

Abstract
Inactivating mutations of axis inhibition protein 1 (AXIN1), a negative regulator of the Wnt/β-Catenin cascade, are among the common genetic events in human hepatocellular carcinoma (HCC), affecting approximately 10% of cases. In the present manuscript, we sought to define the genetic crosstalk between Axin1 mutants and Wnt/β-catenin as well as Notch signaling cascades along hepatocarcinogenesis. We discovered that c-MET activation and AXIN1 mutations occur concomitantly in ~3%–5% of human HCC samples. Subsequently, we generated a murine HCC model by means of CRISPR/Cas9-based gene deletion of Axin1 (sgAxin1) in combination with transposon-based expression of c-Met in the ouse liver (c-Met/sgAxin1). Global gene expression analysis of mouse normal liver, HCCs induced by c-Met/sgAxin1, and HCCs induced by c-Met/ΔN90-β-Catenin revealed activation of the Wnt/β-Catenin and Notch signaling in c-Met/sgAxin1 HCCs. However, only a few of the canonical Wnt/β-Catenin target genes were induced in c-Met/sgAxin1 HCC when compared with corresponding lesions from c-Met/ΔN90-β-Catenin mice. To study whether endogenous β-Catenin is required for c-Met/sgAxin1-driven HCC development, we expressed c-Met/sgAxin1 in liver-specific Ctnnb1 null mice, which completely prevented HCC development. Consistently, in AXIN1 mutant or null human HCC cell lines, silencing of β-Catenin strongly inhibited cell prolif-eration. In striking contrast, blocking the Notch cascade through expression of either the dominant negative form of the recombinant signal-binding protein for immunoglobulin kappa J region (RBP-J) or the ablation of Notch2 did not significantly affect c-Met/sgAxin1-driven hepatocarcinogenesis. Conclusion: We demonstrated here that loss of Axin1 cooperates with c-Met to induce HCC in mice, in a β-Catenin signaling–dependent but Notch cascade–independent way.
Hepatocellular carcinoma (HCC) is the second leading cause of cancer-related deaths in the world.(1) Because of the lack of specific symptoms, most HCCs are diagnosed at late stage. For these patients, locoregional palliative therapies or multikinase inhibitors like sorafenib and regorafenib provide only limited survival benefits.(2) Therefore, there is an urgent need to elucidate the molecular mechanisms underlying HCC pathogenesis for more effective therapeutic options for patients with advanced HCC.
Recently, the comprehensive and integrated analysis of the human HCC genome provided the landscape of genetic alterations occurring along hepatocarcinogenesis.(3) Activation of the Wnt/β-Catenin signaling cascade is one of the most frequent molecular events in HCC. Wnt/β-Catenin pathway is a prominent signaling cascade regulating cell proliferation, differentiation, and tumor development in many organs.(4) In the liver, it has also been implicated in homeostasis, regeneration, and HCC development.(5) In the absence of Wnt ligands, i.e., in the Wnt-off state, β-Catenin protein remains at low level in the cytoplasm because of phosphorylation by a cytoplasmic destruction complex. This destruction complex consists of adenomatous polyposis coli, glycogen synthase kinase-3 beta (GSK3β), casein kinase-1, and the axis inhibition (AXIN) scaffold proteins (AXIN1 and AXIN2). When Wnt ligands combine with their receptor Frizzled and coreceptors LRP5/6, i.e., in the Wnt-on state, disheveled is recruited to the cell membrane, leading to the dissociation of AXIN and GSK3β from the cytoplasmic destruction complex, effectively preventing β-Catenin degradation. Subsequently, β-Catenin accumulates and translocates into the nucleus, where it activates the expression of Wnt target genes through T cell factor (TCF)-mediated transcriptional regulation.(4) AXIN1 is therefore an important negative regulator of Wnt/β-Catenin cascade and functions as a bona fide tumor suppressor gene.(6) Importantly, mutations of AXIN1 have been identified in multiple cancer types. Most of the mutations are truncating mutations or nonsense mutations that prevent the binding of AXIN1 to β-Catenin, resulting in the disruption of the destruction complex and, thus, the activation of the Wnt/β-Catenin pathway.
In human HCCs, AXIN1 mutations occur in approximately 8% of cases.(3,7,8). Based on the COSMIC data-base,(9) AXIN1 mutations were identified in 128 HCC of 1,626 cases examined, thus representing one of the top five mutated genes in human HCC. Intriguingly, despite the fact that AXIN has been implicated as a key negative regulator of the Wnt/β-Catenin pathway, whether AXIN1 mutations lead to activation of Wnt/β-Catenin remains controversial. For instance, a study showed that in human HCCs, catenin beta 1 (CTNNB1) mutation status correlates with the up-regulation of Wnt/β-Catenin pathway genes, but AXIN1 mutation status does not.(7) In addition, it has been found that loss of Axin1 in mouse hepatocytes only leads to the activation of a subset of Wnt target genes.(10) In line with this hypothesis, based on global gene expression patterns, Aritbol et al. recently showed that most AXIN1-mutated human HCCs exhibit no activation of β-Catenin. Gene expression profile analysis suggests rather the enrichment of a Notch signature in AXIN1 mutant HCC samples.(11)
Here, we show that HCC development induced by Axin1 loss requires an intact Wnt/β-Catenin signaling cascade.
Materials and Methods
CONSTRUCTS AND REAGENTS
The plasmids used for mouse injection, including pT3-EF1α, pT3-EF1α-c-Met (human c-Met or hMet), pT3-EF1α-β-CateninS45Y, pT3-EF1α-ΔN90-β-Catenin, pT3-EF1α-dnRBPJ (with N-terminal V5 tag), phosphorylated cytomegalovirus (pCMV), pCMV-Cre, and pCMV/sleeping beauty transposase, have been described in our publications.(12–14) pX330-U6-Chimeric_BB-CBh-hSpCas9 (pX330) and pLentiCRISPRv2_Puro plasmids were obtained from Addgene (#42230 and #98290, respectively). To delete Axin1 in the mouse liver, we constructed pX330 plasmids expressing single-guide RNAs (sgRNAs) against mouse Axin1 (NM_001159598.1). To delete AXIN1 in human or mouse liver tumor cells, we cloned sgRNAs against human AXIN1 (NM_003502.3) or mouse Axin1 into pLentiCRISPRv2_Puro vectors. Additional plasmids, including shβ-Catenin/pLKO.1 (#18803), Super 8x TopFlash (#12456), and Super 8x FopFlash (plasmids #12457), were obtained from Addgene. pRL-CMV Renilla luciferase plasmid was purchased from Promega (Madison, WI). Detailed information about each plasmid is included in Supporting Table S1. All plasmids for in vivo studies were purified using the Endotoxin free Maxi prep kit (Sigma-Aldrich, St. Louis, MO).
MICE AND HYDRODYNAMIC TAIL INJECTION
Wild-type FVB/N, C57BL/6, Ctnnb1fl/fl, and Notch2fl/fl mice were obtained from the Jackson Laboratory (Sacramento, CA). Hydrodynamic injection into 6- to 8-week-old mice was performed as described.(12,13,15) Both male and female mice were used. Detailed information about plasmids used in each experiment is available in Supporting Table S2. Mice were housed, fed, and monitored in accord with protocols approved by the Committee for Animal Research at the University of California, San Francisco (San Francisco, CA).
STATISTICAL ANALYSIS
The Prism 7.0 software (GraphPad, San Diego, CA) was used to analyze the data. Statistical analysis was performed using Student t test and Tukey-Kramer test. P < 0.05 was considered to be statistically significant.
Additional information is available in Supporting Materials and Methods.
Results
c-Met ACTIVATION AND AXIN MUTATIONS OCCUR CONCOMITANTLY IN A HUMAN HCC SUBSET
Because we demonstrated that activated β-Catenin is not oncogenic per se in the mouse liver whereas it synergizes with human c-MET to promote HCC development,(13) we reasoned that AXIN1 mutation alone is insufficient to trigger hepatocarcinogenesis in vivo but may also cooperate with c-MET to induce liver malignant transformation. To assess the validity of this model for the human disease, we investigated whether concomitant AXIN1 mutations and c-MET activation occur in human HCC. First, we analyzed the levels of phosphorylated/activated (p-) MET and c-MET in a panel of human HCC cell lines (Supporting Fig. S1). Three of five cell lines with AXIN1 null expression or mutation showed c-MET activation, as indicated by p-MET expression. c-MET activation levels were similar to those detected in MHCC97-H cells, an HCC cell line with wild-type AXIN1 and c-MET amplification.(16) Next, we retrieved HCC The Cancer Genome Atlas (TCGA) data with AXIN1 and CTNNB1 mutation status. The KAPOSI_LIVER_CANCER_MET_UP gene set was used as c-MET activation signature.(17) Using the FRY test, we found that c-MET_UP signature characterized human AXIN1 mutant HCC (Supporting Fig. S2A,B). Indeed, 11/18 c-MET_UP genes were up-regulated in AXIN1 mutant HCC when compared with surrounding livers (Supporting Fig. S2C). Further analysis demonstrated that ~42% AXIN1 mutant HCC samples in the TCGA data set showed c-MET_UP gene signature (Supporting Fig. S3). Finally, we analyzed c-MET expression using immunohistochemistry in a panel of human HCC samples with known AXIN1 and CTNNB1 mutation status (n = 103; Supporting Fig. S4). Mutations occurred in 10.7% and 19.4% HCC specimens for AXIN1 and CTNNB1 genes, respectively. Noticeably, only one HCC harbored AXIN1 and CTNNB1 mutations, suggesting that the two events are most often mutually exclusive in HCC. Furthermore, we found that 9/11 HCC displaying AXIN1 mutations belonged to the group of HCC with poorer prognosis, whereas CTNNB1 mutations were found in 11/20 HCC with better prognosis (Supporting Table S2), suggesting a stronger role of AXIN1 than CTNNB1 mutations in determining the prognosis of human HCC. No association between AXIN1 or CTNNB1 mutations with other clinicopathological features of the patients, including age, sex, etiology, presence of cirrhosis, alpha-fetoprotein levels, tumor size, or tumor grade, was detected (data not shown).
High c-MET expression was detected in 7/11 HCCs with AXIN1 mutations and 10/20 HCC with CTNNB1 mutations (Supporting Fig. S5A,B). Furthermore, when analyzing the expression of CKS2, a gene in the c-MET_UP gene list, 7/11 of AXIN1 mutant HCC demonstrated high levels of this gene (Supporting Fig. S5C,D).
Altogether, our investigation indicates that ~40%–60% of AXIN1 mutant HCCs exhibit c-MET high expression or activation. Considering that AXIN1 mutant rate is ~8% in all human HCC samples, we estimate that concomitant c-MET activation and AXIN1 mutant represents ~3%−5% of human HCCs.
LOSS OF AXIN1 SYNERGIZES WITH c-Met TO PROMOTE HCC DEVELOPMENT IN MICE
We applied CRISPR-Cas9-mediated gene editing to delete Axin1 in mouse hepatocytes. We designed three independent guide RNA sequence against mouse Axin1 (sgAxin1.1, sgAxin1.2, and sgAxin1.3) (Supporting Fig. S6A). All three sgAxin1 constructs were able to inhibit Axin1 protein expression in two HCC cell lines (Supporting Fig. S6B). Each of the three sgAxin1 sequences was then subcloned into pX330 vector for in vivo gene deletion.
To test our hypothesis that loss of function AXIN1 mutant cooperates with c-Met to induce hepatocarcinogenesis, we injected each of sgAxin1 (in pX330 plasmid) alone or together with c-Met. Additional mice were injected with c-Met alone as control.(18) Hydrodynamic injection of sgAxin1.1, sgAxin1.2, or sgAxin1.3 alone did not lead to liver tumor formation when mice were harvested at 20 weeks after injection (Supporting Fig. S6C), similar to that described for activated β-Catenin alone.(13) Histopathological analysis revealed that liver tissues were completely normal, indistinguishable from uninjected normal mouse liver (Supporting Fig. S6C). Consistent with our report,(18) overexpression of c-Met in the liver also did not induce liver tumor (Supporting Fig. S7). In striking contrast, coexpression of sgAxin1 with c-Met synergized to promote liver tumor formation in mice (Fig. 1A). All three sgAxin1 constructs behaved similarly, with all c-Met/sgAxin1-injected mice developing high tumor burden and requiring euthanasia by 9 to 12 weeks after injection (Fig. 1A–C). Grossly, numerous lesions were observed throughout the mouse liver. Microscopically, liver lesions were indistinguishable depending on the sgAxin1 constructs injected (Fig. 1D). Nine weeks after injection, the liver parenchyma was nearly completely occupied by large hepatocellular tumors up to 10 mm in diameter, most of them consisting of medium-sized hepatocytes with mild to moderate nuclear atypia (Supporting Fig. S8). However, some of these tumors showed severe nuclear atypia, strong mitotic activity, and microscopic foci of necrosis and basophilic cytoplasm as signs of malignancy (Supporting Fig. S8). An intriguing finding in the largest tumors was the development of intratumoral pseudocysts, measuring up to 3 mm in diameter, lined by tumor cells and filled with fibrin and congested blood. Mice injected with c-Met/β-catenin(13) showed similar tumor development 9 weeks after injection, with c-Met/β-catenin tumors being morphologically indistinguishable from c-Met/sgAxin1 counterparts, including the peculiar fibrin pseudocysts (Supporting Fig. S8).
FIG. 1.
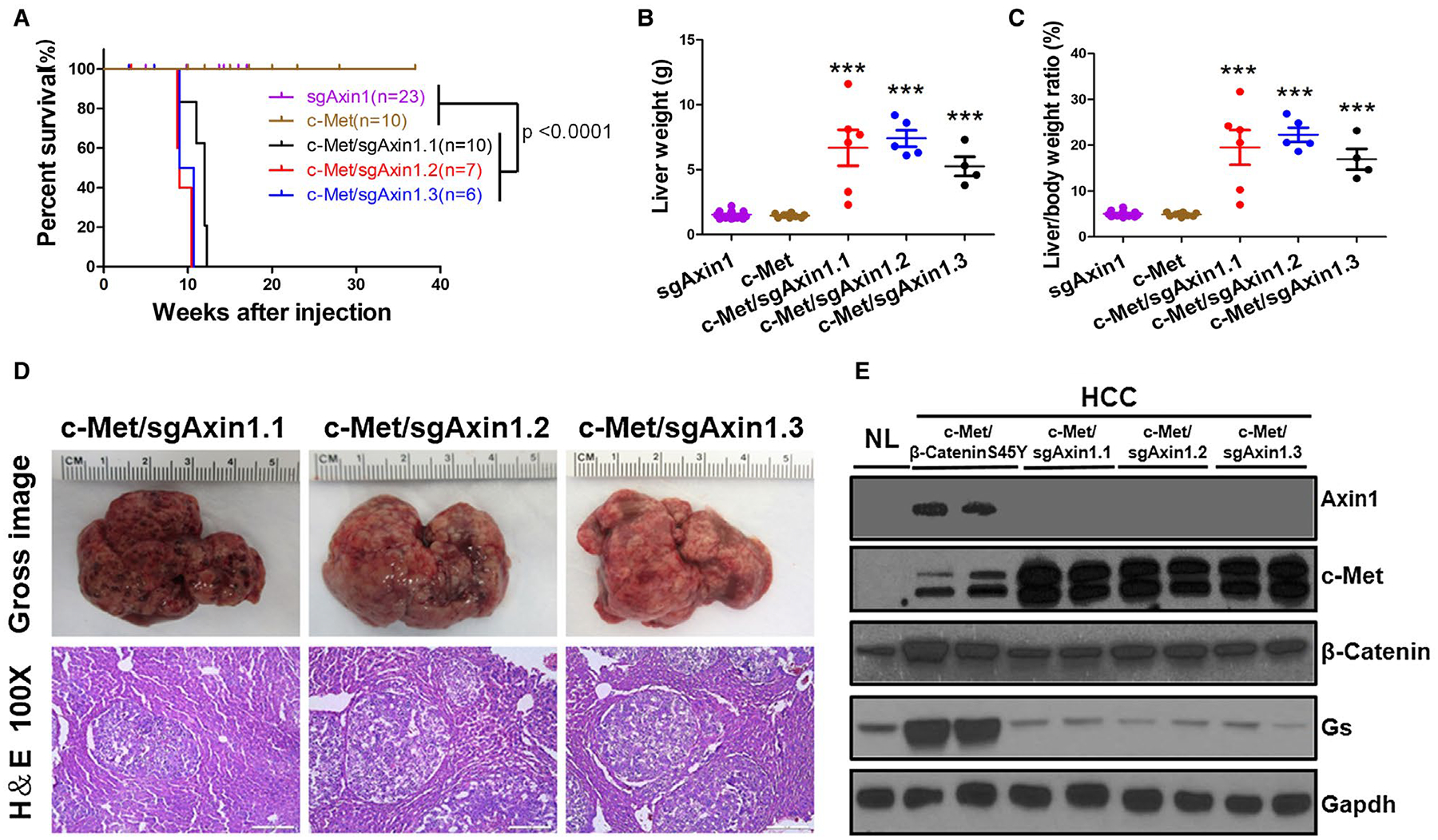
Loss of Axin1 synergizes with c-Met to promote HCC development in mice. (A) Kaplan-Meier curve comparing survival of FVB/N mice injected with sgAxin1.1 (n = 10), sgAxin1.2 (n = 7), sgAxin1.3 (n = 6), c-Met only (n = 10), and combination of sgAxin1.1 (n = 10) or sgAxin1.2 (n = 7) or sgAxin1.3 (n = 6) with pT3-EF1α-c-Met. Mice receiving combined injection exhibited significantly reduced survival compared with sgAxin1 alone and c-Met alone groups. (B,C) Liver weight and liver body weight ratio of sgAxin1 only, c-Met only, and combined injection mice. Data are presented as mean ± SD. ***, P < 0.001. (D) Gross and H&E images of c-Met/sgAxin1.1, c-Met/sgAxin1.2, and c-Met/sgAxin1.3 HCCs. Magnifications: ×100, scale bar: 200 μm. (E) Western blotting of NL, c-Met/β-CateninS45Y, c-Met/sgAxin1.1, c-Met/sgAxin1.2, and c-Met/sgAxin1.3 liver tissues. Gapdh was used as a loading control. Abbreviations: Gapdh, glyceraldehyde 3-phosphate dehydrogenase; H&E, hematoxylin and eosin; NL, normal liver.
At the molecular level, we confirmed the loss of Axin1 and overexpression of human c-Met in c-Met/sgAxin1 induced HCC (Fig. 1E). All tumor cells stained positive for membranous E-Cadherin (Supporting Fig. S9A). Signaling molecules down-stream of c-Met, including phospho-protein kinase B (p-AKT) and phospho-extracellular-signal-regulated kinase (p-ERK), were found to be up-regulated in c-Met/sgAxin1-induced HCC (Supporting Fig. S9B). To further validate sgAxin1-mediated genomic editing, we isolated tumor nodules and performed genomic sequencing on mouse Axin1 alleles. The sequencing results confirmed the mutation of Axin1 on its genomic locus (Supporting Fig. S10). Because all three sgAxin1 constructs demonstrated similar efficacy, we chose to use sgAxin1.2 for the subsequent functional studies.
LACK OF STRONG CANONICAL Wnt/β-Catenin ACTIVATION IN c-Met/sgAxin1 MOUSE HCC
As Axin1 is implicated in Wnt/β-Catenin signaling regulation, we investigated whether the canonical Wnt pathway is activated in c-Met/sgAxin1 HCC. First, we analyzed β-Catenin staining patterns in the liver tumor tissues. Whereas in normal liver β-Catenin immunoreactivity is limited to the hepatocyte membrane, c-Met/β-CateninS45Y and c-Met/ΔN90-β-Catenin mouse HCC lesions exhibited cytoplasmic and/or nuclear β-Catenin staining (Fig. 2A). In contrast, in c-Met/sgAxin1 HCC tissues, only membranous β-Catenin staining was appreciable (Fig. 2A). Furthermore, strong glutamine synthetase (GS) staining characterized activated β-Catenin HCC cells, but not c-Met/sgAxin1 corresponding lesions (Fig. 2A). The lack of up-regulation of GS in c-Met/sgAxin1 was confirmed by western blotting (Figs. 1D, 2B). Because phosphorylation of β-Catenin at various residues is indicative of specific signaling, we next analyzed its phosphorylation status in c-Met/sgAxin1 HCC and compared it with both normal liver tissues and c-Met/β-CateninS45Y HCC tissues. Whereas total β-catenin levels were prominently increased in the c-Met/β-CateninS45Y HCC, modest increase as compared with the normal liver was evident in the c-Met/sgAxin1 HCC (Fig. 2B). No changes occurred in p-β-CateninS552 or p-β-CateninS675, markers of protein kinase A-mediated β-catenin activation (Fig. 2B). p-β-CateninS45 was absent in c-Met/β-CateninS45Y samples, but all other sites relevant to the phosphodegron of β-catenin remained altered between the three groups (Fig. 2B).
FIG. 2.
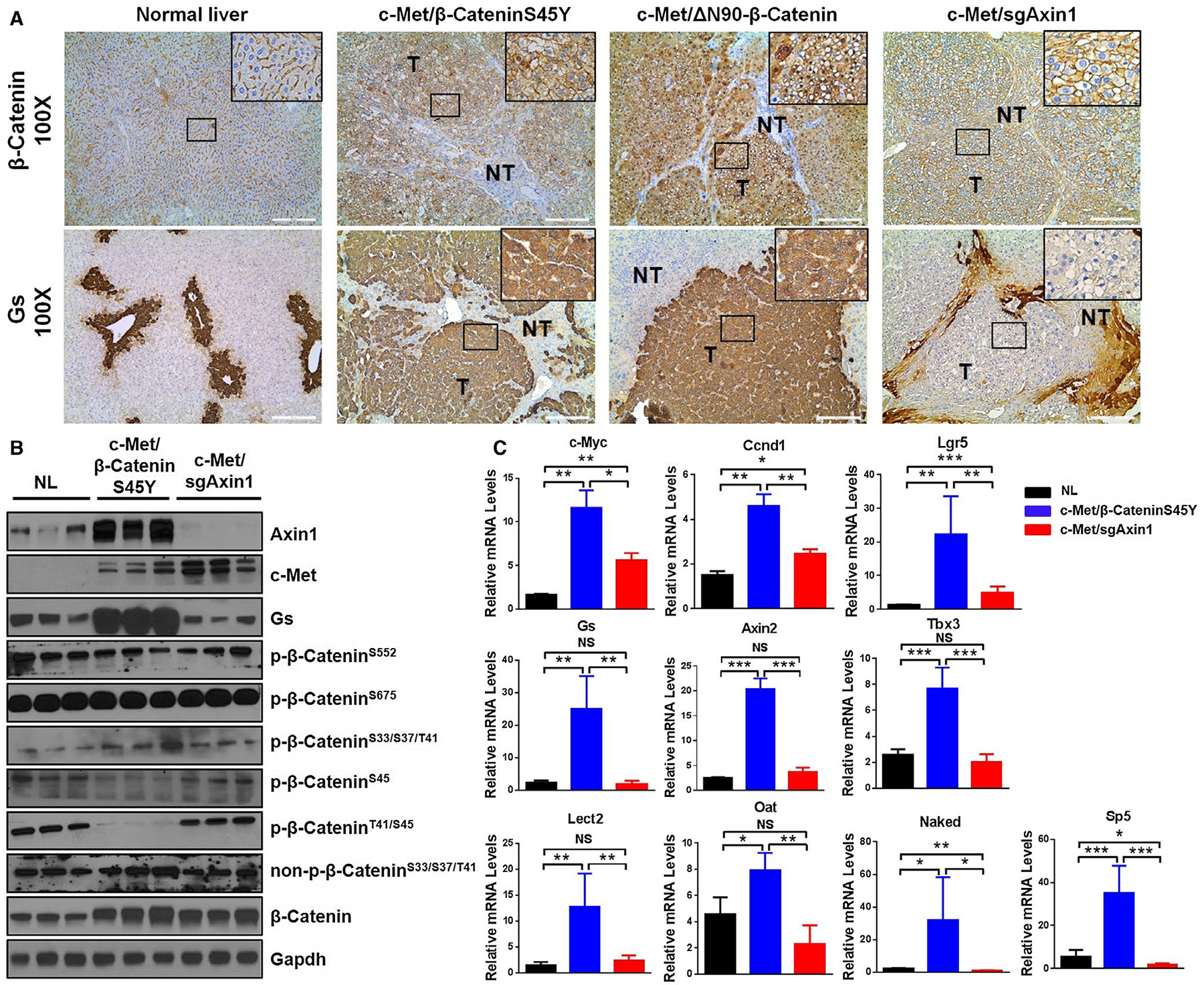
Lack of strong Wnt/β-Catenin activation in c-Met/sgAxin1 tumors. (A) Immunohistochemistry of GS and β-Catenin proteins in normal liver, c-Met/β-CateninS45Y, c-Met/ΔN90β-Catenin, and c-Met/sgAxin1 tumors 9 weeks after hydrodynamic injection. Magnification: ×100, scale bar: 200 μm. Black box displays better visualization at representative sights. (B) Western blotting of normal liver, c-Met/β-CateninS45Y, and c-Met/sgAxin1 samples. Gapdh and β-Actin were used as loading control. (C) Expression of β-Catenin target genes was analyzed by real-time quantitative PCR in normal liver (n = 4), c-Met/β-CateninS45Y (n = 4), and c-Met/sgAxin1 (n = 4) tumors. Data are presented as mean ± SD. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Abbreviations: Gapdh, glyceraldehyde 3-phosphate dehydrogenase; NL, normal liver; NS, not significant.
Subsequently, we analyzed the expression of 11 canonical Wnt/β-Catenin target genes in normal liver, c-Met/β-CateninS45Y HCC, and c-Met/sgAxin1 HCC samples using quantitative real-time PCR. The levels of many of these genes, including GS, Axin2, Tbx3, Lect2, and Oat, were up-regulated in c-Met/β-CateninS45Y HCC but not c-Met/sgAxin1 HCC. Levels of c-Myc, Ccnd1, and Lgr5 were instead up-regulated in c-Met/sgAxin1 HCC (Fig. 2C). Furthermore, in situ hybridization of Gs and Axin2 in HCC samples confirmed the lack of expression of these two genes in c-Met/sgAxin1 HCC lesions (Supporting Fig. S11).
To further substantiate the in vivo findings, we evaluated whether mouse HCC cell lines with Axin1 deletion display molecular features of strong Wnt/β-Catenin activation. For this purpose, we analyzed GS expression in mouse HCC cell lines transfected with sgEgfp and sgAxin1. Notably, none of the cells showed detectable levels of GS (Supporting Fig. S6B). TCF reporter assay revealed that TCF activity was robustly induced by the activated form of β-Catenin, whereas deletion of Axin1 resulted in a very weak activation of the same reporter (Supporting Fig. S6D).
The same question was investigated using human HCC cell lines. Lower levels of TCF reporter activity were detected in all HCC cell lines harboring AXIN1 mutation or lacking AXIN1 expression (Supporting Fig. S12A). In contrast, all hepatoblastoma cell lines demonstrated high levels of TCF reporter activity (Supporting Fig. S12A). Next, we designed three guide RNA against human AXIN1 allele (Supporting Fig. S12B) and found them to be efficient in inhibiting AXIN1 expression in human HCC cell lines (Supporting Fig. S12C). When testing TCF reporter activity in human HCC cell lines with wild-type AXIN1 subjected to AXIN1 knockdown, we found that loss of AXIN1 expression only induces a weak activation of the TCF reporter (Supporting Fig. S12D).
Overall, our study suggests that loss of AXIN1 does not result in classical and strong activation of the Wnt/β-Catenin pathway in mouse and human HCC cells.
GENOMIC PROFILING REVEALS THE ACTIVATION OF WNT SIGNALING CASCADE IN c-Met/sgAxin1 HCC
To further investigate the molecular signatures induced by loss of sgAxin1 in HCC, we performed RNASeq analysis of normal mouse liver (n = 4), c-Met/sgAxin1 HCC (n = 4), and c-Met/ΔN90β-Catenin HCC (n = 4). The multidimensional scaling plot revealed that samples from each of the three groups clustered with each other, although the three groups were well separated (Fig. 3A). We identified 996 genes as up-regulated and 315 genes as down-regulated in the c-Met/ΔN90β-Catenin tumors compared with normal liver, 982 genes as up-regulated and 387 genes as down-regulated in the sgAxin1 tumors compared with normal liver, and 29 up-regulated and 76 down-regulated genes in sgAxin1 compared with c-Met/ΔN90β-Catenin tumors. Gene ontology analysis identified a significant up-regulation of genes involved in cell cycle and mitosis in c-Met/ΔN90β-Catenin tumors versus normal liver (Supporting Fig. S13). Tumors overexpressing sgAxin1 displayed instead down-regulation of genes involved in oxidation-reduction and small molecule metabolic processes (Supporting Fig. S14). Consistent with the role of c-Met as activator of the v-akt murine thymoma viral oncogene homolog (AKT)/mammalian target of rapamycin (mTOR) signaling, gene expression analysis revealed the up-regulation of AKT cascade in both mouse HCC samples (Supporting Fig. S15). To specifically determine whether Wnt signaling pathway genes are differentially regulated in the two tumor models, Fry gene set test was conducted. It showed up-regulation of the Wnt signaling pathway in both c-Met/sgAxin1 and c-Met/ΔN90β-Catenin HCC groups compared with normal liver (Fig. 3B). Further analysis of the differentially expressed genes within the Wnt signaling pathway revealed that of the 46 up-regulated and 8 down-regulated genes in the tumor groups versus normal liver, 25 genes were commonly up-regulated and 3 were commonly down-regulated in c-Met/sgAxin1 and c-Met/ΔN90β-Catenin groups (Fig. 3C). The heatmap of differentially expressed Wnt signaling pathway genes indicated that the well-characterized canonical Wnt/β-Catenin target genes, including Axin2, Lef1, and Sfrp2, were up-regulated only in the c-Met/ΔN90β-Catenin tumors, consistent with our earlier findings (Fig. 3D).
FIG. 3.
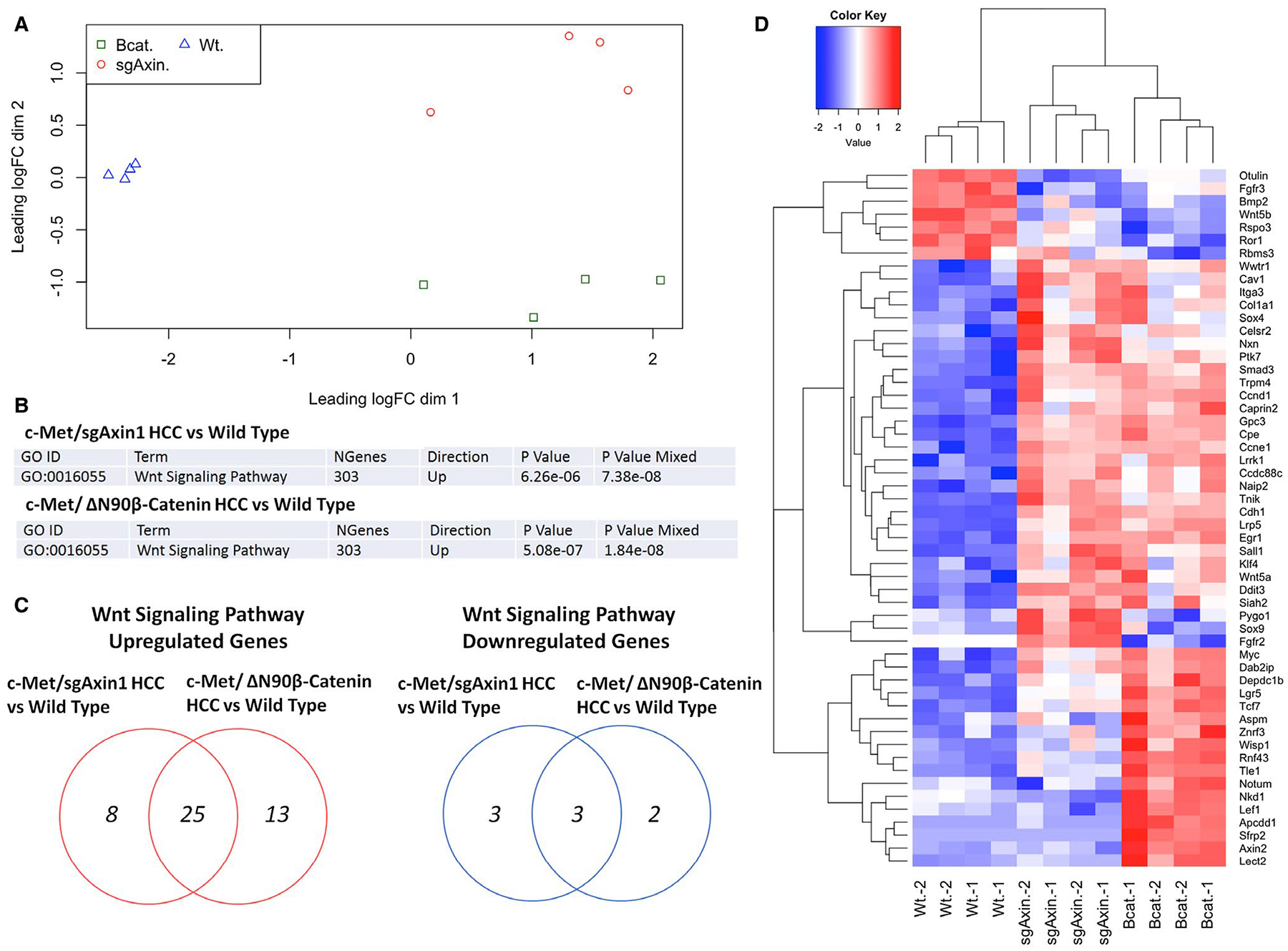
RNA sequencing data show activation of different Wnt target sets in c-Met/sgAxin1 and c-Met/β-Catenin HCC. (A) Multidimensional scaling plot demonstrates the similarity of different sample sets. (B) Fry gene set test of 303 genes of the Wnt signaling pathway in different data sets. (C) Venn diagram showing up-regulated (in red) and down-regulated (in blue) gene number in different data sets. (D) Heatmap of the expression of most-regulated genes in Wnt signaling in the two different HCC models. Red, up-regulated; blue, down-regulated. Abbreviations: Bcat, c-Met/ΔN90β-Catenin HCC; sgAxin, c-Met/sgAxin1 HCC; Wt, wild-type liver.
ABLATION OF Ctnnb1 COMPLETELY INHIBITS c-Met/sgAxin1-DRIVEN MOUSE HCC
The genomic studies suggested the activation of Wnt signaling in c-Met/sgAxin1 mouse HCC. Therefore, we tested the hypothesis that endogenous β-Catenin is required for c-Met/sgAxin1-driven HCC formation in mice. For this purpose, we obtained Ctnnb1fl/fl mice and their Ctnnb1+/+ litter-mates and hydrodynamically injected c-Met, sgAxin1 plasmids together with pCMV-Cre construct into these mice (c-Met/sgAxin1/Cre) (Fig. 4A). Whereas all c-Met/sgAxin1/Cre-injected Ctnnb1+/+ mice developed liver tumors and had to be euthanized by 12 weeks after injection, none of the c-Met/sgAxin1/Cre-injected Ctnnb1fl/fl littermates showed any sign of tumor development. All mice were euthanized by 20 weeks after injection, and all appeared to be healthy with no liver tumors (Fig. 4B–D). Histological evaluation revealed HCC in c-Met/sgAxin1/Cre-injected Ctnnb1+/+ mouse liver, whereas liver tissues were completely normal in c-Met/sgAxin1/Cre-injected Ctnnb1fl/fl mice (Fig. 4D). As another control for the experiment, c-Met, ΔN90β-Catenin, and Cre plasmids were coinjected into Ctnnb1fl/fl mice (Supporting Fig. S16A). c-Met/ΔN90β-Catenin/Cre was able to induce HCC formation in all injected Ctnnb1fl/fl mice (Supporting Fig. S16B). The resulting HCC lesions showed the loss of endogenous β-Catenin protein as demonstrated by western blot analysis (Supporting Fig. S16C).
FIG. 4.
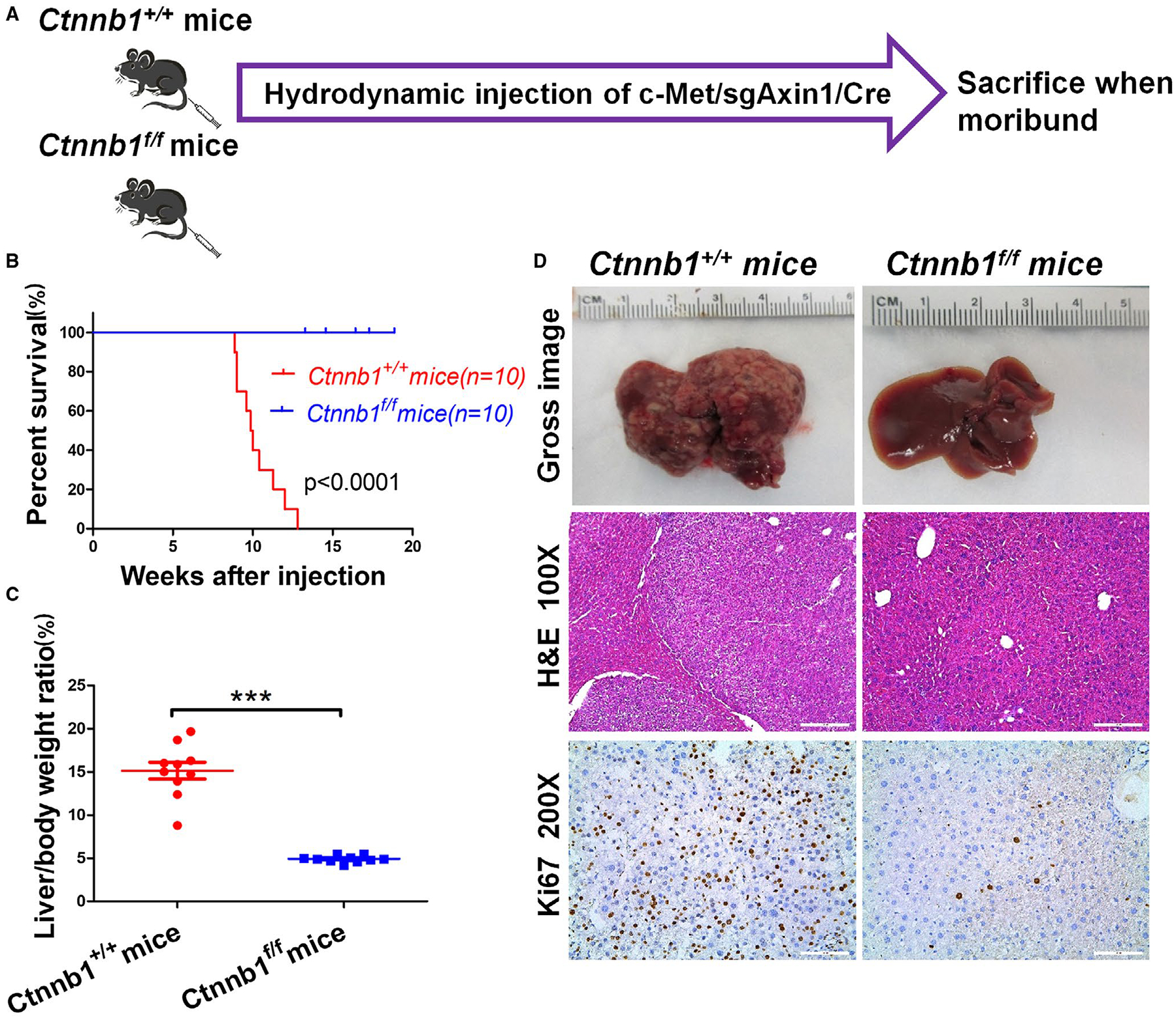
Ablation of Ctnnb1 completely suppresses c-Met/sgAxin1-driven mouse hepatocarcinogenesis. (A) Study design. (B) Survival analysis of Ctnnb1+/+ mice (n = 10) and Ctnnb1f/f mice (n = 10) using the Kaplan-Meier survival method. (C) Liver body weight ratio between two groups. Data are presented as mean ± SD. ***, P < 0.001. (D) Gross images of livers, H&E staining, and Ki-67 staining in Ctnnb1+/+ and Ctnnb1f/f mice livers. Magnifications: ×100, scale bar: 200 μm; ×200, scale bar: 100 μm. Abbreviation: H&E, hematoxylin and eosin.
To complement this approach, we also injected c-Met, sgAxin1 plasmids together with either pCMV-Cre (c-Met/sgAxin1/Cre) or pCMV empty vector (c-Met/sgAxin1/pCMV) into Ctnnb1fl/fl mice (Supporting Fig. S17A). Again, all c-Met/sgAxin1/pCMV-injected Ctnnb1fl/fl mice developed high liver tumor burden by 12 weeks after injection, whereas none of the c-Met/sgAxin1/Cre-injected Ctnnb1fl/fl mice displayed liver tumors at this stage (Supporting Fig. S17B). To rule out that the results were specific for one sgAxin1 construct (sgAxin1.2), we repeated the experiments with sgAxin1.1 construct, and the same results were obtained (Supporting Fig. S17B).
Next, we determined whether β-Catenin is required for AXIN1 mutant or null human HCC cell growth. Thus, β-Catenin was silenced using shβ-Catenin. In accordance with earlier evidence, loss of β-Catenin significantly inhibited AXIN1 mutant or null human HCC cell growth in vitro (Supporting Fig. S18A,B).
In summary, our study demonstrates that c-Met/sgAxin1-induced HCC requires an intact β-Catenin.
NOTCH SIGNALING IS DISPENSABLE FOR c-Met/sgAxin1-DRIVEN HCC FORMATION
A previous study suggests that Notch path way is activated in AXIN1 mutant human HCC.(11) Consistent with this report, our global gene expression analysis revealed the activation of Notch signaling in c-Met/sgAxin1 HCC samples (Fig. 5A,B). To further confirm the results, we analyzed the protein expression of Notch pathway targets, including Notch2, Jag1, and Sox9, in c-Met/sgAxin1 liver lesions (Fig. 5C). Consistently, overall protein levels of Jag1, Notch2, and Sox9 were highest in c-Met/sgAxin1 HCC (Fig. 5C). Importantly, nuclear expression of Notch2 but not Notch1 protein was detected in c-Met/sg-Axin1 mouse HCC (Fig. 5D). Nuclear expression of Sox9 in tumor cells was also detected immunohistochemically (Fig. 5E; Supporting Fig. S19A).
FIG. 5.
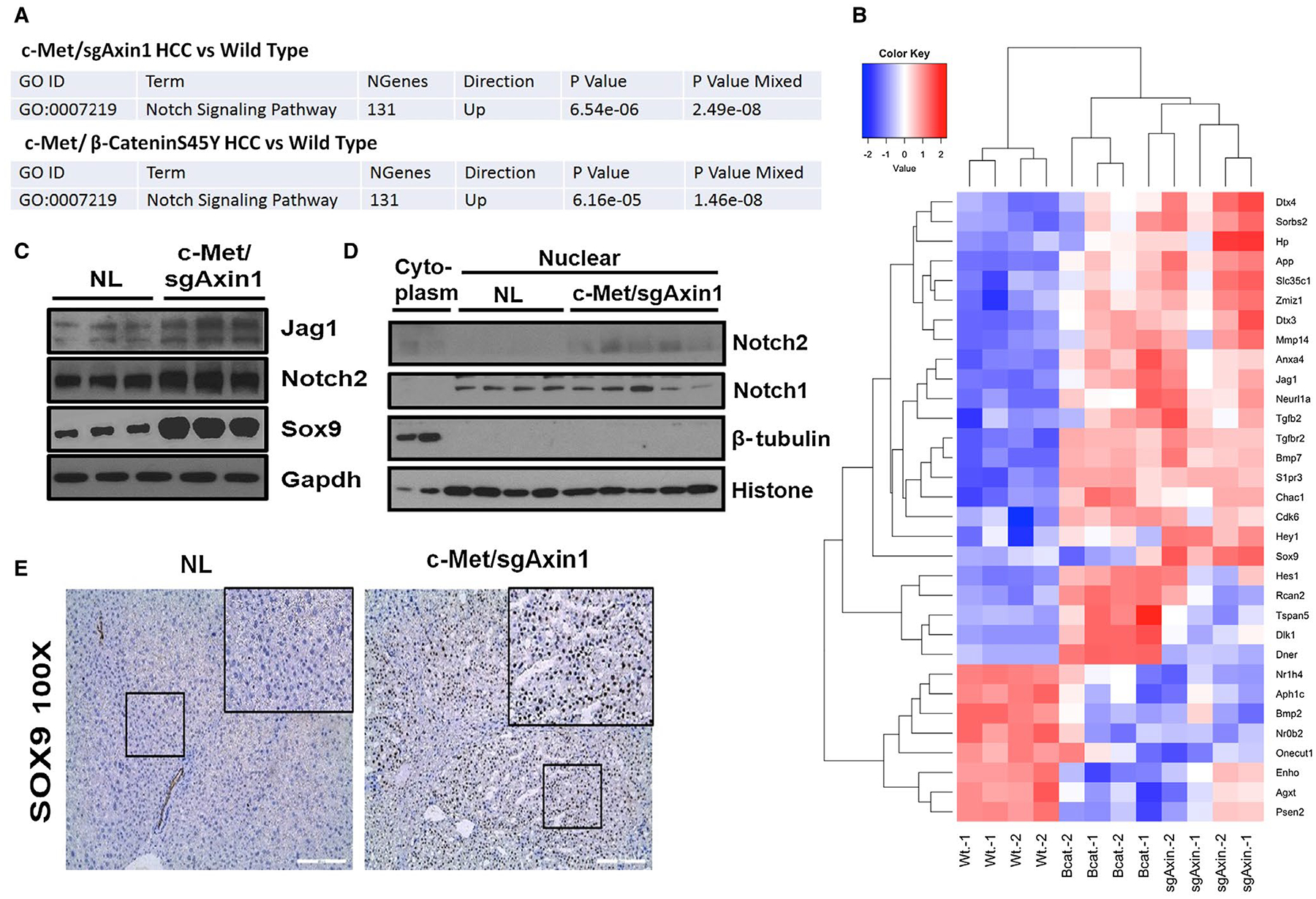
Activation of the Notch pathway in c-Met/sgAxin1 liver tumors. (A) Fry gene set test illustrates the significance of up-regulated genes in Notch signaling pathway in c-Met/sgAxin1 and c-Met/β-CateninS45Y tumors. (B) Heatmap of 24 most-up-regulated genes and 8 most-down-regulated genes in Notch signaling cascades. Red, up-regulated; blue, down-regulated. (C) Western blotting showing elevated protein levels of Notch2, Jag1, and Sox9 in c-Met/sgAxin1 tumors compared with normal liver. Gapdh was used as a loading control. (D) Nuclear protein expression of Notch1 and Notch2. Histone H3 was used as a loading control. (E) Immunohistochemistry staining of Sox9 in normal liver and c-Met/sgAxin1 liver tissues. Magnification: ×100, scale bar: 200 μm. Black square displays better visualization of nuclear staining of Sox9. Abbreviations: Gapdh, glyceraldehyde 3-phosphate dehydrogenase; NL, normal liver; Sox9, SRY (sex determining region Y)-box 9.
To investigate whether Notch2 is required for c-Met/sgAxin1-driven HCC, Notch2fllfl mice were used for HCC induction. Specifically, Notch2fllfl mice were hydrodynamically injected with c-Met/sgAxin1/Cre or c-Met/sgAxin1/pCMV plasmids (Fig. 6A). All injected mice developed liver tumors and were euthanized between 9 and 18 weeks after injection (Fig. 6B). No difference in tumor burden was found between Notch2 wild-type and Notch2-depleted mice (Fig. 6C,D). HCC lesions were identified in both cohorts, and tumor cells were highly proliferative (Fig. 6D). At the biochemical level, loss of Notch2 expression was confirmed in c-Met/sgAxin1/Cre-injected Notch2fllfl mice (Fig. 6E), leading to the decreased expression of Jag1 and Sox 9 (Fig. 6D,E; Supporting Fig. S19B).
FIG. 6.
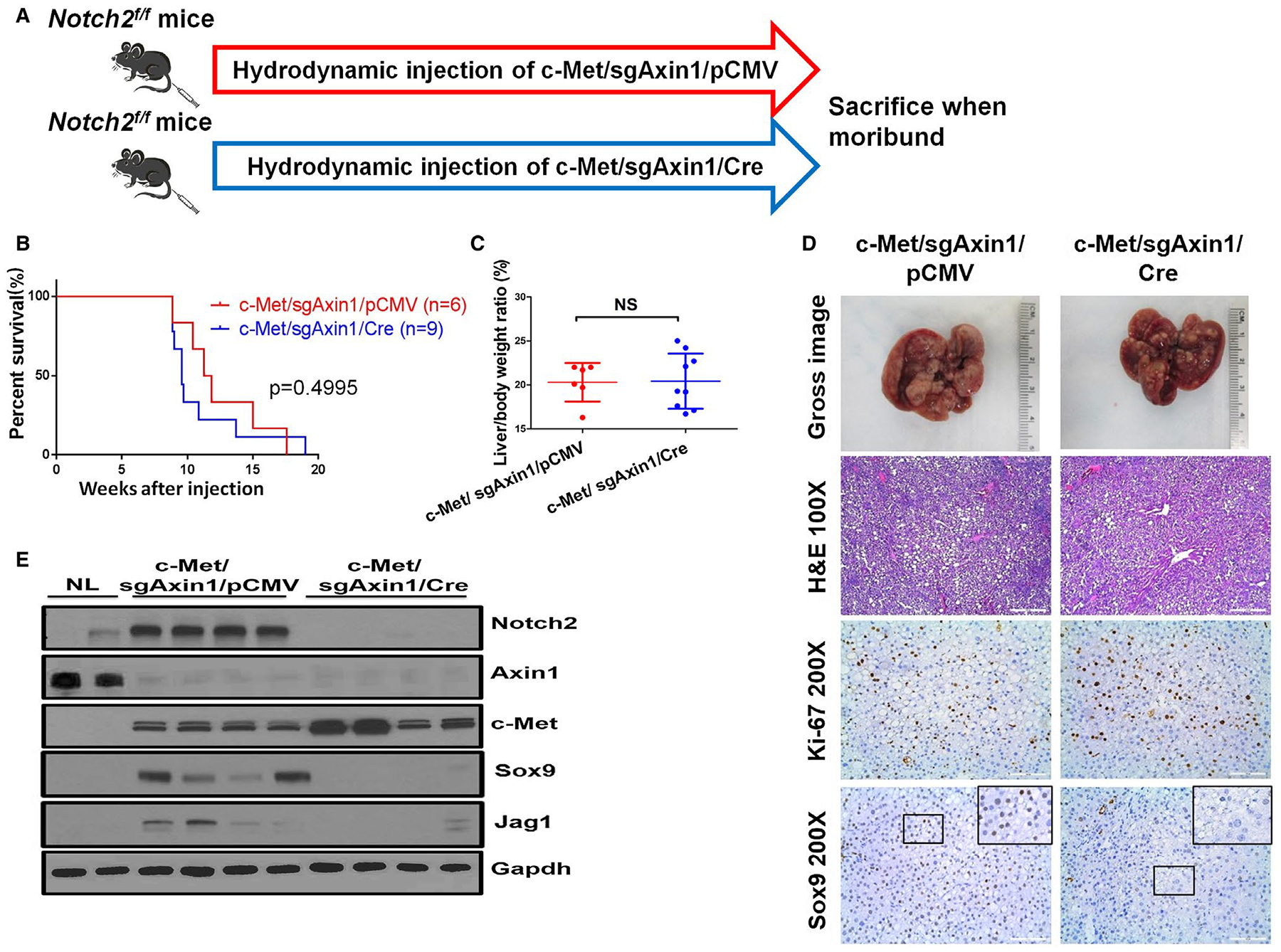
Notch2 signaling is dispensable for c-Met/sgAxin1-driven HCC formation. (A) Study design. (B) Kaplan-Meier survival curve of c-Met/sgAxin1/pCMV Notch2fl/fl mice (n = 6) and c-Met/sgAxin1/Cre Notch2fl/fl mice (n = 9). (C) Liver body weight ratio of the two groups. Data are presented as mean ± SD. (D) Gross images of livers, H&E, Ki-67, and Sox9 staining in c-Met/sgAxin1/pCMV and c-Met/sgAxin1/Cre samples. Magnifications: ×100, scale bar: 200 μm; ×200, scale bar: 100 μm. (E) Western blotting from normal liver, c-Met/sgAxin1/pCMV, and c-Met/sgAxin1/Cre Notch2fl/fl mice liver tissues. Gapdh was used as a loading control. Abbreviations: Gapdh, glyceraldehyde 3-phosphate dehydrogenase; H&E, hematoxylin and eosin; NL, normal liver; NS, not significant; Sox9, SRY (sex determining region Y)-box 9.
To rule out the possible involvement of additional Notch receptors in mediating c-Met/sgAxin1-induced hepatocarcinogenesis, the canonical Notch cascade was blocked with a dominant negative form of the recombinant signal-binding protein for immunoglobulin kappa J region (RBPJ) (dnRBPJ, with V5 tag).(19) Specifically, we coexpressed c-Met and sgAxin1 plasmids together with dnRBPJ vector in FVB/N wild-type mice (c-Met/sgAxin1/dnRBPJ). As control, c-Met and sgAxin1 plasmids were coinjected with pT3-EF1α empty vector (c-Met/sgAxin1/pT3) into mice (Fig. 7A). Notably, mice in both cohorts developed liver tumors at the same latency (Fig. 7B) and showed an equivalent tumor burden (Fig. 7C). HCC lesions were equally appreciable in c-Met/sgAxin1/pT3 and c-Met/sgAxin1/dnRBPJ mouse livers (Fig. 7E). Tumor cells were highly proliferative, with lower expression of the Notch target gene Sox9 (Fig. 7D,E). Importantly, western blotting and immunostaining of V5 tag demonstrated that c-Met/sgAxin1/dnRBPJ lesions expressed ectopically injected dnRBPJ (Fig. 7D,E).
FIG. 7.

c-Met/sgAxin1 induced HCC in the absence of canonical Notch signaling. (A) Study design. (B) Survival curve. (C) Liver body weight ratio of c-Met/sgAxin1/pT3 (n = 6) and c-Met/sgAxin1/dnRBPJ (n = 4). (D) Gross images of livers, H&E, Ki-67, and Sox9 staining in c-Met/sgAxin1/pT3 and c-Met/sgAxin1/dnRBPJ mouse livers. Magnifications: ×100 (H&E), scale bar: 200 μm; ×200 (Ki-67, Sox9, and V5 tag), scale bar: 100 μm. (E) Representative western blotting from NL, c-Met/β-CateninS45Y, c-Met/sgAxin1/pT3, and c-Met/sgAxin1/dnRBPJ mice liver tissues. Data are presented as mean ± SEM. Abbreviations: H&E, hematoxylin and eosin; NS, not significant; NL, normal liver; Sox9, SRY (sex determining region Y)-box 9.
Altogether, our study demonstrates that the canonical Notch signaling is not required for c-Met/sgAxin1-driven hepatocarcinogenesis.
Discussion
Wnt/β-Catenin signaling is an evolutionarily conserved pathway regulating cell fate determination, stem cell renewal, and organogenesis.(4) Aberrant activation of the Wnt/β-Catenin signaling cascade has been implicated in multiple tumor types,(20) including HCC.(21) Deregulated Wnt/β-Catenin cascade might be the consequence of various mechanisms, including epigenetic modifications,(22) microRNA deregulation,(23) and genetic mutations. In human HCC, mutations of CTNNB1 are found in 20%–40% of cases.(3,8) Most of these mutations occur in the exon-3 of CTNNB1 gene, leading to its resistance to degradation, and consequent stabilization and nuclear localization. Besides CTNNB1 mutations, additional genetic events, including AXIN1 mutations(24) or amplification of fibroblast growth factor 19,(25) have been implicated in activating the Wnt/β-Catenin cascades in human HCC. In the case of AXIN1, overall ~8% of human HCC samples harbor this mutation. Most of AXIN1 mutations are frameshift, nonsense mutations or deletions, frequently in combination with loss of heterozygosity at the second allele,(7,26) consistent with the tumor suppressor role of AXIN1.
Because Axin1 functions as a negative regulator of the Wnt/β-Catenin signaling pathway, it has been suggested that AXIN1 mutant HCCs would have high levels of Wnt/β-Catenin activation. Intriguingly, several studies revealed the lack of expression of liver-specific Wnt/β-Catenin target genes, such as GS, in human and mouse HCCs harboring AXIN1 mutations.(7,10) Using gene expression analysis, a recent study by Abitbol et al. suggested that CTNNB1 mutant human HCCs showed strong or weak Wnt/β-Catenin activation, whereas more than 70% of AXIN1 mutant HCCs belonged to the “no activation” sub-group.(11) All these data suggest that AXIN1 might function through Wnt/β-Catenin-independent mechanisms along hepatocarcinogenesis.
In this manuscript, we specifically studied the tumor suppressor role of Axin1 by deleting Axin1 in mouse hepatocytes. Our results indicate that ablation of Axin1 alone does not lead to HCC formation, but cooperates with overexpression of c-Met to promote HCC development in mice. As activated β-Catenin also promotes HCC in combination with c-Met,(13) c-Met/sgAxin1 and c-Met/β-Catenin mouse HCC models were used to compare the gene expression signatures and signaling pathways induced by loss of Axin1 and activated β-Catenin in vivo. We therefore focused on the Wnt/β-Catenin pathway and analyzed the status of Wnt/β-Catenin pathway in normal liver, c-Met/sgAxin1, and c-Met/β-Catenin mouse HCC using immunohistochemistry, real-time quantitative PCR, and RNASeq. Using the β-Catenin activation signature by Abitbol et al.,(11) we found that mouse c-Met/sgAxin1 HCC do not show the activation of this gene signature, whereas c-Met/ΔN90β-Catenin HCC demonstrated a strong induction (Supporting Fig. S20). However, it is important to note that most of the genes in the 23-gene list are known to be induced by strong Wnt/β-Catenin activation, including GS, AXIN2, TBX3, and so on. Based on the global gene expression analysis, the Wnt pathway was found to be up-regulated in c-Met/sgAxin1 HCC as well as c-Met/β-Catenin HCC (Fig. 3A,B). Altogether, our investigation suggests a weak activation of the Wnt/β-Catenin cascade in HCCs where Axin1 is mutated or deleted. Presumably, this weak activation of the Wnt pathway leads to the increased expression of only a subset of β-Catenin target genes, such as c-Myc and Ccnd1, but not others, such as GS and Axin2 (Fig. 8).
FIG. 8.
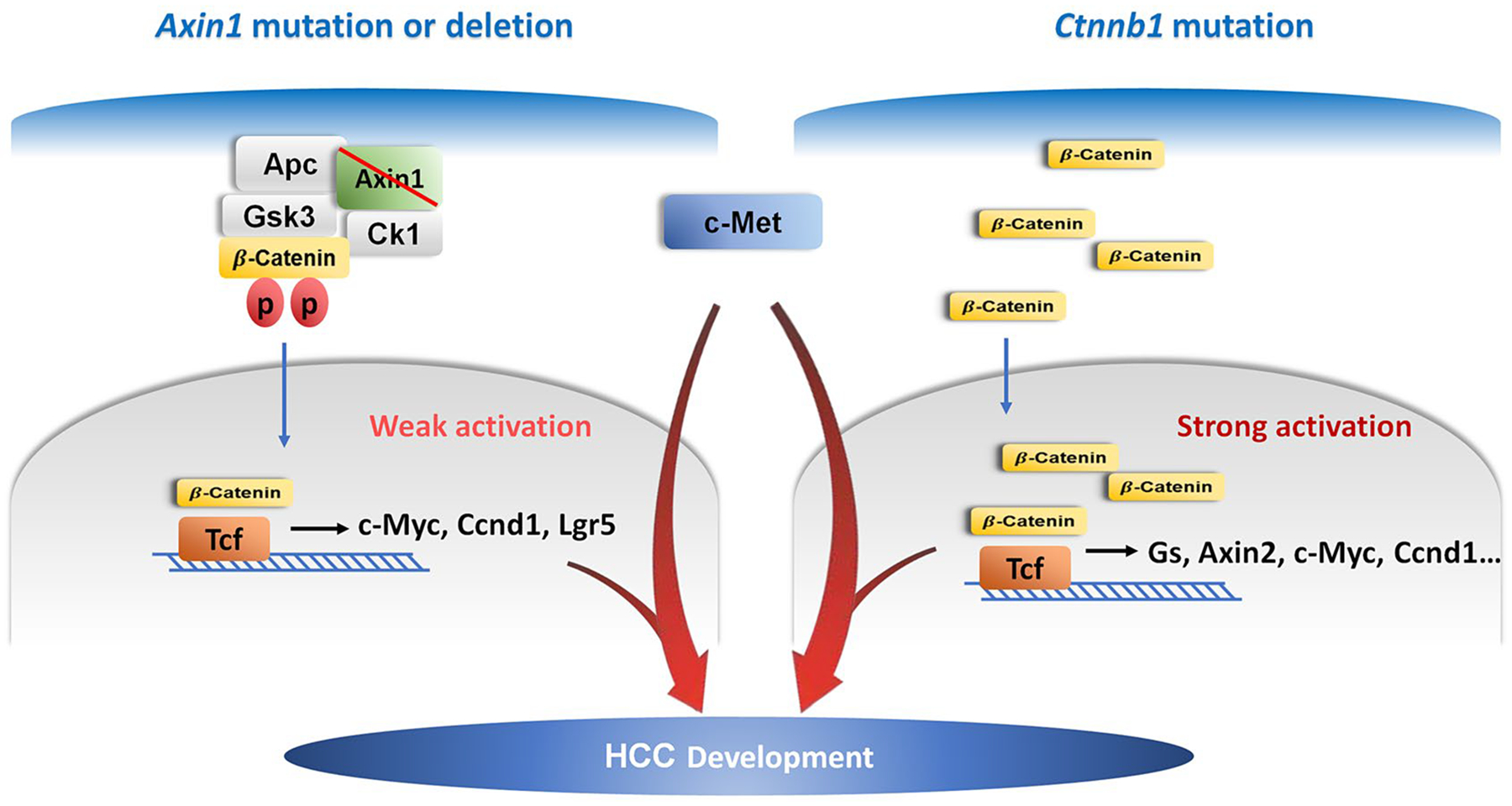
Illustration of c-Met/sgAxin1 and c-Met/β-Catenin model in HCC development.
To investigate whether the endogenous β-Catenin is necessary in mediating c-Met/sgAxin1-driven HCC, genetic approaches were applied, thus deleting Ctnnb1 while expressing c-Met/sgAxin1 in a subset of mouse hepatocytes. Ablation of Ctnnb1 completely prevented c-Met/sgAxin1-driven hepatocarcinogenesis in vivo, whereas it had no effect on c-Met/β-Catenin-driven HCC formation. Our data, therefore, unequivocally prove that loss of Axin1-driven HCC requires an intact β-Catenin. Whether an intact Wnt/β-catenin cascade is also required, including Wnt secretion from nonparenchymal cells like endothelial cells and macrophages or the presence of Wnt coreceptors LRP5/6 on hepatocytes, needs to be investigated in the future.(27,28) Because it was discovered that liver-specific deletion of Axin1 leads to increased hepatocyte cell volume and proliferation in mice,(10) it would be important to determine whether these cellular effects upon loss of Axin1 also require an intact Wnt/β-Catenin cascade. The hypothesis could be tested by means of generating liver-specific Axin1 and Ctnnb1 double knockout (KO) mice. The data will provide additional useful information about the genetic crosstalk between Axin1 and β-Catenin in the normal liver.
The requirement of β-Catenin in loss of Axin1- driven HCC was further supported by findings in human HCC cells with AXIN1 mutations, where silencing of β-Catenin strongly inhibited HCC cell growth (Supporting Fig. S18A,B). It is interesting to note that in two human HCC cell lines with wild-type AXIN1 alleles, silencing of β-Catenin inhibited the growth of MHCC97-H cells, but not HLF cells (Supporting Fig. S18C,D). The specific molecular events in MHCC97-H cells that require an intact Wnt/β-Catenin cascade remain to be determined. Further analysis of additional mouse HCC models using Ctnnb1 conditional KO mice will provide solid data on how this major signaling pathway contributes to hepatocarcinogenesis under different oncogenic stimuli.
An important finding from the study by Abitbol et al. was the activation of Yap and Notch signaling in human and mouse HCCs harboring AXIN1 mutations.(11) However, no functional analysis of Yap or Notch signaling in their study was carried out. In our current study, we focused on the Notch cascade, as the global gene expression analysis that we conducted supported its activation in the c-Met/sgAxin1 mouse HCC (Fig. 5). Using genetic approaches to delete Notch2, the major Notch receptor on the hepatocytes, or blocking the canonical Notch cascade with dnRBPJ, our results demonstrated that despite the activation of Notch signaling, this pathway is completely dispensable for c-Met/sgAxin1-induced HCC formation. Regarding the Yap pathway, our preliminary data revealed an increased nuclear expression of Yap in c-Met/sgAxin1 HCC, suggesting Yap activation in Axin1-deleted mouse HCC. However, contrasting and so far poorly understood results in terms of liver carcinogenesis were obtained when inhibiting Yap by various strategies in c-Met/sgAxin1 mice (Chen X, unpublished data). Thus, further analysis is required to elucidate the functional contribution of the Yap signaling on c-Met/sgAxin1-driven hepatocarcinogenesis.
As AXIN1 mutations are among the most frequent genetic events in human HCC, it would be crucial to develop murine HCC models to study targeted therapies for HCC with AXIN1 mutations. Liver-specific Axin1 KO mice have been generated, and HCC develop in ~40%–50% of these mice over long latency, i.e., 12 months.(10,11) Thus, this model may be difficult to use to study innovative treatments against HCC. Here, we generated a murine HCC model with deletion of Axin1 and coexpression of the c-Met proto-oncogene. Because c-Met and sgAxin1 coexpression promotes HCC formation in 100% of mice within 11 weeks after hydrodynamic injection, these mice represent an excellent preclinical model to study targeted therapy against Axin1 mutant HCC. For instance, a small interfering RNA targeting β-Catenin assembled into lipid nanoparticles (CTNNB1-LNP) has been shown to be effective in inducing regression of HCC induced by KRas/β-Catenin in mice.(28) CTNNB1-LNP could be applied to c-Met/sgAxin1 mouse HCC model to evaluate whether targeting β-Catenin is an effective therapeutic approach against HCC displaying an impaired Axin1.
Supplementary Material
Funding
Supported by R01CA204586 to S.P.M. and X.C., R01DK116993 to S.P.M., and P30DK026743 for UCSF Liver Center.
Abbreviations:
- Axin
axis inhibition protein
- CTNNB1
catenin beta 1
- GS
glutamine synthetase
- HCC
hepatocellular carcinoma
- pCMV
phosphorylated cytomegalovirus
- RBPJ
recombinant signal-binding protein for immunoglobulin kappa J region
- TCF
T cell factor
Footnotes
Potential conflict of interest: Nothing to report.
Supporting Information
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep.30556/suppinfo.
REFERENCES
- 1).Armengol C, Sarrias MR, Sala M. Hepatocellular carcinoma: Present and future. Med Clin (Barc) 2018;150:390–397. [DOI] [PubMed] [Google Scholar]
- 2).Dutta R, Mahato RI. Recent advances in hepatocellular carcinoma therapy. Pharmacol Ther 2017;173:106–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3).Cancer Genome Atlas Research Network. Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell 2017;169:1327–1341 e1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4).Nusse R, Clevers H. Wnt/beta-Catenin signaling, disease, and emerging therapeutic modalities. Cell 2017;169:985–999. [DOI] [PubMed] [Google Scholar]
- 5).Monga SP. β-Catenin signaling and roles in liver homeostasis, injury, and tumorigenesis. Gastroenterology 2015;148:1294–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6).Song X, Wang S, Li L. New insights into the regulation of Axin function in canonical Wnt signaling pathway. Protein Cell 2014;5:186–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7).Zucman-Rossi J, Benhamouche S, Godard C, Boyault S, Grimber G, Balabaud C, et al. Differential effects of inactivated Axin1 and activated beta-catenin mutations in human hepatocellular carcinomas. Oncogene 2007;26:774–780. [DOI] [PubMed] [Google Scholar]
- 8).Schulze K, Imbeaud S, Letouze E, Alexandrov LB, Calderaro J, Rebouissou S, et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat Genet 2015;47:505–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9).Forbes SA, Beare D, Boutselakis H, Bamford S, Bindal N, Tate J, et al. COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res 2017;45:D777–D783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10).Feng GJ, Cotta W, Wei XQ, Poetz O, Evans R, Jarde T, et al. Conditional disruption of Axin1 leads to development of liver tumors in mice. Gastroenterology 2012;143:1650–1659. [DOI] [PubMed] [Google Scholar]
- 11).Abitbol S, Dahmani R, Coulouarn C, Ragazzon B, Mlecnik B, Senni N, et al. AXIN deficiency in human and mouse hepatocytes induces hepatocellular carcinoma in the absence of beta-catenin activation. J Hepatol 2018;68:1203–1213. [DOI] [PubMed] [Google Scholar]
- 12).Lee SA, Ho C, Roy R, Kosinski C, Patil MA, Tward AD, et al. Integration of genomic analysis and in vivo transfection to identify sprouty 2 as a candidate tumor suppressor in liver cancer. Hepatology 2008;47:1200–1210. [DOI] [PubMed] [Google Scholar]
- 13).Tao J, Xu E, Zhao Y, Singh S, Li X, Couchy G, et al. Modeling a human hepatocellular carcinoma subset in mice through co-expression of met and point-mutant beta-catenin. Hepatology 2016;64:1587–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14).Wang J, Dong M, Xu Z, Song X, Zhang S, Qiao Y, et al. Notch2 controls hepatocyte-derived cholangiocarcinoma formation in mice. Oncogene 2018;37:3229–3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15).Xue W, Chen S, Yin H, Tammela T, Papagiannakopoulos T, Joshi NS, et al. CRISPR-mediated direct mutation of cancer genes in the mouse liver. Nature 2014;514:380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16).You H, Ding W, Dang H, Jiang Y, Rountree CB. c-Met represents a potential therapeutic target for personalized treatment in hepatocellular carcinoma. Hepatology 2011;54: 879–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17).Kaposi-Novak P, Lee JS, Gomez-Quiroz L, Coulouarn C, Factor VM, Thorgeirsson SS. Met-regulated expression signature defines a subset of human hepatocellular carcinomas with poor prognosis and aggressive phenotype. J Clin Invest 2006;116: 1582–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18).Lee SA, Ladu S, Evert M, Dombrowski F, De Murtas V, Chen X, et al. Synergistic role of Sprouty2 inactivation and c-Met up-regulation in mouse and human hepatocarcinogenesis. Hepatology 2010;52:506–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19).Che L, Fan B, Pilo MG, Xu Z, Liu Y, Cigliano A, et al. Jagged 1 is a major Notch ligand along cholangiocarcinoma development in mice and humans. Oncogenesis 2016;5:e274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20).Krishnamurthy N, Kurzrock R. Targeting the Wnt/beta-catenin pathway in cancer: Update on effectors and inhibitors. Cancer Treat Rev 2018;62:50–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21).Khalaf AM, Fuentes D, Morshid AI, Burke MR, Kaseb AO, Hassan M, et al. Role of Wnt/beta-catenin signaling in hepatocellular carcinoma, pathogenesis, and clinical significance. J Hepatocell Carcinoma 2018;5:61–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22).Benoit YD, Guezguez B, Boyd AL, Bhatia M. Molecular pathways: epigenetic modulation of Wnt-glycogen synthase kinase-3 signaling to target human cancer stem cells. Clin Cancer Res 2014;20:5372–5378. [DOI] [PubMed] [Google Scholar]
- 23).Peng Y, Zhang X, Feng X, Fan X, Jin Z. The crosstalk between microRNAs and the Wnt/beta-catenin signaling pathway in cancer. Oncotarget 2017;8:14089–14106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24).Salahshor S, Woodgett JR. The links between axin and carcinogenesis. J Clin Pathol 2005;58:225–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25).Pai R, Dunlap D, Qing J, Mohtashemi I, Hotzel K, French DM. Inhibition of fibroblast growth factor 19 reduces tumor growth by modulating beta-catenin signaling. Cancer Res 2008;68:5086–5095. [DOI] [PubMed] [Google Scholar]
- 26).Taniguchi K, Roberts LR, Aderca IN, Dong X, Qian C, Murphy LM, et al. Mutational spectrum of beta-catenin, AXIN1, and AXIN2 in hepatocellular carcinomas and hepatoblastomas. Oncogene 2002;21:4863–4871. [DOI] [PubMed] [Google Scholar]
- 27).Yang J, Mowry LE, Nejak-Bowen KN, Okabe H, Diegel CR, Lang RA, et al. β-catenin signaling in murine liver zonation and regeneration: a Wnt-Wnt situation!. Hepatology 2014;60:964–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28).Tao J, Zhang R, Singh S, Poddar M, Xu E, Oertel M, et al. Targeting beta-catenin in hepatocellular cancers induced by coexpression of mutant beta-catenin and K-Ras in mice. Hepatology 2017;65:1581–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.