

Abstract
Activation of the transcription factor Nrf2 via the Keap1-Nrf2-ARE signaling system regulates the transcription and subsequent expression of cellular cytoprotective proteins and plays a crucial role in preventing pathological conditions exacerbated by the overproduction of oxidative stress. In addition to electrophilic modulators, direct non-covalent inhibitors that interrupt the Keap1-Nrf2 protein-protein interaction (PPI) leading to Nrf2 activation have attracted a great deal of attention as potential preventive and therapeutic agents for oxidative stress-related diseases. Structural studies of Keap1-binding ligands, development of biochemical and cellular assays, and new structure-based design approaches have facilitated the discovery of small molecule PPI inhibitors. This perspective reviews the Keap1-Nrf2-ARE system, its physiological functions, and the recent progress in the discovery and the potential applications of direct inhibitors of Keap1-Nrf2 PPI.
Keywords: Keap1, Nrf2, Keap1-Nrf2 interaction, Protein-protein interaction Inhibitor, Oxidative stress, Structure-activity relationship
GRAPHICAL ABSTRACT
1. INTRODUCTION
All organisms are frequently exposed to endogenous and exogenous sources of oxidative stress during their lifetime, some of which cause deleterious reactive oxidants and electrophiles. Oxidants are generated in the body as a result of natural physiological processes, including various types of cytosolic enzyme systems as well as normal intracellular metabolism in peroxisomes and mitochondria (Finkel and Holbrook 2000). In addition, many environmental stimuli including ultraviolet light (UV) radiation, ionizing radiation, chemotherapeutics, inflammatory cytokines, and environmental toxins can trigger high levels of reactive oxygen species (ROS) and reactive nitrogen species (RNS) that can perturb the normal redox balance between the reactive species and antioxidants, and shift cells into a state of oxidative stress (Finkel and Holbrook 2000). While ROS include a variety of chemical species including superoxide anions, hydroxyl radicals, singlet oxygen and hydrogen peroxide, RNS encompass peroxynitrate and nitric oxide. A rise in the levels of such species results in impaired physiological function, one of which is random oxidative damage to cellular proteins, DNA, and lipids, leading to cell death (Finkel and Holbrook 2000; Abed et al. 2015). Repeated exposure to oxidative stress accelerates pathologic conditions, ultimately leading to a wide range of diseases that include neurodegenerative disorders, inflammatory conditions, respiratory diseases, and cancer (Figure 1) (Finkel and Holbrook 2000; Abed et al. 2015).
Figure 1.

Oxidative stress and antioxidant defense response
The major biological response to oxidative stress is the antioxidant defense system. Cells are equipped with elaborate defense systems, which induce the expression of enzymes that can detoxify the oxidative and electrophilic chemicals (Dinkova-Kostova et al. 2005). The first response involves the expression of phase I and II drug metabolizing enzymes, such as cytochrome P450 enzymes, glutathione S-transferase (GST), NAD(P)H:quinone oxidoreductase I (NQO1), heme-oxygenase-1 (HO-1), and thioredoxin (TRX), and glutamate-cysteine ligase (GCL) (Abed et al. 2015; Dinkova-Kostova et al. 2005). These sophisticated detoxification enzymes change the structure of large hydrophobic organic molecules to increase their polarity, and introduce functionality to allow the conjugation of solubilizing moieties, thereby promoting high clearance (Tonelli et al. 2017). Through Nrf2-mediated antioxidant response, first line defense antioxidants including catalase (CAT), glutathione peroxidase (GPx), and superoxide dismutase (SOD) are also expressed, which counteract and regulate overall ROS levels to maintain physiological homeostasis (Figure 1) (Finkel and Holbrook 2000; Ighodaro and Akinloye 2018). These enzymes are not consumed during the antioxidant process, have relatively long half-lives, and activate chemical detoxification reactions. Some of them are involved in the production of small molecule antioxidants (Abed et al. 2015; Magesh et al. 2012).
In addition to the antioxidant enzymes, other non-enzymatic antioxidants are important in directly reducing a high level of ROS or RNS. They include a scorbate, pyruvate, flavonoids, tocophenols (vitamin E), vitamin K, lipoid acid, ubiquinol, carotenoids, and more importantly, glutathione. These low molecular weight molecules are short-lived, redox-active, and consumed during ROS scavenging, which requires them to be replenished for further prote ction (Abed et al. 2015; Magesh et al. 2012). Consequently, the balance between ROS production and antioxidant defense contributes to redox homeostasis, eventually leading to cell survival (Figure 1) (Fi nkel and Holbrook 2000; Abed et al. 2015; Magesh et al. 2012).
2. REGULATION OF KEAP1-NRF2-ARE PATHWAY
The Keap-Nrf2-ARE pathway is the major regulator of cytoprotective responses that determines the sensitivity of cells to oxidative stress by controlling the inducible expression of detoxification and antioxidant enzymes (Suzuki et al. 2013). The pathway consists of three cellular components, namely Kelch-like ECH-associated protein 1 (Keap1), nuclear factor erythroid 2-related factor 2 (Nrf2) and antioxidant response element (ARE).
2.1. Kelch-like ECH-Associated protein 1 (Keap1).
Keap1 acts as a sensor of chemical and oxidative stresses as well as a negative regulator of Nrf2 (Dinkova-Kostova et al. 2005). With the inhibitory Neh2 domain of Nrf2, Keap1 was identified as a Nrf2-binding protein from yeast two-hybrid screening (Itoh et al. 1999). The complete amino acid sequences of Keap1 from eight species including mouse, pig, and human have been reported, and the four mammalian proteins exhibit a high degree of homology. Keap1 belongs to the superfamily of BTB-Kelch proteins (Dinkova-Kostova et al. 2005). Human Keap1, a 69-kDa protein, contains 627 amino acid residues and consists of five distinct domains: the N-terminal region (NTR), the broad complex, tramtrack and bric-a-brac, (BTB) domain, the intervening region (IVR), double glycine repeats (DGR) or Kelch domain, and the C-terminal region (CTR) (Figure 2A) (Abed et al. 2015; Tkachev et al. 2011). The BTB domain, an evolutionary conserved domain, mediates homo-dimerization and is responsible for the interaction of Keap1 to Cullin 3 (Cul3), a scaffold protein of Nrf2-specific E3 ubiquitin ligases (Lo et al. 2006; Ogura et al. 2010). The DGR domain comprises six repetitive Kelch motifs (KR1–KR6) which form a six-bladed β-propeller structure. The DGR and CTR domains, together named as the DC domain, mediate binding of Keap1 with Nrf2 Neh2 (Li et al. 2004b). The IVR or linker region (LR) domain, located between the BTB and DGR, has a large number of highly reactive cysteine residues that are sensitive to oxidation stress (Lo et al. 2006; Dinkova-Kostova et al. 2002). These residues act as regulators of Keap1 conformation as well as redox stress sensors. More specifically, seven reactive cysteine residues have been identified to be involved in Keap1-mediated repression of Nrf2 activity and Keap1-dependent ubiquitination of Nrf2: Cys151, Cys257, Cys273, Cys288, Cys297, Cys434, and Cys613 (Figure 2A) (Tkachev et al. 2011; Dinkova-Kostova et al. 2002; Zhang and Hannink 2003; Yamamoto et al. 2008).
Figure 2.

(A) Domain structures of the Keap1, Nrf2, and Nrf2 Neh2 proteins (B) Structure of the Keap1-Nrf2 complex.
2.2. Nuclear factor erythroid 2-related factor 2 (Nrf2) and small musculoaponeurotic fibrosarcoma proteins (sMafs).
Nrf2 is a key transcription factor that is critical for cellular homeostasis (Li et al. 2012). In fact, Nrf2 KO mice tend to develop autoimmune diseases and are susceptible to a wide variety of disease conditions (Yoh et al. 2001; Ma et al. 2006). The cloning of Nrf2 cDNA was identified by expression cloning with tandem NFE2 (nuclear factor, erythroid-derived 2)-binding motifs as a screening probe (Moi et al. 1994; Ma 2013). Nrf2 contains a distinct CNC basic leucine zipper (bZip) domain in the C terminal region that mediates DNA binding as well as hetero-dimerization with a small Maf. The CNC (cap ‘n’ collar) protein, named after the Drosophila segmentation protein CNC, shares a high homology among the CNC bZip proteins (Ma 2013). Human Nrf2, containing 605 amino acid residues, consists of seven conserved Neh (Nrf2-ECH homology) domains, namely Neh1 to Neh7 (Figure 2A) (Canning et al. 2015; Abed et al. 2015; Itoh et al. 2010). While Neh1 contains the bZip motif at the C-terminal region, the Neh2 domain at the N-terminus includes DLG and ETGE that are essential for the interactions between Nrf2 and its suppressor Keap1, which negatively regulates transcriptional activity of Nrf2 (Itoh et al. 1997; Katoh et al. 2005). The seven lysine residues, located between the DLG and ETGE motifs, are arranged in an α-helix structure and are involved in Keap1-dependent polyubiquitination as ubiquitin-acceptor sites. The Neh3 domain located at the C-terminus is crucial for the transactivation of ARE-dependent gene (Nioi et al. 2005). The Neh4 and Neh5 domains cooperatively bind to the CH3 motif of the CREB (cAMP responsive element binding protein)-binding protein (CBP), a transcriptional co-activator that regulates Nrf2 transcriptional activity (Katoh et al. 2001). Neh6 has a large number of serine residues in the middle of Nrf2, is involved in redox-insensitive degradation of Nrf2 in a Keap1-independent manner (Abed et al. 2015; McMahon et al. 2004; Wang et al. 2013). The Neh7 domain of Nrf2 was found to be associated with retinoic X receptor alpha (RXRα)-mediated repression of Nrf2 through direct interaction with RXRα (Wang et al. 2013). Small Maf proteins contain a bZip domain for dimerization and DNA binding. There are three small Maf proteins such as MafF, MafG, and MafK that lack a transcriptional activation domain (Fujiwara et al. 1993). The sMaf proteins form heterodimers with large CNC bZip proteins of Nrf2 and bind to the GC dinucleotide of ARE that is classified as a CNC-sMaf-binding element (CsMBE) (Ma 2013).
2.3. Antioxidant response element (ARE).
ARE is a cis-regulatory element located in the enhancer region of many target genes where detoxification enzymes and cytoprotective proteins are encoded (Lee and Johnson 2004). ARE includes a core sequence, 5’-TGACnnnGC-3’ (n = any base) that is later expanded into a 16 base pair sequence, 5’-TMAnnRTGAYnnnGCR-3’ (M = A or C, R = A or G, Y = C or T) (Rushmore et al. 1991; Paul et al. 2003). Under basal conditions, Bach1 (BTB and CNC homology1), a transcriptional repressor of ARE, forms a heterodimer with a sMaf protein, which blocks Nrf2 from binding to DNA. Once oxidative stress occurs, Nrf2 translocates into the nucleus, and Bach1 is degraded by a proteasome (Ma 2013). Nrf2 is capable of forming a heterodimer with sMaf proteins and binding to ARE in the promoter region of Nrf2 target genes, eventually activating the expression of downstream ARE-dependent target genes (Abed et al. 2015; Katsuoka et al. 2005). For example, a Nrf2 KO mouse model identified that Nrf2 regulated the constitutive and inducible expression of ARE-mediated glutathione S-transferase (Gsta1, Gsta2, Gstm1, Gstm2, Gstm3, and Gstm4) genes (Chabas et al. 2002).
2.4. Keap1-Nrf2-ARE pathway.
Under basal conditions, a Keap1 homodimer forms a complex with Nrf2 and two Cul3 where its Kelch domain (also called DGR domain) binds to the Neh2 domain of Nrf2 and its BTB domain binds to the Cul3 N-terminal region (Figure 2B) (Magesh et al. 2012; McMahon et al. 2006). Neh2 interacts with the Keap1 Kelch domains with a ratio of 1:2 through its ETGE and DLG motifs. The ETGE motif has a higher binding affinity than the DLG motif due to more electrostatic interactions occurring between DC and ETGE as compared to DC and DLG (Tong et al. 2006a; Tong et al. 2007). Keap1 recruits the Cul3-Rbx1 (RING-box protein 1) E3 ubiquitin ligase complex and serves as a substrate adaptor to bring Nrf2 into close proximity with E3 complex. The subsequent ubiquitination of Nrf2 marks it for proteasomal degradation by 26S proteasome. Nrf2 is rapidly degraded with a half-life of less than 20 min, resulting in a low level of Nrf2 in many types of cells and thereby maintaining cellular homeostasis (Figure 3) (Ma 2013; Kobayashi et al. 2004; Zhang et al. 2004; Zhang et al. 2005).
Figure 3.
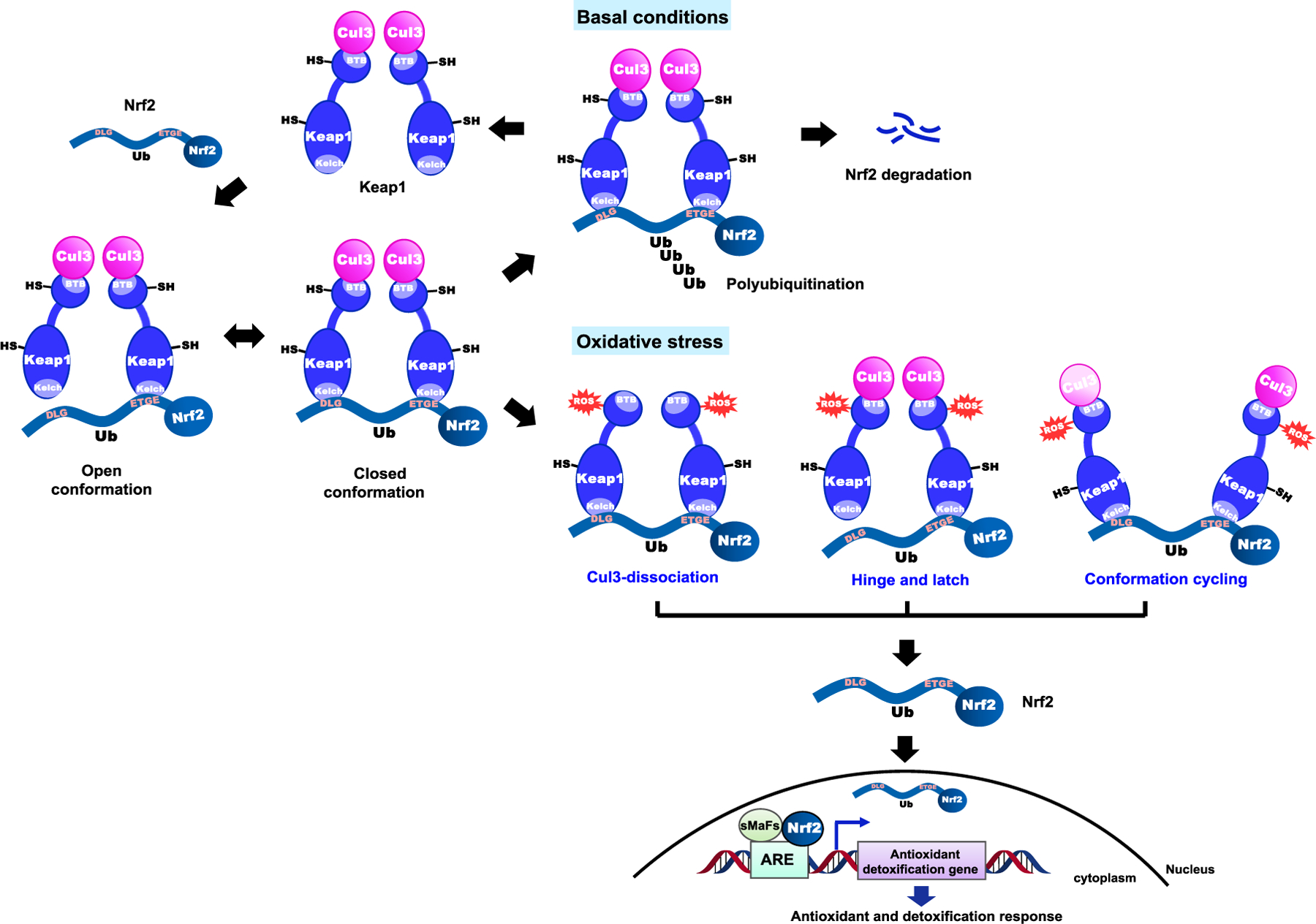
The Keap1-Nrf2-ARE signaling in basal and oxidative stress conditions. There are three proposed mechanism models for dysregulation of the Keap1-Nrf2 complex by cysteine modificat ion in the Keap1 protein: the “Keap1-Cu l3 dissociation model”, the “hinge and latch model”, and the “conformation cy cling model”
Under conditions of oxidative or electrophilic stress, inducers modify cysteine thiols of Keap1 and Nrf2, presumably altering the structure of the Keap1-Nrf2-Cul3 complex to inhibit Nrf2 ubiquitination. Evidence for binding of inducers to Keap1 cysteine thiols was provided using label inducers, stoichiometry, ultraviolet spectroscopy, and mutational studies (Eggler et al. 2005; He and Ma 2010). As a result, free Nrf2, which is stabilized with a half-life of up to 200 min, translocates into the nucleus where it activates expression of antioxidant genes regulated by ARE (Figure 3) (McMahon et al. 2006; Tong et al. 2006a). The exact mechanism of Nrf2 activation by cysteine modification is not known, but there are three representative models that have been proposed: the “Keap1-Cul3 dissociation model”, the “hinge and latch model”, and the “conformation cycling model” (Taguchi et al. 2011).
In the Keap1-Cul3 dissociation model, the binding of Keap1 and Cul3 plays a significant role in the stabilization of Nrf2. Inducers, such as electrophiles, cause thiol modification of cysteine residues in Keap1, leading to disruption of the binding between Keap1 and Cul3 and dissociation of Cul3 from Keap1. As a result, Nrf2 escapes from the ubiquitination system (Figure 3) (Eggler et al. 2009). In this model, it has been reported that Cys151 serves as a key factor, as substitution of Cys151by serine in the BTB domain prevents Keap1 from dissociating with Nrf2 even under oxidative stress (Abed et al. 2015; Eggler et al. 2009; Rachakonda et al. 2008). According to the hinge and latch model, modification of Keap1 cysteine residues, located in the IVR of Keap1, leads to conformational changes in Keap1, resulting in the misalignment of the lysine residues in Nrf2. The Nrf2 DLG motif having lower affinity than ETGE dissociates from Keap1 first, which makes Nrf2 no longer poly-ubiquitinylated (Tong et al. 2007; Fukutomi et al. 2014). Through one of the two suggested models, free or newly synthesized Nrf2 proteins consequently translocate to the nucleus and bind to ARE leading to the expression of Nrf2 target genes, such as HMOX1 (heme oxygenase 1), GCL and GSTs (Figure 3) (Tong et al. 2007; Tong et al. 2006b).
More recently, additional studies using a Förster resonance energy transfer (FRET)-based method have suggested a new conformation cycling model (Baird et al. 2013). Under normal conditions, Keap1-Nrf2 complex exists in two distinct forms, the open or closed conformation, depending on the binding of Nrf2 DLG motif (Figure 3) (Baird et al. 2013; Baird et al. 2014). Although the exact mechanism of the interconversion between these two conformations is not clear, it has been shown that the closed conformation is only involved in the ubiquitination process of Nrf2. The resulting free Keap1 dimer binds to newly translated Nrf2, which consequently generates an open conformation of Keap1-Nrf2 complex, allowing a cycle. Under oxidative stress conditions, the cycle is disrupted, and accumulation of the Keap1-Nrf2 complex in the modified closed conformation is observed without releasing Nrf2. Thus, free Keap1 is not regenerated in this process (Baird et al. 2013; Baird et al. 2014). Ultimately, newly synthesized Nrf2 proteins are stabilized and activate an antioxidant response system (Figure 3).
3. THE PATHOLOGICAL ROLE OF KEAP1-NRF2-ARE PATHWAY IN DISEASES
The continuous exposure to oxidative stress contributes to the development of various diseases, including cancer, neurodegenerative disease, chronic obstructive pulmonary disease (COPD), inflammatory disease, and aging (Rajendran et al. 2014; Abed et al. 2015). The Keap1-Nrf2-ARE pathway plays an important role in regulating the antioxidant defense system in oxidative stress, inflammation, and the development of these diseases.
In cancer, Nrf2 plays conflicting roles depending on the type and stage of the disease (Geismann et al. 2014; Moon and Giaccia 2015; Kansanen et al. 2013). In normal and premalignant cells, Nrf2 plays a part in protecting the body from DNA damaging and carcinogenic effects of ROS by upregulating various cytoprotective enzymes, which consequently prevents cancer initiation and progression. Nrf2 knockout mice are more susceptible to carcinogen exposure and related carcinogenic effects in animal models, and the tumor formation is associated with reduced expression of ARE genes. In addition, inhibition of tumorigenesis in wild-type mice correlated with the increased expression of cytoprotective genes through chemoprevention (Ma 2013). Therefore, Nrf2 activators can be used for cancer chemoprevention (Hayes et al. 2010). In fact, it has been reported that electrophilic Nrf2 activators display broad anti-cancer activity in cellular and animal models, such as vinyl carbamate-induced lung cancer and aflatoxin-induced liver cancer (Wu et al. 2010; Probst et al. 2015a; Probst et al. 2015b; Liby et al. 2007). On the other hand, constitutive hyperactivity of Nrf2, observed in tumor progression, is involved in pathogenic pathways including angiogenesis (Kansanen et al. 2013). The persistent activation of Nrf2 helps tumor cells to be protected against apoptosis from endogenous exposure to high levels of ROS, increases the resistance to chemotherapeutic drugs by activation of metabolic genes, and promotes tumor growth (Taguchi et al. 2011; Kensler and Wakabayashi 2009). However, activation of Nrf2 in tumors is likely to be an outcome of selection during cancer development, rather than a cancer-initiating event. This supports the fact that Keap1 KO mice with activated Nrf2 functions did not develop spontaneous cancer over a 2-year period (Taguchi et al. 2010). Based on these findings, Nrf2 inhibition may be a potential chemotherapeutic strategy for the treatment of cancer. Collectively, it is important to figure out the dual role of Nrf2 in tumor prevention and progression for development of either Nrf2 activator or inhibitor in the management of cancer.
Neurodegenerative diseases, including Huntington’s disease (HD), Parkinson’s disease (PD), Alzheimer’s disease (AD), multiple sclerosis, and amyotrophic lateral sclerosis (ALS), can develop as a result of high levels of oxidative stress, which eventually contribute to neuronal cell death (Simonian and Coyle 1996; Golden and Patel 2008). Several studies have demonstrated that Nrf2 has a protective role against neurodegenerative disorders. Nrf2-deficient cells and Nrf2 KO mice are significantly more vulnerable to mitochondrial complex II inhibitor-mediated neurotoxicity including HD, while preactivation of ARE exhibits a protective effect against neurotoxicity generated by the complex II inhibition (Calkins et al. 2005). In an ALS mouse model, the overexpression of Nrf2 delayed the onset of ALS, leading to extended survival of the mice with ALS (Vargas et al. 2008). In a mouse model of PD, mice lacking Nrf2 are highly more susceptible to MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine)-induced neurotoxicity while astrocyte-specific Nrf2 overexpression is neuroprotective against MPTP toxicity (Dinkova-Kostova et al. 2005; Chen et al. 2009). The absence of Nrf2 exacerbates autoimmune encephalomyelitis (EAE)-induced neuro-inflammation in an autoimmune inflammatory mouse model of MS, which suggests that activation of Nrf2 may be a potential therapeutic strategy in the treatment of multiple sclerosis (MS) (Johnson et al. 2009).
Oxidative stress is a major factor in the pathogenesis of chronic obstructive pulmonary disease (COPD) (Boutten et al. 2011; Kirkham and Barnes 2013). Environmental (e.g., air pollutants, infections and occupational dust) and cellular sources are likely to produce oxidative stress, debilitate respiratory condition, and aggravate COPD (Boutten et al. 2011). Accordingly, Nrf2, a transcription factor expressed in alveolar macrophages, plays a key protective role in the lungs through the activation of ARE-dependent antioxidant and cytoprotective genes. Several animal models and human studies support the importance of Nrf2 and several Nrf2 target genes as a protective system against COPD disease. Results from rodent models of elastase-inducible emphysema indicated that the expression of antioxidants and antiproteases was attenuated in the lungs of Nrf2 KO mice, as compared to wild-type alveolar macrophages (Ishii et al. 2005). Nrf2 KO mice exposed to cigarette smoke are highly sensitive to emphysema with earlier onset and more severe pathology, relative to wild-type mice groups. The mice exhibited elevated oxidative genotoxic stress, apoptosis, and inflammation, as well as decreased function (Rangasamy et al. 2004; Iizuka et al. 2005). Therefore, the activation of the Nrf2-ARE signaling pathway might be a promising strategy to recover antioxidant and detoxifying enzymes, counteracting the effect of cigarette smoke. Moreover, loss of Nrf2 was also shown in lungs of COPD patients (Kirkham and Barnes 2013).
In addition to the representative diseases described above, Nrf2 has been implicated in a wide range of other chronic diseases, including diabetes (Jiménez-Osorio et al. 2014), cardiovascular (Li et al. 2009), gastrointestinal (Khor et al. 2006), liver and autoimmune diseases (Kurzawski et al. 2012; Maicas et al. 2011; Wruck et al. 2011), all of which are characteristically associated with oxidative stress (Abed et al. 2015). In regard to disease development, oxidative damage exacerbates the pathophysiological conditions. Therefore, Nrf2 can participate in multiple protective effects against a variety of chronic diseases by regulating oxidative stress and antioxidant stress response signaling. Thus, the Keap1-Nrf2-ARE pathway is shown to be extensively involved in diverse diseases and biological mechanisms, leading to interest in the pathway as a promising drug target for disease prevention and treatment.
4. INDIRECT INHIBITORS OF KEAP1-NRF2 INTERACTION
Based on the regulatory mechanism of the Keap1-Nrf2-ARE system, Nrf2 activators are divided into two categories: indirect and direct small-molecule inhibitors. Most of the known small molecule inhibitors are indirect inhibitors bearing electrophilic functionality which can covalently bond with sulfhydryl groups of cysteine residues on Keap1. Cysteine thiolate ions, formed under neutral pH, are highly reactive towards ROS, RNS, and other electrophiles; the resulting chemical reactions lead to changes in Keap1 protein conformation and thus disrupting the Keap1-Nrf2 PPI (Abed et al. 2015; Magesh et al. 2012). Unfortunately, these chemical reactions lack specificity and may cause unpredictable off-target side effects.
The covalent cysteine modifiers can be classified into five distinct scaffolds based on their chemical structure (Figure 4) (Magesh et al. 2012; Abed et al. 2015). Isothiocyanates and related derivatives, including sulforaphane (SFN) and phenyl isothiocyanate (PEITC), exhibit protective effects against several diseases, such as cancers, COPD, and neurodegenerative disorders (Robledinos-Anton et al. 2019). More specifically, their central carbon atoms readily react with cysteine groups (e.g., Cys151) in Keap1, preventing the Keap1 protein from interacting with Nrf2 and stabilizing Nrf2. Many studies have shown that isothiocyanates can upregulate enzymes like HO-1 and GST in rodent tissues and in mammalian cells (Hong et al. 2005).
Figure 4.
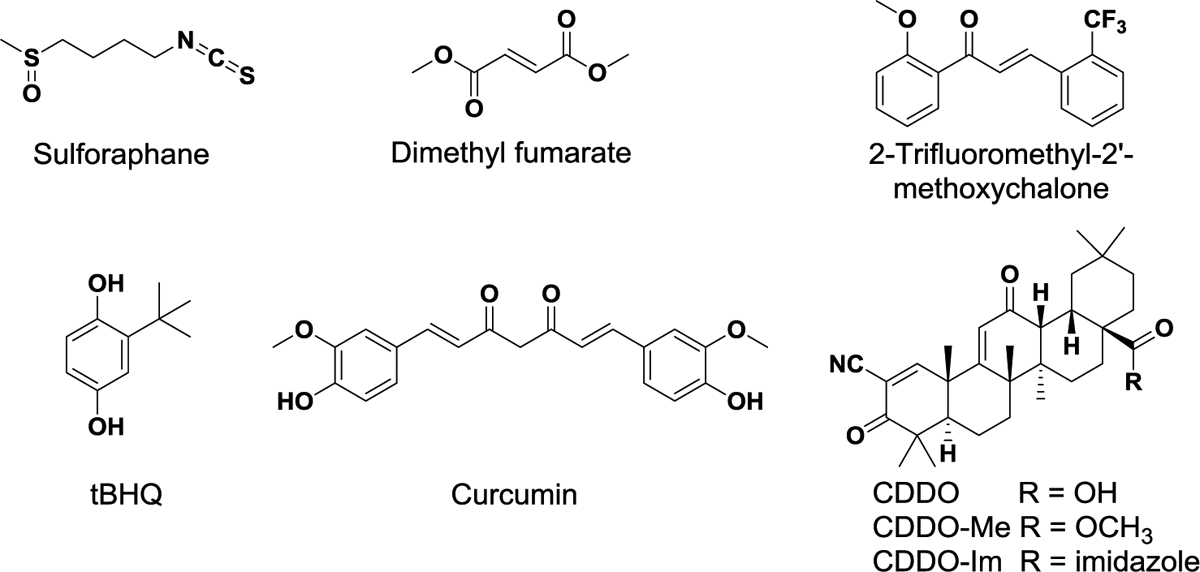
Structures of representative indirect inhibitors of Keap1-Nrf2 PPI with electrophilic functional groups (Magesh et al. 2012)
Fumarates are characterized by Michael acceptors with an α,β-unsaturated carbonyl system, enabling reaction with cysteine thiols of Keap1. The salt mixture of monoethyl fumarate and dimethyl fumarate (Fumaderm®) and dimethyl fumarate (DMF, Tecfidera®) are marketed for the treatment of psoriasis and relapsing multiple sclerosis (MS), respectively (Lee et al. 2012; Mrowietz et al. 2007). These electrophilic modulators mainly make an interaction with Cys151, which consequently inhibits Nrf2 ubiquitination and induces the expression of ARE-mediated antioxidative and cytoprotective enzymes. In Nrf2 KO mice, these anti-inflammatory effects were not observed (Linker et al. 2011).
The main chemical structure of chalcones consists of two phenyl rings and a three-carbon α,β-unsaturated system (Figure 4). Chalcones, including sofalcone and isoliquiritigenin, also act as Michael acceptors to activate Nrf2, indicating a broad range of biological properties including anti-proliferative, anti-infective, and anti-inflammatory activities (Dimmock et al. 1999; Go et al. 2005). These electrophiles are identified as potent activators of the Nrf2 signaling pathway that induces the expression of Nrf2-dependent antioxidant genes including glutamate-cysteine ligase modifier subunit (GCLM), HO-1 and NQO1 in human lung epithelial cells and in mouse small intestines after oral administration (Kumar et al. 2011). In addition to chalcones, vinyl sulfone derivatives have been shown to activate Nrf2 and subsequently enhance the expression of Nrf2-mediated antioxidant enzymes such as NQO1, HO-1, glutamate-cysteine ligase catalytic subunit (GCLC) and GCLM in dopaminergic neuronal cells (Woo et al. 2014).
Quinones (e.g., 1,4-naphthoquinone) and polyphenolic derivatives (e.g., curcumin) also induce Nrf2-mediated ARE genes (Magesh et al. 2012). Different from quinones, catechol, polyphenolic compounds, and hydroquinone (e.g., tert-butylhydroquinone, tBHQ) are oxidized to electrophilic quinones containing Michael acceptors in the presence of sufficient oxygen and copper II or other transition metals, activating ARE-mediated transcription (Dinkova-Kostova and Wang 2011; Sirota et al. 2015; Li et al. 2004a). In addition, catechols undergo cytochrome P450-mediated oxidation, and thus alter redox states in cells or their environment, leading to upregulation of Nrf2. An example of oxidizable polyphenols is curcumin bearing two phenolic functional groups and the β-diketo moiety. Two Michael acceptor groups of the natural product readily modify Cys151 in Keap1 protein (Magesh et al. 2012). Curcumin has been tested in clinical trials for the prevention of colon cancer and the treatment of prediabetes, schizophrenia, and metabolic syndrome (Robledinos-Anton et al. 2019).
Based on natural oleanane triterpenoids which have antitumor and anti-inflammatory effects, CDDO, also known as bardoxolone, was developed as a potent Nrf2 inducer, by introducing highly active α,β-unsaturated α-cyano ketones to the A and C rings of an oleanolic acid scaffold (Figure 4) (Honda et al. 1998). Similar to other covalent modulators, the Michael acceptor groups primarily react with Cys151 in Keap1, which results in the interruption of Keap1-Cul3 interaction (Cleasby et al. 2014). Among its analogs, CDDO-Me (bardoxolone methyl) is one promising Nrf2 modulator (Robledinos-Anton et al. 2019). Interestingly, in vivo study in a transgenic Nrf2 null mouse model suggested that CDDO-Me is highly selective for Nrf2-regulated proteins, resulting in less off-target effects. In Nrf2 KO mice, only altered expression of two proteins was observed, whereas 43 proteins were changed by treatment with CDDO-Me in WT mice (Walsh et al. 2014). CDDO-Me first entered phase II clinical trial to evaluate its efficacy in the treatment of type 2 diabetes mellitus and chronic kidney disease (CKD). Unfortunately, it was withdrawn in phase III clinical trial due to cardiac adverse effects. It was shown that nonspecific interactions of covalent cysteine modifiers contributed to unexpected safety issues (Pergola et al. 2011; De Zeeuw et al. 2013). Recently, additional or new clinical studies for CKD and pulmonary hypertension have been initiated (Robledinos-Anton et al. 2019).
5. DISCOVERY OF DIRECT INHIBITORS OF KEAP1-NRF2 PPI
The discovery of small-molecule PPI inhibitors has been considered a challenging task, even though PPI offers a rich source for novel drug targets. PPI interfaces are generally flat, large and complex, which prevents small molecules from effectively disrupting interactions (Sheng et al. 2015; Blundell et al. 2000). Furthermore, PPI inhibitors are more likely to face pharmacokinetic issues as an orally bioavailable drug due to large molecular weight and poor solubility. However, several potent inhibitors have been successfully developed, and studied in clinical trials through novel biological and chemical technologies and strategies (Sheng et al. 2015; Arkin et al. 2014).
5.1. Characteristics of Keap1-Nrf2 PPI as a drug target of direct inhibitors.
As mentioned previously, the Keap1 homodimer interacts with Nrf2 through the ETGE and DLG motifs, located in the Neh2 domain of Nrf2 (Tong et al. 2006a; Tong et al. 2007). More specifically, the ETGE motif has stronger binding affinity to Keap1 than the DLG motif, which led to the development of ETGE-containing Nrf2 peptide inhibitors. Furthermore, X-ray co-crystal structures of the human and mouse Keap1 Kelch domain along with the Nrf2 peptides have provided a better understanding of the protein-protein interactions (Jiang et al. 2016). One of the crystallographic data (PDB ID: 2FLU) revealed that the Kelch domain forms a symmetric 6-bladed β-propeller structure consisting of four stranded antiparallel β-sheets, and the 16mer Nrf2 peptide has two antiparallel β-strands (β-hairpin conformation) that bind to the top face of the β-propeller (Figure. 5A) (Abed et al. 2015; Lo et al. 2006; Li et al. 2004b). The conformation of Nrf2 peptide is stabilized by intramolecular hydrogen bonds with Asp77 and Thr80. The peptide-domain-mediated PPI is the result of multiple electrostatic interactions between six residues in Keap1 (Ser363, Asn382, Arg380, Arg415, Arg483, and Ser508) and the carboxylate oxygen atoms of Glu79 and Glu82 in the Nrf2 peptides (Figure 5B) (Abed et al. 2015). Accordingly, the peptide-binding site of the Kelch domain is a positively charged region because of the highly conserved Arg residues (Abed et al. 2015).
Figure 5.
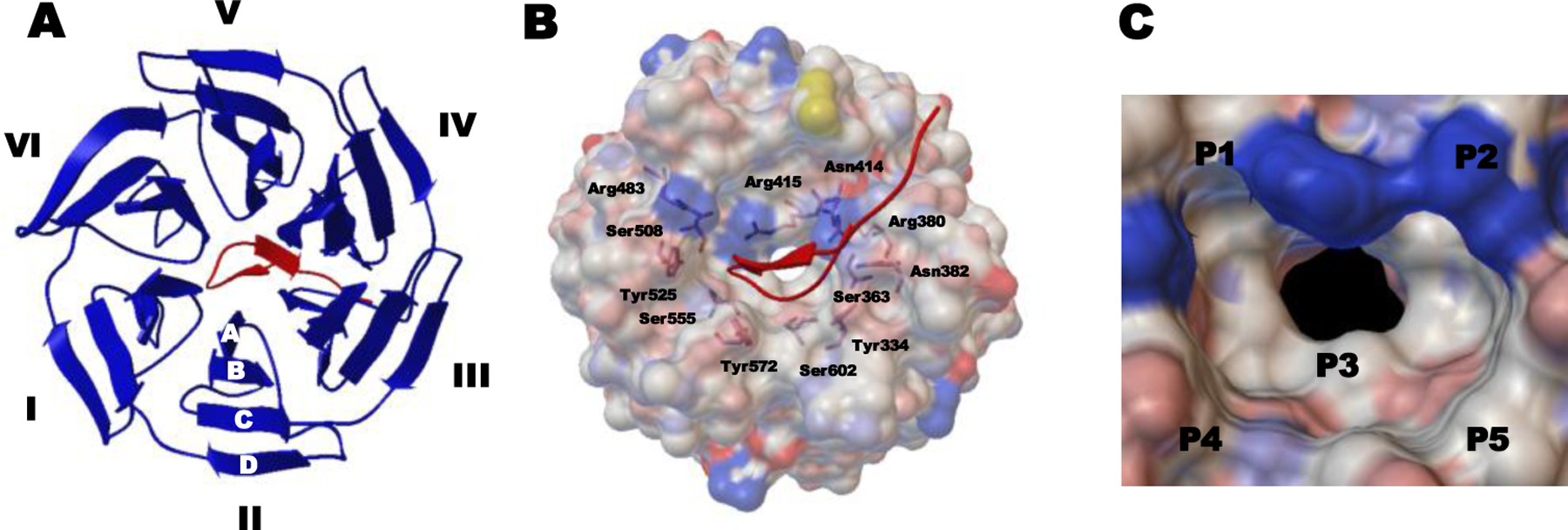
Co-crystal structure of the human Keap1 Kelch domain and the 16mer Nrf2-drived peptide containing the ETGE motif (PDB ID 2FLU). (A) Top view of the binding interaction of the Kelch domain with six blades I-VI and four β-strands A–D (shown as blue ribbon) and the Nrf2 peptide (shown as a red tube). (B) Full view of the interaction between the Keap1 Kelch domain (shown as a gray ele ctrostatic surface) and the Nrf2 peptide (shown as a red tube). Indicated residues are involved in interacting with the 16mer peptide. (C) Structure of the Keap1 binding cavity consisting of five subpockets (hotspots) P1–P5.
Based on the structural analysis of the co-crystal structure of Keap-Nrf2 ETGE motif (PDB ID: 1X2R), You and his colleagues studied the multiple substrate binding determinants of Keap1 using molecular dynamics (MD) simulations combined with mechanics-generalized Born surface area (MM-GBSA) free energy calculations (Jiang et al. 2014a; Jiang et al. 2014b). The computational analysis suggested that the ligand-binding site of the Keap1 Kelch domain is mainly divided into five subpockets (P1-P5), namely five “hot spots”, where different distinct residues are involved in forming significant interactions for high binding affinity (Figure 5c). While P1 and P2 contain polar residues that play an important role in forming electrostatic interactions, P4 and P5 are the non-polar parts that contribute to hydrophobic interactions, and P3 is the central part in the cavity of the Kelch domain (Jiang et al. 2014a; Lu et al. 2015). Consequently, the structural analysis results enabled the discovery of direct inhibitors of Keap1-Nrf2 PPI via structure-based design approaches.
5.2. Assay development for evaluation of Keap1-Nrf2 PPI inhibitors.
At the early drug discovery stage, well-established bioassays play an important role in discovering novel hits through the screening of chemical libraries and in evaluating inhibitory activity against Keap1-Nrf2 PPI for hit-to-lead optimization. Generally, in vitro assays can be divided into two types, Keap1 binding assay and Keap1-Nrf2 inhibition assay, based on the purpose of the assay methods. The binding assay employs a thermodynamic (e.g., isothermal titration calorimetry assay) or kinetic method (e.g., surface plasmon resonance assay) to determine an equilibrium dissociation constant Kd reflecting binding affinity (Jiang et al. 2016). While the ITC assay directly measures the amount of heat exchange depending on binding of a ligand to a target protein, the SPR assay detects the change of refractive index in real time generated by binding of an analyte in solution to a ligand on a sensor chip surface. Our group developed an SPR-based solution competition assay that demonstrated the minimum binding sequence of the 9-mer Nrf2 peptide (LDEETGFEL) (Chen et al. 2011). The SPR assay can also be used to measure binding affinity of small molecules as direct inhibitors of Keap1-Nrf2 PPI. Despite the advantage of label-free detection, the SPR assay cannot be applied to high throughput screening (HTS) of large chemical libraries due to the low throughput (Jiang et al. 2016; Chen et al. 2011).
Other competition assays that measure inhibitory activity of compounds include fluorescence polarization (FP) assay and time-resolved fluorescence resonance energy transfer (TR-FRET) assay. The FP assay is a powerful tool that exhibits substantial tolerance and has been successfully applied for HTS. In 2012, our group first established the FP assay by developing the novel fluorescently-labeled Nrf2 peptide containing the ETGE motif, FITC-9mer Nrf2 peptide amide, as a probe that binds to Keap1 Kelch domain protein in a competition assay for the screening and evaluation of direct inhibitors of Keap1-Nrf2 PPI (Inoyama et al. 2012). The principle of the FP assay is to measure the change of polarization depending on the size of fluorescent molecule or its complex with protein. When the fluorescent probe binds to Keap1 Kelch domain protein, the large complex has slow rotation, leading to high polarization. Once an inhibitor binds to the protein, the probe dissociates from the protein and rapidly rotates, leading to low polarization. Fluorescence intensity is measured in both parallel and perpendicular orientations relative to the polarization plane of excitation light. These measurements can be employed to calculate either fluorescence polarization (FP) or fluorescence anisotropy (FA) (Hall et al. 2016). The FP and FA can then be plotted against the concentration of a test compound to derive IC50 and Ki (Inoyama et al. 2012). In 2017, Sohara et al. reported a new fluorescence correlation spectroscopy (FCS) assay including a fluorescent 6-carboxytetramethylrhodamine (6-TAMRA)-labeled Nrf2 ETGE motif derived peptide, which was utilized in drug-repositioning screening system for Keap1-Nrf2 binding inhibitors(Yoshizaki et al. 2017). FP assay offers several advantages over SPR assay, including simple mix-and-read protocol and lack of separation step, and thus is widely used on HTS platforms (Lea and Simeonov 2011).
Fluorescence resonance energy transfer (FRET) and TR-FRET assays are proximity-based methods that use a distance-dependent energy transfer of excited state energy from a donor to an acceptor fluorophore. In 2013, Wells et al. first developed a steady-state FRET-based assay for identification of Keap1-Nrf2 PPI inhibitors that contains CFP (cyan fluorescent protein)-conjugated Nrf2 peptide and YFP (yellow fluorescent protein)-conjugated Keap1 Kelch domain (Schaap et al. 2013). More recently, TR-FRET competition assay has also been utilized for determining inhibitory activity of potent 1-phenylpyrazole analogs (Callahan et al. 2017b). The TR-FRET assay, which uses a lanthanide chelate, has several advantages over the FRET assay. For instance, the acceptor fluorescence is selectively detected in a time-resolved manner, and other interfering signals generated from background scattering and organic fluorescent dyes are decayed because of their short lifetime in the nanosecond range (Selvin 2002).
To gauge the cellular potency of Nrf2 inducers, there are several types of cell-based assays, including ARE-luciferase reporter assay, NQO1 assay, Nrf2 translocation assay, and gene and protein expression. ARE-luciferase reporter assay has been widely utilized to determine cellular potency of known compounds as Nrf2 activators. In this assay, a luciferase gene under the control of an ARE sequence is involved in monitoring Nrf2 activation in an antioxidant pathway through the expression of the luciferase protein (Boerboom et al. 2006). An NQO1 assay has also been used to identify direct and indirect Nrf2 activators that up-regulate the NQO1 protein, an enzyme under the control of Nrf2. The cellular assay evaluates the NQO1 induction ability of inducers, which is determined by reduction of Menadione with cofactor NAD(P)H (Prochaska and Santamaria 1988). NQO1 induction potency is often expressed as a CD value which is a concentration required to double the NQO1 enzymatic activity after 24-hour incubation. The PathHunter® U2OS Keap1-Nrf2 functional assay (DiscoverX) estimates Nrf2 translocation using β-galactosidase-based enzyme fragment complementation technology (DiscoverX 2011). Upon Nrf2 activation, two β-galactosidase enzyme fragments, which are fused into Nrf2 and located in the nucleus, respectively, reconstitute β-galactosidase enzyme by Nrf2 translocation (DiscoverX 2011). To assess the activation of Nrf2 and its functions at the cellular level, the expression of Nrf2 and its target genes and proteins is quantified using qRT-PCR (quantitative real-time polymerase chain reaction) and western blot (Jiang et al. 2016).
5.3. Small molecule direct inhibitors of Keap1-Nrf2 PPI.
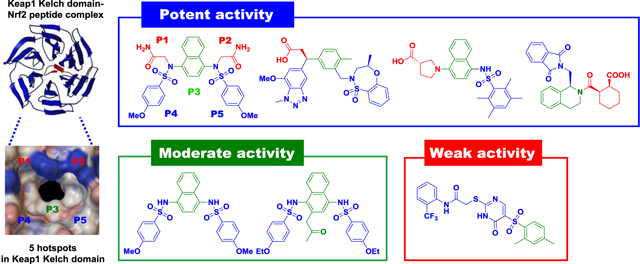
The direct and noncovalent activators of Nrf2 have certain advantages over electrophilic compounds in terms of target selectivity. The binding of the non-electrophilic small molecules to the Keap1 Kelch domain selectively obstructs the protein-protein interaction between the Kelch domain and the ETGE or DLG motif on the Neh2 domain of Nrf2. The potentially better safety profile along with the availability of well-established biochemical binding assays, such as SPR and FP assays, instigated the discovery of small molecule direct inhibitors of Keap1-Nrf2 PPI. The publication of co-crystal structures of Keap1 Kelch domain protein with ligands bound further facilitated the discovery of small molecule inhibitors of Keap1-Nrf2 PPI. Since our group first identified a new series of small molecule direct inhibitors containing a tetrahydroisoquinoline (THIQ) scaffold through high-throughput screening (HTS) in 2013, several other scaffolds have been discovered using drug design strategies including HTS, structure-based virtual screening (SBVS) and fragment-based drug discovery (FBDD), as shown in Figure 6 and 7 (Pallesen et al. 2018).
Figure 6.

Tetrahydroisoquinoline (THIQ) and 1,4-diaminonaphthalene scaffolds
Figure 7.
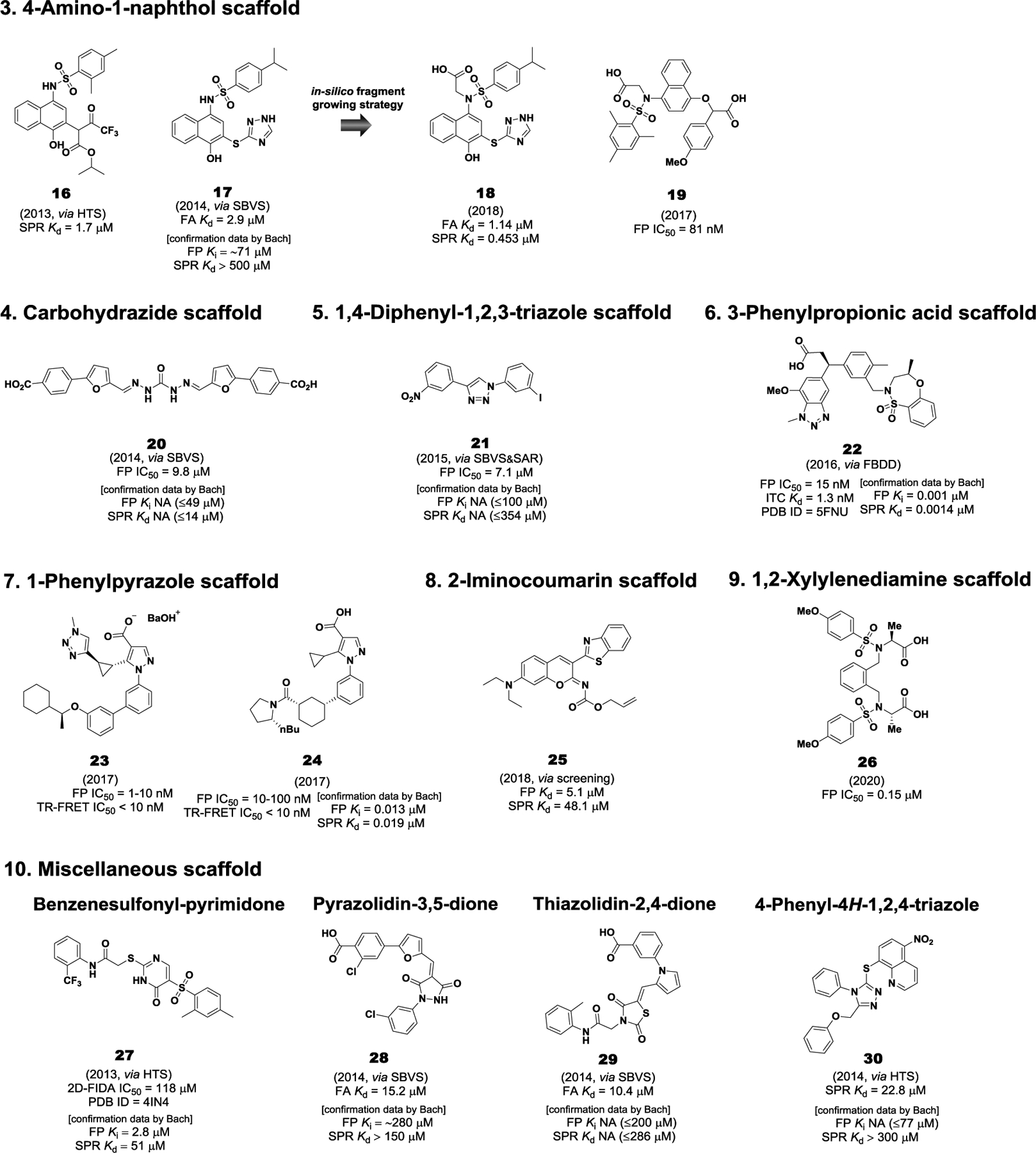
Representative small molecule direct inhibitors of Keap1-Nrf2 PPI. NA: No activity (maximum concentration tested)
5.3.1. Tetrahydroisoquinoline (THIQ) scaffold.
In 2013, our group reported the first small molecule inhibitor of Keap1-Nrf2 PPI that bears a THIQ scaffold using an FP competitive assay in a HTS screening of 337,116 compounds in the NIH MLPCN library (Hu et al. 2013b). Compound 1 (LH601A), the most active (SRS)-isomer obtained from separation of the four stereoisomers through flash silica gel chromatography and chiral HPLC separation, exhibited moderate potency with a Kd value of 1.0 μM in a surface plasmon resonance (SPR) competition assay (Figure 6) (Hu et al. 2013b). We performed a preliminary SAR study of 1, and identified that three moieties of the compound, consisting of the phthalimide, the tetrahydroisoquinoline (THIQ) and the cyclohexanecarboxylic acid, are important for maintaining its activity. In 2014, Courade et al. at UCB confirmed the inhibitory activity of 1 with an IC50 value of 2.3 μM in an FP assay and obtained the co-crystal structure of the Keap1 Kelch domain and 1 (PDB code 4L7B) (Jnoff et al. 2014). Further SAR study indicated that 2, containing an isoindolinone and a 5-methyl substituted THIQ, was the most active with an IC50 value of 0.75 μM (Figure 6) (Jnoff et al. 2014). Cell-based functional assays demonstrated that 1 induces activation of ARE-mediated genes in HepG2 cells with an EC50 value of 18 μM and stimulates nuclear translocation of the Nrf2 protein with an EC50 of 12 μM (Hu et al. 2013b). Furthermore, 1 was shown to upregulate a subset of Nrf2 target genes including HO-1, NQO1 and TRX1, as well as enhance HO-1 (3.5 fold) and TRX1 (2–4 fold) protein expression in cultured human kidney cells (HEK293) at two concentrations of 50 and 100 μM (Wen et al. 2015).
5.3.2. 1,4-Diaminonaphthalene scaffold.
Since 2013, the 1,4-diaminonaphthalene scaffold has been studied by several research groups. Silvian et al. at Biogen first reported a new class of symmetric 1,4-diaminonaphthalene analogs utilizing a homogeneous confocal fluorescence anisotropy (FA) assay (two-dimensional fluorescence intensity distribution analysis, 2D-FIDA), which were discovered by HTS application of the Evotec Lead Discovery Library (267,551 compounds) and 1911 compounds selected from a virtual screening (Marcotte et al. 2013). IC50 value of hit compound 3 was 2.7 μM in the biochemical 2D-FIDA assay (Figure 6). Cell-based experiments indicated that the 1,4-diaminonaphthalene analog was active in a Nrf2 specific ARE-driven luciferase cell-based assay and increased the levels of Nrf2 and its target protein NQO1 in a Western blot analysis. Through structure-based design utilizing MD simulations combined with MM-GBSA free energy calculations, You et al. designed 4 by adding symmetric acetate groups to the sulfonamides of 3, resulting in a 51-fold increase in potency (IC50 = 1.46 μM vs 28.6 nM) in FP assay (Figure 6) (Jiang et al. 2014b). The molecular docking study of 4 suggested that the two acetic acid moieties may be involved in forming multiple strong hydrogen bonds and salt bridges with polar residues in the P1 and P2 subpockets. Due to its potency, 4 has served as a new lead for further optimization process. Indeed, three research groups have conducted SAR studies or designed new analogs based on 4 in efforts to improve both its activity and physiochemical properties, as well as increase the structural diversity of the symmetric 1,4-diaminonaphthalene scaffold.
First, You et al. focused on exploring substituents on the phenyl rings connected to the sulfonamide groups of 4 and demonstrated that 5 with 4-acetamido groups is the most promising Keap1-Nrf2 PPI inhibitor among its derivatives in terms of in vitro inhibitory activity, solubility and cellar potency (Jiang et al. 2015). When compared to 4, compound 5 had improved inhibitory activity of Keap1-Nrf2 PPI with an IC50 of 14.4 nM in the FP assay, and it had better aqueous solubility at pH 7.4 with 5.0 mg/mL (Figure 6). The biological results of cell-based assays in human colon cancer (HCT116) cells revealed that 5 activated Nrf2, up-regulated Nrf2-dependent gene expression and increased the protein level of Nrf2-driven genes including HO-1, NQO1, and GCLM (Jiang et al. 2014b; Jiang et al. 2015). Moreover, this compound dramatically decreased the levels of pro-inflammatory cytokines in a lipopolysaccharide (LPS)-challenged mouse model. Further biological studies of 5 reported in 2019 demonstrated that it had a protective effect against the LPS-induced chronic renal inflammation in both human proximal tubular epithelial HK-2 cells and in vivo mouse model and alleviated kidney damage (Lu et al. 2019b). They also optimized the two acetate groups of 5 through a bioisoteric replacement strategy, and the SAR study indicated that compound 6 with tetrazole groups retained similar potency with an IC50 of 15.8 nM in the FP assay (Figure 6) (Lu et al. 2016). More importantly, the naphthalene analogue 6 displayed improved physicochemical properties, such as logD7.4 (1.02 to 2.31), pKa (4.79 to 5.12), transcellular permeability (Pe value = 0.3 to 42.3 × 103 cm/s at pH =7.4), and more potent cellular activities, including both Nrf2-ARE inducing activity and mRNA or protein levels of the Nrf2 target genes (NOQ1, HO-1 and γ-GCS), as compared to 5 (Lu et al. 2016). Very recently, they reported a series of asymmetric 1,4-diaminonaphthalene analogs by replacing one N-acetic acid substituted sulfonamide with amino acid building blocks possessing various side chains (Lu et al. 2019a). As a result, proline derived 7 was found to be the most potent with an IC50 of 15.8 nM in the FP assay and a Kd value of 53.7 nM in the ITC assay (Figure 6). The Pro analog effectively activated the expression of Nrf2-regulated genes and proteins (HO-1, NQO1, and GCLM) in hepatic L02 cells and exhibited a cytoprotective effect against acetaminophen (APAP)-induced acute live injury in both cellular and in vivo mouse models (Lu et al. 2019a).
Similarly, Moore et al. conducted an SAR study of the diacetate derivative 4, revealing a diamide analog 8 which was the most active with an IC50 of 63 nM in the FP assay and a Kd value of 44 nM in the SPR assay (Figure 6) (Jain et al. 2015). Interestingly, ex vivo studies demonstrated that mono-ethyl ester analog 9 was the best Nrf2 activator among its derivatives in inducing the expression of Nrf2 target genes (HMOX1 and NQO1) and stabilizing Nrf2 levels, which was comparable to the potent electrophilic modulator, sulforaphane (Jain et al. 2015). More recently, they found a new series of 1,4-isoquinoline analogs by replacement of the naphthalene core of 4 with nitrogen-containing heterocycles, one of which is 10 with an IC50 of 60 nM in the FP assay and a Kd value of 102 nM in the SPR assay (Figure 6) (Richardson et al. 2018). When compared to 4, the 1,4-isoquinoline derivative 10 exhibited equivalently high solubility, less metabolism, and good in vitro activities (binding affinity, inhibitory, and cellular potencies) and importantly had a better mutagenic profile in a mini-Ames assay (Richardson et al. 2018). In an attempt to overcome drawbacks of the diacetate derivative 10, such as low lipophilicity and related membrane permeability, they replaced one carboxylate moiety with fluorinated alkyl chains, resulting in 11 (known as PRL-295) with 2,2,2-trifluoroethyl group (Figure 6) (Lazzara et al. 2020). While it showed similar inhibitory activity (IC50 = 73 nM) in FP assay and metabolic stability (t1/2 = 136 min) in human liver microsomes to 10 (IC50 = 60 nM, t1/2 = 104 min), its lipophilicity was improved with a logD7.4 value of −1.5 to 0.5, and there was a remarkable increase in cellular levels of Nrf2 and NQO1 expression. They hypothesized that the lipophilic fluorinated alkyl group of 11 contributed to enhanced cell membrane permeability and eventually greater cellular potencies (Lazzara et al. 2020).
In 2015, Winkel et al discovered naphthylpyrrolidine-3-carboxylic acid analog 12 (RA839) as a Keap1-Nrf2 interaction inhibitor by screening (Winkel et al. 2015). Although RA839 had low metabolic stability in liver microsomes from human, mouse, and rat, it inhibited the interaction between the ETGE motif of Keap1 and Nrf2-derived peptide with an IC50 of 0.14 μM in the FP assay and bound to the Keap1 Kelch domain with a Kd value of 6 μM in the ITC assay (Figure 6). Moreover, the pyrrolidine analog 12 significantly increased the hepatic mRNA levels of the Nrf2 target genes, GCLC and NQO1 in a mouse model, when it was administered to mice together with a cytochrome P450 inhibitor (Winkel et al. 2015). Interestingly, the chemical structure of 12 is quite similar to that of 7 designed by You et al. in 2019, suggesting the pyrrolidine carboxylic acid moiety is favorable for good potency (Winkel et al. 2015; Lu et al. 2019a).
Compound 13, known as K67, was initially identified as a selective PPI inhibitor of phosphorylated p62 (p-p62) and Keap1 using a FP-based HTS method, targeted for the treatment of hepatitis C virus (HCV)-positive hepatocellular carcinoma (HCC) (Saito et al. 2016). They sought to evaluate its inhibitory activities against Keap1-Nrf2 as well as Keap1-p-p62 PPIs, and 13 bearing an acetonyl side chain exhibited moderate activity with an IC50 of 6.2 μM in the FP assay (Figure 6) (Yasuda et al. 2016). Among its derivatives containing diverse side chains on the core naphthalene, compound 14 resulted in a dramatic (41-fold) increase in potency with an IC50 of 0.15 μM (Figure 6) (Yasuda et al. 2016). During the initial discovery process, they found 15 possessing a unique benzo[g]indole skeleton, which displayed similarly strong potency (IC50 = 0.20 μM) to 14 (Figure 6) (Yasuda et al. 2017). The non-covalent inhibitor 15 was also reported to have high metabolic stability (81% remaining after 30 min incubation) and low cytotoxicity.
5.3.3. 4-Amino-1-naphthol scaffold.
There are three different types of small molecule inhibitors, containing a 4-amino-1-naphthol scaffold. The first one was 16 reported by our group in 2013 (Hu et al. 2013a). Besides the THIQ scaffold 1, we confirmed that 16, obtained by HTS of the NIH’s MLPCN small molecule library, has the capacity to disrupt the Keap1-Nrf2 interaction with a Kd value of 1.7 μM in the SPR assay (Figure 7). In 2014, a structure-based virtual screening (SBVS) of a chemical library from Specs database (SPECS, Zoetermeer, Netherlands) followed by the selection of 65 compounds for FA assay identified 17 with a Kd value of 2.9 μM (Figure 7) (Zhuang et al. 2014). Further biological experiments showed that the 4-amino-1-naphthol analog 17 effected Nrf2 nuclear translocation and increased the mRNA levels of Nrf2 downstream genes, HO-1 and NOQ1, suggesting that it is an inhibitor of Keap1-Nrf2 PPI. Recently, compound 18 bearing an acetate group was obtained by an in-silico fragment growing approach based on 17 and subsequent SAR study (Meng et al. 2018). It was predicted that the acetate group of 18 would form additional hydrogen bonding interaction with S363 residue in the P2 subpocket. As expected, 18 showed improved potency by 2.5-fold with a Kd value of 1.14 μM in the FA assay, while its binding activity with the Keap1 protein was confirmed by SPR assay with a Kd value 0.453 μM (Figure 7) (Meng et al. 2018). In cell-based assays, compound 18 also induced Nrf2 nuclear translocation and subsequently the expression of downstream HO-1 and NOQ1 in H9c2 cardiac cells. Furthermore, 18 exhibited a cardioprotective effect against LPS-induced myocarditis in both H9c2 cell and in vivo mouse model by reducing ROS production and the expression of pro-inflammatory cytokines, including TNF-α, IL-1β, and IL-6 (Meng et al. 2018). Importantly, 18 had dramatically reduced cytoprotective effects in siNrf2 RNA transfected cells, thereby providing further support that the 4-amino-1-naphthol derivative 18 activates Nrf2 via direct inhibition of Keap1-Nrf2 PPI (Meng et al. 2018). However, 18 does contain a pan-assay-interference-compounds (PAINS) structure. The last series of asymmetric 4-amino-1-naphthol analogs has been reported in a patent (You et al. 2017). When compared to the 1,4-diaminonaphthalene scaffold, it distinctively includes an ether bond and mono-sulfonamide moiety. SAR studies using an FP competition assay demonstarted that 19 is the most active among 20 compounds, indicating that the methoxy substituent of the phenyl ring containing an oxygen linker plays a significant role for nanomolar activity (Figure 7). In the ARE luciferase reporter assay, compound 19 activated ARE at a concentration of 10 μM, which was better than tert-butylhydroquinone (You et al. 2017).
5.3.4. Carbohydrazide scaffold.
The carbohydrazide derivative 20 was reported as a direct Keap1-Nrf2 PPI inhibitor by structure-based virtual screening (SBVS) of Specs database. 20 was shown to interrupt the Keap1-Nrf2 interaction with an IC50 value of 9.8 μM in an FP assay and exhibit ARE-inducing activity in ARE-luciferase reporter assay (Figure 7) (Sun et al. 2014).
5.3.5. 1,4-Diphenyl-1,2,3-triazole scaffold.
Wells et al. carried out structure-based virtual screening (SBVS) of ZINC database along with rational docking-based structural analysis, designing a 1,4-diphenyl-1,2,3-triazole scaffold (Bertrand et al. 2015). Among 1,2,3-triazole derivatives bearing various substituents on the two phenyl rings, 21 showed the best results in both the FP and NQO1 induction assays. However, its inhibitory activity was not high with an IC50 value of 7.1 μM in the FP assay (Figure 7). Additionally, the iodo-substituted analog 21 up-regulated the expression of the Nrf2-dependent enzymes, HO-1 and NQO1, in Hepa1c1c7 cells (Bertrand et al. 2015).
5.3.6. 3-Phenylpropionic acid scaffold.
In 2016, Astex and GlaxoSmithKline reported a 3-phenylpropionic acid scaffold identified from a fragment-based drug design (FBDD) approach (Davies et al. 2016). Initially, three fragment hits, 4-chlorophenylpropionic acid, 2,6-dimethyl-4H-pyrano[3,4-d]oxazol-4-one, and benzenesulfonamide, were found to occupy hotspots of the Keap1 Kelch domain, consisting of the acid, planar acceptor, and sulfonamide pockets. Starting from the anchor fragment, 4-chlorophenylpropionic acid, located in the acid hotspot, an appropriate functionality designed based on the structures of the other fragment hits was introduced in a stepwise manner for growing toward the planar acceptor and sulfonamide parts, respectively (Heightman et al. 2019). Through SAR studies, 22, possessing the methylated benzoxathiazepine and methoxybenzotriazole moieties, was finally optimized as the most potent small molecule inhibitor of Keap1-Nrf2 PPI in both FP (IC50 = 15 nM) and ITC (Kd = 1.3 nM) assays (Figure 7) (Davies et al. 2016; Heightman et al. 2019). The biological evaluation in cellular models demonstrated that the lead 22 induced the expression of Nrf2-regulated genes, NQO1 and GCLM, in normal or COPD patient-derived bronchial epithelial cells (Davies et al. 2016). Moreover, it was shown to act as an activator of the Keap1-Nrf2 antioxidant response in an in vivo rat model. Importantly, the results of ozone-induced COPD model suggested that 22 had anti-inflammatory effects (Davies et al. 2016; Heightman et al. 2019).
5.3.7. 1-Phenylpyrazole scaffold.
Astex and GlaxoSmithKline reported a novel class of potent Nrf2 regulators containing an 1-phenylprazole scaffold (Callahan et al. 2017a; Callahan et al. 2017b). The biaryl pyrazole analog 23 showed an IC50 value within the range of 1 to 10 nM in the FP assay and an IC50 of less than 10 nM in the TR-FRET assay (Figure 7) (Callahan et al. 2017b). Another arylcyclohexyl pyrazole derivative 24 was also found to be potent with IC50 values of 10–100 nM in FP assay and less than 10 nM in TR-FRET assay (Figure 7) (Callahan et al. 2017a). Importantly, they exhibited strong cellular activities with EC50 values of less than 1 nM and 10–100 nM in BEAS-2B NQO1 MTT assay, respectively (Callahan et al. 2017a; Callahan et al. 2017b).
5.3.8. 2-Iminocoumarin scaffold.
Recently, the 2-iminocoumarin analog 25 (ZJ01) was identified by Zhang et al. via screening of their in-house library (Jiang et al. 2018). Out of 569 compounds tested using an FP assay at 100 μM, further dose-response studies confirmed that ZJ01 was the most potent inhibitor with an IC50 value of 5.1 μM in the FP assay and a Kd value of 48.1 μM in the SPR assay (Figure 7). Despite its moderate activities, 25 led to a significant increase in the mRNA levels of Nrf2 target genes (HO-1 and NQO1) and reduction in LPS-induced production of ROS and pro-inflammatory cytokines (TNF-α, IL-1β, and IL-6) in H9c2 cardiac cells. ZJ101 was found to have a cardiac protective effect in a mouse model of LPS-induced cardiomyopathy (Jiang et al. 2018).
5.3.9. 1,2-Xylylenediamine scaffold.
Most recently, our group identified a new series of 1,2-xylylenediamine analogs via the molecular dissection of 4 in an effort to improve metabolic stability related to 1,4-diamino position of the naphthalene core part (Abed et al. 2020). Subsequent SAR study of the new scaffold provided compound 26 bearing S-methylated acetate groups as a promising Keap1-Nrf2 PPI inhibitor by showing good activity with an IC50 value of 0.15 μM in the FP assay and high metabolic stability in the human liver microsomes with 98.2% remaining after 90 min of incubation (Figure 7).
5.3.10. Miscellaneous inhibitors.
Representative scaffolds with weak inhibitory or binding activity (more than 10 μM in an in vitro assays) include benzenesulfonyl-pyrimidone, pyrazolidin-3,5-dione, thiazolidin-2,4-dione, and 4-phenyl-4H-1,2,4-triazole scaffolds (Figure 7). In addition to 3, the benzenesulfonyl-pyrimidone analog 27 is another hit compound identified by Silvian et al. via HTS (Marcotte et al. 2013). However, it had very low potency with an IC50 value of 118 μM in a biochemical 2D-FIDA assay and no cellular activity in a Nrf2 specific ARE-driven luciferase reporter cell-based assay (Figure 7). Two classes of PPI inhibitors, 28 and 29, were identified from structure-based virtual screening, along with 17 (Zhuang et al. 2014). In the FA assay, compounds 28 and 29 displayed weak Keap1-Nrf2 inhibitory activity with Kd values of 15.2 μM and 10.4 μM, respectively (Figure 7). Although the two scaffolds were further studied employing hit-based substructure search, there was no improvement in activity during SAR studies (Zhuang et al. 2014). The 4-phenyl-4H-1,2,4-triazole analogues 30 (known as MIND4), discovered by screening, had weak binding activity with a Kd value of 22.8 μM in SPR assay and led to the induced expression of Nrf2-responsive proteins, NQO1 and GCLM (Figure 7) (Kazantsev et al. 2014).
6. COMPARISON OF POTENCY AND BINDING MODE OF DIRECT INHIBITORS
Known small molecule inhibitors of Keap1-Nrf2 PPI, containing diverse scaffolds, have shown a wide range of in vitro biological activities. To obtain a better understanding of which compound is more potent and how its chemical structure influences potency in terms of binding to the Keap1 Kelch domain, reliable and comparable data is required. However, the non-covalent inhibitors were separately evaluated in different labs using different types of assays, such as FP, SPR, or ITC. Even if the same assay was used, different assay conditions (e.g., concentrations of protein and type of probes) lead to inconsistent results. Recently, Bach and coworkers carried out side-by-side assessment studies of the reported Keap1-Nrf2 PPI inhibitors employing FP, SPR, TSA, and NQO1 induction cell-based assays, enabling direct comparison of a number of Nrf2 activators (Tran et al. 2019).
As shown in Figures 6 and 7 and Table 1, we summarize the original and reevaluated biological activities of the non-covalent direct inhibitors of Keap1-Nrf2 PPI. In the case of compounds that have reported assay conditions like concentrations of Keap1 Kelch domain protein and a probe used and Kd values of the corresponding probe, we calculated Ki values from the reported inhibitory activities (IC50) in order to easily compare their potencies. With the exception of 21, potent inhibitors (1, 4, 8, 12, 15, 22, and 24) with a Ki or IC50 value of less than 1 μM generally presented a reasonable correlation of inhibitory potencies in the submicromolar or nanomolar range between the different labs. For example, the THIQ analog 1 had comparable binding potencies with Kd values of 1.0 and 2.4 μM in the SPR assay. Interestingly, the Ki values of compounds 3 and 27 estimated by Bach and coworkers exhibited higher potency than originally reported (3, Ki = 0.25 vs 1.27 μM; 27, Ki = 2.8 vs 28.9 μM). Considering their affinity activity in SPR assay (Kd = 0.37 and 51 μM), further confirmation studies are needed. Surprisingly, others (17, 20–21, and 28–30), with moderate affinity (Ki value ranging from 1 to 10 μM) or weak affinity (Ki value of more than 10 μM), were found to inactive in the FP and/or SPR assays carried out by Bach and coworkers. Collectively, the most potent compounds (1, 4, 8, 12, 15, 22, and 24) showed robust biological results.
Table 1.
Comparison of potencies of known small molecule direct inhibitorsa
| compd | FP/FA (IC50, μM)b | FP/FA (Ki μM)c | SPR (Kd, μM)b | Comparative assessment by Bach et. al. | |
|---|---|---|---|---|---|
| FP (Ki μM)d | SPR (Kd, μM)d | ||||
| 1 | 2.3 | 0.32 | 1.0 | 0.56 | 2.4 |
| 2 | 0.75 | 0.11 | - | - | - |
| 3 | 1.46 | 1.27 | - | 0.25 | 0.37 |
| 4 | 0.0286 | 0.0235 | - | 0.0061 | 0.32 |
| 5 | 0.0144 | 0.0111 | - | - | - |
| 6 | 0.0158 | 0.0124 | - | - | - |
| 7 | 0.043 | 0.036 | - | - | - |
| 8 | 0.063 | 0.034 | 0.044 | 0.053 | 0.19 |
| 9 | 0.085 | 0.049 | 0.4 | - | - |
| 10 | 0.060 | 0.051 | 0.102 | - | - |
| 11 | 0.073 | 0.062 | - | - | - |
| 12 | 0.14 | 0.053 | - | 0.048 | 0.47 |
| 13 | 6.2 | - | - | - | - |
| 14 | 0.15 | - | - | - | - |
| 15 | 0.20 | - | - | 0.0021 | 0.026 |
| 16 | - | - | 1.7 | - | - |
| 17 | - | 2.9f | - | ~71 | >500 |
| 18 | - | 1.14f | - | - | - |
| 19 | 0.081 | 0.069 | - | - | - |
| 20 | 9.8 | 8.5 | - | NA (≤49) | NA (≤14) |
| 21 | 7.1 | 0.18 | NA (≤100) | NA (≤354) | |
| 22 | 0.015 | i | - | 0.001 | 0.0014 |
| 23 | 0.001–0.01 | - | - | - | - |
| 24 | 0.01–0.1 | - | - | 0.013 | 0.019 |
| 25 | - | 5.1f | 48.1 | - | - |
| 26 | 0.15 | 0.015 | - | - | - |
| 27 | 118e | 28.9 | - | 2.8 | 51 |
| 28 | 15.2f | - | ~280 | >150 | |
| 29 | - | 10.4f | - | NA (≤200) | NA (≤286) |
| 30 | - | - | 22.8 | NA (≤77) | >300 |
Activities in FP/FA and SPR assays as originally reported and those obtained in a comparative assessment (Tran et al. 2019). For comparison, the Ki values were calculated from IC50 in the FP assay when the binding affinity of the probe is known (Inoyama et al. 2012). “-” indicates data not reported/available.
Activities originally reported.
Ki values derived from the IC50 values using the Kd of a fluorescent Nrf2 peptide probe and concentrations of Keap1 protein and the probe used (Inoyama et al. 2012).
Activities of the FP and SPR assays as reported by Bach et al. when the select inhibitors were tested under the same FP or SPR assay conditions (Tran et al. 2019). NA: No activity (maximum concentration tested)
The value of IC50 was determined by a 2D-FIDA assay.
Kd as reported.
Among 30 known non-covalent inhibitors of the Keap1-Nrf2 PPI that we discussed above, 9 compounds have co-crystal structures with the Keap1 Kelch domain. On the basis of their activity data and co-crystal structures reported, we examined how many subpockets the direct inhibitors of Keap1-Nrf2 PPI occupy in the Keap1 Kelch domain and how their binding modes contribute to inhibitory potency. As mentioned previously, Keap1 Kelch domain primarily consists of 5 subpockets that have high propensity for ligand binding. Accordingly, the number of subpockets occupied by a ligand and its binding affinity would be critical factors to determine potency. The 1,4-diaminonaphthalene derivatives 8 and 11 perfectly support the hypothesis by showing strong activities (Ki = 34 and 62 nM) and fully occupying all five subpockets, as represented in Figure 8. First, the amide groups of 8 form several hydrogen bonds with polar residues (Ser363, Asn414, Arg415, and Ile461) in P1 and P2 subpockets, while the carboxylic acid of 11 has the polar interactions with Arg380, Asn414, and Arg415. Next, the core naphthalene moiety of 8 and 11 occupies the P3 subpocket, fixing their conformations as an anchoring fragment. In the hydrophobic subpockets P4 and P5, the hydroxyl groups of Ser508 and Ser602 were shown to have hydrogen bonding interactions with the sulfonamide oxygen groups of 8, and aromatic amino acids (Tyr334, Tyr525, and Phe577) displayed π-π stacking interactions with the phenyl rings of 8. Similar to 8, the sulfonamide moieties of 11 provided hydrogen bonding interactions with serine residues (Ser508, Ser555, and Ser602). Intriguingly, the naphthalene analogs 3 and 13 without polar functional groups exhibited lower potencies than 8 and 11 in the micromolar range (Ki = 1.27 μM and IC50 = 6.2 μM, respectively) and only occupied three subpockets (P3–P5), as shown in Figure 8. These results support the fact that the polar subpockets, P1 and P2, act as key factors for high binding affinity to the Keap1 Kelch domain via strong polar interactions. For compound 12 containing pyrrolidine-3-carboxylic acid, it still maintained potent activity (Ki = 53 nM) in spite of occupying three subpockets (P1, P3, and P5), as seen in Figure 8. Although it does not have moieties located in P2 and P4 subpockets, the naphthalene analog 12 is well positioned over the three hotspots and its carboxylic acid forms key interactions with Arg483 and Ser508 in the P1 subpocket.
Figure 8.

Co-crystal structures of the Keap1 Kelch domain in complex with a direct small molecule inhibitor of Keap1-Nrf2 PPI. Potency intensity of compounds were classified into potent (Ki or IC50 < 1 μM), moderate (1 μM ≤ K i ≤ 10 μM) and weak (Ki > 10 μM) inhibition, based on the Ki value calculated from the first reported inhibitory act ivity and was represented with the color of a picture border (potent: blue, moderate: green, and weak: red). aThe crystallographic data of 27 indicated two small molecules posed in the Keap1 Kelch cavity. bIf a Ki is not available, IC50 value was used as potency.
Despite the absence of strong polar interactions in the P2 subpocket, the 3-phenylpropionic acid analog 22, which occupies four subpockets (P1 and P3–P5), also had excellent inhibitory activity (IC50 = 15 nM). Similar to the benzenesulfonamide moiety in the P4, the benzotriazole group of 22 showed a π-π stacking interaction with Tyr525 and hydrogen bonding interactions with Gln530 and Ser555 (Figure 8). The THIQ derivatives 1 and 2 demonstrates how important the number of occupied subpockets is for potency. As seen in Figure 8, they partially occupy P2, P3, and P5 subpocket, which may lead to moderate binding affinity to the Keap1 Kelch domain (1, Kd = 1.0 μM) and submicromolar activity. Although the carboxylic acid group of 2 exhibited a polar interaction with Asn414 in the P2 subpocket, its activity was not strong with a Ki value of 0.11 μM, relative to the fully occupied compounds 8 and 11. It appears that the conformation of 2 is almost located on the right side of the Kelch cavity, which makes the P1 and P4 subpockets vacant and hence weakens the binding to the target Keap1 protein (Figure 8). Moreover, compound 1, which has a little different chemical structure from 2, was 3-fold less potent due to a loss of the hydrogen bond in the P2 subpocket. The co-crystal structure of the weak inhibitor 27 bound to the Kelch domain revealed that two small molecules bind side-by-side in the cavity, as represented in Figure 8. The benzenesulfonyl-pyrimidone analog 27 partially occupies right (P3 and P5) or left (P1 and P3–P4) sides, leading to low binding affinity. Additionally, fewer binding interactions, such as hydrogen bonds with Arg415 and Ser602, and a hydrophobic stacking interaction with Tyr334, were observed, in comparison with more active inhibitors.
7. CONCLUSION
The Keap1-Nrf2-ARE signaling system is an attractive drug target for the prevention and treatment of oxidative stress-related diseases including cancer, neurodegenerative disease and COPD, through the induction of the expression of cellular cytoprotective proteins. Indirect inhibitors, which form a covalent bond with cysteine residues in the Keap1 protein, alter the structure of the Keap1-Nrf2-Cul3 complex and consequently activate antioxidant defense system. Although the electrophilic activators of Nrf2 have reached clinical trials, they may interact with multiple targets and have a risk for toxic side effects because of off-target activity. In this respect, direct non-covalent inhibitors of Keap1-Nrf2 PPI have the advantage of target selectivity over the electrophiles. Despite the difficulty in targeting PPI, different types of PPI inhibitors containing diverse chemical scaffolds have been discovered and studied over the past few years. Along with structural studies of the Keap1 protein and Keap1-Nrf2 PPI, the development of diverse assay technologies and drug design approaches have provided a substantial interest in searching for novel small molecule inhibitors that act as Nrf2 activators. To pursue a rational and efficient development process, a thorough understanding of the correlation of known compounds between their potency and binding interactions to the target Keap1 Kelch domain is essential, helping in designing a variety of more potent Nrf2 modulators and thereby increasing the success rate of drug development.
ACKNOWLEDGMENTS
We gratefully acknowledge the financial support of grant CA133791 (to L.H.) from the National Institutes of Health.
ABBREVIATIONS USED
- AD
Alzheimer’s disease
- ALS
amyotrophic lateral sclerosis
- ARE
antioxidant response element
- BTB
broad-complex, Tramtrack and Bric-a-Brac
- bZip
leucine zipper
- CBP
CREB-binding protein
- CDDO-Me
bardoxolone methyl
- CKD
chronic kidney disease
- COPD
chronic obstructive pulmonary disease
- Cul3
cullin3
- 2D-FIDA
two-dimensional fluorescence intensity distribution analysis
- DGR
double glycine repeat
- FBDD
fragment-based drug discovery
- DMF
dimethyl fumarate
- FA
fluorescence anisotropy
- FITC
fluorescein isothiocyanate
- FP
fluorescence polarization
- GCL
glutamate-cysteine ligase
- GCLC
glutamate-cysteine ligase catalytic
- GCLM
glutamate-cysteine ligase modifier
- GPx
glutathione peroxidase
- GST
glutathione S-transferase
- HD
Huntington’s disease
- HMOX1
heme oxygenase 1
- HO-1
heme-oxygenase-1
- HTS
high throughput screening
- ITC
isothermal titration calorimetry assay
- IVR
intervening region
- Keap1
Kelch-like ECH-associated protein 1
- Maf
musculoaponeurotic fibrosarcoma protein
- MD
molecular dynamics
- MM-GBSA
molecular mechanics generalized Born surface area
- MS
multiple sclerosis
- NADPH
Nicotinamide adenine dinucleotide phosphate
- Neh
Nrf2-ECH homology
- NQO1
NAD(P)H:quinone oxidoreductase 1
- NFE2
nuclear factor, erythroid-derived 2
- Nrf2
nuclear factor erythroid 2-related factor 2
- PD
Parkinson’s disease
- PDB
Protein Data Bank
- PPI
protein-protein interaction
- Rbx1
RING-box protein 1
- qRT-PCR
quantitative real-time polymerase chain reaction
- RNS
reactive nitrogen species
- ROS
reactive oxygen species
- SAR
structure–activity relationship
- sMaf
small musculoaponeurotic fibrosarcoma protein
- SOD
superoxide dismutase
- SPR
surface plasmon resonance
- THIQ
tetrahydroisoquinoline
- TR-FRET
time-resolved fluorescence (or Förster) resonance energy transfer
- TRX
thioredoxin
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Declaration of Conflicting Interests
The authors have filed patents on some of the compounds discussed in this paper.
REFERENCS
- Abed DA, Goldstein M, Albanyan H, Jin H, Hu L (2015) Discovery of direct inhibitors of Keap1–Nrf2 protein–protein interaction as potential therapeutic and preventive agents. Acta Pharm Sin B 5:285–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abed DA, Lee S, Hu L (2020) Discovery of disubstituted xylylene derivatives as small molecule direct inhibitors of Keap1-Nrf2 protein-protein interaction. Bioorg Med Chem 28:115343. [DOI] [PubMed] [Google Scholar]
- Arkin Michelle R, Tang Y, Wells James A (2014) Small-molecule inhibitors of protein-protein interactions: progressing toward the reality. Chem Biol 21:1102–1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird L, Llères D, Swift S, Dinkova-Kostova AT (2013) Regulatory flexibility in the Nrf2-mediated stress response is conferred by conformational cycling of the Keap1-Nrf2 protein complex. Proc Natl Acad Sci U S A 110:15259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird L, Swift S, Llères D, Dinkova-Kostova AT (2014) Monitoring Keap1–Nrf2 interactions in single live cells. Biotechnol Adv 32:1133–1144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand HC, Schaap M, Baird L, Georgakopoulos ND, Fowkes A, Thiollier C, Kachi H, Dinkova - Kostova AT, Wells G (2015) Design, synthesis, and evaluation of triazole derivatives that induce Nrf2 dependent gene products and inhibit the Keap1–Nrf2 protein–protein interaction. J Med Chem 58:7186–7194 [DOI] [PubMed] [Google Scholar]
- Blundell TL, Burke DF, Chirgadze D, Dhanaraj V, Hyvonen M, Innis CA, Parisini E, Pellegrini L, Sayed M, Sibanda BL (2000) Protein-protein interactions in receptor activation and intracellular signalling. Biol Chem 381:955–959 [DOI] [PubMed] [Google Scholar]
- Boerboom A-MJF, Vermeulen M, van der Woude H, Bremer BI, Lee-Hilz YY, Kampman E, van Bladeren PJ, Rietjens IMCM, Aarts JMMJG (2006) Newly constructed stable reporter cell lines for mechanistic studies on electrophile-responsive element-mediated gene expression reveal a role for flavonoid planarity. Biochem Pharmacol 72:217–226 [DOI] [PubMed] [Google Scholar]
- Boutten A, Goven D, Artaud-Macari E, Boczkowski J, Bonay M (2011) NRF2 targeting: a promising therapeutic strategy in chronic obstructive pulmonary disease. Trends Mol Med 17:363–371 [DOI] [PubMed] [Google Scholar]
- Calkins MJ, Jakel RJ, Johnson DA, Chan K, Kan YW, Johnson JA (2005) Protection from mitochondrial complex II inhibition in vitro and in vivo by Nrf2-mediated transcription. Proc Natl Acad Sci U S A 102:244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan JF, Kerns JJ, Li T, Nie H, Pero JE, Davies TG, Heightman TD, Woolford AJ, Griffiths -Jones CM, Norton D, Verdonk ML, Howard S (2017a) Arylcyclohexyl pyrazoles as Nrf2 regulators. WO2017060855 [Google Scholar]
- Callahan JF, Kerns JJ, Li TN, H., Pero JE, Davies TG, Heightman TD, Woolford A, Griffiths-Jones CM, Norton D, Willems HMG, Verdonk ML, Carr MG (2017b) Biaryl pyrazoles as Nrf2 regulators. WO2017060854 [Google Scholar]
- Canning P, Sorrell FJ, Bullock AN (2015) Structural basis of Keap1 interactions with Nrf2. Free Radical Biol Med 88:101–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabas SA, Jiang Q, McMahon M, McWalter GK, McLellan LI, Elcombe CR, Henderson CJ, Wolf CR, Moffat GJ, Itoh K, Yamamoto M, Hayes JD (2002) Loss of the Nrf2 transcription factor causes a marked reduction in constitutive and inducible expression of the glutathione S-transferase Gsta1, Gsta2, Gstm1, Gstm2, Gstm3 and Gstm4 genes in the livers of male and female mice. Biochem J 365:405–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P-C, Vargas MR, Pani AK, Smeyne RJ, Johnson DA, Kan YW, Johnson JA (2009) Nrf2-mediated neuroprotection in the MPTP mouse model of Parkinson’s disease: Critical role for the astrocyte. Proc Natl Acad Sci U S A 106:2933–2938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Inoyama D, Kong A-NT, Beamer LJ, Hu L (2011) Kinetic analyses of Keap1–Nrf2 interaction and determination of the minimal Nrf2 peptide sequence required for Keap1 binding using surface plasmon resonance. Chem Biol Drug Des 78:1014–1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleasby A, Yon J, Day PJ, Richardson C, Tickle IJ, Williams PA, Callahan JF, Carr R, Concha N, Kerns JK, Qi H, Sweitzer T, Ward P, Davies TG (2014) Structure of the BTB Domain of Keap1 and Its Interaction with the Triterpenoid Antagonist CDDO. PLoS One 9:e98896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies TG, Wixted WE, Coyle JE, Griffiths-Jones C, Hearn K, McMenamin R, Norton D, Rich SJ, Richardson C, Saxty G, Willems HMG, Woolford AJA, Cottom JE, Kou J-P, Yonchuk JG, Feldser HG, Sanchez Y, Foley JP, Bolognese BJ, Logan G, Podolin PL, Yan H, Callahan JF, Heightman TD, Kerns JK (2016) Monoacidic inhibitors of the Kelch-like ECH-associated protein 1: nuclear factor erythroid 2-related factor 2 (KEAP1:NRF2) protein–protein interaction with high cell potency identified by fragment-based discovery. J Med Chem 59:3991–4006 [DOI] [PubMed] [Google Scholar]
- De Zeeuw D, Akizawa T, Audhya P, Bakris GL, Chin M, Christ-Schmidt H, Goldsberry A, Houser M, Krauth M, Lambers Heerspink HJ (2013) Bardoxolone methyl in type 2 diabetes and stage 4 chronic kidney disease. N Engl J Med 369:2492–2503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimmock JR, Elias DW, Beazely MA, Kandepu NM (1999) Bioactivities of chalcones. Curr Med Chem 6:1125–1149 [PubMed] [Google Scholar]
- Dinkova-Kostova AT, Holtzclaw WD, Cole RN, Itoh K, Wakabayashi N, Katoh Y, Yamamoto M, Talalay P (2002) Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc Natl Acad Sci U S A 99:11908–11913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinkova-Kostova AT, Holtzclaw WD, Kensler TW (2005) The role of Keap1 in cellular protective responses. Chem Res Toxicol 18:1779–1791 [DOI] [PubMed] [Google Scholar]
- Dinkova-Kostova AT, Wang XJ (2011) Induction of the Keap1/Nrf2/ARE pathway by oxidizable diphenols. Chem Biol Interact 192:101–106 [DOI] [PubMed] [Google Scholar]
- DiscoverX (2011) PathHunter® Keap1–Nrf2 functional assay for chemiluminescent detection of activated Nrf2. In. Product Booklet: 93–0821C3, p 12 [Google Scholar]
- Eggler A, Liu G, M Pezzuto J, van Breemen R, D Mesecar A (2005) Modifying specific cysteines of the electrophile-sensing human Keap1 protein is insufficient to disrupt binding to the Nrf2 domain Neh2. Proc Natl Acad Sci U S A 102:10070–10075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggler Aimee L, Small E, Hannink M, Mesecar Andrew D (2009) Cul3-mediated Nrf2 ubiquitination and antioxidant response element (ARE) activation are dependent on the partial molar volume at position 151 of Keap1. Biochem J 422:171–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel T, Holbrook NJ (2000) Oxidants, oxidative stress and the biology of ageing. Nature 408:239–247 [DOI] [PubMed] [Google Scholar]
- Fujiwara KT, Kataoka K, Nishizawa M (1993) Two new members of the maf oncogene family, mafK and mafF, encode nuclear b-Zip proteins lacking putative trans-activator domain. Oncogene 8:2371–2380 [PubMed] [Google Scholar]
- Fukutomi T, Takagi K, Mizushima T, Ohuchi N, Yamamoto M (2014) Kinetic, thermodynamic, and structural characterizations of the association between Nrf2-DLGex degron and Keap1. Mol Cell Biol 34:832–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geismann C, Arlt A, Sebens S, Schäfer H (2014) Cytoprotection “gone astray”: Nrf2 and its role in cancer. Onco Targets Ther 7:1497–1518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go ML, Wu X, Liu XL (2005) Chalcones: an update on cytotoxic and chemoprotective properties. Curr Med Chem 12:481–499 [DOI] [PubMed] [Google Scholar]
- Golden TR, Patel M (2008) Catalytic antioxidants and neurodegeneration. Antioxid Redox Signaling 11:555–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall MD, Yasgar A, Peryea T, Braisted JC, Jadhav A, Simeonov A, Coussens NP (2016) Fluorescence polarization assays in high-throughput screening and drug discovery: a review. Methods Appl Fluoresc 4:022001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes JD, McMahon M, Chowdhry S, Dinkova-Kostova AT (2010) Cancer chemoprevention mechanisms mediated through the Keap1–Nrf2 pathway. Antioxid Redox Signaling 13:1713–1748 [DOI] [PubMed] [Google Scholar]
- He X, Ma Q (2010) Critical cysteine residues of Kelch-like ECH-associated protein 1 in arsenic sensing and suppression of nuclear factor erythroid 2-related factor 2. J Pharmacol Exp Ther 332:66–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heightman TD, Callahan JF, Chiarparin E, Coyle JE, Griffiths-Jones C, Lakdawala AS, McMenamin R, Mortenson PN, Norton D, Peakman TM, Rich SJ, Richardson C, Rumsey WL, Sanchez Y, Saxty G, Willems HMG, Wolfe L, Woolford AJA, Wu Z, Yan H, Kerns JK, Davies TG (2019) Structure–activity and structure–conformation relationships of aryl propionic acid inhibitors of the Kelch-like ECH-associated protein 1/nuclear factor erythroid 2-related factor 2 (KEAP1/NRF2) protein–protein interaction. J Med Chem 62:4683–4702 [DOI] [PubMed] [Google Scholar]
- Honda T, Rounds BV, Gribble GW, Suh N, Wang Y, Sporn MB (1998) Design and synthesis of 2-cyano-3,12-dioxoolean-1,9-dien-28-oic acid, a novel and highly active inhibitor of nitric oxide production in mouse macrophages. Bioorg Med Chem Lett 8:2711–2714 [DOI] [PubMed] [Google Scholar]
- Hong F, Freeman ML, Liebler DC (2005) Identification of sensor cysteines in human Keap1 modified by the cancer chemopreventive agent sulforaphane. Chem Res Toxicol 18:1917–1926 [DOI] [PubMed] [Google Scholar]
- Hu L, Magesh S, Chen L, Lewis T, Munoz B, Wang L (2013a) Direct inhibitors of Keap1-Nrf2 interaction as antioxidant inflammation modulators. WO2013067036 A1 [Google Scholar]
- Hu L, Magesh S, Chen L, Wang L, Lewis TA, Chen Y, Khodier C, Inoyama D, Beamer LJ, Emge TJ, Shen J, Kerrigan JE, Kong AN, Dandapani S, Palmer M, Schreiber SL, Munoz B (2013b) Discovery of a small-molecule inhibitor and cellular probe of Keap1-Nrf2 protein-protein interaction. Bioorg Med Chem Lett 23:3039–3043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ighodaro OM, Akinloye OA (2018) First line defence antioxidants-superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPX): Their fundamental role in the entire antioxidant defence grid. Alexandria J Med 54:287–293 [Google Scholar]
- Iizuka T, Ishii Y, Itoh K, Kiwamoto T, Kimura T, Matsuno Y, Morishima Y, Hegab AE, Homma S, Nomura A, Sakamoto T, Shimura M, Yoshida A, Yamamoto M, Sekizawa K (2005) Nrf2-deficient mice are highly susceptible to cigarette smoke-induced emphysema. Genes Cells 10:1113–1125 [DOI] [PubMed] [Google Scholar]
- Inoyama D, Chen Y, Huang X, Beamer LJ, Kong A-NT, Hu L (2012) Optimization of fluorescently labeled Nrf2 peptide probes and the development of a fluorescence polarization assay for the discovery of inhibitors of Keap1-Nrf2 interaction. J Biomol Screen 17:435–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii Y, Itoh K, Morishima Y, Kimura T, Kiwamoto T, Iizuka T, Hegab AE, Hosoya T, Nomura A, Sakamoto T, Yamamoto M, Sekizawa K (2005) Transcription factor Nrf2 plays a pivotal role in protection against elastase-induced pulmonary inflammation and emphysema. J Immunol 175:6968–6975 [DOI] [PubMed] [Google Scholar]
- Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayama I, Yamamoto M, Nabeshima Y-i (1997) An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun 236:313–322 [DOI] [PubMed] [Google Scholar]
- Itoh K, Mimura J, Yamamoto M (2010) Discovery of the negative regulator of Nrf2, Keap1: a historical overview. Antioxid Redox Signaling 13:1665–1678 [DOI] [PubMed] [Google Scholar]
- Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, Yamamoto M (1999) Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev 13:76–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain AD, Potteti H, Richardson BG, Kingsley L, Luciano JP, Ryuzoji AF, Lee H, Krunic A, Mesecar AD, Reddy SP, Moore TW (2015) Probing the structural requirements of non-electrophilic naphthalene-based Nrf2 activators. Eur J Med Chem 103:252–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C-S, Zhuang C-L, Zhu K, Zhang J, Muehlmann LA, Figueiró Longo JP, Azevedo RB, Zhang W, Meng N, Zhang H (2018) Identification of a novel small-molecule Keap1–Nrf2 PPI inhibitor with cytoprotective effects on LPS-induced cardiomyopathy. J Enzyme Inhib Med Chem 33:833–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Z-Y, Lu M-C, You Q-D (2016) Discovery and development of Kelch -like ECH-associated protein 1. nuclear factor erythroid 2-related factor 2 (KEAP1:NRF2) protein–protein interaction inhibitors: achievements, challenges, and future directions. J Med Chem 59:10837–10858 [DOI] [PubMed] [Google Scholar]
- Jiang Z-Y, Xu L-L, Lu M-C, Pan Y, Huang H-Z, Zhang X-J, Sun H-P, You Q-D (2014a) Investigation of the intermolecular recognition mechanism between the E3 ubiquitin ligase Keap1 and substrate based on multiple substrates analysis. J Comput Aided Mol Des 28:1233–1245 [DOI] [PubMed] [Google Scholar]
- Jiang Z-Y, Xu LL, Lu M-C, Chen Z-Y, Yuan Z-W, Xu X-L, Guo X-K, Zhang X-J, Sun H-P, You Q-D (2015) Structure–activity and structure–property relationship and exploratory in vivo evaluation of the nanomolar Keap1–Nrf2 protein–protein interaction inhibitor. J Med Chem 58:6410–6421 [DOI] [PubMed] [Google Scholar]
- Jiang ZY, Lu MC, Xu LL, Yang TT, Xi MY, Xu XL, Guo XK, Zhang XJ, You QD, Sun HP (2014b) Discovery of potent Keap1-Nrf2 protein-protein interaction inhibitor based on molecular binding determinants analysis. J Med Chem 57:2736–2745 [DOI] [PubMed] [Google Scholar]
- Jiménez-Osorio AS, Picazo A, González-Reyes S, Barrera-Oviedo D, Rodríguez-Arellano ME, Pedraza-Chaverri J (2014) Nrf2 and redox status in prediabetic and diabetic patients. Int J Mol Sci 15:20290–20305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jnoff E, Albrecht C, Barker JJ, Barker O, Beaumont E, Bromidge S, Brookfield F, Brooks M, Bubert C, Ceska T (2014) Binding mode and structure–activity relationships around direct inhibitors of the Nrf2–Keap1 complex. ChemMedChem 9:699–705 [DOI] [PubMed] [Google Scholar]
- Johnson DA, Amirahmadi S, Ward C, Fabry Z, Johnson JA (2009) The absence of the pro-antioxidant transcription factor Nrf2 exacerbates experimen tal autoimmune encephalomyelitis. Toxicol Sci 114:237–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kansanen E, Kuosmanen SM, Leinonen H, Levonen A-L (2013) The Keap1-Nrf2 pathway: mechanisms of activation and dysregulation in cancer. Redox Biol 1:45–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh Y, Iida K, Kang M-I, Kobayashi A, Mizukami M, Tong KI, McMahon M, Hayes JD, Itoh K, Yamamoto M (2005) Evolutionary conserved N-terminal domain of Nrf2 is essential for the Keap1-mediated degradation of the protein by proteasome. Arch Biochem Biophys 433:342–350 [DOI] [PubMed] [Google Scholar]
- Katoh Y, Itoh K, Yoshida E, Miyagishi M, Fukamizu A, Yamamoto M (2001) Two domains of Nrf2 cooperatively bind CBP, a CREB binding protein, and synergistically activate transcription. Genes Cells 6:857–868 [DOI] [PubMed] [Google Scholar]
- Katsuoka F, Motohashi H, Ishii T, Aburatani H, Engel JD, Yamamoto M (2005) Genetic evidence that small maf proteins are essential for the activation of antioxidant response element-dependent genes. Mol Cell Biol 25:8044–8051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazantsev AG, Thompson LM, Abagyan R, Casale M (2014) Small molecule activators of Nrf2 pathway. [Google Scholar]
- Kensler TW, Wakabayashi N (2009) Nrf2: friend or foe for chemoprevention? Carcinogenesis 31:90–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khor TO, Huang M-T, Kwon KH, Chan JY, Reddy BS, Kong A-N (2006) Nrf2-deficient mice have an increased susceptibility to dextran sulfate sodium–induced colitis. Cancer Res 66:11580–11584 [DOI] [PubMed] [Google Scholar]
- Kirkham PA, Barnes PJ (2013) Oxidative stress in COPD. Chest 144:266–273 [DOI] [PubMed] [Google Scholar]
- Kobayashi A, Kang M-I, Okawa H, Ohtsuji M, Zenke Y, Chiba T, Igarashi K, Yamamoto M (2004) Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol 24:7130–7139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V, Kumar S, Hassan M, Wu H, Thimmulappa RK, Kumar A, Sharma SK, Parmar VS, Biswal S, Malhotra SV (2011) Novel chalcone derivatives as potent Nrf2 activators in mice and human lung epithelial cells. J Med Chem 54:4147–4159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurzawski M, Dziedziejko V, Urasińska E, Post M, Wójcicki M, Miętkiewski J, Droździk M (2012) Nuclear factor erythroid 2-like 2 (Nrf2) expression in end-stage liver disease. Environ Toxicol Pharmacol 34:87–95 [DOI] [PubMed] [Google Scholar]
- Lazzara PR, David BP, Ankireddy A, Richardson BG, Dye K, Ratia KM, Reddy SP, Moore TW (2020) Isoquinoline Kelch-like ECH-associated protein 1-nuclear factor (erythroid-derived 2)-like 2 (KEAP1-NRF2) inhibitors with high metabolic stability. J Med Chem ASAP article:DOI: 10.1021/acs.jmedchem.1029b01074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lea WA, Simeonov A (2011) Fluorescence polarization assays in small molecule screening. Expert Opin Drug Discov 6:17–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D-H, Gold R, Linker RA (2012) Mechanisms of oxidative damage in multiple sclerosis an d neurodegenerative diseases: therapeutic modulation via fumaric acid esters. Int J Mol Sci 13:11783–11803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J-M, Johnson JA (2004) An important role of Nrf2-ARE pathway in the cellular defense mechanism. J Biochem Mol Biol 37:139–143 [DOI] [PubMed] [Google Scholar]
- Li J, Ichikawa T, Janicki JS, Cui T (2009) Targeting the Nrf2 pathway against cardiovascular disease. Expert Opin Ther Targets 13:785–794 [DOI] [PubMed] [Google Scholar]
- Li J, Johnson D, Calkins M, Wright L, Svendsen C, Johnson J (2004a) Stabilization of Nrf2 by tBHQ confers protection against oxidative stress-induced cell death in human neural stem cells. Toxicol Sci 83:313–328 [DOI] [PubMed] [Google Scholar]
- Li X, Zhang D, Hannink M, Beamer LJ (2004b) Crystal structure of the Kelch domain of human Keap1. J Biol Chem 279:54750–54758 [DOI] [PubMed] [Google Scholar]
- Li Y, Paonessa JD, Zhang Y (2012) Mechanism of chemical activation of Nrf2. PLoS One 7:e35122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liby KT, Yore MM, Sporn MB (2007) Triterpenoids and rexinoids as multifunctional agents for the prevention and treatment of cancer. Nat Rev Cancer 7:357–369 [DOI] [PubMed] [Google Scholar]
- Linker RA, Lee D-H, Ryan S, van Dam AM, Conrad R, Bista P, Zeng W, Hronowsky X, Buko A, Chollate S, Ellrichmann G, Brück W, Dawson K, Goelz S, Wiese S, Scannevin RH, Lukashev M, Gold R (2011) Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain 134:678–692 [DOI] [PubMed] [Google Scholar]
- Lo SC, Li X, Henzl MT, Beamer LJ, Hannink M (2006) Structure of the Keap1: Nrf2 interface provides mechanistic insight into Nrf2 signaling. EMBO J 25:3605–3617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M-C, Chen Z-Y, Wang Y-L, Jiang Y-L, Yuan Z-W, You Q-D, Jiang Z-Y (2015) Binding thermodynamics and kinetics guided optimization of potent Keap1–Nrf2 peptide inhibitors. RSC Adv 5:85983–85987 [Google Scholar]
- Lu M-C, Tan S-J, Ji J-A, Chen Z-Y, Yuan Z-W, You Q-D, Jiang Z-Y (2016) Polar recognition group study of Keap1-Nrf2 protein–protein interaction inhibitors. ACS Med Chem Lett 7:835–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M-C, Zhang X, Wu F, Tan S-J, Zhao J, You Q-D, Jiang Z-Y (2019a) Discovery of a potent Kelch-like ECH-associated protein 1-nuclear factor erythroid 2-related factor 2 (Keap1–Nrf2) protein–protein interaction inhibitor with natural proline structure as a cytoprotective agent against acetaminophen-induced hepatotoxicity. J Med Chem 62:6796–6813 [DOI] [PubMed] [Google Scholar]
- Lu M-C, Zhao J, Liu Y-T, Liu T, Tao M-M, You Q-D, Jiang Z-Y (2019b) CPUY192018, a potent inhibitor of the Keap1-Nrf2 protein-protein interaction, alleviates renal inflammation in mice by restricting oxidative stress and NF-κB activation. Redox Bio 26:101266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Q (2013) Role of Nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol 53:401–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Q, Battelli L, Hubbs AF (2006) Multiorgan autoimmune inflammation, enhanced lymphoproliferation, and impaired homeostasis of reactive oxygen species in mice lacking the antioxidant-activated transcription factor Nrf2. Am J Pathol 168:1960–1974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magesh S, Chen Y, Hu L (2012) Small molecule modulators of Keap1-Nrf2-ARE pathway as potential preventive and therapeutic agents. Med Res Rev 32:687–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maicas N, Ferrándiz ML, Brines R, Ibáñez L, Cuadrado A, Koenders MI, van den Berg WB, Alcaraz MJ (2011) Deficiency of Nrf2 accelerates the effector phase of arthritis and aggravates joint disease. Antioxid Redox Signaling 15:889–901 [DOI] [PubMed] [Google Scholar]
- Marcotte D, Zeng W, Hus J-C, McKenzie A, Hession C, Jin P, Bergeron C, Lugovskoy A, Enyedy I, Cuervo H, Wang D, Atmanene C, Roecklin D, Vecchi M, Vivat V, Kraemer J, Winkler D, Hong V, Chao J, Lukashev M, Silvian L (2013) Small molecules inhibit the interaction of Nrf2 and the Keap1 Kelch domain through a non-covalent mechanism. Bioorg Med Chem 21:4011–4019 [DOI] [PubMed] [Google Scholar]
- McMahon M, Thomas N, Itoh K, Yamamoto M, Hayes JD (2004) Redox-regulated turnover of Nrf2 is determined by at least two separate protein domains, the redox-sensitive Neh2 degron and the redox-insensitive Neh6 degron. J Biol Chem 279:31556–31567 [DOI] [PubMed] [Google Scholar]
- McMahon M, Thomas N, Itoh K, Yamamoto M, Hayes JD (2006) Dimerization of substrate adaptors can facilitate cullin-mediated ubiquitylation of proteins by a “tethering” mechanism: a two-site interaction model for the Nrf2-Keap1 complex. J Biol Chem 281:24756–24768 [DOI] [PubMed] [Google Scholar]
- Meng N, Tang H, Zhang H, Jiang C, Su L, Min X, Zhang W, Zhang H, Miao Z, Zhang W, Zhuang C (2018) Fragment-growing guided design of Keap1-Nrf2 protein-protein interaction inhibitors for targeting myocarditis. Free Radical Biol Med 117:228–237 [DOI] [PubMed] [Google Scholar]
- Moi P, Chan K, Asunis I, Cao A, Kan YW (1994) Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF -E2/AP1 repeat of the beta-globin locus control region. Proc Natl Acad Sci U S A 91:9926–9930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon EJ, Giaccia A (2015) Dual roles of NRF2 in tumor prevention and progression: Possible implications in cancer treatment. Free Radical Biol Med 79:292–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrowietz U, Altmeyer P, Bieber T, Röcken M, Schopf RE, Sterry W (2007) Treatment of psoriasis with fumaric acid esters (Fumaderm®). J Dtsch Dermatol Ges 5:716–717 [DOI] [PubMed] [Google Scholar]
- Nioi P, Nguyen T, Sherratt PJ, Pickett CB (2005) The carboxy-terminal Neh3 domain of Nrf2 is required for transcriptional activation. Mol Cell Biol 25:10895–10906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogura T, Tong KI, Mio K, Maruyama Y, Kurokawa H, Sato C, Yamamoto M (2010) Keap1 is a forked-stem dimer structure with two large spheres enclosing the intervening, double glycine repeat, and C-terminal domains. Proc Natl Acad Sci U S A 107:2842–2847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallesen JS, Tran KT, Bach A (2018) Non-covalent small-molecule Kelch-like ECH-associated protein 1–nuclear factor erythroid 2-related factor 2 (Keap1–Nrf2) inhibitors and their potential for targeting central nervous system diseases. J Med Chem 61:8088–8103 [DOI] [PubMed] [Google Scholar]
- Paul N, McMahon M, Ken I, Yamamoto M, Hayes JD (2003) Identification of a novel Nrf2 -regulated antioxidant response element (ARE) in the mouse NAD (P) H: quinone oxidoreductase 1 gene: reassessment of the ARE consensus sequence. Biochem J 374:337–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pergola PE, Raskin P, Toto RD, Meyer CJ, Huff JW, Grossman EB, Krauth M, Ruiz S, Audhya P, Chr ist-Schmidt H, Wittes J, Warnock DG (2011) Bardoxolone methyl and kidney function in CKD with type 2 diabetes. N Engl J Med 365:327–336 [DOI] [PubMed] [Google Scholar]
- Probst BL, McCauley L, Trevino I, Wigley WC, Ferguson DA (2015a) Cancer cell growth is differentially affected by constitutive activation of NRF2 by KEAP1 deletion and pharmacological activation of NRF2 by the synthetic triterpenoid, RTA 405. PLoS One 10:e0135257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Probst BL, Trevino I, McCauley L, Bumeister R, Dulubova I, Wigley WC, Ferguson DA (2015b) RTA 408, A novel synthetic triterpenoid with broad anticancer and anti-inflammatory activity. PLoS One 10:e0122942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prochaska HJ, Santamaria AB (1988) Direct measurement of NAD(P)H:quinone reductase from cells cultured in microtiter wells: A screening assay for anticarcinogenic enzyme inducers. Anal Biochem 169:328–336 [DOI] [PubMed] [Google Scholar]
- Rachakonda G, Xiong Y, Sekhar KR, Stamer SL, Liebler DC, Freeman ML (2008) Covalent modification at Cys151 dissociates the electrophile sensor Keap1 from the ubiquitin ligase CUL3. Chem Res Toxicol 21:705–710 [DOI] [PubMed] [Google Scholar]
- Rajendran P, Nandakumar N, Rengarajan T, Palaniswami R, Gnanadhas EN, Lakshminarasaiah U, Gopas J, Nishigaki I (2014) Antioxidants and human diseases. Clin Chim Acta 436:332–347 [DOI] [PubMed] [Google Scholar]
- Rangasamy T, Cho CY, Thimmulappa RK, Zhen L, Srisuma SS, Kensler TW, Yamamoto M, Pe trache I, Tuder RM, Biswal S (2004) Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke–induced emphysema in mice. J Clin Invest 114:1248–1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson BG, Jain AD, Potteti HR, Lazzara PR, David BP, Tamatam CR, Choma E, Skowron K, Dye K, Siddiqui Z, Wang Y-T, Krunic A, Reddy SP, Moore TW (2018) Replacement of a naphthalene scaffold in Kelch-like ECH-associated protein 1 (KEAP1)/nuclear factor (erythroid-derived 2)-like 2 (NRF2) inhibitors. J Med Chem 61:8029–8047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robledinos-Anton N, Fernandez-Gines R, Manda G, Cuadrado A (2019) Activators and Inhibitors of NRF2: A Review of Their Potential for Clinical Development. Oxid Med Cell Longev 2019:9372182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rushmore TH, Morton MR, Pickett CB (1991) The antioxidant responsive element. Activation by oxidative stress and identification of the DNA consensus sequence required for functional activity. J Biol Chem 266:11632–11639 [PubMed] [Google Scholar]
- Saito T, Ichimura Y, Taguchi K, Suzuki T, Mizushima T, Takagi K, Hirose Y, Nagahashi M, Iso T, Fukutomi T, Ohishi M, Endo K, Uemura T, Nishito Y, Okuda S, Obata M, Kouno T, Imamura R, Tada Y, Obata R, Yasuda D, Takahashi K, Fujimura T, Pi J, Lee M-S, Ueno T, Ohe T, Mashino T, Wakai T, Kojima H, Okabe T, Nagano T, Motohashi H, Waguri S, Soga T, Yamamoto M, Tanaka K, Komatsu M (2016) p62/Sqstm1 promotes malignancy of HCV-positive hepatocellular carcinoma through Nrf2-dependent metabolic reprogramming. Nat Commun 7:12030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaap M, Hancock R, Wilderspin A, Wells G (2013) Development of a steady-state FRET-based assay to identify inhibitors of the Keap1-Nrf2 protein–protein interaction. Protein Sci 22:1812–1819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvin PR (2002) Principles and biophysical applications of lanthanide-based probes. Annu Rev Biophys Biomol Struct 31:275–302 [DOI] [PubMed] [Google Scholar]
- Sheng C, Dong G, Miao Z, Zhang W, Wang W (2015) State -of-the-art strategies for targeting protein–protein interactions by small-molecule inhibitors. Chem Soc Rev 44:8238–8259 [DOI] [PubMed] [Google Scholar]
- Simonian NA, Coyle JT (1996) Oxidative stress in neurodegenerative diseases. Annu Rev Pharmacol Toxicol 36:83–106 [DOI] [PubMed] [Google Scholar]
- Sirota R, Gibson D, Kohen R (2015) The role of the catecholic and the electrophilic moieties of caffeic acid in Nrf2/Keap1 pathway activation in ovarian carcinoma cell lines. Redox Biol 4:48–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H-P, Jiang Z-Y, Zhang M-Y, Lu M-C, Yang T-T, Pan Y, Huang H-Z, Zhang X-J, You Q-d (2014) Novel protein–protein interaction inhibitor of Nrf2–Keap1 discovered by structure-based virtual screening. MedChemComm 5:93–98 [Google Scholar]
- Suzuki T, Motohashi H, Yamamoto M (2013) Toward clinical application of the Keap1–Nrf2 pathway. Trends Pharmacol Sci 34:340–346 [DOI] [PubMed] [Google Scholar]
- Taguchi K, Maher JM, Suzuki T, Kawatani Y, Motohashi H, Yamamoto M (2010) Genetic analysis of cytoprotective functions supported by graded expression of Keap1. Mol Cell Biol 30:3016–3026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi K, Motohashi H, Yamamoto M (2011) Molecular mechanisms of the Keap1–Nrf2 pathway in stress response and cancer evolution. Genes Cells 16:123–140 [DOI] [PubMed] [Google Scholar]
- Tkachev V, Menshchikova E, Zenkov N (2011) Mechanism of the Nrf2/Keap1/ARE signaling system. Biochemistry (Moscow) 76:407–422 [DOI] [PubMed] [Google Scholar]
- Tonelli C, Chio IIC, Tuveson DA (2017) Transcriptional Regulation by Nrf2. Antioxid Redox Signaling 29:1727–1745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong KI, Katoh Y, Kusunoki H, Itoh K, Tanaka T, Yamamoto M (2006a) Keap1 recruits Neh2 through binding to ETGE and DLG motifs: characterization of the two-site molecular recognition model. Mol Cell Biol 26:2887–2900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong KI, Kobayashi A, Katsuoka F, Yamamoto M (2006b) Two-site substrate recognition model for the Keap1-Nrf2 system: a hinge and latch mechanism. Biol Chem 387:1311–1320 [DOI] [PubMed] [Google Scholar]
- Tong KI, Padmanabhan B, Kobayashi A, Shang C, Hirotsu Y, Yokoyama S, Yamamoto M (2007) Different electrostatic potentials define ETGE and DLG motifs as hinge and latch in oxidative stress response. Mol Cell Biol 27:7511–7521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran KT, Pallesen JS, Solbak SMØ, Narayanan D, Baig A, Zang J, Aguayo-Orozco A, Carmona RMC, Garcia AD, Bach A (2019) A comparative assessment study of known small-molecule Keap1-Nrf2 protein–protein interaction inhibitors: chemical synthesis, binding properties, and cellular activity. J Med Chem 62:8028–8052 [DOI] [PubMed] [Google Scholar]
- Vargas MR, Johnson DA, Sirkis DW, Messing A, Johnson JA (2008) Nrf2 activation in astrocytes protects against neurodegeneration in mouse models of familial amyotrophic lateral sclerosis. J Neurosci 28:13574–13581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh J, Jenkins RE, Wong M, Olayanju A, Powell H, Copple I, O’Neill PM, Goldring CEP, Kitteringham NR, Park BK (2014) Identification and quantification of the basal and inducible Nrf2-dependent proteomes in mouse liver: Biochemical, pharmacological and toxicological implications. J Proteomics 108:171–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Liu K, Geng M, Gao P, Wu X, Hai Y, Li Y, Li Y, Luo L, Hayes JD, Wang XJ, Tang X (2013) RXRα inhibits the NRF2-ARE signaling pathway through a direct interaction with the Neh7 domain of NRF2. Cancer Res 73:3097–3108 [DOI] [PubMed] [Google Scholar]
- Wen X, Thorne G, Hu L, Joy MS, Aleksunes LM (2015) Activation of NRF2 signaling in HEK293 cells by a first-in-class direct KEAP1-NRF2 inhibitor. J Biochem Mol Toxicol 29:261–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkel AF, Engel CK, Margerie D, Kannt A, Szillat H, Glombik H, Kallus C, Ruf S, Güssregen S, Riedel J, Herling AW, von Knethen A, Weigert A, Brüne B, Schmoll D (2015) Characterization of RA839, a noncovalent small molecule binder to Keap1 and selective activator of Nrf2 signaling. J Biol Chem 290:28446–28455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo SY, Kim JH, Moon MK, Han S-H, Yeon SK, Choi JW, Jang BK, Song HJ, Kang YG, Kim JW, Lee J, Kim DJ, Hwang O, Park KD (2014) Discovery of vinyl sulfones as a novel class of neuroprotective agents toward Parkinson’s disease therapy. J Med Chem 57:1473–1487 [DOI] [PubMed] [Google Scholar]
- Wruck CJ, Fragoulis A, Gurzynski A, Brandenburg L-O, Kan YW, Chan K, Hassenpflug J, Freitag-Wolf S, Varoga D, Lippross S, Pufe T (2011) Role of oxidative stress in rheumatoid arthritis: insights from the Nrf2-knockout mice. Ann Rheum Dis 70:844–850 [DOI] [PubMed] [Google Scholar]
- Wu RP, Hayashi T, Cottam HB, Jin G, Yao S, Wu CCN, Rosenbach MD, Corr M, Schwab RB, Carson DA (2010) Nrf2 responses and the therapeutic selectivity of electrophilic compounds in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A 107:7479–7484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T, Suzuki T, Kobayashi A, Wakabayashi J, Maher J, Motohashi H, Yamamoto M (2008) Physiological significance of reactive cysteine residues of Keap1 in determining Nrf2 activity. Mol Cell Biol 28:2758–2770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda D, Nakajima M, Yuasa A, Obata R, Takahashi K, Ohe T, Ichimura Y, Komatsu M, Yamamoto M, Imamura R (2016) Synthesis of Keap1-phosphorylated p62 and Keap1-Nrf2 protein-protein interaction inhibitors and their inhibitory activity. Bioorg Med Chem Lett 26:5956–5959 [DOI] [PubMed] [Google Scholar]
- Yasuda D, Yuasa A, Obata R, Nakajima M, Takahashi K, Ohe T, Ichimura Y, Komatsu M, Yamamoto M, Imamura R, Kojima H, Okabe T, Nagano T, Mashino T (2017) Discovery of benzo[g]indoles as a novel class of non-covalent Keap1-Nrf2 protein-protein interaction inhibitor. Bioorg Med Chem Lett 27:5006–5009 [DOI] [PubMed] [Google Scholar]
- Yoh K, Itoh K, Enomoto A, Hirayama A, Yamaguchi N, Kobayashi M, Morito N, Koyama A, Yamamoto M, Takahashi S (2001) Nrf2-deficient female mice develop lupus-like autoimmune nephritis. Kidney Int 60:1343–1353 [DOI] [PubMed] [Google Scholar]
- Yoshizaki Y, Mori T, Ishigami-Yuasa M, Kikuchi E, Takahashi D, Zeniya M, Nomura N, Mori Y, Araki Y, Ando F, Mandai S, Kasagi Y, Arai Y, Sasaki E, Yoshida S, Kagechika H, Rai T, Uchida S, Sohara E (2017) Drug-Repositioning Screening for Keap1-Nrf2 Binding Inhibitors using Fluorescence Correlation Spectroscopy. Sci Rep 7:3945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You Q-D, Jiang Z-Y, Lu M-C, Chen Z-Y, Sun H-P, Zhang X-J, Guo X-K, Xu X-L (2017) 1-Sulfonamido-4-aryloxy compound, and preparation method and medicinal application thereof. WO2017124835A1 [Google Scholar]
- Zhang DD, Hannink M (2003) Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol Cell Biol 23:8137–8151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang DD, Lo S-C, Cross JV, Templeton DJ, Hannink M (2004) Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol Cell Biol 24:10941–10953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang DD, Lo S-C, Sun Z, Habib GM, Lieberman MW, Hannink M (2005) Ubiquitination of Keap1, a BTB-Kelch substrate adaptor protein for Cul3, targets Keap1 for degradation by a proteasome-independent pathway. J Biol Chem 280:30091–30099 [DOI] [PubMed] [Google Scholar]
- Zhuang C, Narayanapillai S, Zhang W, Sham YY, Xing C (2014) Rapid identification of Keap1–Nrf2 small-molecule inhibitors through structure-based virtual screening and Hit-based substructure search. J Med Chem 57:1121–1126 [DOI] [PubMed] [Google Scholar]