer Greatly Expands Mass Spectrometry Toolbox. J. Am. Soc. Mass Spectrom 2016, 27, 1929–1936. [DOI] [PubMed] [Google Scholar]
- (46).Ipsen A Efficient Calculation of Exact Fine Structure Isotope Patterns via the Multidimensional Fourier Transform. Anal. Chem 2014, 86, 5316–5322. [DOI] [PubMed] [Google Scholar]
- (47).Jones DP Sequencing the exposome: A call to action. Toxicol Rep. 2016, 3, 29–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Schymanski EL; Jeon J; Gulde R; Fenner K; Ruff M; Singer HP; Hollender J Identifying Small Molecules via High Resolution Mass Spectrometry: Communicating Confidence. Environ. Sci. Technol 2014, 48, 2097–2098. [DOI] [PubMed] [Google Scholar]
- (49).Sumner LW; Amberg A; Barrett D; Beale MH; Beger R; Daykin CA; Fan TWM; Fiehn O; Goodacre R; Griffin JL; Hankemeier T; Hardy N; Harnly J; Higashi R; Kopka J; Lane AN; Lindon JC; Marriott P; Nicholls AW; Reily MD; Thaden JJ; Viant MR Proposed minimum reporting standards for chemical analysis Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics 2007, 3, 211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Bellman R; Bellman RE Adaptive Control Processes: A Guided Tour; Princeton University Press, 1961. [Google Scholar]
- (51).Donoho D High-Dimensional Data Analysis: The Curses and Blessings of Dimensionality; 2000. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.