

Supplemental Digital Content is available in the text.
Keywords: endothelial cell, glycolysis, inflammation, lipoprotein(a), metabolism
Abstract
Rationale:
Patients with elevated levels of lipoprotein(a) [Lp(a)] are hallmarked by increased metabolic activity in the arterial wall on positron emission tomography/computed tomography, indicative of a proinflammatory state.
Objective:
We hypothesized that Lp(a) induces endothelial cell inflammation by rewiring endothelial metabolism.
Methods and Results:
We evaluated the impact of Lp(a) on the endothelium and describe that Lp(a), through its oxidized phospholipid content, activates arterial endothelial cells, facilitating increased transendothelial migration of monocytes. Transcriptome analysis of Lp(a)-stimulated human arterial endothelial cells revealed upregulation of inflammatory pathways comprising monocyte adhesion and migration, coinciding with increased 6-phophofructo-2-kinase/fructose-2,6-biphosphatase (PFKFB)-3–mediated glycolysis. ICAM (intercellular adhesion molecule)-1 and PFKFB3 were also found to be upregulated in carotid plaques of patients with elevated levels of Lp(a). Inhibition of PFKFB3 abolished the inflammatory signature with concomitant attenuation of transendothelial migration.
Conclusions:
Collectively, our findings show that Lp(a) activates the endothelium by enhancing PFKFB3-mediated glycolysis, leading to a proadhesive state, which can be reversed by inhibition of glycolysis. These findings pave the way for therapeutic agents targeting metabolism aimed at reducing inflammation in patients with cardiovascular disease.
Editorial, see p 1360
Meet the First Author, see p 1322
Lipoprotein(a) [Lp(a)] is an LDL (low-density lipoprotein)-like particle characterized by covalently bound apo(a) (apolipoprotein(a)) to apoB (apolipoprotein B-100) of LDL. On an equimolar basis, Lp(a) is considered more atherogenic than LDL due to the presence of phosphocholine containing oxidized phospholipids (OxPLs) bound to apo(a).1,2 OxPLs are recognized as endogenous danger-associated molecular patterns.3 Bound to Lp(a), OxPLs are capable of activating circulating monocytes, rendering them highly inflammatory leading to enhanced monocyte transendothelial migration (TEM).4 Lp(a) elevation (≥50 mg/dL, >125 nmol/L) is a highly prevalent condition5,6 that is associated with a 2- to 4-fold increase in cardiovascular morbidity and mortality.7,8 Lp(a) levels are inversely correlated to the number of kringle repeats found in the apo(a) entity of Lp(a).9,10 The mechanisms by which Lp(a) mediates enhanced arterial inflammation and accelerated atherogenesis may comprise accumulation of Lp(a) in atherosclerotic plaques, enhanced thrombogenic potential, and proinflammatory effects from its content of OxPLs.2 Whereas we and others have focused on the interaction of Lp(a) with immune cells and coagulation,4,11 functional data on the impact of Lp(a) on the endothelium—the first line of defense against atherosclerosis—remain scarce.12–18
In the context of atherosclerosis, it became evident that monocytes rewire their metabolism as a common mechanism required to provide energy and building blocks for inflammatory reactions.4,19–21 In contrast, the impact of Lp(a) on endothelial cells (ECs) with respect to changes in metabolism has not been studied. Since tumor angiogenesis has been found to coincide with EC metabolic alterations,22 we hypothesize that Lp(a) induces EC activation fueled by metabolic alterations to sustain a proinflammatory state.
In the present study, we reveal a novel role of Lp(a) and in particular, Lp(a)-bound OxPLs in driving EC inflammation. Targeting these Lp(a)-induced endothelial metabolic alterations provides a fruitful strategy to reverse EC inflammation, which may eventually help to reduce the atherogenic risk in patients.
Methods
A detailed description of the Methods is available in the Data Supplement.
The RNA sequencing data generated in this study are available at the National Center for Biotechnology Information Gene Expression Omnibus database under accession No. GSE145898.
Results
Lp(a) Elicits a Proinflammatory Response in ECs
To determine whether Lp(a) elicits EC activation leading to increased transmigration of monocytes, we stimulated human aortic ECs for 18 hours with a physiological relevant concentration of 100 mg/dL of Lp(a). Subsequently, healthy monocytes were added to ECs incubated with Lp(a) [Lp(a)-ECs] or unstimulated ECs. The rate of adhesion of monocytes to the endothelium doubled in Lp(a)-ECs compared with control ECs (Figure 1A and 1B), with a concomitant 5-fold increase in TEM of monocytes in the Lp(a)-EC condition (Figure 1A and 1C). To visualize transcriptional changes in Lp(a)-ECs, we incubated human aortic ECs with either low (5 mg/dL) or high Lp(a) (100 mg/dL) and performed RNA sequencing. Heat map analysis of Lp(a)-ECs upregulated transcripts revealed an activated EC phenotype, with clustering of genes involved in leukocyte chemotaxis and migration (Figure 1D; Figure IA in the Data Supplement). Furthermore, 2 distinct groups were separated by principle component analysis (Figure IB in the Data Supplement). Volcano plot analysis showed 270 differentially expressed genes (134 genes upregulated and 136 genes downregulated; Figure IC in the Data Supplement). Validation of key molecules involved in TEM (Figure 1E) by targeted quantitative polymerase chain reaction confirmed significantly increased expression of SELE, ICAM1, and VCAM1 in Lp(a)-ECs (Figure 1F). In addition, expression of MCP1 (monocyte chemoattractant protein 1) showed a 3-fold increase; cytokines IL6 and IL8 revealed a 2- and 20-fold increase, respectively (Figure 1G). Functionally, this resulted in increased secretion of IL (interleukin)-6 and IL-8 (Figure 1H) and a 3.5-fold increase in ICAM (intercellular adhesion molecule)-1 protein (Figure 1I). ICAM-1 is essential for efficient monocyte TEM as a knockdown of ICAM-1 in human aortic ECs led to decreased TEM of monocytes (Figure ID in the Data Supplement). Secretion of MCP-1, IL-6, and IL-8 significantly increased over time as well (Figure IE in the Data Supplement). In addition, the observed effects of Lp(a) were independent of factors present in human serum (Figure IIA in the Data Supplement). Together these data show that Lp(a) induces a proinflammatory signature of ECs.
Figure 1.
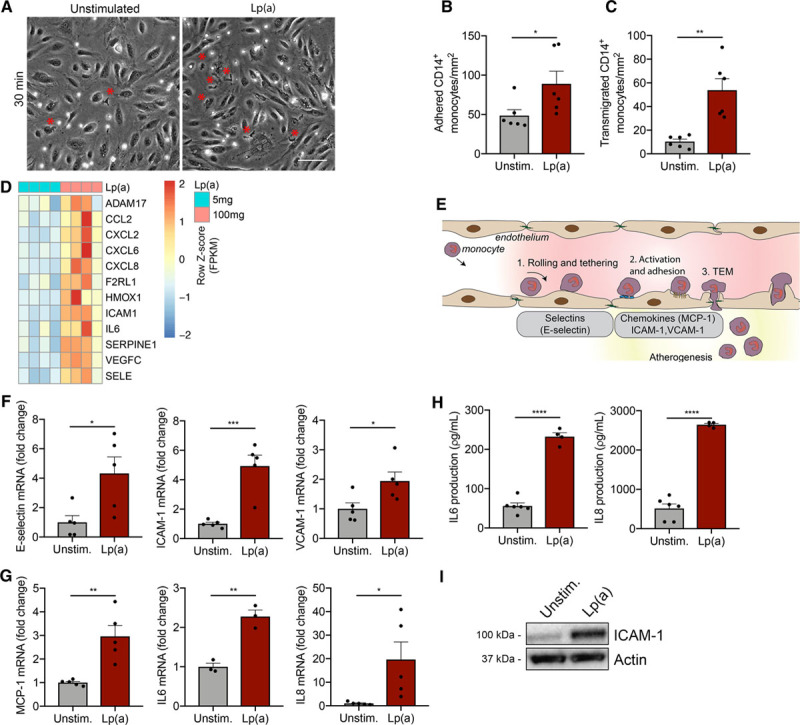
Increased inflammation in Lp(a) (lipoprotein(a))-vs endothelial cells (ECs) facilitates excessive monocyte transmigration. A, Representative differential interference contrast images of transendothelial migration (TEM) in unstimulated ECs (left) compared with Lp(a)-stimulated ECs [Lp(a)-EC] for 18 h. Transmigrated monocytes are visualized as black cells with a red asterisk and adhered monocytes as white cells. White bar=200 μm. B, Quantification of adhered (n=6; P=0.0466) and (C) transmigrated monocytes (n=6; P=0.0014). Data were analyzed using 2-tailed Student unpaired t test. D, Heat map of selected genes involved in TEM and leukocyte chemotaxis of 5 mg/dL Lp(a)-EC compared with 100 mg/dL Lp(a)-EC (6 h stimulation; n=4). E, Schematic overview of the key steps and molecules involved in leukocyte TEM. F, Genes important in rolling and tethering of leukocytes are upregulated in Lp(a)-ECs relative to unstimulated ECs. Data were analyzed using 2-tailed Student unpaired t test (6 h stimulation; n=5; P=0.0253 for E-selectin; P=0.0008 for ICAM1; P=0.0333 for VCAM1). G, Chemotactic gene expression is elevated in Lp(a)-ECs compared with unstimulated ECs. Data were analyzed using 2-tailed Student unpaired t test (6 h stimulation; n=3 for IL6 and rest is n=5; P=0.0030 for MCP1; P=0.0025 for IL6; P=0.0368 for IL8). H, IL (interleukin)-6 and IL-8 cytokine secretion in cell medium increased in Lp(a)-ECs (n=4) vs unstimulated ECs (n=6). Data were analyzed using 2-tailed Student unpaired t test (P<0.0001 for IL-6; P<0.0001 for IL-8; 18 h stimulation). I, Representative immunoblot revealing increased EC ICAM (intercellular adhesion molecule)-1 protein expression after incubation with 100 mg/dL Lp(a) compared with unstimulated ECs (18 h stimulation). All data are mean±SEM. MCP-1 indicates monocyte chemoattractant protein 1; and VCAM-1, vascular adhesion molecule 1. *P<0.05, **P<0.005, ***P<0.0005, ****P<0.00005.
OxPLs Carried by Lp(a) Drive Endothelial Inflammation
In view of the recently reported role of OxPLs in Lp(a) and its effects on immune cells,3 we investigated which moiety of the Lp(a) particle mediates the inflammatory response in the endothelium. To this end, we used either the 17-kringle recombinant apo(a) species 17K r-apo(a), containing OxPLs, or 17KΔLBS r-apo(a), which lacks OxPL-binding capacity due to a mutation in the lysine-binding site (Figure IIB in the Data Supplement).4,23 Gene expression revealed that TEM-associated expression of SELE, MCP1, ICAM1, VCAM1, IL6, and IL8 was increased if ECs were exposed to 17K r-apo(a) treatment (Figure 2A; red bars). In contrast, 17KΔLBS r-apo(a) had no significant effect on inflammatory gene expression (Figure 2A; green bars). To functionally assess these changes, we determined adhesion and migration of healthy monocytes and found that monocyte adhesion was significantly increased when ECs were incubated with 17K apo(a), whereas no significant difference was found between unstimulated and 17KΔLBS-stimulated ECs (Figure 2B and 2C). Furthermore, 17K apo(a) significantly increased TEM 7-fold compared with 17KΔLBS apo(a) (Figure 2B and 2D), underlining the importance of OxPL-Lp(a) in mediating the proinflammatory responses. To validate the importance of OxPLs carried by Lp(a), both OxPL-apoB and OxPL-apo(a) levels were measured in the isolated Lp(a) fraction of used donors. The OxPL-apo(a) content was similar between donors, and correspondingly, the OxPL-apoB levels were comparable in different donors (Figure IIC in the Data Supplement). To confirm whether OxPLs bound to Lp(a) are driving EC activation, we coincubated Lp(a)-ECs with the murine IgM monoclonal antibody E06 (100 μg/mL), which binds the phosphocholine moiety of OxPLs.24,25 Blocking E06-detectable OxPLs abolished Lp(a)-induced ICAM1, VCAM1, IL6, and IL8 gene expression (Figure 2E). Blocking OxPLs by the antibody E06 led to a significant reduction in both monocyte adhesion and TEM (Figure 2F and 2G). In contrast, isolated LDL did not significantly change endothelial phenotype compared with the same concentration of Lp(a) based on apo(a) (Figure IID in the Data Supplement). However, incubation of LDL based on apoB levels did lead to a small, albeit significant increase in IL-8 expression compared with control ECs (Figure IIE and IIF in the Data Supplement). Collectively, these data indicate that the activated state of ECs needed for monocyte TEM is primarily orchestrated by OxPLs present on Lp(a).
Figure 2.
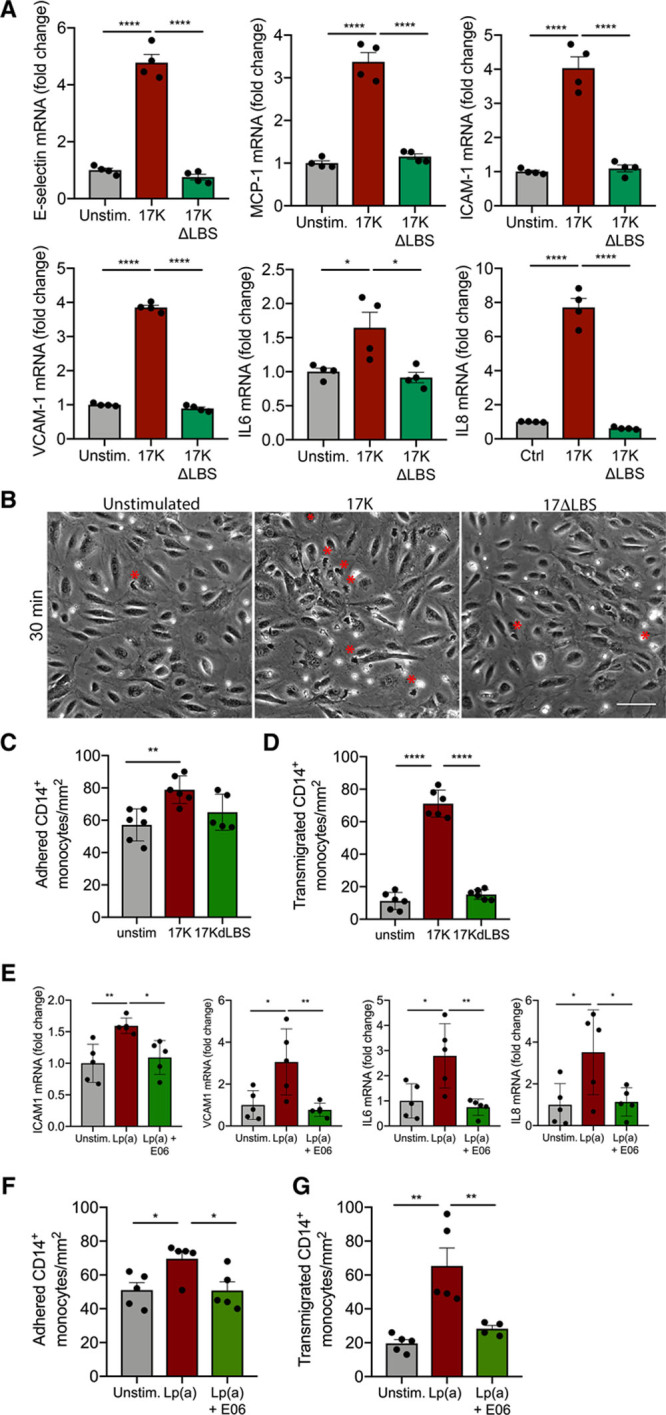
Oxidized phospholipids induce a proinflammatory EC phenotype and thereby facilitate monocyte transendothelial migration (TEM). A, Recombinant 17K apo(a) (apolipoprotein(a)) induces increased expression of TEM-associated genes (50 μg/mL; red bars). No differences were observed in ECs stimulated with 17KΔLBS (50 μg/mL; green bars). Data were analyzed using 1-way ANOVA with Tukey correction (for E-selectin, P<0.0001 for unstimulated vs 17K, P<0.0001 for 17K vs 17KΔLBS; for MCP1, P<0.0001 for unstimulated vs 17K, P<0.0001 for 17K vs 17KΔLBS; for ICAM1, P<0.0001 for unstimulated vs 17K, P<0.0001 for 17K vs 17KΔLBS; for VCAM1, P<0.0001 for unstimulated vs 17K, P<0.0001 for 17K vs 17KΔLBS; for IL6, P=0.037 for unstimulated vs 17K, P=0.0156 for 17K vs 17KΔLBS; for IL8, P<0.0001 for unstimulated vs 17K, P<0.0001 for 17K vs 17KΔLBS 6 h stimulation; n=4). B, Representative differential interference contrast (DIC) images of unstimulated ECs, 17K-stimulated ECs, and 17KΔLBS-stimulated HAECs. Transmigrated monocytes are visualized as black cells with a red asterisk and adhered monocytes as white cells (18 h stimulation; n=6; white bar=200 μm). C, Quantification of adhered monocytes. Data were analyzed using 1-way ANOVA with Tukey correction. P=0.0049 for unstimulated vs 17K (17K, red bars; 17KΔLBS, green bars) and (D) transmigrated monocytes. Data were analyzed using 1-way ANOVA with Tukey correction. P<0.0001 for unstimulated vs 17K, P<0.0001 for 17K vs 17KΔLBS (18 h stimulation; n=6). E, Monoclonal antibody E06 (100 μg/mL), decreased expression of ICAM1, VCAM1, IL6, and IL8 in Lp(a) (lipoprotein(a))-ECs (green bars) vs Lp(a)-ECs (red bars). Data were analyzed using 1-way ANOVA with Tukey correction. For ICAM1, P=0.0059 for unstimulated vs Lp(a), P=0.0173 for Lp(a) vs Lp(a)+E06; for VCAM1, P=0.0187 for unstimulated vs Lp(a), P=0.0099 for Lp(a) vs Lp(a)+E06; for IL6, P=0.0162 for unstimulated vs Lp(a), P=0.0070 for Lp(a) vs Lp(a)+E06; for IL8, P=0.0324 for unstimulated vs Lp(a), P=0.0431 for Lp(a) vs Lp(a)+E06 (6 h stimulation; n=5). F, ECs incubated with 100 mg/dL Lp(a) increased monocyte adhesion (red bars) but coincubation with E06 diminished Lp(a)-induced adhesion (green bars). Data were analyzed using 1-way ANOVA with Tukey correction. P=0.0342 for unstimulated vs Lp(a), P=0.0324 for Lp(a) vs Lp(a)+E06 (18 h stimulation; n=5). G, Monocyte TEM was increased when ECs were incubated with 100 mg/dL Lp(a) (red bars) and decreased after coincubation with E06 (green bars). Data were analyzed using 1-way ANOVA with Tukey correction. P=0.0014 for unstimulated vs Lp(a), P=0.0095 for Lp(a) vs Lp(a)+E06 (18 h stimulation; n=5). All data are mean±SEM. 17K indicates 17K recombinant apolipoprotein(a); 17KΔLBS, 17K recombinant apolipoprotein(a) with a mutation in the lysine-binding site; E06, murine IgM monoclonal antibody E06 that binds the PC moiety of oxidized phospholipids; ICAM-1, intercellular adhesion molecule 1; IL, interleukin 6; MCP-1, monocyte chemoattractant protein 1; and VCAM-1, vascular adhesion molecule 1. *P<0.05, **P<0.005, ***P<0.0005, ****P<0.00005.
PFKFB3-Mediated Glycolysis Drives Lp(a)-Induced Endothelial Inflammation
Next, we evaluated the metabolic changes in ECs after 17K r-apo(a) or Lp(a) stimulation (Figure 3A). Expression of the glycolytic enzymes SLC2A1 (GLUT1 [glucose transporter 1]), 6-phophofructo-2-kinase/fructose-2,6-biphosphatase (PFKFB3), and PFKM increased 2- to 4-fold, whereas HK2 (hexokinase 2) expression remained unaltered after 18-hour incubation with 17K, when compared with EC under control conditions (Figure 3B; red bars). No significant differences were found for these glycolytic genes between unstimulated cells and 17KΔLBS (Figure 3B). Since PFKFB3 acts as an important glycolytic regulator, we investigated the upstream regulators of PFKFB3, HIF1α (hypoxia inducible factor 1α), and KLF2.26 As expected, HIF1α protein expression increased (Figure IIG in the Data Supplement) and the expression of the negative regulator of HIF1<, KLF2,27 was decreased after 17K stimulation (Figure IIH in the Data Supplement). The increase in glycolytic gene expression coincided with enhanced glucose uptake in Lp(a)-ECs, as assessed with labeled 2-(N-[7-nitrobenz-2-oxa-1,3-diazol-4-yl]amino)-2-deoxyglucose (2-NBDG; Figure 3C and 3D), and increased lactate production (Figure 3E). Whereas aforementioned cytokine secretion occurred as early as in 2 hours in Lp(a)-ECs (Figure IE in the Data Supplement), the increase in lactate secretion, observed from 6 hours onward (Figure III in the Data Supplement), coincided with a steep increase in second-wave cytokine secretion. This suggests an increase in glycolysis downstream of inflammation and substantiates the correlation between inflammation and metabolic reprogramming. Seahorse flux analysis corroborated these findings, revealing an increase in both glycolysis and glycolytic capacity following Lp(a) stimulation (Figure 3F). In accordance, 17K r-apo(a)–stimulated ECs induced a similar response in extracellular acidification rate, where the 17KΔLBS r-apo(a) construct showed no significant increase in endothelial glycolysis (Figure 3G). This increase in glycolysis coincided with only minor changes in endothelial respiration, as reflected by the minimal difference in oxygen consumption rate (Figure 3H). The findings were substantiated by stable isotope 13C-glucose tracer experiments (Figure 3I), where an Lp(a)-induced increase in glycolytic metabolic fluxes was observed with a moderate but significant rise in the appearance of tricarboxyl acid cycle intermediates succinate and fumarate (Figure 3J). When ECs migrate and proliferate, they predominantly rely on glycolysis22; therefore, we confirmed that the observed increase in glycolytic activity could not be attributed to increased migration or proliferation of Lp(a)-ECs (Figure IIJ in the Data Supplement).
Figure 3.
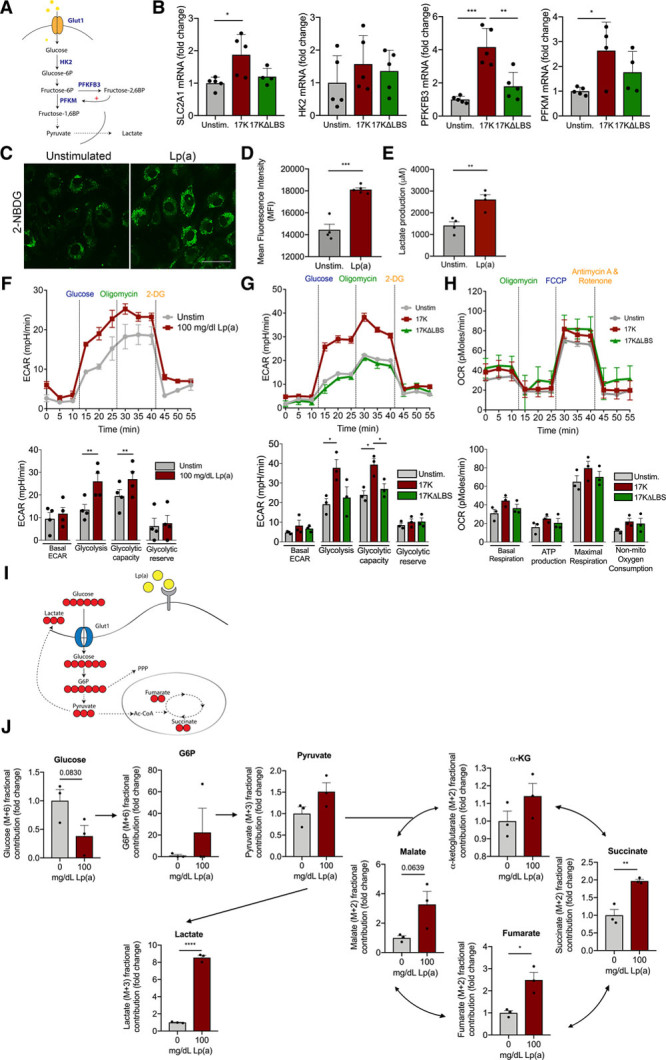
Glycolysis drives inflammation in Lp(a) (lipoprotein(a))-ECs. A, Schematic overview of the EC glycolytic pathway and its key enzymes in blue. B, Glycolytic gene expression profiles of unstimulated EC (gray bars) vs 17K- (red bars) vs 17KΔLBS-stimulated ECs (green bars). Data were analyzed using 1-way ANOVA with Tukey correction. For SLC2A1, P=0.0204 for unstimulated vs 17K; for PFBFB3, P=0.0002 for unstimulated vs 17K, P=0.0013 for 17K vs 17KΔLBS; for PFKM(6-phosphofructokinase, muscle), P=0.0332 for unstimulated vs 17K (18 h incubation; n=5). C, Representative image of unstimulated ECs (left) and Lp(a)-ECs (right), incubated with 50 μM 2-(N-[7-nitrobenz-2-oxa-1,3-diazol-4-yl]amino)-2-deoxyglucose (2-NBDG) for 2 h (18 h incubation with Lp(a); n=4; white bar=200 μm). D, Flow cytometric analysis of 2-NBDG uptake (2 h) of unstimulated (gray bar) and Lp(a)-stimulated ECs (red bar). Data were analyzed using 2-tailed Student unpaired t test, P=0.0001 (18 h incubation with Lp(a); 2 h incubation with 50 μM 2-NBDG; n=4). E, Lactate production of unstimulated ECs (gray bars) compared with Lp(a)-ECs. Data were analyzed using 2-tailed Student unpaired t test, P=0.0052 (red bars; n=4). F, Glycolytic flux measurement by Seahorse Flux Analysis of unstimulated ECs (gray line) and Lp(a)-ECs (red line) by recording extracellular acidification rate (ECAR) after injection of glucose, oligomycin, and 2-deoxyglucose (2-DG). Glycolytic rate plotted in a bar graph comparing unstimulated ECs (green bars) with Lp(a)-ECs (red bars). Data were analyzed using 2-tailed Student unpaired t test. For glycolysis, P=0.0075; for glycolytic capacity, P=0.0043 (18 h incubation; n=4). G, Graph (left) and bar graph of glycolytic flux of unstimulated (gray line; gray bars), 17K (red line; red bars), and 17KΔLBS (green line; green bars). Data were analyzed using 1-way ANOVA with Tukey correction. For glycolysis, P=0.0494 for unstimulated vs 17K; for glycolytic capacity, P=0.0139 for unstimulated vs 17K, P=0.0354 for 17K vs 17KΔLBS (18 h incubation; n=3). H, Oxidative phosphorylation parameters assessed by recording oxygen consumption rate (OCR) after injection of oligomycin, carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP), and rotenone. Unstimulated (gray line; gray bars), 17K (red line; red bars), and 17KΔLBS (green line; green bars; 18 h incubation; n=3). I, Schematic overview of 13C-glucose flux analysis in ECs. J, Bar graphs showing the 13C metabolic flux analysis with and without 100 mg/dL Lp(a) stimulation after 30-min incubation with isotopically labeled glucose. Arrows indicate the flux. Normalized fractional contribution of intracellular glucose, glucose-6-phosphate (G6P), pyruvate, lactate, α-ketoglutarate (α-KG), succinate, malate, and fumarate. Data were analyzed using 2-tailed Student unpaired t test. For succinate, P=0.0047; for fumarate, P=0.0149; for lactate, P<0.0001 (18 h incubation with Lp(a); n=3). All data are mean±SEM. 17K indicates 17K recombinant apolipoprotein(a); 17KΔLBS, 17K recombinant apolipoprotein(a) with a mutation in the lysine-binding site; GLUT1, glucose transporter 1; HK2, hexokinase 2; MFI, mean fluorescent intensity; and PFKFB3, 6-phophofructo-2-kinase/fructose-2,6-biphosphatase. *P<0.05, **P<0.005, ***P<0.0005.
In an ex vivo setting in murine aortas, we subsequently assessed whether Lp(a) affects the enzyme PFKFB3, acting as a key driver in endothelial glycolysis.22 In line with our in vitro data, ex vivo addition of Lp(a) increased aortic EC PFKFB3 protein expression compared with control aortas, whereas ICAM-1 expression was mildly affected (Figure 4A and 4B). To validate this in vivo, we used transgenic mice expressing both apo(a) and human apoB, which assemble to form Lp(a) [Lp(a)-Tg], and mice that express apoB and mutant apo(a) with 2 key point mutations in its lysine-binding site lacking OxPL-binding capacity (LBS-Lp(a)-Tg)23 and analyzed aortic PFKFB3 expression. A significant increase in endothelial PFKFB3 expression was found in Lp(a)-Tg mice compared with LBS-Lp(a)-Tg mice (Figure 4C and 4D), further underlining the importance of PFKFB3 in EC OxPL signaling.
Figure 4.
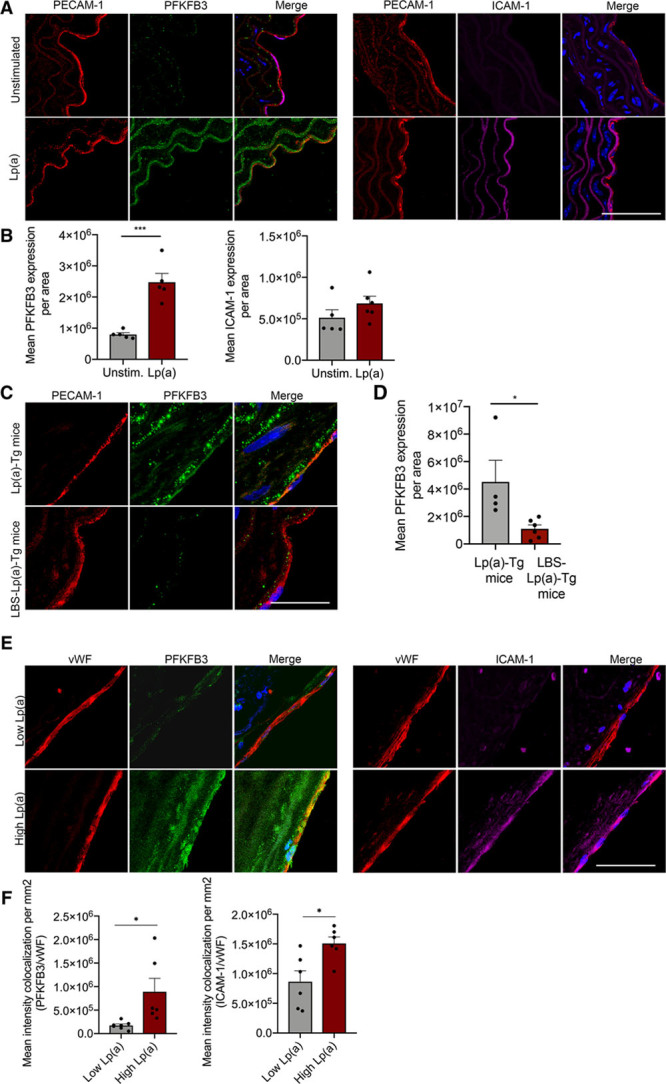
6-Phophofructo-2-kinase/fructose-2,6-biphosphatase (PFKFB3) expression is increased in murine vessels upon Lp(a) (lipoprotein(a)) stimulation. A, Representative images of murine WT (wild type) aortas ex vivo stimulated with (lower images; n=5) and without (upper images; n=6) 100 mg/dL Lp(a) stimulation. Nuclei were stained with DAPI (4′,6-diamidino-2-phenylindole; blue); ECs are stained with PECAM-1 (platelet endothelial cell adhesion molecule 1; red), PFKFB3 (green), and ICAM (intercellular adhesion molecule)-1 (magenta; 18 h incubation; white bar=200 μm). B, Quantification of A. Data were analyzed using 2-tailed Student unpaired t test, P=0.0004. C, Representative images of aortas derived from Lp(a) mice [Lp(a)-Tg; n=4] and mice lacking the lysine-binding site, which, therefore, cannot carry oxidized phospholipids (LBS-Lp(a)-Tg; n=6) stained for PECAM-1 (red) and PFKFB3 (green); nuclei were stained with DAPI (blue; white bar=200 μm). D, Quantification of C; EC PFKFB3 expression in aortas of Lp(a)-Tg and LBS-Lp(a)-Tg mice on a chow diet. Data were analyzed using 2-tailed Student unpaired t test, P=0.0354. E, Images representing human carotid plaques derived from patients with low Lp(a) vs high Lp(a) levels. Nuclei were stained with DAPI (blue); ECs are stained with vWF (von Willebrand factor; red), PFKFB3 (green), and ICAM-1 (magenta; n=6 per group; white bar=200 μm). F, Quantification of E. Data were analyzed using 2-tailed Student unpaired t test; for PFKFB3, P=0.0206; for ICAM-1, P=0.0134. All data are mean±SEM. *P<0.05, ***P<0.0005.
Finally, to also validate this increase in glycolytic activity and inflammatory burden in humans, we investigated human plaques of patients following carotid endarterectomy, included in the Athero-Express Biobank (UMCU [Universitair Medisch Centrum Utrecht], the Netherlands). We measured Lp(a) in 1506 subjects and included subjects with high levels of Lp(a) (>140 mg/dL; n=7) versus low Lp(a) (<20 mg/dL; n=7), matched for body mass index, age, systolic blood pressure, and year of surgery (Table). Subsequently, we analyzed plaques and carotid ECs of Lp(a) patients displayed increased endothelial PFKFB3 expression (Figure 4E and 4F). Moreover, endothelial ICAM-1 expression increased, further substantiating the inflammatory-metabolic axis in patients (Figure 4E and 4F). Together, these data suggest that Lp(a) activates ECs, coinciding with PFKFB3-mediated increased glycolysis.
Table 1.
Clinical Characteristics of Included Patients of Athero-Express Biobank
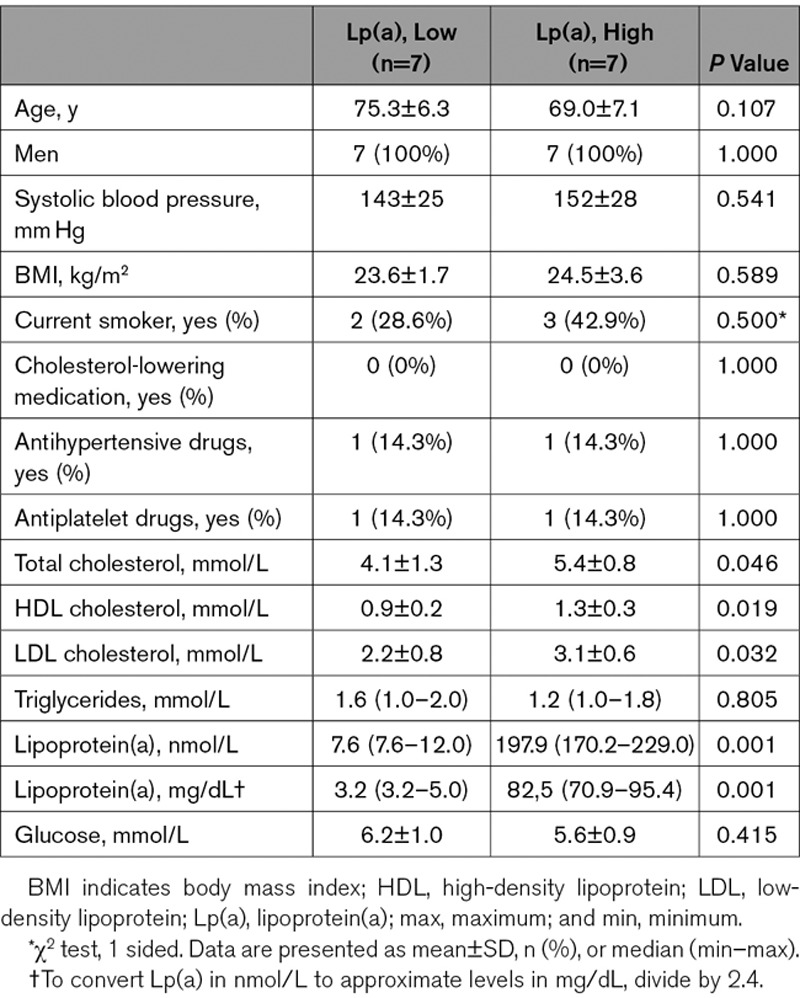
Inhibiting PFKFB3 in Lp(a)-ECs Reverses the Inflammatory Signature
Next, we investigated whether inhibition of glycolysis leads to a decrease in EC inflammation following Lp(a) stimulation. To this end, we tested and used the commercially available glycolysis inhibitor PFK158 (Figure IVA and IVB in the Data Supplement).28 RNA sequencing analysis revealed that PFK158 suppressed transcription of genes in Lp(a)-ECs involved in monocyte TEM (Figure 5A; Figure IIIA and IIIB in the Data Supplement). Validation of EC PFKFB3 knockdown experiments showed that decreased PFKFB3 levels lowered the inflammatory gene expression in Lp(a)-ECs (Figure 5B). PFK158 treatment of Lp(a)-ECs resulted in a decrease of MCP-1 and IL-6 secretion, while IL-8 levels were unaffected (Figure 5C). In agreement, PFK158 decreased GLUT1 and ICAM-1 protein expression compared with control ECs (Figure 5D and 5E). PFK158 treatment did not result in significantly decreased PFKFB3 protein expression as observed both in vitro (Figure 5D and 5E) and ex vivo (Figure 5F). However, PFK158 was able to functionally reverse the increased glycolytic rate observed in Lp(a)-ECs (Figure 5G), leading to decreased lactate production (Figure IVC in the Data Supplement). More importantly, our results demonstrate that inhibition of PFKFB3 activity reverses the Lp(a)-induced increase in monocyte migration by reducing PFKFB3-mediated glycolysis (Figure 5H and 5I). Together, these data imply that inhibiting increased glycolysis by suppressing PFKFB3 activity in Lp(a)-EC leads to a decrease in inflammation and TEM.
Figure 5.
Inhibition with PFK158 suppresses the glycolytic tone of Lp(a) (lipoprotein(a))-ECs and thereby inflammation. A, Heat map of transendothelial migration (TEM)–associated genes in Lp(a)-ECs with (n=3) and without (n=4) inhibition by 5 μM PFK158 (6 h incubation). B, Gene expression of Lp(a)-ECs knocked down for 6-phophofructo-2-kinase/fructose-2,6-biphosphatase (PFKFB3; n=6) and treated with siCtrl (n=7). Data were analyzed using 2-tailed Student unpaired t test. For PFKFB3, P<0.00001; for ICAM1, P<0.00001; for IL6, P<0.00001; for IL8, P<0.0003 (100 mg/dL Lp(a); 6 h incubation). C, Cytokine production of unstimulated (gray bars) and 100 mg/dL stimulated ECs (red bars) with and without 5 μM PFK158. Data were analyzed using 2-way ANOVA with Tukey correction. For MCP-1 (monocyte chemoattractant protein 1), P=0.0004 0 vs 100 mg/dL Lp(a) (−PFK158); P=0.0170 0 (−PFK158) vs 0 (+PFK158) mg/dL Lp(a); P<0.0001 100 (−PFK158) vs 0 (+PFK158) mg/dL Lp(a); P<0.0001 100 (−PFK158) vs 100 (+PFK158) mg/dL Lp(a). For IL (interleukin)-6, P=0.0044 0 vs 100 mg/dL Lp(a) (−PFK158); P=0.0039 100 (−PFK158) vs 0 (+PFK158) mg/dL Lp(a); P=0.0353 100 (−PFK158) vs 100 (+PFK158) mg/dL Lp(a). For IL-8, P<0.0001 0 vs 100 (−PFK158) mg/dL Lp(a); P=0.0476 0 (−PFK158) vs 0 (+PFK158) mg/dL Lp(a); P<0.0001 0 (−PFK158) vs 100 (+PFK158) mg/dL Lp(a); P<0.0001 100 (−PFK158) vs 0 (+PFK158) mg/dL Lp(a); P<0.0001 0 (+PFK158) vs 100 (+PFK158) mg/dL Lp(a) (18 h incubation; n=4 for MCP-1; n=5 for IL-6 and IL-8). D, Representative immunoblots of ICAM (intercellular adhesion molecule)-1, Glut1, PFKFB3, and HIF1α (hypoxia inducible factor 1α) protein expression. E, Quantification of immunoblots of ICAM-1 (upper left graph), GLUT1 (glucose transporter 1; lower left graph), PFKFB3 (upper right graph), and HIF-1α (lower right graph) in Lp(a)-ECs (red bars) and 5 μM PFK158-treated Lp(a)-ECs (light gray bars). Actin was used as loading control (100 mg/dL Lp(a); 18 h incubation; n=3). Data were analyzed using 1-way ANOVA with Tukey correction. For ICAM-1, P=0.0315 unstimulated vs Lp(a); for PFKFB3, P=0.0226 unstimulated vs Lp(a) and P=0.0416 unstimulated vs Lp(a)+PFK158; for GLUT1, P=0.0065 unstimulated vs Lp(a) and P=0.0040 Lp(a) vs Lp(a)+PFK158; for HIF-1α, P=0.0021 unstimulated vs Lp(a) and P=0.0026 Lp(a) vs Lp(a)+PFK158. F, Aortas of WT (wild type) mice ex vivo incubated with 100 mg/dL Lp(a) and 100 mg/dL Lp(a)+5 μM PFK158 (nuclei were stained with DAPI [4′,6-diamidino-2-phenylindole; blue], ECs with PECAM-1 [platelet endothelial cell adhesion molecule 1; red], and PFKFB3 was stained [green]; 18 h incubation; n=5; white bar=100 μm). G, Extracellular acidification rate (ECAR) of unstimulated ECs (gray line/bar), Lp(a)-ECs (red line/bar), Lp(a)-ECs+PFK158 (light gray line/bar), and ECs stimulated with only 5 μM PFK158 (orange line/bar; 100 mg/dL Lp(a); 18 h incubation; n=3). H, Representative DIC images TEM assay with unstimulated ECs (upper left), 100 mg/dL ECs (upper right), Lp(a)+PFK158-stimulated ECs (lower right), and ECs incubated with 5 μM PFK158 (lower left; 18 h incubation; white bar=200 µm). I, Quantification of H; transmigrated monocytes through Lp(a)-ECs coincubated with and without 5 μM PFK158. Gray bars indicate unstimulated ECs, red bar shows Lp(a)-ECs, light gray indicates Lp(a)-ECs coincubated with PFK158, and white bars represent PFK158-stimualted ECs. Data were analyzed using 2-way ANOVA with Tukey correction. P<0.0001 0 vs 100 (−PFK158) mg/dL Lp(a); P<0.0001 100 (−PFK158) vs 0 (+PFK158) mg/dL Lp(a); P<0.0001 100 (−PFK158) vs 100 (+PFK158) vs 100 mg/dL Lp(a) (18 h incubation; n=5). All data are mean±SEM. 2-DG indicates 2-deoxyglucose. *P<0.05, **P<0.005, ***P<0.0005, ****P<0.00005.
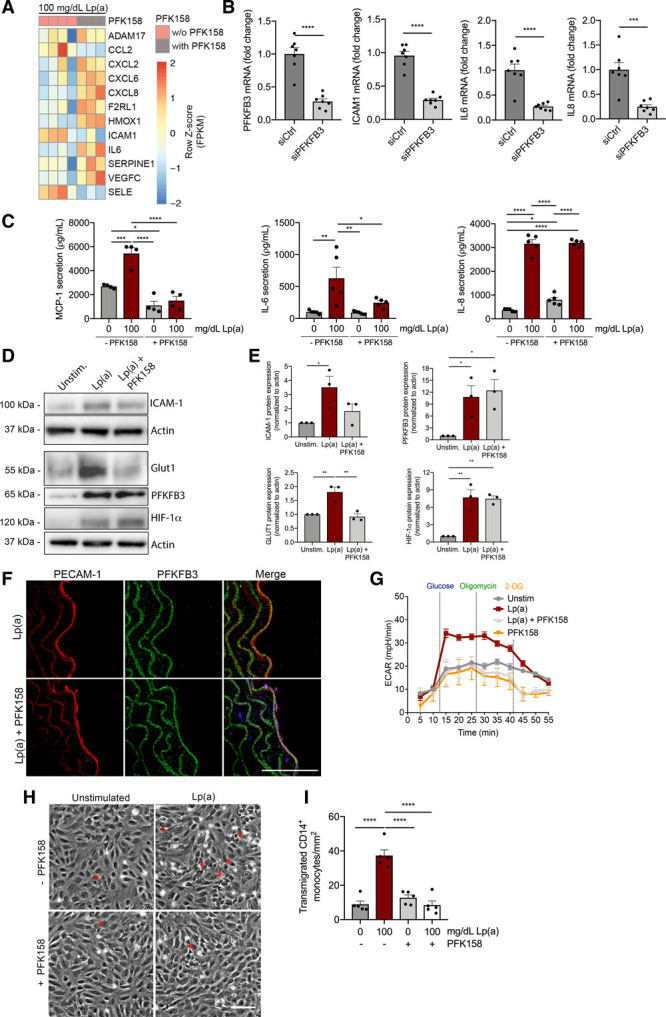
Lp(a) Lowering by Antisense Therapy in Humans Partly Reduces Endothelial Glycolysis and Inflammation
To investigate the potential of Lp(a)-lowering strategies in patients to reduce the proinflammatory effects of Lp(a), serum from participants of the IONIS-APO(a)Rx trial with elevated Lp(a) (mean, 203.5 mg/dL) were collected before, after 12 and 24 weeks of apo(a) antisense administration (100–300 mg dose).29 ECs were incubated with serum samples obtained at baseline, at day 85 of peak drug effect, and after a washout period at day 190 when drug effect was mostly but not completely abolished. Clinically, patients showed a reduction of ≈80% of plasma Lp(a) levels at day 85, which coincided with an overall reduction of 9% to 35% in key EC inflammatory markers compared with baseline. However, VCAM1 and IL8 did not significantly decrease upon treatment (Figure 6A). Expression of key glycolytic genes, including PFKFB3, SLC2A1, and PFKM, decreased at day 85 compared with baseline (Figure 6B). After the washout period (day 190), expression of MCP1 and SLC2A1 returned to baseline levels, whereas others remained downregulated (Figure 6A and 6B). In parallel to the changes in gene expression levels, cytokine secretion of MCP-1 and IL-6 decreased in supernatant of ECs incubated with day 85 samples compared with baseline, whereas no significant difference was observed in IL-8 secretion (Figure 6C). Finally, a concomitant decrease in endothelial lactate secretion was observed (Figure 6D). Collectively, these data support that selective potent Lp(a) lowering in patients reduces increased EC glycolysis and EC inflammation without achieving complete reversal of the inflammatory activation.
Figure 6.
A strong Lp(a) (lipoprotein(a)) decrease in human serum partly reduces inflammatory and glycolytic mediators in ECs. A, Gene expression of ECs incubated with serum derived from subjects with elevated Lp(a) of the IONIS-APO(a)Rx study (1:1 incubation with endothelial growth medium). Data were analyzed using repeated measures 1-way ANOVA with Tukey correction. For ICAM1, P=0.0026 D01 vs D190; for VCAM1, P=0.0338 D01 vs D85; for MCP1, P=0.0026 D01 vs D85 and P=0.0474 D85 vs D190; for IL6, P=0.0234 D01 vs D85 and P=0.0006 D01 vs D190 (black bars, D01; gray bars, D85; anthracite bars, D190; n=6; 6 h incubation). B, Glycolytic gene expression of PFKFB3, SLC2A1, and PFKM (6-phosphofructokinase, muscle) revealed a decrease after Lp(a) lowering in human serum (1:1 incubation with endothelial growth medium). Data were analyzed using repeated measures 1-way ANOVA with Tukey correction. For 6-phophofructo-2-kinase/fructose-2,6-biphosphatase (PFKFB3), P=0.0196 D01 vs D85 and P=0.0276 D01 vs D190 (n=6; 6 h incubation). C, MCP (monocyte chemoattractant protein 1)-1, IL (interleukin)-6, and IL-8 secretion in medium of ECs incubated with human serum before and after Lp(a) lowering. Data were analyzed using repeated measures 1-way ANOVA with Tukey correction. For MCP-1, P=0.0180 D01 vs D85 and P=0.0017 D85 vs D190; for IL-6, P=0.0238 D01 vs D85 (n=6; 18 h incubation). D, Lactate secretion in medium of ECs incubated with human serum before and after Lp(a)-lowering therapy. Data were analyzed using repeated measures 1-way ANOVA with Tukey correction. P=0.0160 D01 vs D85 (n=6; 18 h incubation). All data are mean±SEM. D01 indicates baseline; D85, day 85; D190, day 190; ICAM-1, intercellular adhesion molecule 1; and VCAM-1, vascular adhesion molecule 1. *P<0.05, **P<0.005, ***P<0.0005.
Discussion
Metabolic pathways are not only crucial in controlling energy balance but also act as critical determinants in signaling pathways and hence cellular phenotype.21,30–32 In this study, we demonstrate that Lp(a) and particularly OxPLs induce EC inflammation, leading to increased adhesion and transmigration of monocytes. In parallel, we observed a marked increase of glycolytic activity in ECs exposed to Lp(a), mainly orchestrated via the glycolytic activator PFKFB3. Inhibition of PFKFB3 not only reverses the increased glycolytic activity in Lp(a)-ECs but also dampens the inflammatory response and monocyte migration. Finally, using human serum samples from patients who received apo(a) antisense therapy to lower Lp(a), we find a significantly reduced inflammatory signature in ECs ex vivo.
OxPLs bound to Lp(a) were shown to induce inflammation in ECs and monocytes.4,13 Monocytes derived from patients with elevated levels of Lp(a) showed increased TEM compared with monocytes derived from healthy subjects. Our data indicate that in vivo Lp(a) not only activates monocytes but also ECs.4 Lp(a) consists of different components, comprising an LDL-like particle, apo(a), and variable amounts of OxPLs present in Lp(a), chiefly on the apo(a) and apoB components.1,2 We show that the absence of Lp(a)-OxPL signaling, using either the OxPL-deficient 17KΔLBS apo(a) construct or the E06 OxPL-blocking antibody, markedly decreases the inflammatory potential of Lp(a). In a similar manner, it was previously shown that E06 could block the ability of OxPL on apoptotic cells to similarly activate ECs.24 In line, 17K apo(a) lacking OxPLs does not evoke an inflammatory response, which is similar to LDL alone. These data substantiate that OxPLs, rather than the apo(a) or the apoB moiety of the Lp(a) particle, are the main drivers of Lp(a)’s ability to activate ECs.
Lp(a)-OxPLs also induce a profound glycolytic activation in ECs. This increase in glycolysis was manifested by enhanced expression of glycolytic genes and increased glycolytic flux. Aortas from apo(a)-overexpressing mice and murine aortas incubated with exogenous Lp(a) displayed increased EC PFKFB3 expression; however, Lp(a)-induced PFKFB3 expression was also altered in other cell types (ie, smooth muscle cells), indicating that glycolytic activation is not restricted to only ECs.33 In support, Feng et al34 also reported increased levels of HK2, GLUT1, and PFKFB3 in atheroprone areas in hypercholesterolemic Apoe−/− mice. Mechanistically, we found that Lp(a) triggered HIF1α stabilization, which acts as an upstream regulator of PFKFB3 and GLUT1, suggesting that the HIF1α-PFKFB3 axis fuels inflammation.35,36 Interestingly, our data show that Lp(a) enhances inflammatory cytokine secretion before an increase of lactate secretion, suggesting that the increase in glycolysis is downstream of EC activation. Collectively, this implies that excessive ATP consumption and nucleotide biosynthesis due to EC activation leads to increased ATP generation via glycolysis to maintain a state of inflammation. We observed minor changes in glucose oxidation fluxes, although we mainly focused on glycolysis, as ECs rely predominantly on glycolysis (>200-fold higher glucose fluxes as compared with oxidative phosphorylation fluxes).22
We validated our findings in human carotid plaques obtained from the Athero-Express database. In line with our in vitro and ex vivo data, plaques derived from subjects with high levels of Lp(a) showed a profound increase of EC PFKFB3 and ICAM-1 protein levels, suggesting increased activation, despite an advanced atherosclerotic environment in both low and high Lp(a) groups. Of note, the patients with Lp(a) elevation also had increased levels of LDL cholesterol (LDL-c) compared with low Lp(a) patients (3.1 versus 2.2 mmol/L). However, this increase largely disappears after correction for the Lp(a) elevation, which falsely leads to higher LDL-c laboratory values (LDL-c corrected: ≈2.5 mmol/L).37 Whereas theoretically this minor LDL-c difference may have contributed to a small inflammatory effect, our recent findings in the ANITSCHKOW study make this highly unlikely.38 Thus, in patients with marked Lp(a) elevation, treatment with a PCSK9 (proprotein convertase subtilisin/kexin type 9) antibody resulting in 60% LDL-c reduction and 14% Lp(a) reduction did not result in a reduction of the enhanced inflammatory activity in the arterial wall of these patients. In fact, the persistent inflammatory activation was attributed to the marked residual Lp(a) elevation despite PCSK9 antibody treatment.38 To substantiate the overriding impact of Lp(a) on inflammatory activation as compared with LDL-c, we confirmed the increased inflammatory potency of Lp(a) compared with LDL-c on ECs ex vivo, on equimolar basis.
Several observations in humans underlie the validity of our findings. Thus, Tomas et al found that human unstable atherosclerotic plaques were characterized by an increased glycolytic activity. These unstable plaques displayed increased expression of glycolytic genes HK2, GLUT1, and PFKM, comparable with our findings. The activated metabolic state in patients with elevated Lp(a) was corroborated using 18F-fluorodeoxyglucose/computed tomography scans,4 showing a significantly higher glucose uptake in the arterial wall of subjects with Lp(a) elevation. In vitro data in human umbilical vein ECs show that oxygen-deprived activated ECs show increased HIF1α-mediated glycolysis and accumulate more 18F-fluorodeoxyglucose as compared with control EC.39 This further indicates that ECs are able to accumulate glucose and increase glycolysis when activated as we observed in human carotid plaques of Lp(a) patients.
In our ex vivo incubation experiments, using serum samples of patients with profoundly elevated Lp(a) levels before and after treatment with apo(a) antisense, we showed a marked proinflammatory and glycolytic rise in ECs. Serum obtained from patients after apo(a)-antisense treatment (80% Lp(a) reduction) showed a markedly reduced potential in glycolytic and inflammatory responses in human aortic ECs. This attenuation disappeared using serum obtained after a 185-day washout in these patients, resulting in glycolytic and inflammatory responses comparable to the pretreatment serum samples. These data imply that potent Lp(a) lowering may reduce endothelial inflammation and enhanced glycolytic activity in patients with Lp(a) elevation. However, since Lp(a) lowering only partially inhibited the inflammatory responses, interventions beyond Lp(a) lowering may still be required.
Reducing glycolysis via inhibition of PFKFB3 decreased endothelial secretion of IL-6 and MCP-1, with a concomitant decrease of monocyte TEM. Importantly, endothelial knockdown of PFKFB3 does not result in hypometabolism as ECs generate ATP at a normal rate compared with control.40 Additionally, we show that PFK158 treatment reduces lactate production. Uptake of extracellular lactate stabilizes HIF1α, which in turn increases EC activation in tumor ECs.36 Thus, Lp(a)-induced lactate production could potentially increase glycolysis through HIF1α-PFKFB3 and thereby affect endothelial inflammation. In summary, inhibiting PFKFB3 reduces the Lp(a)-induced glycolytic increase rendering ECs more quiescent.
Conclusions
In the present study, we demonstrate that Lp(a) activates the endothelium, mainly through its OxPL content, thereby facilitating increased monocyte transmigration. Persistent EC activation is induced via PFKFB3-mediated increase of glycolysis. Inhibition of PFKFB3 abolishes the inflammatory potential of OxPLs associated with Lp(a). Since endothelial activation is a hallmark in several proatherogenic disease states, including diabetes mellitus, familial hypercholesterolemia, and rheumatoid arthritis,41–43 selective endothelial targeting of PFKFB3-mediated glycolysis may offer a new target for future anti-inflammatory therapy in patients at increased cardiovascular risk.
Affiliations
From the Experimental Vascular Medicine (J.G.S., L.A., J.C.B., M.V., A.K.G., J.K.), Vascular Medicine (R.M.H., E.S.G.S.), Medical Biochemistry (K.H.M.P., M.P.J.d.W.), and Laboratory Genetic Metabolic Diseases, Amsterdam Gastroenterology and Metabolism (M.v.W., R.H.H.), Amsterdam Cardiovascular Sciences, Amsterdam UMC, University of Amsterdam, the Netherlands; Vascular Surgery, Netherlands (F.W., D.P.V.D.K.), Cardiology (F.W.), and Netherlands Heart Institute (D.P.V.D.K.), UMC Utrecht, University Utrecht, the Netherlands; Core Facility Metabolomics, Amsterdam UMC, University of Amsterdam, the Netherlands (M.v.W.); Robarts Research Institute, Schulich School of Medicine and Dentistry, University of Western Ontario, London, Canada (M.J.B., M.L.K.); Vascular Medicine Program, Division of Cardiology, Department of Medicine, Sulpizio Cardiovascular Center (C.Y., S.T.) and Division of Endocrinology and Metabolism, Department of Medicine (J.L.W.), University of California San Diego, La Jolla; Institute for Cardiovascular Prevention, Munich, Germany (M.P.J.d.W.); and Pediatrics, Laboratory of Metabolic Diseases, University of Groningen, University Medical Center Groningen, the Netherlands (A.K.G.).
Acknowledgments
We would like to thank Dr Han Levels, Stefan Havik, and Hans Janssen for their excellent technical support throughout this study.
Sources of Funding
This work was financially supported by the Netherlands Organization for Scientific Research. J. Kroon received a VENI grant from ZonMW (The Netherlands Organisation for Health Research and Development; 91619098). D.P.V. De Kleijn is supported by the Netherlands Cardiovascular Research Initiative: an initiative with the Netherlands Heart Foundation (CVON 2017-5) and PERSUASIVE EU Horizon 2020 (Taxinomisis 755320). M.L. Koschinsky is supported by the Heart and Stroke Foundation of Canada (G-17-0018740). M.P.J. de Winther is supported by the Netherlands Heart Foundation (CVON 2011/B019 and CVON 2017-20: generating the best evidence-based pharmaceutical targets for atherosclerosis [GENIUS I and II]). M.P.J. de Winther is an established investigator of the Netherlands Heart Foundation (2007T067); is supported by a grant from the Netherlands Heart Foundation and Spark-Holding BV (2015B002), the European Union (ITN grant EPIMAC), Leducq Transatlantic Network Grant; and holds an AMC fellowship. M. van Weeghel and R.H. Houtkooper are supported by the Velux Stiftung (No. 1063). E.S.G. Stroes received funding from the European Union Horizon 2020 research and innovation program REPROGRAM under grant agreement No. 667837.
Disclosures
S. Tsimikas is a consultant to Boston Heart Diagnostics and has a dual appointment at the University of California, San Diego and Ionis Pharmaceuticals. J.L. Witztum is a consultant to Ionis Pharmaceuticals. S. Tsimikas and J.L. Witztum are coinventors and receive royalties from patents owned by the University of California, San Diego on oxidation-specific antibodies and of biomarkers related to oxidized lipoprotein and are cofounders and have an equity interest in Oxitope, Inc, and Kleanthi Diagnostics, LLC. The terms of this arrangement have been reviewed and approved by the University of California, San Diego in accordance with its conflicts of interest policies. The other authors report no conflicts.
Supplemental Materials
Expanded Materials and Methods
Online Figures I–V
Supplementary Material
{kind=link}
Footnotes
Nonstandard Abbreviations and Acronyms
- 2-NBDG
- 2-(N-[7-nitrobenz-2-oxa-1,3-diazol-4-yl]amino)-2-deoxyglucose
- apo(a)
- apolipoprotein(a)
- apoB
- apolipoprotein B-100
- EC
- endothelial cell
- GLUT1
- glucose transporter 1
- HIF1α
- hypoxia inducible factor 1α
- HK2
- hexokinase 2
- ICAM-1
- intercellular adhesion molecule 1
- IL
- interleukin
- LDL
- low-density lipoprotein
- LDL-c
- low-density lipoprotein cholesterol
- Lp(a)
- lipoprotein(a)
- MCP-1
- monocyte chemoattractant protein 1
- OxPL
- oxidized phospholipid
- PFKFB3
- 6-phophofructo-2-kinase/fructose-2,6-biphosphatase
- TEM
- transendothelial migration
For Sources of Funding and Disclosures, see page 1358.
The Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/CIRCRESAHA.119.316206.
Novelty and Significance
What Is Known?
Genetic and observational data have demonstrated that elevated lipoprotein(a) [Lp(a)] is a causal risk factor for cardiovascular disease.
Patients with elevated levels of Lp(a) are noted to have increased metabolic activity in the arterial wall on positron emission tomography/computed tomography.
Glycolysis is an important source of energy for endothelial cells.
What New Information Does This Article Contribute?
Lp(a) and its associated oxidized phospholipids induce endothelial cell inflammation and thereby facilitate leukocyte transendothelial migration—a hallmark of atherosclerosis.
Lp(a) activates the endothelium by enhancing 6-phophofructo-2-kinase/fructose-2,6-biphosphatase (PFKFB3)–mediated glycolysis—the main glycolytic orchestrator of Lp(a)-induced endothelial inflammation.
Carotid endarterectomy patients with elevated levels of Lp(a) show increased endothelial PFKFB3 and ICAM (intercellular adhesion molecule)-1 expression.
Inhibition of PFKFB3 abolishes the inflammatory potential of oxidized phospholipids associated with Lp(a).
Selective endothelial targeting of PFKFB3-mediated glycolysis may offer a new target for future anti-inflammatory therapy in patients at increased cardiovascular risk.
Lp(a) and its associated oxidized phospholipids induce endothelial cell inflammation leading to increased leukocyte transendothelial migration. Endothelial cells must alter their metabolic pathways so as to meet the energy demand required to facilitate an inflammatory state. As a pivotal driver of glycolysis, the glycolytic enzyme PFKFB3 mediates Lp(a)-induced endothelial cell inflammation. Blocking PFKFB3 activity diminishes endothelial inflammation and markedly reduces leukocyte migration through the vessel wall. From a clinical perspective, our findings suggest that selective targeting of endothelial metabolism, in particular, PFKFB3-mediated glycolysis, may offer a new promising strategy for anti-inflammatory therapy in patients at increased cardiovascular risk.
References
- 1.Bergmark C, Dewan A, Orsoni A, Merki E, Miller ER, Shin MJ, Binder CJ, Hörkkö S, Krauss RM, Chapman MJ, et al. A novel function of lipoprotein [a] as a preferential carrier of oxidized phospholipids in human plasma. J Lipid Res. 2008;49:2230–2239. doi: 10.1194/jlr.M800174-JLR200. doi: 10.1194/jlr.M800174-JLR200. [DOI] [PubMed] [Google Scholar]
- 2.Tsimikas S. A test in context: lipoprotein(a): diagnosis, prognosis, controversies, and emerging therapies. J Am Coll Cardiol. 2017;69:692–711. doi: 10.1016/j.jacc.2016.11.042. doi: 10.1016/j.jacc.2016.11.042. [DOI] [PubMed] [Google Scholar]
- 3.Que X, Hung MY, Yeang C, Gonen A, Prohaska TA, Sun X, Diehl C, Määttä A, Gaddis DE, Bowden K, et al. Oxidized phospholipids are proinflammatory and proatherogenic in hypercholesterolaemic mice. Nature. 2018;558:301–306. doi: 10.1038/s41586-018-0198-8. doi: 10.1038/s41586-018-0198-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van der Valk FM, Bekkering S, Kroon J, Yeang C, Van den Bossche J, van Buul JD, Ravandi A, Nederveen AJ, Verberne HJ, Scipione C, et al. Oxidized phospholipids on lipoprotein(a) elicit arterial wall inflammation and an inflammatory monocyte response in humans. Circulation. 2016;134:611–624. doi: 10.1161/CIRCULATIONAHA.116.020838. doi: 10.1161/CIRCULATIONAHA.116.020838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nordestgaard BG, Langsted A. Lipoprotein (a) as a cause of cardiovascular disease: insights from epidemiology, genetics, and biology. J Lipid Res. 2016;57:1953–1975. doi: 10.1194/jlr.R071233. doi: 10.1194/jlr.R071233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nordestgaard BG, Chapman MJ, Ray K, Borén J, Andreotti F, Watts GF, Ginsberg H, Amarenco P, Catapano A, Descamps OS, et al. European Atherosclerosis Society Consensus Panel. Lipoprotein(a) as a cardiovascular risk factor: current status. Eur Heart J. 2010;31:2844–2853. doi: 10.1093/eurheartj/ehq386. doi: 10.1093/eurheartj/ehq386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kamstrup PR, Tybjaerg-Hansen A, Steffensen R, Nordestgaard BG. Genetically elevated lipoprotein(a) and increased risk of myocardial infarction. JAMA. 2009;301:2331–2339. doi: 10.1001/jama.2009.801. doi: 10.1001/jama.2009.801. [DOI] [PubMed] [Google Scholar]
- 8.Langsted A, Kamstrup PR, Nordestgaard BG. High lipoprotein(a) and high risk of mortality. Eur Heart J. 2019;40:2760–2770. doi: 10.1093/eurheartj/ehy902. doi: 10.1093/eurheartj/ehy902. [DOI] [PubMed] [Google Scholar]
- 9.Marcovina SM, Albers JJ, Gabel B, Koschinsky ML, Gaur VP. Effect of the number of apolipoprotein(a) kringle 4 domains on immunochemical measurements of lipoprotein(a). Clin Chem. 1995;41:246–255. [PubMed] [Google Scholar]
- 10.Utermann G. The mysteries of lipoprotein(a). Science. 1989;246:904–910. doi: 10.1126/science.2530631. doi: 10.1126/science.2530631. [DOI] [PubMed] [Google Scholar]
- 11.Caplice NM, Panetta C, Peterson TE, Kleppe LS, Mueske CS, Kostner GM, Broze GJ, Jr, Simari RD. Lipoprotein (a) binds and inactivates tissue factor pathway inhibitor: a novel link between lipoproteins and thrombosis. Blood. 2001;98:2980–2987. doi: 10.1182/blood.v98.10.2980. doi: 10.1182/blood.v98.10.2980. [DOI] [PubMed] [Google Scholar]
- 12.Miller YI, Choi SH, Wiesner P, Fang L, Harkewicz R, Hartvigsen K, Boullier A, Gonen A, Diehl CJ, Que X, et al. Oxidation-specific epitopes are danger-associated molecular patterns recognized by pattern recognition receptors of innate immunity. Circ Res. 2011;108:235–248. doi: 10.1161/CIRCRESAHA.110.223875. doi: 10.1161/CIRCRESAHA.110.223875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pellegrino M, Furmaniak-Kazmierczak E, LeBlanc JC, Cho T, Cao K, Marcovina SM, Boffa MB, Côté GP, Koschinsky ML. The apolipoprotein(a) component of lipoprotein(a) stimulates actin stress fiber formation and loss of cell-cell contact in cultured endothelial cells. J Biol Chem. 2004;279:6526–6533. doi: 10.1074/jbc.M309705200. doi: 10.1074/jbc.M309705200. [DOI] [PubMed] [Google Scholar]
- 14.Ross R. Atherosclerosis--an inflammatory disease. N Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 15.Hahn C, Schwartz MA. The role of cellular adaptation to mechanical forces in atherosclerosis. Arterioscler Thromb Vasc Biol. 2008;28:2101–2107. doi: 10.1161/ATVBAHA.108.165951. doi: 10.1161/ATVBAHA.108.165951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu L, Dai Perrard X, Perrard JL, Yang D, Xiao X, Teng BB, Simon SI, Ballantyne CM, Wu H. Foamy monocytes form early and contribute to nascent atherosclerosis in mice with hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2015;35:1787–1797. doi: 10.1161/ATVBAHA.115.305609. doi: 10.1161/ATVBAHA.115.305609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chistiakov DA, Orekhov AN, Bobryshev YV. Effects of shear stress on endothelial cells: go with the flow. Acta Physiol (Oxf) 2017;219:382–408. doi: 10.1111/apha.12725. doi: 10.1111/apha.12725. [DOI] [PubMed] [Google Scholar]
- 18.Incalza MA, D’Oria R, Natalicchio A, Perrini S, Laviola L, Giorgino F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vascul Pharmacol. 2018;100:1–19. doi: 10.1016/j.vph.2017.05.005. doi: 10.1016/j.vph.2017.05.005. [DOI] [PubMed] [Google Scholar]
- 19.Cheng SC, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, Giamarellos-Bourboulis EJ, Martens JH, Rao NA, Aghajanirefah A, et al. mTOR- and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science. 2014;345:1250684. doi: 10.1126/science.1250684. doi: 10.1126/science.1250684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arts RJ, Novakovic B, Ter Horst R, Carvalho A, Bekkering S, Lachmandas E, Rodrigues F, Silvestre R, Cheng SC, Wang SY, et al. Glutaminolysis and fumarate accumulation integrate immunometabolic and epigenetic programs in trained immunity. Cell Metab. 2016;24:807–819. doi: 10.1016/j.cmet.2016.10.008. doi: 10.1016/j.cmet.2016.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith RL, Soeters MR, Wüst RCI, Houtkooper RH. Metabolic flexibility as an adaptation to energy resources and requirements in health and disease. Endocr Rev. 2018;39:489–517. doi: 10.1210/er.2017-00211. doi: 10.1210/er.2017-00211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.De Bock K, Georgiadou M, Schoors S, Kuchnio A, Wong BW, Cantelmo AR, Quaegebeur A, Ghesquière B, Cauwenberghs S, Eelen G, et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell. 2013;154:651–663. doi: 10.1016/j.cell.2013.06.037. doi: 10.1016/j.cell.2013.06.037. [DOI] [PubMed] [Google Scholar]
- 23.Leibundgut G, Scipione C, Yin H, Schneider M, Boffa MB, Green S, Yang X, Dennis E, Witztum JL, Koschinsky ML, et al. Determinants of binding of oxidized phospholipids on apolipoprotein (a) and lipoprotein (a). J Lipid Res. 2013;54:2815–2830. doi: 10.1194/jlr.M040733. doi: 10.1194/jlr.M040733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chang MK, Binder CJ, Miller YI, Subbanagounder G, Silverman GJ, Berliner JA, Witztum JL. Apoptotic cells with oxidation-specific epitopes are immunogenic and proinflammatory. J Exp Med. 2004;200:1359–1370. doi: 10.1084/jem.20031763. doi: 10.1084/jem.20031763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scipione CA, Sayegh SE, Romagnuolo R, Tsimikas S, Marcovina SM, Boffa MB, Koschinsky ML. Mechanistic insights into Lp(a)-induced IL-8 expression: a role for oxidized phospholipid modification of apo(a). J Lipid Res. 2015;56:2273–2285. doi: 10.1194/jlr.M060210. doi: 10.1194/jlr.M060210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kawanami D, Mahabeleshwar GH, Lin Z, Atkins GB, Hamik A, Haldar SM, Maemura K, Lamanna JC, Jain MK. Kruppel-like factor 2 inhibits hypoxia-inducible factor 1alpha expression and function in the endothelium. J Biol Chem. 2009;284:20522–20530. doi: 10.1074/jbc.M109.025346. doi: 10.1074/jbc.M109.025346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Doddaballapur A, Michalik KM, Manavski Y, Lucas T, Houtkooper RH, You X, Chen W, Zeiher AM, Potente M, Dimmeler S, et al. Laminar shear stress inhibits endothelial cell metabolism via KLF2-mediated repression of PFKFB3. Arterioscler Thromb Vasc Biol. 2015;35:137–145. doi: 10.1161/ATVBAHA.114.304277. doi: 10.1161/ATVBAHA.114.304277. [DOI] [PubMed] [Google Scholar]
- 28.Bartrons R, Rodríguez-García A, Simon-Molas H, Castaño E, Manzano A, Navarro-Sabaté À. The potential utility of PFKFB3 as a therapeutic target. Expert Opin Ther Targets. 2018;22:659–674. doi: 10.1080/14728222.2018.1498082. doi: 10.1080/14728222.2018.1498082. [DOI] [PubMed] [Google Scholar]
- 29.Viney NJ, van Capelleveen JC, Geary RS, Xia S, Tami JA, Yu RZ, Marcovina SM, Hughes SG, Graham MJ, Crooke RM, et al. Antisense oligonucleotides targeting apolipoprotein(a) in people with raised lipoprotein(a): two randomised, double-blind, placebo-controlled, dose-ranging trials. Lancet. 2016;388:2239–2253. doi: 10.1016/S0140-6736(16)31009-1. doi: 10.1016/S0140-6736(16)31009-1. [DOI] [PubMed] [Google Scholar]
- 30.Li X, Kumar A, Carmeliet P. Metabolic pathways fueling the endothelial cell drive. Annu Rev Physiol. 2019;81:483–503. doi: 10.1146/annurev-physiol-020518-114731. doi: 10.1146/annurev-physiol-020518-114731. [DOI] [PubMed] [Google Scholar]
- 31.Koelwyn GJ, Corr EM, Erbay E, Moore KJ. Regulation of macrophage immunometabolism in atherosclerosis. Nat Immunol. 2018;19:526–537. doi: 10.1038/s41590-018-0113-3. doi: 10.1038/s41590-018-0113-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ali L, Schnitzler JG, Kroon J. Metabolism: the road to inflammation and atherosclerosis. Curr Opin Lipidol. 2018;29:474–480. doi: 10.1097/MOL.0000000000000550. doi: 10.1097/MOL.0000000000000550. [DOI] [PubMed] [Google Scholar]
- 33.Kovacs L, Cao Y, Han W, Meadows L, Kovacs-Kasa A, Kondrikov D, Verin AD, Barman SA, Dong Z, Huo Y, et al. PFKFB3 in smooth muscle promotes vascular remodeling in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2019;200:617–627. doi: 10.1164/rccm.201812-2290OC. doi: 10.1164/rccm.201812-2290OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Feng S, Bowden N, Fragiadaki M, Souilhol C, Hsiao S, Mahmoud M, Allen S, Pirri D, Ayllon BT, Akhtar S, et al. Mechanical activation of hypoxia-inducible factor 1alpha drives endothelial dysfunction at atheroprone sites. Arterioscler Thromb Vasc Biol. 2017;37:2087–2101. doi: 10.1161/ATVBAHA.117.309249. doi: 10.1161/ATVBAHA.117.309249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Minchenko A, Leshchinsky I, Opentanova I, Sang N, Srinivas V, Armstead V, Caro J. Hypoxia-inducible factor-1-mediated expression of the 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3 (PFKFB3) gene. Its possible role in the Warburg effect. J Biol Chem. 2002;277:6183–6187. doi: 10.1074/jbc.M110978200. doi: 10.1074/jbc.M110978200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sonveaux P, Copetti T, De Saedeleer CJ, Végran F, Verrax J, Kennedy KM, Moon EJ, Dhup S, Danhier P, Frérart F, et al. Targeting the lactate transporter MCT1 in endothelial cells inhibits lactate-induced HIF-1 activation and tumor angiogenesis. PLoS One. 2012;7:e33418. doi: 10.1371/journal.pone.0033418. doi: 10.1371/journal.pone.0033418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Viney NJ, Yeang C, Yang X, Xia S, Witztum JL, Tsimikas S. Relationship between “LDL-C”, estimated true LDL-C, apolipoprotein B-100, and PCSK9 levels following lipoprotein(a) lowering with an antisense oligonucleotide. J Clin Lipidol. 2018;12:702–710. doi: 10.1016/j.jacl.2018.02.014. doi: 10.1016/j.jacl.2018.02.014. [DOI] [PubMed] [Google Scholar]
- 38.Stiekema LCA, Stroes ESG, Verweij SL, Kassahun H, Chen L, Wasserman SM, Sabatine MS, Mani V, Fayad ZA. Persistent arterial wall inflammation in patients with elevated lipoprotein(a) despite strong low-density lipoprotein cholesterol reduction by proprotein convertase subtilisin/kexin type 9 antibody treatment. Eur Heart J. 2019;40:2775–2781. doi: 10.1093/eurheartj/ehy862. doi: 10.1093/eurheartj/ehy862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Paik JY, Jung KH, Lee JH, Park JW, Lee KH. Reactive oxygen species-driven HIF1α triggers accelerated glycolysis in endothelial cells exposed to low oxygen tension. Nucl Med Biol. 2017;45:8–14. doi: 10.1016/j.nucmedbio.2016.10.006. doi: 10.1016/j.nucmedbio.2016.10.006. [DOI] [PubMed] [Google Scholar]
- 40.De Bock K, Georgiadou M, Carmeliet P. Role of endothelial cell metabolism in vessel sprouting. Cell Metab. 2013;18:634–647. doi: 10.1016/j.cmet.2013.08.001. doi: 10.1016/j.cmet.2013.08.001. [DOI] [PubMed] [Google Scholar]
- 41.Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol. 2011;11:98–107. doi: 10.1038/nri2925. doi: 10.1038/nri2925. [DOI] [PubMed] [Google Scholar]
- 42.Bernelot Moens SJ, Neele AE, Kroon J, van der Valk FM, Van den Bossche J, Hoeksema MA, Hoogeveen RM, Schnitzler JG, Baccara-Dinet MT, Manvelian G, et al. PCSK9 monoclonal antibodies reverse the pro-inflammatory profile of monocytes in familial hypercholesterolaemia. Eur Heart J. 2017;38:1584–1593. doi: 10.1093/eurheartj/ehx002. doi: 10.1093/eurheartj/ehx002. [DOI] [PubMed] [Google Scholar]
- 43.Libby P. Inflammation in atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32:2045–2051. doi: 10.1161/ATVBAHA.108.179705. doi: 10.1161/ATVBAHA.108.179705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alhamdoosh M, Ng M, Wilson NJ, Sheridan JM, Huynh H, Wilson MJ, Ritchie ME. Combining multiple tools outperforms individual methods in gene set enrichment analyses. Bioinformatics. 2017;33:414–424. doi: 10.1093/bioinformatics/btw623. doi: 10.1093/bioinformatics/btw623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schnitzler JG, Bernelot Moens SJ, Tiessens F, Bakker GJ, Dallinga-Thie GM, Groen AK, Nieuwdorp M, Stroes ESG, Kroon J. Nile Red Quantifier: a novel and quantitative tool to study lipid accumulation in patient-derived circulating monocytes using confocal microscopy. J Lipid Res. 2017;58:2210–2219. doi: 10.1194/jlr.D073197. doi: 10.1194/jlr.D073197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schneider M, Witztum JL, Young SG, Ludwig EH, Miller ER, Tsimikas S, Curtiss LK, Marcovina SM, Taylor JM, Lawn RM, et al. High-level lipoprotein [a] expression in transgenic mice: evidence for oxidized phospholipids in lipoprotein [a] but not in low density lipoproteins. J Lipid Res. 2005;46:769–778. doi: 10.1194/jlr.M400467-JLR200. doi: 10.1194/jlr.M400467-JLR200. [DOI] [PubMed] [Google Scholar]
- 50.Verhoeven BA, Velema E, Schoneveld AH, de Vries JP, de Bruin P, Seldenrijk CA, de Kleijn DP, Busser E, van der Graaf Y, Moll F, et al. Athero-express: differential atherosclerotic plaque expression of mRNA and protein in relation to cardiovascular events and patient characteristics. Rationale and design. Eur J Epidemiol. 2004;19:1127–1133. doi: 10.1007/s10564-004-2304-6. doi: 10.1007/s10564-004-2304-6. [DOI] [PubMed] [Google Scholar]
- 51.Sapcariu SC, Kanashova T, Weindl D, Ghelfi J, Dittmar G, Hiller K. Simultaneous extraction of proteins and metabolites from cells in culture. MethodsX. 2014;1:74–80. doi: 10.1016/j.mex.2014.07.002. doi: 10.1016/j.mex.2014.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fernández-Fernández M, Rodríguez-González P, García Alonso JI. A simplified calculation procedure for mass isotopomer distribution analysis (MIDA) based on multiple linear regression. J Mass Spectrom. 2016;51:980–987. doi: 10.1002/jms.3809. doi: 10.1002/jms.3809. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.