

Abstract
Objective:
Individuals with attention deficit hyperactivity disorder (ADHD) show gray matter volume (GMV) reduction in the putamen. KTN1 variants may regulate kinectin 1 expression in the putamen and influence putamen structure and function. We aim to test the hypothesis that the KTN1 variants may represent a genetic risk factor of ADHD.
Methods:
Two independent family-based Caucasian samples were analyzed, including 922 parent-child trios (a total of 2,757 subjects with 924 ADHD children) and 735 parent-child trios (a total of 1,383 subjects with 613 ADHD children). The association between ADHD and a total of 143 KTN1 SNPs was analyzed in the first sample, and the nominally-significant (p<0.05) risk SNPs were classified into independent haplotype blocks. All SNPs, including imputed SNPs within these blocks, and haplotypes across each block, were explored for replication of associations in both samples. The potential biological functions of all risk SNPs were predicted using a series of bioinformatics analyses, their regulatory effects on the putamen volumes were tested, and the KTN1 mRNA expression was examined in three independent human putamen tissue samples.
Results:
Fifteen SNPs were nominally associated with ADHD (p<0.05) in the first sample, and three of them remained significant even after correction for multiple testing (1.3×10−10≤p≤1.2×10−4; α=2.5×10−3). These 15 risk SNPs were located in five haplotype blocks, and 13 SNPs within four of these blocks were associated with ADHD in the second sample. Six haplotypes within these blocks were also significantly (1.2×10−7≤p≤0.009) associated with ADHD in these samples. These risk variants were located in disease-related transposons and/or transcription-related functional regions. Major alleles of these risk variants significantly increased putamen volumes. Finally, KTN1 mRNA was significantly expressed in putamen across three independent cohorts.
Conclusion:
The KTN1 variants were significantly associated with ADHD. KTN1 may play a functional role in the development of ADHD.
Keywords: ADHD, KTN1, putamen, gray matter volume (GMV), transposon, transcription
Introduction
The core symptoms of attention deficit hyperactivity disorder (ADHD) are inattention, impulsivity and hyperactivity. Trait impulsivity is related to smaller putamen GMV [Caravaggio et al. 2017]. The integrity of corticostriatal pathways, including premotor cortical connectivity with the putamen is central to attention and impulse control, and corticostriatal dysfunction manifests in individuals with ADHD [Bush et al. 2005; de Wit et al. 2012]. Catecholaminergic medication treatment aims to restore frontostriatal circuit functions in ADHD [Frank et al. 2007]. Functional connectivity of the putamen with the insula and parietal cortex is disrupted in youth with internet gaming disorder, a condition frequently comorbid with ADHD [Hong et al. 2015]. In particular, ADHD is associated with reduction in gray matter volume (GMV) or lesions in the putamen [Ellison-Wright et al. 2008; Greven et al. 2015; Herskovits et al. 1999; Khadka et al. 2016; Luo et al. 2019; Max et al. 2002; Seidman et al. 2011; Sobel et al. 2010; Valera et al. 2007; Xu et al. 2017]. In healthy subjects, the right putamen is frequently smaller than left putamen [Wellington et al. 2006]; however, children with ADHD more frequently have a smaller left putamen than the right-hemispheric counterpart; this reversal of symmetry may relate to ADHD symptomology [Wellington et al. 2006]. Furthermore, the putamen volume declines with age [McDonald et al. 1991] and the volumetric reduction is independent of age in participants with ADHD and their unaffected siblings, suggesting its link to familial risk for ADHD [Greven et al. 2015]. In summary, the putamen GMV may represent a neural marker of the familial risk for ADHD [Xu et al. 2017].
Recently, a genome-wide association study of putamen GMV shows that a genetic marker at 3’-UTR of KTN1, i.e., rs945270, demonstrates the strongest (p=1.1×10−33), replicable, and specific effects on the putamen GMV [Hibar et al. 2015; Xu et al. 2017], suggesting that the kinectin 1 gene (KTN1) may play a critical regulatory role in determining putamen GMV [Chen et al. 2017; Hibar et al. 2015; Luo et al. 2020; Satizabal et al. 2017; Xu et al. 2017]. KTN1 encodes the kinesin receptor, i.e., kinectin 1, which facilitates vesicle binding to kinesin, regulating crucial developmental processes including axonal guidance and vesicular transport of molecules as well as apoptosis [Hibar et al. 2015; Kumar et al. 1995]. Other studies have shown that kinectin regulates neuronal cell shape and neuronal migration through kinectin–kinesin interactions [Zhang et al. 2010]. Neurons with more kinectin have larger cell bodies [Toyoshima and Sheetz 1996], and kinectin knock-down strongly influences cell shape [Zhang et al. 2010]. Thus, KTN1 variants may regulate kinectin 1 expression in the putamen and thereby influence putamen structure and function. Additionally, the genetic marker rs945270 shows a significant effect on the severity of hyperactivity symptoms of ADHD patients and reward processing in the putamen in girls with ADHD [Xu et al. 2017]. Together, these studies support the proposition that the KTN1 variants may represent a genetic risk factor for ADHD. The present study aims to test this hypothesis.
Materials and Methods
1. Subjects
We examined two independent family-based Caucasian samples in the current study. The first sample (Sample 1) included 922 parent-child trios [2,757 subjects with 924 ADHD children (6–17 years old; mean 10.9±2.9 years; 802 males and 122 females)] from the International Multisite ADHD Genetics (IMAGE) project. The second sample (Sample 2) included 735 parent-child trios [1,383 subjects with 613 ADHD children (6–17 years old; mean 12.3±4.0 years; 472 males and 141 females)] from the PUWMa [Pfizer-funded study from the University of California Los Angeles (UCLA), Washington University (WASH-U), and Massachusetts General Hospital (MGH)] genome-wide association study (GWAS) of ADHD project. One or more sibling(s) in the same age range was included. Both parents or one parent plus two or more siblings were available to provide DNA samples. All children’s IQ scores were above 70.
Children met diagnosis of ADHD on the basis of DSM-IV criteria. The children in the first sample were free of single-gene disorders known to be associated with ADHD (e.g. fragile-X, phenylketonuria, hypercalcaemia, and thyroid hormone resistance), and free of neurological conditions (e.g., hemiplegia, cerebral palsies, epilepsy, hydrocephalus, post-encephalitic syndromes, and sensorimotor handicaps) and psychosis. They did not meet the criteria for autism or Asperger’s syndrome either. The children in the second sample were excluded if they were positive for any of the following: neurological disorder, concussion or other head injuries, lifetime diagnoses of schizophrenia, autism, or mental retardation. The demographic data of the two samples have been described in details earlier [Brookes et al. 2006; Kuntsi et al. 2006; Mick et al. 2010; Neale et al. 2010].
All study procedures were reviewed and approved by the Human Investigation Committee of all institutions. All subjects’ parents or guardians signed written informed consents and children older than 7 years of age signed written assents prior to the study.
2. Genotyping, imputation, ancestry proportion estimation and data cleaning
The first sample was genotyped on PERLEGEN Human600k microarray platform (including 143 KTN1 SNPs), and the second sample was genotyped on Illumina Human1M microarray platform (including 208 KTN1 SNPs). Both samples have 64 overlapping KTN1 SNPs. To render the genetic marker sets consistent across the two samples, we imputed the untyped SNPs across the KTN1 region using the same reference panels of 1000 Genome Project and HapMap3 Project data. This KTN1 region started from Chr14:54995382 (5’-UTR) to Chr14:55550419 (3’-UTR) (Genome Build 36). The program IMPUTE2 [Howie et al. 2009] was applied.
The ancestry proportions for each individual were estimated by integrating the ancestry information content of a set of completely independent ancestry-informative markers (AIMs) using STRUCTURE [Pritchard et al. 2000]. These AIMs were extracted from the whole genome of markers by LD pruning [Purcell et al. 2007] (see details in [Zuo et al. 2012]). The individuals with >50% proportion of European ancestry were classified as Europeans, and others as non-Europeans.
Prior to data analysis, we applied stringent criteria to “clean up” the phenotype and genotype data, as described in detail previously [Zuo et al. 2012]. In brief, subjects with allele discordance, missing race, non-European ethnicity, a mismatch between self-identified and genetically-inferred ethnicity, or a missing genotype call rate ≥ 2% across all SNPs were excluded. Furthermore, we excluded monomorphic SNPs and SNPs with allele discordance, Mendelian errors, or an overall missing genotype call rate ≥ 2%. The SNPs with MAF ≤ 0.01 in either affected or unaffected subjects, or in Hardy-Weinberg disequilibrium (p<0.001) in unaffected subjects were excluded.
3. Allelic and haplotypic association analysis
The first sample served as the discovery sample. A total of 143 KTN1 SNPs were extracted from the array before association analysis. SNP-ADHD association was analyzed using a family-based approach (“--dfam”) as implemented in the program PLINK [Purcell et al. 2007] to derive the p-values. The number (N) of haplotype blocks (D’≥0.8) for these 143 SNPs was calculated using the program Haploview [Barrett et al. 2005]. The significance level (α) for an association was corrected by N in considering multiple independent comparisons (Bonferroni correction). A p < 0.05 indicates a nominally-significant association and p < α indicates a significant association (Table 1). We also used the approach “--adjust” as implemented in PLINK to derive the false discovery rate (FDR) for each p-value. An FDR < 0.05 indicates a significant association.
Table 1.
Bioinformatics of KTN1 SNPs that are significantly associated with ADHD in two independent European-American samples
| Location (haplotype blocks; H) |
Genomic position at 14q | Frequency of risk allele | p-value (α=2.5×10−3) | ENIGMA2 Data (N=13,145) |
PGC sample (20,183 vs. 35,191) |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sample1 (n=924) |
Sample2 (n=613) |
|||||||||||
| SNP | T | U | β | p | OR/Z | p | Function class | Transposons (size; type) | ||||
| rs10140081 | 5’-UTR (H1) |
54995382 | 0.616 (G) | 0.537 | 0.009 | - | - | - | -- | MER117 (155bp; DNA) | ||
| rs8014380 | 55006429 | 0.398 (C) | 0.326 | - | 0.037 | 19.3 | 0.001 | - | - | -- | -- | |
| rs12879661 | 55007883 | 0.607 (G) | 0.529 | 0.011 | - | 14.2 | 0.021 | - | - | -- | L1M5 (523bp; LINE) | |
| rs2880178 | 55024759 | 0.970 (C) | 0.930 | 1.2×10−4 | - | - | - | -- | -- | |||
| rs1610403 | 55027749 | 0.732 (T) | 0.669 | - | 0.042 | 13.3 | 0.050 | - | - | -- | -- | |
| rs2181740 | 5’-UTR (H2.1) |
55052059 | 0.680 (A) | 0.605 | - | 0.028 | 27.0 | 4.5×10−6 | - | - | antisense | -- |
| rs10132888 | 55063703 | 0.693 (A) | 0.608 | - | 0.012 | 26.0 | 1.2×10−5 | - | - | antisense | -- | |
| rs12588935 | 55066650 | 0.693 (C) | 0.607 | - | 0.011 | 25.4 | 2.5×10−5 | 1.98 | 0.047 | antisense | L1MEd (1065bp; LINE) | |
| rs10134019 | 55066745 | 0.695 (G) | 0.613 | - | 0.015 | 23.7 | 1.0×10−4 | - | - | antisense | L1MEd (1065bp; LINE) | |
| rs12434554 | 3’-UTR (H2.2) |
55243858 | 0.633 (G) | 0.574 | 0.042 | - | 27.3 | 2.3×10−5 | - | - | CTCF binding site; enhancer | MIRc (101bp; SINE) |
| rs10150277 | 55257369 | 0.676 (C) | 0.617 | 0.031 | - | 44.9 | 5.3×10−11 | - | - | -- | -- | |
| rs945270 | 55270226 | 0.560 (C) | 0.560 | - | - | -- | -- | - | - | eQTL (p=0.049) | -- | |
| rs12884794 | 3’-UTR (H3) |
55368270 | 0.779 (G) | 0.675 | 1.9×10−5 | - | - | - | 1.04 | 0.028 | -- | MER41B (623bp; LTR) |
| rs945272 | 55406398 | 0.853 (G) | 0.801 | - | 0.028 | - | - | - | - | -- | -- | |
| rs10144952 | 55412322 | 0.851 (C) | 0.803 | 0.014 | - | - | - | - | - | -- | -- | |
| rs11622351 | 3’-UTR (H4) |
55428122 | 0.582 (T) | 0.519 | 0.044 | - | - | - | - | - | lincRNA | L1ME4b (798bp; LINE) |
| rs7148125 | 55428594 | 0.973 (G) | 0.952 | 0.022 | - | - | - | - | - | lincRNA | -- | |
| rs11627062 | 55437408 | 0.674 (T) | 0.609 | 0.020 | - | -- | -- | - | - | lincRNA | Tigger3b (1155bp; DNA) | |
| rs11628495 | 55440678 | 0.618 (G) | 0.554 | 0.033 | - | - | - | 2.18 | 0.029 | lincRNA; chromatin | Tigger3b (1155bp; DNA) | |
| rs7141168 | 55440927 | 0.578 (T) | 0.511 | 0.033 | - | - | - | - | - | lincRNA; chromatin | -- | |
| rs4483776 | 55444918 | 0.762 (T) | 0.711 | 0.033 | - | -- | -- | - | - | lincRNA | -- | |
| rs870887 | 3’-UTR (H5) |
55488583 | 0.845 (T) | 0.768 | - | 2.3×10−3 | - | - | - | - | lincRNA | |
| rs1618626 | 55491518 | 0.815 (C) /0.824 |
0.796/0.755 | 0.042 | 0.009 | - | - | - | - | lincRNA | AluJb (304bp; SINE) | |
| rs12889395 | 55538139 | 0.148 (T) | 0.098 | - | 0.012 | - | - | - | - | -- | L2c (369bp; LINE) | |
| rs12896789 | 55541352 | 0.147 (T) | 0.100 | - | 0.018 | - | - | - | - | -- | -- | |
| rs12893970 | 55544255 | 0.147 (A) | 0.096 | - | 0.011 | - | - | - | - | -- | -- | |
| rs12889979 | 55550419 | 0.943 (C) /0.897 |
0.845 /0.879 |
1.3×10−10 | 0.046 | -- | -- | 1.06 | 0.011 | -- | MSTD (362bp; LTR) | |
H2.1 and H2.2 belong to the same H2 block in structure. n, sample size of affected offspring; N, sample size of ENIGMA2 data; Sample 1, the sample from from the International Multisite ADHD Genetics (IMAGE) project; Sample 2, the sample from the PUWMa project; α, corrected significance level; p < α with FDR< 0.05 is bold; “-”, p>0.05; “--”, missing. T, transmitted; U, untransmitted; risk allele frequencies, corresponding to p<0.05. ENIGMA2 data, imaging genomic data for subcortical structure; β, effect size for SNPs to control putamen volume. PGC sample, the sample deposited in Psychiatric Genomics Consortium (PGC) database (cases vs. controls) (Demontis et al., 2019); OR, odd ratio; Z, z-score. Chromatin, these SNPs are located in an open chromatin region; antisense, located in an antisense lncRNA sequence; lincRNA, located in a long interspersed ncRNA sequence; CTCF binding site, located in a transcriptional repressor CTCF binding site; enhancer, this SNP is located in an enhancer; eQTL, this SNP significantly regulates KTN1 mRNA expression in putamen; transposons, these SNPs are located in transposons that include DNA transposons and RNA retrotransposons (SINE, LINE and LTR).
Multi-locus haplotypes across each block were reconstructed using the program PLINK. Haplotype-ADHD association was analyzed using TDT (“--hap-tdt”) as implemented in PLINK to derive the p-values (Table 2).
Table 2.
Haplotypic associations with ADHD in two samples
| Frequency of haplotype | p-value (α=0.007~0.010) | ||||
|---|---|---|---|---|---|
| Haplotype blocks | Haplotype names | Sample1 | Sample2 | ||
| T | U | (n=924) | (n=613) | ||
| H1 (h=7) | GTGCC | 0.339 | 0.282 | 0.028 | - |
| GCGCT | 0.390 | 0.316 | - | 0.030 | |
| H2 (h=7) | GGATGCC | 0.106 | 0.158 | - | 0.011 |
| AACGGCC | 0.398 | 0.316 | - | 0.016 | |
| H3 (h=6) | GGT | 0.056 | 0.101 | 6.7×10−4 | - |
| AAC | 0.140 | 0.187 | - | 0.041 | |
| AGC | 0.069/0.049 | 0.150/0.084 | 1.2×10−7 | 0.017 | |
| GGC | 0.407 | 0.285 | 8.1×10−6 | ||
| H4 (h=6) | CGTACA | 0.074 | 0.105 | 0.026 | - |
| TGTGTT | 0.512 | 0.401 | 4.1×10−4 | - | |
| H5 (h=5) | TCTTAG | 0.082/0.112 | 0.071/0.067 | 0.032 | 0.009 |
| TCCCGG | 0.016 | 0.054 | 8.4×10−6 | - | |
| TCCCGC | 0.412 | 0.347 | 0.023 | - | |
The haplotype block names, T, U, frequency, Sample1, Sample2, n, bold p values, and “-”, refer to Table 1. α, significance levels corrected by the numbers (h) of haplotypes within each haplotype block.
4. Replication strategy
The second sample served as a replication sample. Briefly, the risk SNPs discovered in the primary sample were classified into haplotype blocks using Haploview first, and then all SNPs (n=1598) including imputed SNPs within these haplotype blocks (Table 1), and haplotypes within each block (Table 2) were tested for SNP-disease and haplotype-disease associations in both discovery and replication samples, in order to examine both SNP- and block- level “replications”. Finally, for the risk variants identified in the present study, we also checked their associations with ADHD in the Psychiatric Genomics Consortium (PGC) database [Demontis et al. 2019]. A replication design would significantly reduce the likelihood of false positives.
5. Bioinformatics analyses
If a risk variant is biologically functional, its statistical association with a disease may be valid. To verify gene-disease associations, we performed functional studies. The potential biological functions of a risk variant can be predicted based on its location on the DNA sequence. For example, an SNP located within a promoter, a transcription factor binding site (TFBS), a methylated CpG island, or an exonic splicing silencer (ESS) or enhancer (ESE) may be able to regulate the transcription; an SNP located in a CTCF binding site may affect the CTCF’s activity and influence gene expression [Chaumeil and Skok 2012; Phillips and Corces 2009]; an SNP located in an open chromatin region is often associated with regulatory factor binding to influence transcription [Song et al. 2011]; an SNP located at a species-conserved element may have functional value [King et al. 2005]; an SNP located with lncRNAs, snRNA or miRNAs may regulate lncRNAs, snRNA and miRNAs and thus influence gene expression and partake in etiological processes; and an SNP located within piRNAs or transposons may affect the phenotypes primarily via transposable element (TE) suppression [Lin et al. 2019; Zuo et al. 2016].
To predict these potential biological functions of the risk SNPs, a series of bioinformatics analyses were conducted. Using the bioinformatics software packages including FuncPred [Xu and Taylor 2009], VE!P [McLaren et al. 2010]), and the UCSC Genome Browser data, the relationship of the risk SNPs with DNA or RNA transposons, long non-coding RNAs (lncRNAs), transcriptional repressor CTCF binding sites, enhancers, and open chromatin regions was explored.
Finally, the potential regulatory effects of the risk SNPs on the KTN1 mRNA expression in human postmortem putamen in an UK European sample (n=129) [Ramasamy et al. 2014] and a European-American cohort (n=111) [The Genotype-Tissue Expression (GTEx) project [GTEx Consortium 2013]] were analyzed using cis-acting expression quantitative trait locus (cis-eQTL) analysis (Table 1).
6. KTN1 mRNA expression in putamen
The KTN1 mRNA expression in putamen was examined in the postmortem brains of three independent cohorts without neurodegenerative disorders (Table 3). The first cohort comprised human putamen tissues extracted from 129 Europeans [UK Brain Expression Consortium (UKBEC); also named BRAINEAC] [Ramasamy et al. 2014]. The second cohort included human putamen tissues extracted from 15 European-Americans (BioGPS) [Zhang et al. 2005]. mRNA expression in these two cohorts was examined using Affymetrix Human ST 1.0 exon arrays (validated by qPCR). The expression levels with normalized intensity > 36, i.e., log2(normalized intensity) > 5.17, were taken as “expressed”. The third cohort included human putamen tissues extracted from 124 European-Americans [The Genotype-Tissue Expression (GTEx) project [GTEx Consortium 2013]. mRNA expression in this cohort was examined using RNA-Seq (validated by qPCR). The expression levels with RPKM values > 1 were taken as “expressed”.
Table 3.
The KTN1 mRNA expression levels in postmortem putamen in three independent cohorts
| UK Europeans (n=129) | European-Americans (n=15) | European-Americans (n=124) | |
|---|---|---|---|
| Databases: | BRAINEAC | BioGPS | GTEx |
| Ages at death (yrs) | 16–102 (59±25) | 54–94 (71±11) | 21–70 (41±14) |
| Brain disorders | No | No | No |
| Experiment methods | Affymetrix Human ST 1.0 exon arrays | Affymetrix Human U133A GeneChip | RNA-Seq |
| Expression Measurement | Log2(normalized intensity) | Log2(normalized intensity) | Transcripts Per Kilobase Million (TPM) |
| Expression threshold | 5.17 | 5.17 | 1 |
| Expression levels | 5.59±0.36 | 9.79±0.62 | 38.70 |
| References | Ramasamy et al. 2014 | Zhang et al. 2005 | GTEx Consortium. 2013 |
n, the sample size. Databases: (1) BRAINEAC and BioGPS: The numbers for expression levels in these columns are log2-transformed normalized intensity values; (2) GTEx: The number for expression level in this column is TPM value.
7. ADHD-risk alleles and putamen volumes
After the risk KTN1 alleles for ADHD were identified from the afore-described association analyses, the potential regulatory effects of these risk alleles on the putamen GMVs were analyzed in a European sample (n=13,145) [Enhancing Neuro Imaging Genetics through Meta-Analysis (ENIGMA2) consortium – GWAS Meta-Analysis of Subcortical Volumes] (http://enigma.ini.usc.edu/) [Hibar et al. 2015] using multiple linear regression analysis. The beta values, a measure of effect size, and the p values, a measure of statistical significance, from regression were reported in Table 1. These subjects were free of neurodegenerative and neuropsychiatric disorders.
Results
1. Significant associations between KTN1 SNPs and ADHD in the first sample (Sample 1; Table 1)
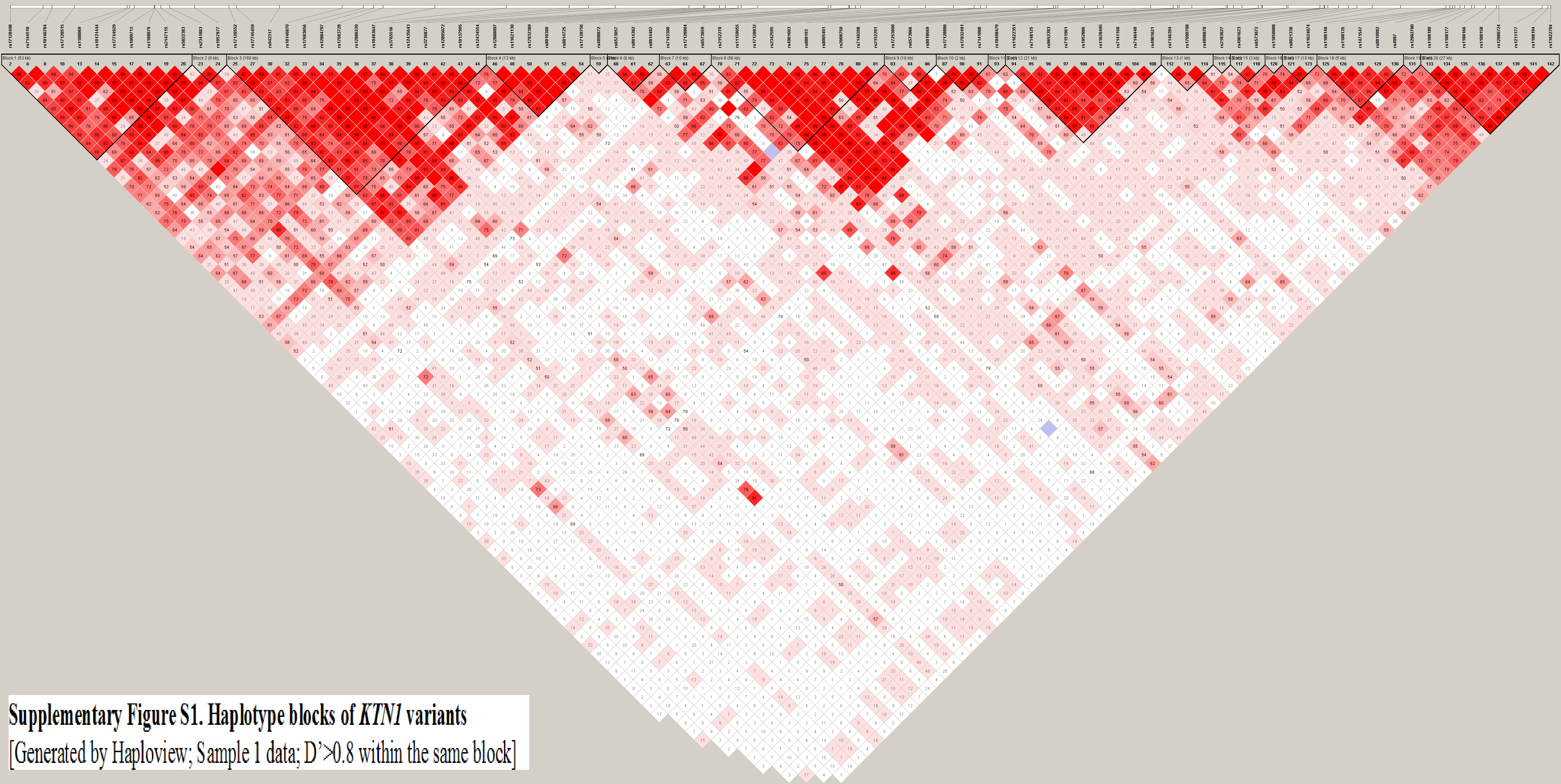
A total of 143 unimputed SNPs were genotyped in KTN1. They were located in 20 haplotype blocks (D’≥0.8; Supplementary Figure S1). Among the 143 SNPs, 15 SNPs were nominally associated with ADHD (p<0.05), and three SNPs were significantly associated with ADHD after both Bonferroni correction for multiple testing (p<corrected α, where α = 0.05/20 blocks = 2.5×10−3) and FDR correction, including rs2880178 (p=1.2×10−4; FDR=0.021), rs12884794 (p=1.9×10−5; FDR=0.004) and rs12889979 (p=1.3×10−10; FDR=6.3×10−8). The 15 nominally-significant SNPs were located in five haplotype blocks (Supplementary Figure S2). The H2 block included the H2.1 block in 5’-UTR and the H2.2 block in 3’-UTR. The three significant SNPs included rs12889979 [p=1.3×10−10; frequency of transmitted risk allele (fT) = 0.943 vs. frequency of untransmitted risk allele (fU) = 0.845], rs12884794 (p=1.9×10−5; fT = 0.779 vs. fU = 0.675), and rs2880178 (p=1.2×10−4; fT = 0.970 vs. fU =0.930). Twelve other SNPs including imputed SNPs located within these five blocks were also tested, but no significant associations were found (p’s>0.05).
2. Associations between KTN1 SNPs and ADHD in the second sample (Sample 2) and the PGC sample (Table 1)
We tested associations in the afore-described five haplotype blocks in the second sample. The results showed nominally-significant associations between ADHD and 13 SNPs in four blocks (p<0.05), H1, H2, H3 and H5, including a significant association between ADHD and rs870887 in H5 (p=2.3×10−3). Two SNPs, i.e., rs1618626 and rs12889979 in H5, which were associated with ADHD in the first sample (p=0.042 and 1.3×10−10, respectively), were also associated with ADHD in the second sample (p=0.009 and 0.046, respectively). Fourteen other SNPs including imputed SNPs located within these five blocks were also tested in the second sample, but no significant associations were found (p’s>0.05). Furthermore, four SNPs, each separately located in four blocks (H2–H5) respectively, were also associated with ADHD in the PGC sample (20,183 ADHD cases vs 35,191 healthy controls), including rs12588935, rs12884794, rs11628495, and rs12889979 (p=0.047, 0.028, 0.029 and 0.011, respectively). Therefore, a total of five SNPs were associated with ADHD across at least two samples, including one SNP (rs12889979) across three samples. The risk alleles, whose frequencies were higher in cases than controls, were consistent across Samples 1, 2 and PGC (see Table 1).
3. Associations between KTN1 haplotypes and ADHD in both samples (Table 2)
Significant haplotypic associations were found in both samples (1.2×10−7≤p≤0.041). Because seven (for H1 haplotype block), seven (H2), six (H3), six (H4) and five (H5) haplotypes were reconstructed within each haplotype block, respectively, the corrected significance levels (α) for haplotypic associations were between 0.007 and 0.010. After correction, haplotypes GGT (p=6.7×10−4), AGC (p=1.2×10−7) and GGC (p=8.1×10−6) within H3 block, TGTGTT (p=4.1×10−4; H4) and TCCCGG (p=8.4×10−6; H5) in the first sample, and TCTTAG (p=0.009; H5) in the second sample remained significantly associated with ADHD. Among these significant risk haplotypes, AGC (p=0.017) was also suggestively associated with ADHD in the second sample (Sample 2) and TCTTAG (p=0.032) was also suggestively associated with ADHD in the first sample (Sample 1).
4. The risk KTN1 SNPs may be biologically functional (Table 1)
Bioinformatic analysis showed that the variants within the same haplotype blocks were not only highly linked but also shared similar biological functions (Table 1). Furthermore, the risk variants were located in many DNA transposons and RNA retrotransposons.
The four variants in the H2.1 block, the six variants in the H4 block, and the two variants in the H5 block were located in an antisense lncRNA and a long intergenic non-coding RNA (lincRNA), respectively. In the H2.2 block, rs12434554, a variant (p=4.7×10–9) that was implicated in cognitive function [Han et al. 2017], was located in a transcriptional repressor CTCF binding site and an enhancer. Two variants in the H4 block; i.e., rs11628495 and rs7141168, were located in an open chromatin region. In the H5, rs12896789 was located in a conserved element conserved across 17 different species [King et al. 2005]. No variants were located at TFBS, promoter, miRNA, snRNA and CpG island. Finally, rs945270 in H2.2, a variant (p=1.1×10−33) associated with putamen GMV [Hibar et al. 2015; Xu et al. 2017], cis-regulated the KTN1 mRNA expression in the putamen (p=0.049), consistent with previous reports [Han et al. 2017; Hibar et al. 2015; Westra et al. 2013]. No other positive eQTL signal was found.
5. The KTN1 mRNA is significantly expressed in putamen across three independent cohorts (Table 3)
In the three independent populations of European origin, the KTN1 mRNA was found to be abundant in the putamen. The expression levels are 5.59 [log2(normalized intensity)], 9.79 [log2(normalized intensity)] and 38.70 [Transcripts Per Kilobase Million (TPM)] in the UK European and two European-American cohorts, respectively, confirming previous reports [Kang et al. 2011].
6. The ADHD-risk alleles potentially increased the putamen volume (Table 1)
The alleles with significantly higher frequencies in the transmitted (T) groups than the untransmitted (U) groups were identified as the risk alleles for ADHD by the afore-mentioned association analyses. All of the risk alleles that increased the risk for ADHD increased the putamen volumes in the ENIGMA2 sample. For example, the risk alleles of three variants in H1 block, including rs8014380 (β=19.3, p=0.001), rs12879661 (β=14.2, p=0.021) and rs1610403 (β=13.3, p=0.050), modestly increased (β>0) the putamen volumes. The risk alleles of four variants in H2.1 block, including rs2181740 (β=27.0, p=4.5×10−6), rs10132888 (β=26.0, p=1.2×10−5), rs12588935 (β=25.4, p=2.5×10−5) and rs10134019 (β=23.7, p=1.0×10−4), significantly increased the putamen volumes; and the risk alleles of two variants in H2.2 block, including rs12434554 (β=27.3, p=2.3×10−5) and rs10150277 (β=44.9, p=5.3×10−11), highly significantly increased the putamen volumes. All the alleles that increased both risk for ADHD and putamen volumes were major alleles (f>0.5). So far only four KTN1 variants have been reported in the literature to significantly regulate the putamen GMVs, and the alleles of these four variants that increased the putamen GMVs all were major alleles. They were rs2181743 (β=43.0, p=4.0×10−8) [Chen et al. 2017] located in H2.1 block, and rs8017172 (Z=15.3, p=1.2×10−52) [Satizabal et al. 2017], rs945270 (β=48.9, p=1.1×10−33) [Hibar et al. 2015] and rs17253792 (β=51.8, p=3.2×10−7) [Chen et al. 2017], located in H2.2 block. Among them, the major allele of rs945270 was reported to most significantly increase the putamen volume by previous GWASs [Hibar et al. 2015] and risk for ADHD as well [Xu et al. 2017].
Discussion
Using a GWAS approach, the largest meta-analysis so far has reported 12 genome-wide significant (p<5×10−8) risk loci for ADHD on chromosomes 1–5, 7–8, 10, 12, 15 and 16 [Demontis et al. 2019]. This study controlled the false positive rates by Bonferroni correction, i.e., setting α at 5×10−8. The use of this stringent but conservative threshold may account for its not identifying KTN1 at chromosome 14. Here, using a candidate gene approach, we targeted the KTN1, controlling the false positive rates by replication with independent samples and verification with a series of functional studies, and demonstrated that KTN1 may be a risk gene for ADHD too.
We found that KTN1 variants and haplotypes were significantly associated with ADHD, with some associations, primarily in the discovery sample, remaining significant after correction for multiple testing. Variants at four haplotype blocks (except for H4), particularly two variants in H5, and haplotypes at three blocks (except for H2 and H4), particularly two haplotypes within H3 and H5, were associated with ADHD across both discovery and replication samples. Many of these risk variants may be biologically functional, regulating the putamen volumes, and KTN1 mRNAs are expressed in the putamen across three independent cohorts, suggesting that KTN1 plays a functional role in the development of ADHD.
Twelve of the 15 risk KTN1 SNPs are located in two major classes of transposable elements (TEs) [Levin and Moran 2011], including RNA transposons (i.e., retrotransposons) and DNA transposons. The retrotransposons include long terminal repeat elements (LTR), long interspersed nuclear element (LINE), and short interspersed nuclear elements (SINE). The primary function of these KTN1-neighboring TEs is to form insertional mutations in the genome that may control the transcription of KTN1, playing a decisive mutagenic role in degenerative pathologies [Li et al. 2013; O’Donnell and Burns 2010; Sturm et al. 2017], most obviously in the brain [De Cecco et al. 2013; Van Meter et al. 2014]. The retrotransposition-mediated events may produce somatic alterations in the brain [Baillie et al. 2011; Muotri et al. 2005; Solyom et al. 2012], such as neuronal somatic mosaics [Singer et al. 2010; Thomas et al. 2012], creating genomic diversity (genetic heterogeneity) across neurons in the same individual. Thus, retrotransposons may play a role in neurogenesis [Coufal et al. 2009; Muotri et al. 2005; Muotri et al. 2010], aging [Li et al. 2013] and the development of neurodegenerative diseases [Li et al. 2012] and other neuropsychiatric conditions such as post-traumatic stress disorder [Ponomarev et al. 2010].
Twelve KTN1 variants in three haplotype blocks located in an antisense lncRNA and a lincRNA may regulate KTN1 mRNA expression. The variant in a transcriptional repressor CCCTC-binding factor (CTCF) binding site or in an enhancer may affect the transcription. rs945270, which has been reported to affect putamen GMV [Hibar et al. 2015; Xu et al. 2017], significantly cis-regulates KTN1 mRNA expression in the putamen. Finally, those variants in an open chromatin region are usually associated with regulatory factor binding. These potential biological functions of risk SNPs along with KTN1 mRNA expression in putamen suggest a functional role of the KTN1 in the development of ADHD.
A very interesting finding was that the ADHD-risk alleles increased the putamen volumes. This finding was consistent with previous findings [Chen et al. 2017; Hibar et al. 2015; Satizabal et al. 2017; Xu et al. 2017] in the following aspects. First, the alleles increasing the putamen GMVs were all major alleles both in the current findings and in the literature. Second, the variants significantly regulating the putamen GMVs were clustered within the H2 block; in particular, the highly significant variants were clustered within the H2.2 block both in the current study and the literature.
Low levels of dopamine have been widely associated with ADHD [Frank et al. 2007], which may result from the lesions in the dopamine-rich ventral putamen [Max et al. 2002] or reduction in putamen GMV [Wellington et al. 2006] that is related to loss of dopaminergic neurons. Primary pharmacological regimens for ADHD, including methylphenidate (Ritalin) and amphetamine (Adderal), increase the dopamine levels [Arnsten 2006]. However, in the unmedicated ADHD patients, a compensatory response to the loss of dopaminergic neurons and dopamine depletion may transpire to maintain dopaminergic transmission [Hulshoff Pol et al. 2000; Krabbe et al. 2005]. This compensatory mechanism may drive the expression of GMV-controlling proteins such as kinectin 1 encoded by KTN1 [Chen et al. 2017; Hibar et al. 2015; Satizabal et al. 2017; Xu et al. 2017]. This could potentially explain the finding that the ADHD-risk alleles increased the putamen volumes in subjects without brain disorders. However, in ADHD patients who have suffered loss of dopaminergic neurons and dopamine depletion in the putamen, this compensatory process does not appear to completely restore the putamen GMVs; as a result, the overall putamen GMVs are diminished in the patients. Thus, the reduction of putamen GMVs in patients with ADHD may not be caused directly by ADHD-risk KTN1 alleles; rather, this reduction may drive the expression of ADHD-risk KTN1 alleles that lead to ADHD but only partially compensate for GMV reduction.
A major limitation of the current study is that the replications of KTN1-ADHD associations were primarily at the haplotype- but not SNP-level. Future research to examine a denser marker set by sequencing may help to verify the SNP-level replications. Secondly, the biological functions of the risk variants were predicted only by bioinformatics analyses, and would warrant confirmation by molecular experiments in the future.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgements:
This work was supported in part by National Institute on Alcohol Abuse and Alcoholism (NIAAA) grants R21 AA021380, R21 AA020319 and R21 AA023237 (XL), and Key Clinical Specialty Discipline Construction Program of Fuzhou, Fujian, P.R.C (JJ) and NIH grant DA023248 (CRL). We thank NIH GWAS Data Repository, the Contributing Investigator(s) who contributed the phenotype and genotype data from his/her original study (e.g., Drs. Faraone, Asherson, Mick, Neale, Kuntsi, Brookes, Nelson, etc.), and the primary funding organization that supported the contributing study. Funding and other supports for phenotype and genotype data were provided through the NIH Grants R13MH059126, R01MH62873, U01MH085518 and R01MH081803 to Stephen Faraone; NINDS grant NS054124 to Sandra Loo; NIMH grant MH01966 to James McGough; NIMH grant MH01805 to James McCracken, NIDA grant K24 DA016264 to Timothy Wilens; NIMH grant MH63706 to Susuan Smalley; and NIMH grants K08MH001503 and R01MH066237 to Janet Wozniak. The datasets used for the analyses described in this manuscript were obtained from dbGaP at http://www.ncbi.nlm.nih.gov/sites/entrez?Db=gap. dbGaP Study Accession: phs000016.v2.p2 and phs000358.v1.p1.
Footnotes
Conflict of Interest: The authors declare no conflicts of interest.
References
- Arnsten AF. (2006). Stimulants: Therapeutic actions in ADHD. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology 31(11):2376–2383. [DOI] [PubMed] [Google Scholar]
- Baillie JK, Barnett MW, Upton KR, Gerhardt DJ, Richmond TA, De Sapio F, … Faulkner GJ. (2011). Somatic retrotransposition alters the genetic landscape of the human brain. Nature 479(7374):534–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett JC, Fry B, Maller J, Daly MJ. (2005). Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21(2):263–265. [DOI] [PubMed] [Google Scholar]
- Brookes K, Xu X, Chen W, Zhou K, Neale B, Lowe N, … Asherson P. (2006). The analysis of 51 genes in DSM-IV combined type attention deficit hyperactivity disorder: association signals in DRD4, DAT1 and 16 other genes. Mol Psychiatry 11(10):934–953. [DOI] [PubMed] [Google Scholar]
- Bush G, Valera EM, Seidman LJ. (2005). Functional neuroimaging of attention-deficit/hyperactivity disorder: a review and suggested future directions. Biological psychiatry 57(11):1273–1284. [DOI] [PubMed] [Google Scholar]
- Caravaggio F, Plitman E, Chung JK, Gerretsen P, Kim J, Iwata Y, … Graff-Guerrero A. (2017). Trait impulsiveness is related to smaller post-commissural putamen volumes in males but not females. The European journal of neuroscience 46(7):2253–2264. [DOI] [PubMed] [Google Scholar]
- Chaumeil J, Skok JA. (2012). The role of CTCF in regulating V(D)J recombination. Curr Opin Immunol 24(2):153–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CH, Wang Y, Lo MT, Schork A, Fan CC, Holland D, … Dale AM. (2017). Leveraging genome characteristics to improve gene discovery for putamen subcortical brain structure. Scientific reports 7(1):15736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coufal NG, Garcia-Perez JL, Peng GE, Yeo GW, Mu Y, Lovci MT, … Gage FH. (2009). L1 retrotransposition in human neural progenitor cells. Nature 460(7259):1127–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Cecco M, Criscione SW, Peckham EJ, Hillenmeyer S, Hamm EA, Manivannan J, … Sedivy JM. (2013). Genomes of replicatively senescent cells undergo global epigenetic changes leading to gene silencing and activation of transposable elements. Aging cell 12(2):247–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit S, Watson P, Harsay HA, Cohen MX, van de Vijver I, Ridderinkhof KR. (2012). Corticostriatal connectivity underlies individual differences in the balance between habitual and goal-directed action control. The Journal of neuroscience : the official journal of the Society for Neuroscience 32(35):12066–12075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demontis D, Walters RK, Martin J, Mattheisen M, Als TD, Agerbo E, … Neale BM. (2019). Discovery of the first genome-wide significant risk loci for attention deficit/hyperactivity disorder. Nature genetics 51(1):63–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellison-Wright I, Ellison-Wright Z, Bullmore E. (2008). Structural brain change in Attention Deficit Hyperactivity Disorder identified by meta-analysis. BMC Psychiatry 8:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank MJ, Santamaria A, O’Reilly RC, Willcutt E. (2007). Testing computational models of dopamine and noradrenaline dysfunction in attention deficit/hyperactivity disorder. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology 32(7):1583–1599. [DOI] [PubMed] [Google Scholar]
- Greven CU, Bralten J, Mennes M, O’Dwyer L, van Hulzen KJ, Rommelse N, … Buitelaar JK. (2015). Developmentally stable whole-brain volume reductions and developmentally sensitive caudate and putamen volume alterations in those with attention-deficit/hyperactivity disorder and their unaffected siblings. JAMA Psychiatry 72(5):490–499. [DOI] [PubMed] [Google Scholar]
- Consortium GTEx. (2013). The Genotype-Tissue Expression (GTEx) project. Nature genetics 45(6):580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han L, Jia Z, Cao C, Liu Z, Liu F, Wang L, … Chen L. (2017). Potential contribution of the neurodegenerative disorders risk loci to cognitive performance in an elderly male gout population. Medicine 96(39):e8195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herskovits EH, Megalooikonomou V, Davatzikos C, Chen A, Bryan RN, Gerring JP. (1999). Is the spatial distribution of brain lesions associated with closed-head injury predictive of subsequent development of attention-deficit/hyperactivity disorder? Analysis with brain-image database. Radiology 213(2):389–394. [DOI] [PubMed] [Google Scholar]
- Hibar DP, Stein JL, Renteria ME, Arias-Vasquez A, Desrivieres S, Jahanshad N, … Medland SE. (2015). Common genetic variants influence human subcortical brain structures. Nature 520(7546):224–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong SB, Harrison BJ, Dandash O, Choi EJ, Kim SC, Kim HH, … Yi SH. (2015). A selective involvement of putamen functional connectivity in youth with internet gaming disorder. Brain research 1602:85–95. [DOI] [PubMed] [Google Scholar]
- Howie BN, Donnelly P, Marchini J. (2009). A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet 5(6):e1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulshoff Pol HE, van der Flier WM, Schnack HG, Tulleken CA, Ramos LM, van Ree JM, Kahn RS. (2000). Frontal lobe damage and thalamic volume changes. Neuroreport 11(13):3039–3041. [DOI] [PubMed] [Google Scholar]
- Kang HJ, Kawasawa YI, Cheng F, Zhu Y, Xu X, Li M, … Sestan N. (2011). Spatio-temporal transcriptome of the human brain. Nature 478(7370):483–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khadka S, Pearlson GD, Calhoun VD, Liu J, Gelernter J, Bessette KL, Stevens MC. (2016). Multivariate Imaging Genetics Study of MRI Gray Matter Volume and SNPs Reveals Biological Pathways Correlated with Brain Structural Differences in Attention Deficit Hyperactivity Disorder. Front Psychiatry 7:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King DC, Taylor J, Elnitski L, Chiaromonte F, Miller W, Hardison RC. (2005). Evaluation of regulatory potential and conservation scores for detecting cis-regulatory modules in aligned mammalian genome sequences. Genome research 15(8):1051–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krabbe K, Karlsborg M, Hansen A, Werdelin L, Mehlsen J, Larsson HB, Paulson OB. (2005). Increased intracranial volume in Parkinson’s disease. J Neurol Sci 239(1):45–52. [DOI] [PubMed] [Google Scholar]
- Kumar J, Yu H, Sheetz MP. (1995). Kinectin, an essential anchor for kinesin-driven vesicle motility. Science 267(5205):1834–1837. [DOI] [PubMed] [Google Scholar]
- Kuntsi J, Neale BM, Chen W, Faraone SV, Asherson P. (2006). The IMAGE project: methodological issues for the molecular genetic analysis of ADHD. Behavioral and brain functions : BBF 2:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin HL, Moran JV. (2011). Dynamic interactions between transposable elements and their hosts. Nature reviews Genetics 12(9):615–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Jin Y, Prazak L, Hammell M, Dubnau J. (2012). Transposable elements in TDP-43-mediated neurodegenerative disorders. PLoS One 7(9):e44099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Prazak L, Chatterjee N, Gruninger S, Krug L, Theodorou D, Dubnau J. (2013). Activation of transposable elements during aging and neuronal decline in Drosophila. Nature neuroscience 16(5):529–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X, Xia Y, Hu D, Mao Q, Yu Z, Zhang H, … Luomicron X. (2019). Transcriptomewide piRNA profiling in human gastric cancer. Oncology reports 41(5):3089–3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X, Guo X, Luo X, Tan Y, Zhang P, Yang K, … Li CR. (2020). Significant, replicable, and functional associations between KTN1 variants and alcohol and drug codependence. Addict Biol:e12888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X, Mao Q, Shi J, Wang X, Li CR. (2019). Putamen gray matter volumes in neuropsychiatric and neurodegenerative disorders. World J Psychiatry Ment Health Res 3(1):pii:1020. [PMC free article] [PubMed] [Google Scholar]
- Max JE, Fox PT, Lancaster JL, Kochunov P, Mathews K, Manes FF, … Lansing AE. (2002). Putamen lesions and the development of attention-deficit/hyperactivity symptomatology. J Am Acad Child Adolesc Psychiatry 41(5):563–571. [DOI] [PubMed] [Google Scholar]
- McDonald WM, Husain M, Doraiswamy PM, Figiel G, Boyko O, Krishnan KR. (1991). A magnetic resonance image study of age-related changes in human putamen nuclei. Neuroreport 2(1):57–60. [DOI] [PubMed] [Google Scholar]
- McLaren W, Pritchard B, Rios D, Chen Y, Flicek P, Cunningham F. (2010). Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics 26(16):2069–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mick E, Todorov A, Smalley S, Hu X, Loo S, Todd RD, … Faraone SV. (2010). Family-based genome-wide association scan of attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry 49(9):898–905 e893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muotri AR, Chu VT, Marchetto MC, Deng W, Moran JV, Gage FH. (2005). Somatic mosaicism in neuronal precursor cells mediated by L1 retrotransposition. Nature 435(7044):903–910. [DOI] [PubMed] [Google Scholar]
- Muotri AR, Marchetto MC, Coufal NG, Oefner R, Yeo G, Nakashima K, Gage FH. (2010). L1 retrotransposition in neurons is modulated by MeCP2. Nature 468(7322):443–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale BM, Medland SE, Ripke S, Asherson P, Franke B, Lesch KP, … Psychiatric GCAS. (2010). Meta-analysis of genome-wide association studies of attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry 49(9):884–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell KA, Burns KH. (2010). Mobilizing diversity: transposable element insertions in genetic variation and disease. Mobile DNA 1(1):21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips JE, Corces VG. (2009). CTCF: master weaver of the genome. Cell 137(7):1194–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponomarev I, Rau V, Eger EI, Harris RA, Fanselow MS. (2010). Amygdala transcriptome and cellular mechanisms underlying stress-enhanced fear learning in a rat model of posttraumatic stress disorder. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology 35(6):1402–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK, Stephens M, Donnelly P. (2000). Inference of population structure using multilocus genotype data. Genetics 155(2):945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, … Sham PC. (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. American journal of human genetics 81(3):559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramasamy A, Trabzuni D, Guelfi S, Varghese V, Smith C, Walker R, … Weale ME. (2014). Genetic variability in the regulation of gene expression in ten regions of the human brain. Nature neuroscience 17(10):1418–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satizabal CL, Adams HHH, Hibar DP, White CC, Stein JL, Scholz M, … Ikram MA. (2017). Genetic Architecture of Subcortical Brain Structures in Over 40,000 Individuals Worldwide. bioRxiv:https://www.biorxiv.org/content/10.1101/173831v173831.article-info.
- Seidman LJ, Biederman J, Liang L, Valera EM, Monuteaux MC, Brown A, … Makris N. (2011). Gray matter alterations in adults with attention-deficit/hyperactivity disorder identified by voxel based morphometry. Biological psychiatry 69(9):857–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer T, McConnell MJ, Marchetto MC, Coufal NG, Gage FH. (2010). LINE-1 retrotransposons: mediators of somatic variation in neuronal genomes? Trends Neurosci 33(8):345–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobel LJ, Bansal R, Maia TV, Sanchez J, Mazzone L, Durkin K, … Peterson BS. (2010). Basal ganglia surface morphology and the effects of stimulant medications in youth with attention deficit hyperactivity disorder. The American journal of psychiatry 167(8):977–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solyom S, Ewing AD, Rahrmann EP, Doucet T, Nelson HH, Burns MB, … Kazazian HH Jr. (2012). Extensive somatic L1 retrotransposition in colorectal tumors. Genome research 22(12):2328–2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L, Zhang Z, Grasfeder LL, Boyle AP, Giresi PG, Lee BK, … Furey TS. (2011). Open chromatin defined by DNaseI and FAIRE identifies regulatory elements that shape cell-type identity. Genome research 21(10):1757–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturm A, Perczel A, Ivics Z, Vellai T. (2017). The Piwi-piRNA pathway: road to immortality. Aging cell 16(5):906–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas CA, Paquola AC, Muotri AR. (2012). LINE-1 retrotransposition in the nervous system. Annu Rev Cell Dev Biol 28:555–573. [DOI] [PubMed] [Google Scholar]
- Toyoshima I, Sheetz MP. (1996). Kinectin distribution in chicken nervous system. Neuroscience letters 211(3):171–174. [DOI] [PubMed] [Google Scholar]
- Valera EM, Faraone SV, Murray KE, Seidman LJ. (2007). Meta-analysis of structural imaging findings in attention-deficit/hyperactivity disorder. Biological psychiatry 61(12):1361–1369. [DOI] [PubMed] [Google Scholar]
- Van Meter M, Kashyap M, Rezazadeh S, Geneva AJ, Morello TD, Seluanov A, Gorbunova V. (2014). SIRT6 represses LINE1 retrotransposons by ribosylating KAP1 but this repression fails with stress and age. Nature communications 5:5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellington TM, Semrud-Clikeman M, Gregory AL, Murphy JM, Lancaster JL. (2006). Magnetic resonance imaging volumetric analysis of the putamen in children with ADHD: combined type versus control. J Atten Disord 10(2):171–180. [DOI] [PubMed] [Google Scholar]
- Westra HJ, Peters MJ, Esko T, Yaghootkar H, Schurmann C, Kettunen J, … Franke L. (2013). Systematic identification of trans eQTLs as putative drivers of known disease associations. Nature genetics 45(10):1238–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B, Jia T, Macare C, Banaschewski T, Bokde ALW, Bromberg U, … Consortium I. (2017). Impact of a Common Genetic Variation Associated With Putamen Volume on Neural Mechanisms of Attention-Deficit/Hyperactivity Disorder. J Am Acad Child Adolesc Psychiatry 56(5):436–444 e434. [DOI] [PubMed] [Google Scholar]
- Xu Z, Taylor JA. (2009). SNPinfo: integrating GWAS and candidate gene information into functional SNP selection for genetic association studies. Nucleic acids research 37(Web Server issue):W600–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Tee YH, Heng JK, Zhu Y, Hu X, Margadant F, … Yu H. (2010). Kinectin-mediated endoplasmic reticulum dynamics supports focal adhesion growth in the cellular lamella. Journal of cell science 123(Pt 22):3901–3912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, James M, Middleton FA, Davis RL. (2005). Transcriptional analysis of multiple brain regions in Parkinson’s disease supports the involvement of specific protein processing, energy metabolism, and signaling pathways, and suggests novel disease mechanisms. Am J Med Genet B Neuropsychiatr Genet 137B(1):5–16. [DOI] [PubMed] [Google Scholar]
- Zuo L, Gelernter J, Zhang CK, Zhao H, Lu L, Kranzler HR, … Luo X. (2012). Genome-wide association study of alcohol dependence implicates KIAA0040 on chromosome 1q. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology 37(2):557–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo L, Wang Z, Tan Y, Chen X, Luo X. (2016). piRNAs and their functions in the brain. Int J Hum Genet 16(1–2):53–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.