

Abstract
Although prion diseases are generally thought to present as rapidly progressive dementias with survival of only a few months, the phenotypic spectrum for genetic prion diseases (gPrDs) is much broader. The majority have rapid decline with short survival but many present as slower progressive ataxic or parkinsonian disorders with progression over a few to several years. A few very rare mutations even present as neuropsychiatric disorders, sometimes with systemic symptoms such as gastrointestinal disorders and neuropathy, progressing over years to decades. Genetic prion diseases are caused by mutations in the prion protein gene (PRNP), and have been historically classified based on their clinicopathological features as genetic Jakob-Creutzfeldt disease (gJCD), Gerstmann-Sträussler-Scheinker (GSS), or Fatal Familial Insomnia (FFI). Mutations in PRNP can be missense, nonsense and octapeptide repeat insertions or a deletion. and present with diverse clinical features, sensitivities of ancillary testing and neuropathological findings. We present the UCSF gPrD cohort, including 129 symptomatic patients referred to and/or seen at UCSF between 2001 and 2016, and compare the features of the 22 mutations identified with the literature, as well as perform a literature review on most other mutations. E200K is the most common mutation worldwide, is associated with gJCD, and was the most common in the UCSF cohort. Among the GSS-associated mutations, P102L is the most commonly reported and was also the most common at UCSF. We also had several octapeptide repeat insertions (OPRI), a rare nonsense mutation (Q160X) and three novel mutations (K194E, E200G, and A224V) in our UCSF cohort.
Keywords: CJD, Creutzfeldt-Jakob disease, prion protein gene, rapidly progressive dementia, octapeptide repeat insertion
Introduction
Prion diseases (PrDs) are neurodegenerative disorders caused by transmissible proteins called prions, which stand for proteinaceous infectious particles[Prusiner 1982]. PrD have been reported not only in humans, but also in some mammals, such as sheep and goats (scrapie) and cattle (bovine spongiform encephalopathy), among others. Prion diseases occur when the prion protein (the normal conformation of the protein) or PrPC, in which C stands for the normal cellular form of the protein, is converted into a misshapen form called the prion, or PrPSc in which Sc stands for scrapie [Prusiner 1998].
Human PrDs are typically classified in three forms based on etiology: sporadic, genetic and acquired. Sporadic PrDs (sPrDs) are the most common form (about 85–90%) comprised primarily of sporadic Jakob-Creutzfeldt-disease (sJCD) which has several subtypes or molecular classifications [Brown and Mastrianni 2010; Parchi et al., 1999; Puoti et al., 2012] as well as a very rare, atypical form, variably protease-sensitive prionopathy (VPSP) [Ladogana et al., 2005; Masters et al., 1979; Zou et al., 2010]. The first cases of JCD were reported by Alfons Jakob in 1921, whereas the patient reported by Hans Creutzfeldt one year prior we now know did not have prion disease [Masters 1989]. As Jakob believed at the time that his patients had similar features to the patient reported by Creutzfeldt, the eponym Jakob-Creutzfeldt disease was created. For many decades the disease was called Jakob’s disease or Jakob-Creutzfeldt disease (JCD), but a prominent researcher in the field C.J. Gibbs began using the term Creutzfeldt-Jakob disease (CJD), so that the acronym would be closer to his own initials [Gibbs 1992]. Thus the proper term should probably be Jakob-Creutzfeldt disease (JCD) or even Jakob’s disease (JD) [Katscher 1998]. Thus, we will use the term Jakob-Creutzfeldt disease (JCD) in this article. It is important that the term JCD, however, not be confused with the JC virus, which is the cause of progressive multifocal leukoencephalopathy and has nothing to do with prion disease.
Genetic PrDs (gPrDs) are caused by mutations in the gene, PRNP, that encodes the prion protein. Genetic PrDs account for about 10–15% of human PrDs [Kovacs et al., 2005; Ladogana et al., 2005; Masters et al., 1979]. Acquired PrDs are the rarest form (<1% of human PrD) and include Kuru, iatrogenic PrD and variant JCD (vJCD) [Brown et al., 2006; Ladogana et al., 2005; Masters et al., 1979; UK National CJD Surveillance Unit 2015]. This paper focuses on gPrDs.
Genetic prion disease background
Historically, gPrDs have been classified into three forms based on clinical and neuropathological features: familial JCD (fJCD, OMIM 123400), Gerstmann-Sträussler-Scheinker disease (GSS, OMIM 137440) and Familial Fatal Insomnia (FFI, OMIM 600072). Familial JCD typically presents similar to sporadic JCD, as a rapidly progressive dementia with motor features resulting in death in about a few months (usually less than a year) from onset. Pathology also is often similar to sJCD with various patterns of PrPSc deposition (synaptic, granular or plaque-like pattern), vacuolation (spongiform changes), neuronal loss and gliosis; the severity of neuronal loss and gliosis might be related to disease duration [Gambetti et al., 2003a]. Gerstmann-Sträussler-Scheinker usually presents as an ataxic disorder often with parkinsonian symptoms and dementia a bit later in the course. Survival is typically about three to 10 years, but some patients have a much shorter course. Pathology usually reveals prominent PrPSc amyloid plaques in the cerebellum and cortex [Gambetti et al., 2003a]. Persons with FFI usually develop prominent insomnia and dysautonomia, with cognitive impairment and motor features occurring later in the disease course. Median survival is about 18 months. The most severe pathology is neuronal loss and gliosis in the thalamus (thalamic atrophy), but also inferior olivary nucleus atrophy and gliosis in the midbrain and hypothalamic gray matter. Vacuolation and PrPSc deposition are sparse or often absent. The cortex is involved with longer duration cases [Gambetti et al., 2003a; Parchi et al., 1995]. This classification scheme of fJCD, GSS and FFI, however, was prior to the discovery of PRNP, and thus is somewhat outdated, as several PRNP mutations do not fit well into one of these three groups, as we discuss later.
Familial aggregation of PrD had been reported since the early 1900s, but it was only in 1989 that mutations in PRNP were shown to be causative of genetic PrD [Goldgaber et al., 1989; Hsiao et al., 1989; Owen et al., 1989]. Familial JCD (fJCD) was first reported by Meggendorfer in 1930, in a German kindred (the Backer family) that presented with rapidly progressive dementia and spongiform changes in the brain, and later was shown to carry a PRNP mutation (D178N-codon 129V; see below) [Kretzschmar et al., 1995]. In 1936, J. Gerstmann, E. Sträussler and I. Scheinker reported an Austrian kindred (the “H” family) that presented with slowly progressive cerebellar ataxia and late dementia, with an autosomal dominant pattern of inheritance, which is now considered the first known GSS family (PRNP P102L mutation identified later ) [Hainfellner et al., 1995]. The term FFI was first used by Lugaresi and colleagues in 1986, in the description of an Italian family with progressive insomnia, dysautonomia, motor signs and thalamic degeneration [Lugaresi et al., 1986]. The neuropathological findings suggested FFI was a PrD, but confirmation of it being genetic did not occur until 1992, when a PRNP D178N-codon 129M mutation was identified in the kindred [Medori et al., 1992]. The fact that the same mutation (D178N) could lead to two different phenotypes (fJCD or FFI) was puzzling at first, but soon later, Goldfarb et al. discovered that the PRNP codon 129 polymorphism found on the mutated (cis) allele could have a strong effect on the clinicopathological phenotype. FFI was typically associated with D178N and methionine at codon 129 and fJCD, with valine at codon 129 [Goldfarb et al., 1992].
The first pathogenic mutations identified in PRNP were missense [Goldgaber et al., 1989; Hsiao et al., 1989] but other types of mutations later were identified. Insertions, mostly octapeptide repeat insertions (OPRI) in the copper binding domain of PRNP, can lead to gJCD or GSS phenotypes and an two octapeptide deletion leads to a gJCD phenotype [Brown and Mastrianni 2010; Hinnell et al., 2011; Owen et al., 1990]. Nonsense or stop-codon mutations in PRNP were more recently identified, and have only been reported in a few kindreds. The clinical presentation of nonsense mutations is highly variable, but overall they are characterized by clinical features that are atypical for most PrDs and the presence of amyloid plaques and/or prion amyloid angiopathy [Fong et al., 2016a; Guerreiro et al., 2014; Mead et al., 2013].
Prion protein (PrP) and the prion protein gene (PRNP)
The PRNP (prion protein) gene is located in chromosome 20p13 and is composed of two exons. The entire open reading frame is in exon 2. The prion protein (PrP) has several isoforms, but the canonical sequence has 253 amino acids and contains an unstable region with repeats in the N-terminal domain, composed of a nonapeptide (Pro-Gln-Gly-Gly-Gly-Gly-Trp-Gly-Gln) followed by four repeats of an octapeptide (Pro-His-Gly-Gly-Gly-Trp-Gly-Gln) (Figure 1). Post-translational modifications include removal of 22 residues from the N-terminal and 23 residues from the C-terminal, attachment of a glycosyl-phosphatidylinositol (GPI) anchor in the C-terminal, and possible glycosylation. PrP can be un-, mono-, or di-glycosylated at asparagine residues 181 and/or 197 [Capellari et al., 2011; Yusa et al., 2012]. Regarding the structure of PrP, a globular domain extends from residues 125 to 228 and the normal protein contains three α-helices (residues 144–154, 173–194 and 200–228) and an anti-parallel β-sheet (residues 128–131 and 161–164) (Figure 1) [Zahn et al., 2000]. The tertiary structure of PrPC is influenced by hydrophobic and aromatic interactions between residues of the globular domain, as well as by the existence of salt bridges and disulfide bridges [Capellari et al., 2011; Giachin et al., 2013; van der Kamp and Daggett 2009].
Figure 1. Schematic of PRNP disease-associated variants.

Mutations are color coded based on clinicopathological classifcation as gJCD, GSS, FFI or nonsense mutations. PRNP mutations present in the UCSF cohort are in bold. Most mutations are shown below the gene schematic; nonsense mutations and polymorphisms associated with prion disease risk are above the gene schematic. Low or intermediate penetrance variants are based on Minikel et al., 2016 (not all low/intermediate penetrance variants are shown)[Minikel et al., 2016]. For the F198V mutation, the clinical presentation was not classifiable as gCJD, GSS or FFI (see Table 3), and neuropathology was not reported [Zheng et al., 2008]. Variants that are probably benign (largely based on Minikel et al., 2016) are not included (e.g., G54S, P39L, E196A, R208C) [Beck et al., 2010; Minikel et al., 2016].
JCD= Jakob-Creutzfeldt disease; GSS= Gerstmann-Sträussler-Scheinker; FFI= Fatal Familial Insomnia. OPRI= Octapeptide repeat insertion; OPRD= Octapeptide repeat deletion.
Cellular PrP (PrPC) is typically attached to the plasma membrane by the GPI anchor, but eventually during its lifecycle, PrPC is internalized in endosomes [Harris 2003]. The repeat region in the N-terminal contains copper binding sites, which suggests PrPC might have a role in the copper homeostasis (Harris 2003). PrP genes are highly conserved across mammals, indicating PrP was important during evolution [Colby and Prusiner 2011]. Nonetheless, the biological function of PrPC is not entirely understood, although there are many putative roles [Bribian et al., 2012; Flechsig and Weissmann 2004; Watts and Westaway 2007].
The prion protein exists in multiple isoforms, two of which are relevant to PrD: the normal cellular isoform (PrPC) and the pathogenic infectious “scrapie” isoform (PrPSc). Both isoforms generally share the same amino acid sequence, but whereas PrPC structure is composed mainly of alpha-helix, PrPSc has a high beta-sheet content [Pan et al., 1993]. This conformational change affects PrP biochemical properties, so that PrPSc becomes insoluble in detergents and relatively resistant to degradation by proteases [Colby and Prusiner 2011; Mastrianni 2010]. According to Stanley Prusiner’s protein-only hypothesis, PrPSc has infectious properties and self-propagates by acting as a template in the misfolding of PrPC into PrPSc (Prusiner 1998). This hypothesis has now been essentially proven and might also apply to other neurodegenerative proteinopathies [Prusiner 2012; Soto 2011; Woerman et al., 2015]. It is not entirely clear where in the cell the conversion of PrPC to PrPSc occurs, but there is evidence suggesting this happens inside cholesterol-rich nonacidic intracellular compartments called caveolae-like domains [Colby and Prusiner 2011]. Many scientists believe the subsequent accumulation of PrPSc is the principal event that leads to neurodegeneration in PrD [Prusiner 2013], whereas others feel that it might be the conversion of PrPC into PrPSc that leads to neuronal injury and loss [Mallucci et al., 2007; Moreno et al., 2013]. PrPSc propagates as oligomers, which might polymerize to form amyloid fibrils [Prusiner 2013]. As we will discuss later, PrP amyloid plaques are sometimes found in brains of patients with PrD, particularly in GSS and stop codon mutations.
There are two major types of PrPSc found in brains of individuals with PrD (particularly in sJCD), termed types 1 and 2 [Puoti et al., 2012]. After brain homogenates are treated with proteinase K (which digests most of PrPC, leaving behind PrPSc, which is generally resistant to proteinase K digestion) and run on a Western blot and stained for PrP, this shows three bands which are the diglycosylated, monoglycosylated and the unglycosylated forms of PrPSc. The unglycosylated bands run at the lowest molecular weight; when the unglycosylated band runs at 21kD, this denotes type 1 PrPSc and at 19kD this denotes type 2 PrPSc [Parchi et al., 1996]. In about two-thirds of cases only one type is found in the brain at pathology, but co-occurrence of types 1 and 2 occurs in about one third of cases [Parchi et al., 2009]. The combination of PrPSc type and PRNP codon 129 polymorphism has been used in the molecular classification of sJCD clinicopathological phenotypes (for excellent reviews, see Puoti et al. 2012 and Brown et al. 2010) [Brown and Mastrianni 2010; Parchi et al., 1999; Puoti et al., 2012].
A few gene risk variants in PRNP have been characterized, and among those, the codon 129 polymorphism (rs1799990) is the most relevant to human PrD, as it influences both disease susceptibility and the phenotypical presentation of the genetic, sporadic and acquired forms of PrD. Codon 129 encodes either methionine or valine, and in the general population, about 55% of persons are homozygous for methionine (MM), 36% are heterozygous (MV) and 9% are homozygous for valine (VV) [1000 Genomes Project Consortium et al., 2012]. Homozygosity at codon 129 is a well-established risk factor for sporadic and acquired PrD [Mead et al., 2012; Palmer et al., 1991]. Among European sJCD cases, for example, 70% are 129MM, 13% are 129MV and 16% are 129VV, which shows there is a clear underrepresentation of codon 129 heterozygosity among sJCD cases in comparison to the normal population [Alperovitch et al., 1999].
In gPrD, the polymorphism located in the same allele as the mutation (cis) has a more significant effect on disease presentation, whereas the trans polymorphism usually has less or even no effect [Capellari et al., 2011]. As previously mentioned, in the PRNP D178N mutation, the codon 129 polymorphism in cis determines whether the phenotype is gJCD (D178N-129V) or FFI (D178N-129M) [Goldfarb et al., 1992]. The phenotype associated with other mutations are also clearly influenced by the codon 129 genotype, which is why some authors suggest the mutations should be reported in combination with the codon 129 genotype [Capellari et al., 2011; Goldfarb et al., 1992].
Common PRNP mutations in various national cohorts
Cohorts of gPrD have been described from most parts of the world, mostly in multi-center studies or from national PrD surveillance centers. More than 60 reportedly pathogenic variants have been published in the literature to date (even though most of them were only reported in single or few patients/families, and several might even be benign variants or mutations with low penetrance) [Kovacs et al., 2005; Minikel et al., 2016]. Among those variants, five are responsible for about 85% of gPrD cases in nine major surveillance centers around the world (in Australia, France, Germany, Italy, Japan, Netherlands, Spain, United Kingdom and US National Prion Disease Pathology Surveillance Center (NPDPSC), namely: E200K, V210I, V180I, P102L, and D178N [Minikel et al., 2016]. The E200K, V210I, V180I and D178N-129V mutations are associated with familial JCD and P102L with GSS. As many cases of familial JCD (fJCD) do not actually have a known history of prion disease (either because of misdiagnosis or low penetrance), we prefer the term genetic JCD (gJCD), which will be used hereon in this paper. A few of the mutations have large regional clusters due to a founder effect, such as the E200K mutation clusters among Slovakians and also Libyan (Sephardic) Jews, many of whom now are in Israel, [Hsiao et al., 1991a; Mitrova and Belay 2002] the V180I mutation most commonly found in Japan, and V210I, which is the most common mutation in Italy [Ladogana et al., 2005; Minikel et al., 2016].
The objectives of this paper are to describe the diversity of prion diseases through literature review of genetic PrD, as well as describing our well-characterized cohort of gPrD subjects referred to a single rapidly progressive dementia (RPD) and PrD research center in the USA.
Methods
Patients in UCSF Cohort
Between August 1, 2001 and May 1, 2016, 1922 subjects with (RPD), including suspected PrD, or unaffected persons from families with gPrD have been referred to the University of California, San Francisco Memory and Aging Center (UCSF MAC) RPD clinical research program. After the initial contact made by a health care provider or patient, caregiver, and/or family member, we requested the patients’ medical records for our review, as well as possible signing of informed consents for the data to be included in our ongoing RPD research. Some research subjects were also evaluated through an ongoing study of persons at risk for gPrD (from an affected family but PRNP status unknown), with a PRNP mutation, or non-carriers (from gPrD families). This study includes symptomatic and asymptomatic subjects. We also have an internal review board (IRB)-approved protocol allowing use of existing referred patient data for research, except when inclusion of data is explicitly declined by the patient or family. Patients with suspected, known or at risk for PrD were offered a research visit at our center. Visits typically included an extensive evaluation including clinical history, neurological examination, neuropsychological and functional assessment, brain magnetic resonance imaging (MRI), genetic counseling including detailed family histories/trees, social work assessment, PRNP gene testing (blood), and blood and cerebrospinal fluid (CSF) collection or research. Subjects found to have a PRNP mutation were included in the descriptive analyses in this paper. Subjects were included in the analyses in this paper if referral or initial contact was made before May 1st, 2016.
Demographic and general clinical data (e.g. age at onset of symptoms and first symptoms) were collected from outside medical records. First symptoms were characterized as previously described [Rabinovici et al., 2006]. Patients or asymptomatic individuals who came to UCSF MAC for a research visit were further characterized in detail, including if appropriate, symptoms present during the course of disease, signs on neurological examination, and cognitive and behavioral assessments. Results of ancillary testing (genetic, CSF and neuroimaging) were included if available in medical records or performed at UCSF. Total disease duration was calculated only if the patient was deceased and date of death was informed to our team. For determining the likelihood of a positive family history (FMH), FMH was rated as 0 when there was no positive FMH suspicious for or known PrD; 1 when there was at least one first-degree relative with dementia, encephalopathy or movement disorder; or 2 in patients who were part of families with known PRNP mutations, or had positive history for clinical or path-proven PrDs.
Laboratory testing
Genetic testing for PRNP mutations and polymorphisms were performed by blood test or frozen brain tissue through the U.S. National Prion Disease Pathology Surveillance Center (NPDPSC, Case Western Reserve), following previously described methods [Zou et al., 2010].
CSF 14-3-3 analysis by Western Blot was also performed at the NPDPSC. CSF total tau (t-tau; either through Athena Diagnostics, Inc. or the NPDPSC) and neuron specific enolase (NSE; Mayo Medical Laboratories, Rochester, MN) results were included when available. Cut-off levels for t-tau were 1150 pg/mL (performed at NPDPSC) or 1200 pg/mL (performed at Athena Diagnostics) and cut-offs for NSE were 30 or 35 ng/mL (Mayo Medical Laboratories cut-off prior to November 20, 2007 was >35 ng/mL and after was changed to >30 ng/mL) [Forner et al., 2015].
Neuroimaging
Brain MRI scans included for analyses in this study were performed at UCSF or outside centers. Images were available from 1.5 and 3T scanners and usually included T1, T2, fluid attenuated inversion recovery (FLAIR), diffusion weighted imaging (DWI ) and attenuation diffusion coefficient (ADC) map sequences. MRI scans were considered positive for PrD if they met the criteria published by Vitali et al [Vitali et al., 2011]. MRIs often were further classified according to cortical or subcortical predominance of DWI abnormalities.
Statistical analyses
Statistical analyses were performed using Stata Release 12 (College Station, TX). Chi-square and Wilcoxon-Mann-Whitney tests were used, according to data type. Significance was set at p≤0.05.
Literature review
We performed a literature review in Pubmed for “genetic prion disease” or “PRNP mutation” as search terms, and also performed searches for each mutation individually (e.g. E200K, P102L, etc.). We also used data on gPrDs from a detailed, but now somewhat dated, book chapter on this subject [Kong et al., 2004]. We also searched for publications from PrD surveillance centers, using “surveillance” and “prion” as search terms.
Results
Patients
Among 1922 subjects referred to our center, 1266 were suspected PrD or were unaffected gPrD family members. In this UCSF cohort, we identified 89 gPrD families, representing 22 different PRNP mutations, with gPrD whose family members have had UCSF visits or with whom we have been in contact (Table 1). In this cohort, we had 129 symptomatic, 64 presymptomatic, 73 non-mutation carriers (controls for our research, and 47 whose PRNP status was unknown. Among the 129 symptomatic gPrD patients, we had clinical data (most of which was extensive) on 79 cases. The mutations in whom we have the largest number of families in our cohort are: E200K (n=31 families), P102L mutations (n=11), D178N-129M (FFI; n=9), D178N-129V (n=6), and three each for 5-OPRI (typically GSS per the literature), A117V (GSS), and F198S (GSS). Among symptomatic cases, the most common individual mutations in our cohort with five or more cases were E200K (n=43), P102L (n=20), D178N-129V (n=10), D178N-129M (FFI, n=9), A117V (n=7), H187R (n=6), F198S (n=6), V210I (n=5), and 6-OPRI (n=5). Three mutations in our cohort (K194E, E200G, A224V) are novel [Kim et al., 2013; Watts et al., 2015] (Table 1). The mutations best represented in our cohort are also the most common in the US and most other countries [Kovacs et al., 2011; Minikel et al., 2016; Webb et al., 2008]. Of the symptomatic cohort of 129 gPrD patients, 67 were classified as gJCD, 40 GSS, and 9 FFI, 12 OPRI (many were GSS-like) and one had a nonsense mutation (Q160X). The demographic and clinical characteristics of each group (except the nonsense mutation) are shown in (Table 2).
Table 1.
| UCSF Genetic Prion Disease Cohort# | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Classification | Family Mutation | All families | All subjects | Mutation positive subjects* | Symptomatic patients | |||||||
| Referred | Seen | Visits | Referred | Seen | Visits | Referred | Seen | Visits | Referred | Seen at UCSF | ||
| fJCD | E200K | 31 | 22 | 22 | 127 | 63 | 52 | 76 | 40 | 35 | 43 | 12 |
| T188R | 2 | 2 | 2 | 11 | 7 | 7 | 6 | 5 | 5 | 2 | 1 | |
| D178N/129V | 6 | 3 | 3 | 18 | 5 | 5 | 9^ | 3 | 3 | 10^ | 3 | |
| V180I | 2 | 2 | 2 | 5 | 3 | 3 | 5 | 3 | 3 | 2 | 1 | |
| E200G | 1 | 1 | 1 | 3 | 3 | 3 | 3 | 3 | 3 | 1 | 1 | |
| A224V | 1 | 1 | 1 | 3 | 3 | 3 | 2 | 2 | 2 | 1 | 1 | |
| V210I | 6 | 1 | 1 | 11 | 1 | 1 | 5 | 0 | 0 | 5 | 0 | |
| K194E | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | |
| R148H | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | |
| G54S | 1 | 0 | 0 | 2 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | |
| SUBTOTAL | 52 | 33 | 33 | 182 | 86 | 75 | 109 | 57 | 52 | 67 | 20 | |
| GSS | A117V | 3 | 3 | 3 | 30 | 25 | 24 | 13 | 11 | 11 | 7 | 6 |
| P102L | 11 | 6 | 5 | 34 | 16 | 15 | 24 | 10 | 10 | 20 | 8 | |
| F198S | 3 | 2 | 2 | 11 | 6 | 5 | 7 | 4 | 4 | 6 | 3 | |
| H187R | 1 | 1 | 1 | 7 | 2 | 2 | 6 | 2 | 2 | 6 | 2 | |
| P105L | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | |
| SUBTOTAL | 19 | 12 | 11 | 83 | 49 | 46 | 51 | 28 | 28 | 40 | 20 | |
| FFI | D178N/129M | 9 | 4 | 4 | 28 | 7 | 6 | 12 | 6 | 5 | 9 | 2 |
| Octapeptide repeat insertions | 5-OPRI | 3 | 3 | 3 | 9 | 5 | 5 | 8 | 4 | 4 | 4 | 1 |
| 6-OPRI | 2 | 1 | 1 | 7 | 4 | 4 | 5 | 2 | 2 | 5 | 2 | |
| 8-OPRI | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | |
| 9-OPRI | 1 | 1 | 1 | 2 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | |
| 365–388dup | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | |
| SUBTOTAL | 8 | 5 | 5 | 20 | 10 | 10 | 16 | 7 | 7 | 12 | 4 | |
| Nonsense Mutation | Q160X | 1 | 1 | 1 | 3 | 3 | 1 | 2 | 2 | 1 | 1 | 1 |
| TOTAL | 22 mutations | 89 | 55 | 54 | 316 | 155 | 138 | 190 | 100 | 93 | 129 | 46 |
For each syndrome, mutations are listed in order of subjects seen and then by mutation codon number. All families include those having any family members who were referred to, seen at or visited UCSF. All subjects include mutation positive subjects (symptomatic and presymptomatic), no-mutation carriers, and subjects at-risk for mutations but not tested for mutations or not knowing the status of mutations.
Mutation positive subjects include symptomatic and presymptomatic cases only. Referred: all subjects; Seen: subjects seen at UCSF through clinic, inpatient, research or any combination of these; Visits: subject had full UCSF research visit
one subject was symptomatic and had a history of fJCD, but did not complete genetic testing or autopsy to confirm diagnosis.
Table 2.
Demographic and clinical features of the symptomatic UCSF gPrD cohort, by gPrD classification.
| Classification | n | gJCD$ | n | GSS$ | n | FFI$ | n | OPRI$ |
|---|---|---|---|---|---|---|---|---|
| Symptomatic cases referred | 67 | 40 | 9 | 12 | ||||
| Number of families with symptomatic cases | 46 | 19 | 5 | 8 | ||||
| Symptomatic cases evaluated at UCSF | 20 | 20 | 2 | 4 | ||||
| Age at onset (years) | 38 | 58±13 (36–84) | 26 | 41±14 (10–66) | 4 | 47±19 (20–64) | 9 | 44±14 (18–70) |
| Onset to UCSF evaluation (months) | 18 | 9.4±7.0 (0.5–25.5) | 16 | 47.0±45.2 (3–168) | 1 | 8 | 5 | 41.6±47.0 (7–120) |
| Sex | 27M:25F | 20M:10F | 4M:4F | 6M:7F | ||||
| Duration of disease (months)^ | 32 | 12.4±15.6 (1–78) | 16 | 56.3±29.7 (27–141) | 4 | 7.7±1.8 (5–9) | 5 | 27.2±25.8 (5–69) |
| First symptom category | ||||||||
| Cognitive | 38 | 53% | 28 | 32% | 3 | 33% | 6 | 83% |
| Cerebellar | 18% | 39% | 0 | 17% | ||||
| Sensory | 16% | 0 | 0 | 17% | ||||
| Behavioral | 10% | 7% | 33% | 17% | ||||
| Constitutional | 10% | 4% | 67% | 0 | ||||
| Visual | 5% | 0 | 0 | 0 | ||||
| Motor | 3% | 18% | 0 | 0 | ||||
| Epileptic | 0 | 4% | 0 | 0 | ||||
| Positive family history§ | 39 | 80% [scores 1=13; 2=18] | 18 | 79% [scores 1=4; 2=10] | 4 | 75% [score 2=3] | 5 | 60% [score 1=2; 2=1] |
| CSF 14-3-3* positive | 15 | 40% | 10 | 10% | 2 | 0 | 5 | 40% (2/5) |
| CSF total tau** | 6 | 50% | 4 | 25% | 0 | - | 2 | 0 |
| CSF NSE*** | 7 | 14% | 8 | 38% | 0 | - | 3 | 33% |
| EEG – PSWC positivity | 23 | 22% | 13 | 0 | 4 | 0 | 4 | 25% |
| MRI DWI positivity# | 28 | 89% | 14 | 7% | 3 | 33% | 6 | 17% |
Data is displayed as mean ± standard deviation (range). Ranges in parentheses. n = Number of patients from whom data was available.
gJCD includes E200K, D178N-129V, V210, V180I, A224V, E200G, E148H, T188R, K194E. GSS includes P102L, A117V, D198S, H187R, P105L and 365–388dup. FFI includes D178N-129M. OPRI include 5-OPRI, 6-OPRI, 8-OPRI and 9-OPRI. “n” refers to the number of cases for a gPrD classification for whom relevant data is available.
Duration of disease in deceased patients only
Family history was scored as (0) Negative; (1) questionable PrD (dementia NOS, encephalopathy, movement disorder, etc…); (2) positive for PrD (confirmed by molecular testing, neuropathological assessment, and/or clinical diagnosis). Positive family history means score 1 or 2.
Inconclusive CSF 14-3-3 was considered negative, based on a previous study [Forner et al., 2015].
Positive CSF total tau >1200pg/ml.
Positive CSF NSE >35ng/ml before November 2007; > 30 ng/ml after November 2007.
According to published UCSF criteria [Vitali et al., 2011].
Abbreviations: CSF= cerebrospinal fluid; DWI=diffusion weighted imaging EEG= electroencephalogram; FFI= familial fatal insomnia; gJCD= genetic Jakob-Creutzfeldt disease; GSS= Gerstmann-Sträussler-Scheinker; MRI= magnetic resonance imaging; NSE= neuron specific enolase; OPRI= octapeptide repeat insertions; PSWC= periodic sharp wave complexes.
Division into Fast vs. Slow presentations
Although it is often very helpful to classify each of the PRNP mutations (missense, nonsense, or octapeptide repeat insertions/deletions) based on clinical and neuropathological characteristics into the historical classificaitons of genetic JCD, GSS or FFI, this division does have some limitations, as nonsense mutations and many octapeptide repeat insertions/deletions do not fit neatly into one of these three clinicopathologial categories. Clinically, we have found it helpful also to dichotomize gPrDs according to the rate of clinical course (decline rate and total disease duration) as Fast vs. Slow types. As we typically define RPD as dementia with rapid decline and less than 3 years of disease duration [Geschwind 2016], we applied this concept to classifying gPrDs and divided gPrDs into Fast and Slow types; Fast type with rapid decline and total disease duration usually less than three years and Slow type with more gradual decline and total disease duration usually greater than three years. This classfication captures OPRD, FFI and almost all gJCD and a few OPRIs (particularly those with <4 repeats) as Fast, and all nonsense mutations, almost all GSS and most OPRIs (some with 5–7 repeats and particularly ≥ 8 repeats) as Slow. Some of the OPRIs and GSS, however, even within the same repeat size can present as Fast or Slow, even within the same family. For OPRIs, this in part might be because there can be variations in the pattern of repeats between the same size of OPRI [Schmitz et al., 2016]. In this review, we present the features of each mutation, including their historical classifcation. We start with Fast types and then present the Slow types and then discuss OPRIs/OPRD seperately as they can present as Fast or Slow. Lastly we discuss the very unusual and rare nonsense mutations, which are Slow types.
Genetic Prion Diseases usually with faster disease progression
Genetic Jakob-Creutzfeldt disease (gJCD)
Specific PRNP mutations have historically been associated with gJCD when the clinical presentation resembled that of sJCD, and spongiform encephalopathy (including spongiform changes, astrocytic gliosis and neuronal loss) and/or PrPSc immunostaining was found in the brains of mutation-carrying patients [Budka et al., 1995]. At least 21 missense variants in PRNP have been reported as causing gJCD in the literature (including P105T, G114V, R148H, D178N (with codon 129 cis V), V180I, T183A, T188A, T188K, T188R, T193I, E196A, E196K, E200K, E200G, V203I, R208H, V210I, E211Q, I215V, M232R, and P238S), but the pathogenicity of some of these variants, especially are questioned by some (see below) [Minikel et al., 2016]. Nine missense mutations were found in the UCSF cohort, namely E200K (n=43 symptomatic cases), D178N-129V (n=10), V210I (n=5), V180I (n=2), A224V (n=1), E200G (n=1), R148H (n=1), T188R (n=2), and K194E (n=1).
The penetrance of PRNP mutations is variable, and will be discussed with each mutation. Importantly, a positive family history of PrD is found in only 50-75% of patients with gJCD, but this percentage varies greatly depending on the mutation [Kovacs et al., 2005]. In many gPrD cases with a reportedly negative family history of PrD, further inspection will usually uncover a family history of dementia or neuropsychiatric illness, however, that was likely PrD, but had likely been misdiagnosed [Goldman et al., 2004].
The mean age of symptom onset in gJCD is about 55 years, and although onset most frequently occur between the fifth and seventh decades of life, there is a great variability in age at onset ranging from the second to ninth decades of life [Kovacs et al., 2002]. The mean duration of disease is about 15 months, and even though 90% of patients die in less than 24 months after disease onset, total duration of disease of more than 8 years has been reported [Kovacs et al., 2002]. Compared to sJCD, the age at onset of gJCD is often lower, and disease duration, longer [Kovacs et al., 2002].
Dementia is very common among patients with gJCD, and has been reported in 95–98% of patients [Kovacs et al., 2002; Meiner et al., 1997]. Cerebellar symptoms are found in approximately 70%, myoclonus in 60–70% [Kovacs et al., 2002; Meiner et al., 1997], extrapyramidal signs in about 50%, and psychiatric symptoms in slightly more than 25% of cases [Kovacs et al., 2002].
Regarding ancillary testing, the accuracies of CSF markers, EEG and brain MRI are overall lower than in sJCD [Kovacs et al., 2005; Kovacs et al., 2002]. In CSF, 14-3-3 positivity has been reported in 85–100% of cases depending greatly on the mutation and the study [Kovacs et al., 2005; Krasnianski et al., 2016], and elevated total tau levels (> 1200 pg/ml), in 75–100% of cases [Breithaupt et al., 2013; Higuma et al., 2013; Krasnianski et al., 2016]. PSWCs in EEG are observed in 11–93% (average ~60–70%) of cases, also depending greatly on the mutation and the study [Breithaupt et al., 2013; Higuma et al., 2013; Kovacs et al., 2005; Krasnianski et al., 2016; Meiner et al., 1997]. The sensitivity of brain MRI varies greatly depending on MRI sequences used, with much lower sensitivity for T2-weighted sequences and very high for diffusion sequences (DWI and ADC). On brain MRIs, basal ganglia and/or cortical hyperintensities in FLAIR or DWI have been found in 25–80% of cases, but there is significant variation across mutations and study [Breithaupt et al., 2013; Kovacs et al., 2005; Krasnianski et al., 2016]. In the UCSF gJCD cohort, we found much lower sensitivity for CSF 14-3-3, total tau and NSE and EEG than the literature, but high DWI sensitivity (89%) (Table 2).
There is sufficient evidence proving that variants such as the E200K and D178N mutation are pathogenic. Some variants, however, are frequently reported as mutations despite there being no cases with any positive or even suggestive family history of PrD and/or beingfound in population data sets, such as the Exome Aggregation Consortium (ExAC), at a frequency higher than expected if they were highly penetrant, Mendelian disease-causing variants. Therefore, some these variants might be considered low-penetrance mutations or risk factors for PrD, and some others might even be benign variants with no influence on PrD risk [Minikel et al., 2016]. For example, the V210I mutation, the most frequent mutation reported in Italy, has penetrance estimated to be about 10%. Two frequently reported mutations in Japan, M232R and V180I, have even lower estimates of penetrance, of 0.1% and 1%, respectively. Other variants were reported only in a few patients (Table 3) and based on the clinical evidence available to date, some of them might be low-penetrance or benign variants, such as R148H, T188R, V203I, R208H [Minikel et al., 2016]. Other variants are too rare in both patients and in control population data sets for a conclusion to be drawn on their pathogenicity.
Table 3.
Missense mutations in PRNP in literature & UCSF cohort*
| PRNP Mutation |
Codon 129 polymorphism |
# of cases in literature or UCSF cohort # |
Clinical phenotypes |
Age at onset (range)^ Y |
Disease Duration (M or Y) |
Pos FHx## |
CSF marker Sensitivity |
EEG PSWC |
MRI c/w JCD§ |
Neuropath | Neuropathl Pheno- type |
References | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 14-3-3 | Total tau^^ | ||||||||||||
| P84S | MV | 1 |
|
(60) | (14) M | 0% (0/1) | N/A | 0% (0/1) | 0% (0/1) | Multic PrP-Plqs w/o NFT No V |
GSS | [Jones et al., 2014] | |
| S97N | Cis M | 1 |
|
(72) | N/A | 0% (0/1) | N/A | N/A | 0% (0/1) | N/A | N/A | [Zheng et al., 2008] | |
| P102L | MM/MV (most cis M) | ~221 |
|
(27–66) | (7–132) M | 84–100% | (15–20% ) | 20% | 0–35% | 25–30% | Multic PrP-Plqs | GSS | [Webb et al., 2008; Higuma et al., 2013; Krasnianski et al., 2015] |
| P102L UCSF | Cis M | 13 | 44±12 (24–57) | 44±13 M (28–60) | FHx Score 0 (n=1) 1 (n=2) 2 (n=6) |
0 % (0/3) | 0% | 0% (0/3) | 33.3% (1/3) | ||||
| P105L | MV | 13 |
|
Mean 44±10 (2nd to 7th decades) | Mean 111±82 M | 37% | 0% | 0% | 0% | 14% | PrP-Pl, diff PrP (deep CLs) | GSS | [Higuma et al., 2013] |
| P105L UCSF | N/A | 1 | (10) | N/A | FHx Score 0 (n=1) |
N/A | N/A | 0% (0/1) | 0% (0/1) | ||||
| P105T | Cis M | 13 |
|
(13–41) | (2–5) Y | 100% (2/2) | 0% | N/A | 0% (0/3) | 25% (1/4) | V, PrP-S (all CLs) Unicentric PrP-Plqs (deep CLs) | JCD | [Rogaeva et al., 2006; Polymenidou et al., 2011] |
| P105S | MV (Cis V) | 1 |
|
(30) | (10)Y | 0% (0/1) | N/A | N/A | 0% (0/1) | 100% (1/1) | Multic PrP-Plqs (HP), punctate aggregates (Cb), V (Pu) | Atypic GSS | [Tunnell et al., 2008] |
| G114V | (MM/MV) | 1 |
|
(18–75) | (1–4)Y | 75% (3 in 4 probands)§§ | 0% | 0% (0/8) | 42.9% (3/7) predom BG | Mod V, G, NL. PrP-S, Type 1 PrPSc (predom monoglycos) | JCD | [Rodriguez et al., 2005; Ye et al., 2008; Liu et al., 2010; Beck et al., 2010] | |
| A117V | Cis V | 33 |
|
(20–64) | (1–11 Y) | 100% (4/4) | N/A | N/A | N/A | 0% (0/2) | Ab PrP-Plqs, F V, NL, G | GSS | [Hsiao et al., 1991; Mastrianni et al., 1995; Kong et al., 2004] |
| A117V UCSF | Cis V | 6 | 34±14 (14–49) | 46±21 M (27–78) | FHx Score 0 (n=1) 2 (n=2) |
0 % | 0 % | 0% (0/3) | 0% (0/5) | ||||
| G131V | MM/MV (cis M) | 3 |
|
(36–42) | (9–16) Y | 50% (1/2) | N/A | N/A | 0% (0/1) | 0% (0/1) | PrP-Plqs, NFT (AH, ERC), No V | GSS | [Panegyres et al., 2001; Jansen et al., 2012] |
| S132I | MM | 2 |
|
(62) | (18) M | 100% (1/1) | N/A | N/A | N/A | N/A | diff unicentric & multic PrP-PLs (neocortex, BG, Cb) Min V |
GSS | [Hilton et al., 2009] |
| A133V | MM | 2 |
|
(62) | (4) M | 0% (0/1) | 0% | 0% (0/1) | 0% (0/1) | diff V, G, NL multic PrP-Plqs in the (Mol), PrP-S (Th) | Atypic GSS | [Rowe et al., 2007] | |
| R148H | (MV/MM) | 3 |
|
(62–82) | (6–18) M | 0% (0 in 2)§§ | 50% (1/2) | 100% (1/1) | 50% (1/2) | 100% (1/1) predom BG | 129MM – Similar to sJCDMM1. V, G, NL (deeper lCLs). PrP-S PrPSc Type 1 129MV - Similar to sJCDMV2 V predom in CLs V & VI, Kuru plaques in Cb & WM, PrP-S PrPSc type 2 (predom monoglycos) | JCD | [Krebs et al., 2005; Pastore et al., 2005] |
| R148H UCSF | MM | 1 | (53) | (2) M | FHx Score 0 (n=1) |
N/A | N/A | N/A | 100% (1/1) | ||||
| D167G | MM | 1 |
|
N/A | N/A | N/A | N/A | N/A | N/A | N/A | sJCD PrP type 1 | JCD | [Bishop et al., 2009] |
| D167N | MM | 1 |
|
(33) | (2) Y | 0% (0/1)§§ | N/A | N/A | 0% (0/1) | 0% (0/1) | N/A | N/A | [Beck et al., 2010] |
| V176G | VV | 1 |
|
(61) | (7) M | 0% (0/1) | 100% (1/1) | 100% (1/1) | 0% (0/1) | 0% (0/1) | Multic PrP-Plqs w/prominent tau | GSS | [Simpson et al., 2013] |
| D178N-129V | MV/V V (Cis V) | 209** |
|
Mean 46 (26–56) | Mean 23 M (7–60) | 100% (12/12) | N/A | N/A | N/A | N/A | Similar to sJCD VV1 | JCD | [Brown et al., 1992; Goldfarb et al., 1992; Kong et al., 2004] |
| D178N-129V UCSF | Cis V | 8 | 45±6 (37–52) | (18–21) M | FHx Score 2 (n=4) |
50% (1/2) | 0% (0/1) | 0% (0/2) | 100% (2/2) | ||||
| V180I | Cis M | 225 |
|
Mean 77 | Mean 25 M | 0.7–6% | 70%; | 78.5% | 11% | 99% | V PrP-S | JCD | [Kong et al., 2004;Higuma et al., 2013] |
| V180I UCSF | Cis M | 2 | (84) | (21) M | FHx Score 1 (n=1) 2 (n=1) |
0% (0/1 | 0% (0/1) | 100% (1/1) | |||||
| T183A | Cis M | 3 |
|
45±4 (42–49) | 4±2 Y (2–9) | 100% (2/2) | N/A | N/A | 0% (0/7) | 0% (0/2) | V & NL (CLs IV, V, VI), PrP (Cb, Pu) Small Plq-like PrP, Predom monoglycos PrPSC | JCD | [Nitrini et al., 1997; Grasbon-Frodl et al., 2004] |
| H187R | MM/MV/VV | 7 |
|
(20–53) | (3–19) Y | 100% (4/4) | 0% (0/2) | 0% (0/4) | 0% (0/4) | G multic-PrP-Plqs in some cases. Curly PrP-G | GSS | [Cervenakova et al., 1999; Butefisch et al., 2000;Hall et al., 2005; Colucci et al., 2006] | |
| H187R UCSF | Cis M | 4 | (30–41) | (12–13) Y | FHx Score 2 (n=1) |
0% (0/1) | 0% (0/1) | 0% (0/1) | 0% (0/1) | ||||
| T188R | MV/VVV (Cis V) | 12 |
|
(55–66) | (14–16) M | 0% (0/1)§§ | 50% (1/2 | 100% (1/1) | 50% (1/2) | 50% (1/2) | V, NL, A PrP-S & plaque-like PrP, Type 1 PrPSc | JCD | [Tartaglia et al., 2010; Roeber et al., 2008] |
| T188K | MM/MV (Cis M) | 3 |
|
Median 58 (39–76) | (2–13) M | 8–37%§§ | 69% | 12% | 69% | SE PrP-S | JCD | [Roeber et al., 2008; Chen et al., 2013; Shi et al., 2015] | |
| T188A | MM | 1 |
|
(82) | (4) M | 0% (0/1) | 100% (1/1) | 100% (1/1) | 100% (1/1) | 0% (0/1) | Sev G, V, Mod NL (predom in OLs) PrP-Neg | JCD | [Collins et al., 2000] |
| T193I | MM |
|
(70) | (10) M | 0% (0/1) | 100% (1/1) | 100% (1/1) | 100% (1/1) | 0% (0/1) | N/A | N/A | [Kotta et al., 2006] | |
| E196K | N/A | 13 |
|
(64–69) | (10–13) M | 100% (1/1) | N/A | N/A | 0% (0/1) | N/A | N/A | N/A | [Peoc’h et al., 2000] |
| F198S | MV/VV | 5 |
|
(40–71) Y | Mean 5 Y (2–12) | 100% (3/3) | N/A | N/A | N/A | 0% (0/1) | Uni- & multic PrP-Plqs | GSS | [Farlow et al., 1989; Dlouhy et al., 1992; Ghetti et al., 1995; Kong et al., 2004] |
| F198S UCSF | Cis V | 5 | 55±8 (46–66) | 67±23 M (34–84) | FHx Score 1 (n=2) 2 (n=1) |
33.3% | 100% (1/1) | 0% (0/4) | 0% (0/3) | ||||
| F198V | MM | 1 |
|
(56) | (4) Y | N/A | N/A | N/A | 0% (0/1) | 0% (0/1) | N/A | N/A | [Zheng et al., 2008] |
| E200K | MM/MV/VV | 571 |
|
Mean 60 (33–84) | (1–18) M | 50% | 85–100% | (80–100%) | 42–85% | 50–88% | Usually sJCD MM1 PrPSc types 1 & 2 | JCD | [Spudich et al., 1995; Meiner et al., 1997; Kovacs et al., 2005; Kovacs et al., 2011; Krasnianski et al., 2015] |
| E200K UCSF | Cis M (n=16) & Cis V (n=1) | 34 | 60±13 (36–84) | 11 ±17 (1–78) | FHx Score 0 (n=2) 1 (n=10) 2 (n=12) |
57.1 % (4/7) | 37.5% (3/8) | 88.9% (16/18) | |||||
| E200G | MV (Cis V) | 1 |
|
(57) | (30) M | 0% (0/1) | 0% (0/1) | 100% (1/1) | 0% (0/1) | 100% (1/1) | SE w/type 2 PrPSc | JCD | [Kim et al., 2013] |
| D202G | MV (cis V) | 1 |
|
(55) | (16) Y | 100% (1/1) | 100% (1/1) | 0% (0/1) | 0% (0/1) | 0% (0/1) | N/A | N/A | [Heinemann et al., 2008] |
| D202N | VV | 1 |
|
(73) | (6) Y | N/A | N/A | N/A | N/A | N/A | PrP-Plqs, NFT | GSS | [Piccardo et al., 1998] |
| V203I | N/A | 17 |
|
(69) | (1) M | 0% (0/1) | N/A | N/A | 100% (1/1) | N/A | N/A | N/A | [Peoc’h, 2000 #12035] |
| R208H | MM/VV | 15 |
|
(58–63) | (3–16) M | 20% (1/5) | 50% (4/8) | 57.1% (4/7) | 25% (2/8) | SE, PrP-S (perineuronal perivacuolar) Type 1 PrP | JCD | [Roeber et al., 2005; Capellari et al., 2005; Matej et al., 2012 Vita et al., 2013; Shi et al., 2015] | |
| V210I | Cis M | 247 |
|
Mean 59 (39–82) | Median 5 (2–20) M | 12–31% | 90–100% | 100% | 44–80% | 15–33% | Similar to sJCD MM1 | JCD | [Kovacs et al., 2005; Kong et al., 2004; Breithaupt et al., 2013 Krasnianski et al., 2015] |
| V210I UCSF | Cis M | 3 | 57±15 (47–74) | (1) M | FHx Score 0 (n=2) 2 (n=1) |
100% (1/1) | 33.3% (1/3) | 100% (2/2) | |||||
| E211Q | MM | 11 |
|
(42–81) | (6–32) M | 100% (2/2) | N/A | N/A | 100% (4/4) | N/A | V, G Mi PrP-S types 1 & 2 PrPSc | JCD | [Peoc’h et al., 2000; Ladogana et al., 2001; Peoc’h K et al. 2012] |
| E211D | VV | 1 |
|
(53–68) | (3–13) Y | 50% (1/2) | N/A | N/A | 0% (0/2) | 0% (0/2) | Multic PrP-Plqs, Dystrophic neurites & NFT | GSS | [Peoc’h et al., 2000; Peoc’h et al., 2012] |
| Q212P | MM | 2 |
|
(60) | (8) Y | N/A | N/A | N/A | N/A | N/A | Mod PrP Mi, PrP-Plqs | GSS | [Piccardo et al., 1998] |
| I215V | MM | 1 |
|
(55–76) | (12–15) M | 0% (0/2) | (1/3) | 100% (2/2) | 50% (1/2) | NL, G, V, PrP-Neg | JCD | ||
| Q217R | VV/MV (Cis V) | 3 |
|
(45–66) | (5–13) Y | 100% (3/3) | N/A | N/A | 0% (0/1) | 0% (0/1) | Uni- & multic PrP-Plqs, NFT (neocortex) | GSS | [Hsiao et al., 1992; Woulfe et al., 2005; Piccardo et al., 1998; Munoz-Nieto et al., 2013] |
| Y218N | VV | 1 |
|
(54–61) | (6) Y | 100% (1/1) | N/A | N/A | 0% (0/2) | 0% (0/2) | Uni & multic PrP-Plqs, NFT w/hyperP tau. | GSS | [Alzualde et al., 2010] |
| A224V | VV (Cis V) | 1 |
|
(48) | (32) M | 0% (0/1)§§ | 100% (1/1) | N/A | 100% (1/1) | Diff V w/PrPSc type 1 | JCD | [Watts et al., 2015] | |
| M232 R | MM | 63 |
|
Mean 64 (15–81) | Mean 8 (0–32) M | ~0% | 55–75% | 55–93% | 20–100% | 85% | sJCD MM1 | JCD | [Shiga et al., 2007; Zheng et al., 2008; Nozaki et al., 2010; Higuma et al., 2013] |
| M232 T | MV |
|
N/A | (6) Y | 0% (0/1) | N/A | N/A | N/A | N/A | Multic PrP-Plqs | GSS | [Bratosiewicz et al., 2000] | |
| P238S | N/A |
|
N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | [Windl et al., 1999] | |
If UCSF cohort differs from the literature, this information is provided in the table.
Including D178N-129V & D178N-129M.
By nine PrD surveillance centers, according to Minikel et al., 2016.
Positive family history of dementia w/similar clinical features (as of the proband) or PrD. For UCSF family history FHx (family history) Score scale: 0 when there was no positive FMH suspicious for or known PrD; 1 when there was at least one first-degree relative w/dementia, encephalopathy or movement disorder; or 2 in patients who were part of families w/known PRNP mutations, or had positive history for clinical or path-proven PrDs.
According to most commonly used European 2009 and UCSF 2011 criteria [Zerr et al., 2009; Vitali et al., 2011].
there is evidence of incomplete penetrance, as asymptomatic older carriers also were identified.
Data on age at onset & duration of disease are shown as mean±SD (range), unless otherwise indicated.
positive if > total tau 1200 pg/mL
Abbreviations: Ab = abundant; AD = Alzheimer-type dementia; AH = Ammon horn; atypic = atypical; Atx = ataxia; BG = basal ganglia; bvFTD = behavioral variant Frontotemporal Dementia; c/w = consistent with; Cb = cerebellum; CBS = corticobasal syndrome; CLs = cortical layers; Cog = cognitive; D = dementia; diff = diffuse; dx = diagnosis; ERC = entorhinal cortex; EP = extrapyramidal; FHx = family history; F = focal; freq = frequent; G = gliosis ; GTC = generalized tonic clonic seizures; HP = hippocampus; HyperP = hyperphosphorylated; LE = lower extremities; LMN = lower motor neuron; M = months; Mi = mild; Min = minimal; Mod = moderate; Mol = molecular layer of the cerebellum; monoglycos = monoglycosylated; multic = multicentric; N/A = not available; Neuropath = Neuropathology; NFT = neurofibrillary tangles; NL = neuronal loss; OLs = occipital lobes; Park = parkinsonism; Pos = positive, prog = progression; predom = predominant; PrP-G = granular PrP deposits; PrP-Neg = negative PrP staining; PrP-Plqs = PrP-amyloid plaques; PrP-S = synaptic PrP deposits; PSP=progressive supranuclear palsy; PSWC = periodic sharp wave complexes; Pu = putamen; Pyram = pyramidal; SE = spongiform (vacuolated) encephalopathy; Sev = severe; SYN = syndrome; Th = thalamus; WM = white matter.; Psych = psychiatric; Sxs = symptoms; V = vacuolation; w/ = with; w/o = without; Y = years
We had 67 symptomatic patients with gJCD (from 52 families) who were referred to the UCSF MAC, and 20 of them were evaluated at our center. The most frequent mutations were E200K (n=43), D178N-129V (n=10), V210I (n=5), and V180I (n=2), as shown in Table 1. We have published our single cases of A224V [Watts et al., 2015], E200G [Kim et al., 2013] and T188R [Tartaglia et al., 2010] mutations. The K194E mutation is a novel mutation that will be reported in a separate manuscript, but is briefly described below. Now, we will present the clinical features of the most common gJCD-related mutations (E200K, D178N-129V, V210I, V180I, and M232R), along with the mutations only reported in the UCSF cohort (E194E, E200G, A224V). The features of the other mutations associated with this phenotype are summarized in Table 3.
E200K
The E200K mutation is the most common mutation worldwide and has been reported in all five continents [Kovacs et al., 2005; Lee et al., 1999; Minikel et al., 2016]. It also was the most common among patients with gJCD (and gPrD) in the UCSF cohort. There are at least four ancestral origins of clusters of patients with this mutation, including the two large ones among Sephardic Jews (and their ancestors) and a Slovakian cohort. There also are two independent cohorts from Germany, Sicily, Austria as well as from Japan [Lee et al., 1999; Meiner et al., 1997; Mitrova and Belay 2002]. The vast majority of reported cases with the E200K mutation have cis codon 129 methionine ( in the same allele as the mutation; E200K-129M), which was also seen in the UCSF cohort (16/17 cases; one case was cis codon 129V).
The penetrance of the mutation is high, but appears to vary by geographic origin. Among Sephardic E200K carriers, penetrance is 70% at age 70 years and close to 100% at age 85 years [Spudich et al., 1995], but even so, approximately 50% of patients have negative family history for PrD [Higuma et al., 2013; Kovacs et al., 2005; Spudich et al., 1995]. In many cases, the family history is kept hidden or there have been misdiagnoses. In Slovakia, penetrance has been reported to be much lower, however, approximately 60% [Mitrova and Belay 2002]. A few reports suggested that anticipation occurred in this mutation [Pocchiari et al., 2013; Rosenmann et al., 1999], even though there was a lack of biological explanation for such phenomenon, but more recently, a large study concluded that the anticipation seen in earlier studies was most likely a false signal due to ascertainment bias [Minikel et al., 2014].
The clinical presentation of gJCD due to E200K mutation is overall similar to that of sJCD [Kovacs et al., 2005; Meiner et al., 1997]. The mean age at onset of symptoms is approximately 60 years of age, but there is a wide range reported in the literature (33–84 years) [Begue et al., 2011; Higuma et al., 2013; Kovacs et al., 2005; Kovacs et al., 2011; Krasnianski et al., 2016; Meiner et al., 1997; Mitrova and Belay 2002]. The median duration of disease is about 5–10 months (range 1–18)[Higuma et al., 2013; Kovacs et al., 2005; Kovacs et al., 2011; Krasnianski et al., 2016; Mitrova and Belay 2002; Shi et al., 2015]. The range of age of onset in the UCSF cohort (36–84 years) was within the range published in the literature, but the duration of disease was longer in some of the patients, as four patients lived longer than 18 months and one outlier had a total disease duration of 78 months (Table 3).
In the literature on E200K of Sephardic origin, onset of symptoms is reported to usually occur gradually over a few weeks to months, but in about 10% of patients, onset is more acute. Cognitive decline is the most common presentation, and is the first symptom in about 50% of patients from Sephardic and European cohorts [Krasnianski et al., 2016; Meiner et al., 1997]. Cerebellar ataxia is the presenting manifestation in 30%, sensory symptoms in 15%, and constitutional/vegetative symptoms (such as hyperthermia, loss of weight, hyperhidrosis), in 5% of patients [Krasnianski et al., 2016]. In the UCSF cohort, 44% of patients presented with cognitive decline, 20% with cerebellar or sensory symptoms, and 16% with constitutional symptoms (such as weight loss or complaints of tiredness).
During the disease course, 95–100% develop dementia, 80–100% develop cerebellar ataxia, and 70–85% develop myoclonus [Kovacs et al., 2011; Krasnianski et al., 2016; Meiner et al., 1997]. Extrapyramidal signs are found in 40–65% of patients, and pyramidal signs, in 60–70%. Parkinsonism occurs in about 15% of patients, and chorea/dystonia in 35% [Kovacs et al., 2011]. Psychiatric symptoms such as hallucinations, depression, delusions, and aggressiveness are also common during the disease course [Krasnianski et al., 2016].
Despite the similarities between gJCD due to E200K and sJCD, there are also differences. Seizures are more common than in sJCD, occurring in as many as 20–35% of patients with E200K [Krasnianski et al., 2016; Meiner et al., 1997]. Peripheral neuropathy and supranuclear gaze palsy – features that are uncommon in sJCD - have been reported in some E200K cases [Bertoni et al., 1992; Kovacs et al., 2011; Meiner et al., 1997; Neufeld et al., 1992]. Headaches are endorsed by 20–30% of patients with this mutation [Krasnianski et al., 2016; Meiner et al., 1997], which is probably higher than for sJCD. Insomnia is reported in about 15% of cases, and insomnia with thalamic degeneration was reported in one case [Kovacs et al., 2011; Meiner et al., 1997].
Whether or not the PRNP codon 129 polymorphism influences the presentation of gJCD due to E200K mutation is somewhat controversial. Our recent paper examining E200K from four large international cohorts found that the codon 129 polymorphism affected disease duration (shorter in 129 MM (cis M) than in 129 MV (cis M) and shorter in 129M haplotype than in 129V haplotype), but not age of onset or death [Minikel et al., 2014]. Most patients reported in the literature have E200K cis 129M, which typically presents similarly to sJCD MM1 [Capellari et al., 2011]. In our UCSF cohort, only one patient had the rare E200K-129V(V) mutation. She developed symptoms at an early age, 36, but had a typically rapid course, dying six months later. Her brain MRI had DWI hyperintensities in the basal ganglia (predominantly) and cortex, and was considered consistent with JCD. The EEG did not show PSWCs, but CSF protein 14-3-3 was positive. In the literature, the mean age at onset of cases with E200K-129V mutation is 55±11 years (range 37–67), and mean disease duration is of 9±4 months (range 2–17) [Kim et al., 2013]. Patients with the E200K-129V mutation tend to have a slight longer disease duration than those with E200K-129M [Kim et al., 2013].
CSF protein 14-3-3 is positive in 85–100% of cases (only 57% within the UCSF cohort, however), whereas CSF tau is positive (>1200 or 1300, depending on the study) in 80–100% [Breithaupt et al., 2013; Higuma et al., 2013; Kovacs et al., 2005; Krasnianski et al., 2016; Shi et al., 2015]. CSF real-time quaking-induced conversion (RT-QUIC) assay positivity was reported as 73–84% [Higuma et al., 2013; Sano et al., 2013]. PSWCs in EEG have been reported in 42–85% of cases with the E200K mutation (37% within the UCSF cohort) [Begue et al., 2011; Breithaupt et al., 2013; Higuma et al., 2013; Kovacs et al., 2005; Kovacs et al., 2011; Krasnianski et al., 2016; Meiner et al., 1997; Shi et al., 2015].
Deep nuclei hyperintensities on DWI or FLAIR MRI are reported in 50–77% of patients with this mutation [Kovacs et al., 2005; Krasnianski et al., 2016; Shi et al., 2015]. If cortical hyperintensities are also considered, the MRI positivity increases to 84–88% [Breithaupt et al., 2013; Higuma et al., 2013] (and was 88% in the UCSF cohort). Most cases (13/18) we had both cortical ribboning and deep nuclei restricted diffusion (bright on DWI, dark on ADC map) (Figure 2A and 2B), whereas the remaining usually had predominantly deep nuclei abnormalities.
Figure 2. Brain MRIs in Fast forms of gPrD.
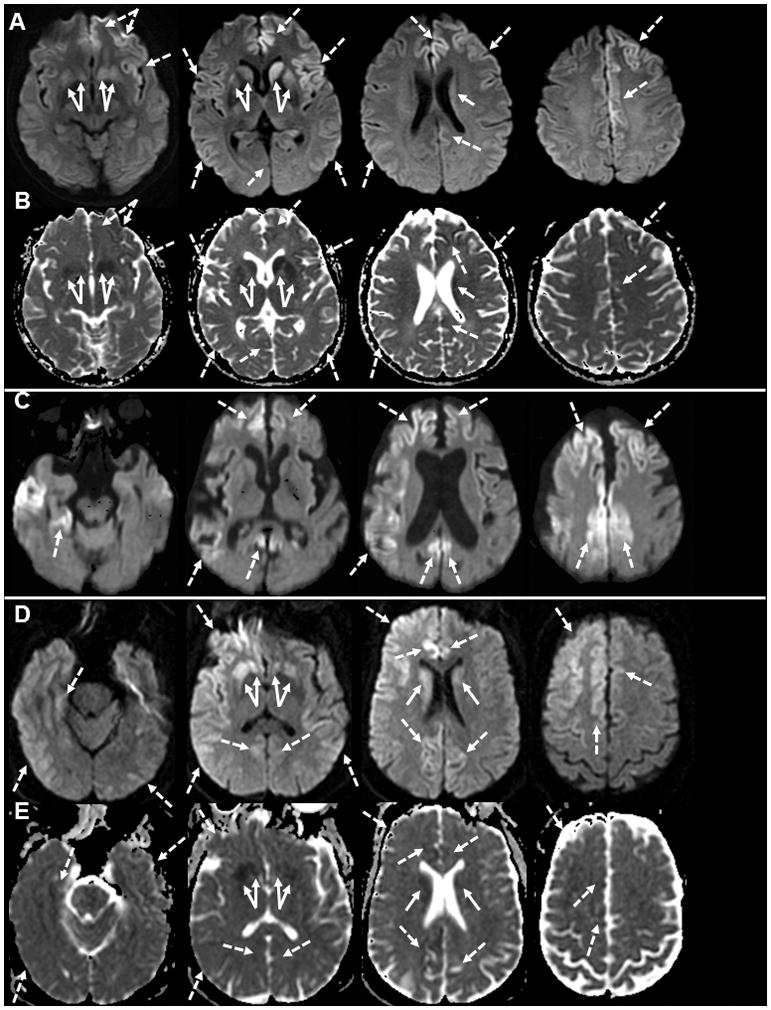
A. DWI MRI of a gJCD 47 year-old E200K patient 3 months after onset shows diffuse cortical hyperintensity (cortical ribboning; dashed arrows) of the bilateral frontal and insula (left > right) and temporo-parietal cortices. There was also DWI hyperintensity in the bilateral striata (left > right, solid arrows). B. The ADC map of the same gJCD E200K case showed hypointensity in most of the regions that were hyperintense on DWI, confirming reduced diffusion, consistent with prion disease. C. DWI MRI of an 85 year old V180I case 11 months after onset shows diffuse cortical atrophy and cortical hyperintensity (cortical ribboning; dashed arrows) of the bilateral frontal and cingulate (right > left) and the right temporal and parietal lobes. There was no clear abnormality in the deep nuclei. D. DWI in a 49 year old gJCD A224V case 11 months after the onset shows diffuse cortical ribboning (dashed arrows) greater on the right than the left and hyperinensity in the bilateral deep nuclei (solid arrows) also greater in the right. E. The ADC map of the same gJCD A224V case showed hypointensity in most of the regions that were bright on DWI, confirming restricted diffusion, consisent with JCD/prion disease. Orientation is radiological (left brain is right side of figure).
The neuropathological findings in E200K include those typical of spongiform encephalopathies, namely vacuolation (spongiform changes), astrocytic gliosis and neuronal loss, which are more severe in the neocortex, basal ganglia and thalami, and are overall similar to those found in sJCD MM1 [Higuma et al., 2013; Kovacs et al., 2011]. The majority of cases with E200K-129M have PrPSc type 1, but co-occurrence of PrPSc types 1 and 2 has been reported. In the few cases with E200K-129V reported in the literature, PrPSc type 2 was found [Kim et al., 2013; Puoti et al., 2000]. The pattern of PrPSc deposition is predominantly synaptic, which is seen in all cases with the E200K mutation, but other types of deposits, such as perineuronal and intraneuronal, can be seen in some cases [Kovacs et al., 2011]. Plaque-like structures are more frequent among those heterozygous at codon 129 (MV) than those homozygous (MM) [Kovacs et al., 2011; Mitrova and Belay 2002]. Regarding PrPSc glycosylation, there seems to be an increase in the diglycosylated forms and decrease of unglycosylated PrP, in comparison to sJCD [Kovacs et al., 2011].
D178N-129V
The D178N mutation with valine at the cis codon 129 is typically associated with the JCD phenotype, whereas (as previously mentioned) methionine at cis codon 129 is associated with the FFI phenotype [Goldfarb et al., 1992]. The But as mentioned in the Introduciton, there are exceptions to this pattern, as D178N-129MM might present clinically and neuropathologically as JCD and even within families, both phenotypes (FFI and JCD) might appear [Zarranz et al., 2005; Zerr et al., 1998].
The mean age at onset is 46 years, and onset ranges from the second to the eight decades [Brown et al., 1992; Goldfarb et al., 1992]. The duration of disease is on average longer than sJCD, with a mean of 23 months (range 7–60 months) [Brown et al., 1992; Goldfarb et al., 1992]. The age at onset and disease duration observed among UCSF cases are similar to those previoulsy reported (Table 3). The codon 129VV genotype appears to be associated with earlier onset and shorter disease duration, in comparison to the 129MV genotype [Goldfarb et al., 1992].
Cognitive decline (particularly memory) is the initial symptom in the majority (95%) of patients, and dementia occurs in virtually all patients, a pattern that was also observed in the UCSF cohort [Brown et al., 1992]. Behavioral changes are present in 30% of patients at disease onset, and cerebellar symptoms, in 21% [Brown et al., 1992]. During the disease course, 86% of patients develop cerebellar symptoms, 67% develop extrapyramidal symptoms and about half develop pyramidal symptoms and signs [Brown et al., 1992]. Myoclonus has been reported in 74% of patients, and seizures in 12% [Brown et al., 1992].
Periodic activity on EEG is infrequent in patients with this mutation [Brown et al., 1992; Nozaki et al., 2010]. We are not aware of cohort data on MRI and CSF findings in these cases. Among the UCSF cases, none of the EEGs showed PSWCs, CSF 14-3-3 was positive in 50% of samples, and total tau was not elevated in the available sample. MRI showed neocortical and subcortical hyperintensities consistent with JCD in 100% of cases (2/2).
The neuropathological findings associated with the D178N-129V mutation are similar to those reported in sJCD VV1 [Gambetti et al., 2003b]. Spongiosis and neuronal loss are more severe in the neocortex, whereas the talamus is only mildly affected and cerebellum is spared of those changes [Gambetti et al., 2003b]. Immunostaining for PrPSc reveal synaptic-type deposits that are weakly stained [Gambetti et al., 2003b]. PrPSc type 1 with an underrepresentation of the unglycosilated form is typically observed in this mutation [Gambetti et al., 2003b].
Low penetrance mutations
For the next three mutations, V180I, V210I and M232R, their role in pathogenicity has been controversial as they often lack a positive family history and they have a higher than expected rate in the general population. As noted above, these three mutations are likely low penetrance mutations [Minikel et al., 2016].
V180I
The V180I mutation, the most commonly identified mutation in Japan [Minikel et al., 2016; Nozaki et al., 2010]. That most cases have no positive family history and the rate in the general population is about 6% suggests this mutation is of very low penetrance, probably approximately 1% [Minikel et al., 2016]. For V180I in Japan, many cases began in late age and progressed relatively slowly; mean age at onset was 76.5 years, rather late onset, and mean duration of disease was 25 months. [Higuma et al., 2013; Qina et al., 2014; Yang et al., 2010]. In the Japanese literature, main clinical features were dementia (99%), extrapyramidal (59%), psychiatric disorder (54%), pyramidal (50%), akinetic mutism (49%), myoclonus (45%) and cerebellar signs (36%). Positive family history was low at 0.7–6%. MRI was positive in 99%; cortical ribboning in 75% and basal ganglia hyperintensity in 22%, and PSWCs on EEG was noted in 11%. CSF 14-3-3 was positive in 70%, total tau in 78.5% (cuf-off > 1260 pg/mL), and PrPSc by RT-QuIC in 39% [Higuma et al., 2013]. Neuropathology revealed vacuolation in cortex but low PrPSc immunopositivity of synaptic type [Higuma et al., 2013].
We evaluated one symptomatic (a rare Caucasian) patient with V180I-129MV (cis M) mutation at UCSF. This patient showed similar clinical features to those from the Asian literature; our case manifested with unsteady gait and memory problems at age 84 years and progressed slowly (relative to classic sJCD), dying 21 months after onset. This patient did not have any family history for prion disease or similar disorder, consistent with the literature suggesting very low penetrance [Minikel et al., 2016]. DWI MRI showed diffuse cortical atrophy and cortical ribboning without deep nuclei hyperintensity at 11 months after the onset (Figure 2C), a pattern consistent with the literature on this disorder. Other biomarkers were unremarkable; background slowing on EEG, inconclusive CSF 14-3-3 and total tau and NSE level mildly elevated at 28.5 ng/mL (intermediate; >30 ng/mL consistent with sJCD). Autopsy demonstrated delicate vacuolization of gray matter of the neocortex, caudate nucleus, putamen, thalamus, and cerebellum but sparing of the brain stem and hippocampus with PrPSc deposits in a finely granular pattern, diagnostic for JCD.
V210I
Similarly to V180I and the M232R mutations, theV210I mutation likely is not fully Mendelian. The frequency of this mutation in the general population is about 2%, too high for a fully penetrant, dominant prion disease–causing variant, but far lower than the corresponding allele frequency in prion disease cases, suggesting that this mutation is either a risk factor or of low penetrance. This mutation is estimated to have about 10% penetrance [Minikel et al., 2016]. It is the most common mutation in Italy [Ladogana et al., 2005; Minikel et al., 2016]. Clinical presentations are similar to sJCD MM1 [Capellari et al., 2011; Kovacs et al., 2005]; The mean age at onset is 59–62 years (range 39 to 82 years) [Kovacs et al., 2005; Krasnianski et al., 2016]. The median duration of disease is 5 months (range 2 to 20 months) [Krasnianski et al., 2016]. Approximately 88% of patients with the V210I mutation have negative family history for PrD or other neurological disorders in one report [Kovacs et al., 2005] and positive family history was reported as about 31% in another [Krasnianski et al., 2016].
First symptoms include dementia (38%), ataxia (15%), vertigo (15%), visual (15%), sleep (8%), sensory (8%) [Krasnianski et al., 2016] ). Clinical symptoms and signs during disease course are as follows; ataxia (100%), dementia (92%), extrapyramidal (92%), myoclonus (92%), visual/oculomotor (85%), pyramidal (71%), sensory (77%), vertigo (61.5%), headache (42%), hemianopsia (38.5%), and seizures (38.5%) [Krasnianski et al., 2016]. MRI positivity was reported in 15–33% of cases, although many of these did not include the more sensitive diffusion sequences (e.g. DWI and ADC map) [Breithaupt et al., 2013; Kovacs et al., 2005; Krasnianski et al., 2016] and PSWCs on EEG were observed in 44–80% [Breithaupt et al., 2013; Kovacs et al., 2005; Krasnianski et al., 2016]. Notably, CSF biomarkers were frequently positive; 14-3-3 positivity in about 90–100% [Breithaupt et al., 2013; Kovacs et al., 2005; Krasnianski et al., 2016] and total tau in 100% [Krasnianski et al., 2016]. We did not evaluate any cases with the V210I mutation in our UCSF cohort, although five cases were referred (Table 1).
M232R
The M232R mutation is one of the four most common mutations identified in the Japanese PrD cohort, and has been reported almost exclusively in patients with JCD from northeastern Asia (Japan, China and Korea) [Choi et al., 2009; Minikel et al., 2016; Nozaki et al., 2010; Zheng et al., 2008]. Its pathogenicity, however, has been questioned by many as there is no functional evidence of pathogenicity, as it was only reported in JCD cases with no family history, and because the M232R variant has been identified in patients with dementia and no neuropathological findings of PrD and also in healthy individuals, with an estimated allelic frequency of more than 3% in the Japanese population [Beck et al., 2012; Higuma et al., 2013; Shiga et al., 2007]. In the ExAC cohort, the allele count of the variant was 10, and its penetrance was estimated to be approximately 0.1%, making it most likely a risk variant with a small effect size [Minikel et al., 2016]. The M232R mutation is almost always found along with MM at codon 129, but it should be noted that 92% of the Japanese population carries that polymorphism (and only 8% are MV) [Shiga et al., 2007].
The clinical presentation is very similar to that of sJCD with a mean age at onset of 64 years (range 15–81) and mean duration of disease of 8 months (range 0–32) [Nozaki et al., 2010], but two different clinical presentations have been reported by Japanese researchers, a rapid type (that represents about 75% of cases) and a slow type (25% of cases) [Shiga et al., 2007]. Both groups have relatively close ages at onset (mean 66.2±7.7 years in the rapid group and 57.6±15.4 years in the slow), but the duration until onset of akinetic-mutism (3.1±1.5 monthsin the rapid group and 20.6±4.4 months in the slow type group) [Shiga et al., 2007] and until death (12.5±10.9 months for rapid and 49.9±53.8 for slow) were quite different [Higuma et al., 2013].
Approximately 85% of patients have MRI scans consistent with JCD (and similar in both rapid and slow groups) [Nozaki et al., 2010; Shiga et al., 2007]. The frequency of PSWCs on EEG is higher in the rapid group (100% vs. 20%), and the positivity of 14-3-3 and tau in CSF are also higher among those with rapid progression (75% and 93% for 14-3-3 and tau respectively in the rapid type group and around 55% for both markers in the slow type group) [Higuma et al., 2013]. RT-Quic positivity was 78% in the rapid type group and 60% in the slow type group [Higuma et al., 2013].
The neuropathological findings in cases with rapid type JCD are similar to those of sJCD MM1, with type 1 PrPSc and synaptic-type deposits, whereas in the slow type group, the findings are similar to sJCD cortical-type MM2, with spongiform changes, perivacuolar PrP deposits and type 2 PrPSc [Higuma et al., 2013; Shiga et al., 2007].
T188R
Only two patients with T188R mutation have been reported, including one from our cohort [Roeber et al., 2008; Tartaglia et al., 2010]. The first was a 66-year-old who presented with visual impairment and developed dementia and mild ataxia several months later [Roeber et al., 2008]. There was no family history for dementia. Brain MRI at 12 months after onset did not show abnormalities consistent with gJCD, although the EEG showed PSWCs, and CSF NSE (38 ng/mL; > 30 consistent with JCD) and CSF 14-3-3 were positive. PRNP testing revealed T188R mutation with codon 129 VV. The patient died at 16 months after the onset without autopy. Our case was somewhat different, possibly because he was codon 129 MV (cis V) and thus had a trans methionine. He developed personality changes at age 55, followed by behavioral changes (poor judgement and delusion), and then rapid cognitive decline. Family history was unclear. Neurological examination showed moderate to severe impairment in memory, frontal-executive function, and language, mild left leg dysmetria and bilateral hand apraxia. MRI at 4 months after the onset showed mild diffuse cortical atrophy with restricted diffusion in the cortex and striatum. EEG showed slowing. CSF 14-3-3 was inconclusive, NSE was intermediate (26 ng/mL), and total tau was elevated at 2236.6 pg/mL (>1150 consistent with JCD). He developed myoclonus and mutism and died 14 months after the onset. The same mutation was found in his asymptomatic 80 year old father (codon 129MV) and 56 year old brother (codon 129 VV). The autopsy of the proband revealed diffuse neuronal loss, gliosis, spongiosis, and synaptic and plaque-like deposits of PrPSc in the cortex and cerebellar molecular layer, compatible with JCD [Tartaglia et al., 2010]. The pathogenic role of T188R mutation is not yet clear [Minikel et al., 2016]. The substitution of threonine to a highly basic amino acid, arginine, might result in structural destabilization, support a pathogenic role of T188R mutation. The cell culture studies demonstrated T188R mutation had increased resistance to proteinase K [Lorenz et al., 2002].
Three novel gJCD mutations in UCSF cohort
K194E
We found a novel K194E PRNP mutation in a 71 year old Caucasian gentlman who initially developed confusion and memory loss, progressing to dementia, profound psychiatric symptoms, parkinsonism, and gait instability over the following 10 months and death at 11 months after the onset. Overall his clinical features and MRI and EEG findings were consistent with JCD. Based on the pdc.org database, the side chain is pointing out into solvent so the change in charge for K194E may not be too significant, but it’s location at the end of helix 2 might suggest that the mutation has an impact on the correct folding of PrPC and thus it might be pathogenic. This is clearly a rare genetic variation as it was not idenfied in the 60,000 exomes in the ExAC database (Eric Minikel, personal communication) [Minikel et al., 2016] or in the 1000 Genomes Project database (http://www.internationalgenome.org/)[1000 Genomes Project Consortium et al., 2012]. To our knowledge, a cell cutlure or mouse model of this mutation has not yet been made, but the K194E variant is predicted to be “possibly damaging” by Polyphen-2 (http://genetics.bwh.harvard.edu/pph2/), “disease causing” by MutationTaster (http://www.mutationtaster.org/), and “damaging” by the SIFT algorithm (http://sift.jcvi.org/).
E200G
We indentified the novel E200G (codon 129VV) mutation in a symptomatic 57 year old Causasian woman who developed gait problems, followed six months later with language problems, and by 25 months, she was wheelchair bound and had difficulty following conversations. Neurological examination 25 months after onset revealed parkinsonism, symmetric decreased sensation in the lower limbs, and cerebellar gait ataxia. Her brain MRI was consistent with JCD, showing restricted diffusion in the cortex (mild cortical ribboning) and striatum. CSF total tau was elevated (1351pg/ml; >1150 consistent with JCD), 14-3-3 was inconclusive and NSE was intermediate (27 pg/ml (>30 consistent with JCD. She died 30 months after symptom onset, and although most of the neuropathological findings on autopsy were consistent with JCD with PrPSc type 2, some neuropathological findings, including the PrPSc Western blot glycosylation pattern, were not typical of sJCD, supporting that the E200G is pathogenic. Although she had known family history of PrD, her father died at age 50 from a rapidly progressive dementia diagnosed, likley incorrectly, as alcohol-related dementia [Kim et al., 2013].
A224V
A novel A224V mutation (cis codon 129V) was identified in a single symptomatic 48 year old woman diagnosed with JCD with rapid onset of short term memory problems, followed by gait and other motor problems. She died 32 months after disease onset. Despite this long disease course, she was in a very advanced disease state, non-ambulatory and non-verbal for at least the last 1.5 plus years, and probably had a prolonged survival due to an extraordinarily high level of aggressive around the clock care. Brain MRI showed classic cortical and subcortical restricted diffusion (Figure 2D), but with very prominent early hippocampal involvement (not shown). Both CSF NSE (39 ng/ml; >30 consistent with JCD) and total tau (6,300 pg/ml; >1200 consistent with JCD) were elevated. PRNP testing found homozygousity for valine at codon 129, and the neuropathological evaluation was consistent with JCD with type 1 PrPSc. The FMH was negative for known prion disease; her father carried the same mutation, but was heterozygous at codon 129 (MV) and at 86 years old has had a slowly progressive neurodegenerative dementia, most consistent with Alzheimer’s disease for at least the last 7 years. Running this mutation through Combined Annotation Dependent Depletion (CADD) program (http://cadd.gs.washington.edu/score) to determine the likelihood of pathogenesis, suggested this variation to be possibly damaging. Furthermore, transmission experiments of prion strains obtained from frozen brain tissue of patients with sJCD (MM1, MV2, VV2) and from the proband into transgenic mice with human PrP containing the A224V cis codon 129V mutation and trans codon 129 either valine or methionine showed very different effects on incubation periods. The A224V mutation clearly had an impact on the incubaton period, and the codon 129 trans also had an effect. That having a trans codon 129M with A224V 129V extended incubation time to that closer to wildtype PRNP mice could explain why the proband’s father with the A224V 129V mutation but trans codon 129M has not developed obvious prion disease [Watts et al., 2015].
Fatal Familial Insomnia (FFI)
D178N-129V
FFI is an unusual form of genetic JCD showing a distinctive phenotype caused by the mutation located at PRNP codon 178 coupled with methionine at cis codon 129 (D178N-129M). FFI is the third most common gPrD worldwide and the most common gPrD in Germany [Gambetti et al., 2003b; Krasnianski et al., 2016]. The term FFI was first used in 1986 to describe the phenotype of an Italian family presenting with progressive insomnia, disturbances in autonomic function and progressive clinical motor deterioration leading to death [Lugaresi et al., 1986]. Subsequently, several years later, sequencing of the PRNP gene in three symptomatic members confirmed the mutation with a cis codon 129M, defining a novel subtype of gPrD [Goldfarb et al., 1992]. As noted above, in D178N, the 129 codon polymorphism has a strong effect on the clinicopathological phenotype, with cis codon V associated more with the JCD phenotype and cis codon 129M associated with an FFI phenotype [Krasnianski et al., 2008].
Diagnosis is made around the fifth decade, whereas the age at presentation varies from 20 to 76 years with illness duration averaging about 13–15 months (range 4–40 months) [Collins et al., 2001; Krasnianski et al., 2016; Reder et al., 1995; Shi et al., 2015; Zarranz et al., 2005]. Interestingly, the disease duration and clinical presentation in FFI is influenced by the trans codon 129 (on the normal PRNP allele), with FFI homozygous (MM) cases have a shorter duraition (~12±4 months) and heterozygous (MV) cases having a longer duraiton (~21±15 months) [Gambetti et al., 2003b]. To date, approximately 30 families have been reported. Positive family history for PrD was reported in 60–88% of cases [Kovacs et al., 2005; Krasnianski et al., 2016; Shi et al., 2015], although based on the rarity of this mutation in large population cohorts, p penetrance is estimated to be close to 100% [Minikel et al., 2016].
The core clinical features of FFI are linked to thalamic dysfunction, and consist of profound disruption of the normal sleep–wake cycle (with complete disorganisation of the electroencephalographic patterns of sleep), sympathetic overactivity, diverse endocrine abnormalities (with attenuation of the normal circadian oscillations) and markedly impaired attention [Gambetti et al., 2003b; Krasnianski et al., 2016]. Nevertheless, clinical diversity is seen across mutation carriers, some of them presenting only moderate insomnia and lacking abnormal EEG sleep studies despite of showing the characteristic thalamic gliosis on their neuropathological exam [Collins et al., 2001; Taniwaki et al., 2000; Zerr et al., 1998]. Autonomic dysfunction is one of the earlier features of the disease in most cohorts, however, psychiatric symptoms and abnormal gait were the most common presenting symptoms in D178N-129M patients from the Basque region [Zarranz et al., 2005]. Hallucinations appear to be more common in FFI than in other gPrDs [Krasnianski et al., 2016]. Moreover, non-specific symptoms such as marked weight loss, tiredness can be the first manifestations of the disease, making the diagnosis challenging [Johnson et al., 1998]. This challenging phenotypic heterogeneity has led to the development of a new diagnostic algorithm with the objective of improve early recognition of FFI patients and allowing diagnosis in the absence of genetic testing (sensitivty up to 91% and specificity of 81%) [Haik et al., 2014]. D178N-129M has been reported in cases with the JCD clinical and neuropathological phenotype [Zarranz et al., 2005; Zerr et al., 1998], and rarely FFI mutations have been related to unusual phenotypes mimicking early onset Alzheimer’s disease [Guerreiro et al., 2009]. It is now realized that the effect of the cis codon 129 in D178N gPrD is not all or none, but that patients present along a spectrum between JCD and FFI [Zarranz et al., 2005].
The clinical diagnosis of FFI usually is based on the careful observation of the clinical course, and only polysomnography and, to some extent, FDG-PET may help with FFI diagnosis before PRNP testing is performed [Cortelli et al., 2006; Haik et al., 2014]. The abnormal sleep patterns may go undetected without a polysomnographic study (showing rapid eye movement and deep sleep reduction) [Krasnianski et al., 2008; Zarranz et al., 2005]. Some endocrine abnormalities such as, persistently elevated levels of cortisol and variable levels of thyroid and gonadotropin hormones also have been described [Collins et al., 2001].
Classic JCD MRI findings (e.g, cortical ribboning and/or basal ganglia hyperintensities in FLAIR/DWI brain MRI sequences) are infrequent (~15%) in [Krasnianski et al., 2016; Shi et al., 2015]. PSWCs on EEGs are rare [Krasnianski et al., 2016; Shi et al., 2015], and CSF markers positivity is also low (positive 14-3-3 in 8–35% and elevated total tau levels in ~8%) [Krasnianski et al., 2016; Sano et al., 2013; Shi et al., 2015]. The sensitivity of RT-QUIC was 83% in one study with 12 patients [Sano et al., 2013].
As discussed in the Introduction, brain neuoropathology in FFI typically shows a characteristic restricted degeneration, largely confined to the thalami, especially the mediodorsal and anteroventral nuclei, as well as the inferior olivary nuclei, with little to no vacuolation or PrPSc deposition [Lugaresi et al., 1986; Medori et al., 1992].
In our UCSF cohort, we had nine symptomatic patients with FFI (from five families) referred, although only two were examined at UCSF (Table 1). Among those with available clinical data, the first symptom was constitutional (such as dry cough or headaches) in 50%. The family history was rated as 0 (no related history) in one proband and as 2 (family history of prion disease; see Methods and Table 2) in three other probands (and hence 75% had positive family history for PrD). CSF protein 14-3-3 was negative in all cases tested and PSWCs were not seen in EEGs. MRI showed DWI/ADC cortical ribboning in one case, but hyperintensities were absent in the other two cases.
Genetic prion diseases usually with slower progression
Gerstmann-Sträussler-Scheinker (GSS)
Gerstmann-Straussler-Scheinker (GSS) disease is defined as an hereditary, autosomal dominant gPrD characterized by the presence of multicentric amyloid plaques, that are immunostained by antibodies against the prion protein (PrP-amyloid plaques) [Budka et al., 1995; Ghetti et al., 1995; Kong et al., 2004]. Therefore, GSS is defined by the neuropathological findings, rather than by the clinical presentation, the latter of which is highly variable. The “classical” GSS phenotype is characterized by slowly progressive cerebellar ataxia, with onset between the third and seventh decades, and late occurrence of cognitive decline [Hsiao et al., 1989; Piccardo et al., 1998]. But onset with cognitive decline, even resembling JCD, has also been reported in numerous cases of GSS, and the heterogeneity in clinical presentation has been recorded even within families [Hainfellner et al., 1995; Hsiao et al., 1989; Piccardo et al., 1998].
Sixteen missense mutations (P84S, P102L, P105L, P105S, A117V, G131V, S132I, V176G, H187R, F198S, D202N, E211D, Q212P, Q217R, Y218N and M232T) have been associated with GSS, but similarly to gJCD, the pathogenicity of some of those variants has not been established with certainty [Minikel et al., 2016]. Among the mutations associated with GSS, P102L is the most commonly reported by surveillance centers (Minikel, Vallabh et al. 2016). Of the 16 missense GSS-like mutations, six were in the UCSF cohort: P102L (n=20), A117V (n=7), F198S (n=6), H187R (n=6), P105L (n=1), as well as a duplication located close to codon 129 (365–388dup, n=1).
The mean age of symptom onset is about 45–50 years, and the age at onset ranges from the third to the ninth decades [Kovacs et al., 2005; Kovacs et al., 2002]. The mean duration of disease close to 60 months, longer than gJCD, but the duration is highly variable, ranging from less than one year to more than ten years in the literature [Kovacs et al., 2005; Kovacs et al., 2002]. Cerebellar ataxia or gait disturbance is observed in 72% of patients. Cognitive decline is common (82%), whereas extrapyramidal signs (36%), psychiatric symptoms (21%) and myoclonus (15%) are less frequently reported in GSS cases. A positive family history is reported in 69%–100% of cases [Kovacs et al., 2005; Krasnianski et al., 2016].
CSF 14-3-3 protein is positive in about 50% of cases, and PSWCs in EEGs are infrequent, and found in less than 10% of cases [Kovacs et al., 2005]. Brain MRI typically shows cerebellar or global atrophy, and FLAIR or DWI hyperintensitites similar to those characteristic of sJCD are uncommon [Krasnianski et al., 2016; Vitali et al., 2011], although the EUROJCD study, found basal ganglia hyperintensities on FLAIR or DWI MRI in 30% of cases [Kovacs et al., 2005]. White matter hyperintensities are occasionally observed, but have also been reported in sJCD and other gPrD (such as gJCD due to the E196K mutation) [Schelzke et al., 2011; Simpson et al., 2013; Webb et al., 2008].
We had 40 symptomatic patients with GSS (from 19 families) referred and 20 patients were seen at UCSF MAC during the inclusion period. The most frequent mutations among referred symptomatic subjects were P102L (n=20), A117V (n=7), F198S (n=6), and H187R (n=6) (Table 1). One patient with the 365–388dup mutation, reported in Hinnell et al [Hinnell et al., 2011], and one patient with the P105L were referred to, but not evaluated at, our center.
P102L
P102L is the most commonly reported mutation in GSS disease and an increasing number of patients from different kindreds had been recognized worldwide. This has allowed a detailed description of the associated phenotypes and the study of the genetic factors that may modulate its expression (see below). Typically, symptoms begin in the fifth decade (with a range of age at onset from 27 to 66 years), with illness durations ranging from 7 to 132 months and a mean duration of 49 months [Higuma et al., 2013; Krasnianski et al., 2016; Webb et al., 2008]. Regarding age and duration of disease, the UCSF cohort of P102L cases is overall in line with the literature, though the earliest age of onset at our cohort occurred at age 24.
Family history is positive in 84–100% of cases, and penetrance is estimated to be complete [Higuma et al., 2013; Krasnianski et al., 2016; Minikel et al., 2016]. Among the nine probands from the UCSF cohort, six had positive family history for PrD, whereas two had family history score of 1 (other dementia) and one proband had family history score of 0. The P102L mutation is most commonly associated with methionine at the cis codon 129 (and this is the combination seen in all UCSF patients from whom we had that information available), and homozygosity at codon 129 (MM) has been linked to earlier disease onset than heterozygosity (MV) [Webb et al., 2008].
A slowly progressive cerebellar syndrome is the most frequent clinical presentation (in 70–90% of patients), and cerebellar signs are almost universal during disease progression [Higuma et al., 2013; Krasnianski et al., 2016; Webb et al., 2008]. Cognitive symptomsare infrequently reported as the first manifestation of the disease, and usually appear or progress later in the disease course, however it is probable that these may be underecognized because of the lack of specific testing [Kong et al., 2004; Krasnianski et al., 2016; Webb et al., 2008]. In a 20% of patients in the literature, neuropsychiatric symptoms with cognitive decline were the most prominent early in the disease course [Higuma et al., 2013; Krasnianski et al., 2016; Webb et al., 2008]. And among those, a small subset develop rapidly progressive dementia similar to sJCD [Webb et al., 2008]. In contrast, within the UCSF cohort, approximately 40% had cognitive (and about the same had cerebellar) symptom onset. Dementia occurred late in some of the patients, up to five years after onset. Motor and constitutional symptoms were other early presentations of P102L at the UCSF cohort.
Lower motor neuron signs, such as areflexia and muscle weakness, are seen in almost 80% of patients [Webb et al., 2008]. In addition, sensory symptoms such as dysaesthesia and are increasingly recognized and may reflect a sensorymotor axonal neuropathy [Webb et al., 2008]. Almost 50% of patients develop parkinsonism, whereas myoclonus, dystonia and apraxia are only ocasionally reported [Krasnianski et al., 2016; Webb et al., 2008]. Irrespective of the clinical presentation, eventually there is a slow progression of cerebellar and pyramidal dysfunction with behavioral symptoms and cognitive decline leading to a akinetic-mutism (Kong, Surewicz et al. 2004, Webb, Poulter et al. 2008). A few rare cases of P102L-cis129V mutation have been reported in patients that presented with psychiatric symptoms and seizures [Bianca et al., 2003; Webb et al., 2008].
Brain MRI reportedly typically shows generalized brain atrophy, or less frequently, focal cerebellar atrophy cases (Figure 3A)[Webb et al., 2008]. White matter lesions have been reported in up to a third of the scans [Webb et al., 2008], and periventricular hyperintensities were present in two out of the three MRIs reviewed at UCSF. Basal ganglia hyperintensities in FLAIR/DWI brain MRI have been reported in 25–30% of patients [Higuma et al., 2013; Krasnianski et al., 2016]. Among patients with a phenotype that resembles JCD, 80% have brain MRI changes that are consistent with JCD [Higuma et al., 2013]. Basal ganglia and cortical hyperintensities on DWI MRI were found in only one of the three UCSF cases.
Figure 3. Brain MRIs in Slow forms of gPrD.
A. FLAIR brain MRI in a 57 year old patient with P102L gPrD possibly 16.5 years after onset (precise age of onset unclear due to co-morbidities) shows mild cerebellar atrophy but no cortical atrophy or typical T2-weighted or DWI hyperintensites (cortical ribboning or deep nuclei hyperintensity), but only several punctate hyperintensities in the white matter consistent with small vessel ischemic vascular disease. The DWI and ADC map sequences showed no restricted diffusion (not shown). B. FLAIR brain MRI of with a 37 year old gPrD 6-OPRI patient showing diffuse cortical atrophy just 7 months after reported clinical onset; the amount of atrophy suggests the disease began years prior to obvious clinical onset. The DWI and ADC map sequences showed no restricted diffusion (not shown). C. Repeat brain MRI on same 6-OPRI patient 25 months later, 32 months after clinical onset, demonstrates progression of atrophy. The DWI and ADC map sequences still showed no restricted diffusion (not shown). Orientation is radiological.
EEG PSWCs are infrequent among patients with this mutation, and have been reported in 0–35% of cases [Higuma et al., 2013; Krasnianski et al., 2016; Webb et al., 2008]. Regarding CSF markers, 14-3-3 positivity has been reported in 15–20% of cases, whereas elevated tau levels (>1260 or 1300 pg/mL) were seen in about 20% of patients [Higuma et al., 2013; Krasnianski et al., 2016]. Likewise, PSWCs, CSF 14-3-3 and tau were usually negative for JCD among UCSF patients. One study reported a RT-QuiC positivity of 80% among patients with the GSS phenotype and 100% among patients with a JCD-like phenotype [Higuma et al., 2013].
Multicentric amyloid plaques immunostained by PrP antibodies are the most common neuropathological finding, whereas spongiform changes and/or synaptic-type PrP deposits are not universally seen [Higuma et al., 2013; Webb et al., 2008].
A117V
A117V mutation were initially described in British-Irish, French-Alsatian, American (one with German descent) and Hungarian families. Subjects with the A117V mutation show great variability in age at onset (20–64 years) and disease duration (1–11 years) [Hsiao et al., 1991b; Kovacs et al., 2001; Mastrianni et al., 1995; Tateishi et al., 1990]. In many cases, ataxia may be rare or absent with sparing of the cerebellum, whereascognitive dysfuction is an early or main clinical presentation [Hsiao et al., 1991b; Mastrianni et al., 1995; Tateishi et al., 1990]. A117V gPrD also has been reported to manifest with progressive hemiparesis or lower motor neuron deficit and parkinsonism, in addtion to dementia and ataxia [Kovacs et al., 2001; Tranchant et al., 1992]. To our knowledge, MRI findings have only been reported in two A117V patients, showing diffuse or asymmetric cortical atrophy. There is little data on EEG and CSF biomarkers in the literature [Kovacs et al., 2001]. Pathology reveals abundant PrP amyloid plaques in the cortex and cerebellum, and focal vacuolation, neuronal loss, and gliosis. Loss and intraneuronal vacuolation of spinal motor neurons with neurogenitc muscular atrophy were also reported in the case with lower motor neuron deficits [Kovacs et al., 2001].
The UCSF cohort had seven patients with A117V; six evaluated at UCSF and one by record review. Clinical features of our patients were quite similar to those from the literature such as varied age at onset and disease duration (mean age at onset, 36±13 years (14–49) and mean disease duration, 46 ± 18 months (27–78)), cognitive dysfunction as early symptoms. Initial symptoms were handwriting change (43%), language impairment (29%), memory loss (14%), and behavioral changes (29%), but no ataxia. Brain MRI showed only cortical atrophy in 67% with MRI (4 of 6 cases) and none of six cases had restricted diffusion. Positivity of other biomarkers was low; only diffuse background slowing on EEG in 1 of 4 cases. Regarding CSF biomarkers, 14-3-3 (n=4) and NSE (n=3) and t-tau (n=1) were all negative. Neuropathology was available in only one patient and showed PrP amyloid positive dystrophic neurites throughout the hippocampus. The cerebellum showed no amyloid, which is unusual for A117V pathology.
F198S
The F198S mutation was first identified in a large kindred from Indiana, USA [Dlouhy et al., 1992]. Symptom onset occurs between the fourth and seventh decades, and codon 129 polymorphism affects the age at onset, as homozygosity (VV) is associated with onset occuring 10 years earlier than heterozygosity (MV) [Dlouhy et al., 1992; Ghetti et al., 1995]. The mean disease duration is five years (range 2–12 years) [Farlow et al., 1989; Ghetti et al., 1995]. Early in the disease course, patients develop memory problems and gait changes. The gait changes are gradualy accompanied by other cerebellar dysfunction, such as eye movement abnormalities, dysarthria and clumsiness. Rigid-akinetic parkinsonism is a common manifestation, and cognitive decline eventually progresses to dementia [Farlow et al., 1989; Ghetti et al., 1995]. Brain MRI findings were only reported in a few cases, in which cerebellar atrophy and decreased T2 signal in the basal ganglia were observed [Farlow et al., 1989], but this was before diffusion MRI was commonly used. The neuropathology is characterized macroscopically by cortical and cerebellar atrophy [Ghetti et al., 1995]. Unicentric and multicentric PrPSc amyloid plaques are observed in multiple regions of the brain along with neuronal loss and gliosis, whereas spongiform changes are minimal [Ghetti et al., 1995].
In the UCSF cohort, six subjects were referred of whom three were evaluated at UCSF (a fourth case was evaluated after May 1, 2016 and mentioned below, but not included as seen/evaluated in the tables). Overall, our F198S cases showed similar findings to those from the literature. Two brothers with a known family history of GSS, were positive for F198S mutation and manifested with gait disturbance at ages 48 and 51. The 48 year old brother developed cognitve impairment two years after onset and died after clinical course of about 6.5 years. His brain MRI was normal at 19 months but his CSF NSE was elevated at 52.9 ng/mL (>35 ng/mL consistent with JCD); CSF14-3-3 was inconclusive and total tau not tested. His brain autopsy was consistent with GSS. The 51 year old brother developed cognitive problems three years after the onset of gait problems and died at six years. His MRI at 48 months after onset showed mild cortical and cerebellar atrophy and in contrast to his brother, all his CSF biomarkers were positive at this time (14-3-3, NSE 94.1 ng/mL and total tau 5133 pg/mL, in which >1150 consistent with JCD). The third case evaluated at UCSF was a 63-year-old gentleman with a four-year history of progressive gait difficulty and cognitive deficits (language, memory, frontal-executive, and visuospatial). His brain MRI at four years showed only symmetric superior parietal and vermian cerebellar atrophy. He was initially suspected to have multiple system atrophy, spinocerebellar ataxia or hereditary spastic paraplegia. His father had died at age 67 from reportedly from idiopathic Parkinson’s disease, but our review of his extensive records revealed atypical features, including ataxia and early dementia. PRNP testing in our proband revealed an F198S mutation. He died at age 66, about seven years after onset, and autopy revealed numerous PrPSc-positive amyloid plaques, typical of GSS. His son was recently evaluated at age 56 for two to three years of mild motor and cogntive symptoms (not included in our counts for symptomatic subjects as he was evaluated after our cut-off of 5/1/16; Table 1).
H187R
The H187R mutation has been reported in a few families, associated with MM, MV or VV at codon 129 [Butefisch et al., 2000; Cervenakova et al., 1999; Colucci et al., 2006; Hall et al., 2005]. Age at onset varied from 20 to 53 years, and disease duration ranged from 3 to 19 years [Butefisch et al., 2000; Cervenakova et al., 1999; Hall et al., 2005]. The clinical presentation is diverse, even within families. Some affected individuals developed cognitive and behavioral changes (such as disinhibition and executive dysfunction) early in the disease course that progressed to dementia, with later onset of cerebellar ataxia, whereas others presented initially with cerebellar ataxia and developed cognitive decline after a few years [Butefisch et al., 2000; Hall et al., 2005]. In one report, neuropsychiatric symptoms characterized as kleptomania, pyromania and/or suicide attempts appeared during adolescence in some of the mutation carriers, with dementia only being diagnosed years laters [Hall et al., 2005]. Pyramidal signs were frequent, and seizures and myoclonus were more common than usually reported in GSS [Butefisch et al., 2000].
Cerebellar atrophy with or without cortical atrophy was the most commonly observed changes in brain MRI, and DWI/FLAIR hyperintensities were not reported [Butefisch et al., 2000]. CSF protein 14-3-3 was typically negative, and EEG changes were nonspecific, with no PSWCs [Butefisch et al., 2000; Hall et al., 2005].
Despite the clinical presentation that resembled GSS, the characteristic multicentric PrPSc amyloid plaques were found in some – but not all – cases with the H187R mutation [Butefisch et al., 2000; Colucci et al., 2006; Hall et al., 2005]. Astrocytic gliosis was more consistently observed, whereas vacuolation was minimal or absent [Butefisch et al., 2000; Colucci et al., 2006; Hall et al., 2005]. Immunohistochemistry for PrPSc revealed granular deposits with a distinct “curly” shape and occasional synaptic-type deposits [Butefisch et al., 2000; Colucci et al., 2006].
We had four affected family members in our UCSF cohort with H187R with similar presentations to those in the literature. We evaluated two relatives at our center; the proband was codon 129 MM and her aunt, was MV. Both had early psychiatric, motor, gait ataxia and cognitive features manifesting with a GSS-like syndrome. The proband (codon 129 MM) had onset about age 29 with a 12 year duration, dying at age 41. Her brain MRI when we evalauted her about 10 years after onset, showed diffuse atrophy but no restircted diffusion or T2-hyperintensties. CSF 14-3-3, total-tau, NSE were all normal or negative. Her brother (referred but not seen at UCSF) had a similar course, dying at age 44. Her mother and uncle died of a similar illness. The proband’s aunt (codon 129 MV) had onset about a decade later at age 41 and died age 53 with about a 12–13 year course. The duration and type of symptoms were similar amongst family members, but ages of onset had some variability.
P105L
The P105L mutation was identified from several Japanese families and one British family. [Beck et al., 2010; Higuma et al., 2013; Mano et al., 2016]. The mean age at onset and mean disease duration of Japanese patients (n=8) [Higuma et al., 2013]were 44 years and 9 years, respectively. Clinical symptoms during the disease course include dementia (100%), extrapyramidal signs (71%), akinetic mutism (71%), pyramidal signs (spastic paraparesis 57%), psychiatric disorders (57%), myoclonus (43%), and cerebellar signs (14%) [Higuma et al., 2013]. Family history was postive in 37.5–75% of cases. Positivity of biomarkers was low; DWI cortical ribboning at 14.3%, PSWCs on EEG at 0%, CSF 14-3-3 and total tau (cuf off > 1260) at 0%. Interestingly, CSF RT-QuIC for PrPSc was positive in 50%. Neuropathology revealed PrP positive amyloid plaques and diffuse PrP deposition in deep layers of the cerebral cortex without vacuolation, compatible with GSS [Higuma et al., 2013]. A recent report on additional Japanese families with P105L (n=9) showed similar mean age at onset (47.2 ± 4.5 years and disease duration of 3 years (n=1)) [Mano et al., 2016] to that of the previously reported Japanese families described above but had more prominent parkinsonism. Brain MRIs revealed diffuse cortical atrophy (n=2) and CSF biomakers were not reported [Mano et al., 2016]. One patient from the British family presented with psychotic depression at age 33, followed by development of gait disturbance (spastic paraparesis) with extrapyramidal features, dyspraxia, short term memory difficulties and frontal lobe dysfunction. The MRI only showed moderate diffuse atrophy and EEG changes were non-specific. CSF S100β was elevated, but 14-3-3 was neagative. The patient was bed-ridden by 10 years after the onset [Beck et al., 2010]. The UCSF cohort has only one referred symptomatic case with P105L mutation, whose clinical features were very distinct - very early age at onset and early cognitive impairment. At 9 years of age, the boy began to decline in school performance, by 10 he had handwriting changes, by 11 he had seizures and a gait ataxia, and by12 he was wheel-chair bound. Extensive work-up was unrevealing until whole exome sequencing revealued a de novo P105L mutation. He had no family history for prion or any other neurodegenerative disorders. His brain MRIs at about 1–2 months after the onset were normal and EEGs showed diffuse background slowing with bursts and spikes or high-amplitude posterior dominant slow wave.
Insertion and Deletion mutations (Mixed, Fast and Slow types)
Octapeptide Repeat Insertions (OPRI) and Deletion (OPRD)
Insertions and deletions of 24 base pair (bp) repeats in the octapeptide repeat region of PRNP can also cause gPrD [Owen et al., 1990]. Wild type PRNP has five 24 bp octapeptide repeats. Octapeptide repeat insertions (OPRIs) and deletions (OPRDs) are usually classified based on the number of additional repeats found, even though the exact position of the insertion/deletion is not always the same and there might be minor nucleotide sequence variations in different reports [Croes et al., 2004; Gambetti et al., 2003a]. OPRIs with two to twelve extra octapeptide repeats have been reported, with significant clinical and neuropathological variability. An insertion or deletion of one octapeptide is not considered pathogenic, as 1-OPRI and 1-OPRD have also been found in healthy individuals [Beck et al., 2010; Yu et al., 2004]. 2-OPRD, on the other hand, was reported in a few cases of JCD and not found in healthy individuals, and is considered pathogenic [Beck et al., 2001; Capellari et al., 2002].
OPRIs do not appear to affect PrP conformation directly, but functional studies suggest that PrPC with two or more extra octapeptide repeats is more protease-resistant and more prone to aggregate [Moore et al., 2006; Priola and Chesebro 1998]. Increased PrPC aggregation is thought to facilitate PrPSc formation, which in turn, leads to neurodegeneration [Moore et al., 2006]. Octapeptide repeat expansions also affects copper binding to PrP, leading to increased interactions between PrPC and PrPSc, and possibly decreasing PrPC stability and resistance to polymerization [Hodak et al., 2009; Leliveld et al., 2006; Stevens et al., 2009].
PrPC binding occurs faster in molecules with more repeats, which might explain why increasing number of OPRIs are associated with earlier onset of symptoms and shorter duration of disease [Croes et al., 2004]. OPRIs with more than eight repeats were also found to be associated with increased PrP amyloid formation, which is consistent with the fact that amyloid plaques are more commonly found in the cerebellum of individuals with 8- or 9-OPRI (in comparison to 4- to 7-OPRI) [Moore et al., 2006; Vital et al., 1998].
Accordind to the literature, insertions of additional one to four octapeptide repeats typically manifest with a phenotype of sJCD, a later age at onset and shorter clinical duration, whereas more than four octapeptide repeats tend to be associated with a longer disease course [Croes et al., 2004]. Insertions of five to seven octapeptide repeats reportedly frequently present as JCD in early to mid-adulthood with a lengthier clinical course; clinical presentation is heterogeneous such as cognitive, psychiatric, and motor dysfunction (cerebellar ataxia, pyramidal dysfunction, parkinsonism, dystonia, chorea, tremor), and seizures. The literature also suggests that OPRIs with eight or more are more frequently associated with the GSS phenotype [Croes et al., 2004; Gambetti et al., 2003b]. Our cases do not necessarily support these above generalizations about the OPRI size and clinical correlation, however, as we detail below. The codon 129 polymorphism does appear to affect phenotype, which is described in detail below.
We had one symptomatic 5-OPRI, two symptomatic 6-OPRI (129 MM, 129MV with cis M), and one symptomatic 9-OPRI cases evaluated at UCSF (Table 4). Medical records of three symptomatic 6-OPRI (129 cis M (trans unknown), 129 VV, 129 cis V (trans unknown)) and one symptomatic 8-OPRI were reviewed at UCSF. Two siblings with the 6-OPRI mutation (129 MM) were previously reported, and had mild in vivo evidence of PrP amyloid deposition using an amyloid binding positron emission tomography agent [Boxer et al., 2007].
Table 4.
Demographic and clinical characteristics of UCSF OPRI cohort
| Mutations | 5-OPRI | 6-OPRI | 8-OPRI | 9-OPRI | |||||
|---|---|---|---|---|---|---|---|---|---|
| Symptomatic cases referred | 4 | 5 | 1 | 1 | |||||
| Symptomatic cases evaluated at UCSF | 1 | 2 | 0 | 1 | |||||
| Number of families with symptomatic cases | 3 | 2 | 1 | 1 | |||||
| 129 polymorphism | MM | MM* | MV*^ | Cis M*^ | VV# | cis V# | MM | VV | |
| PrPSc type | N/A | N/A | N/A | N/A | N/A | N/A | N/A | 1 | |
| sex | M | M | M | M | F | F | M | M | |
| Age at onset (Y) | 39 | 36 | 37 | 32 | 51 | 47 | 22 | 47 | |
| Onset to UCSF evaluation (Mo) | 120 Mo | 7 Mo | 50 Mo | N/A | N/A | N/A | N/A | 21 Mo | |
| Duration of Disease | >19 Y | >3 Y** | 5 Y 9 Mo | ~ 9 Y | 5 Mo | 10 Mo | >5 Y** | 21 Mo | |
| Sex | M | M | M | M | F | F | M | M | |
| First symptom category | Visuosp | Cog Cb | Const, Behav, Cog | Cog | Cog | Sens, Cb | Const, Behav | Visuosp, Cog | |
| Brain MRI (time after the onset) | Diff C atrophy (10 Y) | Diff C atrophy (7 Mo) | Diff C atrophy (2 Mo) | Diff C atrophy (N/A) | Diff C, Cb, and BS atrophy (3 Mo) | P, Fr, Cb atrophy (10 Mo) | C atrophy (5 Y) | C ribboning, Mild diff C atrophy (19 M) | |
| EEG | Normal (@ 10.5 Y) | N/A | N/A | N/A | Bkgnd slowing | norm | N/A | Irregular triphasic waves | |
| CSF | 14-3-3 | N/A | Neg | Neg | N/A | N/A | N/A | N/A | N/A |
| NSE | N/A | Neg | Neg | N/A | N/A | N/A | N/A | Pos | |
| t-tau | N/A | Neg | Neg | N/A | N/A | N/A | N/A | Pos | |
| Neuropathology | N/A | N/A | JCD | JCD | JCD | JCD | N/A | Mixed JCD & GSS | |
three patients from one family,
two patients from another family.
were published in (Boxer et al., 2007)
Some patients still alive, so duration at last follow-up.
Abbreviations: @ = at; behave = behavior; bkgrnd = background; BS = brain stem; C = cortical; Cb = cerebellar; Cog = cognitive impairment; diff = diffuse; dysfx = dysfunction; Fr = frontal; F/E = frontal executive; Mo = months; N/A = not available; Neg = negative; norm = normal; P = parietal; Pos = positive; visuosp = visuospatial; Y = years
2-OPRD
Deletion of two octapeptides from the repeat region was identified in two non-related patients. Beck et al. reported a woman with no family history of neurodegenerative diseases, who developed rapidly progressive dementia at age 86 and died 23 months later [Beck et al., 2001]. She was clinically diagnosed with JCD, but neuropathological assessment was not performed. The 2-OPRD was also reported in a male patient by Capellari et al, who developed at age 62 rapidly progressive dementia with seizures and myoclonus, and died 18 months after the onset of symptoms. His mother had been diagnosed with Alzheimer’s disease and died in her 80s. He was homozygous to methionine at codon 129, and the neuropathological findings were similar to those reported in sJCD MM1 [Capellari et al., 2002].
2-OPRI
Goldfarb et al. reported a woman with neuropathologically-proven JCD and codon 129 MM, who developed rapidly progressive dementia at age 58 and died three months later. His mother also carried the same mutation, but developed a slowly progressive dementia, with onset at age 75 and disease duration longer than 13 years [Goldfarb et al., 1993]. Two apparently non-related Dutch patients were also reported with this mutation [Croes et al., 2004; van Harten et al., 2000]. One (129MV) had onset at age 59 and was mute ten years later [Croes et al., 2004]. The other (129VV) was a woman who developed memory problems at age 61. Her cognitive symptoms progressed gradually, and she developed gait ataxia later in the disease course. Her brain MRI showed extensive white matter abnromalties, and she died 7 years after the onset of symptoms [van Harten et al., 2000]. Both Dutch patients had first-degree relatives diagnosed with dementia, but genetic testing was not performed in those relatives.
3-OPRI
The 3-OPRI mutation coupled with codon 129 VV was identified in a male patient diagnosed with JCD both clinically and neuropathologically at age 69 years, and died 4 months after disease onset. CSF 14-3-3 was positive, but the EEG did not show PSWCs [Grasbon-Frodl et al., 2004b]. Nishida et al. reported a Japanese man diagnosed with probable JCD. Symptom onset was at age 63 years, and he died three years later. He had no family history of PrD. Protein 14-3-3 testing in CSF was negative, but brain MRI 22 months after onset revealed diffuse cortical atrophy and widespread DWI cortical hyperintensities and bilateral striatal hyperintensity. PRNP testing identified 3-OPRI with codon 129MM [Nishida et al., 2004]. The 3-OPRI mutation with 129MM genotype was also identified in a Chinese woman who was asymptomatic in her late thirties, which has led some to question its pathogenicity, although the other cases had onset ~30 years later than her current age [Beck et al., 2010; Yu et al., 2004].
4-OPRI
OPRI with four additional repeats associated with the codon 129 MM genotype at causes gJCD with symptom onset ranging from 39 to 85 years (mean 60 years), and duration of disease ranging from 2 to 77 months (median 13.8 months) [Croes et al., 2004; Kaski et al., 2011]. Age at onset in 4-OPRI on average is later than for 5- and 6-OPRI [Kaski et al., 2011]. The most common clinical presentation is similar to sJCD, with rapidly progressive dementia, myoclonus (80%) and cerebellar ataxia (50%), but the phenotype is variable and even a presentation that clinically resembled Alzheimer’s disease has been reported [Kaski et al., 2011]. CSF 14-3-3 protein positivity in is frequent, and typical JCD changes in EEG are reported in about 30% of cases [Kaski et al., 2011]. Brain MRI scans can be reported as normal, or show global atrophy. Basal ganglia hyperintensities on FLAIR or DWI are infrequent, and only seen in 20% of cases in one series [Kaski et al., 2011]. The neuropathological findings are consistent with a diagnosis of JCD, but with PrPSc deposits in axons and dendrites that were somewhat different from those commonly seen in sJCD, as well as deposits in the dendrites of Punrkinje cells that caused the cerebellum to have a “tigroid” appearance (an appearance also reported in 5-OPRI and 6-OPRI) [Kaski et al., 2011]. Both PrPSc types 1 and 2 (not concomitantly) have been found in brains of patients with this mutation [Kaski et al., 2011].
Laplanche et al. reported a patient 4-OPRI with codon 129 VV who developed rapidly progressive dementia at age 82 and died four months later. Her family history was negative [Laplanche et al., 1995]. Sánchez-Valle et al. reported a patient who carried the 4-OPRI mutation with codon 129 MV genotype (cis V) [Sanchez-Valle et al., 2012]. Onset occurred at age 38 and he died 8 months later. CSF protein 14-3-3 was positive and brain MRI revealed cortical and basal ganglia hyperintensities consistent with JCD. On neuropathological evaluation, a pattern similar to sJCD VV1 was found. The proband’s mother also carried the same mutation and was cognitively normal at age 74, but was homozygous at codon 129 VV,.
5-OPRI
Codon 129 polymorphism was known in nine of the 15 5-OPRI cases previously reported [Beck et al., 2005; Cochran et al., 1996; Goldfarb et al., 1991; Mead et al., 2007; Skworc et al., 1999]. A mean age at onset of 129 MM (n=6) was 42.3 years (range 34–63), whereas 129 MV (cis M, n=3) had a mean age at onset of 58.0 years (range 52–61), possibly suggesting the trans allele V might delay the age of onset. Clinical duration, however, was not affected by codon 129 polymorphism [Beck et al., 2005; Cochran et al., 1996; Goldfarb et al., 1991; Mead et al., 2007; Skworc et al., 1999].
In our cohort, we have had four symptomatic 5-OPRI subjects referred, one of whom was evaluated at UCSF (Table 1); he was codon 129MM and had onset at 39 years old, within the range reported in the literature, but his clinical duration was longer than those cases (>19 years vs 10 months-16 years) (Table 4). Published MRIs of 5 -OPRI cases reportedly showed mild to moderate cerebral and cerebellar atrophy without DWI signal changes [Beck et al., 2005; Cochran et al., 1996; Goldfarb et al., 1991; Mead et al., 2007; Skworc et al., 1999] similar to our 5-OPRI subject’s MRI. EEG findings such as diffuse background slowing, PSWCs, or non-periodic spike and wave complexes were reported in 5-OPRI cases [Mead et al., 2007] whereas our case showed a normal EEG, even at 10.5 years after the onset. We also compared the clinical phenotype of our 5-OPRI case (129 MM, age at onset 39 years, disease duration > 19 years) with a pubished 5-OPRI case with the same codon 129 polymorphism, an early age at onset, and a prolonged disease duration (129 MM, age at onset 30 years, disease duration 16 years). [Depaz et al., 2012]. Both cases began with possible parietal dysfunction (difficulty with parking vs dyscalculia) and gradually deteriorated in cognition and motor function over years. But the 5-OPRI case in the literature had more prominent parietal dysfunction and showed distinct posterior cortical atrophy (PCA) on brain MRI (diffuse cortical atrophy, more pronounced in posterior regions without DWI changes). EEG in the published case demonstrated triphasic periodic sharp wave complexes whereas EEG of our case was normal. Interestingly, autopsy of the literature case showed mixed prion (mild spongiosis and gliosis with PrPSc deposition but no kuru or GSS plaques) and AD pathology, and the father of this published case had dementia at age 55, died at 60 was also found retrospectively to have co-existence of prion disease and AD (the proband was negative genetic testing for mutations in PSEN1, PSEN2, and APP) [Depaz et al., 2012]. The relationship between PrPSc, abeta amyloid and tau is not clear. In an autopsy study of 266 of our sJCD cases, we found Alzheimer’s-like pathological changes in 17% of cases, most of whom were much younger than typical age of onset for AD [Tousseyn et al., 2015]. Another recent study examining pathology in human growth hormone iatrogenic JCD cases found much higher than expeced Alzheimer’s amyloid pathology, which might also suggest prion pathology induced Alzheimer’s pathology [Jaunmuktane et al., 2015]. That nonsense PRNP mutations, discussed below, lead to an amyloid tauopathy also suggests an interaction of these proteins. It is possible that PrPSc might perturb the normal metabolism of beta amyloid or tau protein, possibly in chronic cases. Thus, it would be have been interesting to examine the brain pathology of our case, who has the longest disease duration, but autopsy was declined by the family.
6-OPRI
Two 6-OPRI cases from one French family presented with memory loss followed by cerebellar signs, myoclonic jerks, and seizures. Both had onset at age 38 and disease duration was 4–10 years [Vital et al., 1998]. Another study of a very large and extended family with 6 OPRI in southeast England showed that progressive dementia and frontal lobe dysfunction (aggressive or apathetic behavioral change) were the main clinical features and that cerebellar ataxia and pyramidal tract signs were the most common additional neurological features [Mead et al., 2006]. Other clinical features in patients from the same family, however, was very heterogeneous. The mean age of onset was 34.9 years (range 20–53, n=63) and mean age of death was 45.1 years (range 30–65, n=73). Brain CT or MRI did not show any specific features but only generalized cerebral and cerebellar atrophy. EEG also showed only non-specific abnormalities such as slow background activity or low amplitude. Basic CSF tests were unremarkable (n=3) but total tau, NSE, and 14-3-3 were not tested [Mead et al., 2006]. In this family, age of onset was correlated with duration of disease; patients presenting at an early age have a longer disease duration and it was earlier in patients with 129 MM than in those with 129 MV (cis M) (mean age of onset 31.4 years (n=30) vs 41.7 years (n=10)), suggesting a trans effect of codon 129 on onset age. There was no major difference in a mean disease duration between 129 MM and 129 MV cases, however (11.4 years (n=19) vs 8.9 years (n=8)) [Mead et al., 2006] indicating codon 129 polymorphism affects the age at onset but not disease duration in 6-OPRI cases similarly to those 5-OPRI cases described above.
In our 6-OPRI cases (five referred cases from two families) (Table 4), three relatives with 129 cis M (MM, MV, M?) (the MV and M? cases published in [Boxer et al., 2007]) showed early ages at onset (32, 36 and 37 years) and prolonged clinical courses (> 3 years, ~ 9 years, and 69 months), whereas two cases with 129 cis V was relatively late at age at onset (51 and 47 years) with short clinical courses (5 and 10 months) (Table 4). Even though we could not draw any clear conclusion on the effect of codon 129 polymorphism due to our small sample sizes, the inverse correlation of age of onset with disease duration appeared true in our UCSF cohort. It was interesting to see the presence of atrophy without typical JCD MRI findings but JCD pathology on autopsy (n=4) in our 6-OPRI cases (Figure 3B and 3C). The 6-OPRI cases from the literature also showed variable degrees of PrPSc deposits and spongiosis without amyloid PrPSc plaques [Mead et al., 2006; Vital et al., 1998].
7-OPRI
There is marked phenotypic heterogeneity in 7-OPRI mutation. 7-OPRI in a Dutch pedigree was reported to have GSS phenotype; six family members appeared to be affected, two of whom were tested and found to have an 7-OPRI mutation, one was codon 129VV and the other MV (cis V) [Jansen et al., 2011]. Clinical information was available only in four family members, characterized by slowly progressive cognitive decline, personality change, psychiatric features (depression with anxiety, panic attacks), apraxia, and parkinsonism. Age at onset was in the range of 49–59 years (mean 52.2 years) and disease duration was from 0.6 to 5.4 years (mean 2.4 years). Pathologically (n=3), numerous uni- or multi-centric amyloid plaques throughout the cerebrum and cerebellum were detected in addition to moderate to severe spongiosis, neuronal loss, and gliosis [Jansen et al., 2011]. Other reported 7-OPRI cases with cis M at codon 129 (n=9), however, showed earlier disease onset and longer duration; the age at onset from 18 to 44 years (mean 28.9 years) and disease duration ranging from 4 to 16 years (mean 10.3 years). Interestingly, there were no amyloid plaques detected in these cases [Dermaut et al., 2000; Goldfarb et al., 1991]. It is possible that the different presentations might be due to the PRNP cis codon 129 polymorphism.
8-OPRI
The 8-OPRI mutation was reported in five generations of a French family [Laplanche et al., 1999]. Eleven family members have shown the phenotype of GSS; age at onset was in the range of 21–34 years (mean, 28 years) and disease duration was 1–7 years (mean, 3.8 years). They had prominent psychiatric features with mania or mania-like symptoms being the most frequent, followed by dementia at the later stage of disease. Pathology showed cerebellar atrophy, kuru, and multi-centric PrP plaques with minimal spongiosis. Genetic testing in four family members revealed 8-OPRI mutation with codon 129 cis M. Our cohort has only one 8-OPRI case whose medical records we reviewed (Table 4). Although we do not have complete information on the clinical course of this patient, first symptoms were depression and mild gait ataxia with preserved cognitive function were present about 5 years after onset, suggesting a GSS-like phenotype.
9-OPRI
There were only a few cases reported in the literature with 9-OPRI mutations. The first reported case (129 cis M) presented at age 55 with two years of unsteady gait, self-neglect, and urinary incontinence. She was mildly demented and showed apraxia, myoclonic jerks, and gait ataxia and died several months later [Owen et al., 1992]. CSF, EEG, or brain imaging were not reported. Another case reported by Duchen et al. presented at age 54 with falls and memory loss and had ideomotor apraxia, utilization behavior, gait ataxia, axial rigidity, and myoclonus on exam. CSF and EEG were reported as normal, but it is not clear if any CSF biomakers (e.g. S100B, 14-3-3, total tau, NSE) were tested. Brain CT two years after onset was reported to show diffuse cerebral and cerebellar atrophy. She died after a 2.5 year course, and interestingly the autopsy revealed no spongiosis or astrocytic gliosis but numerous small PrP plaques in the cerebellum, basal ganglia and cerebral cortex, which apparently were not like amyloid plaques typical of GSS [Duchen et al., 1993]. Another patient manifested at age 32 (129 MM) with behavior change (withdrawn) and inability to do household work. She later developed worsening cognitive impairment (inability to recognize bills and dyscalculia), dysarthria, gait disturbances, vision change (concentric narrowing), headache, and agraphia. At age 34, exam revealed moderate dementia, dysarthria, depression, gait ataxia, bilateral dysdiadochokinesis and a postural tremor. Her lab tests were reportledly non-specific; unremarkable CSF test (unclear what was tested), bilateral slow wave activity on EEG, and generalized cortical atrophy on MRI. Her mother, grandmother, and great grandmother all died at their 40s of dementia. She had an 9--OPRI mutation and was still alive when her case was reported [Krasemann et al., 1995].
For these three published cases the age at onset (55, 54, 32 years), clinical duration (about 2.5 years vs greater than 2 years), and initial symptoms (unsteady gait vs behavioral changes) of the reported three 9-OPRI cases appear only mildly heterogeneous. CSF, EEG, and MRI did not show any characteristic features of prion disease. Our symptomatic 9-OPRI mutation (129 VV) case, however, was very distinct from the published cases. Except for somewhat the early age at onset (47 years), he had a typical JCD phenotype including rapid progression (21 months of disease duration), restricted diffusion cortical ribboning on MRI, triphasic waves on EEG, and positive CSF 14-3-3 and NSE. Our 9-OPRI case does not appear to support the notion that the clinical course is slower in longer OPRI cases. Interestinglu, autopsy of our case revealed mixed pathology of JCD and GSS. Vital et al. reported the presence of plaques in cases with an 8- or 9-OPRI and elongated PrP deposits in a 4- to7-OPRI cases [Vital et al., 1998]. It remains to be determined if the mixed JCD and GSS pathology is a unique finding of nine or longer OPRI mutations.
12-OPRI
Insertion of 12 octapeptides was reported in a family with three affected family members in which symptoms started at 43–45 years of age and died 7–10 years later [Kumar et al., 2011]. The clinical presentation started with cognitive decline and behavioral changes (with frontotemporal dementia-like symptoms), and patients developed later gait abnormalities and generalized tonic-clonic seizures. The proband’s brain MRI revealed bilateral frontal atrophy without cortical ribboning or basal ganglia hyperintensities on DWI or T2-weighted MRI. Her CS, total tau levels was increased, but protein 14-3-3 testing was negative. EEG showed generalized spike and sharp-wave discharges, but PSWCs were absent. The neuropathological findings of the proband were consistent with GSS, with multicentric PrPSc amyloid plaques, and were particularly present in the cerebellar cortex, along with tau-positive neurofibrillary tangles in the neocortex [Kumar et al., 2011].
Although previous reports claimed that the longer OPRI length, the earlier age at onset 20–53 years (mean 34.9, n= 63) in a 6-OPRI pedigree vs 31–63 years (mean 45.7, n=15) in the 5-OPRI patients, [Goldfarb et al., 1991; Skworc et al., 1999], our cases did not support it; 32–51 years (mean 41, n=5) in 6-OPRI vs 39 years in 5-OPRI (n=1). It was also thought that the longer OPRI length, the prolonged clinical course, which was not the case with our cohort; 21 months in 9-OPRI (n=1), > 5 years in 8-OPRI (n=1, still alive), 5 months to about 9 years in 6-OPRI (n=4), and greater than 19 years in 5-OPRI (Table 4). Hetegogenous clinical manifestations including early involvement of cognition and pathological findings of JCD (JCD in four 6-OPRI cases and mixed JCD and GSS in one 9-OPRI) did not support the notion that additional OPRIs greater than four manifest with clinical features of GSS.
Non-repeat octapeptide insertion
A 24bp nucleotide insertion located between nucleotides 388 and 389 of PRNP (causing a insertion of an octapeptide between codons 129 and 130) was reported in a single Canadian patient, who developed night terrors at age 28, cognitive decline starting at age 31, and presented at age 34 with status epilepticus [Hinnell et al., 2011]. He later developed gait ataxia, dysarthria and disinhibition. On neurological examination, he had saccadic pursuit, square wave jerks, dysarthria, slight difficulty on heel-to-shin testing, and a wide-based gait, besides myoclonus. His FLAIR brain MRI only revealed mild cortical and cerebellar atrophy without cortical ribboning or deep nuclei hyperintensites (unclear if DWI and ADC map were performed), but CSF protein 14-3-3 was positive and total tau was elevated. EEG showed diffuse slowing and multifocal epileptiform discharges. Brain biopsy showed spongiosis and multicentric plaques immunostained by antibodies against the prion protein, in a pattern consistent with GSS. The parents did not carry the insertion, suggesting a possibie de novo mutation.
PRNP Nonsense mutations
Nonsense mutations are very rare, and have only been reported in a few kindreds, but are very atypical for most prion diseases. The clinical presentations are highly variable, but overall are characterized by atypical clinical features (such as Alzheimer type dementia or behavioral variant Frontotemporal Dementia phenotypes, sensory and autonomic peripheral nervous system involvement, and/or chronic diarrhea), prolonged disease courses, and presence of PrP amyloid plaques and/or PrP cerebral amyloid angiopathy, often combined with tau pathology in the brain [Guerreiro et al., 2014]. Pathogenic nonsense mutations reported to date are located in the C-terminal region, in codon 145 or higher [Guerreiro et al., 2014; Minikel et al., 2016]. In our cohort, e had one subject with a nonsense mutation, Q160X who was evaluated at UCSF, and this case was recently published [Fong et al., 2016b].
A Q160X mutation results in a premature translation stop, thereby production of C-terminally truncated PrP. There are two families with Q160X mutation reported in the literature. Two brothers from one family presented with dementia at age 32 (younger brother, 129 MM) and 48 years (older brother, 129MV, cis M) and were positive for Q160X mutation [Finckh et al., 2000; Owen et al., 1989]. No other clinical information was provided except for their father with dementia onset at age 48 died at age 60. The other family, a daughter and mother, were found to have Q160X mutation [Jayadev et al., 2011]. The daughter (129 MV, cis M), proband, presented at age 39 with short-term memory loss, followed by depression, language impairment, parkinsonism and loss of ADLs over years and died at age 47 with an eight year course and clinical diagnosis of AD. Laboratory tests, EEG, and MRI presumably taken at 3 years after the onset, were reported unremarkable. Her mother (129 MM) developed personality changes and significant memory loss at age 59 accompanied by difficulties with job performance and depression which was followed by severe post-prandial diarrhea. Laboratory testing, brain CT, and EEG, presumably taken at 4 years after onset, were reported as unremarkable. She continued to decline (language impairment, loss of ADLs, chronic diarrhea with weight loss) and died aftter an eight year course at age 67 also with a clinical diagnosis of AD. Interestingly, autopsy brain pathology revealed that the daughter had tau-positive neurofibrillary tangles and PrP amyloid plaques with PrP positive amyloid angiopathy but no spongiosis or beta amyloid plaques. The mother had similar brain tau and PrP pathology to the daughter but had positive beta amyloid plaques, likely having both prion disease and AD. No mention is made as to whether or not the daughter had gastrointestinal symptoms or pathology (presumably she did not), but the mother’s autopsy revealed no gastrointestinal pathology despite her symptoms [Jayadev et al., ].
In comparison to the cases reported in the literature, our UCSF patient had similar clinical features to those reported in the literature such as slowly progressive decline of behavioral, cognitive and motor functions over years. It remains elucidated that peripheral neuropathy (our case) or chronic diarrhea (our case and the mother in the literature) is the characteristic phenotype of Q160X mutation. The is a wide range of age at onset of the Q160X mutation, all of whom are codon 129 cis M; our case 27 (129MM) and others, 32 (129MM), 48 (129MV), 39 (129MV), and 59 (129MM) years, possibly suggesting codon 129 polymorphism might not affect the age at disease onset in this Q160X mutation. It would be interesting to examine neuropathology and other organ pathology in more cases of Q160X to determine the possible role of various pathologies on symptoms (e.g. presence or absence of spongiosis, tau-positive neurofibrillary tangles or PrP amyloid angiopathy in addition to PrP accumulation).
The Y163X-129V mutation was characterized in a large British family, and clinically presents as chronic diarrhea with onset in the fourth decade of life, followed by a predominantly sensory and autonomic axonal polyneuropathy. Cognitive decline and seizures appear about the fifth or sixth decades. Age at onset of symptoms ranged from 40 to 70 years. Neuropathological assessment revealed immunostaining for abnormal prion protein not only in the central and peripheral nervous system, but also in the gut, lymphoreticular system, heart, arteries and veins, liver, kidney and lung. It should be noted that PrP staining in peripheral tissues is not found in sporadic PrD. Neuropathological findings included PrP amyloid plaques, amyloid angiopathy, and tau pathology with activated microglia, as well as mild spongiosis and vacuolation of deeper cortical laminae [Mead et al., 2013].
Other nonsense mutations were reported in fewer cases. A Y145X-129M mutation in an Japanese individual with an Alzheimer-type dementia phenotype and neuropathological examination showing PrP positive amyloid angiopathy and neurofibrillary pathology [Ghetti et al., 1996]. A Y226X-129V mutation was identified in a Dutch patient with dementia and hallucinations who also had PrP cerebral amyloid angiopathy. Another Dutch patient, diagnosed clinically with bvFTD and parkinsonism, had a Q227X-129V mutation and brain pathology showing PrP positive amyloid plaques and severe neurofibrillary pathology [Jansen et al., 2010]. Lastly, a 2bp deletion at codon 178 leading to a frameshift mutation with premature stop codon was found in a Japanese kindred presenting with severe dysautonomia, sensory neuropathy and cognitive impairment [Matsuzono et al., 2013].
Discussion
As shown in this paper, the phenotypes associated with PRNP mutations are very diverse. The historical clinicalpathological classification of gPrD into gJCD, GSS and FFI no longer encompass the diversity of clinicalpathological features, particularly for the octapeptide repeat insertions/deletions and stop codon mutations. Nevertheles, vven within mutations associated with gJCD, GSS or FFI, the spectrum of clinical manifestations is wide. Not only the clinical presentation is variable in regards to age at disease onset, disease duration, presence of family history for PrD, and other features, but also the phenotype might occasionally resemble those associated with other neurodegenerative diseases, such as Alzheimer’s disease and behavioral variant frontotemporal dementia. The nonsense mutations are frequently diagnostic challenges, as they present as atypical forms of early onset dementia, and sometimes are characterized by prominent symptoms not usually associated with PrD, such as peripheral neuropathy or chronic diarrhea.
A positive family history is frequently used as a sign of genetically determined disease. About 50% of patients with pathogenic PRNP mutations, however, are seemingly sporadic and have negative family history for prion diseases or other neurodegenerative disorders, and hence absence of family history alone should not be used as an evidence against the need of genetic testing [Goldman et al., 2004; Kovacs et al., 2005]. We and many specialists feel that all parients diagnosed with PrD should be offered genetic counseling and testing for mutations in PRNP, when available. The diagnosis of gPrD is also made more challenging by the fact that patients might have a longer disease course than typically expected for PrD, and onset might occur at earlier ages than sJCD. Also, certain biomarkers in CSF, EEG and MRI are more commonly negative in gPrDs than in sJCD and might lead to erroneous diagnoses. Thus, diagnosing gPrD frequently depends on consideringprion disease on a diagnostic differential and knowledge that even apparently sporadic disease can be caused by a genetic mutation. Moreover, recent advances in diagnostic testing might be helpful to establish a correct diagnosis of gPrD. The real-time quaking induced conversion (RT-QuIC) assay was developed to detect abnormal prion protein in CSF and has shown high sensitivity and specificity in certain types of gPrD (87–100%) [Sano et al., 2013], but this test needs to be assessed in more cases and PRNP mutations. The progressive decrease in cost and increase in availability of genetic testing for dementia with next generation sequencing technologies has facilitated the diagnosis of genetic forms of dementia. Nowadays, dementia-associated gene panels or exome sequencing can be used in the clinical setting to diagnose neurodegenerative dementias of genetic origin [Beck et al., 2014]. Next generation sequencing technologies are particularly useful in the genetic assessment of atypical forms of dementia, such as those caused by nonsense PRNP mutations.
There are great advantages to studying the relatively rare gPrDs, as they can be a good research model for more common PrDs such as sJCD. Indeed, much of the knowledge on the pathogenesis of PrDs has come from genetic models. Research on gPrD (and other monogenic neurodegenerative dementias) has also focused on presymptomatic individuals, particularly regarding the identification of early symptoms and disease biomarkers, which will be important for the future disease-modifying treaments.
Diagnosing a patient with gPrD has profound implications for biological relatives who might be at risk of carrying the same mutation and therefore developing a disease to which there is no cure available. Once a pathogenic mutation has been identified, the relatives might inquire about predictive genetic testing to find out whether he or she also carries the same mutation. Predictive testing should only be ordered once the individual has undergone genetic counselling to be able to understand the possible positive and negative implications of being diagnosed with a disease-causing mutation [Goldman 2015]. Following the Huntington’s protocol for genetic testing, psychiatric and neurological assessments are also recommended prior to testing, so to decrease the possibility of negative outcomes (such as psychological distress or depression), and to certify that the individual is indeed asymptomatic [Goldman 2015; MacLeod et al., ]. Predictive testing not only allows individuals to make career-related and financial decisions, but also – if they are considering to have children – to decide for a preimplantation genetic diagnosis and in vitro fertilization in order to prevent passing on the mutation [Uflacker et al., 2014]. As is the trend in Alzheimer’s disease and many other neurodegenerative disorders, future clinical trials on PrD will probably focus on presymptomatic individuals, particularly those with PRNP mutations.
The characterization of our UCSF gPrD cohort has its limitations. Even though it is a large cohort from a single research center, only a portion of mutations were represented and for some mutations we have few subjects. Also, not all patients were evaulated at UCSF, thereby clinical information was not complete in some cases. Nevertheles, it is important that this population of those with and at-risk for gPrD be studied so that we can learn more about these devasting disorders in order to develop treatments for them and the more common sporadic forms of PrD.
Acknowledgments
We thank the patients and their families for allowing us to learn from them and share this information. The authors would like to thank Stacy Metcalf for assistance in data collection and tabulation.
Footnotes
This work was performed at University of California, San Francisco (UCSF)
Disclosures:
Dr. Geschwind has the following disclosures: Dr Geschwind serves on the board of directors for San Francisco Bay Area Physicians for Social Responsibility, on the editorial board of Dementia & Neuropsychologia, and serves or has served as a consultant for Best Doctors, Inc; Advance Medical; the Gerson Lehrman Group, Inc; MEDACorp; Franciscan Hospitals; Kendall Brill Kelly; Guidepoint Global, LLC; Lewis Brisbois Bisgaard & Smith LLP; Biohaven Pharmaceuticals Inc.; Lundbeck Inc; NeuroPhage Pharmaceuticals; and Quest Diagnostics. Related to his work on this paper, he receives research support for his work on prion diseases from the Michael J. Homer Family Fund and the National Institute on Aging (R01 AG AG031189). He also receives research support from CurePSP, Quest Diagnostics, Alliance BioSecure and the Tau Consortium. The other authors have no disclosures.
References
- 1000 Genomes Project Consortium. Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491(7422):56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alperovitch A, Zerr I, Pocchiari M, Mitrova E, de Pedro Cuesta J, Hegyi I, Collins S, Kretzschmar H, van Duijn C, Will RG. Codon 129 prion protein genotype and sporadic Creutzfeldt-Jakob disease. Lancet. 1999;353(9165):1673–1674. doi: 10.1016/s0140-6736(99)01342-2. [DOI] [PubMed] [Google Scholar]
- Alzualde A, Indakoetxea B, Ferrer I, Moreno F, Barandiaran M, Gorostidi A, Estanga A, Ruiz I, Calero M, van Leeuwen FW, Atares B, Juste R, Rodriguez-Martinez AB, Lopez de Munain A. A novel PRNP Y218N mutation in Gerstmann-Straussler-Scheinker disease with neurofibrillary degeneration. J Neuropathol Exp Neurol. 2010;69(8):789–800. doi: 10.1097/NEN.0b013e3181e85737. [DOI] [PubMed] [Google Scholar]
- Beck G, Kawano T, Naba I, Nishimura T, Sawada J, Hazama T. A case with a 120 base pair insertional mutation in the prion protein gene: the first case in Japan. J Neurol Neurosurg Psychiatry. 2005;76(5):756–757. doi: 10.1136/jnnp.2004.048553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck J, Collinge J, Mead S. Prion protein gene M232R variation is probably an uncommon polymorphism rather than a pathogenic mutation. Brain : a journal of neurology. 2012;135(Pt 2):e209. doi: 10.1093/brain/awr294. author reply e210. [DOI] [PubMed] [Google Scholar]
- Beck J, Pittman A, Adamson G, Campbell T, Kenny J, Houlden H, Rohrer JD, de Silva R, Shoai M, Uphill J, Poulter M, Hardy J, Mummery CJ, Warren JD, Schott JM, Fox NC, Rossor MN, Collinge J, Mead S. Validation of next-generation sequencing technologies in genetic diagnosis of dementia. Neurobiol Aging. 2014;35(1):261–265. doi: 10.1016/j.neurobiolaging.2013.07.017. [DOI] [PubMed] [Google Scholar]
- Beck JA, Mead S, Campbell TA, Dickinson A, Wientjens DP, Croes EA, Van Duijn CM, Collinge J. Two-octapeptide repeat deletion of prion protein associated with rapidly progressive dementia. Neurology. 2001;57(2):354–356. doi: 10.1212/wnl.57.2.354. [DOI] [PubMed] [Google Scholar]
- Beck JA, Poulter M, Campbell TA, Adamson G, Uphill JB, Guerreiro R, Jackson GS, Stevens JC, Manji H, Collinge J, Mead S. PRNP allelic series from 19 years of prion protein gene sequencing at the MRC Prion Unit. Hum Mutat. 2010;31(7):E1551–1563. doi: 10.1002/humu.21281. [DOI] [PubMed] [Google Scholar]
- Begue C, Martinetto H, Schultz M, Rojas E, Romero C, D’Giano C, Sevlever G, Somoza M, Taratuto AL. Creutzfeldt-Jakob disease surveillance in Argentina, 1997–2008. Neuroepidemiology. 2011;37(3–4):193–202. doi: 10.1159/000331907. [DOI] [PubMed] [Google Scholar]
- Bertoni JM, Brown P, Goldfarb LG, Rubenstein R, Gajdusek DC. Familial Creutzfeldt-Jakob disease (codon 200 mutation) with supranuclear palsy. JAMA. 1992;268(17):2413–2415. [PubMed] [Google Scholar]
- Bianca M, Bianca S, Vecchio I, Raffaele R, Ingegnosi C, Nicoletti F. Gerstmann-Straussler-Scheinker disease with P102L-V129 mutation: a case with psychiatric manifestations at onset. Annales de genetique. 2003;46(4):467–469. doi: 10.1016/s0003-3995(03)00017-0. [DOI] [PubMed] [Google Scholar]
- Bishop MT, Pennington C, Heath CA, Will RG, Knight RS. PRNP variation in UK sporadic and variant Creutzfeldt Jakob disease highlights genetic risk factors and a novel non-synonymous polymorphism. BMC Med Genet. 2009;10:146. doi: 10.1186/1471-2350-10-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boxer AL, Rabinovici GD, Kepe V, Goldman J, Furst AJ, Huang SC, Baker SL, O’Neil JP, Chui H, Geschwind MD, Small GW, Barrio JR, Jagust W, Miller BL. Amyloid imaging in distinguishing atypical prion disease from Alzheimer disease. Neurology. 2007;69(3):283–290. doi: 10.1212/01.wnl.0000265815.38958.b6. [DOI] [PubMed] [Google Scholar]
- Bratosiewicz J, Barcikowska M, Cervenakowa L, Brown P, Gajdusek DC, Liberski PP. A new point mutation of the PRNP gene in Gerstmann-Straussler-Scheinker case in Poland. Folia Neuropathol. 2000;38(4):164–166. [PubMed] [Google Scholar]
- Breithaupt M, Romero C, Kallenberg K, Begue C, Sanchez-Juan P, Eigenbrod S, Kretzschmar H, Schelzke G, Meichtry E, Taratuto A, Zerr I. Magnetic resonance imaging in E200K and V210I mutations of the prion protein gene. Alzheimer Dis Assoc Disord. 2013;27(1):87–90. doi: 10.1097/WAD.0b013e31824d578a. [DOI] [PubMed] [Google Scholar]
- Bribian A, Fontana X, Llorens F, Gavin R, Reina M, Garcia-Verdugo JM, Torres JM, de Castro F, del Rio JA. Role of the cellular prion protein in oligodendrocyte precursor cell proliferation and differentiation in the developing and adult mouse CNS. PLoS One. 2012;7(4):e33872. doi: 10.1371/journal.pone.0033872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown K, Mastrianni JA. The prion diseases. J Geriatr Psychiatry Neurol. 2010;23(4):277–298. doi: 10.1177/0891988710383576. [DOI] [PubMed] [Google Scholar]
- Brown P, Brandel JP, Preece M, Sato T. Iatrogenic Creutzfeldt-Jakob disease: the waning of an era. Neurology. 2006;67(3):389–393. doi: 10.1212/01.wnl.0000231528.65069.3f. [DOI] [PubMed] [Google Scholar]
- Brown P, Goldfarb LG, Kovanen J, Haltia M, Cathala F, Sulima M, Gibbs CJ, Jr, Gajdusek DC. Phenotypic characteristics of familial Creutzfeldt-Jakob disease associated with the codon 178Asn PRNP mutation. Ann Neurol. 1992;31(3):282–285. doi: 10.1002/ana.410310309. [DOI] [PubMed] [Google Scholar]
- Budka H, Aguzzi A, Brown P, Brucher JM, Bugiani O, Gullotta F, Haltia M, Hauw JJ, Ironside JW, Jellinger K, et al. Neuropathological diagnostic criteria for Creutzfeldt-Jakob disease (CJD) and other human spongiform encephalopathies (prion diseases) Brain Pathol. 1995;5(4):459–466. doi: 10.1111/j.1750-3639.1995.tb00625.x. [DOI] [PubMed] [Google Scholar]
- Butefisch CM, Gambetti P, Cervenakova L, Park KY, Hallett M, Goldfarb LG. Inherited prion encephalopathy associated with the novel PRNP H187R mutation: a clinical study. Neurology. 2000;55(4):517–522. doi: 10.1212/wnl.55.4.517. [DOI] [PubMed] [Google Scholar]
- Capellari S, Cardone F, Notari S, Schinina ME, Maras B, Sita D, Baruzzi A, Pocchiari M, Parchi P. Creutzfeldt-Jakob disease associated with the R208H mutation in the prion protein gene. Neurology. 2005;64(5):905–907. doi: 10.1212/01.WNL.0000152837.82388.DE. [DOI] [PubMed] [Google Scholar]
- Capellari S, Parchi P, Wolff BD, Campbell J, Atkinson R, Posey DM, Petersen RB, Gambetti P. Creutzfeldt-Jakob disease associated with a deletion of two repeats in the prion protein gene. Neurology. 2002;59(10):1628–1630. doi: 10.1212/01.wnl.0000035533.86833.28. [DOI] [PubMed] [Google Scholar]
- Capellari S, Strammiello R, Saverioni D, Kretzschmar H, Parchi P. Genetic Creutzfeldt-Jakob disease and fatal familial insomnia: insights into phenotypic variability and disease pathogenesis. Acta Neuropathol. 2011;121(1):21–37. doi: 10.1007/s00401-010-0760-4. [DOI] [PubMed] [Google Scholar]
- Cervenakova L, Buetefisch C, Lee HS, Taller I, Stone G, Gibbs CJ, Jr, Brown P, Hallett M, Goldfarb LG. Novel PRNP sequence variant associated with familial encephalopathy. Am J Med Genet. 1999;88(6):653–656. doi: 10.1002/(sici)1096-8628(19991215)88:6<653::aid-ajmg14>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- Chen C, Shi Q, Zhou W, Zhang XC, Dong JH, Hu XQ, Song XN, Liu AF, Tian C, Wang JC, Gao C, Zhang J, Han J, Dong XP. Clinical and familial characteristics of eight Chinese patients with T188K genetic Creutzfeldt-Jakob disease. Infection, genetics and evolution : journal of molecular epidemiology and evolutionary genetics in infectious diseases. 2013;14:120–124. doi: 10.1016/j.meegid.2012.11.019. [DOI] [PubMed] [Google Scholar]
- Choi BY, Kim SY, Seo SY, An SS, Kim S, Park SE, Lee SH, Choi YJ, Kim SJ, Kim CK, Park JS, Ju YR. Mutations at codons 178, 200–129, and 232 contributed to the inherited prion diseases in Korean patients. BMC Infect Dis. 2009;9:132. doi: 10.1186/1471-2334-9-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochran EJ, Bennett DA, Cervenakova L, Kenney K, Bernard B, Foster NL, Benson DF, Goldfarb LG, Brown P. Familial Creutzfeldt-Jakob disease with a five-repeat octapeptide insert mutation. Neurology. 1996;47(3):727–733. doi: 10.1212/wnl.47.3.727. [DOI] [PubMed] [Google Scholar]
- Colby DW, Prusiner SB. Prions. Cold Spring Harbor perspectives in biology. 2011;3(1):a006833. doi: 10.1101/cshperspect.a006833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins S, Boyd A, Fletcher A, Byron K, Harper C, McLean CA, Masters CL. Novel prion protein gene mutation in an octogenarian with Creutzfeldt-Jakob disease. Arch Neurol. 2000;57(7):1058–1063. doi: 10.1001/archneur.57.7.1058. [DOI] [PubMed] [Google Scholar]
- Collins S, McLean CA, Masters CL. Gerstmann-Straussler-Scheinker syndrome, fatal familial insomnia, and kuru: a review of these less common human transmissible spongiform encephalopathies. J Clin Neurosci. 2001;8(5):387–397. doi: 10.1054/jocn.2001.0919. [DOI] [PubMed] [Google Scholar]
- Colucci M, Moleres FJ, Xie ZL, Ray-Chaudhury A, Gutti S, Butefisch CM, Cervenakova L, Wang W, Goldfarb LG, Kong Q, Ghetti B, Chen SG, Gambetti P. Gerstmann-Straussler-Scheinker: a new phenotype with ‘curly’ PrP deposits. J Neuropathol Exp Neurol. 2006;65(7):642–651. doi: 10.1097/01.jnen.0000228198.81797.4d. [DOI] [PubMed] [Google Scholar]
- Cortelli P, Perani D, Montagna P, Gallassi R, Tinuper P, Provini F, Avoni P, Ferrillo F, Anchisi D, Moresco RM, Fazio F, Parchi P, Baruzzi A, Lugaresi E, Gambetti P. Pre-symptomatic diagnosis in fatal familial insomnia: serial neurophysiological and 18FDG-PET studies. Brain. 2006;129(Pt 3):668–675. doi: 10.1093/brain/awl003. [DOI] [PubMed] [Google Scholar]
- Croes EA, Theuns J, Houwing-Duistermaat JJ, Dermaut B, Sleegers K, Roks G, Van den Broeck M, van Harten B, van Swieten JC, Cruts M, Van Broeckhoven C, van Duijn CM. Octapeptide repeat insertions in the prion protein gene and early onset dementia. J Neurol Neurosurg Psychiatry. 2004;75(8):1166–1170. doi: 10.1136/jnnp.2003.020198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depaz R, Haik S, Peoc’h K, Seilhean D, Grabli D, Vicart S, Sarazin M, DeToffol B, Remy C, Fallet-Bianco C, Laplanche JL, Fontaine B, Brandel JP. Long-standing prion dementia manifesting as posterior cortical atrophy. Alzheimer Dis Assoc Disord. 2012;26(3):289–292. doi: 10.1097/WAD.0b013e318231e449. [DOI] [PubMed] [Google Scholar]
- Dermaut B, Cruts M, Backhovens H, Lubke U, Van Everbroeck B, Sciot R, Dom R, Martin JJ, Van Broeckhoven C, Cras P. Familial Creutzfeldt-Jakob disease in a patient carrying both a presenilin 1 missense substitution and a prion protein gene insertion. J Neurol. 2000;247(5):364–368. doi: 10.1007/s004150050603. [DOI] [PubMed] [Google Scholar]
- Dlouhy SR, Hsiao K, Farlow MR, Foroud T, Conneally PM, Johnson P, Prusiner SB, Hodes ME, Ghetti B. Linkage of the Indiana kindred of Gerstmann-Straussler-Scheinker disease to the prion protein gene. Nat Genet. 1992;1(1):64–67. doi: 10.1038/ng0492-64. [DOI] [PubMed] [Google Scholar]
- Duchen LW, Poulter M, Harding AE. Dementia associated with a 216 base pair insertion in the prion protein gene. Clinical and neuropathological features. Brain. 1993;116(Pt 3):555–567. doi: 10.1093/brain/116.3.555. [DOI] [PubMed] [Google Scholar]
- Farlow MR, Yee RD, Dlouhy SR, Conneally PM, Azzarelli B, Ghetti B. Gerstmann-Straussler-Scheinker disease. I. Extending the clinical spectrum. Neurology. 1989;39(11):1446–1452. doi: 10.1212/wnl.39.11.1446. [DOI] [PubMed] [Google Scholar]
- Finckh U, Muller-Thomsen T, Mann U, Eggers C, Marksteiner J, Meins W, Binetti G, Alberici A, Sonderegger P, Hock C, Nitsch RM, Gal A. High frequency of mutations in four different disease genes in early-onset dementia. Ann N Y Acad Sci. 2000;920:100–106. doi: 10.1111/j.1749-6632.2000.tb06910.x. [DOI] [PubMed] [Google Scholar]
- Flechsig E, Weissmann C. The role of PrP in health and disease. Curr Mol Med. 2004;4(4):337–353. doi: 10.2174/1566524043360645. [DOI] [PubMed] [Google Scholar]
- Fong J, Rojas JC, Bang J, Legati A, Rankin KJ, Forner S, Miller ZA, Karydas AM, Coppola G, Grouse CK, Ralph J, Miller BL, Geschwind MD. Genetic prion disease caused by PRNP Q160X mutation presenting with an orbitofrontal syndrome, cyclic diarrhea and peripheral neuropathy. J Alz Disease. 2016b doi: 10.3233/JAD-160300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forner SA, Takada L, Bettcher BM, Lobach IV, Tartaglia CM, Torres-Chae CC, Haman A, Thai J, Vitali P, Neuhaus J, Bostrom A, Miller BL, Rosen HJ, Geschwind MD. Comparing CSF biomarkers and brain MRI in the diagnosis of sporadic Creutzfeldt-Jakob disease. Neurology Clinical practice. 2015;5(2):116–125. doi: 10.1212/CPJ.0000000000000111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambetti P, Kong Q, Zou W, Parchi P, Chen SG. Sporadic and familial CJD: classification and characterisation. Br Med Bull. 2003a;66(1):213–239. doi: 10.1093/bmb/66.1.213. [DOI] [PubMed] [Google Scholar]
- Gambetti P, Parchi P, Chen SG. Hereditary Creutzfeldt-Jakob disease and fatal familial insomnia. Clinics in laboratory medicine. 2003b;23(1):43–64. doi: 10.1016/s0272-2712(02)00065-3. [DOI] [PubMed] [Google Scholar]
- Geschwind MD. Rapidly Progressive Dementia. Continuum (Minneap Minn) 2016;22(2 Dementia):510–537. doi: 10.1212/CON.0000000000000319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghetti B, Dlouhy SR, Giaccone G, Bugiani O, Frangione B, Farlow MR, Tagliavini F. Gerstmann-Straussler-Scheinker disease and the Indiana kindred. Brain Pathol. 1995;5(1):61–75. doi: 10.1111/j.1750-3639.1995.tb00578.x. [DOI] [PubMed] [Google Scholar]
- Ghetti B, Piccardo P, Spillantini MG, Ichimiya Y, Porro M, Perini F, Kitamoto T, Tateishi J, Seiler C, Frangione B, Bugiani O, Giaccone G, Prelli F, Goedert M, Dlouhy SR, Tagliavini F. Vascular variant of prion protein cerebral amyloidosis with tau-positive neurofibrillary tangles: the phenotype of the stop codon 145 mutation in PRNP. Proc Natl Acad Sci U S A. 1996;93(2):744–748. doi: 10.1073/pnas.93.2.744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giachin G, Biljan I, Ilc G, Plavec J, Legname G. Probing early misfolding events in prion protein mutants by NMR spectroscopy. Molecules. 2013;18(8):9451–9476. doi: 10.3390/molecules18089451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs CJ., Jr . Spongiform encephalopathies - slow, latent, and temperate virus infections - in retrospect. In: Prusiner SB, Collinge J, Powell J, Anderton B, editors. Prion Diseases of Humans and Animals. London: Ellis Horwood; 1992. pp. 53–62. [Google Scholar]
- Goldfarb LG, Brown P, Little BW, Cervenakova L, Kenney K, Gibbs CJ, Jr, Gajdusek DC. A new (two-repeat) octapeptide coding insert mutation in Creutzfeldt-Jakob disease. Neurology. 1993;43(11):2392–2394. doi: 10.1212/wnl.43.11.2392. [DOI] [PubMed] [Google Scholar]
- Goldfarb LG, Brown P, McCombie WR, Goldgaber D, Swergold GD, Wills PR, Cervenakova L, Baron H, Gibbs CJ, Jr, Gajdusek DC. Transmissible familial Creutzfeldt-Jakob disease associated with five, seven, and eight extra octapeptide coding repeats in the PRNP gene. Proc Natl Acad Sci U S A. 1991;88(23):10926–10930. doi: 10.1073/pnas.88.23.10926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfarb LG, Petersen RB, Tabaton M, Brown P, LeBlanc AC, Montagna P, Cortelli P, Julien J, Vital C, Pendelbury WW, et al. Fatal familial insomnia and familial Creutzfeldt-Jakob disease: disease phenotype determined by a DNA polymorphism. Science. 1992;258(5083):806–808. doi: 10.1126/science.1439789. [DOI] [PubMed] [Google Scholar]
- Goldgaber D, Goldfarb LG, Brown P, Asher DM, Brown WT, Lin S, Teener JW, Feinstone SM, Rubenstein R, Kascsak RJ, et al. Mutations in familial Creutzfeldt-Jakob disease and Gerstmann-Straussler-Scheinker’s syndrome. Exp Neurol. 1989;106(2):204–206. doi: 10.1016/0014-4886(89)90095-2. [DOI] [PubMed] [Google Scholar]
- Goldman JS. Genetic testing and counseling in the diagnosis and management of young-onset dementias. Psychiatr Clin North Am. 2015;38(2):295–308. doi: 10.1016/j.psc.2015.01.008. [DOI] [PubMed] [Google Scholar]
- Goldman JS, Miller BL, Safar J, de Tourreil S, Martindale JL, Prusiner SB, Geschwind MD. When Sporadic Disease Is Not Sporadic: The Potential for Genetic Etiology. Arch Neurol. 2004;61(2):213–216. doi: 10.1001/archneur.61.2.213. [DOI] [PubMed] [Google Scholar]
- Grasbon-Frodl E, Lorenz H, Mann U, Nitsch RM, Windl O, Kretzschmar HA. Loss of glycosylation associated with the T183A mutation in human prion disease. Acta Neuropathol. 2004a;108(6):476–484. doi: 10.1007/s00401-004-0913-4. [DOI] [PubMed] [Google Scholar]
- Grasbon-Frodl E, Schmalzbauer R, Weber P, Krebs B, Windl O, Zerr I, Kretzschmar HA. A novel three extra-repeat insertion in the prion protein gene (PRNP) in a patient with Creutzfeldt-Jakob disease. Neurogenetics. 2004b;5(4):249–250. doi: 10.1007/s10048-004-0196-x. [DOI] [PubMed] [Google Scholar]
- Guerreiro R, Bras J, Wojtas A, Rademakers R, Hardy J, Graff-Radford N. Nonsense mutation in PRNP associated with clinical Alzheimer’s disease. Neurobiol Aging. 2014;35(11):2656, e2613–2656. doi: 10.1016/j.neurobiolaging.2014.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerreiro RJ, Vaskov T, Crews C, Singleton A, Hardy J. A case of dementia with PRNP D178Ncis-129M and no insomnia. Alzheimer Dis Assoc Disord. 2009;23(4):415–417. doi: 10.1097/WAD.0b013e3181ae3a76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haik S, Marcon G, Mallet A, Tettamanti M, Welaratne A, Giaccone G, Azimi S, Pietrini V, Fabreguettes JR, Imperiale D, Cesaro P, Buffa C, Aucan C, Lucca U, Peckeu L, Suardi S, Tranchant C, Zerr I, Houillier C, Redaelli V, Vespignani H, Campanella A, Sellal F, Krasnianski A, Seilhean D, Heinemann U, Sedel F, Canovi M, Gobbi M, Di Fede G, Laplanche JL, Pocchiari M, Salmona M, Forloni G, Brandel JP, Tagliavini F. Doxycycline in Creutzfeldt-Jakob disease: a phase 2, randomised, double-blind, placebo-controlled trial. Lancet neurology. 2014;13(2):150–158. doi: 10.1016/S1474-4422(13)70307-7. [DOI] [PubMed] [Google Scholar]
- Hainfellner JA, Brantner-Inthaler S, Cervenakova L, Brown P, Kitamoto T, Tateishi J, Diringer H, Liberski PP, Regele H, Feucht M, et al. The original Gerstmann-Straussler-Scheinker family of Austria: divergent clinicopathological phenotypes but constant PrP genotype. Brain Pathol. 1995;5(3):201–211. doi: 10.1111/j.1750-3639.1995.tb00596.x. [DOI] [PubMed] [Google Scholar]
- Hall DA, Leehey MA, Filley CM, Steinbart E, Montine T, Schellenberg GD, Bosque P, Nixon R, Bird T. PRNP H187R mutation associated with neuropsychiatric disorders in childhood and dementia. Neurology. 2005;64(7):1304–1306. doi: 10.1212/01.WNL.0000156911.70131.06. [DOI] [PubMed] [Google Scholar]
- Harris DA. Trafficking, turnover and membrane topology of PrP. Br Med Bull. 2003;66:71–85. doi: 10.1093/bmb/66.1.71. [DOI] [PubMed] [Google Scholar]
- Heinemann U, Krasnianski A, Meissner B, Grasbon-Frodl EM, Kretzschmar HA, Zerr I. Novel PRNP mutation in a patient with a slow progressive dementia syndrome. Med Sci Monit. 2008;14(5):CS41–43. [PubMed] [Google Scholar]
- Higuma M, Sanjo N, Satoh K, Shiga Y, Sakai K, Nozaki I, Hamaguchi T, Nakamura Y, Kitamoto T, Shirabe S, Murayama S, Yamada M, Tateishi J, Mizusawa H. Relationships between clinicopathological features and cerebrospinal fluid biomarkers in Japanese patients with genetic prion diseases. PLoS One. 2013;8(3):e60003. doi: 10.1371/journal.pone.0060003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilton DA, Head MW, Singh VK, Bishop M, Ironside JW. Familial prion disease with a novel serine to isoleucine mutation at codon 132 of prion protein gene (PRNP) Neuropathol Appl Neurobiol. 2009;35(1):111–115. doi: 10.1111/j.1365-2990.2008.00964.x. [DOI] [PubMed] [Google Scholar]
- Hinnell C, Coulthart MB, Jansen GH, Cashman NR, Lauzon J, Clark A, Costello F, White C, Midha R, Wiebe S, Furtado S. Gerstmann-Straussler-Scheinker disease due to a novel prion protein gene mutation. Neurology. 2011;76(5):485–487. doi: 10.1212/WNL.0b013e31820a0ab2. [DOI] [PubMed] [Google Scholar]
- Hodak M, Chisnell R, Lu W, Bernholc J. Functional implications of multistage copper binding to the prion protein. Proc Natl Acad Sci U S A. 2009;106(28):11576–11581. doi: 10.1073/pnas.0903807106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao K, Baker HF, Crow TJ, Poulter M, Owen F, Terwilliger JD, Westaway D, Ott J, Prusiner SB. Linkage of a prion protein missense variant to Gerstmann-Straussler syndrome. Nature. 1989;338(6213):342–345. doi: 10.1038/338342a0. [DOI] [PubMed] [Google Scholar]
- Hsiao K, Dlouhy SR, Farlow MR, Cass C, Da Costa M, Conneally PM, Hodes ME, Ghetti B, Prusiner SB. Mutant prion proteins in Gerstmann-Straussler-Scheinker disease with neurofibrillary tangles. Nat Genet. 1992;1(1):68–71. doi: 10.1038/ng0492-68. [DOI] [PubMed] [Google Scholar]
- Hsiao K, Meiner Z, Kahana E, Cass C, Kahana I, Avrahami D, Scarlato G, Abramsky O, Prusiner SB, Gabizon R. Mutation of the prion protein in Libyan Jews with Creutzfeldt-Jakob disease. N Engl J Med. 1991a;324(16):1091–1097. doi: 10.1056/NEJM199104183241604. [DOI] [PubMed] [Google Scholar]
- Hsiao KK, Cass C, Schellenberg GD, Bird T, Devine-Gage E, Wisniewski H, Prusiner SB. A prion protein variant in a family with the telencephalic form of Gerstmann-Straussler-Scheinker syndrome. Neurology. 1991b;41(5):681–684. doi: 10.1212/wnl.41.5.681. [DOI] [PubMed] [Google Scholar]
- Jansen C, Parchi P, Capellari S, Ibrahim-Verbaas CA, Schuur M, Strammiello R, Corrado P, Bishop MT, van Gool WA, Verbeek MM, Baas F, van Saane W, Spliet WG, Jansen GH, van Duijn CM, Rozemuller AJ. Human prion diseases in the Netherlands (1998–2009): clinical, genetic and molecular aspects. PLoS One. 2012;7(4):e36333. doi: 10.1371/journal.pone.0036333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen C, Parchi P, Capellari S, Vermeij AJ, Corrado P, Baas F, Strammiello R, van Gool WA, van Swieten JC, Rozemuller AJ. Prion protein amyloidosis with divergent phenotype associated with two novel nonsense mutations in PRNP. Acta Neuropathol. 2010;119(2):189–197. doi: 10.1007/s00401-009-0609-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen C, Voet W, Head MW, Parchi P, Yull H, Verrips A, Wesseling P, Meulstee J, Baas F, van Gool WA, Ironside JW, Rozemuller AJ. A novel seven-octapeptide repeat insertion in the prion protein gene (PRNP) in a Dutch pedigree with Gerstmann-Straussler-Scheinker disease phenotype: comparison with similar cases from the literature. Acta Neuropathol. 2011;121(1):59–68. doi: 10.1007/s00401-010-0656-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaunmuktane Z, Mead S, Ellis M, Wadsworth JD, Nicoll AJ, Kenny J, Launchbury F, Linehan J, Richard-Loendt A, Walker AS, Rudge P, Collinge J, Brandner S. Evidence for human transmission of amyloid-beta pathology and cerebral amyloid angiopathy. Nature. 2015;525(7568):247–250. doi: 10.1038/nature15369. [DOI] [PubMed] [Google Scholar]
- Jayadev S, Nochlin D, Poorkaj P, Steinbart EJ, Mastrianni JA, Montine TJ, Ghetti B, Schellenberg GD, Bird TD, Leverenz JB. Familial prion disease with Alzheimer disease-like tau pathology and clinical phenotype. Ann Neurol. 2011;69(4):712–720. doi: 10.1002/ana.22264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson MD, Vnencak-Jones CL, McLean MJ. Fatal familial insomnia: clinical and pathologic heterogeneity in genetic half brothers. Neurology. 1998;51(6):1715–1717. doi: 10.1212/wnl.51.6.1715. [DOI] [PubMed] [Google Scholar]
- Jones M, Odunsi S, du Plessis D, Vincent A, Bishop M, Head MW, Ironside JW, Gow D. Gerstmann-Straussler-Scheinker disease: novel PRNP mutation and VGKC-complex antibodies. Neurology. 2014;82(23):2107–2111. doi: 10.1212/WNL.0000000000000500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaski DN, Pennington C, Beck J, Poulter M, Uphill J, Bishop MT, Linehan JM, O’Malley C, Wadsworth JD, Joiner S, Knight RS, Ironside JW, Brandner S, Collinge J, Mead S. Inherited prion disease with 4-octapeptide repeat insertion: disease requires the interaction of multiple genetic risk factors. Brain : a journal of neurology. 2011;134(Pt 6):1829–1838. doi: 10.1093/brain/awr079. [DOI] [PubMed] [Google Scholar]
- Katscher F. It’s Jakob’s disease, not Creutzfeldt’s. Nature. 1998;393(6680):11. doi: 10.1038/29862. [DOI] [PubMed] [Google Scholar]
- Kim MO, Cali I, Oehler A, Fong JC, Wong K, See T, Katz JS, Gambetti P, Bettcher BM, Dearmond SJ, Geschwind MD. Genetic CJD with a novel E200G mutation in the prion protein gene and comparison with E200K mutation cases. Acta neuropathologica communications. 2013;1(1):80. doi: 10.1186/2051-5960-1-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong Q, Surewicz WK, Petersen RB, Zou W, Chen SG, Gambetti P, Parchi P, Capellari S, Goldfarb L, Montagna P, Lugaresi E, Piccardo P, Ghetti B. Inherited prion diseases. In: Prusiner SB, editor. Prion Biology and Diseases. 2. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; 2004. pp. 673–775. [Google Scholar]
- Kotta K, Paspaltsis I, Bostantjopoulou S, Latsoudis H, Plaitakis A, Kazis D, Collinge J, Sklaviadis T. Novel mutation of the PRNP gene of a clinical CJD case. BMC Infect Dis. 2006;6:169. doi: 10.1186/1471-2334-6-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs GG, Ertsey C, Majtenyi C, Jelencsik I, Laszlo L, Flicker H, Strain L, Szirmai I, Budka H. Inherited prion disease with A117V mutation of the prion protein gene: a novel Hungarian family. J Neurol Neurosurg Psychiatry. 2001;70(6):802–805. doi: 10.1136/jnnp.70.6.802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs GG, Puopolo M, Ladogana A, Pocchiari M, Budka H, van Duijn C, Collins SJ, Boyd A, Giulivi A, Coulthart M, Delasnerie-Laupretre N, Brandel JP, Zerr I, Kretzschmar HA, de Pedro-Cuesta J, Calero-Lara M, Glatzel M, Aguzzi A, Bishop M, Knight R, Belay G, Will R, Mitrova E. Genetic prion disease: the EUROCJD experience. Human genetics. 2005;118(2):166–174. doi: 10.1007/s00439-005-0020-1. [DOI] [PubMed] [Google Scholar]
- Kovacs GG, Seguin J, Quadrio I, Hoftberger R, Kapas I, Streichenberger N, Biacabe AG, Meyronet D, Sciot R, Vandenberghe R, Majtenyi K, Laszlo L, Strobel T, Budka H, Perret-Liaudet A. Genetic Creutzfeldt-Jakob disease associated with the E200K mutation: characterization of a complex proteinopathy. Acta Neuropathol. 2011;121(1):39–57. doi: 10.1007/s00401-010-0713-y. [DOI] [PubMed] [Google Scholar]
- Kovacs GG, Trabattoni G, Hainfellner JA, Ironside JW, Knight RS, Budka H. Mutations of the prion protein gene phenotypic spectrum. J Neurol. 2002;249(11):1567–1582. doi: 10.1007/s00415-002-0896-9. [DOI] [PubMed] [Google Scholar]
- Krasemann S, Zerr I, Weber T, Poser S, Kretzschmar H, Hunsmann G, Bodemer W. Prion disease associated with a novel nine octapeptide repeat insertion in the PRNP gene. Brain Res Mol Brain Res. 1995;34(1):173–176. doi: 10.1016/0169-328x(95)00175-r. [DOI] [PubMed] [Google Scholar]
- Krasnianski A, Bartl M, Sanchez Juan PJ, Heinemann U, Meissner B, Varges D, Schulze-Sturm U, Kretzschmar HA, Schulz-Schaeffer WJ, Zerr I. Fatal familial insomnia: Clinical features and early identification. Ann Neurol. 2008;63(5):658–661. doi: 10.1002/ana.21358. [DOI] [PubMed] [Google Scholar]
- Krasnianski A, Heinemann U, Ponto C, Kortt J, Kallenberg K, Varges D, Schulz-Schaeffer WJ, Kretzschmar HA, Zerr I. Clinical findings and diagnosis in genetic prion diseases in Germany. Eur J Epidemiol. 2016;31(2):187–196. doi: 10.1007/s10654-015-0049-y. [DOI] [PubMed] [Google Scholar]
- Krebs B, Lederer RM, Windl O, Grasbon-Frodl EM, Zerr I, Kretzschmar HA. Creutzfeldt-Jakob disease associated with an R148H mutation of the prion protein gene. Neurogenetics. 2005;6(2):97–100. doi: 10.1007/s10048-004-0208-x. [DOI] [PubMed] [Google Scholar]
- Kretzschmar HA, Neumann M, Stavrou D. Codon 178 mutation of the human prion protein gene in a German family (Backer family): sequencing data from 72-year-old celloidin-embedded brain tissue. Acta Neuropathol. 1995;89(1):96–98. doi: 10.1007/BF00294264. [DOI] [PubMed] [Google Scholar]
- Kumar N, Boeve BF, Boot BP, Orr CF, Duffy J, Woodruff BK, Nair AK, Ellison J, Kuntz K, Kantarci K, Jack CR, Jr, Westmoreland BF, Fields JA, Baker M, Rademakers R, Parisi JE, Dickson DW. Clinical characterization of a kindred with a novel 12-octapeptide repeat insertion in the prion protein gene. Arch Neurol. 2011;68(9):1165–1170. doi: 10.1001/archneurol.2011.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladogana A, Almonti S, Petraroli R, Giaccaglini E, Ciarmatori C, Liu QG, Bevivino S, Squitieri F, Pocchiari M. Mutation of the PRNP gene at codon 211 in familial Creutzfeldt-Jakob disease. Am J Med Genet. 2001;103(2):133–137. doi: 10.1002/ajmg.1511. [DOI] [PubMed] [Google Scholar]
- Ladogana A, Puopolo M, Poleggi A, Almonti S, Mellina V, Equestre M, Pocchiari M. High incidence of genetic human transmissible spongiform encephalopathies in Italy. Neurology. 2005;64(9):1592–1597. doi: 10.1212/01.WNL.0000160118.26865.11. [DOI] [PubMed] [Google Scholar]
- Laplanche JL, Delasnerie-Laupretre N, Brandel JP, Dussaucy M, Chatelain J, Launay JM. Two novel insertions in the prion protein gene in patients with late-onset dementia. Hum Mol Genet. 1995;4(6):1109–1111. doi: 10.1093/hmg/4.6.1109. [DOI] [PubMed] [Google Scholar]
- Laplanche JL, Hachimi KH, Durieux I, Thuillet P, Defebvre L, Delasnerie-Laupretre N, Peoc’h K, Foncin JF, Destee A. Prominent psychiatric features and early onset in an inherited prion disease with a new insertional mutation in the prion protein gene. Brain. 1999;122(Pt 12):2375–2386. doi: 10.1093/brain/122.12.2375. [DOI] [PubMed] [Google Scholar]
- Lee HS, Sambuughin N, Cervenakova L, Chapman J, Pocchiari M, Litvak S, Qi HY, Budka H, del Ser T, Furukawa H, Brown P, Gajdusek DC, Long JC, Korczyn AD, Goldfarb LG. Ancestral origins and worldwide distribution of the PRNP 200K mutation causing familial Creutzfeldt-Jakob disease. Am J Hum Genet. 1999;64(4):1063–1070. doi: 10.1086/302340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leliveld SR, Dame RT, Wuite GJ, Stitz L, Korth C. The expanded octarepeat domain selectively binds prions and disrupts homomeric prion protein interactions. The Journal of biological chemistry. 2006;281(6):3268–3275. doi: 10.1074/jbc.M510606200. [DOI] [PubMed] [Google Scholar]
- Liu Z, Jia L, Piao Y, Lu D, Wang F, Lv H, Lu Y, Jia J. Creutzfeldt-Jakob disease with PRNP G114V mutation in a Chinese family. Acta Neurol Scand. 2010;121(6):377–383. doi: 10.1111/j.1600-0404.2009.01236.x. [DOI] [PubMed] [Google Scholar]
- Lorenz H, Windl O, Kretzschmar HA. Cellular phenotyping of secretory and nuclear prion proteins associated with inherited prion diseases. The Journal of biological chemistry. 2002;277(10):8508–8516. doi: 10.1074/jbc.M110197200. [DOI] [PubMed] [Google Scholar]
- Lugaresi E, Medori R, Montagna P, Baruzzi A, Cortelli P, Lugaresi A, Tinuper P, Zucconi M, Gambetti P. Fatal familial insomnia and dysautonomia with selective degeneration of thalamic nuclei. N Engl J Med. 1986;315(16):997–1003. doi: 10.1056/NEJM198610163151605. [DOI] [PubMed] [Google Scholar]
- MacLeod R, Tibben A, Frontali M, Evers-Kiebooms G, Jones A, Martinez-Descales A, Roos RA Editorial C, Working Group ‘Genetic Testing Counselling’ of the European Huntington Disease N. Recommendations for the predictive genetic test in Huntington’s disease. Clin Genet. 2013;83(3):221–231. doi: 10.1111/j.1399-0004.2012.01900.x. [DOI] [PubMed] [Google Scholar]
- Mallucci GR, White MD, Farmer M, Dickinson A, Khatun H, Powell AD, Brandner S, Jefferys JG, Collinge J. Targeting cellular prion protein reverses early cognitive deficits and neurophysiological dysfunction in prion-infected mice. Neuron. 2007;53(3):325–335. doi: 10.1016/j.neuron.2007.01.005. [DOI] [PubMed] [Google Scholar]
- Mano KK, Matsukawa T, Mitsui J, Ishiura H, Tokushige S, Takahashi Y, Sato NS, Nakamoto FK, Ichikawa Y, Nagashima Y, Terao Y, Shimizu J, Hamada M, Uesaka Y, Oyama G, Ogawa G, Yoshimura J, Doi K, Morishita S, Tsuji S, Goto J. Atypical parkinsonism caused by Pro105Leu mutation of prion protein: A broad clinical spectrum. Neurology Genetics. 2016;2(1):e48. doi: 10.1212/NXG.0000000000000048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masters CL. Creutzfeldt-Jakob disease: its origins. Alzheimer Dis Assoc Disord. 1989;3(1–2):46–51. doi: 10.1097/00002093-198903010-00006. [DOI] [PubMed] [Google Scholar]
- Masters CL, Harris JO, Gajdusek DC, Gibbs CJ, Jr, Bernoulli C, Asher DM. Creutzfeldt-Jakob disease: patterns of worldwide occurrence and the significance of familial and sporadic clustering. Ann Neurol. 1979;5(2):177–188. doi: 10.1002/ana.410050212. [DOI] [PubMed] [Google Scholar]
- Mastrianni JA. The genetics of prion diseases. Genet Med. 2010;12(4):187–195. doi: 10.1097/GIM.0b013e3181cd7374. [DOI] [PubMed] [Google Scholar]
- Mastrianni JA, Curtis MT, Oberholtzer JC, Da Costa MM, DeArmond S, Prusiner SB, Garbern JY. Prion disease (PrP-A117V) presenting with ataxia instead of dementia. Neurology. 1995;45(11):2042–2050. doi: 10.1212/wnl.45.11.2042. [DOI] [PubMed] [Google Scholar]
- Matej R, Kovacs GG, Johanidesova S, Keller J, Matejckova M, Novakova J, Sigut V, Keller O, Rusina R. Genetic Creutzfeldt-Jakob disease with R208H mutation presenting as progressive supranuclear palsy. Mov Disord. 2012;27(4):476–479. doi: 10.1002/mds.24002. [DOI] [PubMed] [Google Scholar]
- Matsuzono K, Ikeda Y, Liu W, Kurata T, Deguchi S, Deguchi K, Abe K. A novel familial prion disease causing pan-autonomic-sensory neuropathy and cognitive impairment. Eur J Neurol. 2013;20(5):e67–69. doi: 10.1111/ene.12089. [DOI] [PubMed] [Google Scholar]
- Mead S, Gandhi S, Beck J, Caine D, Gallujipali D, Carswell C, Hyare H, Joiner S, Ayling H, Lashley T, Linehan JM, Al-Doujaily H, Sharps B, Revesz T, Sandberg MK, Reilly MM, Koltzenburg M, Forbes A, Rudge P, Brandner S, Warren JD, Wadsworth JD, Wood NW, Holton JL, Collinge J. A novel prion disease associated with diarrhea and autonomic neuropathy. N Engl J Med. 2013;369(20):1904–1914. doi: 10.1056/NEJMoa1214747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mead S, Poulter M, Beck J, Webb TE, Campbell TA, Linehan JM, Desbruslais M, Joiner S, Wadsworth JD, King A, Lantos P, Collinge J. Inherited prion disease with six octapeptide repeat insertional mutation--molecular analysis of phenotypic heterogeneity. Brain. 2006;129(Pt 9):2297–2317. doi: 10.1093/brain/awl226. [DOI] [PubMed] [Google Scholar]
- Mead S, Uphill J, Beck J, Poulter M, Campbell T, Lowe J, Adamson G, Hummerich H, Klopp N, Ruckert IM, Wichmann HE, Azazi D, Plagnol V, Pako WH, Whitfield J, Alpers MP, Whittaker J, Balding DJ, Zerr I, Kretzschmar H, Collinge J. Genome-wide association study in multiple human prion diseases suggests genetic risk factors additional to PRNP. Human molecular genetics. 2012;21(8):1897–1906. doi: 10.1093/hmg/ddr607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mead S, Webb TE, Campbell TA, Beck J, Linehan JM, Rutherfoord S, Joiner S, Wadsworth JD, Heckmann J, Wroe S, Doey L, King A, Collinge J. Inherited prion disease with 5-OPRI: phenotype modification by repeat length and codon 129. Neurology. 2007;69(8):730–738. doi: 10.1212/01.wnl.0000267642.41594.9d. [DOI] [PubMed] [Google Scholar]
- Medori R, Tritschler HJ, LeBlanc A, Villare F, Manetto V, Chen HY, Xue R, Leal S, Montagna P, Cortelli P, et al. Fatal familial insomnia, a prion disease with a mutation at codon 178 of the prion protein gene. N Engl J Med. 1992;326(7):444–449. doi: 10.1056/NEJM199202133260704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiner Z, Gabizon R, Prusiner SB. Familial Creutzfeldt-Jakob disease. Codon 200 prion disease in Libyan Jews. Medicine (Baltimore) 1997;76(4):227–237. doi: 10.1097/00005792-199707000-00001. [DOI] [PubMed] [Google Scholar]
- Minikel EV, Vallabh SM, Lek M, Estrada K, Samocha KE, Sathirapongsasuti JF, McLean CY, Tung JY, Yu LP, Gambetti P, Blevins J, Zhang S, Cohen Y, Chen W, Yamada M, Hamaguchi T, Sanjo N, Mizusawa H, Nakamura Y, Kitamoto T, Collins SJ, Boyd A, Will RG, Knight R, Ponto C, Zerr I, Kraus TF, Eigenbrod S, Giese A, Calero M, de Pedro-Cuesta J, Haik S, Laplanche JL, Bouaziz-Amar E, Brandel JP, Capellari S, Parchi P, Poleggi A, Ladogana A, O’Donnell-Luria AH, Karczewski KJ, Marshall JL, Boehnke M, Laakso M, Mohlke KL, Kahler A, Chambert K, McCarroll S, Sullivan PF, Hultman CM, Purcell SM, Sklar P, van der Lee SJ, Rozemuller A, Jansen C, Hofman A, Kraaij R, van Rooij JG, Ikram MA, Uitterlinden AG, van Duijn CM, Daly MJ, MacArthur DG Exome Aggregation C. Quantifying prion disease penetrance using large population control cohorts. Science translational medicine. 2016;8(322):322ra329. doi: 10.1126/scitranslmed.aad5169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minikel EV, Zerr I, Collins SJ, Ponto C, Boyd A, Klug G, Karch A, Kenny J, Collinge J, Takada LT, Forner S, Fong JC, Mead S, Geschwind MD. Ascertainment bias causes false signal of anticipation in genetic prion disease. Am J Hum Genet. 2014;95(4):371–382. doi: 10.1016/j.ajhg.2014.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitrova E, Belay G. Creutzfeldt-Jakob disease with E200K mutation in Slovakia: characterization and development. Acta Virol. 2002;46(1):31–39. [PubMed] [Google Scholar]
- Moore RA, Herzog C, Errett J, Kocisko DA, Arnold KM, Hayes SF, Priola SA. Octapeptide repeat insertions increase the rate of protease-resistant prion protein formation. Protein science : a publication of the Protein Society. 2006;15(3):609–619. doi: 10.1110/ps.051822606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno JA, Halliday M, Molloy C, Radford H, Verity N, Axten JM, Ortori CA, Willis AE, Fischer PM, Barrett DA, Mallucci GR. Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Science translational medicine. 2013;5(206):206ra138. doi: 10.1126/scitranslmed.3006767. [DOI] [PubMed] [Google Scholar]
- Munoz-Nieto M, Ramonet N, Lopez-Gaston JI, Cuadrado-Corrales N, Calero O, Diaz-Hurtado M, Ipiens JR, Ramon y Cajal S, de Pedro-Cuesta J, Calero M. A novel mutation I215V in the PRNP gene associated with Creutzfeldt-Jakob and Alzheimer’s diseases in three patients with divergent clinical phenotypes. J Neurol. 2013;260(1):77–84. doi: 10.1007/s00415-012-6588-1. [DOI] [PubMed] [Google Scholar]
- Neufeld MY, Josiphov J, Korczyn AD. Demyelinating peripheral neuropathy in Creutzfeldt-Jakob disease. Muscle Nerve. 1992;15(11):1234–1239. doi: 10.1002/mus.880151103. [DOI] [PubMed] [Google Scholar]
- Nishida Y, Sodeyama N, Toru Y, Toru S, Kitamoto T, Mizusawa H. Creutzfeldt-Jakob disease with a novel insertion and codon 219 Lys/Lys polymorphism in PRNP. Neurology. 2004;63(10):1978–1979. doi: 10.1212/01.wnl.0000144196.43430.e1. [DOI] [PubMed] [Google Scholar]
- Nitrini R, Rosemberg S, Passos-Bueno MR, da Silva LS, Iughetti P, Papadopoulos M, Carrilho PM, Caramelli P, Albrecht S, Zatz M, LeBlanc A. Familial spongiform encephalopathy associated with a novel prion protein gene mutation. Ann Neurol. 1997;42(2):138–146. doi: 10.1002/ana.410420203. [DOI] [PubMed] [Google Scholar]
- Nozaki I, Hamaguchi T, Sanjo N, Noguchi-Shinohara M, Sakai K, Nakamura Y, Sato T, Kitamoto T, Mizusawa H, Moriwaka F, Shiga Y, Kuroiwa Y, Nishizawa M, Kuzuhara S, Inuzuka T, Takeda M, Kuroda S, Abe K, Murai H, Murayama S, Tateishi J, Takumi I, Shirabe S, Harada M, Sadakane A, Yamada M. Prospective 10-year surveillance of human prion diseases in Japan. Brain. 2010;133(10):3043–3057. doi: 10.1093/brain/awq216. [DOI] [PubMed] [Google Scholar]
- Owen F, Poulter M, Collinge J, Leach M, Lofthouse R, Crow TJ, Harding AE. A dementing illness associated with a novel insertion in the prion protein gene. Brain Res Mol Brain Res. 1992;13(1–2):155–157. doi: 10.1016/0169-328x(92)90056-h. [DOI] [PubMed] [Google Scholar]
- Owen F, Poulter M, Lofthouse R, Collinge J, Crow TJ, Risby D, Baker HF, Ridley RM, Hsiao K, Prusiner SB. Insertion in prion protein gene in familial Creutzfeldt-Jakob disease. Lancet. 1989;1(8628):51–52. doi: 10.1016/s0140-6736(89)91713-3. [DOI] [PubMed] [Google Scholar]
- Owen F, Poulter M, Shah T, Collinge J, Lofthouse R, Baker H, Ridley R, McVey J, Crow TJ. An in-frame insertion in the prion protein gene in familial Creutzfeldt-Jakob disease. Brain Res Mol Brain Res. 1990;7(3):273–276. doi: 10.1016/0169-328x(90)90038-f. [DOI] [PubMed] [Google Scholar]
- Palmer MS, Dryden AJ, Hughes JT, Collinge J. Homozygous prion protein genotype predisposes to sporadic Creutzfeldt–Jakob disease. Nature. 1991;352(6333):340–342. doi: 10.1038/352340a0. [DOI] [PubMed] [Google Scholar]
- Pan KM, Baldwin M, Nguyen J, Gasset M, Serban A, Groth D, Mehlhorn I, Huang Z, Fletterick RJ, Cohen FE, et al. Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proc Natl Acad Sci U S A. 1993;90(23):10962–10966. doi: 10.1073/pnas.90.23.10962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panegyres PK, Toufexis K, Kakulas BA, Cernevakova L, Brown P, Ghetti B, Piccardo P, Dlouhy SR. A new PRNP mutation (G131V) associated with Gerstmann-Straussler-Scheinker disease. Arch Neurol. 2001;58(11):1899–1902. doi: 10.1001/archneur.58.11.1899. [DOI] [PubMed] [Google Scholar]
- Parchi P, Castellani R, Capellari S, Ghetti B, Young K, Chen SG, Farlow M, Dickson DW, Sima AA, Trojanowski JQ, Petersen RB, Gambetti P. Molecular basis of phenotypic variability in sporadic Creutzfeldt-Jakob disease. Ann Neurol. 1996;39(6):767–778. doi: 10.1002/ana.410390613. [DOI] [PubMed] [Google Scholar]
- Parchi P, Castellani R, Cortelli P, Montagna P, Chen SG, Petersen RB, Manetto V, Vnencak-Jones CL, McLean MJ, Sheller JR, et al. Regional distribution of protease-resistant prion protein in fatal familial insomnia. Ann Neurol. 1995;38(1):21–29. doi: 10.1002/ana.410380107. [DOI] [PubMed] [Google Scholar]
- Parchi P, Giese A, Capellari S, Brown P, Schulz-Schaeffer W, Windl O, Zerr I, Budka H, Kopp N, Piccardo P, Poser S, Rojiani A, Streichemberger N, Julien J, Vital C, Ghetti B, Gambetti P, Kretzschmar H. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol. 1999;46(2):224–233. [PubMed] [Google Scholar]
- Parchi P, Strammiello R, Notari S, Giese A, Langeveld JP, Ladogana A, Zerr I, Roncaroli F, Cras P, Ghetti B, Pocchiari M, Kretzschmar H, Capellari S. Incidence and spectrum of sporadic Creutzfeldt-Jakob disease variants with mixed phenotype and co-occurrence of PrPSc types: an updated classification. Acta Neuropathol. 2009;118(5):659–671. doi: 10.1007/s00401-009-0585-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastore M, Chin SS, Bell KL, Dong Z, Yang Q, Yang L, Yuan J, Chen SG, Gambetti P, Zou WQ. Creutzfeldt-Jakob disease (CJD) with a mutation at codon 148 of prion protein gene: relationship with sporadic CJD. Am J Pathol. 2005;167(6):1729–1738. doi: 10.1016/S0002-9440(10)61254-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peoc’h K, Levavasseur E, Delmont E, De Simone A, L-PI, Privat N, Chebaro Y, CC, Bedoucha P, Brandel JP, Laquerriere A, Kemeny JL, Hauw JJ, Borg M, RH, Derreumaux P, Laplanche JL, Haïk S. Substitutions at residue 211 in the prion protein drive a switch between CJD & GSS syndrome, a new mechanism governing inherited neurodegenerative disorders. Hum Mol Genet. 2012;21(26):5417–5428. doi: 10.1093/hmg/dds377. [DOI] [PubMed] [Google Scholar]
- Peoc’h K, Manivet P, Beaudry P, Attane F, Besson G, Hannequin D, Delasnerie-Laupretre N, Laplanche JL. Identification of three novel mutations (E196K, V203I, E211Q) in the prion protein gene (PRNP) in inherited prion diseases with Creutzfeldt-Jakob disease phenotype. Hum Mutat. 2000;15(5):482. doi: 10.1002/(SICI)1098-1004(200005)15:5<482::AID-HUMU16>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Piccardo P, Dlouhy SR, Lievens PM, Young K, Bird TD, Nochlin D, Dickson DW, Vinters HV, Zimmerman TR, Mackenzie IR, Kish SJ, Ang LC, De Carli C, Pocchiari M, Brown P, Gibbs CJ, Jr, Gajdusek DC, Bugiani O, Ironside J, Tagliavini F, Ghetti B. Phenotypic variability of Gerstmann-Straussler-Scheinker disease is associated with prion protein heterogeneity. J Neuropathol Exp Neurol. 1998;57(10):979–988. doi: 10.1097/00005072-199810000-00010. [DOI] [PubMed] [Google Scholar]
- Pocchiari M, Poleggi A, Puopolo M, D’Alessandro M, Tiple D, Ladogana A. Age at Death of Creutzfeldt-Jakob disease in subsequent family generation carrying the E200K mutation of the prion protein gene. PLoS One. 2013;8(4):e60376. doi: 10.1371/journal.pone.0060376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymenidou M, Prokop S, Jung HH, Hewer E, Peretz D, Moos R, Tolnay M, Aguzzi A. Atypical prion protein conformation in familial prion disease with PRNP P105T mutation. Brain Pathol. 2011;21(2):209–214. doi: 10.1111/j.1750-3639.2010.00439.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priola SA, Chesebro B. Abnormal properties of prion protein with insertional mutations in different cell types. The Journal of biological chemistry. 1998;273(19):11980–11985. doi: 10.1074/jbc.273.19.11980. [DOI] [PubMed] [Google Scholar]
- Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216(4542):136–144. doi: 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- Prusiner SB. Prions. Proc Natl Acad Sci U S A. 1998;95(23):13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusiner SB. Cell biology. A unifying role for prions in neurodegenerative diseases. Science. 2012;336(6088):1511–1513. doi: 10.1126/science.1222951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusiner SB. Biology and genetics of prions causing neurodegeneration. Annu Rev Genet. 2013;47:601–623. doi: 10.1146/annurev-genet-110711-155524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puoti G, Bizzi A, Forloni G, Safar JG, Tagliavini F, Gambetti P. Sporadic human prion diseases: molecular insights and diagnosis. Lancet Neurol. 2012;11(7):618–628. doi: 10.1016/S1474-4422(12)70063-7. [DOI] [PubMed] [Google Scholar]
- Puoti G, Rossi G, Giaccone G, Awan T, Lievens PM, Defanti CA, Tagliavini F, Bugiani O. Polymorphism at codon 129 of PRNP affects the phenotypic expression of Creutzfeldt-Jakob disease linked to E200K mutation. Ann Neurol. 2000;48(2):269–270. [PubMed] [Google Scholar]
- Qina T, Sanjo N, Hizume M, Higuma M, Tomita M, Atarashi R, Satoh K, Nozaki I, Hamaguchi T, Nakamura Y, Kobayashi A, Kitamoto T, Murayama S, Murai H, Yamada M, Mizusawa H. Clinical features of genetic Creutzfeldt-Jakob disease with V180I mutation in the prion protein gene. BMJ open. 2014;4(5):e004968. doi: 10.1136/bmjopen-2014-004968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovici GD, Wang PN, Levin J, Cook L, Pravdin M, Davis J, DeArmond SJ, Barbaro NM, Martindale J, Miller BL, Geschwind MD. First symptom in sporadic Creutzfeldt-Jakob disease. Neurology. 2006;66(2):286–287. doi: 10.1212/01.wnl.0000196440.00297.67. [DOI] [PubMed] [Google Scholar]
- Reder AT, Mednick AS, Brown P, Spire JP, Van Cauter E, Wollmann RL, Cervenakova L, Goldfarb LG, Garay A, Ovsiew F, et al. Clinical and genetic studies of fatal familial insomnia. Neurology. 1995;45(6):1068–1075. doi: 10.1212/wnl.45.6.1068. [DOI] [PubMed] [Google Scholar]
- Rodriguez MM, Peoc’h K, Haik S, Bouchet C, Vernengo L, Manana G, Salamano R, Carrasco L, Lenne M, Beaudry P, Launay JM, Laplanche JL. A novel mutation (G114V) in the prion protein gene in a family with inherited prion disease. Neurology. 2005;64(8):1455–1457. doi: 10.1212/01.WNL.0000158618.39527.93. [DOI] [PubMed] [Google Scholar]
- Roeber S, Grasbon-Frodl EM, Windl O, Krebs B, Xiang W, Vollmert C, Illig T, Schroter A, Arzberger T, Weber P, Zerr I, Kretzschmar HA. Evidence for a pathogenic role of different mutations at codon 188 of PRNP. PLoS ONE. 2008;3(5):e2147. doi: 10.1371/journal.pone.0002147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roeber S, Krebs B, Neumann M, Windl O, Zerr I, Grasbon-Frodl EM, Kretzschmar HA. Creutzfeldt-Jakob disease in a patient with an R208H mutation of the prion protein gene (PRNP) and a 17-kDa prion protein fragment. Acta Neuropathol. 2005;109(4):443–448. doi: 10.1007/s00401-004-0978-0. [DOI] [PubMed] [Google Scholar]
- Rogaeva E, Zadikoff C, Ponesse J, Schmitt-Ulms G, Kawarai T, Sato C, Salehi-Rad S, St George-Hyslop P, Lang AE. Childhood onset in familial prion disease with a novel mutation in the PRNP gene. Arch Neurol. 2006;63(7):1016–1021. doi: 10.1001/archneur.63.7.1016. [DOI] [PubMed] [Google Scholar]
- Rosenmann H, Kahana E, Korczyn AD, Kahana I, Chapman J, Gabizon R. Preliminary evidence for anticipation in genetic E200K Creutzfeldt-Jakob disease. Neurology. 1999;53(6):1328–1329. doi: 10.1212/wnl.53.6.1328. [DOI] [PubMed] [Google Scholar]
- Rowe DB, Lewis V, Needham M, Rodriguez M, Boyd A, McLean C, Roberts H, Masters CL, Collins SJ. Novel prion protein gene mutation presenting with subacute PSP-like syndrome. Neurology. 2007;68(11):868–870. doi: 10.1212/01.wnl.0000256819.61531.98. [DOI] [PubMed] [Google Scholar]
- Sanchez-Valle R, Yague J, Turon A, Arostegui JI, Nos C, Rey MJ, Ferrer I, Gelpi E. Inherited prion disease with 4-octapeptide repeat insertion linked to valine at codon 129. Brain. 2012;135(Pt 4):e212. doi: 10.1093/brain/awr358. [DOI] [PubMed] [Google Scholar]
- Sano K, Satoh K, Atarashi R, Takashima H, Iwasaki Y, Yoshida M, Sanjo N, Murai H, Mizusawa H, Schmitz M, Zerr I, Kim YS, Nishida N. Early detection of abnormal prion protein in genetic human prion diseases now possible using real-time QUIC assay. PLoS One. 2013;8(1):e54915. doi: 10.1371/journal.pone.0054915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schelzke G, Eigenbrod S, Romero C, Varges D, Breithaupt M, Taratuto AL, Kretzschmar HA, Zerr I. Genetic prion disease with codon 196 PRNP mutation: clinical and pathological findings. Neurobiology of aging. 2011;32(4):756, e751–759. doi: 10.1016/j.neurobiolaging.2010.11.023. [DOI] [PubMed] [Google Scholar]
- Schmitz M, Dittmar K, Llorens F, Gelpi E, Ferrer I, Schulz-Schaeffer WJ, Zerr I. Hereditary Human Prion Diseases: an Update. Mol Neurobiol. 2016 doi: 10.1007/s12035-016-9918-y. [DOI] [PubMed] [Google Scholar]
- Shi Q, Zhou W, Chen C, Zhang BY, Xiao K, Zhang XC, Shen XJ, Li Q, Deng LQ, Dong JH, Lin WQ, Huang P, Jiang WJ, Lv J, Han J, Dong XP. The Features of Genetic Prion Diseases Based on Chinese Surveillance Program. PLoS One. 2015;10(10):e0139552. doi: 10.1371/journal.pone.0139552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiga Y, Satoh K, Kitamoto T, Kanno S, Nakashima I, Sato S, Fujihara K, Takata H, Nobukuni K, Kuroda S, Takano H, Umeda Y, Konno H, Nagasato K, Satoh A, Matsuda Y, Hidaka M, Takahashi H, Sano Y, Kim K, Konishi T, Doh-ura K, Sato T, Sasaki K, Nakamura Y, Yamada M, Mizusawa H, Itoyama Y. Two different clinical phenotypes of Creutzfeldt-Jakob disease with a M232R substitution. J Neurol. 2007;254(11):1509–1517. doi: 10.1007/s00415-007-0540-9. [DOI] [PubMed] [Google Scholar]
- Simpson M, Johanssen V, Boyd A, et al. Unusual clinical and molecular-pathological profile of gerstmann-sträussler-scheinker disease associated with a novel prnp mutation (V176G) JAMA Neurology. 2013;70(9):1180–1185. doi: 10.1001/jamaneurol.2013.165. [DOI] [PubMed] [Google Scholar]
- Skworc KH, Windl O, Schulz-Schaeffer WJ, Giese A, Bergk J, Nagele A, Vieregge P, Zerr II, Poser S, Kretzschmar HA. Familial Creutzfeldt-Jakob disease with a novel 120-bp insertion in the prion protein gene. Ann Neurol. 1999;46(5):693–700. [PubMed] [Google Scholar]
- Soto C. Prion hypothesis: the end of the controversy? Trends Biochem Sci. 2011;36(3):151–158. doi: 10.1016/j.tibs.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spudich S, Mastrianni JA, Wrensch M, Gabizon R, Meiner Z, Kahana I, Rosenmann H, Kahana E, Prusiner SB. Complete penetrance of Creutzfeldt-Jakob disease in Libyan Jews carrying the E200K mutation in the prion protein gene. Mol Med. 1995;1(6):607–613. [PMC free article] [PubMed] [Google Scholar]
- Stevens DJ, Walter ED, Rodriguez A, Draper D, Davies P, Brown DR, Millhauser GL. Early onset prion disease from octarepeat expansion correlates with copper binding properties. PLoS Pathog. 2009;5(4):e1000390. doi: 10.1371/journal.ppat.1000390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniwaki Y, Hara H, Doh-Ura K, Murakami I, Tashiro H, Yamasaki T, Shigeto H, Arakawa K, Araki E, Yamada T, Iwaki T, Kira J. Familial Creutzfeldt-Jakob disease with D178N-129M mutation of PRNP presenting as cerebellar ataxia without insomnia. J Neurol Neurosurg Psychiatry. 2000;68(3):388. doi: 10.1136/jnnp.68.3.388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tartaglia MC, Thai JN, See T, Kuo A, Harbaugh R, Raudabaugh B, Cali I, Sattavat M, Sanchez H, DeArmond SJ, Geschwind MD. Pathologic evidence that the T188R mutation in PRNP is associated with prion disease. J Neuropathol Exp Neurol. 2010;69(12):1220–1227. doi: 10.1097/NEN.0b013e3181ffc39c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tateishi J, Kitamoto T, Doh-ura K, Sakaki Y, Steinmetz G, Tranchant C, Warter JM, Heldt N. Immunochemical, molecular genetic, and transmission studies on a case of Gerstmann-Straussler-Scheinker syndrome. Neurology. 1990;40(10):1578–1581. doi: 10.1212/wnl.40.10.1578. [DOI] [PubMed] [Google Scholar]
- Tousseyn T, Bajsarowicz K, Sanchez H, Gheyara A, Oehler A, Geschwind M, DeArmond B, DeArmond SJ. Prion Disease Induces Alzheimer Disease-Like Neuropathologic Changes. J Neuropathol Exp Neurol. 2015;74(9):873–888. doi: 10.1097/NEN.0000000000000228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tranchant C, Doh-ura K, Warter JM, Steinmetz G, Chevalier Y, Hanauer A, Kitamoto T, Tateishi J. Gerstmann-Straussler-Scheinker disease in an Alsatian family: clinical and genetic studies. J Neurol Neurosurg Psychiatry. 1992;55(3):185–187. doi: 10.1136/jnnp.55.3.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tunnell E, Wollman R, Mallik S, Cortes CJ, Dearmond SJ, Mastrianni JA. A novel PRNP-P105S mutation associated with atypical prion disease and a rare PrPSc conformation. Neurology. 2008;71(18):1431–1438. doi: 10.1212/01.wnl.0000330237.94742.fa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uflacker A, Doraiswamy PM, Rechitsky S, See T, Geschwind M, Tur-Kaspa I. Preimplantation genetic diagnosis (PGD) for genetic prion disorder due to F198S mutation in the PRNP gene. JAMA Neurol. 2014;71(4):484–486. doi: 10.1001/jamaneurol.2013.5884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UK National CJD Surveillance Unit. Variant Creutzfeldt-Jakob disease Worldwide Current Data. Edinburgh: Western General Hospital; 2015. Apr, [Google Scholar]
- van der Kamp MW, Daggett V. The consequences of pathogenic mutations to the human prion protein. Protein engineering, design & selection : PEDS. 2009;22(8):461–468. doi: 10.1093/protein/gzp039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Harten B, van Gool WA, Van Langen IM, Deekman JM, Meijerink PH, Weinstein HC. A new mutation in the prion protein gene: a patient with dementia and white matter changes. Neurology. 2000;55(7):1055–1057. doi: 10.1212/wnl.55.7.1055. [DOI] [PubMed] [Google Scholar]
- Vita MG, Gaudino S, Di Giuda D, Sauchelli D, Alboini PE, Gangemi E, Bizzarro A, Scaricamazza E, Capellari S, Parchi P, Masullo C. R208H-129VV haplotype in the prion protein gene: phenotype and neuroimaging of a patient with genetic Creutzfeldt-Jakob disease. J Neurol. 2013;260(10):2650–2652. doi: 10.1007/s00415-013-7078-9. [DOI] [PubMed] [Google Scholar]
- Vital C, Gray F, Vital A, Parchi P, Capellari S, Petersen RB, Ferrer X, Jarnier D, Julien J, Gambetti P. Prion encephalopathy with insertion of octapeptide repeats: the number of repeats determines the type of cerebellar deposits. Neuropathol Appl Neurobiol. 1998;24(2):125–130. doi: 10.1046/j.1365-2990.1998.00098.x. [DOI] [PubMed] [Google Scholar]
- Vitali P, Maccagnano E, Caverzasi E, Henry RG, Haman A, Torres-Chae C, Johnson DY, Miller BL, Geschwind MD. Diffusion-weighted MRI hyperintensity patterns differentiate CJD from other rapid dementias. Neurology. 2011;76(20):1711–1719. doi: 10.1212/WNL.0b013e31821a4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts JC, Giles K, Serban A, Patel S, Oehler A, Bhardwaj S, Guan S, Greicius MD, Miller BL, DeArmond SJ, Geschwind MD, Prusiner SB. Modulation of Creutzfeldt-Jakob disease prion propagation by the A224V mutation. Ann Neurol. 2015;78(4):540–553. doi: 10.1002/ana.24463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts JC, Westaway D. The prion protein family: diversity, rivalry, and dysfunction. Biochimica et biophysica acta. 2007;1772(6):654–672. doi: 10.1016/j.bbadis.2007.05.001. [DOI] [PubMed] [Google Scholar]
- Webb TE, Poulter M, Beck J, Uphill J, Adamson G, Campbell T, Linehan J, Powell C, Brandner S, Pal S, Siddique D, Wadsworth JD, Joiner S, Alner K, Petersen C, Hampson S, Rhymes C, Treacy C, Storey E, Geschwind MD, Nemeth AH, Wroe S, Collinge J, Mead S. Phenotypic heterogeneity and genetic modification of P102L inherited prion disease in an international series. Brain. 2008;131(Pt 10):2632–2646. doi: 10.1093/brain/awn202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windl O, Giese A, Schulz-Schaeffer W, Zerr I, Skworc K, Arendt S, Oberdieck C, Bodemer M, Poser S, Kretzschmar HA. Molecular genetics of human prion diseases in Germany. Human genetics. 1999;105(3):244–252. doi: 10.1007/s004399900124. [DOI] [PubMed] [Google Scholar]
- Woerman AL, Stohr J, Aoyagi A, Rampersaud R, Krejciova Z, Watts JC, Ohyama T, Patel S, Widjaja K, Oehler A, Sanders DW, Diamond MI, Seeley WW, Middleton LT, Gentleman SM, Mordes DA, Sudhof TC, Giles K, Prusiner SB. Propagation of prions causing synucleinopathies in cultured cells. Proceedings of the National Academy of Sciences of the United States of America. 2015;112(35):E4949–4958. doi: 10.1073/pnas.1513426112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woulfe J, Kertesz A, Frohn I, Bauer S, George-Hyslop PS, Bergeron C. Gerstmann-Straussler-Scheinker disease with the Q217R mutation mimicking frontotemporal dementia. Acta Neuropathol. 2005;110(3):317–319. doi: 10.1007/s00401-005-1054-0. [DOI] [PubMed] [Google Scholar]
- Yang TI, Jung DS, Ahn BY, Jeong BH, Cho HJ, Kim YS, Na DL, Geschwind MD, Kim EJ. Familial Creutzfeldt-Jakob disease with V180I mutation. Journal of Korean medical science. 2010;25(7):1097–1100. doi: 10.3346/jkms.2010.25.7.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Han J, Shi Q, Zhang BY, Wang GR, Tian C, Gao C, Chen JM, Li CJ, Liu Z, Li XZ, Zhang LZ, Dong XP. Human prion disease with a G114V mutation and epidemiological studies in a Chinese family: a case series. Journal of medical case reports. 2008;2:331. doi: 10.1186/1752-1947-2-331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu SL, Jin L, Sy MS, Mei FH, Kang SL, Sun GH, Tien P, Wang FS, Xiao GF. Polymorphisms of the PRNP gene in Chinese populations and the identification of a novel insertion mutation. Eur J Hum Genet. 2004;12(10):867–870. doi: 10.1038/sj.ejhg.5201245. [DOI] [PubMed] [Google Scholar]
- Yusa S, Oliveira-Martins JB, Sugita-Konishi Y, Kikuchi Y. Cellular prion protein: from physiology to pathology. Viruses. 2012;4(11):3109–3131. doi: 10.3390/v4113109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahn R, Liu A, Luhrs T, Riek R, von Schroetter C, Lopez Garcia F, Billeter M, Calzolai L, Wider G, Wuthrich K. NMR solution structure of the human prion protein. Proc Natl Acad Sci U S A. 2000;97(1):145–150. doi: 10.1073/pnas.97.1.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarranz JJ, Digon A, Atares B, Rodriguez-Martinez AB, Arce A, Carrera N, Fernandez-Manchola I, Fernandez-Martinez M, Fernandez-Maiztegui C, Forcadas I, Galdos L, Gomez-Esteban JC, Ibanez A, Lezcano E, Lopez de Munain A, Marti-Masso JF, Mendibe MM, Urtasun M, Uterga JM, Saracibar N, Velasco F, de Pancorbo MM. Phenotypic variability in familial prion diseases due to the D178N mutation. J Neurol Neurosurg Psychiatry. 2005;76(11):1491–1496. doi: 10.1136/jnnp.2004.056606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerr I, Giese A, Windl O, Kropp S, Schulz-Schaeffer W, Riedemann C, Skworc K, Bodemer M, Kretzschmar HA, Poser S. Phenotypic variability in fatal familial insomnia (D178N-129M) genotype. Neurology. 1998;51(5):1398–1405. doi: 10.1212/wnl.51.5.1398. [DOI] [PubMed] [Google Scholar]
- Zheng L, Longfei J, Jing Y, Xinqing Z, Haiqing S, Haiyan L, Fen W, Xiumin D, Jianping J. PRNP mutations in a series of apparently sporadic neurodegenerative dementias in China. American journal of medical genetics Part B, Neuropsychiatric genetics : the official publication of the International Society of Psychiatric Genetics. 2008;147B(6):938–944. doi: 10.1002/ajmg.b.30761. [DOI] [PubMed] [Google Scholar]
- Zou WQ, Puoti G, Xiao X, Yuan J, Qing L, Cali I, Shimoji M, Langeveld JP, Castellani R, Notari S, Crain B, Schmidt RE, Geschwind M, Dearmond SJ, Cairns NJ, Dickson D, Honig L, Torres JM, Mastrianni J, Capellari S, Giaccone G, Belay ED, Schonberger LB, Cohen M, Perry G, Kong Q, Parchi P, Tagliavini F, Gambetti P. Variably protease-sensitive prionopathy: a new sporadic disease of the prion protein. Ann Neurol. 2010;68(2):162–172. doi: 10.1002/ana.22094. [DOI] [PMC free article] [PubMed] [Google Scholar]