

A robust d3-methylating agent DMTT has been developed, and its selectivity in d3-methylation of complex molecules evaluated.
Abstract
Methods to incorporate deuterium atoms into organic molecules are valuable for the pharmaceutical industry. The introduction of deuterium atoms by a synthetic method enables the direct tracing of the drug molecule without substantially altering its structure or function. The methyl group is one of the most commonly occurring carbon fragments in biologically active molecules. Here, a biomimetic design reagent, 5-(methyl-d3)-5H-dibenzo[b,d]thiophen-5-ium trifluoromethane sulfonate (DMTT), as an analog of S-adenosylmethionine (SAM), has been developed for the selective d3-methylation of complex molecules bearing several possible reactive sites with excellent selectivity and high-level deuterium incorporation. A series of d3-methylated organic molecules and deuterated pharmaceuticals were synthesized under the mild system with excellent functional group compatibility.
INTRODUCTION
The methyl group is the simplest organic substituent, yet it is not only important for synthetic and medicinal chemistry (1) but also plays a central role in a wide range of biological processes, including DNA replication, protein modification, lipid biosynthesis, and various other metabolic pathways (2–5). Most biological methylation uses members of the S-adenosylmethionine (SAM) superfamily, commonly known as radical methyltransferases, in which SAM is selectively cleaved by its C─S bond under the reaction with various nucleophiles to generate stable S-adenosyl-l-homocysteine, leading to the methylation of complex molecules such as nucleic acids, proteins, and natural products in a SN2 manner (Fig. 1A) (6–7). Inspiration by SAM, Baran and coworkers (8) developed an elegant methylating agent, zinc bis(phenylsulfonylmethanesulfinate), for the methylation of heteroarenes. Very recently, other methylating reagents such as 1,1-diborylmethane (9), α-iodosulfones (10), trimethylphosphate (11), 1-methoxy-2,2,6,6-tetramethylpiperidine (TEMPO-Me) (12), MeZnCl (13), Lewis base BH3/DMF (14), and [Me2Cl][Al(OTeF5)4] (15) exhibit unique reactivity during the research of methylation. Despite these notable advances, the corresponding d3-methylation has not been well developed.
Fig. 1. Bioinspired invention of DMTT as a robust d3-methylating agent.
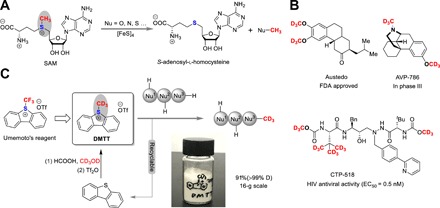
(A) SAM methyl transferase mediated nucleophilic reactions. (B) Deuterium-labeled drugs and candidates. (C) Invention of d3-methylating reagent DMTT. Reaction conditions: (1) HCOOH (3.0 equiv, 88 wt % aqueous solution), CD3OD (10.0 equiv, >99% D), H2SO4 (11.0 equiv, 98 wt %), reflux at 60°C for 4 hours to afford methyl-d3 formate; (2) dibenzothiophene (1.0 equiv), and above methyl-d3 formate, heat up to 60°C until the mixture became a clear solution. Photo credit: Yunfei Zhao, Nanjing University. EC50, half median effective concentration.
Deuterium atoms can be used as tracer atoms to elucidate metabolic pathways in medicinal chemistry (16). Owing to the higher bond dissociation energy than the C─H bond, the incorporation of deuterium can impart marked improvements to drug candidates, related to absorption, distribution, and metabolism in organisms (17). Numerous efficient strategies of deuterium labeling techniques have been developed recently including direct H/D exchange (18–19), dehalogenative deuterium mediated by transition metal catalysis (20), bases (21–22), or radicals (23), and so on (24–25). A substantial number of deuterium-labeled drug candidates have been synthesized and submitted to clinical trials in the past few decades (26–27). Austedo with two OCD3 groups, as a treatment for symptoms of Huntington’s disease, was finally approved by the U.S. Food and Drug Administration in 2017 as the first deuterated drug (Fig. 1B) (28). In general, the d3-methylated group incorporation significantly affected the ADME (absorption, distribution, metabolism, and excretion)–related processes and attenuated metabolism of certain pharmacologically active compounds (29–30). From the viewpoint of synthetic chemistry, d3-methylation is the most ideal approach because it has the capability to directly introduce the D-labeled methyl group into structurally diverse molecules (31–33). Because of the presence of multiple nucleophilic sites in a complex molecule with subtle differences in the activation barriers, controlling the positional selectivity represents a key challenge. Traditionally, CD3I (34), (CD3)2SO4, and CD3OD have been widely used as deuterium methylating reagents, albeit with volatility, corrosion, toxicity, and carcinogenicity concerns (35–36). However, these highly reactive reagents usually lead to poor chemoselectivity and overmethylation. Reductive deuteriation of carboxylic acids (37), amides (38), and nitriles (39–41) provides protocols to incorporate d2-methyleneation. Nevertheless, the invention of a reagent to accomplish direct, scalable, and predictably selective d3-methylation under mild and environmentally friendly conditions is in high demand. Umemoto’s reagent is a commercially available and widely used electrophilic trifluoromethylating agent (42). Bioinspired by the reactivity of SAM, together with the design principle of Umemoto’s reagent, we report herein the invention of DMTT (5-(methyl-d3)-5H-dibenzo[b,d]thiophen-5-ium trifluoromethane sulfonate) (43), a d3-methylating agent using the dibenzo[b,d]thiophene framework to simplify the SAM structure, successfully blending high reactivity and selectivity with a safety profile that is particularly attractive for medicinal and pharmaceutical chemistry (Fig. 1C). DMTT could be simply prepared from dibenzo[b,d]thiophene (44–45) and CD3OD in 91% yield on a large scale, and the compound is very stable when stored as a solid at ambient temperature. Significantly, the d3-methylation of DMTT with substrates proceeds in a biomimetic process, thus exhibiting excellent chemoselectivity under extremely mild conditions analogous to SAM chemistry in a living organism.
RESULTS
Reaction condition optimization
Trideuteromethylation of carboxylic acids is ubiquitously used in chemical research. Initially, we chose the carboxylic acid 1a with a carboxyl and a N,N-dimethyl group to check the reactivity and selectivity of the DMTT agent (Table 1). Compared to the NMe2 substituent, the carboxylic group preferentially reacted with DMTT, forming the desired d3-methylation product 1b in 90% isolated yield with deuterium retained completely in the presence of K2CO3 (entry 1). This d3-methylation process cannot proceed in the absence of base (entry 2). Screening of other bases did not provide higher yields (entries 3 and 4). When increasing the amount of DMTT to 1.5 equiv, the overmethylation product 1c was formed in 20% yield (entry 5). Using CD3I as the deuterium source led to a different chemoselectivity. Using 1.0 equiv of CD3I proved to prefer quaternization of N,N-dimethyl group (entry 7), and a larger excess of CD3I generated a by-product 1d with CD3/CH3 exchange (entry 7). In addition, the selection of (CD3)2SO4 provided the desired product 1b in a much lower efficiency with 27% yield of by-product 1c (entry 8). These results indicate that DMTT exhibited significantly different reactivity from CD3I and (CD3)2SO4 depending on the different sterically hindered leaving groups. In addition, other sulfur-based reagents were also examined. Dimethyl sulfoxide (DMSO)–d6 cannot promote the methylation-d3 process under our developed conditions (entry 9), while the reagent [CD3Ph2S]+OTf− was proved to be less effective (entry 10).
Table 1. Reaction development.
Reaction conditions: 1a (0.2 mmol), DMTT (0.2 mmol), and base (0.4 mmol) in 2 ml of MeCN for 12 hours.
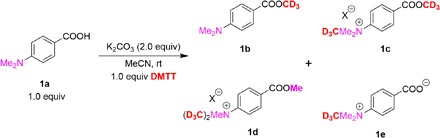
| Entry |
Variation from the standard conditions |
1b/1c/1d/1e* |
| 1 | None | 90 (99% D) †/0/0/0 |
| 2 | Without K2CO3 | 0/0/0/0 |
| 3 | Using KOH | 35 (99% D)/0/0/0 |
| 4 | Using DBU | 50 (99% D)/16/0/0 |
| 5 | 1.5 equiv of DMTT | 62 (99% D)/20/0/0 |
| 6 | CD3I (1.0 equiv.) instead of DMTT | 2/0/0/8 |
| 7‡ | CD3I (5.0 equiv) instead of DMTT | 7/0/43/10 |
| 8 | (CD3)2SO4 (1.0 equiv) instead of DMTT |
31/27/0/0 |
| 9 | DMSO-d6 instead of DMTT | 0/0/0/0 |
| 10 | [CD3Ph2S]+OTf− instead of DMTT | 64/0/0/0 |
*1H NMR yields were presented using CH2Br2 as an internal standard.
†Isolated yield.
‡At 80°C.
On the basis of the tested conditions, 1.0 equiv of DMTT furnished the desired methyl-d3 esterification product in excellent yield and suppressed the generation of by-products. To further illustrate this selectivity, substrate 2a with three nucleophilic sites was treated with DMTT under the above conditions (Fig. 2). To our delight, compound 2a reacted with DMTT also tends to form the esterification product 2b in 76% yield. Again, treatment of CD3I and (CD3)2SO4 with 2a under the same conditions showed poor selectivity. Therefore, DMTT should be used as a very important, predictable, and reliable reagent for trideuteromethylation.
Fig. 2. Investigations of compound 2a with DMTT agent.
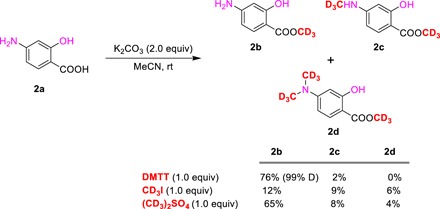
Reaction conditions: 2a (0.2 mmol), DMTT (0.2 mmol), and base (0.4 mmol) in 2 ml of MeCN for 12 hours, 1H NMR yields were presented using CH2Br2 as an internal standard. rt, room temperature.
DISCUSSION
Application in late-stage modification and deuterated drug synthesis
Following identification of the optimized d3-methylation with DMTT under developed mild conditions, various nucleophilic reagents bearing several possible reactive sites were examined (Table 2). Notably, the substrates with tertiary alcohol hydroxyl (3a) and amino (4a) groups were well tolerated. The reaction showed excellent selectivity for esterification (3-4b), in which alcohol elimination and N-methylation byproducts were not observed. When indole 5a was used, the esterification product 5b was obtained in 84% yield with the N─H bond preserved. α-Amino acid 6a was also suitable for d3-methylation, resulting in 89% yield (6b). Numerous pharmaceuticals and natural products were also investigated, and various d3-methylated biomolecules were generated in good to excellent yields. As an important immune inhibitor, mycophenolic acid (7a) with multiple hydroxyl groups also reacted smoothly with sole selectivity. The carboxylic acid preferentially reacted with DMTT in the presence of hydroxyl groups and showed excellent selectivity (8b-12b) under practical mild conditions, avoiding the formation of intermolecular esterification and overmethylation using traditional CD3 reagents. When substrates had oxoimidazolidine and tetrahydrothiophene motifs, d3-methylation reacted exclusively at the carboxylic acid group (13b) in 65% yield with 99% D incorporation. Furthermore, oligopeptides were tested under standard conditions; 14b and 15b were obtained in 92 and 48% yields, respectively. Among the substrates, the reactions of 12a and 14a with DMTT were conducted on a gram scale without the loss of the yield or selectivity.
Table 2. Substrate scope, late-stage modification, and deuterated drug synthesis.
Reaction conditions: substrates a (1.0 equiv), DMTT (1 to 2 equiv), and K2CO3 (2 to 4 equiv) in 2 ml of MeCN for 12 hours. Isolated yields were presented.
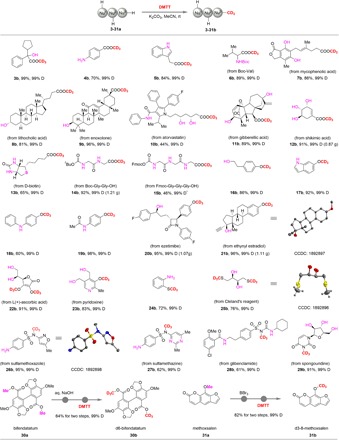
*Reaction conditions: substrates a (1.0 equiv), DMTT (1.0 equiv), and KOH (2.0 equiv) in 2 ml of MeCN/H2O (v:v = 1/1) for 12 hours. Isolated yields were presented.
Next, the phenols, which have higher pKa (where Ka is the acid dissociation constant) values than the corresponding carboxylic acids, were also investigated. The phenols containing hydroxyl (16b), indole N─H (17b), amine (18b), and amide (19b) substituents were well compatible, with excellent selectivity. The drug molecules 20-21a, which have sensitive functional groups, including 2-azetidinone, aliphatic hydroxyl, and terminal alkynyl frameworks, could be late stage modified by DMTT at the phenolic hydroxyl selectively, and the products 20-21b were generated on a gram scale without notable decreases in yields (95 to 96%) or selectivity. The structure of 21b was also determined by x-ray crystallographic analysis. Notably, for the reaction of DMTT with (+)-ascorbic acid (22a), the two aliphatic hydroxyl groups were also tolerated, and two phenol ether groups were preferentially produced, generating product 22b in 91% yield. Trideuteromethylation of pyridoxine (23a) formed the desired product 23b in 83% yield, and side reactions such as pyridine N- and alcohol O-methylation were completely inhibited.
In addition, DMTT was compatible with many common biologically active functional groups. When 2-aminobenzenethiol (24a) is used, only S atom d3-methylation (24b) is observed. Cleland’s reagent bearing two mercapto and two hydroxyl groups is used as a reducing or “deprotecting” agent for thiolated DNA (46), and it can undergo selective d3-methylation with DMTT at the mercapto group, leading to product 25b in 76% yield. Using sulfonamides 26-28a as substrates having amino and amide substituents, the reaction of DMTT shows excellent selectivity for the sulfonamide NH group, in which the N─H bond connected to the sulfone group has the lowest pKa value. Last, spongouridine 28a bearing three hydroxyl groups was also tested with DMTT, and the NH d3-methylated compound 29b was obtained in 91% yield with excellent deuterium incorporation. Among the products, the molecular structures of 25-26b were confirmed by x-ray analysis. In the presence of K2CO3, the methylation-d3 took place selectively on the carboxylic acids, phenols, and thiols with their NH2, NH, and alkyl-OH intact. The order of reactivity in increasing order is amines and alkyl-OH < phenols < carboxylic acids. In particular, N-methylated-d3 products were only observed when the NH connected with strong electron-withdrawing groups, such as N-sulfonyl or N,N-diacetyl groups.
H/D exchange at the C─H bonds of the precursors is a common method for the preparation of deuterium-labeled drug molecules (31). However, highly efficient and regioselective H/D exchange of a methyl group in a complex molecule remains challenging. With the DMTT reagent in hand, we can provide a reliable route to convert commercially available drugs to the corresponding d3-deuterated variants. For example, bifendatatum (30a), an antihepatitis drug, was hydrolyzed to form a carboxylic acid intermediate and then underwent d3-methylation with DMTT, producing the d3-bifendataum (30b) with high-level deuterium incorporation. Methoxsalen (31a) is a commercial drug used to treat psoriasis, eczema, vitiligo, and some cutaneous lymphomas in conjunction with exposing the skin to ultraviolet A light from lamps or sunlight. This substrate was demethylated in the presence of BBr3 and subsequently reacted with DMTT to generate d3-methoxsalen (31b) in 82% yield.
Application in Ni-catalyzed dimethylation-d6 of amines
The direct methylation of a primary amine with methyl iodide (47) is usually not a feasible method for the preparation of secondary amines since overmethylation occurs to form quaternary ammonium salts. To further investigate the superiority of DMTT, dimethylation of primary amines was carried out smoothly in the presence of 10 mol% Ni(OAc)2·4H2O, leading to the N,N-dimethyl-d3 products in modern to good yields (Table 3). Compared with these developed approaches, DMTT showed excellent selectivity under mild conditions, and the overmethylation quaternary ammonium salts did not observe. The hydroxyl group is compatible in this system to deliver 33c in 71% yield with 99% D incorporation. The aryl amines containing other dialkylamino groups, such as 33d and 33e, are also well tolerated to furnish the corresponding methylation-d3 products in 61 and 74% yield, respectively. When the substrate 32f with active amino group was treated with DMTT under standard conditions, the 33f was isolated in 46% yield with active amino intact. The reaction of alkyl amines with DMTT gave the corresponding methylation-d3 products in moderate yields. What is more, the functional molecules with amines groups (33j and 33k) were also investigated. Using the developed method, the d6-aminophenazone 33l, as important antipyretic analgesics, could be easily be synthesized from the available 4-aminoantipyrine 32l. Last, the secondary amines such as diphenylaniline 32m can also be easily d3-methylated in the presence of nickel catalyst affording the N-(methyl-d3)-N-phenylaniline 33m in 74% yield.
Table 3. Dimethylation-d6 of primary amines.
Reaction conditions: substrates 32 (1.0 equiv), DMTT (2 equiv), Ni(OAc)2·4H2O (10 mol%), and KF (2 equiv) in 2 ml of DCM/H2O (v:v = 20/1) for 12 hours. Isolated yields were presented.
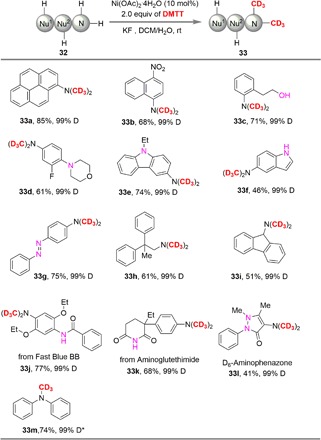
*1.0 equiv of DMTT was used.
Application in C─H bond activation and d3-methylation
DMTT has also played an important role in the transition metal-catalyzed C─H bond activation (Fig. 3) (45). Using pyridyl as directing group, the ortho-methylation-d6 was achieved in the presence of palladium catalyst, leading to the dimethylation-d6 product 35a in 62% isolated yield with 99% D value. In the same condition, CD3I and (CD3)2SO4 failed to accelerate the methylation process. Increasing the steric hindrance of directing group and reducing the loading of DMTT, the ortho-monomethylation-d3 product 35b was observed without any deuterium loss. Using acetylamino as directing group, monomethylation-d3 was detected in the presence of 1.2 equivalents of DMTT under standard condition, whereas only trace amount of ortho-bismethylation-d3 was observed. The drug molecules were also modified using the developed palladium catalyzed C─H bond methylation-d3. Phenacetin, which was used to treat fever, headache, and neuralgia, can be methylated-d3 smoothly to change pharmacological activity and avoid some side effects, leading the methyl-d3 phenacetin 35d in 75% yield with 99% deuterium incorporation.
Fig. 3. Pd-catalyzed C─H bond methylation-d3.
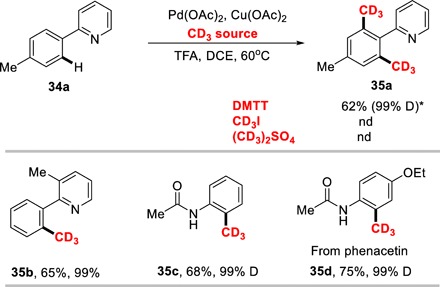
Reaction conditions: substrates 34 (1.0 equiv), DMTT (3.0 to 1.2 equiv), Pd(OAc)2 (10 mol %), Cu(OAc)2 (1.2 equiv), and trifluoroacetate (TFA) (5.0 equiv) in 2 ml of dichloroethane (DCE) for 12 hours at 60°C. Isolated yields were presented. *3.0 equivalents of DMTT were used. nd, not detected.
Application in d2-methyleneation of cyclopropanes
In addition to a d3-methylating agent, DMTT also showed the intrinsic reactivity of a sulfur ylide (Fig. 4). For example, in the presence of strong base, DMTT underwent deprotonation to afford methylene intermediate, which captured by trifluoromethyl substituted alkenes 36 to furnish the deuterated cyclopropanes 37 under mild conditions. Various aryl substituted cyclopropanes were obtained in the yields of 50 to 72%, which provide a powerful evidence to prove the widely application value of DMTT.
Fig. 4. Methyleneation of trifluoromethyl-substituted alkenes.
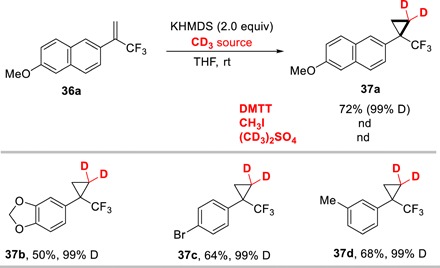
Reaction conditions: substrates 36 (1.0 equiv), DMTT (2.0 equiv), and KHMDS (2.0 equiv) in 2 ml of tetrahydrofuran (THF) for 12 hours at room temperature. Isolated yields were presented.
CONCLUSION
In conclusion, we have developed a new d3-methylating reagent, DMTT, and applied it in the many methylation processes, serving as a powerful approach to install deuterium in organic molecules and providing a new diagnostic tool in drug development. The advantage of this transformation is the simple experimental protocols, excellent selectivity, and high-level deuterium incorporation. We believe that this strategy will be used in drug modification in the pursuit of new deuterated therapeutics.
MATERIALS AND METHODS
General information
Unless otherwise noted, all the reaction was performed under air atmosphere. All new compounds were fully characterized. 1H, 13C, and 19F nuclear magnetic resonance (NMR) spectra were recorded on a Bruker Avance III 400 or 500 MHz spectrometer. 1H NMR spectra data were reported as δ values in parts per million relative to CDCl3 (δ = 7.26), CD3CN (δ = 1.94), CD3OD (δ = 3.31), or DMSO-d6 (δ = 2.50). 13C NMR spectra data were reported as δ values in parts per million relative to CDCl3 (δ = 77.00), CD3CN (δ = 118.30), CD3OD (δ = 49.15), or DMSO-d6 (δ = 39.52). Mass spectra were conducted at Micromass Q-Tof instrument electron spray ionization (ESI) and Agilent Technologies 5973N electron ionization (EI). Infrared spectra were recorded on a Fourier transform infrared spectrometer. All materials obtained from commercial suppliers were used without further purification.
General procedure for synthesis of trideuteromethyl reagent DMTT
According to our patent (43), to a 50-ml round flask was added formic acid (7.8 ml, 150 mmol, 3 equiv, 88 wt % aqueous solution), CD3OD (20.2 ml, 500 mmol, 10 equiv, d = 0.89 g/ml, 99% D). Then, sulfuric acid (8.8 ml, 165 mmol, 11 equiv, 98%) was added dropwise to the above solution with stirring. The reaction was heated at 60°C and stirred for 4 hours. The methyl-d3 formate containing a small amount of deuterated methanol was directly distilled at atmospheric pressure (33° to 38°C). The methyl-d3 formate was used without further purification.
A 50-ml round flask was charged with dibenzothiophene (9.2 g, 50 mmol, 1 equiv) and above distilled methyl-d3 formate. The mixture was stirred at 0°C, and Tf2O (20 ml) was added dropwise with stirring. Then, the reaction was heated up to 60°C. When the mixture became a clear solution, the reaction was poured into water (100 ml), extracted with CH2Cl2 , dried with MgSO4, and concentrated in vacuo. The obtained crude product was washed with Et2O and dried under vacuum to afford the trideuteromethyl reagent DMTT (16.0 g, 91%, 99% D).
General procedure for synthesis of 3b-31b
Carboxylic acid a (0.2 mmol, 1.0 equiv), DMTT (0.2 to 0.4 mmol, 1 to 2 equiv), K2CO3 (0.4 to 0.8 mmol, 2 to 4 equiv), and MeCN (2 ml) were added to a Schlenk tube. The reaction was stirred at room temperature for 12 hours. Then, the resulting mixture was concentrated in vacuo, and the residue was purified by column chromatography to afford the corresponding product b.
General procedure for synthesis of 33a-33 l
Ni(acac)2 (0.02 mmol, 0.1 equiv), amines (0.2 mmol, 1.0 equiv), potassium fluoride (KF) (0.4 mmol, 2.0 equiv), DMTT (0.4 mmol, 2.0 equiv), and dichloromethane (DCM)/H2O (2 ml, v:v = 20:1) were added to a 25-ml Schlenk tube. The reaction was stirred at room temperature for 12 hours. Then, the resulting mixture was concentrated in vacuo, and the residue was purified by column chromatography to afford the corresponding products 33.
General procedure for synthesis of 35a-35d
Pd(OAc)2 (0.02 mmol, 0.1 equiv), 34 (0.2 mmol, 1.0 equiv), Cu(OAc)2 (0.24 mmol, 1.2 equiv), DMTT (0.24 to 0.6 mmol, 1.2 to 3.0 equiv), trifluoroacetate (TFA; 1.0 mmol, 5.0 equiv), and dichloroethane (DCE; 2 ml) were added to a 25-ml Schlenk tube. The reaction was stirred at 60°C for 12 hours. Then, the resulting mixture was concentrated in vacuo, and the residue was purified by column chromatography to afford the corresponding products 35.
General procedure for synthesis of 37a-37d
36 (0.2 mmol, 1.0 equiv), DMTT (0.4 mmol, 2.0 equiv), potassium bis(trimethylsilyl)amide (KHMDS) (0.4 mmol, 2.0 equiv), and tetrahydrofuran (THF; 2 ml) were added to a 25-ml Schlenk tube. The reaction was stirred at room temperature for 12 hours. Then, the resulting mixture was concentrated in vacuo, and the residue was purified by column chromatography to afford the corresponding products 37.
Supplementary Material
Acknowledgments
We thank C. Yuan in this group for reproducing the results of 10b, 18b, and 27b in Table 2. Funding: This study was supported by the “1000-Youth Talents Plan”, the National Natural Science Foundation of China (grant numbers 21972064 and 21901111), and the National Natural Science Foundation of Jiangsu Province (grant BK20170632). Author contributions: Z.S. supervised the project and revised the manuscript. M.W. developed the reaction, conducted the experiments, and wrote the manuscript. Y.F.Z. developed the reaction and expanded the substrate scopes. Y.Z. helped for the x-ray crystallographic analysis. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/19/eaba0946/DC1
REFERENCES AND NOTES
- 1.Barreiro E. J., Kümmerle A. E., Fraga C. A. M., The methylation effect in medicinal chemistry. Chem. Rev. 111, 5215–5246 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Jones P. A., Takai D., The role of DNA methylation in mammalian epigenetics. Science 293, 1068–1070 (2001). [DOI] [PubMed] [Google Scholar]
- 3.Zhang L., Ding X., Cui J., Xu H., Chen J., Gong Y.-N., Hu L., Zhou Y., Ge J., Lu Q., Liu L., Chen S., Shao F., Cysteine methylation disrupts ubiquitin-chain sensing in NF-κB activation. Nature 481, 204–208 (2012). [DOI] [PubMed] [Google Scholar]
- 4.Roundtree I. A., Evans M. E., Pan T., He C., Dynamic RNA modifications in gene expression regulation. Cell 169, 1187–1200 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harcourt E. M., Kietrys A. M., Kool E. T., Chemical and structural effects of base modifications in messenger RNA. Nature 541, 339–346 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vey J. L., Drennan C. L., Structural insights into radical generation by the radical SAM superfamily. Chem. Rev. 111, 2487–2506 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Broderick J. B., Duffus B. R., Duschene K. S., Shepard E. M., Radical S-adenosylmethionine enzymes. Chem. Rev. 114, 4229–4317 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gui J., Zhou Q., Pan C.-M., Yabe Y., Burns A. C., Collins M. R., Ornelas M. A., Ishihara Y., Baran P. S., C–H methylation of heteroarenes inspired by radical SAM methyl transferase. J. Am. Chem. Soc. 136, 4853–4856 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jo W., Kim J., Choi S., Cho S. H., Transition-metal-free regioselective alkylation of pyridine N-oxides using 1,1-diborylalkanes as alkylating reagents. Angew. Chem. Int. Ed. 55, 9690–9694 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Filippini G., Silvi M., Melchiorre P., Enantioselective formal α-methylation and α-benzylation of aldehydes by means of photo-organocatalysis. Angew. Chem. Int. Ed. 56, 4447–4451 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.He Z.-T., Li H., Haydl A. M., Whiteker G. T., Hartwig J. F., Trimethylphosphate as a methylating agent for cross coupling: A slow-release mechanism for the methylation of arylboronic esters. J. Am. Chem. Soc. 140, 17197–17202 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Norcott P. L., Hammill C. L., Noble B. B., Robertson J. C., Olding A., Bissember A. C., Coote M. L., TEMPO–Me: An electrochemically activated methylating agent. J. Am. Chem. Soc. 141, 15450–15455 (2019). [DOI] [PubMed] [Google Scholar]
- 13.Serpier F., Pan F., Ham W. S., Jacq J., Genicot C., Ritter T., Selective methylation of arenes: A radical C−H functionalization/cross-coupling sequence. Angew. Chem. Int. Ed. Engl. 57, 10697–10701 (2018). [DOI] [PubMed] [Google Scholar]
- 14.Xia H.-M., Zhang F.-L., Ye T., Wang Y.-F., Selective α-monomethylation by an amine-borane/N,N-dimethylformamide system as the methyl source. Angew. Chem. Int. Ed. Engl. 57, 11770–11775 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Hämmerling S., Thiele G., Steinhauer S., Beckers H., Müller C., Riedel S., A very strong methylation agent: [Me2Cl][Al(OTeF5)4]. Angew. Chem. Int. Ed. Engl. 58, 9807–9810 (2019). [DOI] [PubMed] [Google Scholar]
- 16.Gant T. G., Using deuterium in drug discovery: Leaving the label in the drug. J. Med. Chem. 57, 3595–3611 (2014). [DOI] [PubMed] [Google Scholar]
- 17.Zhu Y., Zhou J., Jiao B., Deuterated clopidogrel analogues as a new generation of antiplatelet agents. ACS Med. Chem. Lett. 4, 349–352 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Elmore C. S., Bragg R. A., Isotope chemistry; A useful tool in the drug discovery arsenal. Bioorg. Med. Chem. Lett. 25, 167–171 (2015). [DOI] [PubMed] [Google Scholar]
- 19.Atzrodt J., Derdau V., Fey T., Zimmermann J., The renaissance of H/D exchange. Angew. Chem. Int. Ed. Engl. 46, 7744–7765 (2007). [DOI] [PubMed] [Google Scholar]
- 20.Atzrodt J., Derdau V., Kerr W. J., Reid M., C−H functionalisation for hydrogen isotope exchange. Angew. Chem. Int. Ed. Engl. 57, 3022–3047 (2018). [DOI] [PubMed] [Google Scholar]
- 21.Alonso F., Beletskaya I. P., Yus M., Metal-mediated reductive hydrodehalogenation of organic halides. Chem. Rev. 102, 4009–4092 (2002). [DOI] [PubMed] [Google Scholar]
- 22.Yus M., Arene-catalysed lithiation reactions. Chem. Soc. Rev. 25, 155–161 (1996). [Google Scholar]
- 23.Wang X., Zhu M.-H., Schuman D. P., Zhong D., Wang W.-Y., Wu L.-Y., Liu W., Stoltz B. M., Liu W.-B., General and practical potassium methoxide/disilane-mediated dehalogenative deuteration of (Hetero)arylhalides. J. Am. Chem. Soc. 140, 10970–10974 (2018). [DOI] [PubMed] [Google Scholar]
- 24.Soulard V., Villa G., Vollmar D. P., Renaud P., Radical deuteration with D2O: Catalysis and mechanistic insights. J. Am. Chem. Soc. 140, 155–158 (2018). [DOI] [PubMed] [Google Scholar]
- 25.Koniarczyk J., Hesk D., Overgard A., Davies I. W., McNally A., A general strategy for site-selective incorporation of deuterium and tritium into pyridines, diazines, and pharmaceuticals. J. Am. Chem. Soc. 140, 1990–1993 (2018). [DOI] [PubMed] [Google Scholar]
- 26.Zhang M., Yuan X.-A., Zhu C., Xie J., Deoxygenative deuteration of carboxylic acids with D2O. Angew. Chem. Int. Ed. Engl. 58, 312–316 (2019). [DOI] [PubMed] [Google Scholar]
- 27.Mullard A., Deuterated drugs draw heavier backing. Nat. Rev. Drug Discov. 15, 219–221 (2016). [DOI] [PubMed] [Google Scholar]
- 28.Schmidt C., First deuterated drug approved. Nat. Biotechnol. 35, 493–494 (2017). [DOI] [PubMed] [Google Scholar]
- 29.Mullard A., FDA approves first deuterated drug. Nat. Rev. Drug Discov. 16, 305 (2017). [DOI] [PubMed] [Google Scholar]
- 30.Pirali T., Serafini M., Cargnin S., Genazzani A. A., Applications of deuterium in medicinal chemistry. J. Med. Chem. 62, 5276–5297 (2019). [DOI] [PubMed] [Google Scholar]
- 31.Caporaso R., Manna S., Zinken S., Kochnev A. R., Lukyanenko E. R., Kurkin A. V., Antonchick A. P., Radical trideuteromethylation with deuterated dimethyl sulfoxide in the synthesis of heterocycles and labelled building blocks. Chem. Commun. 52, 12486–12489 (2016). [DOI] [PubMed] [Google Scholar]
- 32.Zhang R., Yu H., Li Z., Yan Q., Li P., Wu J., Qi J., Jiang M., Sun L., Iron-mediated azidomethylation or azidotrideuteromethylation of active alkenes with azidotrimethylsilane and dimethyl sulfoxide. Adv. Synth. Catal. 360, 1384–1388 (2018). [Google Scholar]
- 33.Shen Z., Zhang S., Geng H., Wang J., Zhang X., Zhou A., Yao C., Chen X., Wang W., Trideuteromethylation enabled by a sulfoxonium metathesis reaction. Org. Lett. 21, 448–452 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hu L., Liu X., Liao X., Nickel-catalyzed methylation of aryl halides with deuterated methyl iodide. Angew. Chem. Int. Ed. 55, 9743–9747 (2016). [DOI] [PubMed] [Google Scholar]
- 35.Adamczyk M., Fishpaugh J. R., Johnson D., Synthesis of [N-trideuteromethyl] labelled E-doxepin. J. Labelled. Comp. Radiopharm. 33, 153–155 (1993). [Google Scholar]
- 36.Komarapuri S., Krishnan K., Covey D. F., Synthesis of 19-trideuterated ent-testosterone and the GABAA receptor potentiators ent-androsterone and ent-etiocholanolone. J. Labelled. Compd. Radiopharm. 51, 430–434 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Szostak M., Spain M., Procter D. J., Selective synthesis of α,α-dideuterio alcohols by the reduction of carboxylic acids using SmI2 and D2O as deuterium source under SET conditions. Org. Lett. 16, 5052–5055 (2014). [DOI] [PubMed] [Google Scholar]
- 38.Zhang B., Li H., Ding Y., Yan Y., An J., Reduction and reductive deuteration of tertiary amides mediated by sodium dispersions with distinct proton donor-dependent chemoselectivity. J. Org. Chem. 83, 6006–6014 (2018). [DOI] [PubMed] [Google Scholar]
- 39.Ding Y., Luo S., Adijiang A., Zhao H., An J., Reductive deuteration of nitriles: The synthesis of α,α-dideuterio amines by sodium-mediated electron transfer reactions. J. Org. Chem. 83, 12269–12274 (2018). [DOI] [PubMed] [Google Scholar]
- 40.Mészáros R., Peng B.-J., Ötvös S. B., Yang S.-C., Fülöp F., Continuous-flow hydrogenation and reductive deuteration of nitriles: A simple access to α,α-dideutero amines. ChemPlusChem 84, 1508–1511 (2019). [DOI] [PubMed] [Google Scholar]
- 41.Ding Y., Luo S., Weng C., An J., Reductive deuteration of nitriles using D2O as a deuterium source. J. Org. Chem. 84, 15098–15105 (2019). [DOI] [PubMed] [Google Scholar]
- 42.Umemoto T., Zhang B., Zhu T., Zhou X., Zhang P., Hu S., Li Y., Powerful, thermally stable, one-pot-preparable, and recyclable electrophilic trifluoromethylating agents: 2,8-Difluoro- and 2,3,7,8-tetrafluoro-S-(trifluoromethyl)dibenzothiophenium salts. J. Org. Chem. 82, 7708–7719 (2017). [DOI] [PubMed] [Google Scholar]
- 43.Z. Shi, Y. Zhao, M. Wang, China Patent CN 2018-10770921.
- 44.Vasu D., Yorimitsu H., Osuka A., Palladium-assisted “Aromatic Metamorphosis” of dibenzothiophenes into triphenylenes. Angew. Chem. Int. Ed. 54, 7162–7166 (2015). [DOI] [PubMed] [Google Scholar]
- 45.Simkó D. C., Elekes P., Pázmándi V., Novák Z., Sulfonium salts as alkylating agents for palladium-catalyzed direct ortho alkylation of anilides and aromatic ureas. Org. Lett. 20, 676–679 (2018). [DOI] [PubMed] [Google Scholar]
- 46.Cleland W. W., Dithiothreitol, a new protective reagent for SH Groups. Biochemistry 3, 480–482 (1964). [DOI] [PubMed] [Google Scholar]
- 47.Chiappe C., Piccioli P., Pieraccini D., Selective N-alkylation of anilines in ionic liquids. Green Chem. 8, 277–281 (2006). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/19/eaba0946/DC1