

Summary
Resistance breeding is crucial for a sustainable control of leaf rust (Puccinia triticina) in wheat (Triticum aestivum L.) while directly targeting functional variants is the Holy Grail for efficient marker‐assisted selection and map‐based cloning. We assessed the limits and prospects of exome association analysis for severity of leaf rust in a large hybrid wheat population of 1574 single‐crosses plus their 133 parents. After imputation and quality control, exome sequencing revealed 202 875 single‐nucleotide polymorphisms (SNPs) covering 19.7% of the high‐confidence annotated gene space. We performed intensive data mining and found significant associations for 2171 SNPs corresponding to 50 different loci. Some of these associations mapped in the proximity of the already known resistance genes Lr21, Lr34‐B, Lr1 and Lr10, while other associated genomic regions, such as those on chromosomes 1A and 3D, harboured several annotated genes putatively involved in resistance. Validation with an independent population helped to narrow down the list of putative resistance genes that should be targeted by fine‐mapping. We expect that the proposed strategy of intensive data mining coupled with validation will significantly influence research in plant genetics and breeding.
Keywords: leaf rust, hybrid wheat, exome capture, association mapping, candidate genes, independent validation
Introduction
Wheat (Triticum aestivum L.) is the world’s second most cultivated cereal after maize and provides one‐fifth of the calories intake of human population (FAO, 2019). Leaf rust, caused by Puccinia triticina f. sp. tritici, is one of the most widespread wheat diseases, which can cause up to 40% loss of wheat yield mainly by reducing kernel weight and decreasing the number of kernels per spike (Khan et al., 2013). Resistance breeding, an economic and environment friendly approach, is critical for the sustainable control of wheat leaf rust (Oliver, 2014). Many researchers have studied the genetic architecture of leaf rust in order to efficiently increase the resistance level of cultivars. For instance, more than 70 leaf rust resistance genes (Lr) have been identified in wheat (Kassa et al., 2017) and a few of them have been cloned, including the seedling stage resistance genes Lr1, Lr10 and Lr21, as well as the adult plant resistance Lr34 and Lr67 loci (Cloutier et al., 2007; Feuillet et al., 2003; Huang et al., 2009; Krattinger et al., 2011; Moore et al., 2015). Most of the discovered Lr genes confer all‐stage resistance and are race‐specific, with only a few exceptions like Lr34, Lr46 and Lr67 which confer non–race‐specific resistance during adult plant stages (da Silva et al., 2018). Race‐specific resistance proves to be ineffective after a few years of introduction because of the high mutation rate or virulence dynamics of pathogen populations (Lowe et al., 2011; McCallum et al., 2016). Thus, researchers and breeders are trying to understand the diversity of resistance genes currently used in elite breeding populations and are continuously searching for novel Lr genes.
Genome‐wide association mapping is often used to dissect the genetic architecture of important agronomic traits (Gong et al., 2017) such as leaf rust resistance of wheat (Gao et al., 2016; Juliana et al., 2018; Kertho et al., 2015; Maccaferri et al., 2010). Association mapping can provide a high mapping resolution, particularly in genetically diverse populations, and in some cases facilitates the detection of functional quantitative trait nucleotides (Yano et al., 2016). The high resolution, however, requires the characterization of the mapping population with a high density of markers. Given the advances in next‐generation sequencing techniques and reduced sequencing costs, whole genome sequencing (WGS) has been suggested as a method to characterize the genetic variants of mapping populations (Schneeberger, 2014). Nevertheless, the price of WGS in wheat remains high due to the large genome and its allohexaploid nature, making it necessary to have enough coverage to distinguish homologs and homeologs (Appels et al., 2018). Therefore, resistance gene enrichment sequencing (RenSeq) focusing on genes exclusively encoding intracellular nucleotide‐binding/leucine‐rich repeat immune receptor proteins has been suggested. In fact, combining RenSeq with association genetics allowed to clone four stem rust resistance genes in wheat (Arora et al., 2019). Exome capture sequencing is an alternative solution to dramatically reduce sequencing costs by focusing on gene coding regions (Mo et al., 2018). The potential of using exome sequencing has been demonstrated in a pioneering study in wheat where genes underlying wheat improvement and environmental adaptation could be identified (He et al., 2019).
Our study is based on a large elite wheat population (Longin et al., 2013; Zhao et al., 2013) including ~1800 single‐cross hybrids and their 135 parental lines adapted to the growing conditions of Central Europe: The population was phenotyped in multi‐environmental field trials for leaf rust resistance and fingerprinted using exome capture sequencing. Extensive data mining facilitated to broaden our insights into the diversity of leaf rust resistance genes currently used in wheat breeding in Central Europe. Resistance genes Lr1, Lr10, Lr21 and Lr34‐B are already exploited by breeders but also novel candidate regions on chromosome 1A or 3D were detected. The latter were validated in an independent population of 128 single‐cross hybrids, which facilitated to narrow down the list of putative resistance genes. We expect that the outcomes will benefit marker‐assisted selection of leaf rust resistance and represent promising targets to clone novel resistance genes.
Results
Exome sequencing revealed a broad nucleotide and haplotype diversity
Since two female parents failed to produce meaningful read coverage during exome capture sequencing and enough seed was available for 1604 of the potential 120 × 15 = 1800 single‐cross hybrid combinations, our study is based on 1574 hybrids generated by crossing 118 female and 15 male elite winter wheat lines. The 133 wheat lines were selected to cover a broad range of diversity currently exploited in elite breeding in Central Europe (Zhao et al., 2015). We performed exome capture sequencing of the 133 parental lines based on the NimbleGen array (Winfield et al., 2012) and using an Illumina HiSeq 2500 platform. This resulted in 10.6 billion 100 bp reads (10.4 billion paired‐end and 200 million single‐end) that were mapped against the reference genome of ‘Chinese Spring’ (Appels et al., 2018), unravelling 7 253 398 single‐nucleotide polymorphism (SNP) sites. Only 0.37% (about 40 million reads) of the reads could not be mapped to the reference sequence. The mean coverage of called sites amounted to 1.5 (Figure S1). After imputation using FILLIN (Swarts et al., 2014), we selected SNPs with minor allele frequency (MAF) larger or equal than 0.05 and missing rate smaller than 0.05, resulting in 202 875 SNPs used for subsequent analyses. Although rare resistance/susceptible loci may be underdetected by using a MAF threshold of 0.05, this filtering criteria should avoid the increased false‐positive rate expected for rare variants in large‐scale exome association studies of human diseases (Akle et al., 2015). Prediction of the functional effect revealed that 22 166 SNPs induced non‐synonymous variants and 144 687 out of the 202 875 SNPs (71.32%) were located in genic regions flanked by an upstream and downstream window of 1kb (Table S1). We identified SNPs for 21 249 genes (about 19.7% of all 107 891 high‐confidence genes, Figure S2), with 13 399 genes (63.1%) exhibiting at least two SNPs.
Principal coordinate analysis (PCoA) on the pairwise Rogers’ distances suggested an absence of obvious subpopulations among the 133 parents (Figure 1d), which is in line with previous findings based on a 90k SNP array (Wurschum et al., 2013). These results were further supported by the tight positive correlation (Pearson’s r = 0.911, P < 0.001 using a Mantel test, Figure S3) between the genetic distances estimated using the exome sequencing and those estimated based on the 90k SNP array.
Figure 1.
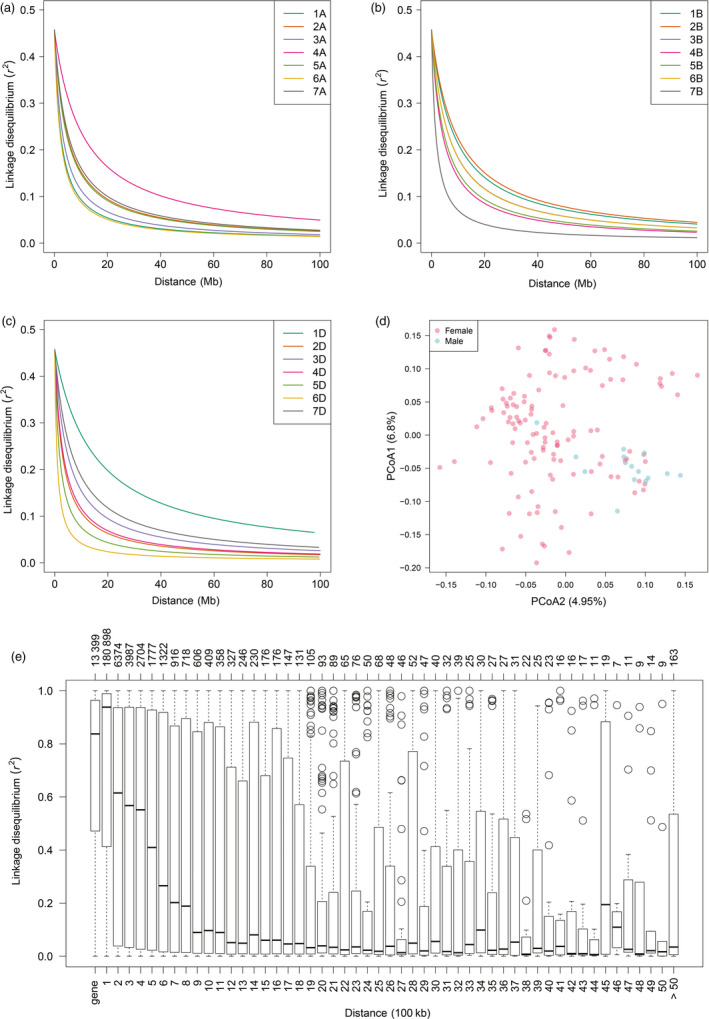
Linkage disequilibrium decay and diversity analysis in a wheat population composed by 1574 hybrids plus their 118 female and 15 male parent lines. Linkage disequilibrium (LD, as r 2) decay plots as a function of physical distance (Mb) within each chromosome for subgenomes A (a), B (b) and D (c). (d) Diversity among the 133 parent lines of the studied hybrid wheat population portrayed in a biplot of the first two principal coordinates from a principal coordinate analysis on the pairwise Rogers’ distance matrix calculated using exome capture single‐nucleotide polymorphisms (SNPs) profiles. (e) Boxplot charts showing the distributions of average LD within a gene and of LD between adjacent SNPs. Bins in the lower x‐axis correspond to the region defined by a gene or to regions defined by adjacent SNPs separated by certain physical distance (100 Kb). The upper x‐axis shows the number of SNP pairs belonging to each corresponding bin in the lower x‐axis.
Molecular and nucleotide diversity was greater for the B than for the A genome and these two genomes, in turn, have a much higher diversity than the D genome (Figures S4 and S5). Analysis of linkage disequilibrium (LD) showed that the LD decay varied among subgenomes and chromosomes. On average, the estimated LD decayed to an r 2 = 0.2 at a distance of 7 Mb (Figure 1 and Table S2). LD decayed faster in the A genome than in B and D genomes, while LD curves were virtually the same for B and D genomes (Figure S6). Even though most studies have reported that LD decays slower for the D genome (e.g. Chen et al., 2012; Liu et al., 2017; Lopes et al., 2015; Sukumaran et al., 2015), there have been also some studies showing LD decay values for the D genome that are similar to or even lower than those of the A and/or B genomes (Sehgal et al., 2017; Zhang et al., 2013). One possible explanation for this discrepancy are the differences in number of marker pairs as a function of physical distance observed in our study (Figure S7). In this sense, a great proportion (~27%) of marker pairs in the D genome is located within very short physical distances, whereas this percentage is clearly smaller for the A and B genomes. This higher density of marker pairs at short physical distances for the D genome can cause a leverage effect that artificially forces the LD curve to be fitted towards the origin of the plot. Therefore, LD comparisons among genomes should take this issue into consideration. Chromosomes 1D and 6D presented the slowest and fastest decay, respectively, and fell below the r 2 = 0.2 threshold at 1 and 20 Mb, correspondingly. This rapid decay revealed the broad haplotype diversity in the underlying mapping population. Considering that a gene is the basic physical and functional unit of heredity, we estimated the LD within genes and found that the LD varied between SNPs within the same gene. In this respect, about 25% of the estimated LD values were less than r 2 = 0.5 (Figure 1e) while some marker pairs even approached linkage equilibrium.
Bimodal distribution of the hybrid performance for leaf rust severity
The 1574 hybrids and their 133 parents were phenotyped for leaf rust severity in 5 environments, that is year × location combinations (Table 1). Correlations between the performances of different environments ranged between 0.37 and 0.89, which suggests the existence of genotype × environment interaction effects influencing leaf rust severity. According to these estimates, the most abrupt changes in phenotype rankings are expected for HHOF2012 when compared with ROS2012 and ROS2013. We adjusted for the effects of environments and obtained best linear unbiased estimations (BLUEs) for the 1707 genotypes. The BLUEs were widely distributed ranging from 0.5 to 7.4 for leaf rust severity assessed by using a 1 (fully resistant) to 9 (fully susceptible) scoring scale (Figure 2a). The heritability amounted to 0.81 for hybrids and 0.82 for parent lines, while variation due to general combining abilities (GCAs) was 16.12 times the variance attributed to specific combining abilities (SCAs) (Table S4). Overall, the male parents were more susceptible than the female parents, while the severity of hybrids followed a bimodal distribution (Figure 2a). We decided to look in more detail into this bimodal distribution and split the hybrids into two subpopulations according to the resistance of the female parents: The hybrids based on crosses involving the top 25% resistant female parents form the Top25% subpopulation and the hybrids from the remaining 75% female parents formed the Inferior75% subpopulation. In this regard, the BLUEs of these two subpopulations followed two different normal distributions with a mean of 1.42 and 3.61, respectively (Figure 2b). This trend was also visible – but less pronounced – when splitting the population into a Top50% and Inferior50% subpopulation. Interestingly, we observed much more pronounced average midparent heterosis for Top25% with −39.5% compared to 4.69% for Inferior75% (Figure 2c). Hierarchical cluster analysis was used to search for genetic similarities among the parents of the Top25% and Inferior75% population. The phylogenetic tree revealed a tendency for female parents from Top25% to cluster together, with the exceptions of lines F004, F011, F021, F029, F059, F086, F098, F100, F101 and F112 (Figure 2d).
Table 1.
Correlations among environment‐specific and across‐environment best linear unbiased estimations of leaf rust severity scores of a hybrid population (1574 hybrids plus their 118 female and 15 male parent lines) tested in five environments
| Correlation | HAD2012 | HHOF2012 | ROS2012 | ROS2013 | BLUEs |
|---|---|---|---|---|---|
| BOH2012 | 0.66 | 0.45 | 0.71 | 0.71 | 0.89 |
| HAD2012 | 0.42 | 0.52 | 0.52 | 0.75 | |
| HHOF2012 | 0.38 | 0.37 | 0.59 | ||
| ROS2012 | 0.70 | 0.87 | |||
| ROS2013 | 0.85 |
Figure 2.
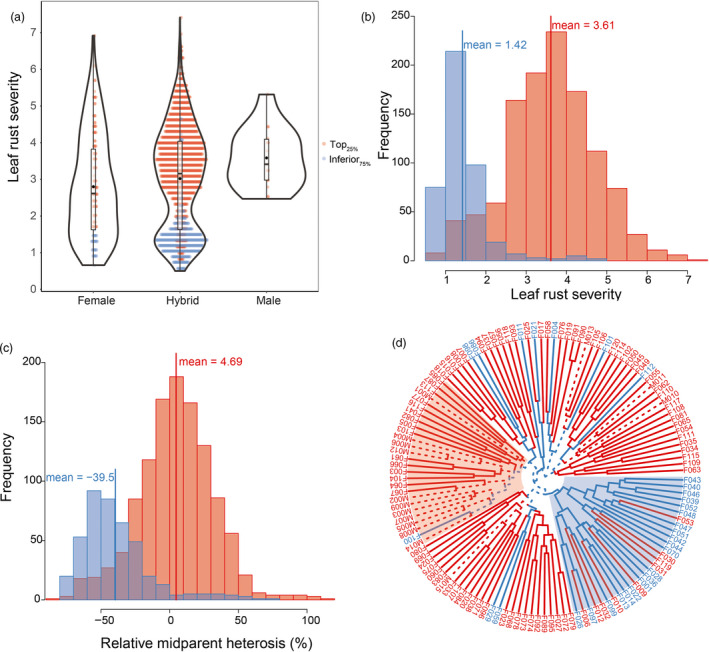
Bimodal distribution of leaf rust severity in a wheat population of 1574 hybrids plus their 118 female and 15 male parent lines. Genotypes from Top25% and Inferior75% subpopulations are indicated in blue and red, respectively. (a) Leaf rust severity scores (1 = fully resistant and 9 = fully susceptible) shown according to the different groups (females, hybrids and males). (b) Histogram of leaf rust severity scores of hybrids. (b) Histogram of midparent heterosis. (d) Dendrogram constructed by performing hierarchical clustering based on the pairwise Rogers’ distance matrix among 133 parent genotypes calculated using exome capture single‐nucleotide polymorphisms profiles. Solid and dashed lines represent the female and male parents, respectively.
Detection of subpopulation specific marker‐trait associations pointing to known resistance genes
Using a mixed linear model to correct for population stratification, we firstly performed association mapping for leaf rust in the total population (Figure 3a, d). In this scan, 1565 SNPs exceeded the significant threshold of −log10(P‐value) = 5.44, defined by applying the multiple‐test correction suggested by Gao (Gao et al., 2008; Figure S8). The 1565 SNPs trace back to 45 independent loci. The most significant SNPs were located on chromosome 4A and mapped as close as 7.2 Mb from the previously described resistance gene Lr34‐B; a homolog of Lr34 (Krattinger et al., 2011). Lr34 maps on chromosome 7D (Dakouri et al., 2010; Dyck, 1987) and functions in adult plants by encoding an ATP‐binding cassette (ABC) transporter. Interestingly, Lr34‐B does not map on chromosome 7B as it would be expected due to chromosome homology, which is explained by a translocated segment from 7BS to the 4A chromosome in Chinese Spring (Krattinger et al., 2011). In addition to Lr34‐B, several significantly associated loci on chromosome 1D mapped approximately 0.4Mb away from another previously described causal gene: Lr21 (Huang et al., 2009; Figure 3d, Table 3).
Figure 3.
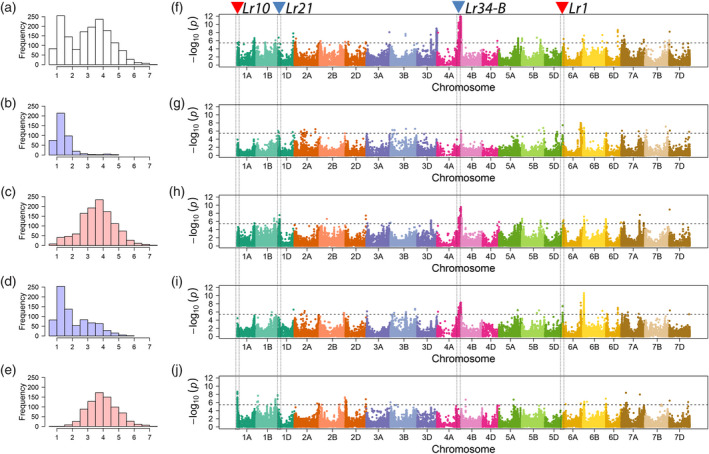
Genome‐wide exome association scans for additive effects underlying leaf rust severity in a hybrid wheat population and its different subpopulations. Left panel: Histogram of leaf rust severity in: (a) a hybrid wheat population of 1574 hybrids plus their 118 female and 15 male parent lines and its subpopulations (b) Top25%, (c) Inferior75%, (d) Top50% and (e) Inferior50%. Right panel: Manhattan plots of genome‐wide exome association scans for additive effects underlying leaf rust severity in: (f) total population, (g) subpopulation Top25%, (h) Inferior75%, (i) Top50% and (j) Inferior50%. –log10(P‐value)s of the significance test are plotted against physical positions on chromosome. Black horizontal dashed lines indicate the genome‐wide multiple test corrected significance threshold for association analysis. The candidate region of Lr10, Lr21, Lr1 and Lr34‐B homologous gene of Lr34 is marked with vertical dashed lines and triangles. Blue triangles mean that these loci were detected in total population and subpopulations, while red triangles mean those are only significant in subpopulations.
Table 3.
Significantly associated single‐nucleotide polymorphisms (SNP)s from exome capture sequencing that map closest to already known leaf rust resistant genes and located within a 10 Mb window away from the known candidate gene
| Population | SNP | Chromosome | Position (bp) | P‐value a | Geneb | Distance (Mb) |
|---|---|---|---|---|---|---|
| Total | S15_2077073 | 1D | 2 077 073 | 1.88E‐07 | Lr21 | 0.4 |
| Total | S4_669444522 | 4A | 669 444 522 | 3.86E‐11 | Lr34‐B | 7.2 |
| Top25% | S15_2077073 | 1D | 2 077 073 | 1.95E‐06 | Lr21 | 0.4 |
| Top25% | S19_554396794 | 5D | 554 396 794 | 3.76E‐08 | Lr1 | 7.5 |
| Inferior75% | S15_99767 | 1D | 99 767 | 2.80E‐06 | Lr21 | 2.4 |
| Inferior75% | S4_669441424 | 4A | 669 441 424 | 1.09E‐07 | Lr34‐B | 7.1 |
| Inferior75% | S19_560500848 | 5D | 560 500 848 | 1.94E‐06 | Lr1 | 1.4 |
| Inferior75% | S7_55412966 | 7A | 55 412 966 | 3.21E‐06 | Lr34‐B | 5.4 |
| Top50% | S4_669444182 | 4A | 669 444 182 | 7.17E‐08 | Lr34‐B | 7.2 |
| Top50% | S19_554396794 | 5D | 554 396 794 | 3.46E‐08 | Lr1 | 7.5 |
| Inferior50% | S1_3668532 | 1A | 3 668 532 | 2.45E‐09 | Lr10 | 5.9 |
We explored whether the phenotypic structure of our mapping population – as indicated by the bimodal distribution of the phenotypic values – has an effect on our association mapping results (Figure 3a). To do so, we divided the total population into resistant (Top25%, Top50%) and susceptible subpopulations (Inferior75%, Inferior50%) as outlined in detail above (Figure 3). The amount of markers differed in the subpopulations owing to the quality control of minor allele frequency (MAF, Table 2) with following ranking: Total > Inferior75% > Top50% ≈ Inferior50% > Top25%. Among the significant SNPs that were detected in the total population, 429 (27.4%) were detected again as significantly associated in at least one of the four subpopulations (Figure S8). In particular, SNP S15_2077073, which is located proximal to Lr21, was significant again in the subpopulation Top25% while significant associations for loci mapping as close as 7.1 Mb away from Lr34‐B were also successfully identified in subpopulation Inferior75% and Top50% (Table 3 and Figure 3h, j). Interestingly, for both of the resistant subpopulations Top25% and Top50%, we detected new marker‐trait associations mapping as close as 1.4 Mb away from the known leaf rust resistance gene Lr1 (Cloutier et al., 2007), which is located on chromosome 5D (Figure 3j, i). Moreover, a strong marker‐trait association on chromosome 1A was exclusively observed in the subpopulation Inferior50%. The SNPs of this region were located 5.9 Mb away from the CC‐NBS‐LRR type resistant gene Lr10 (Feuillet et al., 2003; Table 3).
Table 2.
Composition, size and number of informative exome capture sequencing single‐nucleotide polymorphisms (SNPs) of each population/subpopulation for genome‐wide association analysis
| Female | Male | Hybrids | SNPs | |
|---|---|---|---|---|
| Total population | 118 | 15 | 1574 | 202 875 |
| Top25% subpopulation | 30 | 15 | 425 | 162 327 |
| Inferior75% subpopulation | 88 | 15 | 1149 | 191 377 |
| Top50% subpopulation | 59 | 15 | 788 | 180 942 |
| Inferior50% subpopulation | 59 | 15 | 786 | 184 498 |
| Validation population | 24 | 16 | 128 | 112 587 |
Validation of marker‐trait associations in an independent population
We used an independent wheat population, further denoted as validation set that comprised 128 hybrids from crosses between 24 female and 16 male parents to validate the detected marker‐trait associations. Genomic data for the validation set were obtained again by exome capture sequencing, resulting in 129 818 SNPs with MAF larger or equal than 0.05 and missing rate smaller than 0.05. Out of the 1565 SNPs presenting significant marker‐trait associations in the mapping population comprising the 1707 wheat genotypes, 466 were polymorphic in the independent validation set. These polymorphic SNPs reflect 15 independent loci. From the 466 SNPs, 23 were significant in the validation set at a threshold of P < 0.01 after applying the method for correction for multiple testing suggested by Gao et al. (2008). Marker effects for the 23 SNPs were estimated in the population of the 1,707 hybrids and used to predict the leaf rust severity of the hybrids in the validation set. The predicted and observed phenotypic values were significantly (P‐value = 2.676 × 10−7) correlated with a Pearson correlation of 0.384.
Independent validation facilitates to narrow down the list of putative resistance genes
We detected in the population of 1707 genotypes, a pronounced peak spanning a 25 Mb region on chromosome 3D (590–615 Mb). The SNPs were in high LD, which makes the identification of the underlying candidate gene difficult (Figure S9a). Interestingly, the diversity and pattern of LD among SNPs in this region were different in the validation set (Figure S9b), which allowed us to narrow down the list of candidate SNPs to 6 SNPs that were significant in the validation set. Within the 1Mb candidate region (597.1–598.1 Mb), four protein‐coding genes (TraesCS3D01G513000, TraesCS3D01G513200, TraesCS3D01G513400 and TraesCS3D01G513500) were annotated as potential disease resistance‐related genes and each of them encoded the NB‐ARC domain, an important part of many plant resistance proteins (Van Ooijen et al., 2008). These four genes are a promising target for further functional validation strategies such as virus‐induced gene silencing or overexpression.
Another example was the region around 532.5–533.2 Mb on chromosome 1A. We identified 14 SNPs significantly associated with leaf rust severity in the population of 1707 genotypes. In total, 24 genes were located in this candidate region and the LD among SNPs was very high (r 2> 0.65). Interestingly, the extent of LD decreased in the validation set and only 5 out of the 14 SNPs surpassed the significance threshold in the validation set (Figure 4). These SNPs were located at 532.7Mb and were linked with each other (r 2 > 0.84). We detected three genes in this region, which are putatively related with disease resistance. Two of them (TraesCS1A01G345400 and TraesCS1A01G345500) were annotated with protein kinase activity and the remaining one (TraesCS1A01G345600) encodes the LRR receptor‐like serine/threonine‐protein kinase domain. In more detail, annotated genes TraesCS1A01G345400 and TraesCS1A01G345500 presented SNPs with significant associations in both populations, while TraesCS1A01G345600 has no significant SNPs in any of the studied populations (Figure S18). Similarly, as for the narrowed‐down region on chromosome 3D, this region on chromosome 1A carries promising targets for further detailed validation studies.
Figure 4.
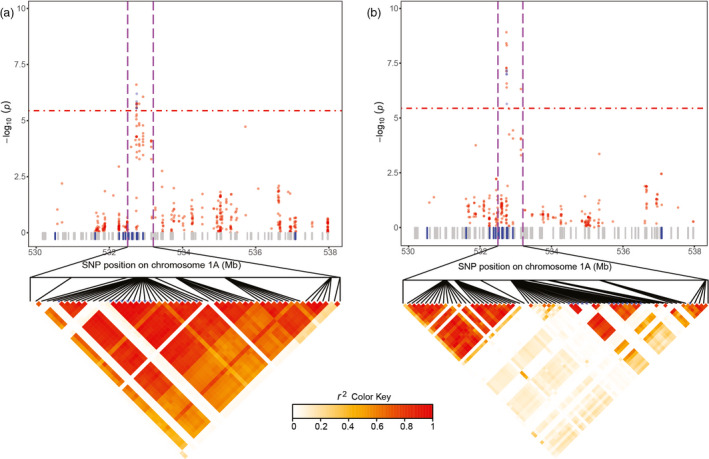
Candidate region associated with leaf rust resistance on chromosome 1A in a hybrid wheat population and narrowed down using an independent validation population. Manhattan plots showing the significant exome associations for additive effects underlying leaf rust within a candidate region on chromosome 1A found in: (a) a hybrid wheat population of 1574 hybrids plus their 118 female and 15 male parent lines and (b) a validation population of 128 hybrids plus their 24 female and 16 male parent lines. –log10(P‐value)s of the significance test of additive effects are plotted against physical positions on chromosome 1A. Red horizontal dot‐dashed lines indicate the multiple test‐corrected significance thresholds for association analysis. Single‐nucleotide polymorphisms (SNPs) significantly associated in the two data sets are shown as blue points and other SNPs are shown as red points. The genes with annotated resistance function and others are shown as vertical boxes in blue and grey, respectively. The upper‐triangular halves of the linkage disequilibrium (LD, as r 2) matrices between SNPs within the candidate region are shown as heat maps below Manhattan plots. Blue stars in LD plots indicate the physical positions of SNPs with significant associations.
Discussion
Exploiting environmentally stable QTL for durable resistance breeding
Our study showed that the phenotypic variation of leaf rust resistance is influenced by major and minor‐effect loci (Figure S10) with the most important major effect loci located on chromosome 4A proximal to Lr34‐B. Thus and as often suggested by several authors (Nelson et al., 2018), a strategy combining major‐ and minor‐effect genes could provide durable resistance. Moreover, resistance loci, especially those with major effects, should ideally provide race nonspecific resistance, because this type of resistance has proven to be longer lasting as compared to the race‐specific one (Nelson et al., 2018). For instance, the Lr34 gene has provided resistance against several pathogens, including leaf rust, stem rust, stripe rust and powdery mildew, for over 100 years (Moore et al., 2015). Obviously, whether or not these loci would provide durable resistance will be determined by the evolution of leaf rust populations in the field, which is, to a greater extent, determined by good and bad practices of integrated pest management used by wheat growers (Mundt, 2014). Nonetheless, even though the term durability can only be defined in a retrospective fashion (Nelson et al., 2018), analysing the environmental stability of marker‐trait associations may give some insights into it. At this stage, loci whose associations are environmentally unstable are obviously too risky to be used in marker‐assisted selection. Thus, we fitted a multiple linear regression model for each marker on the environmental BLUEs of genotypes that included main environment and marker effects plus their interactions. Interestingly, these analyses revealed that only for 3.4 % of the associated loci, the marker by environment interaction components explained an equal or higher amount of variation on leaf rust severity as compared to the main locus effects (Figure S12). On the other hand, the most environmentally stable locus was located in the 4A QTL region, with main effects explaining 13.6‐fold the amount of variation explained by the interaction components. Moreover, this QTL region harboured 6 additional highly environmentally stable loci, with main effects explaining at least 10 times the amount of leaf rust severity variation attributed to their interactions with the environment. Considering that we could not find significant associations for SNPs directly targeting Lr34‐B in our population, our results point to potentially new sources of resistance mapping on chromosome 4A. In fact, some of these strongly associated loci were located within annotated genes with putative disease resistance activity (Table S5). Interestingly, by sorting SNPs according to the physical map positions of the reference sequence of Chinese Spring, associated loci portrayed two regions on chromosome 4A influencing leaf rust resistance, whose peaks were separated by 43 Mb (Figure S11). This observation was surprising considering the very high LD (r 2 = 0.9) between both peaks. One plausible explanation for this discrepancy is a lack of structural collinearity between the reference genome and those of our studied population due to insertions and deletions, translocations, among other genome rearrangements (Dvorak et al., 2018; Helguera et al., 2015; Thind et al., 2018). Nonetheless, reverse and forward genetic techniques would be necessary to elucidate if these highly promising loci mapping on chromosome 4A confer new sources of resistance against leaf rust or, to the contrary, belong actually to Lr34‐B.
Data mining broadened insights into the gene portfolio currently used in wheat breeding in Central Europe
Factors influencing the statistical power for QTL detection in association mapping are very well known (Myles et al., 2009). Although the size of the association mapping population is certainly the key factor influencing statistical power, the ability to detect true QTL signals can also be improved by a decreased correlation between genetic and phenotypic similarity as well as by increased frequencies of rare alleles at functional loci. In this respect, our strategy of subdividing the total hybrid population into four different subpopulations based on the bimodal leaf rust severity distribution of parents decreased the correlation between genetic and phenotypic distances (rRD,PD) in subpopulations Inferior75%, Top50% and Inferior50% as compared to that of the total population (Table S3). Therefore, a general increment in QTL detection power was expected for these three subpopulations, obviously, at expenses of the general power achieved by an increased size in the total population. Moreover, some associations found in the proximity of already known resistance loci such as Lr1 and Lr10 were only detected in subpopulations (Figure 3, Table 3), which further highlights the advantage of our strategy. In addition, analysing subpopulations improved QTL detection power by increasing MAF in some cases. For instance and compared to the total population, −log10(P‐value)s of loci on chromosome 6A were higher in subpopulations Top25% and Top50% (Figure 3g, i and Figure S15), while MAF of most of these loci was concomitantly higher in both subpopulations. A similar observation was done for loci on chromosome 6B in the Top50% subpopulation (Figures 3i and S14). Nonetheless, there were some increases in QTL detection power in subpopulations whose causal factors could not be elucidated. For example, associations in the proximity of Lr10 were only detected in the Inferior50% subpopulation. However, neither the differences in MAF (Figures 3j and S17), nor the changes in rRD,PD nor the presence/absence of a confounding genetic background (i.e. when genetic distances are more/less correlated with those distances portrayed by associated loci; Table S3), can explain the improved detection ability for this QTL in this particular subpopulation (Figure 3f‐j). Although some driving forces underlying the improved QTL detection ability remain hidden for some subpopulations, the combined analysis of a large population plus its subpopulations increased the number of associations by ~37% compared to the total population (Figure S8), thus providing a robust strategy for QTL detection in our study.
Exploiting dominance effects through hybrid wheat breeding
The quantitative inheritance of leaf rust resistance is predominantly of additive nature, although past studies have shown that dominance effects at some loci also contribute to the genetic variation (Ahamed et al., 2004; Jacobs and Broers, 1989; Navabi et al., 2003). The additive nature is also supported by the associations for leaf rust severity detected by our approach, with 2151 (45 independent loci, 90%) being of additive type (Figure 3f‐j). Despite this, 20 (5 independent loci, 10%) prominent dominance association effects were detected. Among them, a locus on chromosome 5A appeared as highly significant (−log10(P‐value) = 8) in the Top25% subpopulation (Figure S13). In this respect, hybrid breeding provides a straightforward manner for their exploitation. This is in particular attractive considering that midparent heterosis reached desired negative values of up to −82.89% (Figures 2c, S14).
Prospects of exome sequencing association studies in resistance breeding: a critical view
Association mapping has gained much popularity in the plant breeding community because it provides a very straightforward and cheap way to discover new marker‐trait associations that could be exploited by means of marker‐assisted selection. Nevertheless, true genetic linkage to the functional causal variant(s) underlying trait(s) is not always the cause of marker‐trait association(s). In this respect, linkage disequilibrium decay between associated marker(s) and functional variant(s) in the material under selection will reduce the efficiency of marker‐assisted selection (Lande and Thompson, 1990). Theoretically, one way to overcome this limitation is to focus on protein‐coding regions (Hayes and Szucs, 2006), that is genes, by relying on targeted sequence methods such as exome capture. In this sense, whole‐exome association mapping has proven to be beneficial for dissecting human diseases in the past (Carter et al., 2014; Guo et al., 2018; Kim et al., 2012) and lately, exome association mapping has been also conducted using plant populations (He et al., 2019; Henry et al., 2014; Looseley et al., 2017; Pont et al., 2019; Russell et al., 2016). For instance, exome capture revealed regions containing genes that are associated to traits involved in adaptation and also subjected to selection due to domestication and plant breeding in a population composed of 487 genotypes of wheat and related species (Pont et al., 2019). Taking into account the size of our association mapping population, we expected to find associations that were narrowed down to the level of true functional associations. It is important to consider that due to the presence/absence nature of genetic variants conferring resistance (Arora et al., 2019), insertions and deletions may play a central role when detecting candidate resistance genes based on a reference genome. In this sense, our candidate search was confined to those resistance genes annotated for Chinese Spring. In a first step, we considered already known Lr genes (Table 3) as a kind of proof‐of‐concept and in the best case; we were able to approach Lr genes as close as 0.4 Mb. As discussed for the Lr34‐B on chromosome 4A; a lack of structural collinearity with the reference genome can explain an imprecise mapping of known leaf rust resistance loci and their associations in our mapping population. In addition, limited allelic variation and recombination rates are also plausible causes for this restricted ability. For instance, even though 10 SNPs were found and tested for significant associations within Lr1, their low MAF values (MAF = 0.05–0.08) limited our detection ability. Particularly, this gene was presumably almost fixed in all parent lines due to selection and breeding. We were able to overcome some of these challenges by using an independent population as a way to narrow down and statistically validate new associations. This strategy allowed us to increase mapping precision by, for example, narrowing down an extensive 25‐Mb region on chromosome 3D found as associated in the population of 1707 genotypes to a 1‐Mb region in the validation population (Figure S8). Despite this and partly because regions harbouring SNPs in strong LD and with similar allele frequency result in similar P‐values in association mapping, we were not able to validate and narrow down associations to the level of one single gene. This last ability may also be limited in our study by the overall low gene coverage of exome capture (Figure S2, Table S1). On the other hand and due to the evolution of resistance genes, many of them lie together within clusters of highly linked resistance genes (Dilbirligi et al., 2004; Liu et al., 2015); an issue that clearly challenges the detection of single functional variants. At this stage, we expect that deep next‐generation sequencing approaches or whole genome sequencing can shed some light into this issue. Alternatively, if the goal is the detection of new functional genes with resistance activity against biotrophic fungi, RenSeq approaches (Steuernagel et al., 2018) targeting sequences such as nucleotide‐binding site–leucine‐rich repeats (NB‐LRRs) with known participation in plant resistance should be also appropriate.
Experimental procedures
Plant material and phenotypic data analyses
The phenotypic data are based on 1749 wheat genotypes including 10 checks, 1604 F1 factorial hybrids, and their 120 female and 15 male parental lines (Longin et al., 2013; Zhao et al., 2013). Leaf rust disease severities were evaluated based on natural infection or deliberate inoculation in four locations (Böhnshausen, Hadmersleben, Harzhof and Rosenthal) in 2012 as described in detail elsewhere (Gowda et al., 2014; Longin et al., 2013). An additional field trial was conducted based on natural infection in Rosenthal during 2013 using the same experimental procedures. The leaf rust disease severities were scored at the date of flowering on the flag leaf using a scale from 1 (fully resistant) to 9 (fully susceptible) referring to the guidelines of the German Federal Plant Variety Office (Bundessortenamt, 2000).
Best linear unbiased estimations (BLUEs) of genotypes, variance components, and broad‐sense heritabilities for parent lines and hybrids were estimated as outlined in detail elsewhere (Zhao et al., 2015).
Genotypic data and diversity analyses
The 135 elite parental lines were genotyped with exome capture sequencing using an Illumina HiSeq 2500 platform. Sequencing data were mapped to the reference genome of Chinese Spring (Appels et al., 2018). This landrace is susceptible against leaf rust at the seedling stage (Li et al., 2010), but carries adult plant resistance (Dyck, 1991; Kerber and Aung, 1999). Details of the bioinformatics pipelines used for read mapping and variant calling are described in a previous study (Milner et al., 2019). Briefly, BWA‐MEM (Li, 2013) and SAMtools (Li et al., 2009) were used to align reads to the reference sequence and convert them to binary format (BAM), respectively. GATK (DePristo et al., 2011; version v3.8) was applied to realign reads near indels. Variant calling was performed with the SAMtools/BCFtools pipeline (version 1.6; Li, 2011).
A custom awk script was used for gentle filtering of variants, retaining VCF file entries with a minimum number of reads set to one for homozygous and two for heterozygous calls, respectively. Minimum SNP quality was set to 40. The resulting VCF file was imported into R statistical environment (version 3.4.3) for further filtering. Applying the SeqArray package (Zheng et al., 2017) we set polymorphisms detected on chrUn to missing and filtered remaining SNPs for a minor allele count of at least one and a minimum number of present calls of 0.05. Missing genotype calls were imputed with FILLIN (Swarts et al., 2014) from the TASSEL5 (Bradbury et al., 2007) software suit.
After imputation with FILLIN (Swarts et al., 2014), only bi‐allelic variants with MAF ≥ 0.05 and missing rate < 0.05 were used for subsequent analyses. Following quality control, two female parental lines were excluded, resulting in SNP profiles for 118 female and 15 male parental lines. The genotypes of 1574 hybrids were derived from the genotypes of their parental lines. Prediction of the functional effect was performed with the tool SnpEff version 4.3 (Cingolani et al., 2012) based on the IWGSC_v1.1 gene models of high confidence. Nucleotide diversity π was calculated with 1Mb non‐overlapping sliding window using the software vcftools version 0.1.12b (Danecek et al., 2011). The linkage disequilibrium (LD) of each chromosome was calculated using the r 2 statistic (Hill and Robertson, 1968). We applied a non‐linear regression model described by Hill and Weir (Hill and Weir, 1988) to estimate the LD decay. We used the average r 2 of all SNPs within the same gene to represent LD of genes. LD within a specific genomic region was calculated based on parental lines and visualized with the R package LDheatmap (Shin et al., 2006). Principal coordinate analysis and hierarchical cluster analysis were performed based on pairwise Rogers’ distances among genotypes (Reif et al., 2005). In a previous study (Wurschum et al., 2013), parents were characterized by using a 90K Infinium SNP chip (Wang et al., 2014) and pairwise Rogers’ distances based on these marker profiles were also considered for comparative purposes. These analyses were performed within R environment (version 3.4.3).
Genome‐wide association mapping
Genome‐wide association mapping was implemented in R environment (version 3.4.3) using a linear mixed model considering additive and dominance effects (Zhao et al., 2013). In brief, the model can be described as follows:
| (1) |
where y are the observed phenotypic values, 1 n corresponds to a n‐length vector of ones, µ denotes a common intercept term, a and d represent the additive and dominance effect of the tested SNP, respectively, while A and D stand for the design matrices relating y to a an d, correspondingly, g is a vector of genotypic effects or polygenic background effects and e indicates the error term of the model. For each tested SNP, genotypes homozygous for the first allele, heterozygous and homozygous for the alternative allele were coded as −1, 0 and 1, respectively, in the case of the A matrix. In the D matrix, homozygous and heterozygous genotypes were coded as 0 and 1, respectively. For the model (1), we assumed that µ, a and d are fixed factors, while g and e were considered random, with and , where K is a marker‐derived kinship matrix, I is an identity matrix, and are the corresponding variance components. Each kinship coefficient between two parent genotypes within K was computed as twice the difference of one minus the corresponding Rogers’ distance (Reif et al., 2005) between them. In the case of hybrids, the additive polygenic background is decomposed as the sum of the general combining abilities of female () and male () parents, thus, the kinship matrices for hybrids model the covariance among GCA effects of the respective female and male parents (Bernardo, 1994). Linear mixed models for the phenotypic data analyses as well as for association mapping were solved using the ASReml‐R package (Butler et al., 2009).
Validation of marker‐trait associations in an independent population
The validation population is a fraction of another large hybrid wheat population, which consists of 41 male lines, 189 female lines and their 1815 single‐cross hybrids produced using an incomplete factorial mating design. The 230 parental lines were tested together with 1815 hybrids and 11 commercial check varieties in 7 environments in un‐replicated field trials based on an alpha design with block size equals to 11 plots. Infection of genotypes with leaf rust occurred naturally and was scored at the date of flowering on the flag leaf as described in detail above.
Across environments, BLUEs of lines and hybrids from validation population were obtained as outlined in detail elsewhere (Zhao et al., 2015). For 24 female and 16 male parental lines, exome capture data were obtained as already detailed in the ‘Genotypic data and diversity analyses’ section. The genotypes of hybrids were deduced according to the genotypes of their parents. The 40 lines served as parents for 128 hybrids, which were denoted in the following as the validation population.
To validate the significant SNPs found in the population of the 1707 genotypes (133 parental lines and 1574 hybrids), we first identified common markers between the two populations. Then, all the common markers were used to predict the phenotype of lines in the validation population using a linear model, in which the markers were sorted by physical position on the chromosome. Finally, we calculated the Pearson correlation coefficient between the predicted and observed phenotypic values.
Narrow down candidate regions combining information of the two populations and identification of candidate genes
For the novel candidate regions, we used all the markers in those regions that were found in the validation population and performed association tests based on a linear regression model. The detected potential disease resistance‐related genes (R genes) were annotated with the pipeline RGAugury (Li et al., 2016). R genes were classified as CC (coiled‐coil), NBS (nucleotide‐binding site), CN (CC‐NBS), NL (NBS‐LRR), CNL (CC‐NBS‐LRR), RLK (receptor‐like kinase), RLP (receptor‐like protein) and TM‐CC (Transmembrane‐CC). Moreover, we double‐checked the functional annotation of these genes in the candidate region IWGSC (Appels et al., 2018).
Funding
FL was supported with funds from China Scholarship Council (CSC; Grant no. 201606760065). The experimental work of the validation set as well as YJ and PT were supported by the German Federal Ministry of Food and Agriculture within the ZUCHTWERT Project (Grant FKZ0103010). The Federal Ministry of Education and Research of Germany is acknowledged for funding AWS (Grant no. FKZ031B0184B).
Conflict of interest
The authors declare that they have no conflict of interest and the experiments comply with the current laws of Germany.
Author contributions
YZ and JCR designed the study; MG generated genomic data and SB processed it; PT, YZ and FL curated phenotypic and genomic data; FL performed the analyses with the support of YZ and YJ; FL and AWS wrote the paper with input from all co‐authors.
Supporting information
Figure S1 Histogram of the mean depth (number of unique reads) of exome capture sequencing.
Figure S2 Histogram of the number of single‐nucleotide polymorphisms per gene captured by exome sequencing.
Figure S3 Comparison between Rogers’ distances calculated using exome capture sequencing (RDEC) and the 90K SNP array data set (RD90K_SNP) for the 133 parents of the population composed by 1574 wheat hybrids.
Figure S4 Number of single‐nucleotide polymorphisms per wheat chromosome captured by exome sequencing.
Figure S5 Genome‐wide distribution of average exome nucleotide diversity.
Figure S6 Linkage disequilibrium (r 2) as a function of physical distance (Mb) for the three different wheat subgenomes.
Figure S7 Percentual distribution of marker pairs in each of the three wheat genomes as a function of the physical distance (Mb).
Figure S8 Venn diagram showing the amount of overlapping associations for leaf rust severity found by genome‐wide exome association scans in a hybrid wheat population and its corresponding subpopulations.
Figure S9 Extensive genomic region associated with leaf rust resistance on chromosome 3D detected by a genome‐wide exome association scan for additive effects in a vast hybrid wheat population and narrowed down to a smaller candidate region using an independent validation population.
Figure S10 Histogram of the percentages of genetic variance (R2/h) explained by single‐nucleotide polymorphisms found as associated with leaf rust severity in a hybrid wheat population of 1574 hybrids plus their 118 female and 15 male parent lines.
Figure S11 Close‐up view of the two association peaks within a candidate region on chromosome 4A found by a genome‐wide exome association scan for additive and dominance effects underlying leaf rust severity in a hybrid wheat population of 1574 hybrids plus their 118 female and 15 male parents.
Figure S12 Histogram of the ratio of phenotypic variation explained by main additive and dominance effects () over that explained by interaction effects with environments () of single‐nucleotide polymorphisms found as associated with leaf rust severity in a hybrid population (1574 hybrids plus their 118 female and 15 male parent lines) tested in five environments.
Figure S13 Genome‐wide exome association scans for dominance effects underlying leaf rust severity in a vast hybrid wheat population and its different subpopulations.
Figure S14 Distribution of leaf rust severity scores in the validation population.
Figure S15 Some general statistics in a vast hybrid wheat population and its different subpopulations regarding associations mapping on chromosome 6A.
Figure S16 Some general statistics in a vast hybrid wheat population and its different subpopulations regarding associations mapping on chromosome 6B.
Figure S17 Some general statistics in a vast hybrid wheat population and its different subpopulations regarding associations mapping on chromosome 1A.
Figure S18 Candidate genes underlying significant associations on chromosome 1A.
Table S1 Functional location and type of substitution (synonymous and non‐synonymous) within the coding sequence for single‐nucleotide polymorphisms (SNPs) present in a hybrid wheat population of 1574 hybrids plus their 118 female and 15 male parent lines.
Table S2 Estimated average physical distance (Mb) according to different wheat chromosomes where the linkage disequilibrium (r 2) fell below r 2 = 0.2 in a hybrid wheat population of 1574 hybrids plus their 118 female and 15 male parent lines
Table S3 Expected impact of population stratification during genome‐wide exome association scans for leaf rust severity on associations of single‐nucleotide polymorphisms (SNPs) mapping close to Lr10 in a hybrid population of 1574 hybrids plus their 118 female and 15 male parents and its subpopulations Top25%, Inferior75%, Top50% and Inferior50%.
Table S4 Estimates of variances components and heritabilities (h 2) for leaf rust severity in a hybrid wheat population composed by 1,574 hybrids produced by factorial crosses between 118 female (F) and 15 male (M) parents.
Table S5 Putative genes underlying significant regions on chromosome 4A.
Acknowledgements
We would like to thank Martin Mascher and Beat Keller for their valuable comments that helped us to improve the manuscript. We also want to thank Peter Morrell for his advices for the population genomic analyses.
Liu, F. , Zhao, Y. , Beier, S. , Jiang, Y. , Thorwarth, P. , H. Longin, C. F. , Ganal, M. , Himmelbach, A. , Reif, J. C. and Schulthess, A. W. (2020) Exome association analysis sheds light onto leaf rust (Puccinia triticina) resistance genes currently used in wheat breeding (Triticum aestivum L.). Plant Biotechnol J, 10.1111/pbi.13303
References
- Ahamed, M.L. , Singh, S.S. , Sharma, J.B. and Ram, R.B. (2004) Evaluation of inheritance to leaf rust in wheat using area under disease progress curve. Hereditas, 141, 323–327. [DOI] [PubMed] [Google Scholar]
- Akle, S. , Chun, S. , Jordan, D.M. and Cassa, C.A. (2015) Mitigating false‐positive associations in rare disease gene discovery. Hum. Mutat. 36, 998–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appels, R. , Eversole, K. , Feuillet, C. , Keller, B. , Rogers, J. , Stein, N. , Pozniak, C.J. et al. (2018) Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science, 361, 661. [DOI] [PubMed] [Google Scholar]
- Arora, S. , Steuernagel, B. , Gaurav, K. , Chandramohan, S. , Long, Y. , Matny, O. , Johnson, R. et al. (2019) Resistance gene cloning from a wild crop relative by sequence capture and association genetics. Nat. Biotechnol. 37, 139–143. [DOI] [PubMed] [Google Scholar]
- Bernardo, R. (1994) Prediction of maize single‐cross performance using Rflps and information from related hybrids. Crop Sci 34, 20–25. [Google Scholar]
- Bradbury, P.J. , Zhang, Z. , Kroon, D.E. , Casstevens, T.M. , Ramdoss, Y. and Buckler, E.S. (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics, 23, 2633–2635. [DOI] [PubMed] [Google Scholar]
- Bundessortenamt (2000) Richtlinien für die Durchführung von landwirtschaftlichen Wertprüfungen und Sortenversuchen. Hannover, Germany: Landbuch Verlagsgesellschaft mbH. [Google Scholar]
- Butler, D. , Cullis, B.R. , Gilmour, A. and Gogel, B. (2009) ASReml‐R reference manual. Brisbane: The State of Queensland, Department of Primary Industries and Fisheries. [Google Scholar]
- Carter, S.L. , Brastianos, P.K. , McFadden, D.G. , Papagiannakopoulos, T. , Taylor‐Weiner, A. , Cibulskis, K. , Jacks, T. et al. (2014) Modes of metastasis evolution in human and murine cancer revealed by whole exome sequencing. Cancer Res. 74, 3992. [Google Scholar]
- Chen, X.J. , Min, D.H. , Yasir, T.A. and Hu, Y.G. (2012) Genetic diversity, population structure and linkage disequilibrium in elite Chinese winter wheat investigated with SSR markers. Plos One, 7, e44510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani, P. , Platts, A. , Wang, L.L. , Coon, M. , Nguyen, T. , Wang, L. , Land, S.J. et al. (2012) A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w(1118); iso‐2; iso‐3. Fly, 6, 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloutier, S. , McCallum, B.D. , Loutre, C. , Banks, T.W. , Wicker, T. , Feuillet, C. , Keller, B. et al. (2007) Leaf rust resistance gene Lr1, isolated from bread wheat (Triticum aestivum L.) is a member of the large psr567 gene family. Plant Mol. Biol. 65, 93–106. [DOI] [PubMed] [Google Scholar]
- Dakouri, A. , McCallum, B.D. , Walichnowski, A.Z. and Cloutier, S. (2010) Fine‐mapping of the leaf rust Lr34 locus in Triticum aestivum (L.) and characterization of large germplasm collections support the ABC transporter as essential for gene function. Theoret. Appl. Genet. 121, 373–384. [DOI] [PubMed] [Google Scholar]
- Danecek, P. , Auton, A. , Abecasis, G. , Albers, C.A. , Banks, E. , DePristo, M.A. , Handsaker, R.E. et al. (2011) The variant call format and VCFtools. Bioinformatics, 27, 2156–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo, M.A. , Banks, E. , Poplin, R. , Garimella, K.V. , Maguire, J.R. , Hartl, C. , Philippakis, A.A. et al. (2011) A framework for variation discovery and genotyping using next‐generation DNA sequencing data. Nat. Genet. 43, 491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dilbirligi, M. , Erayman, M. , Sandhu, D. , Sidhu, D. and Gill, K.S. (2004) Identification of wheat chromosomal regions containing expressed resistance genes. Genetics, 166, 461–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvorak, J. , Wang, L. , Zhu, T. , Jorgensen, C.M. , Luo, M.C. , Deal, K.R. , Gu, Y.Q. et al. (2018) Reassessment of the evolution of wheat chromosomes 4A, 5A, and 7B. Theoret. Appl. Genet. 131, 2451–2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyck, P.L. (1987) The association of a gene for leaf rust resistance with the chromosome‐7d suppressor of stem rust resistance in common wheat. Genome, 29, 467–469. [Google Scholar]
- Dyck, P.L. (1991) Genetics of adult‐plant leaf rust resistance in Chinese spring and sturdy wheats. Crop Sci. 31, 309–311. [Google Scholar]
- FAO (2019) Food and agriculture organisation of the United Nations, FAOSTAT, http://www.fao.org/faostat/en/#data
- Feuillet, C. , Travella, S. , Stein, N. , Albar, L. , Nublat, A. and Keller, B. (2003) Map‐based isolation of the leaf rust disease resistance gene Lr10 from the hexaploid wheat (Triticum aestivum L.) genome. Proc. Natl. Acad. Sci. USA, 100, 15253–15258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, X.Y. , Stamier, J. and Martin, E.R. (2008) A multiple testing correction method for genetic association studies using correlated single nucleotide polymorphisms. Genet. Epidemiol. 32, 361–369. [DOI] [PubMed] [Google Scholar]
- Gao, L.L. , Turner, M.K. , Chao, S.M. , Kolmer, J. and Anderson, J.A. (2016) Genome wide association study of seedling and adult plant leaf rust resistance in elite spring wheat breeding lines. PLoS ONE, 11, e0148671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong, J.Y. , Miao, J.S. , Zhao, Y. , Zhao, Q. , Feng, Q. , Zhan, Q.L. , Cheng, B.Y. et al. (2017) Dissecting the genetic basis of grain shape and chalkiness traits in hybrid rice using multiple collaborative populations. Mol. Plant, 10, 1353–1356. [DOI] [PubMed] [Google Scholar]
- Gowda, M. , Zhao, Y. , Wurschum, T. , Longin, C.F. , Miedaner, T. , Ebmeyer, E. , Schachschneider, R. et al. (2014) Relatedness severely impacts accuracy of marker‐assisted selection for disease resistance in hybrid wheat. Heredity (Edinb), 112, 552–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, W. , Zhu, X.H. , Yan, L.Y. and Qiao, J. (2018) The present and future of whole‐exome sequencing in studying and treating human reproductive disorders. J. Genet. Genom. 45, 517–525. [DOI] [PubMed] [Google Scholar]
- Hayes, P. and Szucs, P. (2006) Disequilibrium and association in barley: Thinking outside the glass. Proc. Natl. Acad. Sci. USA, 103, 18385–18386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, F. , Pasam, R. , Shi, F. , Kant, S. , Keeble‐Gagnere, G. , Kay, P. , Forrest, K. et al. (2019) Exome sequencing highlights the role of wild‐relative introgression in shaping the adaptive landscape of the wheat genome. Nat. Genet. 51, 896–904. [DOI] [PubMed] [Google Scholar]
- Helguera, M. , Rivarola, M. , Clavijo, B. , Martis, M.M. , Vanzetti, L.S. , Gonzalez, S. , Garbus, I. et al. (2015) New insights into the wheat chromosome 4D structure and virtual gene order, revealed by survey pyrosequencing. Plant Sci. 233, 200–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry, I.M. , Nagalakshmi, U. , Lieberman, M.C. , Ngo, K.J. , Krasileva, K.V. , Vasquez‐Gross, H. , Akhunova, A. et al. (2014) Efficient genome‐wide detection and cataloging of EMS‐induced mutations using exome capture and next‐generation sequencing. Plant Cell, 26, 1382–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill, W.G. and Robertson, A. (1968) Linkage disequilibrium in finite populations. Theoret. Appl. Genet. 38, 226–231. [DOI] [PubMed] [Google Scholar]
- Hill, W.G. and Weir, B.S. (1988) Variances and covariances of squared linkage disequilibria in finite populations. Theor. Popul. Biol. 33, 54–78. [DOI] [PubMed] [Google Scholar]
- Huang, L. , Brooks, S. , Li, W.L. , Fellers, J. , Nelson, J.C. and Gill, B. (2009) Evolution of new disease specificity at a simple resistance locus in a crop‐weed complex: reconstitution of the Lr21 gene in wheat. Genetics, 182, 595–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs, T. and Broers, L.H.M. (1989) The inheritance of host plant effect on latency period of wheat leaf rust in spring wheat. 1. Estimation of gene‐action and number of effective factors in F1, F2 and backcross generations. Euphytica, 44, 197–206. [Google Scholar]
- Juliana, P. , Singh, R.P. , Singh, P.K. , Poland, J.A. , Bergstrom, G.C. , Huerta‐Espino, J. , Bhavani, S. et al. (2018) Genome‐wide association mapping for resistance to leaf rust, stripe rust and tan spot in wheat reveals potential candidate genes. Theoret. Appl. Genet. 131, 1405–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassa, M.T. , You, F.M. , Hiebert, C.W. , Pozniak, C.J. , Fobert, P.R. , Sharpe, A.G. , Menzies, J.G. et al. (2017) Highly predictive SNP markers for efficient selection of the wheat leaf rust resistance gene Lr16. BMC Plant Biol. 17, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerber, E.R. and Aung, T. (1999) Leaf rust resistance gene lr34 associated with nonsuppression of stem rust resistance in the wheat cultivar canthatch. Phytopathology, 89, 518–521. [DOI] [PubMed] [Google Scholar]
- Kertho, A. , Mamidi, S. , Bonman, J.M. , McClean, P.E. and Acevedo, M. (2015) Genome‐wide association mapping for resistance to leaf and stripe rust in winter‐habit hexaploid wheat landraces. PLoS ONE, 10, e0129580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan, M.H. , Bukhari, A. , Dar, Z.A. and Rizvi, S.M. (2013) Status and strategies in breeding for rust resistance in wheat. Agric. Sci. 4, 292. [Google Scholar]
- Kim, J.J. , Park, Y.M. , Baik, K.H. , Choi, H.Y. , Yang, G.S. , Koh, I. , Hwang, J.A. et al. (2012) Exome sequencing and subsequent association studies identify five amino acid‐altering variants influencing human height. Hum. Genet. 131, 471–478. [DOI] [PubMed] [Google Scholar]
- Krattinger, S.G. , Lagudah, E.S. , Wicker, T. , Risk, J.M. , Ashton, A.R. , Selter, L.L. , Matsumoto, T. et al.(2011) Lr34 multi‐pathogen resistance ABC transporter: molecular analysis of homoeologous and orthologous genes in hexaploid wheat and other grass species. Plant J. 65, 392–403. [DOI] [PubMed] [Google Scholar]
- Lande, R. and Thompson, R. (1990) Efficiency of marker‐assisted selection in the improvement of quantitative traits. Genetics, 124, 743–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. (2011) A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics, 27, 2987–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. (2013) Aligning sequence reads, clone sequences and assembly contigs with BWA‐MEM. arXiv preprint arXiv:1303.3997. [Google Scholar]
- Li, Z.F. , Xia, X.C. , He, Z.H. , Li, X. , Zhang, L.J. , Wang, H.Y. , Meng, Q.F. et al. (2010) Seedling and slow rusting resistance to leaf rust in Chinese wheat cultivars. Plant Dis. 94, 45–53. [DOI] [PubMed] [Google Scholar]
- Li, P. , Quan, X. , Jia, G. , Xiao, J. , Cloutier, S. and You, F.M. (2016) RGAugury: a pipeline for genome‐wide prediction of resistance gene analogs (RGAs) in plants. BMC Genom. 17, 852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Handsaker, B. , Wysoker, A. , Fennell, T. , Ruan, J. , Homer, N. , Marth, G. , Abecasis, G. and Durbin, R. and Genome Project Data Processing, S. (2009) The sequence alignment/map format and SAMtools. Bioinformatics, 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Y.Q. , Wu, H. , Chen, H. , Liu, Y.L. , He, J. , Kang, H.Y. , Sun, Z.G. et al. (2015) A gene cluster encoding lectin receptor kinases confers broad‐spectrum and durable insect resistance in rice. Nat. Biotechnol. 33, 301. [DOI] [PubMed] [Google Scholar]
- Liu, J.D. , He, Z.H. , Rasheed, A. , Wen, W.E. , Yan, J. , Zhang, P.Z. , Wan, Y.X. et al. (2017) Genome‐wide association mapping of black point reaction in common wheat (Triticum aestivum L.). BMC Plant Biol. 17, 220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longin, C.F. , Gowda, M. , Muhleisen, J. , Ebmeyer, E. , Kazman, E. , Schachschneider, R. , Schacht, J. et al. (2013) Hybrid wheat: quantitative genetic parameters and consequences for the design of breeding programs. Theoret. Appl. Genet. 126, 2791–2801. [DOI] [PubMed] [Google Scholar]
- Looseley, M.E. , Bayer, M. , Bull, H. , Ramsay, L. , Thomas, W. , Booth, A. , Canto, C.D. et al. (2017) Association mapping of diastatic power in UK Winter and spring barley by exome sequencing of phenotypically contrasting variety sets. Front. Plant Sci. 8, 1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes, M.S. , Dreisigacker, S. , Pena, R.J. , Sukumaran, S. and Reynolds, M.P. (2015) Genetic characterization of the wheat association mapping initiative (WAMI) panel for dissection of complex traits in spring wheat. Theor. Appl. Genet. 128, 453–464. [DOI] [PubMed] [Google Scholar]
- Lowe, I. , Cantu, D. and Dubcovsky, J. (2011) Durable resistance to the wheat rusts: integrating systems biology and traditional phenotype‐based research methods to guide the deployment of resistance genes. Euphytica, 179, 69–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maccaferri, M. , Sanguineti, M.C. , Mantovani, P. , Demontis, A. , Massi, A. , Ammar, K. , Kolmer, J.A. et al. (2010) Association mapping of leaf rust response in durum wheat. Mol. Breed. 26, 189–228. [Google Scholar]
- McCallum, B.D. , Hiebert, C.W. , Cloutier, S. , Bakkeren, G. , Rosa, S.B. , Humphreys, D.G. , Marais, G.F. et al. (2016) A review of wheat leaf rust research and the development of resistant cultivars in Canada. Can. J. Plant Path. 38, 1–18. [Google Scholar]
- Milner, S.G. , Jost, M. , Taketa, S. , Mazon, E.R. , Himmelbach, A. , Oppermann, M. , Weise, S. et al. (2019) Genebank genomics highlights the diversity of a global barley collection. Nat. Genet. 51, 319. [DOI] [PubMed] [Google Scholar]
- Mo, Y.J. , Howell, T. , Vasquez‐Gross, H. , de Haro, L.A. , Dubcovsky, J. and Pearce, S. (2018) Mapping causal mutations by exome sequencing in a wheat TILLING population: a tall mutant case study. Mol. Genet. Genom. 293, 463–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore, J.W. , Herrera‐Foessel, S. , Lan, C.X. , Schnippenkoetter, W. , Ayliffe, M. , Huerta‐Espino, J. , Lillemo, M. et al. (2015) A recently evolved hexose transporter variant confers resistance to multiple pathogens in wheat. Nat. Genet. 47, 1494–1498. [DOI] [PubMed] [Google Scholar]
- Mundt, C.C. (2014) Durable resistance: A key to sustainable management of pathogens and pests. Infect. Genet. Evol. 27, 446–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myles, S. , Peiffer, J. , Brown, P.J. , Ersoz, E.S. , Zhang, Z.W. , Costich, D.E. and Buckler, E.S. (2009) Association mapping: critical considerations shift from genotyping to experimental design. Plant Cell, 21, 2194–2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navabi, A. , Singh, R.P. , Tewari, J.P. and Briggs, K.G. (2003) Genetic analysis of adult‐plant resistance to leaf rust in five spring wheat genotypes. Plant Dis. 87, 1522–1529. [DOI] [PubMed] [Google Scholar]
- Nelson, R. , Wiesner‐Hanks, T. , Wisser, R. and Balint‐Kurti, P. (2018) Navigating complexity to breed disease‐resistant crops. Nat. Rev. Genet. 19, 21–33. [DOI] [PubMed] [Google Scholar]
- Oliver, R.P. (2014) A reassessment of the risk of rust fungi developing resistance to fungicides. Pest Manag. Sci. 70, 1641–1645. [DOI] [PubMed] [Google Scholar]
- Van Ooijen, G. , Mayr, G. , Kasiem, M.M.A. , Albrecht, M. , Cornelissen, B.J.C. and Takken, F.L.W. (2008) Structure‐function analysis of the NB‐ARC domain of plant disease resistance proteins. J. Exp. Bot. 59, 1383–1397. [DOI] [PubMed] [Google Scholar]
- Pont, C. , Leroy, T. , Seidel, M. , Tondelli, A. , Duchemin, W. , Armisen, D. , Lang, D. et al. (2019) Tracing the ancestry of modern bread wheats. Nat. Genet. 51, 905–911. [DOI] [PubMed] [Google Scholar]
- Reif, J.C. , Melchinger, A.E. and Frisch, M. (2005) Genetical and mathematical properties of similarity and dissimilarity coefficients applied in plant breeding and seed bank management. Crop Sci. 45, 1–7. [Google Scholar]
- Russell, J. , Mascher, M. , Dawson, I.K. , Kyriakidis, S. , Calixto, C. , Freund, F. , Bayer, M. et al. (2016) Exome sequencing of geographically diverse barley landraces and wild relatives gives insights into environmental adaptation. Nat. Genet. 48, 1024. [DOI] [PubMed] [Google Scholar]
- Schneeberger, K. (2014) Using next‐generation sequencing to isolate mutant genes from forward genetic screens. Nat. Rev. Genet. 15, 662–676. [DOI] [PubMed] [Google Scholar]
- Sehgal, D. , Autrique, E. , Singh, R. , Ellis, M. , Singh, S. and Dreisigacker, S. (2017) Identification of genomic regions for grain yield and yield stability and their epistatic interactions. Sci. Rep. 7, 41578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin, J.‐H. , Blay, S. , McNeney, B. and Graham, J. (2006) LDheatmap: an R function for graphical display of pairwise linkage disequilibria between single nucleotide polymorphisms. J. Stat. Softw. 16, 1–10. [Google Scholar]
- da Silva, G.B.P. , Zanella, C.M. , Martinelli, J.A. , Chaves, M.S. , Hiebert, C.W. , McCallum, B.D. and Boyd, L.A. (2018) Quantitative trait loci conferring leaf rust resistance in hexaploid wheat. Phytopathology, 108, 1344–1354. [DOI] [PubMed] [Google Scholar]
- Steuernagel, B. , Witek, K. , Krattinger, S.G. , Ramirez‐Gonzalez, R.H. , Schoonbeek, H.‐J. , Yu, G. , Baggs, E. et al. (2018) Physical and transcriptional organisation of the bread wheat intracellular immune receptor repertoire. bioRxiv, 339424. [Google Scholar]
- Sukumaran, S. , Dreisigacker, S. , Lopes, M. , Chavez, P. and Reynolds, M.P. (2015) Genome‐wide association study for grain yield and related traits in an elite spring wheat population grown in temperate irrigated environments. Theoret. Appl. Genet. 128, 353–363. [DOI] [PubMed] [Google Scholar]
- Swarts, K. , Li, H.H. , Navarro, J.A.R. , An, D. , Romay, M.C. , Hearne, S. , Acharya, C. et al. (2014) Novel Methods to Optimize Genotypic Imputation for Low‐Coverage, Next‐Generation Sequence Data in Crop Plants. Plant Genome 7, 3. [Google Scholar]
- Thind, A.K. , Wicker, T. , Muller, T. , Ackermann, P.M. , Steuernagel, B. , Wulff, B.B.H. , Spannagl, M. et al. (2018) Chromosome‐scale comparative sequence analysis unravels molecular mechanisms of genome dynamics between two wheat cultivars. Genome Biol. 19, 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, S.C. , Wong, D.B. , Forrest, K. , Allen, A. , Chao, S.M. , Huang, B.E. , Maccaferri, M. et al. (2014) Characterization of polyploid wheat genomic diversity using a high‐density 90 000 single nucleotide polymorphism array. Plant Biotechnol. J. 12, 787–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winfield, M.O. , Wilkinson, P.A. , Allen, A.M. , Barker, G.L.A. , Coghill, J.A. , Burridge, A. , Hall, A. et al. (2012) Targeted re‐sequencing of the allohexaploid wheat exome. Plant Biotechnol. J. 10, 733–742. [DOI] [PubMed] [Google Scholar]
- Wurschum, T. , Langer, S.M. , Longin, C.F.H. , Korzun, V. , Akhunov, E. , Ebmeyer, E. , Schachschneider, R. et al. (2013) Population structure, genetic diversity and linkage disequilibrium in elite winter wheat assessed with SNP and SSR markers. Theoret. Appl. Genet. 126, 1477–1486. [DOI] [PubMed] [Google Scholar]
- Yano, K. , Yamamoto, E. , Aya, K. , Takeuchi, H. , Lo, P.C. , Hu, L. , Yamasaki, M. et al. (2016) Genome‐wide association study using whole‐genome sequencing rapidly identifies new genes influencing agronomic traits in rice. Nat. Genet. 48, 927. [DOI] [PubMed] [Google Scholar]
- Zhang, K.P. , Wang, J.J. , Zhang, L.Y. , Rong, C.W. , Zhao, F.W. , Peng, T. , Li, H.M. et al. (2013) Association analysis of genomic loci important for grain weight control in elite common wheat varieties cultivated with variable water and fertiliser supply. PLoS ONE, 8, e57853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, Y.S. , Gowda, M. , Wurschum, T. , Longin, C.F.H. , Korzun, V. , Kollers, S. , Schachschneider, R. et al. (2013) Dissecting the genetic architecture of frost tolerance in Central European winter wheat. J. Exp. Bot. 64, 4453–4460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, Y.S. , Li, Z. , Liu, G.Z. , Jiang, Y. , Maurer, H.P. , Wurschum, T. , Mock, H.P. et al. (2015) Genome‐based establishment of a high‐yielding heterotic pattern for hybrid wheat breeding. Proc. Natl. Acad. Sci. USA, 112, 15624–15629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng, X.W. , Gogarten, S.M. , Lawrence, M. , Stilp, A. , Conomos, M.P. , Weir, B.S. , Laurie, C. et al. (2017) SeqArray‐a storage‐efficient high‐performance data format for WGS variant calls. Bioinformatics, 33, 2251–2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Histogram of the mean depth (number of unique reads) of exome capture sequencing.
Figure S2 Histogram of the number of single‐nucleotide polymorphisms per gene captured by exome sequencing.
Figure S3 Comparison between Rogers’ distances calculated using exome capture sequencing (RDEC) and the 90K SNP array data set (RD90K_SNP) for the 133 parents of the population composed by 1574 wheat hybrids.
Figure S4 Number of single‐nucleotide polymorphisms per wheat chromosome captured by exome sequencing.
Figure S5 Genome‐wide distribution of average exome nucleotide diversity.
Figure S6 Linkage disequilibrium (r 2) as a function of physical distance (Mb) for the three different wheat subgenomes.
Figure S7 Percentual distribution of marker pairs in each of the three wheat genomes as a function of the physical distance (Mb).
Figure S8 Venn diagram showing the amount of overlapping associations for leaf rust severity found by genome‐wide exome association scans in a hybrid wheat population and its corresponding subpopulations.
Figure S9 Extensive genomic region associated with leaf rust resistance on chromosome 3D detected by a genome‐wide exome association scan for additive effects in a vast hybrid wheat population and narrowed down to a smaller candidate region using an independent validation population.
Figure S10 Histogram of the percentages of genetic variance (R2/h) explained by single‐nucleotide polymorphisms found as associated with leaf rust severity in a hybrid wheat population of 1574 hybrids plus their 118 female and 15 male parent lines.
Figure S11 Close‐up view of the two association peaks within a candidate region on chromosome 4A found by a genome‐wide exome association scan for additive and dominance effects underlying leaf rust severity in a hybrid wheat population of 1574 hybrids plus their 118 female and 15 male parents.
Figure S12 Histogram of the ratio of phenotypic variation explained by main additive and dominance effects () over that explained by interaction effects with environments () of single‐nucleotide polymorphisms found as associated with leaf rust severity in a hybrid population (1574 hybrids plus their 118 female and 15 male parent lines) tested in five environments.
Figure S13 Genome‐wide exome association scans for dominance effects underlying leaf rust severity in a vast hybrid wheat population and its different subpopulations.
Figure S14 Distribution of leaf rust severity scores in the validation population.
Figure S15 Some general statistics in a vast hybrid wheat population and its different subpopulations regarding associations mapping on chromosome 6A.
Figure S16 Some general statistics in a vast hybrid wheat population and its different subpopulations regarding associations mapping on chromosome 6B.
Figure S17 Some general statistics in a vast hybrid wheat population and its different subpopulations regarding associations mapping on chromosome 1A.
Figure S18 Candidate genes underlying significant associations on chromosome 1A.
Table S1 Functional location and type of substitution (synonymous and non‐synonymous) within the coding sequence for single‐nucleotide polymorphisms (SNPs) present in a hybrid wheat population of 1574 hybrids plus their 118 female and 15 male parent lines.
Table S2 Estimated average physical distance (Mb) according to different wheat chromosomes where the linkage disequilibrium (r 2) fell below r 2 = 0.2 in a hybrid wheat population of 1574 hybrids plus their 118 female and 15 male parent lines
Table S3 Expected impact of population stratification during genome‐wide exome association scans for leaf rust severity on associations of single‐nucleotide polymorphisms (SNPs) mapping close to Lr10 in a hybrid population of 1574 hybrids plus their 118 female and 15 male parents and its subpopulations Top25%, Inferior75%, Top50% and Inferior50%.
Table S4 Estimates of variances components and heritabilities (h 2) for leaf rust severity in a hybrid wheat population composed by 1,574 hybrids produced by factorial crosses between 118 female (F) and 15 male (M) parents.
Table S5 Putative genes underlying significant regions on chromosome 4A.