

Abstract
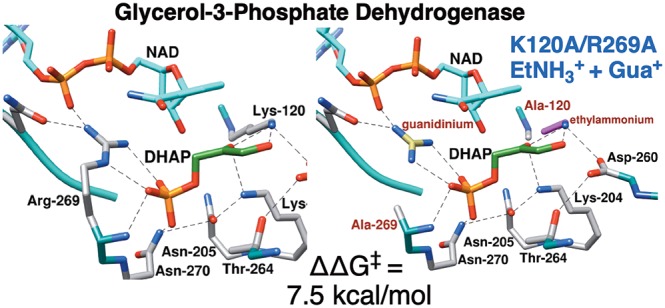
A comparison of the values of kcat/Km for reduction of dihydroxyacetone phosphate (DHAP) by NADH catalyzed by wild type and K120A/R269A variant glycerol-3-phosphate dehydrogenase from human liver (hlGPDH) shows that the transition state for enzyme-catalyzed hydride transfer is stabilized by 12.0 kcal/mol by interactions with the cationic K120 and R269 side chains. The transition state for the K120A/R269A variant-catalyzed reduction of DHAP is stabilized by 1.0 and 3.8 kcal/mol for reactions in the presence of 1.0 M EtNH3+ and guanidinium cation (Gua+), respectively, and by 7.5 kcal/mol for reactions in the presence of a mixture of each cation at 1.0 M, so that the transition state stabilization by the ternary E·EtNH3+·Gua+ complex is 2.8 kcal/mol greater than the sum of stabilization by the respective binary complexes. This shows that there is cooperativity between the paired activators in transition state stabilization. The effective molarities (EMs) of ∼50 M determined for the K120A and R269A side chains are ≪106 M, the EM for entropically controlled reactions. The unusually efficient rescue of the activity of hlGPDH-catalyzed reactions by the HPi/Gua+ pair and by the Gua+/EtNH3+ activator pair is due to stabilizing interactions between the protein and the activator pieces that organize the K120 and R269 side chains at the active site. This “preorganization” of side chains promotes effective catalysis by hlGPDH and many other enzymes. The role of the highly conserved network of side chains, which include Q295, R269, N270, N205, T264, K204, D260, and K120, in catalysis is discussed.
There are many examples of the rescue of the activity of truncated variant enzymes and of truncated alternative substrates by small molecule analogues of the deleted enzyme or substrate piece.1−9 However, such rescue is not universally observed, and there has been relatively little consideration of the structural requirements for the observation of efficient small molecule rescue of the catalytic activity of truncated enzymes and substrates. These studies provide insight into the mechanism for small molecule activation of enzyme activity that is analogous to allosteric activation, while enabling practical uses of chemical rescue in the activation of enzymes for catalysis.1,10 A parameter that reports on the efficiency of chemical rescue is the effective molarity (EM) of the truncated amino acid side chain,11,12 which is calculated as the ratio of kcat/Km for the wild type enzyme-catalyzed reaction and k′cat/KdKX for the reaction catalyzed by the variant enzyme with the missing enzyme piece X, at a standard state of 1 M. The entropic advantage to reaction of the whole enzyme compared to the enzyme in pieces is ≈106.13,14 The EMs of ≪106 that we have reported for rescue of variant enzyme-catalyzed reactions are consistent with an attenuation of the entropically controlled EM due to stabilization of the complex with the missing piece by interactions with the protein catalyst.3,12,15,16
Glycerol-3-phosphate dehydrogenase (GPDH) catalyzes the reduction of dihydroxyacetone phosphate (DHAP) by NADH to form l-glycerol 3-phosphate [G3P (Scheme 1A)].17−20 We have examined the activation of wild type human liver GPDH-catalyzed (hlGPDH) reduction of glycolaldehyde (GA) by phosphite dianion (HPi) and determined an EM value of 290 M for the phosphodianion of DHAP at the Michaelis complex with the wild type enzyme.3,4 The R269 side chain of hlGPDH interacts with the phosphodianion of DHAP and provides a 9.1 kcal/mol stabilization of the hydride transfer transition state (Figure 1).16 We determined an EM value of 60 M for this side chain in a study of the rescue of the R269A variant by the guanidine cation (Gua+).16 We next examined the activity of the R269A variant toward catalysis of reduction of the truncated substrate GA and characterized the assembly of GA, NADH, Gua+, HPi, and the variant enzyme into an active catalyst of reduction of GA (Scheme 1B).3 These data gave an EM2 value of 13500 M2 for the substrate phosphodianion and R269 side chain at the Michaelis complex of DHAP with wild-type hlGPDH.
Scheme 1. Reactions Catalyzed by (A) Wild Type hlGPDH and by (B) R269A and (C) K120A/R269A Variants of the Wild Type Enzyme.
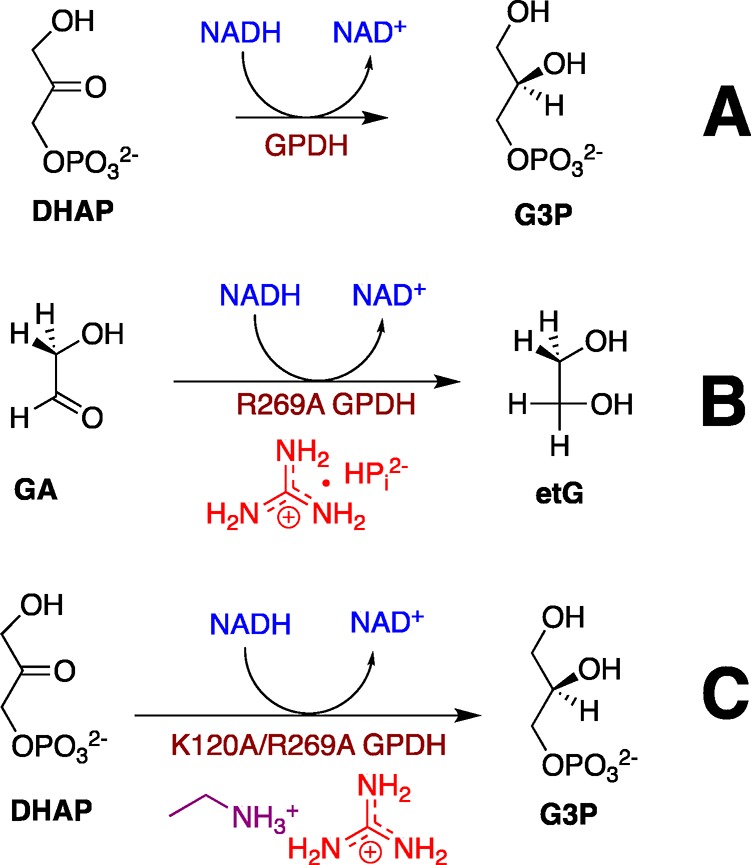
Figure 1.
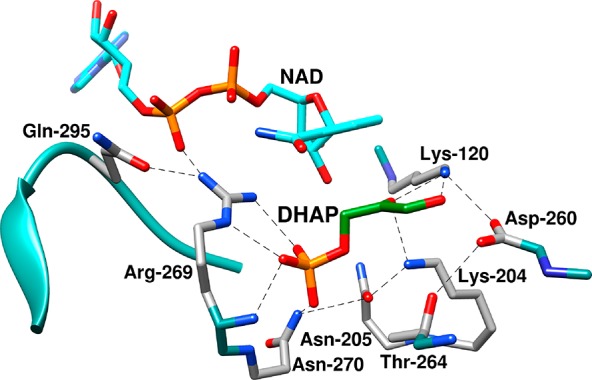
Representation of the X-ray crystal structure of the nonproductive ternary complex of hlGPDH, DHAP, and NAD (PDB entry 6E90), which shows the following highly conserved amino acid side chains:23,24 the side chains of R269 and N270 that interact with the substrate phosphodianion; the side chain of Q295, from a flexible enzyme loop, which interacts with R269; the cationic side chains from K120 and K204; and the side chains from N205, T264, and D260, which are part of a network of hydrogen-bonded side chains that connect the catalytic and dianion activation sites of hlGPDH.25
The side chain cation of K120 is positioned to stabilize the negative charge that develops at the C-2 oxygen of DHAP in the transition state for hydride transfer (Figure 1). The loss of this interaction for the K120A variant results in a 5.3 kcal/mol increase in the barrier to reduction of DHAP by NADH, while the loss of the K120 and R269 side chain cation interactions in the K120A/R269A variant results in a 12 kcal/mol barrier increase.21 These data are consistent with the conclusion that the focused transition state stabilization by the K120 and R269 side chain cations is responsible for a large fraction of the ∼15 kcal/mol stabilization of the hydride transfer transition state.21,22 The K120 and R269 side chains are part of a network of highly conserved amino acid side chains that extend from Q295 to K120 and that includes N270, N205, K204, T264, D260, and K120 (Figure 1).23,24 The importance of this network is highlighted by the large effects of N270A25 and D260G21 substitutions on the activity of wild type GPDH, but the network’s full role in catalysis of hydride transfer has not been determined.
We report here the results of characterization of the efficiency of the rescue of K120A/R269A variant-catalyzed reduction of DHAP by the combined action of Gua+ and ethylammonium (EtNH3+) cations (Scheme 1C) and the reduction of GA by the combined action of the phosphite dianion, Gua+, and EtNH3+. The first set of experiments shows the efficient rescue by these two cations and gave an EM2 value of 2400 M2 for the product of the effective molarity of the K120 and R269 side chains at the wild type enzyme. We conclude that the K120A/R269A variant provides a good template for binding of the excised cationic enzyme pieces and for organization of the K120A and R269A side chains at the active site of wild type hlGPDH. By contrast, we did not detect the fifth-order K120A/R269A variant-catalyzed reduction of GA in the presence of HPi, Gua+, and EtNH3+ activators. This sets a limit on the capacity of hlGPDH to usefully assemble small molecule activators at the enzyme active site.
Experimental Section
Materials
Water was obtained from a Milli-Q Academic purification system. Q-Sepharose and Sephacryl S-200 were purchased from GE Healthcare. Nicotinamide adenine dinucleotide, reduced form (NADH, disodium salt), glycolaldehyde dimer, 2-(N-morpholino)ethanesulfonic acid sodium salt (MES, ≥99.5%), triethanolamine hydrochloride (≥99.5%), guanidinium chloride, and sodium phosphite dibasic pentahydrate were purchased from Sigma-Aldrich. Ethylammonium chloride, d,l-dithiothreitol (DTT), sodium hydroxide (1.0 N), and hydrochloric acid (1.0 N) were purchased from Fisher Scientific. All other chemicals were reagent grade or better and were used without further purification. The solution pH was determined at 25 °C using an Orion model 720A pH meter equipped with a Radiometer pHC4006-9 combination electrode that was standardized at pH 4.00, 7.00, and 10.00 at 25 °C. Stock solutions of NADH were prepared by dissolving the disodium form of the coenzyme in water and then stored at 4 °C. The concentration of NADH in these solutions was determined from the absorbance at 340 nm using the extinction coefficient ε of 6220 M–1 cm–1. Stock solutions of DHAP were prepared by dissolving the lithium salt of DHAP in water, adjusting the pH to 7.5 with 1.0 NaOH, and storing the solution at −20 °C. The concentration of DHAP was determined as the concentration of NADH consumed during an hlGPDH-catalyzed reduction. Published procedures were used to prepare stock solutions of the guanidine cation,3 ethylammonium cation,15 and phosphite dianion6 at pH 7.5. Triethanolamine (TEA) buffers were prepared by addition of 1 M NaOH or 1 M HCl and solid NaCl to give the desired acid/base ratio and final ionic strength. Stock solutions of glycolaldehyde dimer (200 mM monomer) were prepared by dissolving the dimer in water and waiting for 3 days at room temperature to allow for quantitative breakdown of the dimer to the monomer.6
The K120A/Q295A and K120A/R269A variants of hlGPDH were expressed and purified by published procedures.21 Concentrated solutions of these variants were dialyzed exhaustively against 20 mM TEA buffer (pH 7.5) at 4 °C. When necessary, these solutions were diluted with 20 mM TEA buffer (pH 7.5) that contained 10 mM DTT and 0.1 mg/mL bovine serum alumin (BSA). The concentration of these enzyme variants was calculated from the absorbance at 280 nm using the extinction coefficient ε of 18450 M–1 cm–1 and a subunit molecular mass of 37500 Da that were determined using the ProtParam tool available on the ExPASy server.26,27
Enzyme Assays
All enzyme assays were conducted at an ionic strength I of 0.12 (NaCl) in a volume of 1.0 mL at 25 °C. hlGPDH was assayed by monitoring the oxidation of NADH (0.2 mM) by DHAP. Initial velocities of NADH oxidation over ≤10% reaction of DHAP were calculated from the change in absorbance at 340 nm using a molar extinction coefficient of 6220 M–1 cm–1 for NADH. Published procedures were used to assay the activity of K120A/Q295A and K120A/R269A variant-catalyzed reduction of DHAP by NADH at 25 °C, pH 7.5 (20 mM TEA), and I = 0.12 (NaCl) and for the K120A/R269A variant-catalyzed reduction of GA under the same conditions.3,21,28
Activation of Variant hlGPDH-Catalyzed Reactions
(A) Ethylammonium Cation. The assay mixtures at 25 °C and pH 7.5 (20 mM TEA) contained 0.1 mg/mL BSA and 0.2 mM NADH. For the K120A/Q295A variant, the assay mixture contained 1–3 mM DHAP, 0–80 mM activator, and 2.1 μM K120A/Q295A variant hlGPDH. For the K120A/R269A variant, the reaction mixture contained 0.800 mM DHAP and 22 μM K120A/R269A variant hlGPDH. The reactions were monitored for 20 min for the K120A/Q295A variant and for 720 min for the K120A/R269A variant.
(B) Guanidinium Cation. The assay mixtures at 25 °C and pH 7.5 (20 mM TEA) contained 0.1 mg/mL BSA, 0.2 mM NADH, 0.4–0.8 mM DHAP, 0–60 mM activator, and 20 μM K120A/R269A variant. The initial velocities for oxidation of NADH were calculated from the change in absorbance at 340 nm for a 20–40 min reaction time.
Activation of the K120A/R269A Variant by Mixtures of Guanidinium and Ethylammonium Cations
The assay mixtures for the K120A/R269A variant-catalyzed reduction of DHAP by NADH at I = 0.12 (NaCl) and 25 °C in the presence of mixtures of guanidinium and ethylammonium ions contained 20 mM TEA buffer (pH 7.5), 0.1 mg/mL BSA, 0.2 mM NADH, 0.4–0.8 mM DHAP, 0–60 mM guanidinium and ethylammonium cation activators (total concentration of the mixture), and 20 μM K120A/R269A variant hlGPDH. The initial velocities for oxidation of NADH were calculated from the change in absorbance at 340 nm for a 20 min reaction time.
Results
Slow GPDH-catalyzed reactions may be monitored for at least 24 h, during which time there is no detectable (<10%) loss of the activity of the wild type or K120A/R269A variant enzyme. There was no detectable reduction of 1.8 mM GA in the presence of 0.2 mM NADH catalyzed by 30 μM K120A/R269A variant hlGPDH (ΔA340 < 0.004 for a 40–60 min reaction time). This sets a limit for kcat/Km of ≤0.003 M–1 s–1 for this enzymatic reaction.3 There was likewise no detectable reduction of 1.8 mM GA at 0.2 mM NADH catalyzed by 30 μM K120A/R269A variant hlGPDH in the presence of single activator HPi (20 mM), Gua+ (20 mM), or EtNH3+ (20 mM), and in the presence of a mixture of 20 mM HPi, 20 mM Gua+, and 20 mM EtNH3+. There is no detectable effect (<5%) of 60 mM Gua+ or 60 mM EtNH3+ on v/[E] for reduction of DHAP by 0.2 mM NADH catalyzed by the K120A or R269A variant, respectively, so that small molecule rescue of these variant enzymes is specific for the cation analogue of the excised side chain.16,21
Figure 2 shows plots of v/[E] against [DHAP], with slopes (kcat/Km)obs, for K120A/Q295A hlGPDH-catalyzed reduction of DHAP by 0.2 mM NADH (saturating)21 at different fixed EtNH3+ concentrations. The inset of Figure 2 shows the plot of (kcat/Km)obs against [EtNH3+], with slope kcat/KmKam of 880 ± 20 M–2 s–1 (Table 1), for rescue by EtNH3+. Figure 3A shows the plot of v/[E] against [EtNH3+], with slope (kcat/Kam)obs, for K120A/R269A hlGPDH-catalyzed reduction of 0.8 mM DHAP by 0.2 mM NADH (saturating).21 Combining the slope of this correlation (kcat/Kam)obs (2.8 × 10–5 M–1 s–1) with a [DHAP] of 8 × 10–4 M gives a kcat/KmKam value of 0.035 M–2 s–1 (Table 1) for rescue of the K120A/R269A variant by EtNH3+. Figure 3B shows the related plots of v/[E] against [Gua+], with slopes (kcat/KGua)obs, for K120A/R269A hlGPDH-catalyzed reduction of 0.8 or 0.4 mM DHAP by 0.2 mM NADH. The inset of Figure 3B shows the plot of (kcat/KGua)obs against [DHAP], with a slope kcat/KmKGua of 3.9 ± 0.1 M–2 s–1 (Table 1) for rescue of the K120A/R269A variant by Gua+ (Table 1).
Figure 2.

Effect of increasing [EtNH3+] on v/[E] (s–1) for K120A/Q295A variant hlGPDH-catalyzed reduction of DHAP by NADH for reactions at pH 7.5 (20 mM TEA buffer), 25 °C, saturating [NADH] = 0.2 mM, and I = 0.12 (NaCl): (●) 80 mM cation, (■) 60 mM cation, (▼) 40 mM cation, (▲) 20 mM cation, and (◆) 10 mM cation. The inset shows the plot of (kcat/Km)obs against [EtNH3+], where (kcat/Km)obs values are the slopes of the correlations from the main panel.
Table 1. Kinetic Parameters and Derived Gibbs Free Energy Terms for Reactions of the Substrate and Pieces Catalyzed by hlGPDH at 25 °C, pH 7.5 (20 mM TEA), and I = 0.12 (NaCl).
| For Wild Type hlGPDH, kcat/Km = (4.6 ± 0.3) × 106 M–1 s–1;a for Q295A hlGPDH, kcat/Km = (6.3 ± 1.0) × 104 M–1 s–1b | ||||||
|---|---|---|---|---|---|---|
| variant | kcat/Km (M–1 s–1)c | activator | kcat/KmKX (M–2 s–1)d | ΔΔG⧧e (kcal/mol) | ΔGAct⧧f (ΔGAct/ΔΔG⧧) (kcal/mol) | ΔGS⧧g (kcal/mol) (EM)h |
| K120A | 550 ± 30 | EtNH3+ | (8.5 ± 0.4) × 104 | 5.3 ± 0.1 | 3.0 ± 0.1 (0.57) | 2.3 (50 M) |
| K120A/Q295A | 2.0 ± 0.1 | EtNH3+ | 880 ± 20 | 6.2 ± 0.1i | 3.6 ± 0.1 (0.58) | 2.5 (70 M) |
| R269A | 1.0 ± 0.2 | Gua+ | (8.0 ± 0.5) × 104 | 9.1 ± 0.1 | 6.7 ± 0.1 (0.74) | 2.4 (60 M) |
| K120A/R269A | (6.2 ± 0.4) × 10–3 | EtNH3+ | (3.5 ± 0.4) × 10–2 | 2.9 ± 0.1j | 1.0 ± 0.1l (0.33) | 2.0 (30 M) |
| Gua+ | 3.9 ± 0.1 | 6.7 ± 0.1k | 3.8 ± 0.1m (0.56) | 2.9 (140 M) | ||
| EtNH3+·Gua+ | (1.9 ± 0.1) × 103 M–3 s–1 | 12.0 ± 0.1 | 7.5 ± 0.1n (0.62) | 4.6o (2400 M2) | ||
From ref (4).
From ref (28).
Second-order rate constant for variant hlGPDH-catalyzed reduction of DHAP.
Third- or fourth-order rate constant for rescue of the activity of variant enzyme-catalyzed reduction of DHAP by the given activator(s).
Effect of the amino acid substitution on the stability of the transition state for wild type hlGPDH-catalyzed reduction of DHAP, unless stated otherwise.
Effect of 1.0 M activator on the stability of the transition state for the variant hlGPDH-catalyzed reaction.3
Transition stabilization obtained from the covalent connection between the enzyme pieces (ΔGS⧧ = ΔΔG⧧ – ΔGAct).
Effective molarity, of wild type or variant hlGPDH, of the deleted amino acid side chain [(kcat/Km)/kcat/KmKX].
Effect of the K120A substitution on Q295A hlGPDH.
Effect of the K120A substitution on R269A hlGPDH.
Effect of the R269A substitution on K120 hlGPDH.
Figure 3.

(A) Effect of increasing [EtNH3+] on v/[E] (s–1) for K120A/R269A variant hlGPDH-catalyzed reduction of 0.8 mM DHAP by saturating 0.2 mM NADH for reactions at pH 7.5 (20 mM TEA buffer), 25 °C, [NADH] = 0.2 mM, and I = 0.12 (NaCl). (B) Effect of increasing [Gua+] on v/[E] (s–1) for K120A/R269A variant hlGPDH-catalyzed reduction of DHAP by 0.2 mM NADH for reactions at pH 7.5 (20 mM TEA buffer), 25 °C, saturating [NADH] = 0.2 mM, and I = 0.12 (NaCl): (●) 0.8 mM DHAP and (■) 0.4 mM DHAP. The inset shows the plot of (kcat/KGua)obs against [DHAP], where (kcat/KGua)obs values are the slopes of the correlations from the main panel. The empty symbols show the agreement of data obtained for two different preparations of the K120A/R269A variant.
Figure 4A ([DHAP] = 0.4 mM) and Figure 4B ([DHAP] = 0.8 mM) show plots of v/[E] against [Gua+], with slopes (kcat/KGua)obs, for K120A/R269A hlGPDH-catalyzed reduction of DHAP by 0.2 mM NADH at different fixed concentrations of EtNH3+. Figure 5 shows plots of (kcat/KGua)obs from panels A and B of Figure 4 against [EtNH3+], with slopes (kcat/KGuaKam)obs, for K120A/R269A hlGPDH-catalyzed reduction of 0.4 or 0.8 mM DHAP by 0.2 mM NADH. The inset of Figure 5 shows the plot of (kcat/KGuaKam)obs against [DHAP], with a slope (kcat/KmKGuaKam) of 1900 M–3 s–1 (Scheme 2 and Table 1).
Figure 4.

Effect of increasing [Gua+] and [EtNH3+] on v/[E] for K120A/R269A variant hlGPDH-catalyzed reduction of DHAP by NADH at pH 7.5 (20 mM TEA buffer), 25 °C, saturating [NADH] = 0.2 mM, and I = 0.12 (NaCl). (A) Increase in v/[E], with increasing [Gua+], for the reaction of 0.4 mM DHAP at different fixed EtNH3+ concentrations. (B) Increase in v/[E], with increasing [Gua+], for the reaction of 0.8 mM DHAP at different fixed EtNH3+ concentrations: (◆) 5 mM EtNH3+, (▲) 10 mM EtNH3+, (■) 20 mM EtNH3+, and (●) 30 mM EtNH3+. The empty symbols show the agreement of data obtained for two different preparations of the K120A/R269A variant hlGPDH.
Figure 5.

Effect of increasing [EtNH3+] on (kcat/KGua)obs for K120A/R269A variant hlGPDH-catalyzed reduction of DHAP by NADH at pH 7.5 (20 mM TEA buffer), 25 °C, saturating [NADH] = 0.2 mM, and I = 0.12 (NaCl). The slope of these correlations is (kcat/KGuaKam)obs for variant hlGPDH-catalyzed reduction activated by the combined action of Gua+ and EtNH3+: (■) 0.4 mM DHAP and (●) 0.8 mM DHAP. The inset shows the plot of (kcat/KGuaKam)obs against [DHAP], with a slope (kcat/KmKGuaKam) of 1900 M–3 s–1 (Scheme 2 and Table 1).
Scheme 2. Rescue of the K120A/R269A Variant hlGPDH by EtNH3 and Gua+.

Discussion
The failure to detect reduction of GA catalyzed by 20 μM R269A hlGPDH results in a (kcat/Km)obs of ≤0.003 M–1 s–1 for the variant hlGPDH-catalyzed reaction.3 The failure to observe activation of the reduction of GA catalyzed by 30 μM K120A/R269A hlGPDH by 20 mM HPi, Gua+, or EtNH3+ activators or by a mixture of 20 mM HPi, 20 mM Gua+, and 20 mM EtNH3+ shows that (kcat/Km)obs ≤ 0.003 M–1 s–1 for the K120A/R269A variant-catalyzed reaction in the presence of these activators. Equation 1 gives the relationship between this limit for (kcat/Km)obs and the fifth-order rate constant kQ for the reaction catalyzed by the quaternary complex of the K120A/R269A variant, GA, HPi, Gua+, and EtNH3+. These data set an upper limit for kQ of 375 M–4 s–1 (eq 2). This shows that two small molecules may act together to give detectable activation of GPDH under our experimental conditions [HPi and Gua+ for R269A variant hlGPDH-catalyzed reduction of GA3 and Gua+ and EtNH3+ for K120A/R269A variant hlGPDH-catalyzed reduction of DHAP (this work, Table 1)] but that it is not possible to detect activation by the combined action of HPi, Gua+, and EtNH3+.
| 1 |
| 2 |
Rescue of the K120A/Q295A Variant-Catalyzed Reactions of DHAP
The K120A substitution results in similar 5.3 and 6.2 kcal/mol increases in the activation barrier for wild type- and Q295A variant-catalyzed reduction of DHAP, respectively (Table 1).21 The efficiency of rescue of the activity of the K120A/Q295A variant by EtNH3+ was characterized in this work (Figure 2) and compared with the efficiency of rescue of the K120A variant (Table 1).21 The transition states for the K120A and K120A/Q295A hlGPDH-catalyzed reductions of DHAP show similar stabilizations of 3.0 and 3.6 kcal/mol, respectively, for reactions in the presence of 1.0 M EtNH3+, while the EMs of 50 and 70 M determined for the K120 side chain of the wild type and the Q295A variant of hlGPDH, respectively, are similar (Table 1). These data provide additional support for the conclusion that the Q295A substitution only slightly impairs the transition state stabilization by the K120 side chain.21
K120A/R269A Variant-Catalyzed Reactions of DHAP
The position of side chains at the ternary hlGPDH·NAD·DHAP complex is shown in Figure 1, while panels A–C of Figure 6 show the side chains as representations of the surface of the binary hlGPDH·NAD complex (Figure 6A), the binary hlGPDH·NAD complex with DHAP inserted at the position of the ternary complex (Figure 6B), and the ternary hlGPDH·NAD·DHAP complex (Figure 6C). The K120 and R269 side chains lie on opposite sides of the active site cavity, with the K120 side chain positioned to interact with the carbonyl group and the R269 side chain positioned to interact with the phosphodianion of the substrate. The side chains lie at the two ends of a network of highly conserved side chains (Figure 1), which extends from Q295 to K120 and includes R269, N270, N205, K204, D260, and K120. A comparison of the structures shown in panels A–C of Figure 6 shows that the ligand-driven enzyme conformational change results in the folding of a flexible protein loop (292-LNGQKL-297) over the DHAP and NAD cofactor that is facilitated by formation of a hydrogen bond between the Q295 and R269 side chains. Loop closure traps the substrate in a solvent-occluded cage, where the K120 and R269 side chains are optimally placed for catalysis, and where electrostatic catalysis is presumably enhanced by the decrease in the effective dielectric constant of the closed compared to the open form of the complex (Figure 6A–C).29−31
Figure 6.

Representations of the protein surface for binary and ternary complexes of hlGPDH: (A) binary complex with NAD (PDB entry 6E8Z), (B) binary complex with NAD with substrate DHAP inserted at the position observed for the ternary hlGPDH·NAD·DHAP complex, and (C) ternary hlGPDH·NAD·DHAP complex (PDB entry 6E90). Color key: NAD, cyan; flexible protein loop (292-LNGQKL-297), navy blue; Q295, olive; R269, red; K120, magenta; DHAP, dark green for the phosphodianion and khaki for the remainder of the substrate. Substrate binding is accompanied by a movement of the navy blue flexible protein loop that covers the substrate and cofactor in the closed enzyme, formation of a hydrogen bond between the Q295 (olive) and R269 (red) side chains, and movement of the R269 side chain toward the substrate phosphodianion (green) and the cofactor pyrophosphate (cyan). The buried K120 side chain and substrate phosphodianion are hidden from view in panel C.
The consecutive K120 and R269 substitutions result in a total 12 kcal/mol increase in the activation barrier ΔG⧧ for hydride transfer (Scheme 3). The efficient rescue of the K120A/R269A variant of hlGPDH by Gua+ and EtNH3+ (Figures 4 and 5) shows that the wild type and variant enzymes proceed through similar transition states, which are strongly stabilized by interactions with the K120 and R269 side chains (wild type hlGPDH) or with bound Gua+ and EtNH3+. The larger sum of the effects of individual K120A and R269A substitutions on ΔG⧧ (5.3 + 9.1 = 14.4 kcal/mol) shows that the interaction energies of the single side chains of wild type hlGPDH are higher than the total 12 kcal/mol interaction determined by deleting the two side chains.
Scheme 3. Cycle That Shows the Effect of Consecutive K120A and R269A Substitutions on ΔΔG⧧ for Wild Type hlGPDH-Catalyzed Reduction of DHAP by NADH.
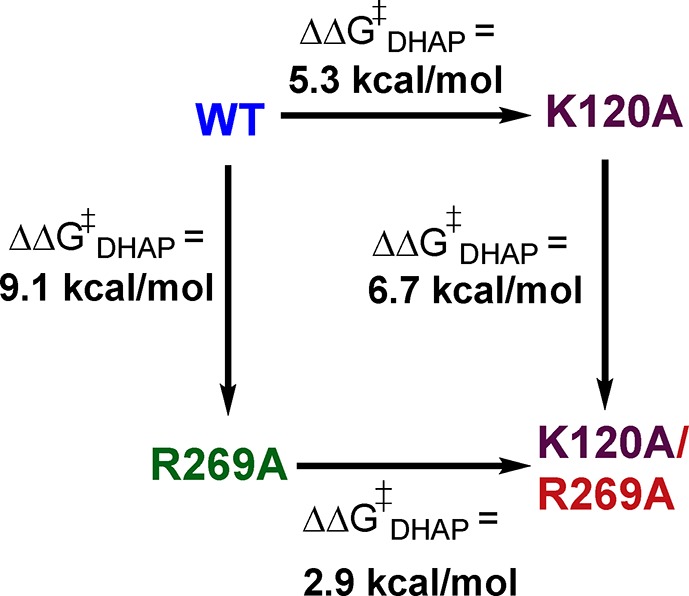
The 2.4 kcal/mol difference between the total side chain interaction estimated when the remaining side chain is preserved and the interaction determined by deleting both side chains (Scheme 3) represents the stronger side chain interactions of the tight, organized, conformation of wild type hlGPDH, compared to interactions of the K120A or R269A variant. We propose that this difference is due to effects of the first substitution, which reduce the transition state stabilization by the second side chain, such as an increase in the side chain conformational flexibility. This proposal is consistent with the notion that wild type hlGPDH derives a catalytic advantage from the high degree of organization of the catalytic side chains at the wild type active site and that the effect on ΔG⧧ of substitutions that erode this organization is greater than the effect of the lost interaction between the transition state and excised side chain.
Rescue of K120A/R269A hlGPDH by EtNH3+ and Gua+
The kinetic data for rescue of the K120A/R269A variant of hlGPDH by Gua+ and EtNH3+ give a (ΔΔGAct⧧)EtNH3·Gua of 7.5 kcal/mol (eq 3, Table 1) for stabilization of the transition state by interaction with the bound cations that is only 4.5 kcal/mol smaller than the transition state stabilization by the K120 and R269 side chains of wild type hlGPDH (ΔΔG⧧ = 12.0 kcal/mol, eq 4). The difference corresponds to a 12.0 – 7.5 = (ΔGS)E+EtNH3+Gua = 4.5 kcal/mol advantage to the reaction catalyzed by wild type hlGPDH compared to catalysis by the K120A/R269A variant in the presence of 1.0 M Gua+ and EtNH3+ (eq 5). By comparison, the binding of 1.0 M EtNH3+ or 1.0 M Gua+ to the K120A/R269A variant provides a (ΔGAct⧧)EtNH3 = 1.0 (eq 6) or (ΔGAct)Gua = 3.8 kcal/mol (eq 7) stabilization, respectively, of the transition state for the reaction catalyzed by this variant, so that the sum of transition state stabilization by the individual cations in binary complexes (1.0 + 3.8 = 4.8 kcal/mol) is 2.7 kcal/mol smaller than the 7.5 kcal/mol stabilization observed for the cations in the ternary complex.
| 3 |
| 4 |
| 5 |
| 6 |
| 7 |
These results are illustrated by Scheme 4, which partitions the total 12.0 kcal/mol effect of the K120A and R269A substitutions on transition state stability into the 7.5 kcal/mol interaction recovered in the EtNH3+·Gua+ complex and the 4.5 kcal/mol advantage to the reaction of the intact enzyme. The 7.5 kcal/mol recovered interaction is then partitioned into the 1.0 and 3.8 kcal/mol transition state stabilization of the individual binary complexes and the 2.7 kcal/mol advantage for cation activation of the ternary E′·EtNH3+·Gua+ complex. By comparison, the sum of the effect of single K120 and R269 substitutions in the K120A/R269A variant of hlGPDH (6.7 + 3.0 = 9.7 kcal/mol) on the stability of the transition state for enzyme-catalyzed reduction of DHAP is 2.3 kcal/mol smaller than the overall 12 kcal/mol stabilization by consecutive K120 and R269 substitutions (Scheme 3). We conclude that substitution of a single cationic side chain, or binding of a small molecule side chain analogue, in the K120A/R269A variant enhances the interaction of the second side chain or cation. We propose that this is due to utilization of the binding energy of the first bound cation in the organization of the active site, which enhances the transition state stabilization by the second bound cation. It is interesting that these cooperative interactions are expressed between side chains that are separated by ∼9 Å.
Scheme 4. Comparison between the Total Transition State Stabilization for Wild Type hlGPDH by Interactions with the K120 and R269 Side Chains and the Transition State Stabilization for the K120A/R269A Variant by Interactions with Exogenous Cations.

The 4.5 kcal/mol “connection energy” (Scheme 4) is an estimate for the advantage to connecting the Gua+ and EtNH3+ cations to the protein at the K120A/R269A variant of hlGPDH. This defines the effective concentration or effective molarity (EM)11 of the side chains of wild type hlGPDH compared to the value of 1.0 M of the free side chain in water (Table 1). EMs of 50 and 60 M were determined for the K120 and R269 side chains from the efficiency of rescue of the K120A and R269A variants, respectively.3,21 The product of the EMs from studies of single variants (50 M)(60 M) = 3000 M2 is similar to the value of 2400 M2 determined from rescue studies of the K120A/R269A variant (Table 1). This is consistent with similar stabilizing interactions between the catalyst and rescue agents of the K120A and R269A single variants and the K120A/R269A double variant.
The EMs from Table 1 are clustered between 30 and 140 M and correspond to an ∼2.5 kcal/mol advantage in ΔGS⧧ for reactions catalyzed by wild type hlGPDH compared to the reactions catalyzed by the complex between the variant enzyme and the missing piece. These values of EMs and ΔGS are smaller than the values of ∼106 M and ≈8 kcal/mol predicted for cases in which the advantage for unimolecular compared with bimolecular reaction is wholly entropic.14,32 The low EMs from Table 1 reflect the effective stabilization of the complexes to the pieces by interactions with the protein catalyst (Figure 1),15 where the K120 chain is locked into place by an ion pair to the D260 side chain,21 and the R269 side chain is held by interactions with the phosphodianion of DHAP, the pyrophosphate anion of NAD, and the Q295 side chain (Figure 1). These same interactions stabilize the EtNH3+ and Gua+ pieces bound to the K120A/R269A variant of hlGPDH (Figure 7A).
Figure 7.

(A) Representations of the X-ray crystal structures of the following complexes with variant hlGPDH. (A) Complex of K120A/R269A hlGPDH with NAD, DHAP, Gua+, and EtNH3+. (B) Complex of R269A hlGPDH with NAD, GA, HPi, and Gua+. The complexes were generated in silico, starting with Figure 1 for the X-ray crystal structure of the nonproductive complex of wild type hlGPDH, DHAP, and NAD+ (PDB entry 6E90) with deletion of the relevant covalent linkage(s) while maintaining a fixed position for the remaining atoms of the hypothetical Michaelis complexes.
Efficiency of Hydride Transfer Catalyzed by hlGPDH
The large transition state stabilization for hlGPDH-catalyzed reduction of DHAP is achieved largely through strong, focused, stabilizing interactions with the K120 and R269 side chains.21 The recovery of these interactions through the robust rescue of the K120A/R269A variant by Gua+ and EtNH3+ enzyme pieces reflects the strong stabilization of complexes with Gua+ and EtNH3+ (Figure 7A) by interaction with the neighboring amino acid side chains discussed above. Similarly, there is efficient activation of R269A variant hlGPDH-catalyzed reduction of glycolaldehyde by the combined action of the exogenous phosphite dianion and guanidine cation.3 This is a consequence of the stabilization of the enzyme-bound HPi·Gua+ ion pair (Figure 7B) by hydrogen bonding and ionic interactions, respectively, of Gua+ with the Q295 side chain and the cofactor pyrophosphate oxygen and by hydrogen bonds between HPi and the A269 backbone amide and the N270 amide side chain.
The side chain interactions, which promote efficient rescue of the K120A and R269A variants, serve to organize (or to preorganize)33−35 the K120 and R269 side chain cations of wild type hlGPDH (Figure 1). We propose that this preorganization enables the large, focused, 12 kcal/mol transition state stabilization from interactions with the K120 and R269 side chains by minimizing the energetic price for side chain immobilization that occurs on proceeding from the Michaelis complex to the hydride transfer transition state.30 Additional examples of side chain preorganization at enzyme active sites, such as the catalytic triad found in serine proteases,36,37 have been discussed by Warshel and co-workers.34,35,38−40
There are surprising similarities between the mechanisms for hydride transfer from NADH to DHAP catalyzed by hlGPDH and for isomerization of DHAP, with proton transfer, catalyzed by triosephosphate isomerase (TIM). Each enzyme shows strong phosphite dianion (HPi) activation of catalysis of the reaction of the common truncated substrate glycolaldehyde. The K12 side chain at TIM sits near the substrate dianion, and the K12G variant shows efficient rescue by alkyl ammonium cations. However, there is no rescue of K12G-catalyzed deprotonation of GA by the combined action of HPi and RNH3+. The stronger stabilization of the transition state for R269A variant-catalyzed hydride transfer to GA by the HPi·Gua+ ion pair compared with the transition state for K12G variant-catalyzed deprotonation of GA by the HPi·EtNH3+ ion pair is consistent with a higher degree of preorganization of the ion pair at the active site of hlGPDH. This reflects, at least in part, the presence of an intervening water molecule between the substrate phosphodianion and the TIM K12 side chain.
A Conserved Network of Amino Acid Side Chains
The highly conserved side chains from Q295, R269, N270, N205, T264, K204, D260, and K120 form a continuous chain of hydrogen bonds that stretch from the bound cofactor to the carbonyl group of DHAP (Figures 1 and 7).24,25 This side chain conservation suggests that the network operates as a unit, with K120, K204, and R269 providing direct transition state stabilization; N205, D260, and Q295 functioning directly to immobilize the catalytic side chains; and N270, N205, and T264 playing secondary roles in maintaining the network’s structural integrity. We suggest that this tight network of side chain interactions promotes effective catalysis and that the preorganization of this network by intra-side chain interactions with K120 and R269 provides a mechanism for the expression of cooperative interactions between the two cations, which are separated by 9 Å (Figure 1).
The D260 side chain shows no direct stabilizing interaction with the hydride transfer transition state, and there should be only a weak interaction with the N270 side chain. The large 6.5 and 5.6 kcal/mol effects of D260G21 and N270A25 substitutions, respectively, on the stability of the transition state for wild type hlGPDH-catalyzed reduction of DHAP by NADH (neither of which is subject to small molecule rescue) are therefore consistent with a substantial reorganization of the extended side chain network at the D260G and N270A variants, which results in barriers to formation of the preorganized, catalytically active closed conformation of variant hlGPDH. Finally, the observation that the N270A substitution results in an ∼40-fold increase in kcat/Km for enzyme-catalyzed reduction of GA by NADH is consistent with a reorganization of the active site at this variant, which favors binding of GA in a reactive conformation. This unusual observation shows that there is still much to be learned about the exact role of this extended network of interactions in the organization of active site side chains that provides for optimal catalysis of the reactions of whole and truncated substrates.
Conclusions
The R269 side chain of GPDH functions to trap the substrate DHAP in a tight cage,16,29 which provides strong stabilization of the transition state for hydride transfer, while the K120 side chain acts to stabilize negative charge at the C-2 oxygen, which develops in this transition state.21 The K120A and R269A substitutions at GPDH result in a total 7 × 108-fold decrease in kcat/Km for reduction of DHAP by NADH, which corresponds to an enormous 12 kcal/mol destabilization of the transition state for hydride transfer: the K120A/R269A variant provides an excellent template for association of exogenous Gua+ and EtNH3+ activators, which afford a 7.5 kcal/mol transition state stabilization at standard states of 1.0 M Gua+ and EtNH3+. The transition state stabilization from single R269 or K120 side chains, or from single Gua+ or EtNH3+ activators, is enhanced by the presence of the second side chain or activator. These results provide compelling support for the conclusion that the network of conserved side chains at GPDH (Figure 1) functions in the preorganization of the K120 and R269 side chains into positions that provide optimal stabilization of the transition state for hydride transfer. This preorganization promotes cooperativity in the expression of the strongly stabilizing interactions of the K120 and R269 side chains across a separation distance of 9 Å. We propose that the side chains at enzyme active sites often function organically and as a unit to provide for optimal stabilizing interactions between the transition state and a few key catalytic side chains.
Glossary
Abbreviations
- ScOMPDC
orotidine 5′-monophosphate decarboxylase from yeast
- TIM
triosephosphate isomerase
- GPDH
glycerol-3-phosphate dehydrogenase
- hlGPDH
glycerol-3-phosphate dehydrogenase from human liver
- DHAP
dihydroxyacetone phosphate
- etG
ethylene glycol
- GA
glycolaldehyde
- NADH
nicotinamide adenine dinucleotide, reduced form
- NAD
nicotinamide adenine dinucleotide, oxidized form
- MES
2-(N-morpholino)ethanesulfonic acid
- TEA
triethanolamine
- DTT
d,l-dithiothreitol
- BSA
bovine serum albumin.
Accession Codes
Human glycerol-3-phosphate dehydrogenase [NAD+], cytoplasmic, UniProt accession code P21695.
The authors acknowledge National Institutes of Health Grants GM116921 and GM134881 for generous support of this work.
The authors declare no competing financial interest.
References
- Peracchi A. (2008) How (and why?) to revive a dead enzyme: The power of chemical rescue. Curr. Chem. Biol. 2, 32–49. 10.2174/187231308783334162. [DOI] [Google Scholar]
- Olucha J.; Meneely K. M.; Lamb A. L. (2012) Modification of Residue 42 of the Active Site Loop with a Lysine-Mimetic Side Chain Rescues Isochorismate-Pyruvate Lyase Activity in Pseudomonas aeruginosa PchB. Biochemistry 51, 7525–7532. 10.1021/bi300472n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes A. C.; Amyes T. L.; Richard J. P. (2016) Enzyme Architecture: Self-Assembly of Enzyme and Substrate Pieces of Glycerol-3-Phosphate Dehydrogenase into a Robust Catalyst of Hydride Transfer. J. Am. Chem. Soc. 138, 15251–15259. 10.1021/jacs.6b09936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes A. C.; Zhai X.; Morgan K. T.; Reinhardt C. J.; Amyes T. L.; Richard J. P. (2015) The Activating Oxydianion Binding Domain for Enzyme-Catalyzed Proton Transfer, Hydride Transfer and Decarboxylation: Specificity and Enzyme Architecture. J. Am. Chem. Soc. 137, 1372–1382. 10.1021/ja5123842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang W.-Y.; Amyes T. L.; Richard J. P. (2008) A Substrate in Pieces: Allosteric Activation of Glycerol 3-Phosphate Dehydrogenase (NAD+) by Phosphite Dianion. Biochemistry 47, 4575–4582. 10.1021/bi8001743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amyes T. L.; Richard J. P. (2007) Enzymatic catalysis of proton transfer at carbon: activation of triosephosphate isomerase by phosphite dianion. Biochemistry 46, 5841–5854. 10.1021/bi700409b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amyes T. L.; Richard J. P.; Tait J. J. (2005) Activation of orotidine 5′-monophosphate decarboxylase by phosphite dianion: The whole substrate is the sum of two parts. J. Am. Chem. Soc. 127, 15708–15709. 10.1021/ja055493s. [DOI] [PubMed] [Google Scholar]
- Toney M. D.; Kirsch J. F. (1989) Directed Bronsted analysis of the restoration of activity to a mutant enzyme by exogenous amines. Science 243, 1485–1488. 10.1126/science.2538921. [DOI] [PubMed] [Google Scholar]
- Hung J. E.; Fogle E. J.; Garg N.; Chekan J. R.; Nair S. K.; van der Donk W. A. (2014) Chemical Rescue and Inhibition Studies to Determine the Role of Arg301 in Phosphite Dehydrogenase. PLoS One 9, e87134–e87139. 10.1371/journal.pone.0087134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamba V.; Yabukarski F.; Herschlag D. (2017) An Activator–Blocker Pair Provides a Controllable On–Off Switch for a Ketosteroid Isomerase Active Site Mutant. J. Am. Chem. Soc. 139, 11089–11095. 10.1021/jacs.7b03547. [DOI] [PubMed] [Google Scholar]
- Kirby A. (1980) Effective Molarities for Intramolecular Reactions. Adv. Phys. Org. Chem. 17, 183–278. 10.1016/S0065-3160(08)60129-X. [DOI] [Google Scholar]
- Barnett S. A.; Amyes T. L.; McKay Wood B.; Gerlt J. A.; Richard J. P. (2010) Activation of R235A Mutant Orotidine 5′-Monophosphate Decarboxylase by the Guanidinium Cation: Effective Molarity of the Cationic Side Chain of Arg-235. Biochemistry 49, 824–826. 10.1021/bi902174q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jencks W. P. (1981) On the attribution and additivity of binding energies. Proc. Natl. Acad. Sci. U. S. A. 78, 4046–4050. 10.1073/pnas.78.7.4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page M. I.; Jencks W. P. (1971) Entropic contributions to rate accelerations in enzymic and intramolecular reactions and the chelate effect. Proc. Natl. Acad. Sci. U. S. A. 68, 1678–1683. 10.1073/pnas.68.8.1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go M. K.; Amyes T. L.; Richard J. P. (2010) Rescue of K12G mutant TIM by NH4+ and alkylammonium cations: The reaction of an enzyme in pieces. J. Am. Chem. Soc. 132, 13525–13532. 10.1021/ja106104h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes A. C.; Koudelka A. P.; Amyes T. L.; Richard J. P. (2015) Enzyme Architecture: Optimization of Transition State Stabilization from a Cation–Phosphodianion Pair. J. Am. Chem. Soc. 137, 5312–5315. 10.1021/jacs.5b02202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley P.; Dickinson F. M. (1974) Coenzyme-binding characteristics of rabbit muscle L-glycerol 3-phosphate dehydrogenase. Biochem. J. 143, 11–17. 10.1042/bj1430011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley P.; Dickinson F. M.; Jones I. G. (1973) Purification and properties of rabbit muscle L-glycerol 3-phosphate dehydrogenase. Biochem. J. 135, 853–859. 10.1042/bj1350853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fondy T. P.; Ross C. R.; Sollohub S. J. (1969) Structural studies on rabbit muscle glycerol 3-phosphate dehydrogenase and a comparison of chemical and physical determinations of its molecular weight. J. Biol. Chem. 244, 1631–1644. [PubMed] [Google Scholar]
- Fondy T. P.; Levin L.; Sollohub S. J.; Ross C. R. (1968) Structure of L-glycerol-3-phosphate:NAD oxidoreductase isolated from rat skeletal muscle. J. Biol. Chem. 243, 3148–3160. [PubMed] [Google Scholar]
- Mydy L. S.; Cristobal J.; Katigbak R.; Bauer P.; Reyes A. C.; Kamerlin S. C. L.; Richard J. P.; Gulick A. M. (2019) Human Glycerol 3-Phosphate Dehydrogenase: X-Ray Crystal Structures that Guide the Interpretation of Mutagenesis Studies. Biochemistry 58, 1061–1073. 10.1021/acs.biochem.8b01103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukiji S.; Pattnaik S. B.; Suga H. (2004) Reduction of an Aldehyde by a NADH/Zn 2+-Dependent Redox Active Ribozyme. J. Am. Chem. Soc. 126, 5044–5045. 10.1021/ja0495213. [DOI] [PubMed] [Google Scholar]
- Choe J.; Guerra D.; MICHELS P. A. M.; Hol W. G. J. (2003) Leishmania mexicana glycerol-3-phosphate dehydrogenase showed conformational changes upon binding a bi-substrate adduct. J. Mol. Biol. 329, 335–349. 10.1016/S0022-2836(03)00421-2. [DOI] [PubMed] [Google Scholar]
- Ou X.; Ji C.; Han X.; Zhao X.; Li X.; Mao Y.; Wong L.-L.; Bartlam M.; Rao Z. (2006) Crystal structures of human glycerol 3-phosphate dehydrogenase 1 (GPD1). J. Mol. Biol. 357, 858–869. 10.1016/j.jmb.2005.12.074. [DOI] [PubMed] [Google Scholar]
- Reyes A. C.; Amyes T. L.; Richard J. P. (2016) Enzyme Architecture: A Startling Role for Asn270 in Glycerol 3-Phosphate Dehydrogenase-Catalyzed Hydride Transfer. Biochemistry 55, 1429–1432. 10.1021/acs.biochem.6b00116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasteiger E.; Hoogland C.; Gattiker A.; Duvaud A.; Wilkins M. R.; Appel R. D.; Bairoch A. (2005) Protein Identification and Analysis Tools on the ExPASy Server. Proteomics Protocols Handbook 571–607. 10.1385/1-59259-890-0:571. [DOI] [Google Scholar]
- Gasteiger E.; Gattiker A.; Hoogland C.; Ivanyi I.; Appel R. D.; Bairoch A. (2003) ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 31, 3784–3788. 10.1093/nar/gkg563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He R.; Reyes A. C.; Amyes T. L.; Richard J. P. (2018) Enzyme Architecture: The Role of a Flexible Loop in Activation of Glycerol-3-phosphate Dehydrogenase for Catalysis of Hydride Transfer. Biochemistry 57, 3227–3236. 10.1021/acs.biochem.7b01282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard J. P.; Amyes T. L.; Goryanova B.; Zhai X. (2014) Enzyme architecture: on the importance of being in a protein cage. Curr. Opin. Chem. Biol. 21, 1–10. 10.1016/j.cbpa.2014.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard J. P. (2019) Protein Flexibility and Stiffness Enable Efficient Enzymatic Catalysis. J. Am. Chem. Soc. 141, 3320–3331. 10.1021/jacs.8b10836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amyes T. L.; Malabanan M. M.; Zhai X.; Reyes A. C.; Richard J. P. (2017) Enzyme activation through the utilization of intrinsic dianion binding energy. Protein Eng., Des. Sel. 30, 159–168. 10.1093/protein/gzw064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jencks W. P. (2006) Binding energy, specificity, and enzymic catalysis: the Circe effect. Adv. Enzymol. Relat. Areas Mol. Biol. 43, 219–410. 10.1002/9780470122884.ch4. [DOI] [PubMed] [Google Scholar]
- Jindal G.; Warshel A. (2017) Misunderstanding the preorganization concept can lead to confusions about the origin of enzyme catalysis. Proteins: Struct., Funct., Genet. 85, 2157–2161. 10.1002/prot.25381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warshel A.; Sharma P. K.; Kato M.; Parson W. W. (2006) Modeling electrostatic effects in proteins. Biochim. Biophys. Acta, Proteins Proteomics 1764, 1647–1676. 10.1016/j.bbapap.2006.08.007. [DOI] [PubMed] [Google Scholar]
- Warshel A. (1998) Electrostatic Origin of the Catalytic Power of Enzymes and the Role of Preorganized Active Sites. J. Biol. Chem. 273, 27035–27038. 10.1074/jbc.273.42.27035. [DOI] [PubMed] [Google Scholar]
- Blankenship E.; Vukoti K.; Miyagi M.; Lodowski D. T. (2014) Conformational flexibility in the catalytic triad revealed by the high-resolution crystal structure of Streptomyces erythraeus trypsin in an unliganded state. Acta Crystallogr., Sect. D: Biol. Crystallogr. 70, 833–840. 10.1107/S1399004713033658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedstrom L. (2002) Serine Protease Mechanism and Specificity. Chem. Rev. 102, 4501–4524. 10.1021/cr000033x. [DOI] [PubMed] [Google Scholar]
- Kamerlin S. C. L.; Sharma P. K.; Chu Z. T.; Warshel A. (2010) Ketosteroid isomerase provides further support for the idea that enzymes work by electrostatic preorganization. Proc. Natl. Acad. Sci. U. S. A. 107, 4075–4080. 10.1073/pnas.0914579107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamerlin S. C. L.; Chu Z. T.; Warshel A. (2010) On catalytic preorganization in oxyanion holes: highlighting the problems with the gas-phase modeling of oxyanion holes and illustrating the need for complete enzyme models. J. Org. Chem. 75, 6391–6401. 10.1021/jo100651s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warshel A.; Sharma P. K.; Kato M.; Xiang Y.; Liu H.; Olsson M. H. M. (2006) Electrostatic basis for enzyme catalysis. Chem. Rev. 106, 3210–3235. 10.1021/cr0503106. [DOI] [PubMed] [Google Scholar]