

Abstract
The neuropeptide substance P can induce degranulation of cardiac mast cells at high concentrations. Herein, we seek to further understand substance P activation of cardiac mast cells in the context of other neuropeptides as well as modulation by non-neuropeptides. This is important given the increasingly recognized role of both cardiac mast cells and substance P in adverse cardiac remodeling. To address this, we isolated cardiac mast cells and compared their response to substance P as well as other members from the tachykinin family of peptides, including neurokinin A and hemokinin-1. We also tested the ability of other factors to manipulate the cardiac mast cell response to substance P. We found that while neurokinin A did not induce cardiac mast cell degranulation, both substance P and hemokinin-1 induced a concentration-dependent release of histamine; the maximal response to hemokinin-1 was greater than to substance P. Neurokinin-1 receptor blockade prevented substance P-induced histamine release, while only partially attenuating hemokinin-1-induced histamine release. The antioxidant N-acetylcysteine attenuated histamine release in response to hemokinin-1 and had no effect on substance P-induced histamine release. Selective PPAR-γ agonists attenuated histamine release in response to substance P. These data indicate that substance P activates cardiac mast cells via the neurokinin-1 receptor, and that the activation response is different to other tachykinins. That the response to substance P is receptor mediated and can be modulated by activation of other receptors (PPAR-γ), argues that substance P activation of cardiac mast cells has potential biological significance.
INTRODUCTION
In the heart, the tachykinin neuropeptide substance P (SP) is primarily found in sensory nerves projecting to the coronary perivascular space, and in a small population of coronary endothelial cells (Dalsgaard et al., 1986; Milner et al., 1989; Reinecke et al., 1980; Wharton et al., 1981). Similarly, the perivascular space is also a common location for cardiac mast cells (MCs), and MCs and nerves are known to physically interact in many organs, including the heart. Interestingly, this includes SP-containing nerves (Morrey et al., 2010). Presumably this interaction allows for communication between sensory nerves and MCs, whereby sensory nerves can utilise MCs to promote a signal at the local tissue level. In support of this concept, Suzuki et al., (Suzuki et al., 1999) demonstrated that SP mediated communication between cultured superior cervical ganglia neurites and the RBL-2H3 MC line.
Neuropeptides, including SP, are being increasingly recognized as important factors in the development of adverse cardiac remodelling and heart failure (D’Souza et al., 2007; Dehlin and Levick, 2014; Mak et al., 2011; Mak et al., 2015; Morrey et al., 2010; Ng et al., 2014; Robinson et al., 2009; Robinson et al., 2016; Weglicki et al., 1994; Widiapradja et al., 2017), and MCs are also important in the development of numerous cardiac pathologies (Batlle et al., 2007; Brower et al., 2002; Brower and Janicki, 2005; Frangogiannis et al., 1998; Levick et al., 2009; Marone et al., 1995; McLarty et al., 2011; Olivetti et al., 1989; Stewart et al., 2003). Therefore, it is important to better understand neuropeptide interactions with cardiac MCs. Because of their relatively low cardiac density, MCs are often overlooked inflammatory cells capable of many functions due to the large amounts, and vast array of pre-stored (e.g. chymase, tryptase, TNF-α and de novo synthesized mediators (e.g. cytokines, growth factors, and phospholipid metabolites) that they produce (Theoharides et al., 2007). Some examples of documented MC contributions to cardiac remodeling are as follows: 1) chymase inhibition reduced cardiac fibrosis and improved left ventricular (LV) diastolic performance in cardiomyopathic hamsters (Takai et al., 2003); 2) chymase inhibition prevented fibrosis and improved LV end diastolic pressures in a dog model of tachycardia-induced heart failure (Matsumoto et al., 2003); 3) MC stabilization with nedocromil or MC-deficiency prevented matrix metalloproteinase (MMP) activation, fibrillar collagen degradation, ventricular dilatation, and heart failure in a rat model of sustained ventricular volume overload (Brower et al., 2002; Brower and Janicki, 2005; Levick et al., 2008); 4) nedocromil prevented cardiac fibrosis in spontaneously hypertensive rats (SHR) (Levick et al., 2009); 5) tryptase activation of protease activated receptor-2 (PAR-2) on cardiac fibroblasts promotes myofibroblast conversion and collagen production via ERK, and PAR-2 blockade prevents cardiac fibrosis in SHR (McLarty et al., 2011); 6) nedocromil reduced adverse remodeling in STZ diabetic mouse hearts (Huang et al., 2013); and 7) crossing TNF-α overexpressing mice with MC-deficient mice prevented the cardiac fibrosis normally observed in TNF-α overexpressing mice, as well as improved the LV pressure-volume relationship and reduced TGF-β1 levels (Zhang et al., 2011).
Traditionally, it was believed that cardiac MCs did not degranulate in response to SP (Patella et al., 1995; Sperr et al., 1994), however, we demonstrated that SP is capable of inducing cardiac MC degranulation in vitro, when the cells are isolated by a method not involving harsh enzymatic digestion (Melendez et al., 2011; Morgan et al., 2008). However, activation did require high concentrations of SP (30–100 μM). Despite this requirement, evidence does exist for functional effects of SP and MC interactions in the heart in vivo. For example, SP caused MC renin release in guinea pig hearts stimulated with capsaicin (Morrey et al., 2010). Further, blockade of the neurokinin-1 receptor (NK-1R), the receptor for SP, reduced reperfusion arrhythmias in an ex vivo guinea pig heart preparation. The effect of NK-1R blockade was likely via the prevention of MC release of renin (Morrey et al., 2010). We demonstrated in a mouse model of cardiac volume overload that deletion of SP prevented MMP activation, collagen degradation, and ventricular dilatation (Melendez et al., 2011); all of which are events known to be mediated by MCs (Brower et al., 2002; Brower and Janicki, 2005). Further, we showed in a rat model of cardiac volume overload that NK-1R blockade prevented increases in MC density (Melendez et al., 2011). Interestingly, neurokinin A (NKA), another member of the tachykinin family, does not activate cardiac MCs (Melendez et al., 2011). However, whether hemokinin-1 (HK-1), another tachykinin that activates the NK-1R similarly to SP, has stimulatory effects on cardiac MCs is unknown. Furthermore, despite the evidence presented above, our understanding of SP interactions with MCs is still limited, including whether SP regulation of cardiac MC function is direct, whether it can be modulated by other receptors, and whether it elicits a greater response in MCs from diseased hearts. Herein, we seek to gain additional insight into SP regulation of cardiac MC function by: 1) comparing SP induction of cardiac MC activation with activation by select members of the tachykinin family of neuropeptides, including NKA and HK-1; and 2) by examining the ability of non-tachykinin mediators to modulate the effects of SP activation on cardiac MCs. To explore SP/MC interactions in a way that removed possible complications of indirect activation that could occur in an in vivo setting, we used isolated cardiac MCs that were obtained using our non-enzymatic digestion technique.
METHODS
All experiments were performed using 8-week-old, male Sprague Dawley (Hsd:SD, n = 15) rats, spontaneously hypertensive rats (SHR, n = 5), or normotensive Wistar Kyoto (WKY, n = 4) housed under standard environmental conditions and maintained on commercial rodent chow and tap water ad libitum. These studies conformed to the principles of the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and the protocol was approved by the University’s Institutional Animal Care and Use Committee. Rats were deeply anesthetized with sodium pentobarbital (50 mg/kg) administered by intra-peritoneal injection before surgical procedures. Proper analgesia for euthanasia was evaluated by palpebral reflex, toe pinch reflex, and corneal reflex. At the experimental endpoint, euthanasia was accomplished by removal of the heart.
Cardiac Mast Cell Isolation and Treatment Procedures
Cardiac MCs were isolated from rat hearts as previously described (Morgan et al., 2008). Briefly, a thoracotomy was performed to expose the intact pericardial sac. The sac was then pierced with a teflon catheter sleeve attached to a 10 cc syringe and Hanks balanced salt solution (HBSS, 7.4 pH) composed of: 1) Hanks calcium and magnesium free salt solution; 2) HEPES (13 mM); 3) 607 units/mL of deoxyribonuclease I (Sigma Chemical Co., St. Louis, MO); and 4) an antibiotic-antimycotic mixture of penicillin G sodium, 10,000 mg/mL of streptomycin sulfate, and 25 mg/mL of amphotericin B (Gibco-BRL, Life Technologies, Grand Island, NY) was injected onto the surface of the heart and the pooled buffer gently aspirated from the pericardial sac. This process was repeated several times. The resultant preparation consists of a mixed population of MCs, CD4+ T cells, and monocyte/macrophages (Levick et al., 2010). The isolated cells were pelleted and resuspended in Hyclone buffer (HBSSMC; HBSS containing magnesium sulphate (1.1 mM), calcium chloride (1.3 mM), and phenol red; Hyclone, Logan, UT) before undergoing treatment.
To examine the responsiveness of cardiac MCs to select tachykinins, 4 × 103 cardiac MCs per treatment tube were incubated with: 1) SP (0, 3, 10, 30, 100 and 300 μM); 2) HK-1 (0, 1, 3, 10, 30, and 100 μM); or 3) NKA (0, 3, 10, 30, and 100 μM). The samples were incubated in a shaking water bath at 37 °C for 20 min at 60 agitations per min and the post-treatment supernatants and pellets separated for subsequent analysis of histamine as a specific marker of MC degranulation. Histamine concentration was measured using a Neogen® Veratox Histamine ELISA kit (Lexington, KY), in accordance with Neogen’s protocol manual and read on a microplate analyzer using a double wavelength of 450 nm and 620 nm. The percent histamine released was determined by dividing the histamine value from the supernatant by total histamine (supernatant plus pellet). Based on the results obtained from the concentration-response curves, a concentration of 100 μM was selected for subsequent cardiac MC experiments involving SP and HK-1. To determine whether SP and HK-1 stimulation of cardiac MCs was mediated by the NK-1R or NK-2R, an additional group of isolated cells were pre-incubated for 20 min with the NK-1R antagonist, L 732 138 (20 μM), or the NK-2R antagonist GR 159 897 (10 μM) prior to treatment with 100 μM of SP or HK-1. To determine the contribution of reactive oxygen species to cardiac MC degranulation in response to 100 μM of SP and HK-1, cells were pre-incubated for 20 min with the antioxidant, N-acetylcystein (NAC, 200 μM).
To determine whether peroxisome proliferator-activated receptor-γ (PPAR-γ) agonists could inhibit SP-induced cardiac MC degranulation, isolated cardiac MCs were pre-treated with rosiglitazone (10 μM), ciglitazone (20 and 50 μM), pioglitazone (20 and 50 μM) or 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2, 10 μM) for 20 min prior to stimulation with SP. To further examine whether the effects of rosiglitazone and 15d-PGJ2 were mediated via PPAR-γ, isolated cells were pre-treated with the selective PPAR-γ antagonist, GW 9662 (1 μM), for 20 min prior to incubation with rosiglitazone or 15d-PGJ2 and then SP. The MC secretagogue, compound 48/80 (20 μg/mL) served as a positive control.
Histology
To identify MCs in the rat heart, 5 μm sections of LV were dehydrated with increasing concentrations of ethanol, before staining with toluidine blue in 1% NaCl for 30 seconds. The LV sections were then briefly rinsed in water. Cardiac MCs stained with toluidine blue appear purple in color.
Identification of the NK-1 Receptor by Immunofluroescence
Cardiac MCs were isolated as described above. Isolated cells were washed and blocked against non-specific labeling with PBS/1% BSA for 30 min before incubation for 1 h at 37 °C with AR32AA4 (mast cell, FcεRI; mouse monoclonal, 1:100; BD Pharmingen), followed by conjugation for 1 h at 37 °C with Alexa Fluor 488 secondary antibody (rabbit, 1:1000; Abcam). After washing, the cells were fixed with 4% paraformaldehyde. The cells were then incubated with anti-neurokinin-1 receptor antibody (rabbit monoclonal, 1:500; Abcam) and conjugated with PE secondary antibody. Cell preparations were imaged using fluorescence microscopy.
T Cells and Activation of Mast Cells
Athymic nude rats (RH-FOXN1RNU/RNU, n = 3), which are devoid of T cells, were used to evaluate whether T cells are important in tachykinin-induced degranulation of cardiac MCs in the isolated cell preparations. The wild type counterpart served as controls (RH-FOXN1+/+, n = 4). Briefly, MCs were isolated, as described above, from the hearts of athymic nude rats and incubated with SP (0, 3, 10, 30, 100, 300 μM) for 20 min at 37°C. Cell pellets and supernatant were separated and analyzed for histamine as described above.
Real-Time PCR
LV tissue was homogenized and dispersed cells lysed in 1 ml of PurZOLTM with RNA isolated according to manufacturer’s instructions (Aurum Total RNA Fatty and Fibrous Tissue Kit, Bio-Rad). 1 μg/μl of total RNA was used for each cDNA reaction (iScript cDNA synthesis kit, Bio-Rad) and 1 μl of cDNA was used in each 20 μl RT-qPCR reaction (SsoAdvanced SYBR Green, Bio-Rad and Bio-Rad cycler). Each reaction was initiated with a 30 sec denaturation step at 95ᵒC followed by 39 cycles of a 10 sec denature step at 95ᵒC and a 30 sec anneal/extension step at 60ᵒC. The annealing temperature for each primer pair was calculated from the mean melting temperature of the forward and reverse primer. The sequences for rat β-actin primers are: forward, 5’-CCATGTACCCAGGCATTGCT-3’, reverse, 5’-AGCTCAGTAACAGTCCGCCTA-3’, and rat Tac4: forward, 5’-TGATGCCACGCCCATCTCGC-3’, reverse, 5’-TGSCTTAGCCCACGGAGCCA-3’. The averaged Cq values were analyzed using the ΔΔCq method with actin as the reference gene and values were plotted relative to zero.
Drugs and Chemical Reagents
SP, HK-1, and NKA were purchased from Sigma Chemical Company. L732138 and GR159897 were purchased from Tocris. Rosiglitazone, ciglitazone, pioglitazone, 15d-PGJ2 and GW 9662 were purchased from Cayman Chemicals.
Statistics
All data were expressed as mean ± standard error of the mean (SEM). Grouped data comparisons were made by one-way ANOVA with Tukey’s post hoc testing for multi-group comparisons with one independent variable. A two-way ANOVA with Tukey’s post hoc testing was used for multi-group comparisons with two independent variables. Significance was set at p ≤ 0.05.
RESULTS
Cardiac Mast Cells Possess the NK-1R.
As has been described previously (Marone et al., 2014; Morrey et al., 2010; Silver et al., 2004), cardiac MCs are present in the heart both interstitially (Figure 1A) and in close proximity to coronary vessels (Figure 1B). To demonstrate the existence of the NK-1R on cardiac MCs, isolated cardiac MCs were labelled with antibody against FcεRI (IgE receptor, green), while the NK-1R was labelled red. Figure 2A, B, and D shows the presence of the NK-1R on cardiac MCs. While most cardiac MCs labelled positive for the NK-1R, there were occasional cells that appeared to be negative for the receptor (Figure 2C). Since our isolation procedure yields a mixed population of cardiac inflammatory cells (Levick et al., 2010), non-MCs also labeled positive for the NK-1R. We have previously characterized this mixed cardiac inflammatory cell population as being approximately 12% MCs, 68% T cells, and 12% macrophages (Levick et al., 2010).
Figure 1.
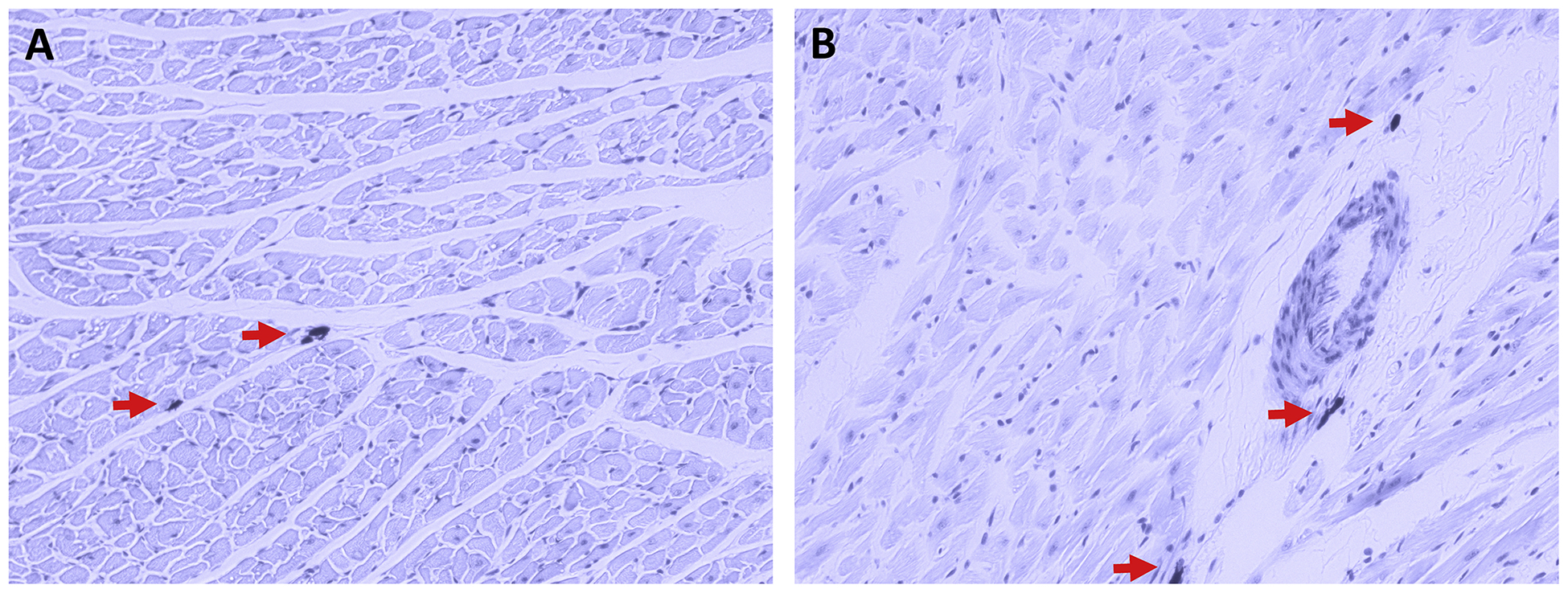
Rat left ventricular tissue sections stained with Toluidine blue, where MCs appear dark purple in colour with the surrounding tissue appearing light purple. The staining demonstrates that cardiac MCs (indicated by red arrows) can be found interstitially (A) as well as perivascularly (B).
Figure 2.
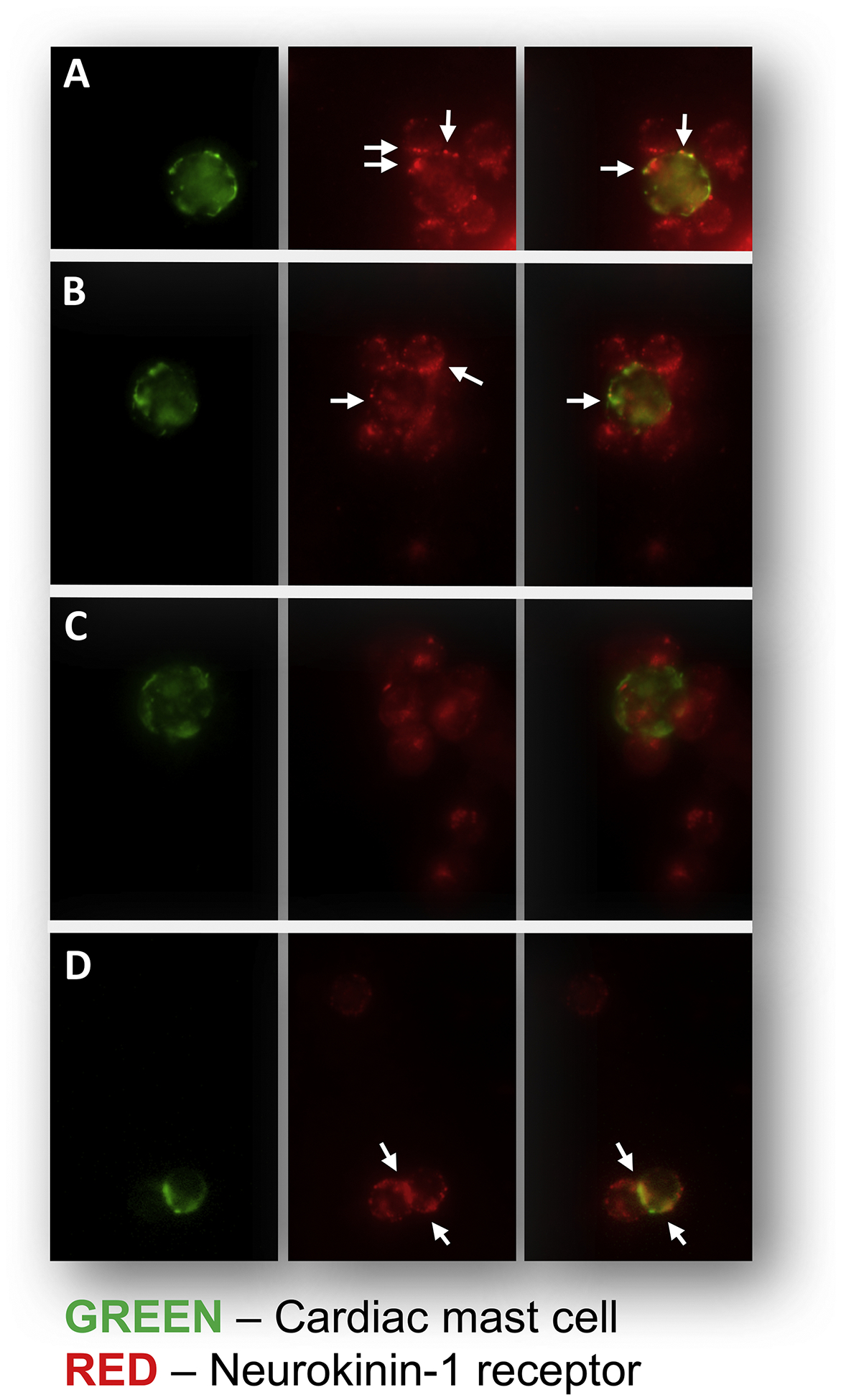
Immunofluorescent analysis of the isolated cardiac inflammatory cell preparation. The left panel of A-D shows examples of cardiac MCs which were identified by antibody to FcεRI (IgE receptor) and labelled green. The middle panel of A-D shows examples of cells (MCs and other inflammatory cells) that labelled positively for the NK-1R (red, arrows). The right hand panel of A-D are the merged images that indicate MCs that also labelled positive for the NK-1R. Panels A, B, and D show cardiac MCs that possess the NK-1R (arrows). Panel C shows an example of a cardiac MC that does not possess the NK-1R.
Tachykinin-Induced Degranulation of Cardiac Mast Cells.
We examined the ability of select tachykinins (SP, HK-1, and NKA) to induce cardiac MC degranulation. Data reported herein for the NKA concentration-response curve has been published previously (Melendez et al., 2011). SP (3 to 300 μM) induced cardiac MC degranulation was reflected by the concentration-response curve for histamine release displayed in Figure 3A. SP elicited a concentration-dependent secretagogue effect with maximum histamine release (66.2 ± 2.7%) comparable to that induced by the MC secretagogue, compound 48/80 (70.3 ± 4.3%). HK-1 elicited a greater maximal histamine release (85 ± 7%) and a leftward shift of the concentration-response curve indicating increased potency compared to SP. NKA did not induce activation. The selective NK-1R antagonist, L732138, was used to determine whether SP and HK-1-induced release of histamine was NK-1R-mediated. Data reported herein for the effects of L732138 and the NK-2R antagonist GR159897 on SP have been published previously (Melendez et al., 2011). Pre-treatment of the isolated cell preparation with L732138 prior to stimulation, completely prevented the release of histamine from cardiac MCs in response to SP (Figure 3B), however, L732138 only attenuated histamine release in response to HK-1. The selective NK-2R antagonist, GR159897 did not alter histamine release in response to either SP or HK-1 (Figure 3B). Additionally, we pretreated cells with the antioxidant NAC to determine whether activation of MCs by SP and HK-1 involved oxidative stress. NAC did not alter histamine release induced by SP, however, it did cause a small, but significant, reduction in histamine release in response to HK-1 (Figure 3C). Since histamine release in response to SP was completely NK-1R-mediated and HK-1-induced activation was only partly NK-1R-mediated, we focused on the effects of SP in subsequent experiments.
Figure 3. (A).
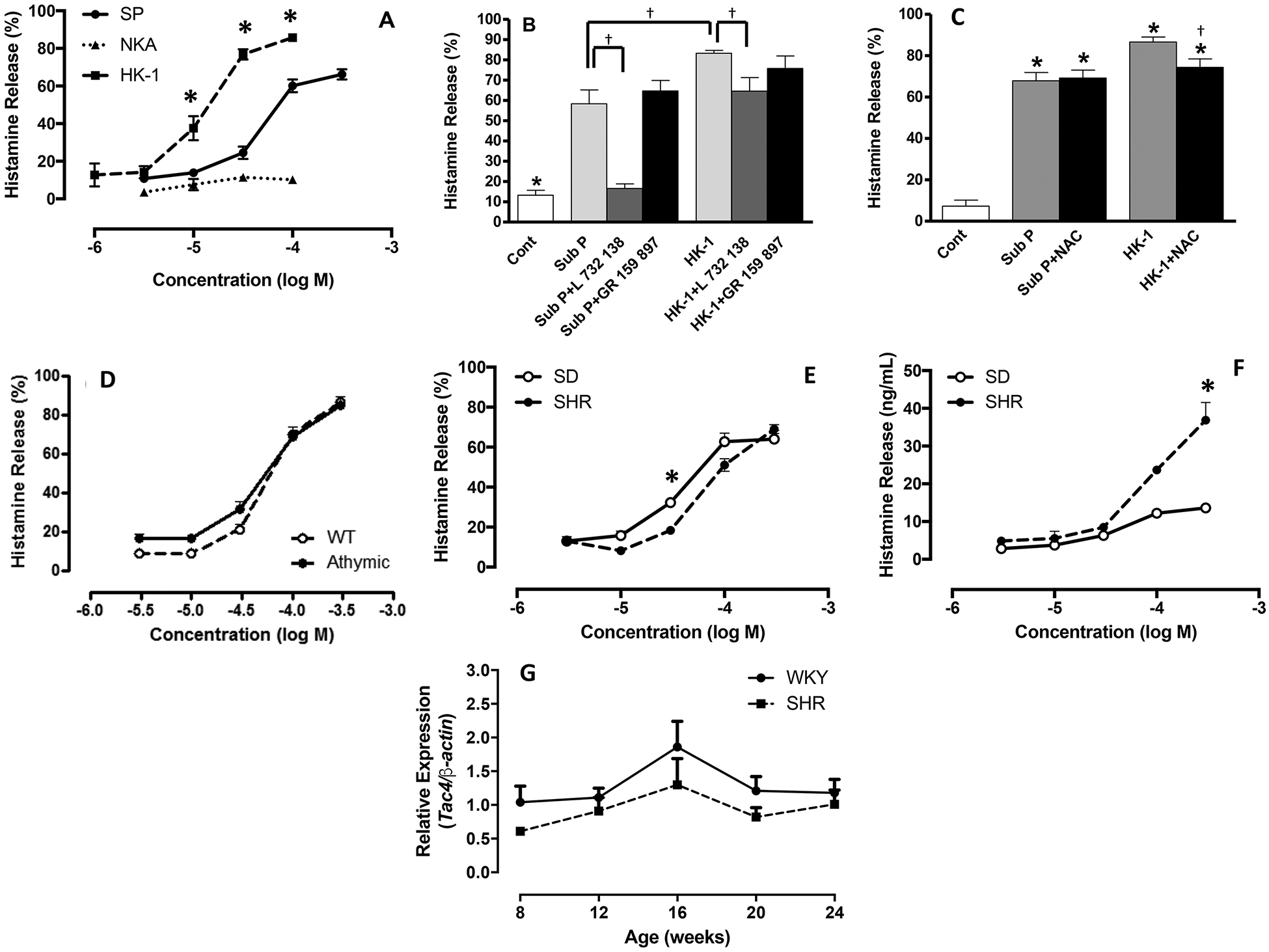
Concentration-response curves for percentage histamine release from cardiac MCs in response to stimulation with SP, HK-1, and NKA. Values are mean ± SEM. * = p<0.05 compared to substance P; (B) Histamine release from cardiac MCs that were untreated (cont), SP-treated (100 μM), SP-treated (100 μM) pre-incubated with the NK-1R antagonist, L 732 138 (20 μM), SP-treated (100 μM) pre-incubated with the NK-2R antagonist, GR 159 897 (10 μM), HK-1-treated (100 μM), HK-1-treated (100 μM) pre-incubated with L 732 138 (20 μM), and HK-1-treated pre-incubated with the NK-2R antagonist, GR 159 897 (10 μM).Values are mean ± SEM. * = p<0.05 control vs substance P and HK-1, † = p<0.05 compared to substance P and HK-1; (C) Histamine release from cardiac MCs that were untreated (cont), SP-treated (100 μM), SP-treated (100 μM) pre-incubated with the antioxidant, NAC (200 μM), HK-1-treated (100 μM), and HK-1-treated pre-incubated with NAC (200 μM); (D) Concentration-response curves for percentage histamine release from cardiac MCs isolated from wild type (WT, n=4/concentration) and athymic nude rats (n=3/concentration) in response to substance P; (E) Concentration-response curves for percentage histamine release from cardiac MCs isolated from normotensive Sprague-Dawley (SD, n=5/concentration) and spontaneously hypertensive rats (SHR, n=5/concentration). Values are mean ± SEM. * = p<0.05 compared to corresponding concentration; (F) Concentration-response curves for absolute histamine release from cardiac MCs isolated from SD rats (n=5/concentration) and SHR (n=5/concentration); (G) expression levels of Tac4 mRNA from SHR and WKY left ventricles (n=4/time-point). Values are mean ± SEM. * = p<0.05 compared to corresponding concentration.
It was clear from Figure 2 that other inflammatory cells in the isolated cell preparation possessed the NK-1R. Approximately 70% of the inflammatory cells in our isolated preparation are T cells (Levick et al., 2010). To understand whether the NK-1R-mediated effects of SP on MC degranulation were via a direct interaction between SP and cardiac MC NK-1Rs, or were indirect and required T cells, we examined SP-induced release of histamine from cardiac MCs isolated from athymic nude rats devoid of T cells. We observed no difference in histamine release from MCs obtained from T cell-deficient athymic nude rats or the wild type counterpart in response to SP (Figure 3D). Therefore, cardiac MC degranulation in response to SP is not dependent on the presence of T cells, and SP likely interacts directly with cardiac MCs to cause their activation.
Histamine Release from Hypertensive Cardiac Mast Cells.
MCs from SHR and normotensive SD rats were treated with SP to ascertain if there were any differences in MC response from diseased versus normal hearts. Maximal percent release of histamine in response to SP was similar between cardiac MCs from both SHR and SD rats, although percent histamine release was slightly greater through the mid-point of the concentration-response curve for SD MCs (Figure 3E). Interestingly, while the percent release of histamine was similar, the absolute amount of histamine released was greater from SHR MCs indicating a greater amount of histamine stored in those cells (Figure 3F). We did not assess SHR MC responses to HK-1 because Tac4 (Figure 3G), the gene that encodes HK-1, was found not to be elevated at the same time-points at which we had previously reported Tac1 to be dramatically increased in the SHR (Dehlin et al., 2013).
Inhibition of SP-induced Histamine Release by Activation of PPAR-γ.
To further test the functionality of SP and the NK-1R on cardiac MCs, we examined the effect of other mediators on the NK-1R-mediated actions of SP. Three synthetic PPAR-γ agonists, rosiglitazone, pioglitazone and ciglitazone, as well as the endogenous ligand 15d-PGJ2, were tested for their possible inhibitory effect on SP-induced histamine release. Figure 4A shows that rosiglitazone (10 μM) and 15d-PGJ2 (10 μM) significantly attenuated SP-induced histamine release. Neither rosiglitazone nor 15d-PGJ2 had an effect when administered alone (Figure S1) or following treatment with compound 48/80 (Figure 4B). The inhibitory effects of rosiglitazone and 15d-PGJ2 were mediated via PPAR-γ since the selective PPAR-γ antagonist, GW 9662 (1 μM), prevented their inhibitory actions (Figure 4A). Interestingly, neither pioglitazone (20 and 50 μM) nor ciglitazone (20 and 50 μM) showed any inhibitory effect on SP-induced histamine release (Figure 4C).
Figure 4. (A).
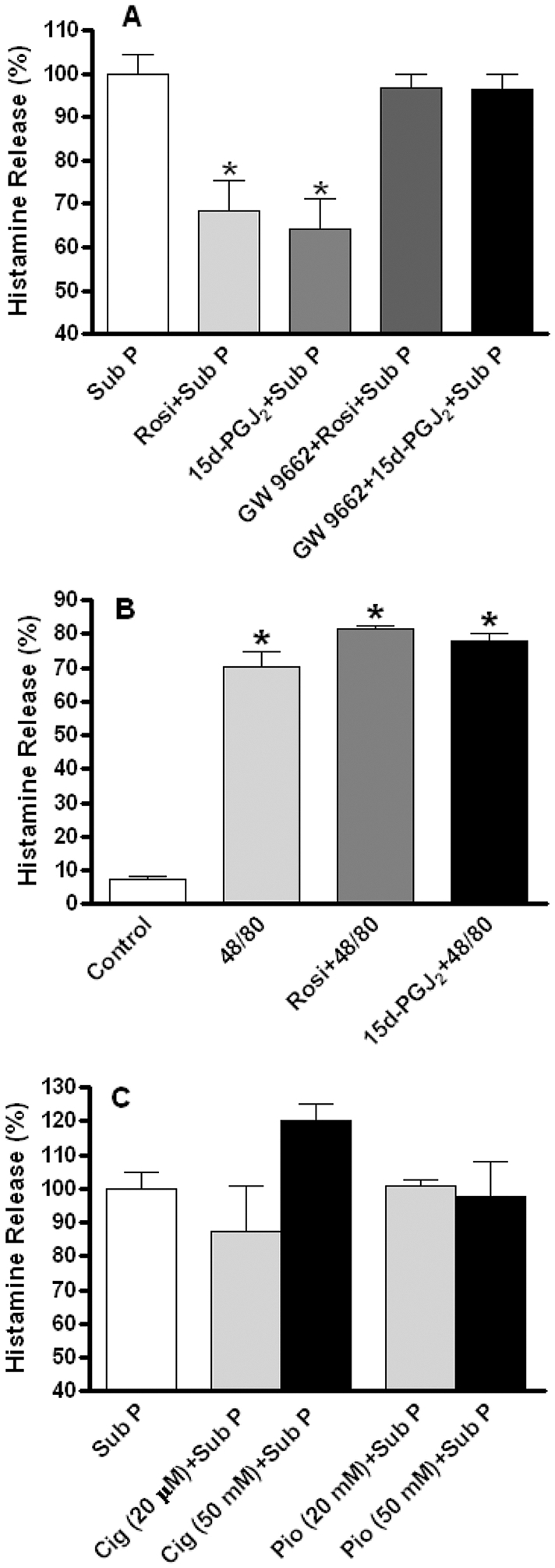
Shows the inhibitory effect of rosiglitazone (Rosi, 10 μM) and 15d-PGJ2 (10 μM) on substance P-induced histamine release. The selective PPAR-γ receptor antagonist, GW 9662 (1 μM) prevented the inhibitory effects of rosiglitazone and 15d-PGJ2. Percent histamine release is displayed relative to substance P-induced effects, which were set at 100%. (B) Shows that both rosiglitazone and 15d-PGJ2 had no effect on histamine release induced by compound 48/80. (C) Shows that ciglitazone and pioglitazone had no effect on histamine release induced by substance P. Values are mean ± SEM. * = p<0.05 versus substance P (A) or versus control (B). Note that SP release is set at 100% in (A) and (C).
DISCUSSION
An as yet not fully answered question is what is the function of the NK-1R on cardiac MCs? We had previously shown that isolated cardiac MCs from rats could degranulate (histamine release) in response to SP, albeit at very high concentrations (Melendez et al., 2011; Morgan et al., 2008). These high concentrations of SP required for cardiac MC activation raised the question of whether this indicated a physiologic receptor-mediated response or artifact. Herein, we sought to further explore SP activation of cardiac MCs by comparing MC responses to SP with other select members of the tachykinin family of neuropeptides, of which SP is a member, and determining whether other molecules could modulate the activation of cardiac MCs by SP. To do this we used an in vitro approach to remove possible indirect effects on MC activation that could occur in vivo.
Firstly, we determined whether other select members of the tachykinin family could activate cardiac MCs similarly to SP. As we previously reported, while NKA does not cause histamine release, HK-1 induced a greater release of histamine than SP. HK-1 has been shown to act in an autocrine manner to cause TNF-α and IL-6 secretion in response to FcεRI activation in bone marrow-derived MCs (Sumpter et al., 2015). It should be noted that we did not test breakdown products of SP, which include SP3–11, SP5–11, SP8–11, and SP1–7, that could be biologically active.
We identified that most cardiac MCs possessed the NK-1R, and that while SP-induced histamine release could be completely abolished by the NK-1R antagonist L732138, NK-1R blockade only attenuated the response to HK-1. Further, inhibition of oxidative stress attenuated HK-1-induced histamine release, while having no effect on histamine release in response to SP. Therefore, important differences exist in the way that SP and HK-1 activate cardiac MCs. The small effect of NK-1R blockade on the HK-1 response was somewhat surprising given that HK-1 is a full agonist at the NK-1R (Borbely and Helyes, 2017). One possibility is that HK-1 functions through other neurokinin receptors on cardiac MCs. HK-1 is also a full agonist at the NK-2R and NK-3R albeit with much lower affinity (Borbely and Helyes, 2017). To our knowledge, it is not known if there are other neurokinin receptors present on cardiac MCs. The lack of a response to NKA would suggest that they do not express the NK-2R, but it is possible that they may have the NK-3R, however, this remains to be determined. Our finding that NK-2R blockade did not reduce HK-1-induced histamine release also argues against an effect at the NK-2R even if these receptors are present. There is also some evidence of a separate HK-1R (Borbely and Helyes, 2017). In this regard there are many examples in different systems where SP and HK-1 elicit different effects, including NK-1R-independent actions of HK-1 (Borbely and Helyes, 2017).
Since our isolated cell preparation is not a pure MC population, we determined whether SP-induced histamine release involved other cell types. That is, could SP cause activation of other inflammatory cells that then release factors that induce MC activation. T cells comprise roughly 70% of the cells in our isolated cell preparation. Thus, using the same isolation method, we extracted cells from athymic nude rats that are T cell-deficient, resulting in an isolated cell preparation that contained no T cells. SP elicited a histamine release response in this cell preparation almost identical to the wild type controls, indicating that T cells did not mediate MC degranulation in response to SP.
Given the role of SP in cardiovascular disease (Dehlin and Levick, 2014; Widiapradja et al., 2017), we were interested in whether cardiac MCs from diseased hearts were more responsive to SP. Accordingly, we isolated MCs from SHR hearts and compared SP-induced histamine release with SD normotensive MCs. Interestingly, the percentage release of histamine was the same between groups, however, there was a greater release of histamine in absolute terms in response to SP by SHR MCs. This indicates that SHR MCs contained more histamine than cells from normotensive controls. Thus, potentially SP-induced activation of cardiac MCs could have increased effects in diseased hearts via increased mediator release. We did not assess SHR MC responses to HK-1 because Tac4, the gene that encodes HK-1, was found not to be elevated at the same time-points at which we had previously reported that Tac1 was increased in the SHR (Dehlin et al., 2013). Thus, unlike SP, HK-1 appears unlikely to contribute to myocardial remodeling, at least in the hypertensive heart.
To further evaluate SP degranulation of cardiac MCs, we sought to determine if we could alter the response to SP by manipulating other receptors. For this task, we investigated interactions between SP and PPARγ since PPAR-γ is known to have multiple actions on MCs. Zhang et al., (Zhang et al., 2017) demonstrated in bone marrow-derived MCs that the PPAR-γ agonist pioglitazone inhibited MC maturation as assessed by granule formation and expression of MC protease 6, the gene that encodes tryptase. Additionally, the human MC line, HMC-1, showed blunted capacity for migration and adhesion when exposed to rosiglitazone (Zhang et al., 2014). Of relevance to our study, PPAR-γ is also reported to have inhibitory actions on MC activation and degranulation. PPAR-γ agonists have been reported to inhibit IgE-mediated release of GM-CSF, as well as histamine and leukotriene C4 in cultured MCs (Shinmura et al., 2000). To our knowledge, there have been no descriptions of the actions of PPAR-γ on cardiac MCs until now. Accordingly, we pretreated cardiac MCs with the endogenous PPAR-γ agonist 15d-PGJ2 and the synthetic PPAR-γ agonist rosiglitazone. Both agonists reduced histamine release in response to SP by more than 30%. The PPAR-γ antagonist GW9662 prevented the actions of 15d-PGJ2 and rosiglitazone, confirming that they acted via PPAR-γ. However, neither 15d-PGJ2 nor rosiglitazone act as a MC stabilizer in the traditional sense like compounds such as nedocromil, since they failed to prevent MC degranulation invoked by compound 48/80. Thus, this was an effect specific to NK-1R activation. As far as we are aware, this is the first report of functional interactions between the NK-1R and PPAR-γ in MCs. SP had previously been shown to increase PPAR-γ levels in human monocytes and macrophages, with the effect being greater in cells derived from smokers (Amoruso et al., 2008). This was mediated by the NK-1R. Further, PPAR-γ activation opposed SP-mediated release of TNF-α from human monocytes. Currently, the mechanisms of the interactions between the NK-1R and PPAR-γ are not known. Interestingly, we found that ciglitazone and pioglitazone were not able to reduce SP-induced histamine release. Therefore, interactions between SP/NK-1R and PPAR-γ are agonist specific. In a previous study, 15d-PGJ2 inhibited the release of GM-CSF, leukotriene C4, and histamine following IgE stimulation, while the synthetic agonists troglitazone and ciglitazone only inhibited GM-CSF and leukotriene C4 release, but not histamine. Inhibition of IgE-mediated stimulation of the RBL-2H3 MC line by ciglitazone has been shown to be PPAR-γ independent (Okuyama et al., 2005). To date there are no reports regarding interactions between HK-1 and PPAR-γ.
The experiments contained herein strongly argue that SP-induced histamine from cardiac MCs is not an artifact of the high concentrations of SP required for activation, but is a receptor-mediated event that can be modulated by other molecules, including PPAR-γ agonists. The selective inhibitory effects of PPAR-γ agonists were specific for SP and not the general MC secretogogue, 48/80. The need now is to identify the role of SP activation of cardiac MCs in vivo. There are some specific scenarios where this interaction may be important. MCs mediate MMP activation and collagen degradation in the volume overloaded heart (Brower et al., 2002). Deletion of SP also prevents MMP activation and collagen degradation following volume overload (Melendez et al., 2011), suggesting a possible link between SP and MCs. Adding to this, NK-1R blockade also prevents increased MC density in the heart under volume overload conditions (Melendez et al., 2011). Whether these are direct effects of SP on cardiac MCs is still unknown. The heart can also participate in hypersensitivity reactions. IgE sensitization of human cardiac MCs does occur (Assem and Ghanem, 1988), and initiates a complex cardiac anaphylactic response. This includes tachycardia, acute increased ventricular contraction, chronic reduced ventricular contraction, decreased A-V conduction, ventricular fibrillation, and reduced coronary flow (Assem, 1989; Denizot et al., 1990; Levi et al., 1978; Silva Machado et al., 1985). Paralleling this are increased histamine levels, with experiments using right atrial appendages from human hearts indicating that the tachycardic and positive inotropic effects of IgE are mediated by histamine H2 receptors (Assem, 1989; Denizot et al., 1990; Graver et al., 1986). SP has been shown to potentiate IgE-induced degranulation of rat peritoneal MCs (Lau et al., 2001), although SP has also been shown to downregulate FcεRI levels, and hence IgE binding, in human LAD2 MCs (McCary et al., 2010). Interestingly, though, SP potentiates IL-31 release from LAD2 MCs when combined with IL-33 and anti-IgE (Petra et al., 2018). Thus, SP/NK-1R interactions could participate in the hearts anaphylactic response to IgE. Accordingly, the next step is to identify the role of NK-1Rs on cardiac MCs in vivo.
In summary, we used an in vitro approach to investigate interactions between tachykinins and cardiac MCs, thereby, avoiding possible confounding indirect action that could occur in vivo. We report that tachykinins have differential effects on cardiac MC degranulation. SP activates cardiac MCs via the NK-1R, however, this does not appear to be the sole mechanism of activation in response to HK-1. Further, SP-induced cardiac MC degranulation is opposed by activation of PPARγ. The differential effects of SP versus other tachykinins, the prevention of the effects of SP by NK-1R blockade, and the ability of PPARγ agonists to reduce the effects of SP all argue that SP activation of cardiac MCs is a physiological event with biological significance. The next step is to determine the role of SP activation of cardiac MCs in disease states.
Supplementary Material
Acknowledgements
This work was supported in part by the National Heart, Lung and Blood Institute at the National Institutes of Health (HL093215 to SPL).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Amoruso A, Bardelli C, Gunella G, Ribichini F, Brunelleschi S, 2008. A novel activity for substance P: stimulation of peroxisome proliferator-activated receptor-gamma protein expression in human monocytes and macrophages. British journal of pharmacology 154, 144–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assem ES, 1989. Anaphylactic reactions affecting the human heart. Agents Actions 27, 142–145. [DOI] [PubMed] [Google Scholar]
- Assem ES, Ghanem NS, 1988. Demonstration of IgE-sensitized mast cells in human heart and kidney. Int. Arch. Allergy Appl. Immunol 87, 101–104. [DOI] [PubMed] [Google Scholar]
- Batlle M, Perez-Villa F, Lazaro A, Garcia-Pras E, Ramirez J, Ortiz J, Orus J, Roque M, Heras M, Roig E, 2007. Correlation between mast cell density and myocardial fibrosis in congestive heart failure patients. Transplantation proceedings 39, 2347–2349. [DOI] [PubMed] [Google Scholar]
- Borbely E, Helyes Z, 2017. Role of hemokinin-1 in health and disease. Neuropeptides 64, 9–17. [DOI] [PubMed] [Google Scholar]
- Brower GL, Chancey AL, Thanigaraj S, Matsubara BB, Janicki JS, 2002. Cause and effect relationship between myocardial mast cell number and matrix metalloproteinase activity. American journal of physiology. Heart and circulatory physiology 283, H518–525. [DOI] [PubMed] [Google Scholar]
- Brower GL, Janicki JS, 2005. Pharmacologic inhibition of mast cell degranulation prevents left ventricular remodeling induced by chronic volume overload in rats. Journal of cardiac failure 11, 548–556. [DOI] [PubMed] [Google Scholar]
- D’Souza M, Garza MA, Xie M, Weinstock J, Xiang Q, Robinson P, 2007. Substance P is associated with heart enlargement and apoptosis in murine dilated cardiomyopathy induced by Taenia crassiceps infection. J Parasitol 93, 1121–1127. [DOI] [PubMed] [Google Scholar]
- Dalsgaard CJ, Franco-Cereceda A, Saria A, Lundberg JM, Theodorsson-Norheim E, Hokfelt T, 1986. Distribution and origin of substance P- and neuropeptide Y-immunoreactive nerves in the guinea-pig heart. Cell Tissue Res 243, 477–485. [DOI] [PubMed] [Google Scholar]
- Dehlin HM, Levick SP, 2014. Substance P in heart failure: the good and the bad. Int. J. Cardiol 170, 270–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehlin HM, Manteufel EJ, Monroe AL, Reimer MH Jr., Levick SP, 2013. Substance P acting via the neurokinin-1 receptor regulates adverse myocardial remodeling in a rat model of hypertension. Int. J. Cardiol 168, 4643–4651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denizot Y, Boudet J, Burtin C, Marro I, Benveniste J, 1990. Monoclonal IgE-mediated cardiac hypersensitivity reactions in the guinea-pig. Agents Actions 29, 167–171. [DOI] [PubMed] [Google Scholar]
- Frangogiannis NG, Lindsey ML, Michael LH, Youker KA, Bressler RB, Mendoza LH, Spengler RN, Smith CW, Entman ML, 1998. Resident Cardiac Mast Cells Degranulate and Release Preformed TNF-{alpha}, Initiating the Cytokine Cascade in Experimental Canine Myocardial Ischemia/Reperfusion. Circulation 98, 699–710. [DOI] [PubMed] [Google Scholar]
- Graver LM, Robertson DA, Levi R, Becker CG, Weksler BB, Gay WA, 1986. IgE-mediated hypersensitivity in human heart tissue: histamine release and functional changes. J. Allergy Clin. Immunol 77, 709–714. [DOI] [PubMed] [Google Scholar]
- Huang ZG, Jin Q, Fan M, Cong XL, Han SF, Gao H, Shan Y, 2013. Myocardial remodeling in diabetic cardiomyopathy associated with cardiac mast cell activation. PLoS. One 8, e60827. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Lau AHY, Chow SSM, Ng YS, 2001. Immunologically induced histamine release from rat peritoneal mast cells is enhanced by low levels of substance P. European Journal of Pharmacology 414, 295–303. [DOI] [PubMed] [Google Scholar]
- Levi R, Zavecz JH, Ovary Z, 1978. IgE-mediated cardiac hypersensitivity reactions. An experimental model. Int. Arch. Allergy Appl. Immunol 57, 529–534. [DOI] [PubMed] [Google Scholar]
- Levick SP, Gardner JD, Holland M, Hauer-Jensen M, Janicki JS, Brower GL, 2008. Protection from adverse myocardial remodeling secondary to chronic volume overload in mast cell deficient rats. Journal of Molecular and Cellular Cardiology 45, 56–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levick SP, McLarty JL, Murray DB, Freeman RM, Carver WE, Brower GL, 2009. Cardiac mast cells mediate left ventricular fibrosis in the hypertensive rat heart. Hypertension (Dallas, Tex. : 1979) 53, 1041–1047. [DOI] [PubMed] [Google Scholar]
- Levick SP, Murray DB, Janicki JS, Brower GL, 2010. Sympathetic nervous system modulation of inflammation and remodeling in the hypertensive heart. Hypertension (Dallas, Tex. : 1979) 55, 270–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak IT, Chmielinska JJ, Kramer JH, Spurney CF, Weglicki WB, 2011. Loss of neutral endopeptidase activity contributes to neutrophil activation and cardiac dysfunction during chronic hypomagnesemia: Protection by substance P receptor blockade. Exp. Clin. Cardiol 16, 121–124. [PMC free article] [PubMed] [Google Scholar]
- Mak IT, Kramer JH, Chmielinska JJ, Spurney CF, Weglicki WB, 2015. EGFR-TKI, erlotinib, causes hypomagnesemia, oxidative stress, and cardiac dysfunction: attenuation by NK-1 receptor blockade. J. Cardiovasc. Pharmacol 65, 54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marone G, Genovese A, Varricchi G, Granata F, 2014. Human heart as a shock organ in anaphylaxis. Allergo journal international 23, 60–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marone G, Patella V, de CG, Genovese A, Adt M, 1995. Human heart mast cells in anaphylaxis and cardiovascular disease. Int. Arch. Allergy Immunol 107, 72–75. [DOI] [PubMed] [Google Scholar]
- Matsumoto T, Wada A, Tsutamoto T, Ohnishi M, Isono T, Kinoshita M, 2003. Chymase Inhibition Prevents Cardiac Fibrosis and Improves Diastolic Dysfunction in the Progression of Heart Failure. Circulation 107, 2555–2558. [DOI] [PubMed] [Google Scholar]
- McCary C, Tancowny BP, Catalli A, Grammer LC, Harris KE, Schleimer RP, Kulka M, 2010. Substance P downregulates expression of the high affinity IgE receptor (FcepsilonRI) by human mast cells. Journal of neuroimmunology 220, 17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLarty JL, Melendez GC, Brower GL, Janicki JS, Levick SP, 2011. Tryptase/Protease-activated receptor 2 interactions induce selective mitogen-activated protein kinase signaling and collagen synthesis by cardiac fibroblasts. Hypertension (Dallas, Tex. : 1979) 58, 264–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melendez GC, Li J, Law BA, Janicki JS, Supowit SC, Levick SP, 2011. Substance P Induces Adverse Myocardial Remodeling via a Mechanism Involving Cardiac Mast Cells. Cardiovasc. Res 92, 420–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner P, Ralevic V, Hopwood AM, Feher E, Lincoln J, Kirkpatrick KA, Burnstock G, 1989. Ultrastructural localisation of substance P and choline acetyltransferase in endothelial cells of rat coronary artery and release of substance P and acetylcholine during hypoxia. Experientia 45, 121–125. [DOI] [PubMed] [Google Scholar]
- Morgan LG, Levick SP, Voloshenyuk TG, Murray DB, Forman MF, Brower GL, Janicki JS, 2008. A novel technique for isolating functional mast cells from the heart. Inflamm Res 57, 1–6. [DOI] [PubMed] [Google Scholar]
- Morrey C, Brazin J, Seyedi N, Corti F, Silver RB, Levi R, 2010. Interaction between sensory C-fibers and cardiac mast cells in ischemia/reperfusion: activation of a local renin-angiotensin system culminating in severe arrhythmic dysfunction. J Pharmacol. Exp. Ther 335, 76–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng LL, Sandhu JK, Narayan H, Quinn PA, Squire IB, Davies JE, Struck J, Bergmann A, Maisel A, Jones DJ, 2014. Pro-Substance P for Evaluation of Risk in Acute Myocardial Infarction. J. Am. Coll. Cardiol 64, 1698–1707. [DOI] [PubMed] [Google Scholar]
- Okuyama K, Yamashita M, Kitabatake Y, Kawamura S, Takayanagi M, Ohno I, 2005. Ciglitazone inhibits the antigen-induced leukotrienes production independently of PPAR[gamma] in RBL-2H3 mast cells. European Journal of Pharmacology 521, 21–28. [DOI] [PubMed] [Google Scholar]
- Olivetti G, Lagrasta C, Ricci R, Sonnenblick EH, Capasso JM, Anversa P, 1989. Long-term pressure-induced cardiac hypertrophy: capillary and mast cell proliferation. AJP - Heart and Circulatory Physiology 257, H1766–H1772. [DOI] [PubMed] [Google Scholar]
- Patella V, de CG, Ciccarelli A, Marino I, Adt M, Marone G, 1995. Human heart mast cells: a definitive case of mast cell heterogeneity. Int. Arch. Allergy Immunol 106, 386–393. [DOI] [PubMed] [Google Scholar]
- Petra AI, Tsilioni I, Taracanova A, Katsarou-Katsari A, Theoharides TC, 2018. Interleukin 33 and interleukin 4 regulate interleukin 31 gene expression and secretion from human laboratory of allergic diseases 2 mast cells stimulated by substance P and/or immunoglobulin E. Allergy and asthma proceedings 39, 153–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinecke M, Weihe E, Forssmann WG, 1980. Substance P-immunoreactive nerve fibers in the heart. Neurosci. Lett 20, 265–269. [DOI] [PubMed] [Google Scholar]
- Robinson P, Garza A, Moore J, Eckols TK, Parti S, Balaji V, Vallejo J, Tweardy DJ, 2009. Substance P is required for the pathogenesis of EMCV infection in mice. Int. J. Clin. Exp. Med 2, 76–86. [PMC free article] [PubMed] [Google Scholar]
- Robinson P, Kasembeli M, Bharadwaj U, Engineer N, Eckols KT, Tweardy DJ, 2016. Substance P Receptor Signaling Mediates Doxorubicin-Induced Cardiomyocyte Apoptosis and Triple-Negative Breast Cancer Chemoresistance. Biomed. Res. Int 2016, 1959270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinmura K, Tang XL, Wang Y, Xuan YT, Liu SQ, Takano H, Bhatnagar A, Bolli R, 2000. Cyclooxygenase-2 mediates the cardioprotective effects of the late phase of ischemic preconditioning in conscious rabbits. Proceedings of the National Academy of Sciences of the United States of America 97, 10197–10202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva Machado FR, Assem ES, Ezeamuzie CI, 1985. Cardiac anaphylaxis: the role of different mediators. Part I: Histamine. Allergol. Immunopathol. (Madr.) 13, 259–272. [PubMed] [Google Scholar]
- Silver RB, Reid AC, Mackins CJ, Askwith T, Schaefer U, Herzlinger D, Levi R, 2004. Mast cells: a unique source of renin. Proceedings of the National Academy of Sciences of the United States of America 101, 13607–13612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperr WR, Bankl HC, Mundigler G, Klappacher G, Grossschmidt K, Agis H, Simon P, Laufer P, Imhof M, Radaszkiewicz T, 1994. The human cardiac mast cell: localization, isolation, phenotype, and functional characterization. Blood 84, 3876–3884. [PubMed] [Google Scholar]
- Stewart JA, Wei CC, Brower GL, Rynders PE, Hankes GH, Dillon AR, Lucchesi PA, Janicki JS, Dell’Italia LJ, 2003. Cardiac mast cell- and chymase-mediated matrix metalloproteinase activity and left ventricular remodeling in mitral regurgitation in the dog. Journal of Molecular and Cellular Cardiology 35, 311–319. [DOI] [PubMed] [Google Scholar]
- Sumpter TL, Ho CH, Pleet AR, Tkacheva OA, Shufesky WJ, Rojas-Canales DM, Morelli AE, Larregina AT, 2015. Autocrine hemokinin-1 functions as an endogenous adjuvant for IgE-mediated mast cell inflammatory responses. The Journal of allergy and clinical immunology 135, 1019–1030.e1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki R, Furuno T, McKay DM, Wolvers D, Teshima R, Nakanishi M, Bienenstock J, 1999. Direct Neurite-Mast Cell Communication In Vitro Occurs Via the Neuropeptide Substance P. The Journal of Immunology 163, 2410–2415. [PubMed] [Google Scholar]
- Takai S, Jin D, Sakaguchi M, Katayama S, Muramatsu M, Sakaguchi M, Matsumura E, Kim S, Miyazaki M, 2003. A novel chymase inhibitor, 4-[1-([bis-(4-methyl-phenyl)-methyl]-carbamoyl)3-(2-ethoxy-benzyl)-4-oxo-azetidin e-2-yloxy]-benzoic acid (BCEAB), suppressed cardiac fibrosis in cardiomyopathic hamsters. The Journal of pharmacology and experimental therapeutics 305, 17–23. [DOI] [PubMed] [Google Scholar]
- Theoharides TC, Kempuraj D, Tagen M, Conti P, Kalogeromitros D, 2007. Differential release of mast cell mediators and the pathogenesis of inflammation. Immunol Rev 217, 65–78. [DOI] [PubMed] [Google Scholar]
- Weglicki WB, Mak IT, Phillips TM, 1994. Blockade of cardiac inflammation in Mg2+ deficiency by substance P receptor inhibition. Circ. Res 74, 1009–1013. [DOI] [PubMed] [Google Scholar]
- Wharton J, Polak JM, McGregor GP, Bishop AE, Bloom SR, 1981. The distribution of substrate P-like immunoreactive nerves in the guinea-pig heart. Neuroscience 6, 2193–2204. [DOI] [PubMed] [Google Scholar]
- Widiapradja A, Chunduri P, Levick SP, 2017. The role of neuropeptides in adverse myocardial remodeling and heart failure. Cellular and molecular life sciences : CMLS 74, 2019–2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Yang J, Li P, Cao J, Nie H, 2014. Rosiglitazone inhibits HMC-1 cell migration and adhesion through a peroxisome proliferator-activated receptor gamma-dependent mechanism. Iranian journal of allergy, asthma, and immunology 13, 11–18. [PubMed] [Google Scholar]
- Zhang W, Chancey AL, Tzeng HP, Zhou Z, Lavine KJ, Gao F, Sivasubramanian N, Barger PM, Mann DL, 2011. The development of myocardial fibrosis in transgenic mice with targeted overexpression of tumor necrosis factor requires mast cell-fibroblast interactions. Circulation 124, 2106–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Li X, Fang S, Zhu Z, Yao M, Ying L, Zhu L, Ma Z, Wang W, 2017. Peroxisome proliferator-activated receptor gamma agonist suppresses mast cell maturation and induces apoptosis. Molecular medicine reports 16, 1793–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.