

Summary
To understand the role of the extensive senescence-associated 3D genome reorganization, we generated genome-wide chromatin interaction maps, epigenome, replication-timing, whole-genome bisulfite sequencing, and gene expression profiles from cells entering replicative senescence (RS) or upon oncogene-induced senescence (OIS). We identify senescence-associated heterochromatin domains (SAHDs). Differential intra- versus inter-SAHD interactions lead to the formation of senescence-associated heterochromatin foci (SAHFs) in OIS but not in RS. This OIS-specific configuration brings active genes located in genomic regions adjacent to SAHDs in close spatial proximity and favors their expression. We also identify DNMT1 as a factor that induces SAHFs by promoting HMGA2 expression. Upon DNMT1 depletion, OIS cells transition to a 3D genome conformation akin to that of cells in replicative senescence. These data show how multi-omics and imaging can identify critical features of RS and OIS and discover determinants of acute senescence and SAHF formation.
Keywords: senescence, replicative senescence, oncogene-induced senescence, 3D genome architecture, Hi-C, chromatin compartments, gene regulation, DNMT1
Graphical Abstract
Highlights
-
•
Deep multi-omics characterization of replicative and oncogene-induced senescence
-
•
Senescence-associated heterochromatin domains (SAHDs) form SAHFs via 3D changes
-
•
DNMT1 is required for SAHF formation via regulation of HMGA2 expression
-
•
SAHF formation leads to expression of SAHF-adjacent genes via 3D chromatin contacts
Sati et al. studied 3D chromatin organization in different types of cellular senescence. They have identified DNMT1 and HMGA2-mediated changes in the structural organization of senescence-associated heterochromatin domains (SAHDs) and architecture-associated gene-expression changes as the key difference among different senescent systems.
Introduction
Senescence was first described in normal human fibroblasts as a permanent proliferation arrest due to the exhaustion of the cellular replicative potential, termed replicative senescence (RS) (Hayflick, 1965, Hayflick and Moorhead, 1961). Subsequently, it was observed in primary cells in response to oncogene activation, and, in that case, it was named oncogene-induced senescence (OIS) (Serrano et al., 1997). It was then further extended to processes leading to cell-cycle arrest in response to a variety of insults, including DNA damage, oxidative stress, and chemotherapeutic drugs (Campisi and d’Adda di Fagagna, 2007, Di Micco et al., 2011, Kuilman et al., 2008, Narita et al., 2003, Schmitt et al., 2002), as well as to stress signals in embryo development during morphogenesis, in wound healing, and regeneration (Muñoz-Espín et al., 2013, Storer et al., 2013, Yun et al., 2015).
RS and OIS cells display several common features, like accumulation of senescence-associated beta-galactosidase (SA-β-gal), activation of Rb/p16 and p53/p21 pathways, morphological changes, and induction of the senescence-associated secretory phenotype (SASP) (Campisi, 2013, Gorgoulis et al., 2019). However, they differ markedly in their nuclear architecture. OIS nuclei display heterochromatin bodies, called senescence-associated heterochromatin foci (SAHF), enriched in H3K9me3 and other core heterochromatin marks in different human cell types and pathologic conditions (Chandra et al., 2012, Narita et al., 2003, Sun et al., 2018, Xu et al., 2014). RS nuclei on the other hand are enlarged and display a variety of features, ranging from compaction of individual chromosome arms to distension of peri-centromeric regions (Cruickshanks et al., 2013, De Cecco et al., 2013, Swanson et al., 2013).
3D genome organization regulates cellular processes such as DNA replication, transcription, DNA repair, and gene expression (Bonev and Cavalli, 2016). Recent studies employing microscopy (Cremer et al., 2015) and chromosome conformation capture based methods like Hi-C (Sati and Cavalli, 2017) have improved our understanding of genome organization. Hi-C methods have revealed a hierarchy in genome organization, where the basic units of genome folding, represented by TADs (topologically associating domains), are organized into two compartments: the early replicating or active (A) compartment and the late replicating or inactive (B) compartment. These compartments then coalesce into chromosome territories (Bonev et al., 2017, Dixon et al., 2012, Nora et al., 2012, Sexton et al., 2012). Likewise, microscopy approaches have identified chromatin domains (CDs), which might represent the microscopical counterpart of TADs. CDs form clusters that form chromosome territories. Microscopy also identified a so-called interchromatin compartment (IC), and active genes and soluble chromatin regulatory components tend to locate at the CD surface and within the IC (Cremer et al., 2015, Nagashima et al., 2019).
While all these approaches have advanced our understanding of 3D genome and nuclear organization, our knowledge about higher-order chromatin changes during different types of senescence is limited. One study, employing low resolution Hi-C on early stages of OIS identified no changes in TAD borders (Chandra et al., 2015), whereas another study performed on RS cells displayed changes in TAD borders and compartmental switching of some TADs from active to inactive compartments and vice versa (Criscione et al., 2016, Zirkel et al., 2018). These studies found a shift in the ratio between short- and long-range chromatin contacts, but they were not in agreement on the direction of this shift. In addition, the relationship between the 3D genome architecture, its underlying epigenome, and gene expression has never been compared between RS and OIS, and differential interactions among the H3K9me3 marked heterochromatin domains, that form SAHFs in OIS but not in RS, might be of major importance in these regulations.
In this study, we have addressed these issues by utilizing human fibroblast-based RS and OIS systems to understand similarities and differences in their 3D genome organization. We performed Hi-C, chromatin immunoprecipitation sequencing (ChIP-seq) for key heterochromatin and euchromatin marks, replication timing experiments, whole-genome bisulfite sequencing, RNA sequencing in OIS and RS, and used oligopaint-based 3D DNA fluorescence in situ hybridization (FISH) assay to delineate the interplay between 3D architecture and transcriptional changes in these conditions.
Results
Differences in 3D Genome Organization in OIS and RS Are Based on Distinct Interactions among Chromatin Compartments
WI38 primary fibroblasts (RS-Proliferative) and their serial passaging-induced replicative senescent stage (RS-Senescent) were used as a RS system (Figure 1A). For OIS, we used the WI-38hTERT/GFP-RAF1-ER (referred to as “RAF cells”) model system (Jeanblanc et al., 2012), where OIS was induced by adding 4-hydroxytamoxifen into the culture media. Following RAF induction, the cells progress into senescence, triggering cell-cycle arrest by day 2 (OIS-D2), and form SAHFs in almost all cells by day 4 (OIS-D4) (Figure 1A). In contrast, a few bright DAPI foci appear in RS, but no SAHFs are observed (Figure 1A). To track cell senescence, we performed a classic SA-βGal assay in OIS and RS. From OIS-D4 onward, all cells display SAHFs. By OIS-D6 all cells are senescent, and they remain in senescence when followed up to OIS-D10 (Figures S1A and S1B). We further confirmed the senescence status by bromodeoxyuridine (BrdU) incorporation in both RS and OIS (Figure S1C) and, as expected, both RS-Proliferative and RAF cells were found to be karyotypically normal (Figure S1D). To get a homogeneous population for Hi-C, cells were fluorescence-activated cell sorted (FACS), selecting for the G1 phase (Figure S1E), and Hi-C was performed in replicates on non-induced cells (OIS-D0), RAF-induced cells after 2, 4, 6, and 10 days of induction (OIS-D2 to OIS-D10), RS-Proliferative, and RS-Senescent cells. In total, 4.5 billion Hi-C contacts were generated and analyzed in this study (Table S1).
Figure 1.

Genome Organization in OIS and RS Based on Distinct Chromatin Compartment Interactions
(A) Schematic representation (DAPI staining) of the WI-38 hTERT/GFP-RAF1-ER cell-specific OIS and WI-38 primary cell-dependent RS systems. Only OIS cells show SAHF bodies (D4 onward). Scale bar, 5 μm.
(B) Normalized Hi-C contact maps for chromosome 5 at 500 kb resolution. The bottom left of individual Hi-C plots represents control cells (D0 or RS-Proliferative), and the top-right part displays senescence conditions. The respective eigenvectors are above the Hi-C plot. c, controls; e, senescent conditions. The maximum intensity for each panel is indicated in the bottom-left corner.
(C) Contact probability in logarithmic bins. Lines: mean values from biological replicates.
(D) Representative images of chromosome territories from OIS-D0/OIS-D6 and RS-Proliferative/senescent cells mapped via whole-chromosome paint assay. Chromosome territories were delineated with Cy5 (green)-labeled whole-chromosome painting probes, and the nuclei were counterstained with DAPI (blue). Bar, 10 μm.
(E) The distributions of areas of chromosome territories are shown as boxplots. Statistical significance was calculated using the Mann-Whitney test.
(F) Intra- and inter-compartment contact enrichment from OIS and RS samples. Data represented as bar plots show the mean ± SD.
(G) Average contact enrichment between pairs of 250 kb loci arranged by their eigenvalue (shown on top). The green bar at the bottom depicts the trend in compartmentalization. See also Figure S1.
The normalized Hi-C matrices displayed a progressive global reorganization in both OIS and RS nuclei. OIS displayed a huge gain in long-range contacts, which became prominent by OIS-D4 (by which time SAHFs are formed) (Figure 1B). Hi-C matrices of OIS-D6 and OIS-D10 are visually similar regarding long-range contacts, suggesting that a steady state in chromatin architecture is reached starting from OIS-D6 onward. Strikingly, the mean intra-chromosomal contact probability over distance displays a gradual shift from close-cis (<260 kb) to far-cis (> 30Mb) in both OIS and RS (Figure 1C). For intermediate distances (260 kb to 2 Mb), corresponding to most of the large CDs or TADs, RS nuclei display more cis contacts that their proliferative counterparts (Figure 1C; Figure S1F). The gain in long-range contact suggests chromosome compaction, which was confirmed in DNA-FISH experiments by a decrease in whole-chromosome area in OIS-D6 versus OIS-D0, as well as RS-Senescent versus RS-Proliferative nuclei (Figures 1D and 1E; Figure S1G). To compare this increase in long-range contacts with other cell types, we also generated Hi-C maps from BJ-hTERT-B-RAF-V600E (henceforth called BJ-raf) and from skin fibroblasts form a 74-year-old human (HSF74). Since HSF74 cells were still growing, they were passaged into RS and Hi-C maps were produced from both HSF74-Proliferative and HSF74-Senescent cells (Figures S1G–S1L). We further compared our chromosomal contact probability profiles with previously published OIS and RS datasets (Figures S1K–S1L). Except for human umbilical vein endothelial cells (HUVECs), changes in long-range contacts observed in our OIS and RS system were in concordance with previously studied cell types (Figure S1L) (Chandra et al., 2015, Zirkel et al., 2018).
Since the previous studies were performed with different cell types, for downstream comparisons we focused on the WI38-based OIS and RS systems, which share the same cell background. Analysis of Hi-C profiles shows that, in OIS, the regions gaining long-range interactions are in the B compartment (Figure 1B; Figures S1M and S1N). To quantify the changes in compartment interactions, we identified TADs and classified them into two compartments: active (A) and inactive (B) (see STAR Methods). Plotting the log ratio of observed versus expected contacts revealed a progressive increase in compartmentalization during OIS, with a preferential gain of B-B interactions and loss of A-B interactions (Figure 1F). In contrast, RS leads to a loss of A-A interactions and a preferential gain of A-B interactions that highlights a decrease in genome compartmentalization (Figure 1F). We confirmed these results by using an alternative approach based on eigenvector values (see STAR Methods) (Figure 1G).
In conclusion, senescence is associated with a major 3D genome reorganization, with progressive loss of close-cis and gain of far-cis contacts. However, RS and OIS differ markedly in their organization of genomic compartments. RS cells reduce compartmentalization due to a relative decrease in A compartment interactions, whereas in OIS, the genome organization is dominated by heterochromatin or B-B interactions leading to strong genome compartmentalization, which is evident in the form of SAHFs.
Long-Range Interactions among Senescence-Associated Heterochromatin Domains Generate SAHFs
To identify the regions involved in SAHF formation, we combined epigenome profiling with diffHiC analysis (Lun and Smyth, 2015). The systematic overlay of differentially interacting regions from diffHiC with 27 different histone modifications from IMR90 cells (NIH roadmap to epigenomics) in the WashU browser (Lun and Smyth, 2015) highlighted broad H3K9me3-enriched regions showing a relative gain in long-range interactions in both RS and OIS (Figure 2A) (Zhou et al., 2013, Zhou et al., 2011). We thus performed ChIP-seq experiments for H3K9me3, H3K27me3 and H3K4me3 on OIS and RS cells. Data analysis showed these identified regions to be selectively enriched for H3K9me3 but not for other marks, both in OIS and in RS (Figures S2A and S2B). We defined these conserved OIS H3K9me3 regions as senescence-associated heterochromatin domains (SAHDs) (Table S2). We found SAHDs to be gene poor, enriched in constitutive lamina-associated domains (cLADs), L1 and L2 isochores, long interspersed nuclear elements (LINE) / long terminal repeat (LTR) elements, poor in SINE elements/simple repeats, and enriched in late replication timing regions (S4 and G2) (Figures S2C–S2G).
Figure 2.

Differential Intra- versus Inter-SAHD Interactions Constitute SAHF
(A) Genome browser shot of differential interactions. Top: histone modification tracks of H4 (K5ac, K8ac, K20me1, K91ac), H3 (K4me1, K4me2, K4me3, K4ac, K9me1, K9ac, K14ac, K18ac, K23ac, K27me2, K27ac, K36me3, K56ac, K79me1, K79me2), and H2 (A.Z, BK5ac, AK9ac, BK12ac, BK15ac, Bk20ac, BK120ac) modifications from IMR90 cells. Middle: H3K9me3, mC, and Repli-seq tracks from IMR90 cells. Bottom: differential interaction (from diffHiC) tracks from OIS-D0 versus OIS-D6 and RS-Proliferative versus RS-Senescent at 100 kb bins. Red rectangles highlight SAHDs. The color scale bar represents statistically significant differential scores in 100 kb bins. The blue arcs are gains in interaction while purple arcs mean losses.
(B) Quantification of contacts within and between SAHDs in OIS and RS. Data are represented as a scatter dot plot showing the mean ± SD.
(C) Schematic representation of the location of FISH probes on chromosome 5.
(D) Representative 3D-DNA FISH images (z-slice) from OIS-D0 and OIS-D6 samples with indicated probes. Scale bar, 5 μm.
(E) Representative 3D-DNA FISH images (z-slice) from RS-Proliferative and RS-Senescent samples with indicated probes. Scale bar, 5 μm.
(F) Percentage of the respective FISH probes localized within SAHFs in individual nuclei in OIS-D6 samples.
(G) Boxplot quantification of the SAHD (A, B, and C) and non-SAHD (D) probe distance from the nuclear periphery.
(H) Boxplot of inter-probe distances in OIS-D0 and OIS-D6.
(I) Quantification of the changes in the diameter of the individual probes in OIS-D0 and D6.
(J) Quantification showing the distances of the probe from the nuclear periphery in RS-Proliferative and RS-Senescent.
(K) Boxplot of inter-probe distances in RS-Proliferative and RS-Senescent.
(L) Quantification of the changes in the diameter of the individual probes in RS-Proliferative and RS-Senescent.
(M) Representative 3D-DNA FISH images (z-slice) from BJ raf D0 and BJ raf D6 samples with A–D probes. Scale bar, 5 μm.
(N) Percentage of the respective FISH probes, localized within SAHFs in individual nuclei, in BJ raf D6 samples.
(O) Quantification showing the distance of the SAHD (A) and non-SAHD (D) probes from the nuclear periphery. Data are represented as boxplots.
(P) Boxplot of inter-probe distances in BJ raf D0 and BJ raf D6.
(Q) Quantification of the changes in the diameter of the individual probes in BJ raf D0 and BJ raf D6. Statistical significance in (G)–(L) and (O)–(Q) was calculated using the Mann-Whitney test.
The quantification of inter-SAHD interactions showed that OIS cells display a much larger gain compared to RS cells. Furthermore, OIS cells massively lose intra-SAHD interactions, while the effect is moderate in RS (Figures 2B and S2H). Interestingly, SAHDs also gain inter-SAHD interactions in trans in OIS conditions (Figure S2I). Thus, SAHDs are a general feature of senescent cells and the formation of SAHFs only in OIS cells correlates with a shift from intra- to inter-SAHD interactions that is more prominent than in RS. To test this hypothesis, we performed 3D FISH in a selected SAHD subset. Since the regions of interest (SAHDs) were large in terms of genomic size (1–4 Mb), we employed an oligopaint-based approach for the FISH assay (Figures 2C–2E; Figure S2J; Table S3). Image analysis of OIS-D0 and OIS-D6 FISH data showed that SAHDs (probes A, B, and C) localize within SAHFs upon OIS (STAR Methods) (Figures 2D and 2F). The appearance of SAHFs in OIS-D6 occurred concurrently with the relocation of SAHD probes away from the nuclear periphery, resulting in strong shortening of the inter-SAHD 3D distances (Figures 2G and 2H). Size normalized inter-SAHD distances were consistent with these observations (Figure S2K). Furthermore, SAHDs display an increase in the signal diameter in OIS, consistent with internal chromatin decompaction and reduction of intra-SAHD contacts (Figure 2I). In contrast, image analysis of RS-Proliferative and RS-Senescent FISH data indicates partial displacement of SAHD probes from the nuclear periphery and no significant change in the inter-SAHD distances (Figures 2E, 2J, and 2K). However, size normalization in RS-Senescent condition displays a significant decrease in all inter-probe distances, which is in concordance with our previous observation of massive chromosome compaction in RS-Senescent cells (Figures 1E and S2K). Moreover, not all SAHD probes display an increase in area in RS cells and only SAHD probes that move away from the periphery (B and C) display an increase in signal diameter (Figures 2J and 2L), suggesting that movement from the nuclear periphery toward the interior might be associated with SAHD decompaction. Finally, we found SAHDs to be conserved in BJ cells and, like in WI38 OIS cells, SAHDs in BJ cells also lose intra-SAHD and gain inter-SAHD interactions under OIS conditions (Figures S2L–S2M). FISH on one of the SAHDs (probe A) confirms localization into SAHFs in raf-induced OIS in BJ cells (Figures 2M and 2N). Furthermore, SAHDs were displaced from the nuclear periphery and displayed an increase in signal diameter in OIS (Figures 2O–2Q).
Previous work has shown that Hi-C matrices can be analyzed by the TADbit modeling tool to derive 3D chromosome folding models that are in good agreement with in vivo chromosome architecture (STAR Methods) (Baù et al., 2011, Mas et al., 2018, Serra et al., 2017). We thus used TADbit to model all chromosomes from OIS-D0 and OIS-D6 conditions at 100 kb resolution. We then quantified the distance distribution between SAHD and non-SAHD regions on chromosome 5 and compared them with 3D FISH data. This analysis shows that the distance distribution from our 3D models is in good agreement with experimental 3D FISH data and provide means of visually inspect the interaction data in 3D (Figures S2N and S2O). In conclusion, deep Hi-C sequencing leads to the genome-wide identification of the chromosomal domains, called SAHDs, that form SAHFs upon oncogene induction. These regions display unique architecture in terms of epigenetic marks, replication timing, sequence composition, and 3D organization. Their 3D models along with the Hi-C maps and relative ChIP-seq tracks can be visualized in the MuGVRE browser. (https://vre.multiscalegenomics.eu/data_repositories/data_senescence.php).
Detachment from the Nuclear lamina and Weakening of Inter-SAHD Interactions Can Lead to SAHF Formation
Previous studies suggested that SAHF formation occurs in OIS due to lamina degradation, causing release of heterochromatin from the lamina which, in turn, was hypothesized to lead to heterochromatin aggregation (Sadaie et al., 2013, Shah et al., 2013). Detachment of cLADs in OIS has been recently confirmed but, surprisingly, knockout of the lamin B receptor (LBR) does not induce detachment of cLADs (Lenain et al., 2017). Since we found that the movement of SAHDs away from the periphery was correlated with SAHD decompaction, we decided to quantitatively assess the relative roles of SAHD chromatin decompaction and release from the nuclear periphery in SAHF formation. To this aim, we built a polymer model of an 80 Mbp region of the q-arm of chromosome 5, which encompasses FISH probes B, C, and D used in this study. Chromatin was modeled as a self-avoiding block copolymer moving in a cubic box (STAR Methods) (Figure 3A). We considered that SAHDs may self-attract at short range with strength and that cLAD regions may transiently interact with the nuclear membrane with strength . We first inferred model parameters to quantitatively describe SAHD organization in cycling/D0 cells (STAR Methods) with the formation of medium-size droplets spread in the simulation box, typical of micro-phase separation (Ghosh and Jost, 2018, Jost et al., 2014) (Figures 3B–3D and S3A–S3C). This model was then used to test the effects of different hypotheses for how chromatin alterations could induce the observed 3D genome organization. Initially, we tested the two following cases: (1) a time-relaxation (TR) scenario, in which we simulated 6 days of real time without modifying model parameters; (2) a membrane release (MR) scenario: after 24 h of real time, we imposed , i.e., we arrested the preferential interaction of LADs with the nuclear membrane, and we followed the dynamics of chromosome reorganization until D6. In the TR scenario, we observed a global increase in the relative SAHD contacts at all genomic scales by 30% at OIS-D6 (Figures 3E–3G and S3D–S3F). Indeed, SAHD monomers have time to encounter and when they do they form large clusters that tend to locate at the membrane periphery (Figures 3E–3G and S3D–S3F). These predictions are not consistent with the experimental data where SAHFs locate internally and SAHD contacts at OISD6 show a weak decrease (∼10%) at small scales (<1–5 Mbp) and a stronger increase (∼20%–30%) at larger scales (>10–20 Mbp), compared to OISD0. In the MR scenario, we observed a very weak decrease in relative contacts (∼1%) at small scales and a strong increase at intermediate and large scales (∼35%–50%) at OISD6 compared to OISD0 (Figures 3H–3J and S3G–S3I). SAHDs now formed large, SAHF-like, compartments localized away from the membrane (Figures 3H–3J and S3G–S3I). The formation of SAHF in the MR scenario is more compatible with experimental observations than the TR scenario. However, the predicted rise in relative SAHD contacts is too strong, compared with the experimental Hi-C data. Since both Hi-C and DNA FISH show a decondensation of SAHDs during senescence (Figures 2B and 2I), we tested the combination of the MR scenario with a weakening of the interaction strength (reducing the value of ) between heterochromatin monomers (called MR-WI scenario). We found that lowering to −0.035 kT leads to consistent predictions at all length and timescales, with a detachment of SAHDs from the membrane and formation of SAHFs (Figures 3K–3M and S3J–S3L). Furthermore, a comparison of the MR-WI scenario with DNA FISH data showed that the model can reproduce quantitatively the changes in distance between SAHD (B and C) and non-SAHD (D) probes (Figure 3N). Finally, we compared the top-down, constraint-based chromosome model (produced by TADbit) with the bottom-up approach using the MR-WI scenario. The two models show a positive correlation, both in global chromosome conformation and in specific 3D folding of SAHD regions (Figures S3N and S3O).
Figure 3.

Detachment from the Lamina Partially Explains 3D Genome Reorganization in OIS
(A) A two-parameter polymer model of heterochromatin organization and positioning. Each 10 kbp monomer is characterized by an epigenetic and lamina-interacting state. SAHD monomers exhibit pairwise contact attraction and cLAD-like loci may interact preferentially with the nuclear membrane. An example of configuration evolving in a cubic box (one of its face representing the membrane) is given at the bottom-right corner of the figure.
(B–D) The inference of a polymer model for cycling/OISD0 cells. (B) Ratio between the average contact probability between SAHD regions and the total average contact probability between any pairs of loci (expected probability), as a function of the genomic distance. (C) Example of SAHD compartment. (D) Distributions of the number of small, intermediate, and large SAHF-like SAHD compartments per simulated configuration.
(E–G) Display the predictions for the time-release (TR) scenario. (E) Ratio between the observed versus expected ratio for SAHD regions at OIS-D6 and at OIS-D0. (F) Example of SAHD compartment. (G) Distributions of the number of small, intermediate, and large SAHF-like SAHD compartments per simulated configuration.
(H)–(J) Predictions for the membrane-release (MR) scenario. (H) Ratio between the observed versus expected ratio for SAHD regions at OIS-D6 and at D0. (I) Example of SAHD compartment. (J) Distributions of the number of small, intermediate, and large SAHF-like SAHD compartments per simulated configuration.
(K–M) Predictions for the membrane-release with weakening of SAHD attraction (MR-WI) scenario. (K) Ratio between the observed versus expected ratio for SAHD regions at OIS-D6 and at OIS-D0. (L) Example of SAHD compartment. (M) Distributions of the number of small, intermediate, and large SAHF-like SAHD compartments per simulated configuration.
(N) Distance distribution between B, C, and D probes from the polymer model.
See also Figure S3.
Taken together, these data suggest that local decompaction of SAHDs and their detachment from the nuclear lamina can explain quantitatively SAHF formation observed in OIS senescence and the genome-wide changes in Hi-C contacts.
DNMT1 Is Required for SAHF Formation
Since SAHDs correspond to genomic regions that replicate late in proliferating cells, we tested whether replication timing is perturbed upon OIS. For this, we performed replication timing sequencing (Repli-seq) on un-induced RAF cells (OIS-D0) and RAF cells after 12, 24, and 36 h of RAF induction. The Repli-seq profile during OIS progression displays very good correlation between SAHDs and late replicating regions (Figures 4A and S4A). Globally, replication timing was very similar across all samples. Only few regions shifted from late replicating to early replicating or vice versa (Figure 4A; Figure S4B) and the very few late replicating regions that switch to early replication were gene poor and displayed no change in chromatin contacts (Figures S4C–S4E).
Figure 4.

DNMT1 Knockdown Prevents SAHF Formation
(A) Replication-timing (RT) profiles of WI38 RAF cells at early stages of OIS. The data are displayed as log2 ratios of signals from early and late S-phase fractions. The positive scale corresponds to an early replication, and the negative scale corresponds to late replication timing.
(B) Washington University Genome browser shot. Top: mC tracks from IMR90 cells. The blue color indicates methylation levels and the gray background indicates coverage. Data are from NCBI roadmap to epigenomics. Middle: replication timing track of uninduced WI38 RAF cells (G1 phase to G2 phase). The classification of G1 to G2 phase is mentioned in the STAR Methods. Bottom: SAHDs falling in the displayed region.
(C) Schematic overview of the knockdown experiment. OISi, oncogene induction; RNAi, administration of siRNA. Day 0 (D0) to day 5 (D5).
(D) Representative DAPI staining images (z-slice) from oncogene-induced (D5) and control (NT) and upon depletion of HMGA1 + HMGA2, DNMT1, DNMT3A, DNMT3B, SUV420H1, SUV420H2, LRWD1, RIF1, GAPDH. Scale bar, 10 μm.
(E) Quantification of the changes in the SAHF score (details in STAR Methods) of the cells displayed in (D). Statistical significance was calculated using the Mann-Whitney test.
See also Figure S4.
These data suggest that changes in replication timing are unlikely to play a role in SAHF formation. We then tested selected candidate proteins for their role in SAHF formation. RIF1 and LRWD1, which are known to bind late replicating regions and to affect their 3D localization and replication timing (Foti et al., 2016, Giri et al., 2015), have no effect on SAHF formation. Likewise, depletion of Suv420H1 and Suv420H2, which deposit H4K20me3 in H3K9me3-enriched regions in OIS (Nelson et al., 2016) had no effect (Figures 4C–4E).
One unifying feature of SAHDs is a low level of CpG DNA methylation (Figure 4B), and DNA methylation levels are highly correlated with replication timing in IMR90 human fibroblasts (Figure S4F). We therefore tested whether the maintenance DNMT1, or the de novo DNMT3A or DNMT3B DNA methyltransferases are involved in SAHF formation. We depleted these proteins at the onset of OIS formation (Figure 4C) and compared the results with small interfering RNAs (siRNAs) against firefly luciferase gene (henceforth called non-targeted or NT) as a negative control and with siRNAs against HMGA1 and HMGA2 proteins as positive controls, since depletion of these proteins is known to prevent SAHF formation (Narita et al., 2006). These experiments showed that DNMT1 knockdown prevents SAHF formation in OIS (Figures 4D and 4E), although senescence-like cell-cycle arrest is maintained, as shown by cell-cycle analysis following BrdU incorporation (Figure S4G). Therefore, DNMT1 is a new essential factor for SAHF formation.
DNMT1 Is Associated with OIS-Dependent 3D Genome Rewiring
To study whether the absence of SAHFs upon depletion of DNMT1 corresponds to restoration of a normal 3D genome architecture, we performed Hi-C on DNMT1 and control (NT)-depleted OIS cells. The normalized Hi-C matrices from siDNMT1-treated cells display a global loss of long-range contacts when compared to control NT cells (Figure 5A). Hierarchical clustering of pairwise Pearson’s correlations between all Hi-C samples (at 250 kb resolution and considering contacts in the 100 kb to 3 Mb range) revealed three main clusters, namely, the proliferative condition, the OIS condition, and the replicative senescent condition (Figure S5A). As expected, the control siRNA-treated cells cluster with OIS samples. Surprisingly, however, siDNMT1-treated samples cluster with the replicative senescent condition rather than with the OIS-D0 or the OIS-D6 conditions (Figure S5A).
Figure 5.

DNMT1 Is Associated with OIS-Dependent 3D Genome Rewiring
(A) Normalized Hi-C contact maps shown for chromosome 5 at 500 kb resolution. The maximum intensity for each data point is indicated in the bottom-left corner.
(B) Average contact enrichment between pairs of 250 kb loci arranged by their eigenvalue (shown on top).
(C) The left panel is representative 3D-DNA FISH images (z-slice) from NT and DNMT1-depleted samples with A-C, B-C, B-D, and C-D probes. Scale bar, 5 μm. The right panel displays the percentage of the respective FISH probes, localized within SAHFs in individual nuclei, in NT samples.
(D) Boxplot of inter-probe distances in NT and DNMT1-depleted samples.
(E) Quantification showing the distance of the SAHD (A, B, and C) and non-SAHD (D) probe from the nuclear periphery. Data are represented as boxplots.
(F) Quantification of the changes in the diameter of the individual probes in NT- and DNMT1-depleted samples. Statistical significance in (D)–(F) was calculated using the Mann-Whitney test.
(G) BRCA1, ZFP350, and RBBP8 expression represented as the mean ± SD of two biological replicates of RNA sequencing (RNA-seq) experiments for NT and DNMT1-depleted samples.
(H) IGV (Integrative Genomics Viewer) snapshot of chromosome 10 with WGBS tracks from siNT- and siDNMT1-treated OIS samples.
(I) Scatterplot comparing the mC levels (percentage of methylation) in siNT and siDNMT1 samples. The correlation of the individual datasets is indicated on the bottom-right panel.
(J) IGV snapshot of the -BRCA-1, ZNF350 and RBBP8 genes, along with WGBS tracks from NT- and DNMT1-depleted OIS cells. The red box highlights the differentially methylated promoter region.
(K) Quantification of the changes in the SAHF score (details in STAR Methods). Statistical significance was calculated using the Mann-Whitney test.
(L) An immunofluorescence experiment on OISD5 cells in DNMT1-deleted and HMGA2 overexpression condition using an antibody against HMGA2. The zoomed cell displays a single nucleus with SAHF bodies and HMGA2 overexpression.
See also Figure S5.
Plotting the mean intra-chromosomal contact probability over distance shows an increase in contacts over the range of 500 kb to 16 Mb in the siDNMT1 samples when compared to controls, similar to RS-Senescent samples (Figures S5B and 1C). An increase in chromatin contacts in this range might indicate a reduced genome compartmentalization. Consistent with this hypothesis, we observed a decrease in compartmentalization in siDNMT1 samples, which resembles the shift observed from the RS-Proliferative to the RS-Senescent state (Figures 5B and S5C). Furthermore, we observed a mild gain in intra-SAHD interactions and a mild loss in inter-SAHD interactions in siDNMT1 versus siNT samples (Figure S5D). Next, we performed 3D FISH using SAHD (A, B, and C) and non-SAHD (D) probe in siDNMT1 and siNT nuclei. Image analysis shows that the SAHD probes localize within SAHFs in siNT samples (Figure 5C), as expected for OIS cells. This leads to the shortening of the distance between the most distant A and D probes in siNT (Figure 5D). In contrast, no SAHFs and no distance shortening were observed in siDNMT1 samples (Figure 5D). The A-D probe distance remains significantly shorter in siNT versus siDNMT1 even after nuclear size normalization (Figure S5E). Further, DNMT1 KD prevents SAHD displacement from the nuclear periphery (Figure 5E) in comparison to siNT samples. Finally, except for one SAHD region (probe A), the other SAHD regions (probes B and C) did not undergo decompaction in siDNMT1 (i.e., signal diameters were larger in the siNT condition compared to siDNMT1, see Figure 5F).
Together, these data show that DNMT1 plays an important role in SAHF formation and OIS-specific 3D genome rewiring, which is also consistent with the transcriptional induction of DNMT1 at early stages of OIS induction but not in RS conditions (Figure S5F). An important question is thus how does DNMT1 lead to SAHF formation. Among possible candidates, we found that gene expression of lamin B and the lamin B receptor are reduced during OIS. However, they were also reduced in RS-Senescent and DNMT1 knockdown cells, which do not form SAHFs (Figure S5G). This suggests that repression of the lamin B receptor might not be the mechanism through which DNMT1 induces SAHFs. Displacement from the nuclear periphery might be required to form SAHFs, however. To validate our observation, we performed DNA FISH on OIS-D6 cells depleted of HMGA1 and HMGA2 proteins, which are known to be required for SAHF formation (Figure 4B). Consistent with the requirement for SAHD displacement from the nuclear periphery in SAHF positive cells, FISH analysis showed the absence of displacement of a SAHD (probe A) from the periphery in HMGA-depleted OIS cells (Figure S5H). We then tested whether HMGA proteins might be possible downstream mediators of DNMT1 function. The mRNA levels of HMGA1 and HMGA2 were strongly induced upon OIS but not in RS and siDNMT1-treated cells (Figure S5I). Since HMGA2 is known to be critical for SAHF formation, these data suggest that the downregulation of HMGA2 in siDNMT1-treated cells might be the reason for the blockade of SAHF formation.
DNMT1 is known to act as a transcriptional repressor. Since its levels are induced upon OIS, the most plausible hypothesis is that DNMT1 might repress an HMGA repressor in OIS. HMGA2 is known to be repressed by the BRCA1/ZNF350/RBBP8 repressor complex (West et al., 2019). We found that these three genes were upregulated in DNMT1-depleted samples (Figures 5G and S5J). In order to analyze whether DNMT1 might downregulate BRCA1/ZNF350/RBBP8 repressor complex via DNA methylation, we performed whole-genome bisulfite sequencing (WGBS) of DNMT1 and NT-depleted OIS cells. Globally, mC levels in both siDNMT1 and siNT samples were well correlated, with SAHDs being hypomethylated (Figures 5H and 5I). However, we found that BRCA1 and ZNF350 have reduced CpG methylation levels around their promoter regions (±3 kb from transcription start sites [TSSs]) (Figure 5J). In order to test whether DNMT1 might induce SAHFs via derepression of HMGA2, we overexpressed HMGA2 in OIS cells treated with DNMT1 siRNA (Figure S5K). HMGA2 overexpression rescued SAHFs (Figures 5K–5L), demonstrating that HMGA2 acts downstream of DNMT1 in OIS cells. Together, these data suggest that OIS-mediated induction of DNMT1 might repress the BRCA1 and ZNF350 genes via DNA hypermethylation, leading to derepression of HMGA2 in order to induce SAHF formation.
Identification of Genes Associated with SAHF-Mediated Chromatin Remodeling
To analyze the effect of SAHFs on gene expression in senescence, we took advantage of multi-omics profiling to identify 3D genome architectural changes at active genes, both at the global scale as well as selectively associated with SAHF formation. First, we found that both OIS and RS cells partially lose insulation during the onset of senescence, particularly at TAD borders (Figures 6A and S6A). We then focused on active TSSs, since they were shown to induce local insulation and undergo long-distance 3D interactions (Bonev et al., 2017). Surprisingly, active TSSs lose insulation in OIS samples and in control siRNA-treated OIS samples, whereas the RS-Senescent and siDNMT1-treated cells maintain high insulation at active TSSs (Figure 6B). Furthermore, we found an increase in the active TSS-TSS interactions upon OIS induction but not in the RS-Senescent and siDNMT1-treated OIS nuclei (Figures 6C and S6B). This suggests that SAHF formation is concomitant with a global increase in interaction among active TSSs.
Figure 6.

Gene Expression Changes Associated with SAHF Formation
(A) Average insulation score over the 200 kb region around TAD boundaries in OIS, RS, and siRNA-treated OIS cells. Lines show mean values, while dark and light shaded ribbons represent SD and 95% confidence interval (CI), respectively.
(B) Average insulation score centered on gene promoters in OIS, RS, and siRNA-treated OIS cells. Lines show mean values; dark and light shaded ribbons represent SD and 95% CI, respectively.
(C) Long-range (2- to 10-Mb) inter-TAD aggregate Hi-C contact maps around pairs of active transcription start sites (TSS) in OIS-D0, OIS-D2, OIS-D4, RS-Proliferative, RS-Senescent, NT-depleted, and DNMT1-depleted samples within ±40 kb.
(D) Quantification of gene expression of the SAHF-specific genes following the STAR Methods. The data are displayed as boxplot of log 2(FPKM +1) values.
(E) Boxplot quantification (showing the mean ± SD) of the contact strength between pairs of TSS from SAHF-specific genes represented in (D).
(F) Quantification of gene expression of the random ADJ control genes (random gene set). The data are shown as a boxplot of log 2(FPKM +1) values.
(G) Boxplot quantification (showing the mean ± SD) of the contact strength between pairs of TSS from random gene-set category.
(H) SASP gene expression represented as the mean ± SD of two biological replicates of RNA-seq experiments for NT and DNMT1-depleted OIS cells.
(I) Schematic representation of the location of FISH probes on chromosome 2.
(J) Representative 3D-DNA FISH images (z-slice) from OIS-D0 and OIS-D6 samples with set 1 (RHOB and CXCR4) and set 2 (OTOF and GPR39) probes. Scale bar, 5 μm.
(K) Boxplot quantification showing the distance of the within each probe set. Statistical significance was calculated using the Mann-Whitney test.
See also Figure S6.
We reasoned that TSSs located close to SAHDs along the linear genome might be brought in spatial proximity upon SAHF formation and that their physical interaction might reinforce transcriptional changes (Figure S6C). In order to test this hypothesis, we selected genes specifically upregulated in the presence of SAHFs (STAR Methods). Among these genes, we then further selected genes adjacent to SAHDs (ADJ) (STAR Methods). The final result was a set of 330 genes that we called SAHF-specific genes (Figures 6D and S6D; Table S4). To test whether SAHF-specific genes would have a greater chance of interacting with each other when SAHDs interact to form SAHFs, we mapped active-TSS-TSS interactions in SAHD-adjacent regions (Figure 6E). We found that SAHF-specific genes are expressed only when their TSSs interact at OIS-D4, due to SAHF formation, and both their interactions and their expression levels decrease when SAHF formation is prevented in DNMT1 knockout cells (Figure 6E). For comparison, we performed this analysis on a set of control regions, obtained by randomly shuffling SAHD domains intra-chromosomally. This operation defined a set of “pseudo-SAHDs” with an interdomain distance distribution similar to that of original SAHDs. The regions adjacent to these randomly placed pseudo-SAHDs were called random adjacent SAHDs (RDM-ADJ) and genes in the RDM-ADJ regions were called “random set genes.” Although the gene expression profiles of random set genes were matched to those of the SAHF-specific gene set (Figure 6F) and active TSS-TSS interactions in the random gene set increased during OIS, these interactions remained high in siDNMT1-treated cells, unlike for SAHD-specific genes, which lost TSS-TSS interactions (cf. Figure 6E with Figure 6G). This indicates that SAHF-specific genes are specifically regulated by DNMT1-dependent chromatin interactions that are mediated by SAHF formation. In order to validate TSS-TSS interactions among SAHD-specific genes in SAHF positive cells, we used 3D FISH assays. As a positive set, we choose TSSs of the RHOB and CXCR gene from the SAHD-specific category. As a negative control, we chose the TSS of OTOF and GPR39 genes, which are adjacent to RHOB and CXCR, respectively (Figure 6I). 3D FISH and gene expression data demonstrate that SAHD-specific genes (RHOB and CXCR) become upregulated and come significantly closer than the control set of genes (OTOF and GPR39), in SAHF positive cells (Figures 6J, 6K, and S6E). Together, these data indicate that SAHF-specific genes are transcriptionally upregulated upon SAHF-dependent induction of spatial proximity.
The gene ontology analysis of our OIS and RS system identifies key pathways, such as notch signaling, nuclear factor κB (NF-κB) pathway, DNA repair, histone modifications pathway, etc., which are concordant with previously published GO analysis (Table S5) (Hernandez-Segura et al., 2017). The gene ontology analysis of SAHF-specific gene set showed an enrichment of genes for cancer, organismal injury and abnormalities, and inflammatory responses. (Figure S6F). Since most of the SASP genes fall in the immune response pathways and play a vital role in senescence, we mapped their expression profiles across all samples. We found most of these genes in the ADJ region. Nevertheless, despite being present in the ADJ region, we found no significant decrease in SASP gene expression upon SAHF inhibition (Figure 6H), suggesting that SASP genes are regulated independently of SAHF formation.
Finally, while our findings suggest that SAHFs are made up of SAHD heterochromatin regions, they also suggest that both transcriptionally active and inactive TSSs might be present in the vicinity of SAHFs. To test this scenario, we designed two oligopaint FISH probes spanning the TSS of two genes, one active (IGF2R) and one inactive (PNLDC1), both adjacent to SAHDs (Figure S6G). The aim of the assay was to test whether a repressed gene might locate closer to SAHFs in 3D space than a neighboring active gene. FISH results demonstrate that both genes co-localize in the SAHF periphery and are equidistant from the closest SAHFs (Figures S6H–S6J). These observations along with our previous results suggests a model in which SAHFs represent clustering of SAHDs, harboring H3K9me3 marked silent regions, with both active and inactive genes in their vicinity. Interestingly, the genes within the SAHD regions matches with genes that are downregulated in lung tissue and were upregulated in human brain tissue (Table S6), suggesting that SAHDs might be reprogrammable upon cell differentiation.
In summary, we provide here a detailed characterization of epigenome and 3D genome changes during senescence. We identified chromosomal domains that form SAHF upon oncogene induction and uncovered DNMT1 as a component that induces SAHFs by stimulation of HMGA2 expression. Finally, we revealed transcriptional changes accompanying SAHF formation that might be relevant for the senescent cell state (Figure 7).
Figure 7.

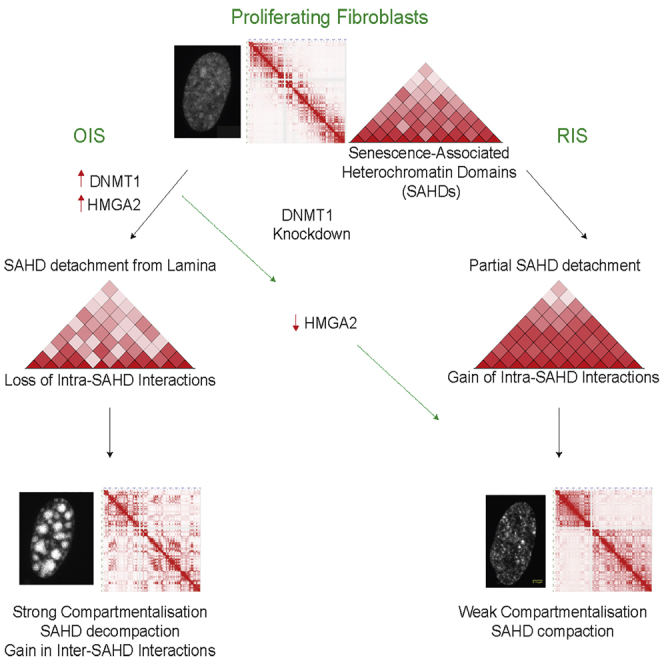
Unified Model of Chromatin Organization in Different Senescence Conditions
OIS leads to DNMT1 overexpression and a simultaneous release of SAHDs from the periphery. SAHDs decondense as they move away from periphery and gain inter-SAHD interactions to form SAHFs. Conversely, in RS conditions the SAHDs remain at the periphery with no major changes in inter- or intra-SAHD interactions. However, in cells undergoing OIS, DNMT1 knockdown leads to a partial rescue of SAHDs detachment from the lamin and prevents HMGA2 overexpression. These conditions prevent SAHF formation in DNMT1-depleted OIS nuclei, and the new chromatin architecture resembles the Replicative-Senescent state.
Discussion
Although previous studies in OIS and RS have suggested that TADs and compartments change in a subtle way during OIS (Chandra et al., 2015, Criscione et al., 2016), the OIS time course shown here identified a massive change in compartmentalization, with a decrease in RS compartmentalization and a reverse increase in OIS, driven by long-range B-compartment interactions that leads to SAHF formation. SAHFs are formed by coalescence of conserved H3K9me3 and late replicating heterochromatin domains that we called senescence-associated heterochromatin domains (SAHDs). DAPI-dense SAHF-like foci have also been observed in human as well as in mouse neurons overexpressing GFP tagged histones (Ito et al., 2014, Sun et al., 2018, Xu et al., 2014). SAHF-like structures with gene-rich regions shifting to the nuclear periphery and DAPI foci corresponding to individual chromosome territories were also observed in fibroblasts treated with chaetocin (Illner et al., 2010), suggesting that these structures may be relevant landmarks of senescent states induced by various stimuli, including in vivo situations.
Our Hi-C 3D modeling and FISH results highlighted a fundamental difference in SAHD organization between RS and OIS nuclei. While SAHDs remain partially peripheral and compacted in RS, they are displaced from the periphery and lose internal compaction in OIS. This is in agreement with a recent report demonstrating loss in compaction when heterochromatin domains move away from the periphery in mouse embryonic stem cells (ESCs) (Zheng et al., 2018) and with previous observations suggesting that SAHFs might arise because of release of heterochromatic lamina-associating domains from the lamina (Chandra et al., 2015, Sadaie et al., 2013, Shah et al., 2013). Our polymer modeling shows that the release of SAHDs from the lamina is sufficient to form large foci of heterochromatin (MR scenario), consistent with a recent independent polymer model (Chiang et al., 2019). However, by accounting precisely for the large-scale, out-of-equilibrium relaxation of chromosomes and by making detailed comparisons with Hi-C and FISH data, our approach suggests in addition (MR-WI scenario) that the weakening of intra-SAHD interactions is one key factor which, together with lamin release, required for SAHF formation.
A key question is thus what might detach SAHDs from the lamina and decondenses SAHDs during OIS. Here, we identified DNMT1 as a key component in SAHF formation. Surprisingly, OIS cells treated with siDNMT1 have 3D structural features similar to RS cells, including a loss of SAHFs that depends on the downregulation of HMGA2, a component which was previously shown to be required for SAHF formation (Narita et al., 2006). The induction of DNMT1 induces SAHFs via activation of HMGA2 expression. HMGA2 might decondense chromatin through competition with histone H1 (Catez et al., 2006, Ozturk et al., 2014) while sustaining long-range heterochromatin interactions, possibly helped by HMGA dimer formation (Frost et al., 2015). Local heterochromatin decondensation, combined with the strong reduction in lamins and lamin B receptor expression upon induction of senescence, might be responsible for triggering the detachment from the nuclear periphery in OIS. This scenario is consistent with our microscopy-based observation of a strong correlation between local SAHD decondensation and SAHD detachment from the lamina. Finally, OIS is also known to activate the DNA Damage response pathway and recent studies suggest that DDR proteins are also involved in the structural stabilization of the genome (Ochs et al., 2019). Thus, in future work it will be interesting to study the role of DDR in OIS with respect to DNMT1.
Interestingly, SAHF formation has not only structural but also functional consequences. Our data show that active genes adjacent to SAHDs are brought in 3D physical proximity and establish more frequent Hi-C contacts upon induction of SAHFs. Many of these genes function in senescence associated processes, such as changes in cell adhesion and the activation of cancer-related genes. Since senescence has been described in vivo in certain physiological conditions, it will be interesting to study in vivo cell senescence (Evangelou et al., 2017) and to compare them with our results.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| H3K4me3, rabbit, monoclonal | Millipore | Cat# 04-745; RRID: AB_1163444 |
| H3K9me3, rabbit, polyclonal | Abcam | Cat# ab8898; RRID: AB_306848 |
| H3K27me3, rabbit, | Active Motif | Cat# 39155; RRID: AB_2561020 |
| Anti 5-bromodeoxyuridine, mouse, monoclonal | Exbio | Cat# 11-286-C100; RRID: AB_10732986 |
| Anti-HMGA2 | Abcam | Cat# ab97276; RRID: AB_10679322 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| siRNA-DNMT1 | GE Healthcare Dharmacon | M-004605-01-0010 |
| siRNA-DNMT3A | GE Healthcare Dharmacon | M-006672-03-0005 |
| siRNA-DNMT3B | GE Healthcare Dharmacon | M-006395-01-0005 |
| siRNA-NT4 | GE Healthcare Dharmacon | D-0012-10-04-20 |
| siRNA-HMGA1 | GE Healthcare Dharmacon | MQ-004597-02-0010 |
| siRNA-HMGA2 | GE Healthcare Dharmacon | M-01349-5-02-0010 |
| siRNA-SUV420H1 | GE Healthcare Dharmacon | D-013366-01-0002 |
| siRNA-SUV420H1 | GE Healthcare Dharmacon | D-013366-02-0002 |
| siRNA-SUV420H2 | GE Healthcare Dharmacon | D-018622-23-0002 |
| siRNA-LRWD1 | GE Healthcare Dharmacon | D-016934-01-0002 |
| siRNA-LRWD1 | GE Healthcare Dharmacon | D-016934-02-0002 |
| siRNA-RIF1 | GE Healthcare Dharmacon | MQ-027983-01-0002 |
| siRNA-GAPDH | GE Healthcare Dharmacon | D-001140-01-20 |
| Lipofectamine RNAiMAX | ThermoFisher Scientific | 13778075 |
| Cell Line Nucleofector kit R | Lonza Bioscience | VCA-1001 |
| 4-Hydroxy-Tamoxifen | Sigma-Aldrich | H7904 |
| Senescence Beta Galactosidae Staining Kit | Cell Signaling Technology | 9806 |
| Critical Commercial Assays | ||
| FxCycle, Far Red | ThermoFisher | F10348 |
| TruSeq ChIP Sample Preparation Kit | Illumina | IP-202-1012 |
| TruSeq Stranded Total RNA Library Kit | Illumina | RS-122-2301 |
| NEBNext Ultra DNA Library Prep Kit for Illumina | NEB | E7370 |
| TruSeq DNA Methylation Kit | Illumina | EGMK81312 |
| Deposited Data | ||
| Raw Hi-C data generated from WI38 and BJ OIS system | This study | GSE130306 |
| Raw Hi-C data generated from RS system, HSF74 and HSF92 cells | This study | GSE130306 |
| Raw Hi-C data generated from DNMT1 and scramble siRNA treated samples | This study | GSE130306 |
| H3K4me3 ChIP-Seq in OISD0, OISD2, OISD6 and OISD10 cells | This study | GSE130306 |
| H3K4me3 ChIP-Seq in RS-Proliferative and RS- Senescent cells | This study | GSE130306 |
| H3K9me3 ChIP-Seq in OISD0, OISD2, OISD6 and OISD10 cells | This study | GSE130306 |
| H3K9me3 ChIP-Seq in RS-Proliferative and RS- Senescent cells | This study | GSE130306 |
| H3K27me3 ChIP-Seq in OISD0, OISD2, OISD6 and OISD10 cells | This study | GSE130306 |
| H3K27me3 ChIP-Seq in RS-Proliferative and RS- Senescent cells | This study | GSE130306 |
| Strand Specific toatal RNASeq from OISD0, OISD2, OISD4, OISD6 and OISD10 cells | This study | GSE130306 |
| Strand Specific toatal RNASeq from RS-Proliferative and RS- Senescent cells | This study | GSE130306 |
| WGBS DNMT1 depleted OIS cells | This study | GSE130306 |
| WGBS NT depleted OIS cells | This study | GSE130306 |
| Replication timing from OISD0 cells | This study | https://www2.replicationdomain.com/ |
| Replication timing from oncogene induced raf cells at 12 hours, 24 hours and 36 hours | This study | https://www2.replicationdomain.com/ |
| Image data from microscopy images | Mendeley Data, https://doi.org/10.17632/sj8yykm6bn.1 | |
| Datasets Reanalyzed | ||
| WGBS IMR90 | UCSD Human reference Epigenome Mapping Project | GEO GSM432687 |
| WGBS H1 | UCSD Human reference Epigenome Mapping Project | GEO GSM429321 |
| WGBS K562 | ENCODE | GEO GSE86747 |
| Hi-C data HUVEC Proliferative Cells | Zirkel et al., Molecular Cell, VOLUME 70, ISSUE 4, P730-744.E6, MAY 17, 2018 | GEO GSE98448 |
| Hi-C data IMR90 Proliferative Cells | Zirkel et al., Molecular Cell, VOLUME 70, ISSUE 4, P730-744.E6, MAY 17, 2018 | GEO GSE98448 |
| Hi-C data MSC Proliferative Cells | Zirkel et al., Molecular Cell, VOLUME 70, ISSUE 4, P730-744.E6, MAY 17, 2018 | GEO GSE98448 |
| Hi-C data HUVEC Proliferative Cells | Zirkel et al., Molecular Cell, VOLUME 70, ISSUE 4, P730-744.E6, MAY 17, 2018 | GEO GSE98448 |
| Hi-C data IMR90 Proliferative Cells | Zirkel et al., Molecular Cell, VOLUME 70, ISSUE 4, P730-744.E6, MAY 17, 2018 | GEO GSE98448 |
| Hi-C data MSC Proliferative Cells | Zirkel et al., Molecular Cell, VOLUME 70, ISSUE 4, P730-744.E6, MAY 17, 2018 | GEO GSE98448 |
| Hi-C dataWI38 raf Proliferative Cells | Chandra et al., Cell Reports, VOLUME 10, ISSUE 4, P471-483, FEBRUARY 03, 2015 | ENA PRJEB8073 |
| Hi-C dataWI38 raf oncogene senescent Cells | Chandra et al., Cell Reports, VOLUME 10, ISSUE 4, P471-483, FEBRUARY 03, 2015 | ENA PRJEB8073 |
| BJ H3K9me3 ChIP-Seq | GSE87039 | |
| BJ Repli-Seq data | GSM923444 | |
| Experimental Models: Cell Lines | ||
| WI-38hTERT/GFP-RAF1-ER | Jeanblanc et al., 2012 | N/A |
| WI38 | From Jean Marc Lemaitre Lab | N/A |
| BJ-hTERT-B-RAF-V600E | Carvalho et al., 2019 | N/A |
| Skin Fibroblats from 74 year old Human | From Jean Marc Lemaitre used in Genes Dev. 2011 Nov 1; 25(21): 2248–2253 | N/A |
| Software and Algorithms | ||
| Shaman R package for a-parametric Hi-C normalization | N/A | https://bitbucket.org/tanaylab/shaman |
| imageJ | N/A | https://imagej.nih.gov/ij |
| Bowtie2 v2.2.0 | N/A | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Samtools v0.1.19 | N/A | http://samtools.sourceforge.net/ |
| DeepTools2 | N/A | https://deeptools.readthedocs.io/en/latest/ |
| CSAW | N/A | bioconductor |
| STAR v2.5 | N/A | https://github.com/alexdobin/STAR |
| DESeq2 | N/A | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| MACS2 | N/A | https://github.com/taoliu/MACS/ |
| SICER2 | N/A | https://github.com/bioinf/SICER2 |
| methpipe | Andrew D Smith | http://smithlabresearch.org/software/methpipe/ |
| C-world (Hi-C analysis software) | Job Dekker lab | https://github.com/dekkerlab/cworld-dekker |
| GraphPad Prism | N/A | https://www.graphpad.com/ |
| MuG VRE | N/A | https://www.multiscalegenomics.eu/MuGVRE/ |
| methpipe | N/A | http://smithlabresearch.org/software/methpipe/ |
Lead Contact and Materials Availability
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Giacomo Cavalli (giacomo.cavalli@igh.cnrs.fr). This study did not generate new unique reagents.
Experimental Model and Subject Details
Cell Lines
WI-38hTERT/GFP-RAF1-ER and BJ-hTERT-B-RAF-V600E cells were kindly provided by Carl Mann (Carvalho et al., 2019, Jeanblanc et al., 2012). The HSF74 cells are from Lapasset et al. (Lapasset et al., 2011). The cells were grown under conditions described by Jeanblanc et al. (Jeanblanc et al., 2012). Briefly, WI-38hTERT cultured in modified Eagle’s medium (Thermo-Fisher Scientific, Cat.N: 12492013), supplemented with 10% fetal bovine serum (Thermo-Fisher Scientific, Cat.N: 10270106), 1mM sodium pyruvate (Thermo-Fisher Scientific, Cat.N: 11360070), 1mM of glutamax (Thermo-Fisher Scientific, Cat.N: 35050-038), 50U of penicillin and streptomycin (Thermo-Fisher Scientific, Cat.N: 15070-063) and 0.1mM MEM non- essential amino acids (Thermo-Fisher Scientific, Cat.N: 11140-035). Cells were cultured under 5% carbon dioxide and 5% oxygen conditions. Cells become senescent in 72 hours post 4-hydroxytamoxifen induction (Sigma-Aldrich, Cat.N: H7904). The WI-38 primary cells were bought from ATCC.
Method Details
BrdU FACS for Cell cycle analysis
Cell cycle progression was assessed by flow cytometry. Cells were pulsed 2h with 100 μM of Bromodeoxyuridine (BrdU). After tryspinization (Thermo-Fisher Scientific, Cat.N: 25200056) and wash in PBS 1% fetal bovine serum (FBS), cells were fixed in 75% ice cold ethanol. Fixed cells were washed, collected by centrifugation, treated with 2N HCl-0.5% Triton X-100 30 min at room temperature, washed and neutralized in 0.1M Na2B4O7.10H2O, pH 8.5. After centrifugation, cells were incubated overnight at 4°C with primary anti-BrdU antibody (Exbio, Cat.N:11286-c100) diluted in PBS-0.5% Tween 20%–1% BSA, washed in PBS 1% CFS and incubated 1h at RT with Alexa 488 labeled secondary antibody (Abcam, Cat.N: ab150113). Then, cells were washed and resuspend in PBS 1% FBS containing 50 μg/ml of Propidium Iodide (Thermo-Fisher Scientific, Cat.N: P3566) and RNase A 200μg/ml (Thermo-Fisher Scientific, Cat.N: 12091021) for 30 min at room temperature. Data acquisition were done in Gallios FACS apparatus (Beckman Coulter) and analyzed using Kaluza software (Beckman Coulter). Relative cell cycle phase distributions were performed by using bivariate dot plot analysis.
siRNA transfection
siRNA transfection for image analysis was performed on 100 thousand cells. For Hi-C 1 million WI-38hTERT/GFP-RAF1-ER cells were transfected. In both cases, we use RNAiMax with a 30 nM final concentration of predesigned siRNAs form Dharmacon (product details in Key Resources Table)
Hi-C Library Preparation
The cells were dissociated using 0.25% Trypsin-EDTA (ThermoFisher, Cat.N: 25200056). After fixation for 10 min at room temperature with freshly prepared 1% formaldehyde in PBS (ThermoFisher, Cat.N: 28908), the reaction was quenched for 5 minutes by adding 0.2M glycine solution (final). Cells were permeabilized using 0.1% saponin (in PBS) and concentration was adjusted to 1x106 cells / mL. 1 μL/mL of fxCycle far red dye (ThermoFisher, Cat.N: F10348) and 5 μL/mL RNase A (20mg/mL) were added and samples were incubated for 30 min at room temperature protected from light with slight agitation. After washing once with cold 1x PBS, samples were resuspended in cold 0.5% BSA in PBS at a concentration of 107 cells/mL and immediately processed for FACS. FACS was performed using BD FACSAria and appropriate gates were set based on the relative levels of fxCycle in order to isolate G0G1 cells. After FACS sorting, cell were pelleted and were frozen in liquid nitrogen and stored at −80°C. The RS-Senescent cells were not FACS sorted as replicative senescent cells are mostly in G1 phase. Hi-C and library preparation was carried out using the in situ method as described previously (Rao et al., 2014) with minor modifications. In brief, cells either FACS-purified or RS-Senescent were digested overnight at 37°C using 500U of DpnII. After biotin filling, proximity ligation was carried out for 4 hours at 18°C with 2000U T4 DNA Ligase. After reverse-crosslinking, DNA was purified using ethanol precipitation and sheared to 300-400bp fragments using Covaris S220 sonicator. Ligation fragments containing biotin were immobilized on MyOne Streptavidin T1 beads (ThermoFisher Cat.N: 65602), end-repaired and a-tailed as described. NEXTflex adaptors (Bioo Scientific, Cat.N: 514101) were then ligated and fragments were PCR amplified using KAPA HiFi Library Amplification Kit (Kapa Biosystems, Cat.N: KK2620) for 8 cycles. DNA was then double-size selected using AMPure XP beads (Agencourt, Cat.N: A63881) to isolate fragments between 300 and 800bp.
Strand-Specific Total RNA Library Preparation
Cells were rinsed with PBS once and then lysed immediately on plate by adding TRIzol (ThermoFisher, Cat.N: 15596026) and scraped with cell scraper. The lysate was transferred to the DNase-RNase free tubes and incubated at room temperature for 5 min. Thereafter samples were vortexed for 20 s, 0.2x volumes of chloroform was added, tubes were mixed by inverting and samples were centrifuged at 13,000 rpm at 4°C for 15 min. The aqueous phase was then processed using the RNeasy Mini Kit (QIAGEN, Cat.N: 74104) with DNase treatment, according to the manufacturer’s instructions. Purified RNA (maximum of 1ug) was then used for library generation using the TruSeq Stranded Total RNA Library Kit according to the manufacturer’s instructions.
ChIP and Library Preparation
The cells were rinsed with PBS once and then fixed immediately by adding freshly prepared 1% formaldehyde in PBS (ThermoFisher, Cat.N: 28908). The reaction was then quenched by adding glycine solution (final 0.125M) for 5min. After rinsing the cells twice with 10 mL ice cold PBS. Cells were scraped thoroughly with a cell scraper (in 10 mL ice cold PBS with 1xEDTA-free Protease Inhibitors) and transfer into 50 mL tube. The cells were pelleted immediately. The pelleted cells were then frozen in liquid nitrogen and stored at −80°C until further use. We used ∼2x106 cells per IP for chromatin marks. The pellets were thawed on ice, resuspended in cold cell lysis buffer (10mM Tris pH 8, 10mM NaCl, 0.2% NP-40) + 1xEDTA-free Protease Inhibitors. Cells were lysed for 30 min at 4°C, washed once with cold lysis buffer and resuspended in cold nuclei lysis buffer (50mM Tris pH8, 10mM EDTA, 1% SDS) + 1xProtease Inhibitors (20μl per 1x106 cells). Nuclei were lysed for 30 min at 4°C with rotation and then sonicated for 30sec on/ 30 s off per cycle for 18 cycles using Bioruptor (Diagenode). After sonication, 10x volumes of IP dilution buffer (20mM Tris pH8, 2mM EDTA, 150mM NaCl, 1% Triton X-100, 0.01% SDS + protease inhibitors) was added, chromatin was precleared using 20μl Protein A dynabeads (ThermoFisher, Cat.N: 10002D) / 1mL for 2hrs at 4°C with rotation. Meanwhile, 25ul beads / IP were washed once with cold 0.5% BSA in PBS, and incubated with the antibody for 4-5hrs at 4°C in 0.5ml 0.5% BSA in PBS. Beads were then washed once with 0.5% BSA in PBS, added to the precleared chromatin and incubated overnight at 4°C with rotation. Beads were then washed once with cold IP wash buffer 1 (20mM Tris pH8, 2mM EDTA, 50mM NaCl, 1% Triton X-100, 0.1% SDS), twice with high salt wash buffer (20mM Tris pH8, 2mM EDTA, 500mM NaCl, 1% Triton X-100,0.1% SDS), once with cold IP wash buffer 2 (10mM Tris pH8, 1mM EDTA, 250mM LiCl, 1% NP40, 1% sodium deoxycholate) and twice with cold TE buffer (1mM Tris pH8, 1mM EDTA). DNA: protein complexes were then eluted twice for 30 min at 65°C in 100 μl elution buffer (100mM NaHCO3, 1%SDS) each time. 16 μl 5M NaCl was then added and samples + inputs were reverse cross-linked at 65°C, RNase A and proteinase K treated and purified using ultrapure phenol/chloroform (ThermoFisher, Cat.N: 15593-049). Libraries were prepared using Illumina’s TruSeq ChIP Sample Preparation Kit, according to the manufacturer’s instructions with two exceptions: libraries were PCR amplified using KAPA HiFi Library Ampification Kit for 10-12 cycles to maximize complexity and the gel-based size-selection was performed after PCR amplification.
Replication Timing Sequencing (Repli-Seq)
Genome-wide Replication timing (RT) profiles were constructed as previously described (Marchal et al., 2018, Ryba et al., 2012). Briefly, cells were pulse labeled with BrdU and separated into early and late S-phase fractions by flow cytometry and processed by Repli-Seq. Sequencing libraries of BrdU-substituted DNA from early and late fractions were prepared by NEBNext Ultra DNA Library Prep Kit for Illumina (E7370). 50bp single end sequencing was performed on Illumina-HiSeq 2500.
Oligopaint Probe design and synthesis
Oligopaint libraries were constructed following the procedures described in Beliveau et al. (2015); see the Oligopaints website (https://oligopaints.hms.harvard.edu) for further details. Libraries were ordered from CustomArray (Bothell, WA) in the 12K Oligo pool format. Genomic regions A, B, C, D used in this study were located on human chromosome 5. Hg19 coordinates, size, number and density of probes for the libraries are given in Table S3.
Gene-poor and gene-rich oligopaint libraries were discovered using the “stringent” parameter set of OligoMiner (Beliveau et al., 2015), consisting of 40-46-mer genomic sequences throughout the regions of interest. TSS oligopaint libraries were discovered using the “balanced” parameter set, consisting of 35-41-mer genomic sequences. These oligonucleotide probes can now be retrieved from the Oligopaints website (https://oligopaints.hms.harvard.edu). Each library contains a universal primer pair followed by a specific primer pair hooked to the 40-46-mer or 35-41-mer genomic sequences (124-130-mers or 119-125 in total, respectively). Oligopaint libraries were produced by emulsion PCR amplification from oligonucleotide pools followed by a “two-step PCR” procedure and the lambda exonuclease method described in Beliveau et al. (2015). The “two-step PCR” leads to secondary oligonucleotide-binding sites for signal amplification with a secondary oligonucleotide (Sec1 or Sec6) containing two additional fluorochromes, each oligonucleotide carrying three fluorochromes in total. All Oligonucleotides used for Oligopaint production were purchased from Integrated DNA Technologies (IDT, Leuven, Belgium). All oligonucleotide sequences (5′ - > 3′) used in this study are listed below (all probe details are in Table S3).
Emulsion PCR with universal primers
BB297-FWD GACTGGTACTCGCGTGACTTG
BB299-REV GTAGGGACACCTCTGGACTGG
Two-step PCR with specific primers
PCR1 with FWD 5′ phosphorylation and REV 53-mer primers
A (red) BB298-FWD: /5Phos/CGTCAGTACAGGGTGTGATGC
Sec6-BB187-REV: /Sec6BS/TTGATCTTGACCCATCGAAGC
B (green) BB82-FWD: /5Phos/GTATCGTGCAAGGGTGAATGC
Sec1-BB278-REV: /Sec1BS/GAGCAGTCACAGTCCAGAAGG
B (red) BB82-FWD: /5Phos/GTATCGTGCAAGGGTGAATGC
Sec6-BB278-REV: /Sec6BS/GAGCAGTCACAGTCCAGAAGG
C (green) BB81-FWD: /5Phos/ATCCTAGCCCATACGGCAATG
Sec1-BB281-REV: /Sec1BS/GGACATGGGTCAGGTAGGTTG
C (red) BB81-FWD: /5Phos/ATCCTAGCCCATACGGCAATG
Sec6-BB281-REV: /Sec6BS/GGACATGGGTCAGGTAGGTTG
D (green) BB293-FWD: /5Phos/CCGAGTCTAGCGTCTCCTCTG
Sec1-BB294-REV: /Sec1BS/AACAGAGCCAGCCTCTACCTG
Secondary Binding Sequences (Sec1BS and Sec6BS):
Sec1BS: CACCGACGTCGCATAGAACGGAAGAGCGTGTG
Sec6BS: CACACGCTCTCCGTCTTGGCCGTGGTCGATCA
PCR2 with the labeled REV ‘back primer’
BB506-Alexa488 /5Alex488N/CACCGACGTCGCATAGAACGG
BB511-Cy3 /5Cy3/CACACGCTCTCCGTCTTGGC
Secondary Oligos carrying two fluorochromes
Sec1-Alexa488-X2: /
5Alex488N/CACACGCTCTTCCGTTCTATGCGACGTCGGTGagatgttt
/3AlexF488N/
Sec6-Cy3-X2:
/5Cy3/TGATCGACCACGGCCAAGACGGAGAGCGTGTGagatgttt
/3Cy3Sp/
FISH
3D FISH was adapted from Bantignies et al. (2011). Briefly, cells were fixed with 4% PFA in PBS at room temperature for 15 min. Cells were treated with PBS, 0.5% Triton for 10 minutes, washed in PBS, incubated with 0.1M HCl for 10 minutes, washed in 2XSSCT (2XSSC, 0.1% Tween20) and incubated for 60 minutes in 50% Formamide, 2XSCCT. Probe mixture contains 20 pmol of each probe with 20 pmol of their complementary secondary, 0.8 μl of ribonuclease A (10 mg/ml), and FISH hybridization buffer [FHB; 50% formamide, 10% dextran sulfate, 2 × SSC, and salmon sperm DNA (0.5 mg/ml)], in a total mixture volume of approximately 20 to 25 μl, keeping at least a 3:4 ratio of FHB/total volume. Probe mixture was added to the coverslip before sealing on a glass slide with rubber cement. Cell DNA was denatured at 80°C for 20 min, and hybridization was performed at 37°C overnight in a humid dark chamber. Cells were then washed 3 × 5 min at 37°C in 2 × SSC, 3 × 5 min at 45°C in 0.1 × SSC, and 2 × 5 min in PBS before DNA counterstaining with DAPI (final concentration at 0.3 mg/ml in PBS). After final washing in PBS, coverslips were mounted on slides with Vectashield (CliniSciences) and sealed with nail polish.
Image acquisition
Images were acquired using confocal laser scanning microscopy (Leica SP8) equipped with x63/1.4 NA Plan Apochromat oil immersion objective (pixel size of 59 nm, z-step of 300 nm).
Images for SAHF screening were acquired using wide field microscopy (Zeiss Axioimager Apotome) equipped with x63/1.4 NA Plan Apochromat oil immersion objective (Pixel size of 102 nm, z-step of 240 nm).
Whole genome bisulfite sequencing (WGBS)
Genomic DNA was isolated from the cell using QIAGEN DNeasy Blood & Tissue Kit (QIAGEN, Valencia, USA) following manufacturers’ recommendations. The WGBS libraries were constructed following Illumina’s recommendations using Illumina TruSeq DNA Methylation Kit and EZ DNA Methylation-Gold kit.
Quantification and Statistical Analysis
ChIP-Seq Analysis
Fastq files were aligned to the hg19 reference genome, PCR duplicates were removed using Samtools and normalized genome coverage tracks were generated from uniquely mapping reads (mapq > 30) using deepTools2. In order to avoid double counting for paired-end data, reads with mates were extended to match the fragment size defined by the two read mates, and the second read mate was discarded. Single-end reads, unmated reads, mate reads that map too far apart (> 4x fragment length) were extended by 200 bp. Biological replicates were pooled and coverage was then calculated as average reads per million of mapped reads (RPM) in 10bp bins. To determine the peaks for ChIPs with narrow binding profiles (H3K4me3), datasets were uniformly processed using the MACS2 with default parameters. For broad histone marks, the peaks (or regions) we employed SICER2 with default parameters. We have called peaks in both individual replicates and in the pooled dataset. Then we used bedtools intersect to get common peaks between replicates and finally these peaks were intersected with the pooled peaks data to get the final peak coordinate and score. This method was employed on both MACS2 and SICER2 generated peaks.
RNA-Seq Analysis
Fastq files were aligned to the hg19 reference genome using STAR_2.4.0i in basic two Pass mode using the ‘‘Encode’’ options as specified in the manual. PCR duplicates were removed and RPM normalized strand-specific coverage tracks based on uniquely aligned reads were also produced using STAR in ‘‘inputAlignmentsFromBAM’’ runMode. Reads overlapping with annotated genes (Ensembl build hg19) were counted using the summarizeOverlaps function from the R package ‘‘GenomicAlignments’’ in strand-specific, paired – end mode. FPKM (Fragments per kilobase per million mapped fragments) counts and differential expression was estimated using DESeq2. Genes with FPKM > = 1 were considered expressed for subsequent analysis.
Hi-C Data Analysis
The Hi-C data analysis was performed according to Bonev et al. (Bonev et al., 2017, Olivares-Chauvet et al., 2016). Briefly, raw sequencing reads were mapped to hg19 reference genome using Bowtie2, and uniquely mapped (MAPQ > 30) reads being translated into a pair of fragment-ends (fends) by associating each read with its downstream fend. Library statistics are presented in Table S1 and include total reads sequenced and number of valid interactions after filtering (PCR duplications, no digestion etc). Downstream analysis was then performed at fragment end resolution with no binning unless otherwise stated. To generate an expected model, we shuffled the observed Hi-C contacts using the Shaman R package (https://bitbucket.org/tanaylab/shaman) with default parameters (Olivares-Chauvet et al., 2016). The trans contacts are calculated as ratio of sum of observed contacts between the regions in trans versus sum of their marginal contacts (the contacts between region of interest and all other chromosomes in trans). Related to Figure S2I.
Contact Probability
Contact probability as a function of the genomic distance was calculated as previously described (Bonev et al., 2017, Olivares-Chauvet et al., 2016). We calculated the distribution of the Hi-C contacts either as a log10 contact probability in log10 genomic distance bins, or in order to better visualize differences between conditions, as a simple contact probability (sum of the observed counts per log2 bin, divided by the all of the observed contacts, without normalizing for the bin size). We measured the ‘‘contact probability scaling’’ exponent as the slope of the best-fit line of the cis-decay curve when plotted on log-log axes, within a chosen range of distances. In addition to the genome-wide contact probability curves which were aggregated across individual chromosomes, we also generated contact probability maps where we considered only contacts that are located completely within a TAD (intra-TAD cis-decay curve) or inter-TAD. For Figure 1F we also took into consideration whether the interaction is across compartments (A-B or B-A) or within a compartment (A-A, B-B).
Insulation, TAD, and TAD Boundary Calling
Calculation of insulation scores and boundary calling was performed as described (Bonev et al., 2017, Nagano et al., 2017, Olivares-Chauvet et al., 2016). Briefly insulation was based on observed contacts the definition of insulation score was taken from Nagano et al. and Olivares-Chauvet et al. (Nagano et al., 2017, Olivares-Chauvet et al., 2016). The insulation score was computed individually on replicates and on the pooled contact map at 1Kb resolution within a region of ± 250Kb and is multiplied by (−1) so that high insulation score represents strong insulation. To account for any genome-wide changes in the insulation score, we further normalized it by multiplying with a factor defined as the average insulation score across all 1Kb genomic bins in each cell type, divided by the mean of all cell types. These insulation scores were termed as normalized insulation scores. Domain boundaries were then defined as the local 2Kb maxima in regions, where the insulation score is above the 90% quantile of the genome-wide distribution. Boundaries within 10Kb of each other were merged and only boundaries, which were detected in at least 2 biological replicates, were retained. Domains shorter than 50Kb were discarded. Related to Figure 6.
Identification of A and B Compartments
TADs were assigned to either the A or the B compartment as described (Bonev et al., 2017, Nagano et al., 2017). In brief, we used k-means clustering (K = 2) on the log2 ratio of observed and expected (based on genomic length) inter-TAD contacts in trans. Domains in each cluster exhibit distinct signature based on the enrichment of replication timing (Figure S1G), prompting us to assign cluster 1 to the B, inactive compartment and cluster 2 to the A, active compartment. To determine the compartment strength, we calculated the log2 ratio of observed versus expected contacts (intrachromosomal separated by at least 10Mb) either between domains of the same (A-A, B-B) or different type (A-B). As another measure of compartmentalization which relies on the intra-chromosomal contacts, we calculated the dominant eigenvector of the contact matrices binned at 100Kb as described (Lieberman-Aiden et al., 2009) using scripts available at (https://github.com/dekkerlab/cworld-dekker). Related to Figures 1 and 5).
Image Analysis
3D-FISH analysis was performed using Imaris software and its XT module. After Background substraction and Gaussian filter (sigma = 1), distances between FISH probes were calculated between the nearest mutual neighbor centers of mass of segmented FISH. For distance to nuclear periphery, nuclei were segmented in 3D using DAPI staining and the distances between FISH centers of mass and closest DAPI segmentation contour were calculated. SAHF bodies were segmented using DAPI staining, and FISH centers of mass present within SAHF bodies were considered as colocalized. Distances between FISH spots and SAHF bodies were calculated between FISH and SAHF centers of mass. Diameters of FISH signals were calculated using ImageJ software from Gaussian filtered (sigma = 1) and maximum projected images, using the Full Width at Half Maximum (FWHM) of Gaussian fitted curves to the intensity measured along scan-lines passing through FISH loci. Areas of chromosome paints and SAHF scores were calculated using MATLAB (Related to Figure 2). For chromosome paints areas, channels were smoothed using Gaussian filters (sigma = 3 for DAPI; sigma = 1 for chromosome paints) and segmented in 2D; only nuclei displaying 2 chromosome paints segmented objects were considered for analysis (Related to Figure 1).
For SAHF score quantification, images were first maximum projected along the z axis. Nuclei were then segmented from Gaussian filtered (sigma = 3) DAPI staining. SAHF score was calculated using the coefficient of variation (CV) of DAPI intensity (intensity values smoothed using a Gaussian filter with sigma = 1). Briefly, the standard deviation and the mean DAPI intensity of each nucleus were calculated. CV were then calculated as the ratio of the standard deviation over the mean intensity (Related to Figures 4 and 5) (Contrepois et al., 2012).
SAHF specific gene expression analysis
To identify genes that might be regulated by the formation of SAHFs, we selected the genes that i) do not show significant changes in transcription at OISD2 (OISD2 versus OISD0, padj > 0.05), ii) are significantly upregulated upon SAHF formation at OISD4 (OISD4 versus OISD0, log2FC > 1, padj ≤ 0.05). Then, the subset of these genes that either overlap with SAHD domains or are located at ± 3Mb from the closest SAHD (Adjacent SAHD: ADJ) were selected and their transcription was quantified. This genes set was termed as SAHF-Specific genes. The GEO was performed using DAVID Bioinformatics resources (Huang et al., 2009a, Huang et al., 2009b). Related to Figure 5.
Polymer Modeling
We modeled the genomic region from 100 to 180.94 Mbp of chromosome 5 by a semi-flexible self-avoiding block copolymer, consisting of 8094 beads of 10kbp, each of size b, moving within a face-centered cubic lattice of size SxSxS (Olarte-Plata et al., 2016). We used rigid wall condition to mimic the confinement of the region inside a chromosome territory, one face of the cubic box (z = 0) representing the nuclear membrane (Figure 3A).
For the region of interest, we collected the position of the SAHDs (Table S2) and of the constitutive LADs (Lenain et al., 2017). To each 10kbp bead i, we assigned a state (ei,li) regarding its epigenetic (ei) and lamina-interacting (li) status: ei = 1 if the bead is inside a SAHD ( = 0 otherwise),and li = 1 if the bead is inside a cLAD ( = 0 otherwise). The total energy of a given configuration of the polymer is then given by:
With k the bending rigidity of the polymer, θi the angle between the bond vectors i and i+1, ε(< 0) the contact energy between SAHD beads, δ(i,j) = 1 if beads i and j occupy nearest-neighbor sites on the lattice ( = 0 otherwise), γ(< 0) the contact energy between a cLAD bead and the membrane, and Δi = 1 if bead i occupy a site at the membrane ( = 0 otherwise). ε accounts for the capacity of proteins bound to heterochromatic regions like HP1 to oligomerize (Canzio et al., 2013, Larson et al., 2017, Strom et al., 2017), while γ reflects still unknown molecular mechanism that lead to the preferential location of cLAD at the nuclear periphery. The dynamics of the chain follows a simple kinetic Monte-Carlo scheme with local moves using a Metropolis criterion applied to H, as described in (Ghosh and Jost, 2018). The values of k( = 2.25kT), b( = 95 nm) and S( = 2 μm) were fixed using the coarse-graining strategy developed in (Ghosh and Jost, 2018) for a 10-nm fiber model and a volumic density ρ = 0.01 bp/nm3 (estimated by dividing the size of a diploid human genome 6 Gbp by the typical volume of cycling WI-38 nuclei (600 μm3)).
Time mapping between the simulation and experimental time were roughly performed by considering the dynamics of relaxation of the long-range contacts between OISD0 and OISD6 (Figure S3M). Such mapping leads to a time-evolution of the mean squared displacement MSD(t)(in μm2)≈0.003 t1/2 with time t given in seconds, in agreement with typical experimental estimation of MSD in human cells (Lucas et al., 2014). Considering the simplicity of the model, estimated times should be viewed as orders of magnitude of real times. For a given parameter set, we simulate 200 independent trajectories starting from random, compact, knot-free initial configurations as in (Ghosh and Jost, 2018).
We estimated ε and γ parameters for OISD0 cells by comparing model predictions with various experimental data. ε = −0.055 kT was estimated by adjusting the predicted evolution of the “observed vs expected” ratio Pe(s)/Ptot(s) as a function of the genomic distance s to the corresponding experimental values (∼2.2 fold), with Pe(s) the average contact probability between SAHD regions and Ptot(s) the total average contact probability between any pairs of loci, sometimes called the expected contact probability (Figure 3B). γ≈-0.6 kT was estimated by assuming that the median distance to nuclear periphery of LAD monomers was ∼700nm (see the position of B & C probes that are in LADs in Figure 2G). Interestingly, the resulting LAD organization is fully consistent with independent experimental measurements of LAD positioning inside mammalian cells performed by Kind et al. (Kind et al., 2013) (Figure S3B). In particular, we retrieved that ∼30% of the LADs remains close to the periphery in every cell but that the identity of the regions bound to the membrane is very stochastic and changes at every cell cycle.
For a given polymer configuration, dense SAHD compartments (Figures 3 and S3) were determined by
-
(1)
convoluting the position of each SAHD monomer on the lattice by a 3D Gaussian of width λ = 270 nm to create a 3D matrix of intensity is occupied by a SAHD bead;
-
(2)
by thresholding this matrix to half its maximal value to focus on dense clusters (M←(M ≥ 30))
TADbit Models
We binned at 200 kb the normalized Hi-C matrices of chromosome 5. These matrices of interactions were further normalized through Z-score transformation, and used as input to establish the restraints of the modeling. The modeling parameters of max_dist, up_freq and low_freq were optimized as described (Baù and Marti-Renom, 2012). From this optimization step we selected the set of parameters that globally maximized the satisfaction of restraints in all the ensemble of time points considered (max_dist = 3000, up_freq = 1.0, low_freq = −1.0). We then generated four thousand models from which we kept the best one thousand for the analysis. Each of these one thousand models is meant to represent the chromatin conformation of a subpopulation of cells within the total pull used in the Hi-C experiment. The general methodology follows each of the steps defined in (Baù and Marti-Renom, 2012, Serra et al., 2017) and was automatized using TADbit (Serra et al., 2017). The 3D models were finally scaled to match sizes observed by FISH. To do so, we computed the median of the distances between the A, B, C, and D coordinates in the OIS sample 3D models and compared it with the median of the same distances observed by FISH. We used the ratio of these medians to correct the sizes of the 3D models (they were overall 3.72 times larger). Distances were extracted independently in each of the one thousand model of each time point. Finally, to visually inspect the subpopulations of models generated at each time point, we clustered them by structural similarity. Each cluster of 3D models representing a major conformational state of the chromatin within the subpopulation of cells. The clustered models were then uploaded to TADkit (https://github.com/3DGenomes/TADkit) in the MuG VRE (http://multiscalegenomics.eu) for visual inspection and alignment with other genomic features, and chromatin marks. Related to Figures 2 and 3.
WGBS Analysis
Raw reads were trimmed using trim galore. The reads that passed trim galore were analyzed using methpipe package (version 3.4.3). Briefly the reads were mapped to hg19 genome reference using “walt,” and PCR duplicates were removed with “duplicate-remover” in methpipe package. The output files were then analyzed using “methcounts” to obtain methylation levels at each cytosine site for both strands. Then “merge-methcounts” was used to merge all replicates. Since CpG existence is symmetrical in nature we employed “symmetric-cpgs” to produce a single value for each CpG pair. CpG sites which were covered with at least 10 reads were taken for downstream analysis. Library statistics are presented in Table S1 and include total reads sequenced. These tracks were then used in Figures 5I and 5J.
Repli-Seq Analysis
Reads of quality scores above 30 were mapped to hg19 reference genome using bowtie2. Approximately 7 million uniquely mapped reads were obtained from each library. Read counts were binned into 5kb non-overlapping windows, and log2 ratios of read-counts between early and late fractions were calculated. For classifying the replication timing into different replication timing regions, we follow the following cutoff on the Log2 ratio between early and late fractions. The following definition taken in Figure 4B
G1 phase: Log 2 ratio from 1 and above
S1 phase: Log 2 ratio from 0.5 to 1
S2 phase: Log 2 ratio from 0 to 0.5
S3 phase: Log 2 ratio from −0.5 to 0
S4 phase: Log 2 ratio from −1 to −0.5
G2 phase: Log 2 ratio from −1 and below
Data and Code Availability
The accession number for the data reported in this paper is GEO: GSE130306
Software
The R package to compute the expected tracks and the Hi-C scores is freely available at: https://bitbucket.org/tanaylab/shaman. Processing of the raw-reads is done using https://bitbucket.org/tanaylab/schic2 as described above.
Data Resources
The accession number for the data reported in this paper is GEO: GSE130306
The 3D model of OIS and RS are available at https://vre.multiscalegenomics.eu/data_repositories/data_senescence.php
Acknowledgments
We thank Dr. Carl Mann for generously providing the WI-38-RAF and BJ-RAF cells used in this study, Genotoul facility for providing high computing resources, and MRI-IGH for microscopy support. We thank Dr. Guy Nir and Ting Wu for their support in designing oligopaints used in the study. We thank Max James Fritz for proofreading our manuscript. S.S. was supported by LabEx EPIGENMED postdoctoral fellowship and MuG project. Research in the laboratory of G.C. was supported by the CNRS, the University of Montpellier, and by grants from the European Research Council (ERC-2008-AdG No 232947), the European Union’s Horizon 2020 research and innovation programme under grant agreement No 676556 (MuG), the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 813327 (ChromDesign), the Agence Nationale de la Recherche (N. ANR-15-CE12-006-01), the Fondation pour la Recherche Médicale (Grant N. DEI20151234396), the French National Cancer Institute (INCa, Projects N. PLBIO16-222 and PLBIO18-362), the Laboratory of Excellence EpiGenMed (N. ANR-10-LABX-12), and the MSD Grant “GENE-IGH.” Research in the laboratory of J.M.-L. was supported by INSERM, the University of Montpellier, the CHRU Montpellier/SAFE-iPSC facility (INGESTEM consortium Infrastructure en Biology Santé), the French National Cancer Institute (INCa Project N. PLBIO12-103), and by grant from Ligue National Centre le Contre le cancer “Equipe Labellisée (2015–2019).” F.B. was supported by CNRS. Q.S. was supported by the French Ministry of Higher Education and Research and La Ligue Nationale Contre le Cancer. D.J. was supported by CNRS Mission Interdisciplinarité, Agence Nationale de la Recherche (ANR-15-CE12-006-03) and the Fondation pour la Recherche Médicale (DEI20151234396) and acknowledges computational resources from CIMENT infrastructure (supported by the Rhone-Alpes region, Grant CPER07 13 CIRA). Work at the M.A.M.-R. lab was supported by the European Research Council under the 7th Framework Program FP7/2007-2013 (ERC grant agreement 609989), the European Union’s Horizon 2020 research and innovation programme (grant agreement 676556), the Ministry of Economy and Competitiveness (BFU2017-85926-P), and the Agència de Gestió d'Ajuts Universitaris i de Recerca, AGAUR (SGR468). Work at CRG, BIST, and UPF was in part funded by the Spanish Ministry of Economy and Competitiveness, “Centro de Excelencia Severo Ochoa 2013-2017” (SEV-2012-0208), and “Centro de Excelencia María de Maeztu 2016-2019.” This article/publication is based upon work from COST Action CA18127, supported by COST (European Cooperation in Science and Technology).
Author Contributions
Conceptualization, S.S., J.-M.L., and G.C.; Methodology, S.S.; Investigation, S.S., P. Bensadoun, P. Bouret, L.F., F.P., and J.-C.R.-M.; Software, S.S., B.B., Q.S., D.J., F.S., G.L.P., C.V., and V.L.; Resources, D.M.G., M.A.M.-R., J.L.G., M.O., and F.B.; Review and Editing, S.S., J.-M.L., and G.C.; Supervision, J.-M.L. and G.C.
Declaration of Interests
The authors declare no competing interests.
Published: March 26, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.molcel.2020.03.007.
Contributor Information
Jean-Marc Lemaitre, Email: jean-marc.lemaitre@inserm.fr.
Giacomo Cavalli, Email: giacomo.cavalli@igh.cnrs.fr.
Supporting Citations
The following references appear in the Supplemental Information: Becker et al. (2017).
Supplemental Information
References
- Bantignies F., Roure V., Comet I., Leblanc B., Schuettengruber B., Bonnet J., Tixier V., Mas A., Cavalli G. Polycomb-dependent regulatory contacts between distant Hox loci in Drosophila. Cell. 2011;144:214–226. doi: 10.1016/j.cell.2010.12.026. [DOI] [PubMed] [Google Scholar]
- Baù D., Marti-Renom M.A. Genome structure determination via 3C-based data integration by the Integrative Modeling Platform. Methods. 2012;58:300–306. doi: 10.1016/j.ymeth.2012.04.004. [DOI] [PubMed] [Google Scholar]
- Baù D., Sanyal A., Lajoie B.R., Capriotti E., Byron M., Lawrence J.B., Dekker J., Marti-Renom M.A. The three-dimensional folding of the α-globin gene domain reveals formation of chromatin globules. Nat. Struct. Mol. Biol. 2011;18:107–114. doi: 10.1038/nsmb.1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker J.S., McCarthy R.L., Sidoli S., Donahue G., Kaeding K.E., He Z., Lin S., Garcia B.A., Zaret K.S. Genomic and Proteomic Resolution of Heterochromatin and Its Restriction of Alternate Fate Genes. Mol. Cell. 2017;68:1023–1037. doi: 10.1016/j.molcel.2017.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beliveau B.J., Boettiger A.N., Avendaño M.S., Jungmann R., McCole R.B., Joyce E.F., Kim-Kiselak C., Bantignies F., Fonseka C.Y., Erceg J. Single-molecule super-resolution imaging of chromosomes and in situ haplotype visualization using Oligopaint FISH probes. Nat. Commun. 2015;6:7147. doi: 10.1038/ncomms8147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonev B., Cavalli G. Organization and function of the 3D genome. Nat. Rev. Genet. 2016;17:661–678. doi: 10.1038/nrg.2016.112. [DOI] [PubMed] [Google Scholar]
- Bonev B., Mendelson Cohen N., Szabo Q., Fritsch L., Papadopoulos G.L., Lubling Y., Xu X., Lv X., Hugnot J.P., Tanay A., Cavalli G. Multiscale 3D Genome Rewiring during Mouse Neural Development. Cell. 2017;171:557–572. doi: 10.1016/j.cell.2017.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campisi J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 2013;75:685–705. doi: 10.1146/annurev-physiol-030212-183653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campisi J., d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007;8:729–740. doi: 10.1038/nrm2233. [DOI] [PubMed] [Google Scholar]
- Canzio D., Liao M., Naber N., Pate E., Larson A., Wu S., Marina D.B., Garcia J.F., Madhani H.D., Cooke R. A conformational switch in HP1 releases auto-inhibition to drive heterochromatin assembly. Nature. 2013;496:377–381. doi: 10.1038/nature12032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho C., L’Hôte V., Courbeyrette R., Kratassiouk G., Pinna G., Cintrat J.C., Denby-Wilkes C., Derbois C., Olaso R., Deleuze J.F. Glucocorticoids delay RAF-induced senescence promoted by EGR1. J. Cell Sci. 2019;132 doi: 10.1242/jcs.230748. Published online August 22, 2019. [DOI] [PubMed] [Google Scholar]
- Catez F., Ueda T., Bustin M. Determinants of histone H1 mobility and chromatin binding in living cells. Nat. Struct. Mol. Biol. 2006;13:305–310. doi: 10.1038/nsmb1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra T., Kirschner K., Thuret J.Y., Pope B.D., Ryba T., Newman S., Ahmed K., Samarajiwa S.A., Salama R., Carroll T. Independence of repressive histone marks and chromatin compaction during senescent heterochromatic layer formation. Mol. Cell. 2012;47:203–214. doi: 10.1016/j.molcel.2012.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra T., Ewels P.A., Schoenfelder S., Furlan-Magaril M., Wingett S.W., Kirschner K., Thuret J.Y., Andrews S., Fraser P., Reik W. Global reorganization of the nuclear landscape in senescent cells. Cell Rep. 2015;10:471–483. doi: 10.1016/j.celrep.2014.12.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang M., Michieletto D., Brackley C.A., Rattanavirotkul N., Mohammed H., Marenduzzo D., Chandra T. Polymer Modeling Predicts Chromosome Reorganization in Senescence. Cell Rep. 2019;28:3212–3223. doi: 10.1016/j.celrep.2019.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contrepois K., Thuret J.Y., Courbeyrette R., Fenaille F., Mann C. Deacetylation of H4-K16Ac and heterochromatin assembly in senescence. Epigenetics Chromatin. 2012;5:15. doi: 10.1186/1756-8935-5-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremer T., Cremer M., Hübner B., Strickfaden H., Smeets D., Popken J., Sterr M., Markaki Y., Rippe K., Cremer C. The 4D nucleome: Evidence for a dynamic nuclear landscape based on co-aligned active and inactive nuclear compartments. FEBS Lett. 2015;589(20 Pt A):2931–2943. doi: 10.1016/j.febslet.2015.05.037. [DOI] [PubMed] [Google Scholar]
- Criscione S.W., De Cecco M., Siranosian B., Zhang Y., Kreiling J.A., Sedivy J.M., Neretti N. Reorganization of chromosome architecture in replicative cellular senescence. Sci. Adv. 2016;2:e1500882. doi: 10.1126/sciadv.1500882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruickshanks H.A., McBryan T., Nelson D.M., Vanderkraats N.D., Shah P.P., van Tuyn J., Singh Rai T., Brock C., Donahue G., Dunican D.S. Senescent cells harbour features of the cancer epigenome. Nat. Cell Biol. 2013;15:1495–1506. doi: 10.1038/ncb2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Cecco M., Criscione S.W., Peckham E.J., Hillenmeyer S., Hamm E.A., Manivannan J., Peterson A.L., Kreiling J.A., Neretti N., Sedivy J.M. Genomes of replicatively senescent cells undergo global epigenetic changes leading to gene silencing and activation of transposable elements. Aging Cell. 2013;12:247–256. doi: 10.1111/acel.12047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Micco R., Sulli G., Dobreva M., Liontos M., Botrugno O.A., Gargiulo G., dal Zuffo R., Matti V., d’Ario G., Montani E. Interplay between oncogene-induced DNA damage response and heterochromatin in senescence and cancer. Nat. Cell Biol. 2011;13:292–302. doi: 10.1038/ncb2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon J.R., Selvaraj S., Yue F., Kim A., Li Y., Shen Y., Hu M., Liu J.S., Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–380. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evangelou K., Lougiakis N., Rizou S.V., Kotsinas A., Kletsas D., Muñoz-Espín D., Kastrinakis N.G., Pouli N., Marakos P., Townsend P. Robust, universal biomarker assay to detect senescent cells in biological specimens. Aging Cell. 2017;16:192–197. doi: 10.1111/acel.12545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foti R., Gnan S., Cornacchia D., Dileep V., Bulut-Karslioglu A., Diehl S., Buness A., Klein F.A., Huber W., Johnstone E. Nuclear Architecture Organized by Rif1 Underpins the Replication-Timing Program. Mol. Cell. 2016;61:260–273. doi: 10.1016/j.molcel.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost L., Baez M.A., Harrilal C., Garabedian A., Fernandez-Lima F., Leng F. The Dimerization State of the Mammalian High Mobility Group Protein AT-Hook 2 (HMGA2) PLoS ONE. 2015;10:e0130478. doi: 10.1371/journal.pone.0130478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S.K., Jost D. How epigenome drives chromatin folding and dynamics, insights from efficient coarse-grained models of chromosomes. PLoS Comput. Biol. 2018;14:e1006159. doi: 10.1371/journal.pcbi.1006159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giri S., Aggarwal V., Pontis J., Shen Z., Chakraborty A., Khan A., Mizzen C., Prasanth K.V., Ait-Si-Ali S., Ha T., Prasanth S.G. The preRC protein ORCA organizes heterochromatin by assembling histone H3 lysine 9 methyltransferases on chromatin. eLife. 2015;4 doi: 10.7554/eLife.06496. Published online April 29, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorgoulis V., Adams P.D., Alimonti A., Bennett D.C., Bischof O., Bishop C., Campisi J., Collado M., Evangelou K., Ferbeyre G. Cellular Senescence: Defining a Path Forward. Cell. 2019;179:813–827. doi: 10.1016/j.cell.2019.10.005. [DOI] [PubMed] [Google Scholar]
- Hayflick L. The Limited in Vitro Lifetime of Human Diploid Cell Strains. Exp. Cell Res. 1965;37:614–636. doi: 10.1016/0014-4827(65)90211-9. [DOI] [PubMed] [Google Scholar]
- Hayflick L., Moorhead P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961;25:585–621. doi: 10.1016/0014-4827(61)90192-6. [DOI] [PubMed] [Google Scholar]
- Hernandez-Segura A., de Jong T.V., Melov S., Guryev V., Campisi J., Demaria M. Unmasking Transcriptional Heterogeneity in Senescent Cells. Curr. Biol. 2017;27:2652–2660. doi: 10.1016/j.cub.2017.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W., Sherman B.T., Lempicki R.A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Illner D., Zinner R., Handtke V., Rouquette J., Strickfaden H., Lanctôt C., Conrad M., Seiler A., Imhof A., Cremer T., Cremer M. Remodeling of nuclear architecture by the thiodioxoxpiperazine metabolite chaetocin. Exp. Cell Res. 2010;316:1662–1680. doi: 10.1016/j.yexcr.2010.03.008. [DOI] [PubMed] [Google Scholar]
- Ito S., Magalska A., Alcaraz-Iborra M., Lopez-Atalaya J.P., Rovira V., Contreras-Moreira B., Lipinski M., Olivares R., Martinez-Hernandez J., Ruszczycki B. Loss of neuronal 3D chromatin organization causes transcriptional and behavioural deficits related to serotonergic dysfunction. Nat. Commun. 2014;5:4450. doi: 10.1038/ncomms5450. [DOI] [PubMed] [Google Scholar]
- Jeanblanc M., Ragu S., Gey C., Contrepois K., Courbeyrette R., Thuret J.Y., Mann C. Parallel pathways in RAF-induced senescence and conditions for its reversion. Oncogene. 2012;31:3072–3085. doi: 10.1038/onc.2011.481. [DOI] [PubMed] [Google Scholar]
- Jost D., Carrivain P., Cavalli G., Vaillant C. Modeling epigenome folding: formation and dynamics of topologically associated chromatin domains. Nucleic Acids Res. 2014;42:9553–9561. doi: 10.1093/nar/gku698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kind J., Pagie L., Ortabozkoyun H., Boyle S., de Vries S.S., Janssen H., Amendola M., Nolen L.D., Bickmore W.A., van Steensel B. Single-cell dynamics of genome-nuclear lamina interactions. Cell. 2013;153:178–192. doi: 10.1016/j.cell.2013.02.028. [DOI] [PubMed] [Google Scholar]
- Kuilman T., Michaloglou C., Vredeveld L.C., Douma S., van Doorn R., Desmet C.J., Aarden L.A., Mooi W.J., Peeper D.S. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008;133:1019–1031. doi: 10.1016/j.cell.2008.03.039. [DOI] [PubMed] [Google Scholar]
- Lapasset L., Milhavet O., Prieur A., Besnard E., Babled A., Aït-Hamou N., Leschik J., Pellestor F., Ramirez J.M., De Vos J. Rejuvenating senescent and centenarian human cells by reprogramming through the pluripotent state. Genes Dev. 2011;25:2248–2253. doi: 10.1101/gad.173922.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson A.G., Elnatan D., Keenen M.M., Trnka M.J., Johnston J.B., Burlingame A.L., Agard D.A., Redding S., Narlikar G.J. Liquid droplet formation by HP1α suggests a role for phase separation in heterochromatin. Nature. 2017;547:236–240. doi: 10.1038/nature22822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenain C., de Graaf C.A., Pagie L., Visser N.L., de Haas M., de Vries S.S., Peric-Hupkes D., van Steensel B., Peeper D.S. Massive reshaping of genome-nuclear lamina interactions during oncogene-induced senescence. Genome Res. 2017;27:1634–1644. doi: 10.1101/gr.225763.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman-Aiden E., van Berkum N.L., Williams L., Imakaev M., Ragoczy T., Telling A., Amit I., Lajoie B.R., Sabo P.J., Dorschner M.O. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas J.S., Zhang Y., Dudko O.K., Murre C. 3D trajectories adopted by coding and regulatory DNA elements: first-passage times for genomic interactions. Cell. 2014;158:339–352. doi: 10.1016/j.cell.2014.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun A.T., Smyth G.K. diffHic: a Bioconductor package to detect differential genomic interactions in Hi-C data. BMC Bioinformatics. 2015;16:258. doi: 10.1186/s12859-015-0683-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchal C., Sasaki T., Vera D., Wilson K., Sima J., Rivera-Mulia J.C., Trevilla-García C., Nogues C., Nafie E., Gilbert D.M. Genome-wide analysis of replication timing by next-generation sequencing with E/L Repli-seq. Nat. Protoc. 2018;13:819–839. doi: 10.1038/nprot.2017.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mas G., Blanco E., Ballaré C., Sansó M., Spill Y.G., Hu D., Aoi Y., Le Dily F., Shilatifard A., Marti-Renom M.A., Di Croce L. Promoter bivalency favors an open chromatin architecture in embryonic stem cells. Nat. Genet. 2018;50:1452–1462. doi: 10.1038/s41588-018-0218-5. [DOI] [PubMed] [Google Scholar]
- Muñoz-Espín D., Cañamero M., Maraver A., Gómez-López G., Contreras J., Murillo-Cuesta S., Rodríguez-Baeza A., Varela-Nieto I., Ruberte J., Collado M., Serrano M. Programmed cell senescence during mammalian embryonic development. Cell. 2013;155:1104–1118. doi: 10.1016/j.cell.2013.10.019. [DOI] [PubMed] [Google Scholar]
- Nagano T., Lubling Y., Várnai C., Dudley C., Leung W., Baran Y., Mendelson Cohen N., Wingett S., Fraser P., Tanay A. Cell-cycle dynamics of chromosomal organization at single-cell resolution. Nature. 2017;547:61–67. doi: 10.1038/nature23001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagashima R., Hibino K., Ashwin S.S., Babokhov M., Fujishiro S., Imai R., Nozaki T., Tamura S., Tani T., Kimura H. Single nucleosome imaging reveals loose genome chromatin networks via active RNA polymerase II. J. Cell Biol. 2019;218:1511–1530. doi: 10.1083/jcb.201811090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita M., Nũnez S., Heard E., Narita M., Lin A.W., Hearn S.A., Spector D.L., Hannon G.J., Lowe S.W. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell. 2003;113:703–716. doi: 10.1016/s0092-8674(03)00401-x. [DOI] [PubMed] [Google Scholar]
- Narita M., Narita M., Krizhanovsky V., Nuñez S., Chicas A., Hearn S.A., Myers M.P., Lowe S.W. A novel role for high-mobility group a proteins in cellular senescence and heterochromatin formation. Cell. 2006;126:503–514. doi: 10.1016/j.cell.2006.05.052. [DOI] [PubMed] [Google Scholar]
- Nelson D.M., Jaber-Hijazi F., Cole J.J., Robertson N.A., Pawlikowski J.S., Norris K.T., Criscione S.W., Pchelintsev N.A., Piscitello D., Stong N. Mapping H4K20me3 onto the chromatin landscape of senescent cells indicates a function in control of cell senescence and tumor suppression through preservation of genetic and epigenetic stability. Genome Biol. 2016;17:158. doi: 10.1186/s13059-016-1017-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nora E.P., Lajoie B.R., Schulz E.G., Giorgetti L., Okamoto I., Servant N., Piolot T., van Berkum N.L., Meisig J., Sedat J. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature. 2012;485:381–385. doi: 10.1038/nature11049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochs F., Karemore G., Miron E., Brown J., Sedlackova H., Rask M.B., Lampe M., Buckle V., Schermelleh L., Lukas J., Lukas C. Stabilization of chromatin topology safeguards genome integrity. Nature. 2019;574:571–574. doi: 10.1038/s41586-019-1659-4. [DOI] [PubMed] [Google Scholar]
- Olarte-Plata J.D., Haddad N., Vaillant C., Jost D. The folding landscape of the epigenome. Phys. Biol. 2016;13:026001. doi: 10.1088/1478-3975/13/2/026001. [DOI] [PubMed] [Google Scholar]
- Olivares-Chauvet P., Mukamel Z., Lifshitz A., Schwartzman O., Elkayam N.O., Lubling Y., Deikus G., Sebra R.P., Tanay A. Capturing pairwise and multi-way chromosomal conformations using chromosomal walks. Nature. 2016;540:296–300. doi: 10.1038/nature20158. [DOI] [PubMed] [Google Scholar]
- Ozturk N., Singh I., Mehta A., Braun T., Barreto G. HMGA proteins as modulators of chromatin structure during transcriptional activation. Front. Cell Dev. Biol. 2014;2:5. doi: 10.3389/fcell.2014.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao S.S., Huntley M.H., Durand N.C., Stamenova E.K., Bochkov I.D., Robinson J.T., Sanborn A.L., Machol I., Omer A.D., Lander E.S., Aiden E.L. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159:1665–1680. doi: 10.1016/j.cell.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryba T., Battaglia D., Chang B.H., Shirley J.W., Buckley Q., Pope B.D., Devidas M., Druker B.J., Gilbert D.M. Abnormal developmental control of replication-timing domains in pediatric acute lymphoblastic leukemia. Genome Res. 2012;22:1833–1844. doi: 10.1101/gr.138511.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadaie M., Salama R., Carroll T., Tomimatsu K., Chandra T., Young A.R., Narita M., Pérez-Mancera P.A., Bennett D.C., Chong H. Redistribution of the Lamin B1 genomic binding profile affects rearrangement of heterochromatic domains and SAHF formation during senescence. Genes Dev. 2013;27:1800–1808. doi: 10.1101/gad.217281.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sati S., Cavalli G. Chromosome conformation capture technologies and their impact in understanding genome function. Chromosoma. 2017;126:33–44. doi: 10.1007/s00412-016-0593-6. [DOI] [PubMed] [Google Scholar]
- Schmitt C.A., Fridman J.S., Yang M., Lee S., Baranov E., Hoffman R.M., Lowe S.W. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell. 2002;109:335–346. doi: 10.1016/s0092-8674(02)00734-1. [DOI] [PubMed] [Google Scholar]
- Serra F., Baù D., Goodstadt M., Castillo D., Filion G.J., Marti-Renom M.A. Automatic analysis and 3D-modelling of Hi-C data using TADbit reveals structural features of the fly chromatin colors. PLoS Comput. Biol. 2017;13:e1005665. doi: 10.1371/journal.pcbi.1005665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano M., Lin A.W., McCurrach M.E., Beach D., Lowe S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- Sexton T., Yaffe E., Kenigsberg E., Bantignies F., Leblanc B., Hoichman M., Parrinello H., Tanay A., Cavalli G. Three-dimensional folding and functional organization principles of the Drosophila genome. Cell. 2012;148:458–472. doi: 10.1016/j.cell.2012.01.010. [DOI] [PubMed] [Google Scholar]
- Shah P.P., Donahue G., Otte G.L., Capell B.C., Nelson D.M., Cao K., Aggarwala V., Cruickshanks H.A., Rai T.S., McBryan T. Lamin B1 depletion in senescent cells triggers large-scale changes in gene expression and the chromatin landscape. Genes Dev. 2013;27:1787–1799. doi: 10.1101/gad.223834.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storer M., Mas A., Robert-Moreno A., Pecoraro M., Ortells M.C., Di Giacomo V., Yosef R., Pilpel N., Krizhanovsky V., Sharpe J., Keyes W.M. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell. 2013;155:1119–1130. doi: 10.1016/j.cell.2013.10.041. [DOI] [PubMed] [Google Scholar]
- Strom A.R., Emelyanov A.V., Mir M., Fyodorov D.V., Darzacq X., Karpen G.H. Phase separation drives heterochromatin domain formation. Nature. 2017;547:241–245. doi: 10.1038/nature22989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y., Zheng Y., Wang C., Liu Y. Glutathione depletion induces ferroptosis, autophagy, and premature cell senescence in retinal pigment epithelial cells. Cell Death Dis. 2018;9:753. doi: 10.1038/s41419-018-0794-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson E.C., Manning B., Zhang H., Lawrence J.B. Higher-order unfolding of satellite heterochromatin is a consistent and early event in cell senescence. J. Cell Biol. 2013;203:929–942. doi: 10.1083/jcb.201306073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West R.C., McWhorter E.S., Ali A., Goetzman L.N., Russ J.E., Gonzalez-Berrios C.L., Anthony R.V., Bouma G.J., Winger Q.A. HMGA2 is regulated by LIN28 and BRCA1 in human placental cells. Biol. Reprod. 2019;100:227–238. doi: 10.1093/biolre/ioy183. [DOI] [PubMed] [Google Scholar]
- Xu X., Lu Z., Qiang W., Vidimar V., Kong B., Kim J.J., Wei J.J. Inactivation of AKT induces cellular senescence in uterine leiomyoma. Endocrinology. 2014;155:1510–1519. doi: 10.1210/en.2013-1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun M.H., Davaapil H., Brockes J.P. Recurrent turnover of senescent cells during regeneration of a complex structure. eLife. 2015;4 doi: 10.7554/eLife.05505. Published online May 5, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X., Hu J., Yue S., Kristiani L., Kim M., Sauria M., Taylor J., Kim Y., Zheng Y. Lamins Organize the Global Three-Dimensional Genome from the Nuclear Periphery. Mol. Cell. 2018;71:802–815.e7. doi: 10.1016/j.molcel.2018.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X., Maricque B., Xie M., Li D., Sundaram V., Martin E.A., Koebbe B.C., Nielsen C., Hirst M., Farnham P. The Human Epigenome Browser at Washington University. Nat. Methods. 2011;8:989–990. doi: 10.1038/nmeth.1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X., Lowdon R.F., Li D., Lawson H.A., Madden P.A., Costello J.F., Wang T. Exploring long-range genome interactions using the WashU Epigenome Browser. Nat. Methods. 2013;10:375–376. doi: 10.1038/nmeth.2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zirkel A., Nikolic M., Sofiadis K., Mallm J.P., Brackley C.A., Gothe H., Drechsel O., Becker C., Altmuller J., Josipovic N. HMGB2 Loss upon Senescence Entry Disrupts Genomic Organization and Induces CTCF Clustering across Cell Types. Mol. Cell. 2018;70:730–744. doi: 10.1016/j.molcel.2018.03.030. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the data reported in this paper is GEO: GSE130306
Software
The R package to compute the expected tracks and the Hi-C scores is freely available at: https://bitbucket.org/tanaylab/shaman. Processing of the raw-reads is done using https://bitbucket.org/tanaylab/schic2 as described above.
Data Resources
The accession number for the data reported in this paper is GEO: GSE130306
The 3D model of OIS and RS are available at https://vre.multiscalegenomics.eu/data_repositories/data_senescence.php