

Abstract
Blockade of programmed death-ligand 1 (PD-L1) by therapeutic antibodies has shown to be a promising strategy in cancer therapy, yet clinical response in many types of cancer, including prostate cancer (PCa), is limited. Tumor cells secrete PD-L1 through exosomes or splice variants, which has been described as a new mechanism for the resistance to PD-L1 blockade therapy in multiple cancers, including PCa. This suggests that cutting off the secretion or expression of PD-L1 might improve the response rate of PD-L1 blockade therapy in PCa treatment. Here we report that p300/CBP inhibition by a small molecule p300/CBP inhibitor dramatically enhanced the efficacy of PD-L1 blockade treatment in a syngeneic model of PCa by blocking both the intrinsic and interferon gamma (IFN-γ)-induced PD-L1 expression. Mechanistically, p300/CBP could be recruited to the promoter of CD274 (encoding PD-L1) by the transcription factor IRF-1, which induced the acetylation of Histone H3 at CD274 promoter followed by the transcription of CD274. A485, a p300/CBP inhibitor, abrogated this process and cut off the secretion of exosomal PD-L1 by blocking the transcription of CD274, which combined with the anti-PD-L1 antibody to reactivate T cells function for tumor attack. This finding reports a new mechanism of how cancer cells regulate PD-L1 expression through epigenetic factors and provides a novel therapeutic approach to enhance the efficacy of immune checkpoint inhibitors treatment.
Keywords: PD-L1, p300, HDAC, PCa treatment
INTRODUCTION
Based on Cancer statistics, 2019, prostate cancer (PCa) is the most common cancer expected to occur in American men, accounting for 20% of new diagnoses. It is the secondary leading cause of cancer death for men, only after lung cancer [1]. The standard treatment for PCa usually begins with local therapy, followed by androgen deprivation therapy (ADT). ADT is effective initially, but unfortunately, most of the patients eventually become resistant and the tumor progresses to castration-resistant prostate cancer (CRPC) [2]. Enzalutamide, the second-generation of androgen receptor (AR) antagonist, approved by FDA in 2012 for the treatment of metastatic CRPC (mCRPC), significantly improved the survival of patients with mCRPC. However, just as ADT, enzalutamide is only effective for a period of time and cancer cells become resistant in the majority of patients [3]. So far, no effective treatment is available for enzalutamide-resistant mCRPC. New therapeutic agents or approaches are urgently needed for these and future patients.
Cancer immunotherapies through an immune checkpoint blockade with anti-programmed cell death protein 1 (PD-1)/programmed death-ligand 1 (PD-L1) antibodies have shown significant clinical benefits in multiple cancer types, including melanoma, non-small cell lung carcinoma and renal cell carcinomas [4, 5]. Meanwhile, a large number of clinical trials are currently underway for other cancers. It is now clear that tumor cells escape immune attack by upregulating the expression of programmed death-ligand 1 (PD-L1), which could bind with programmed death-1 (PD-1) receptor on T cells to block the function of T cells and induce T cell exhaustion [6]. Antibodies against PD-1/PD-L1 inhibit the function of immune checkpoints and lead to the reactivation of T cells to attack cancer cells. While millions of patients with cancer would benefit from this new approach for therapy, not all types of cancers well respond to this treatment; PCa is one of them.
It was previously reported that tumor cells could release the exosomes carrying PD-L1 in an interferon gamma (IFN-γ) dependent manner, consequently suppressing T cell function and antagonizing anti-PD-1 response [7]. More recently, two unique tumor-secreted PD-L1 splicing variants without the transmembrane domain were identified, providing another mechanism for resistance to anti-PD-L1 blockade therapy [8]. More interestingly, it was found that CD274 (encoding PD-L1) mRNA was actually much higher in PCa cells, even compared with that of melanoma cells, a well responded cancer type to PD-L1 blockade. Surprisingly, most of the translated PD-L1 was secreted extracellularly by exosomes instead of transporting to the cell surface, consequently inhibiting T cell function and contributing to resistance to PD-L1 blockade treatment. And, removal of exosomal PD-L1 could overcome the resistance of PCa to PD-L1 blockade [9]. These findings suggest that cell-to-cell interaction is not required for PD-L1 to inhibit T cell function and cause immune evasion. A strategy that could control exosomal PD-L1 level might enhance the efficacy of PD-L1 blockade treatment in PCa.
Recently, epigenetics has broadened our knowledge in complex human diseases, including cancer. At least four different DNA modifications and 16 histone modifications have been reported [10]. These events are carefully regulated and tightly controlled in cells. It has been well-documented that the deregulation of these epigenetic events contributes to cancer progression, including PCa [11]. As expected, androgen receptor (AR), the well-known driver of PCa progression, and its downstream signaling events are closely regulated by epigenetic modifications [12, 13]. Of note, targeting the epigenetics factors like p300, bromodomain-containing protein 4 (BRD4) and enhancer of zeste homolog 2 (EZH2) has shown promising therapeutic avenue in PCa treatment [13–15]. Thus, we aimed to test whether epigenetic modification could regulate PD-L1 expression in PCa, and whether this regulation would affect the response of PCa to the immune checkpoint blockade therapy.
RESULTS
Class I HDAC inhibition increased PD-L1 level in metastatic PCa cells
To search for potential epigenetic modulators that are involved in the regulation of PD-L1 expression, we used multiple inhibitors that target DNA methylation or histone modifications in metastatic PCa cell line DU145. As shown in Figure 1A, SAHA, the inhibitor of histone deacetylases (HDACs), significantly increased the level of PD-L1. This increase was comparable with the IFN-γ treatment, a well-known cytokine inducing PD-L1 expression. To further validate this finding, we treated DU145 and PC-3, another metastatic PCa cell line, with SAHA and another HDAC inhibitor, LBH589 (LBH), respectively. As indicated in Figure 1B, both SAHA and LBH589 strikingly increased PD-L1 expression in PC-3 and DU145 cells. These observations were further confirmed by the flow cytometry analysis (Fig. 1C), indicating that inhibition of HDACs could also increase the surface PD-L1. SAHA and LBH589 could target both class I and II HDACs. Next, to exclude the off-target of inhibitors and further explore which HDAC was responsible for the regulation of PD-L1 expression, we used shRNAs to deplete class I and II HDACs, respectively. As shown in Figure 1D, only depletion of class I HDACs (HDAC1, 2, 3) was responsible for the up-regulation of PD-L1, but not depletion of class II (HDAC4 and 6, Supp. Fig. 1A). Interestingly, knockdown of HDAC6 actually decreased the level of PD-L1.
Figure 1. Class I HDACs inhibition increased PD-L1 expression in metastatic PCa cells.
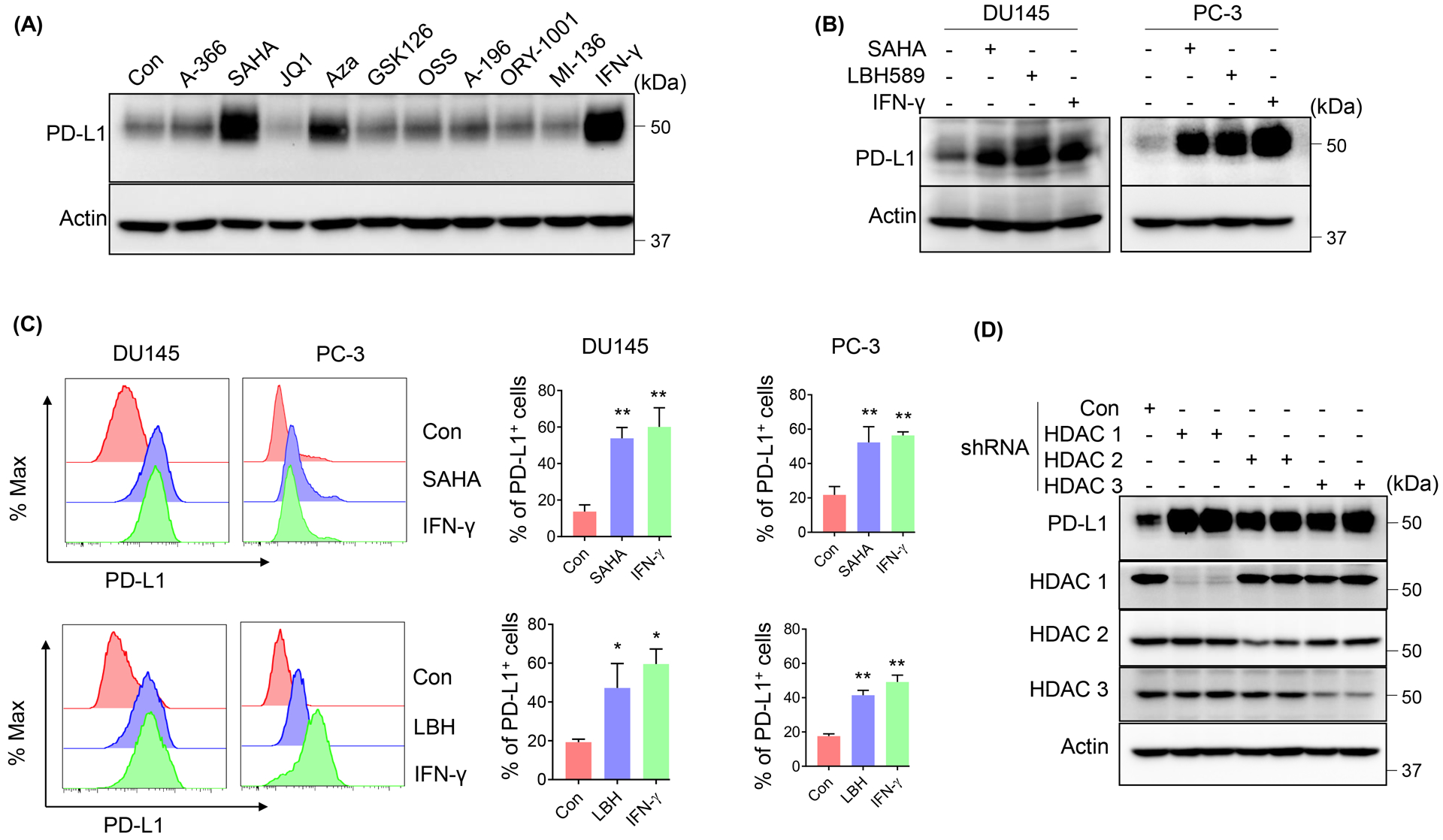
(A) IB analysis of whole cell lysates (WCL) derived from DU145 cells treated with the indicated inhibitors (concentrations and targets were shown in Supplemental Table 1) or 10 ng/ml IFN-γ for 24 h. PD-L1 expression was determined by IB (B) or fluorescence-activated cell sorting (FACS) (C) after DU145/PC-3 cells were treated with 1 μM SAHA or 50 nM LBH589 or 10 ng/ml IFN-γ for 24 h. The statistical data were shown as mean values ± SD (n ≥ 3), *p < 0.05 **p<0.01. (D) IB analysis of WCL derived from DU145 cells infected with lentivirus expressing the indicated shRNAs.
Upregulated PD-L1 by class I HDACs inhibition was due to the increased CD274 transcription
It was reported that the acetylation of protein could regulate protein stability [16]. To test whether class I HDAC inhibition induced the acetylation of PD-L1 protein, we treated DU145 cells with SAHA and performed an IP assay for PD-L1 followed by IB with an anti-Acetylated-Lysine antibody. As shown in Supplemental Figure 1B, no acetylated signal was detected on PD-L1. Next, we determined the expression level of CD274 (encoding PD-L1), after SAHA, LBH589 or IFN-γ treatment. As shown in Figure 2A, HDACs inhibition significantly increased the expression of CD274. Consistent with this, the protein synthesis inhibitor cycloheximide (CHX) abrogated HDACs inhibition induced PD-L1 expression (Fig. 2B). Moreover, overexpression of HDACs significantly reduced IFN-γ-induced PD-L1 both in mRNA (Fig. 2C) and protein levels (Fig. 2D). And the deacetylase activity was required for this process, as the deacetylase-dead mutant could not attenuate IFN-γ-induced PD-L1 (Supp. Fig. 1C). Together, these results suggested that class I HDACs inhibition-induced PD-L1 elevation was due to the increased transcription of CD274. It’s well known that HDACs suppress genetic transcription by removing the acetyl groups from the histone of chromatin. We next determined the histone acetylation level of CD274 promoter after cells were treated with SAHA. As indicated in Figure 2E, SAHA treatment significantly increased Histone H3 acetylation of CD274 promoter. And, consistent with SAHA-induced CD274 transcription, RNA polymerase II (Pol II) was also enriched at the CD274 promoter after SAHA treatment (Fig. 2E). Recently, BRD4 was reported as an acetylation reader for PD-L1 transcription [17]. As shown in Figure 2F, the inhibition of BRD4 by its inhibitor JQ1 or knockdown by siRNA significantly abrogated SAHA-induced PD-L1 expression as well. And, BRD4 was enriched at the CD274 promoter upon SAHA treatment (Fig. 2G). This further confirmed that the increase of PD-L1 by HDAC inhibition was due to the gene transcription, not the protein stability.
Figure 2. Upregulated PD-L1 by HDAC inhibition was due to the increased CD274 transcription.
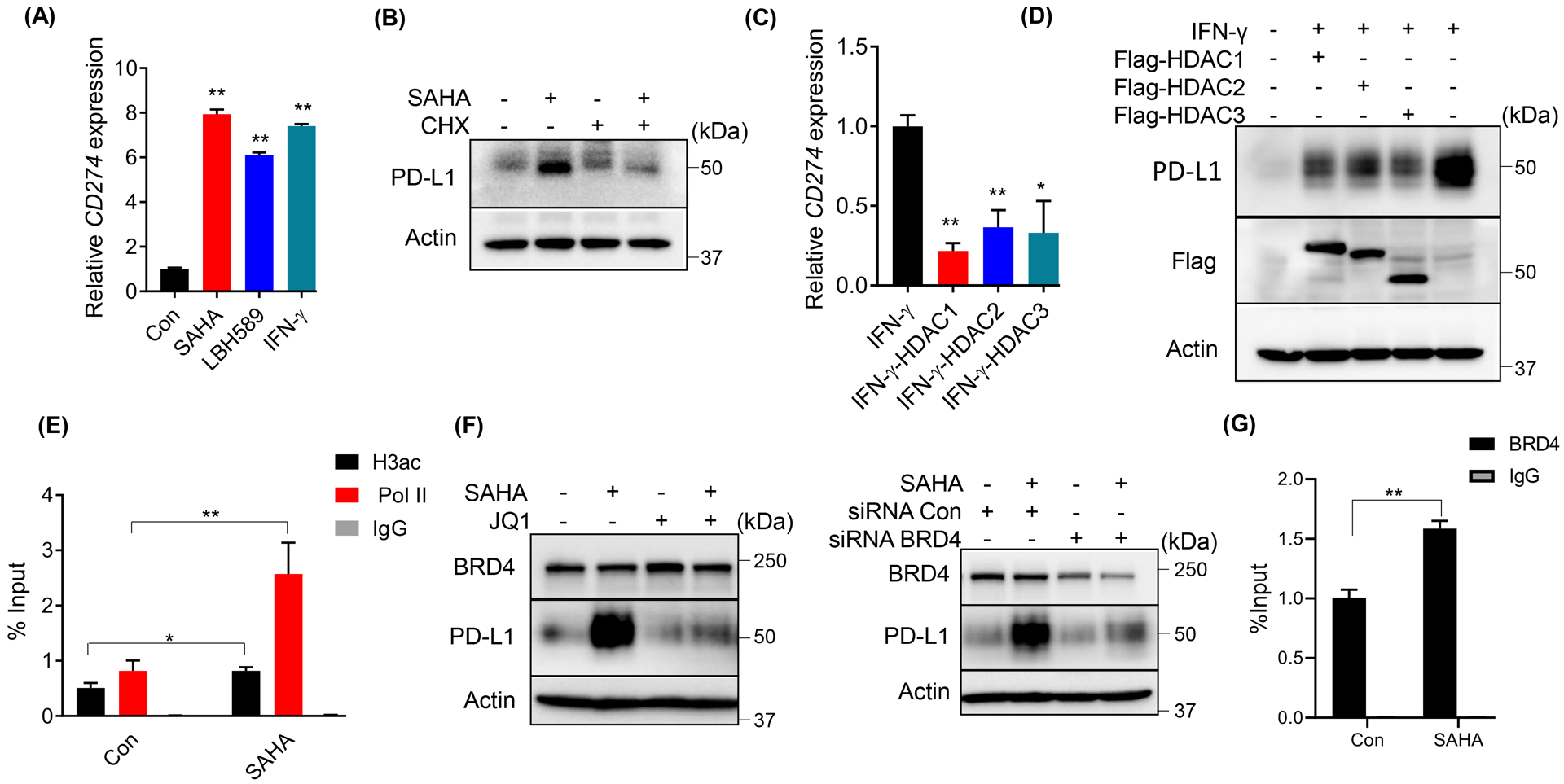
(A) Expression of CD274 was analyzed by RT-qPCR after DU145 cells were treated with 1 μM SAHA or 50 nM LBH589 or 10 ng/ml IFN-γ for 24 h. (B) IB analysis of WCL derived from DU145 cells as indicated treated (100 μg/ml CHX or 1 μM SAHA) for 12 h. (C, D) DU145 cells were transfected with the indicated constructs, followed with 10 ng/ml IFN-γ, and harvested to measure mRNA levels (C) or protein levels of PD-L1 (D). (E, G) ChIP-qPCR analysis of H3ac, BRD4 and Pol II binding at CD274 promotor in DU145 cells treated with 1 μM SAHA. (F) IB analysis of WCL derived from DU145 cells treated with 1 μM SAHA and 5 μM JQ1 (left panel) or transfected with siRNA to deplete BRD4, then treated with 1 μM SAHA (right panel). The statistical data were shown as mean values ± SD (n ≥ 3), *p < 0.05 **p<0.01.
p300/CBP was involved in HDACs inhibition-induced PD-L1 expression in PCa cells
Next, we asked which histone acetyltransferase (HAT) cooperated with class I HDACs to modulate the chromatin structure of CD274 promoter and regulates its transcription. As p300 and its highly homologous HAT CBP were critical for PCa progression [12, 18], we first tested whether they were the HATs responsible for the PD-L1 regulation. As shown in Figure 3A, inhibition of p300/CBP by a potent inhibitor A485 (with H3K27ac and H3K18ac as the markers for p300/CBP activity), or knockdown by shRNA/siRNA significantly abrogated SAHA-induced PD-L1 expression, while knockdown of other acetyltransferases Tip60 or PCAF could not attenuate SAHA-induced PD-L1 expression (Supp. Fig. 1D). In line with this, overexpression of p300 or CBP dramatically increased the expression of PD-L1 both in mRNA (Fig. 3B) and protein levels (Fig. 3C), while the acetylase-deficient mutant of p300 could not induce PD-L1 expression (Supp. Fig. 1E). Moreover, inhibition of p300 by the inhibitor or knockdown with the shRNA also attenuated IFN-γ-induced PD-L1 expression (Fig. 3D). As mentioned above, most of the translated PD-L1 in PCa cells was secreted extracellularly by exosomes, which inhibits T cells function and induces the resistance to PD-L1 blockade treatment. We next asked whether the reduction of PD-L1 induced by the p300/CBP inhibition also affected the exosomal PD-L1 level. As shown in Figure 3E, the inhibition of p300/CBP by A485 remarkably reduced both the intrinsic and IFN-γ induced PD-L1 in the exosomes (CD9 is as a marker for exosomes). These findings indicated that p300/CBP was involved in the regulation of PD-L1 expression in PCa cells, and the inhibition of p300/CBP could block both the cellular and exosomal PD-L1. Next, we evaluated whether p300 could directly bind to the promoter of CD274 as a HAT. As the ChIP assay of p300 indicated, an obvious enrichment of p300 was observed on the promoter of CD274, especially at the location from 5450650 to 5450800 (Fig. 3F and Supp. Fig. 1F). Furthermore, SAHA or IFN-γ treatment significantly increased the enrichment of p300 and its target H3K27ac at the CD274 promoter (Fig. 3G). Of note, consistent with our findings, the ENCODE database [19] also showed a chromatin IP peak for p300 (EP300) at the upstream of CD274 promoter around 5450500 (Fig. 3H), which overlaps with the binding site of transcription factor, IRF-1 (IRF1). Moreover, in accordance with class I HDACs downregulating PD-L1 expression, HDAC1 and HDAC2 also showed enrichment at this location (Fig. 3H). These findings suggested that p300, as a histone acetyltransferase, directly binds to the promoter of CD274 and regulates its expression.
Figure 3. p300 was involved in PD-L1 expression in PCa cells.
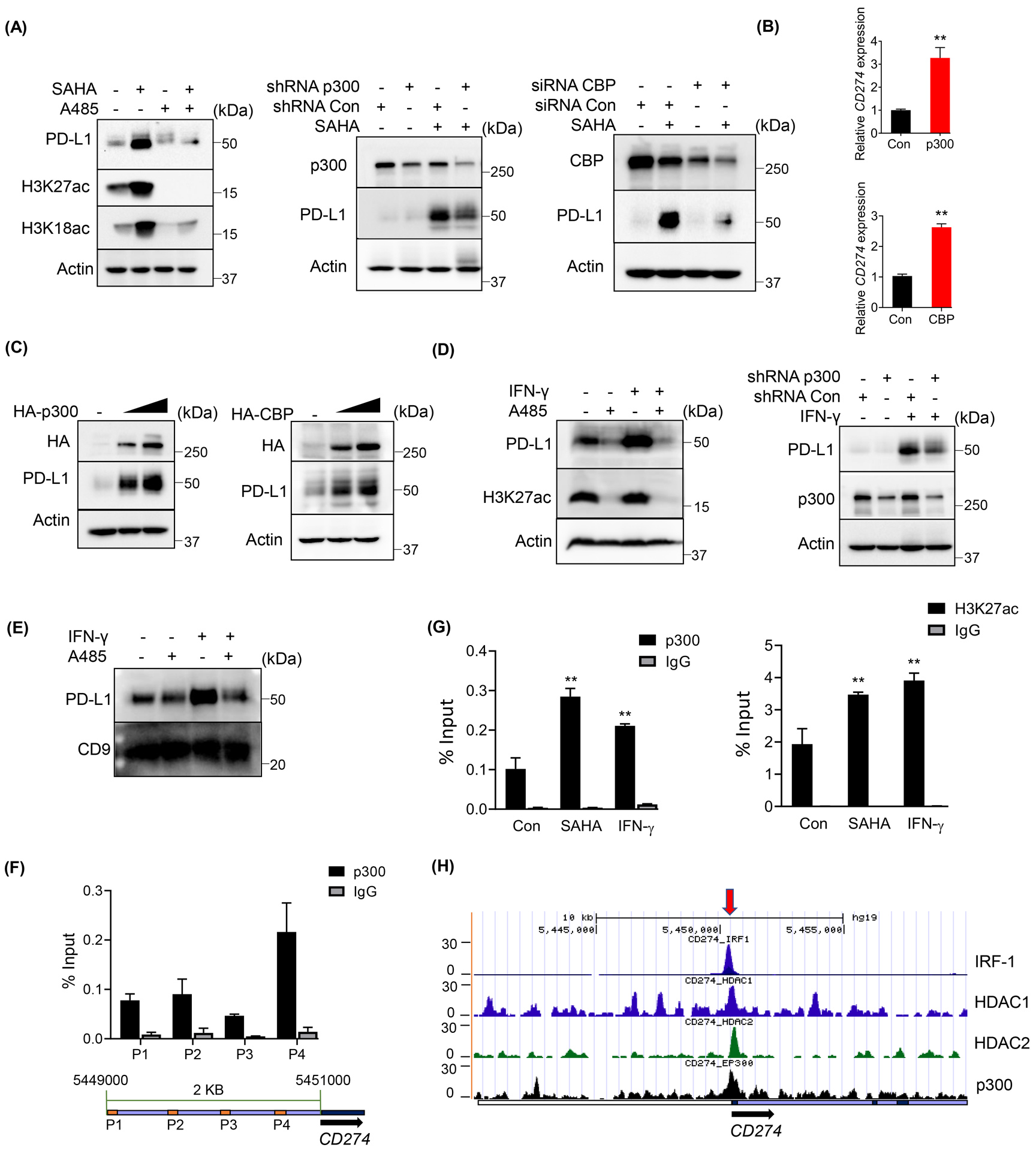
(A, C and D) IB analysis of WCL derived from DU145 cells treated with the indicated inhibitors (1 μM SAHA, 5 μM A485) or 10 ng/ml IFN-γ, or transfected with the indicated constructs or shRNA/siRNA. (B) Expression of CD274 was analyzed by RT-qPCR after DU145 cells transfected with the indicated constructs. (E) IB analysis of exosomal lysate derived from DU145 cells with the indicated treatment (5 μM A485 or 10 ng/ml IFN-γ. (F) ChIP-qPCR analysis of p300 binding at CD274 promoter in DU145 cells with the indicated primers shown on CD274 promoter (the sequences were shown in Supplemental Table 2). (G) ChIP-qPCR analysis of p300 or H3K27ac binding at CD274 promoter with primer P4 in DU145 cells treated with 1 μM SAHA or 10 ng/ml IFN-γ. (H) Depiction of p300, HDAC1, HDAC2 and IRF-1 binding peaks at CD274 promoter from ENCODE database. All the statistical data were shown as mean values ± SD (n ≥ 3), *p < 0.05 **p<0.01.
IRF-1 was involved in p300-regulated PD-L1 in PCa cells
Usually, HATs carry out their function of activating transcription by binding to transcription factors and the transcription machinery. Recently, several studies have identified IRF-1 as an important transcription factor for PD-L1 regulation [20, 21], and the binding peak analysis with ENCODE database indicated a strong overlap between p300 and IRF-1 on the promoter of CD274 (Fig. 3H). Moreover, IRF-1 showed a strong positive correlation with PD-L1 expression (Fig. 4A) in prostate cancer cells. To validate whether IRF-1 is the transcription factor bound by p300 in the regulation of PD-L1, we depleted IRF-1 with shRNA followed by overexpression of p300 and CBP in DU145 cells. As shown in Figure 4B, expression of p300 or CBP could not enhance PD-L1 expression when IRF-1 was depleted. Consistent with this result, IRF-1 knockdown also attenuated SAHA-induced PD-L1 expression (Fig. 4C), while knockdown of other interferon regulatory transcription factors, IRF-3 or IRF-7, could not attenuate SAHA-induced PD-L1 expression (Supp. Fig. 1G). Next, we determined whether IRF-1 could directly bind with p300 or CBP with an IP assay. As shown in Figure 4D, both overexpressed and endogenous p300/CBP could interact with IRF-1 directly. Moreover, knockdown of IRF-1 dramatically decreased the enrichments of p300 and CBP on CD274 promoter and abrogated SAHA-induced enrichment of p300 and CBP on the promoter of CD274 (Fig. 4E). Also, knockdown of IRF-1 significantly decreased the H3K27ac level on CD274 promoter and the binding of BRD4 to CD274 promoter (Fig. 4F). In addition, SAHA treatment could not enrich H3K27ac and BRD4 on CD274 promotor when IRF-1 was depleted (Fig. 4F). These findings indicate that IRF-1 is required for p300/CBP binding to CD274 promoter and their regulation on PD-L1 expression.
Figure 4. IRF-1 was involved in p300/CBP-induced PD-L1 expression in PCa cells.
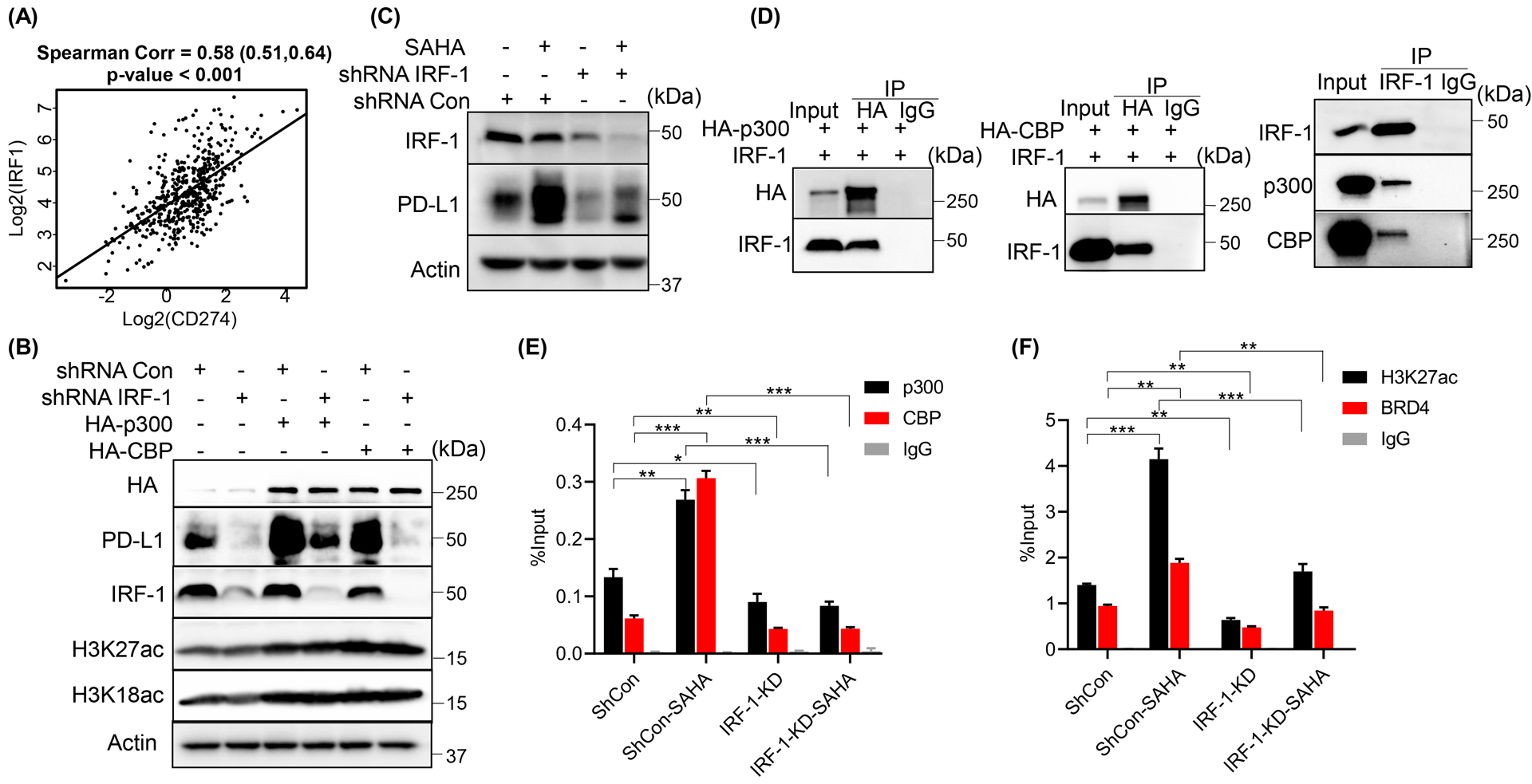
(A)Scatter plot showed the correlation between (log2-transformed) expression of CD274 and IRF1 in prostate adenocarcinoma. IB analysis of WCL derived from DU145 cells infected with the indicated shRNA lentivirus followed by transfected with the indicated constructs (B) or 1 μM SAHA treatment (C). (D) IB of immunoprecipitate or WCL derived from DU145 cells transfected with the indicated constructs or not. (E, F) ChIP-qPCR analysis of p300, CBP, BRD4 and H3K27ac binding at CD274 promoter with primer P4 in DU145 cells infected with the indicated shRNA lentivirus followed by the treatment with 1 μM SAHA. All the statistical data were shown as mean values ± SD (n ≥ 3), *p < 0.05 **p<0.01 ***p<0.001.
PD-L1 expression was correlated with p300/CBP in patients with PCa
Next, to validate the associations between PD-L1 and p300/CBP in human patients’ samples, we interrogated the 495 samples from The Cancer Genome Atlas (TCGA, https://www.cancer.gov/tcga) provisional prostate adenocarcinoma data set by using the non-parametric Spearman Correlation analysis. As shown in Figure 5A–C, EP300 (encoding p300), CREBBP (encoding CBP) and their associated factor KAT2B (encoding PCAF) showed strong positive correlations with CD274 expression (p values < 0.001), which were comparable with IRF1 (Fig. 4A). Interestingly, CBP showed stronger correlation with PD-L1 expression than p300. And, this is consistent with the previous findings in Figures 3A and 4E, that the transcription of PD-L1 was more dependent on CBP than on p300 under SAHA treatment. In addition, consistent with our finding, BRD4 was positively correlated with CD274 expression (Fig. 5D, p value <0.001), while HDAC1 was negatively and significantly correlated with CD274 expression (Fig. 5E) (p value < 0.001). There was no significant correlation between HDAC2/3 with CD274 (Fig. 5F and 5G) (p values > 0.05). Moreover, HDAC1 protein level was also negatively correlated with PD-L1 level in different cell lines (Fig. 6A), while there was no correlation showed between HDAC2/3 and PD-L1. These findings suggest that HDAC1 might be the major histone deacetylase for PD-L1 regulation in human.
Figure 5. PD-L1 expression was correlated with p300/CBP in PCa patients.
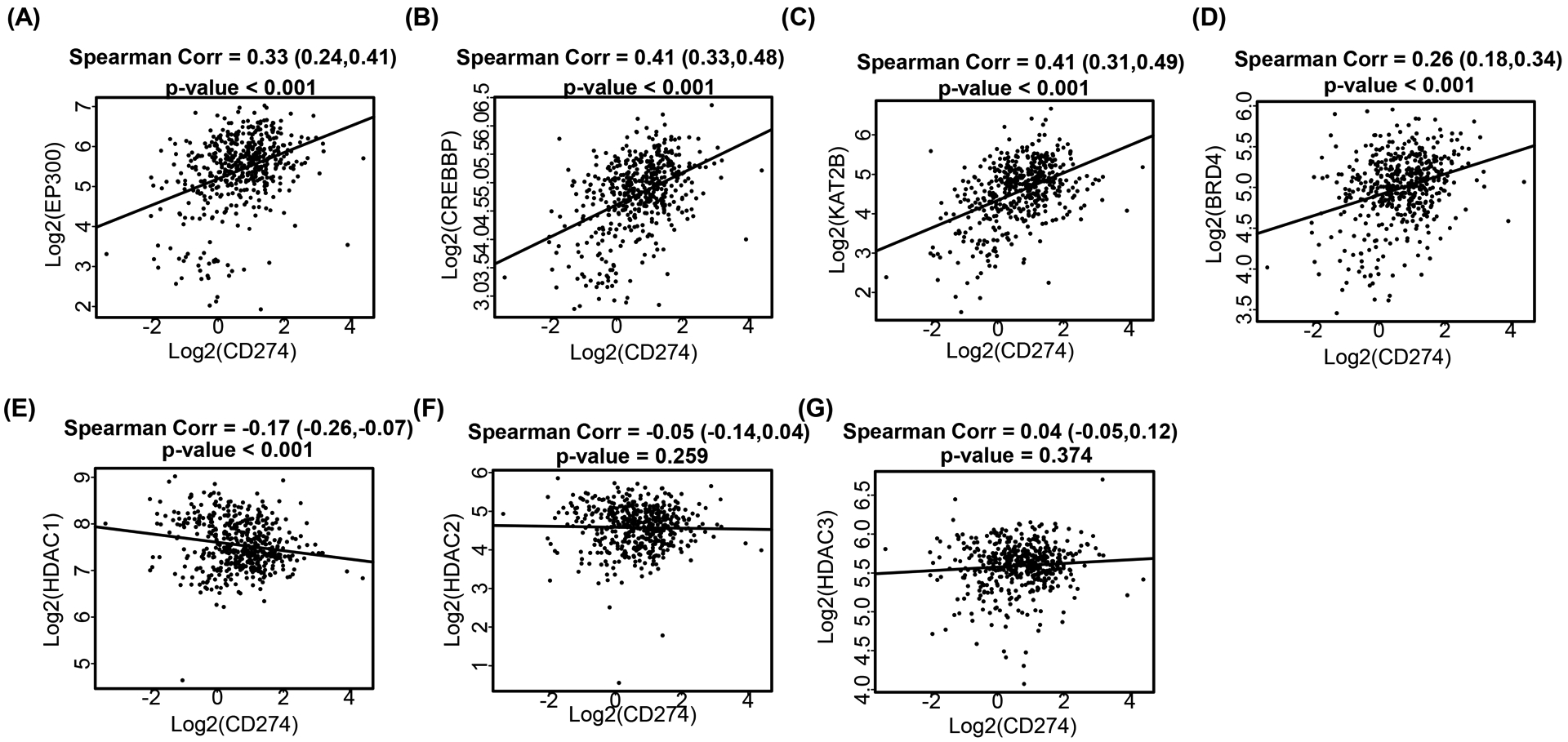
Scatter plot showed the correlations between (log2-transformed) expressions of CD274 and EP300 (A), CREBBP (B), KAT2B (C), BRD4 (D), HDAC1 (E), HDAC2 (F) and HDAC3 (G) in prostate adenocarcinoma. A linear regression line was included for better visualization.
Figure 6. PD-L1 was correlated with tumor progression and tumor purity in prostate adenocarcinoma.
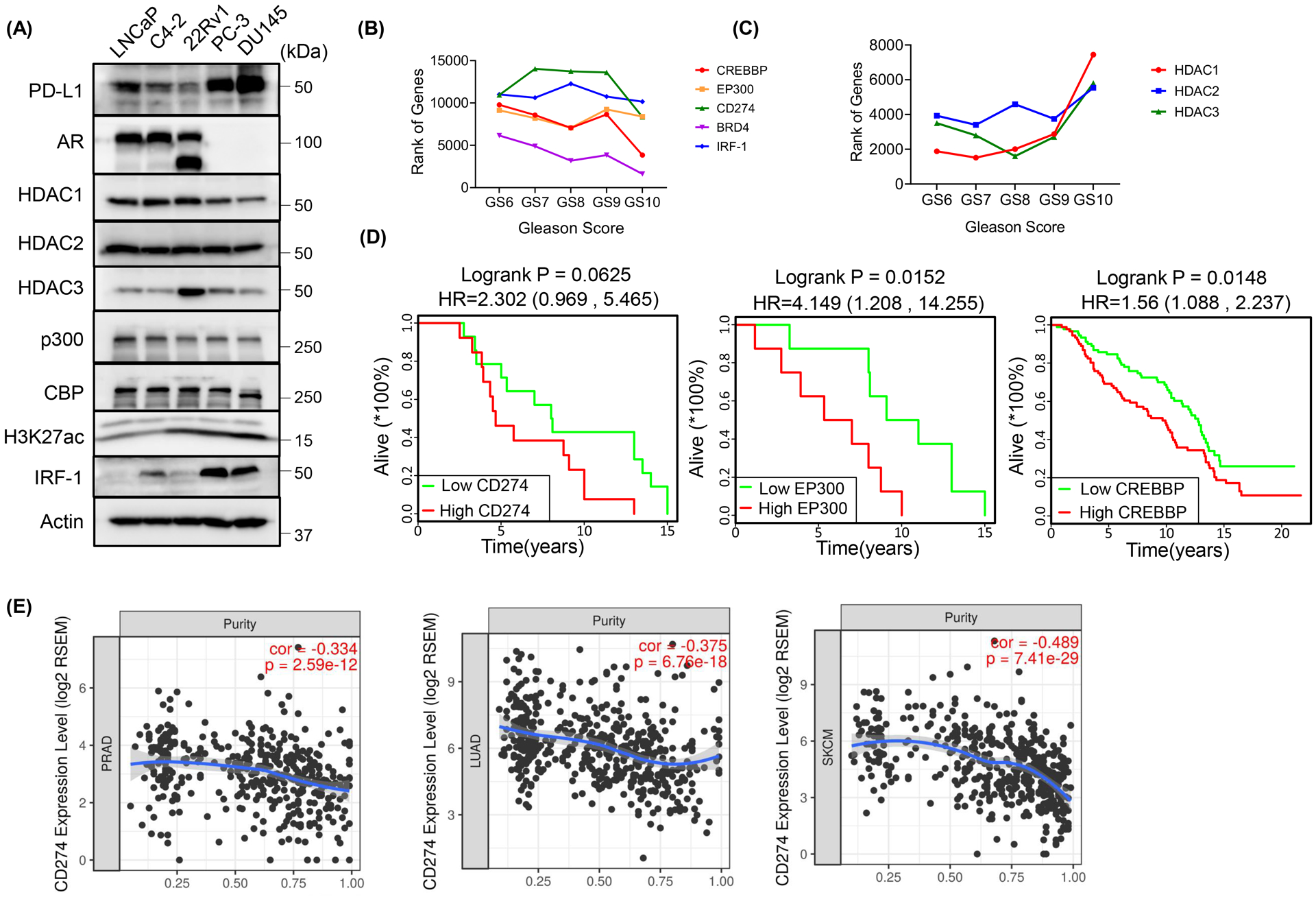
(A) IB analysis of WCL derived from the indicated prostate cancer cell lines. (B, C) The line charts showed correlations of the indicated genes’ rank and the Gleason Score. (D) Survival plots showed the correlations between prostate adenocarcinoma patients’ overall survival and genes of interest. (E) The correlations of tumor purity with CD274 expressions in prostate adenocarcinoma (PRAD), lung adenocarcinoma (LUAD) or skin cutaneous melanoma (SKCM) were estimated by TIMER algorithm.
PD-L1 was correlated with tumor progression and tumor purity in prostate adenocarcinoma
A recent study showed that a higher level of PD-L1 mRNA was found in PCa cells but most of the translated PD-L1 protein was secreted extracellularly by exosomes instead of remaining on the cell surface, consequently resulting in resistance to PD-L1 blockade [9]. Consistent with this finding, we found that most patients with PCa showed negative (163/165) or weak (<10%) (1/165) PD-L1 staining on the cell surface (Supp. Fig. 2A–C). While intrinsic cellular or exosomal PD-L1 was observed in advanced PCa cell lines, DU145 and PC-3 (Fig. 3E and 6A), which are AR-null cells and do not respond to the current CRPC treatment with AR antagonists. Next, to further explore the roles of PD-L1 in the progression of PCa, we performed a gene functional rank analysis at various Gleason Scores with the RNA sequence database from TCGA. We found that the ranks of CD274 and its positive regulators EP300, CREBBP, IRF1 and BRD4 were negatively correlated with the Gleason Score (Fig. 6B), suggesting that these genes’ activity or function increased during cancer progression. In contrast, the negative regulators for PD-L1 expression, HDAC1, HDAC2 and HDAC3 seemed to pale into insignificance during the cancer progression (Fig. 6C). Furthermore, expression levels of CD274, EP300 and CREBBP were negatively correlated with overall survival in PCa patients (Fig. 6D). Next, we evaluated whether PD-1/PD-L1 pathway was also involved in the tumor-immune interactions in PCa patients using the Tumor Immune Estimation Resource (TIMER), an algorithm developed to analyze the abundance of tumor-infiltrating immune cells in a comprehensive and flexible manner [22]. As shown in Figure 6E and Supplemental Figures 2D and 2E, the expressions of CD274 (encoding PD-L1) and PDCD1 (encoding PD-1) were inversely correlated with tumor purity in prostate adenocarcinoma (PRAD), which were comparable with lung adenocarcinoma (LUAD) and skin cutaneous melanoma (SKCM), two cancer types well responding to PD-1/PD-L1 blockade treatment. And, consistent with this, expression levels of CD274 and PDCD1 were also associated with increased tumor infiltrating immune cells in PRAD (Supp. Fig. 2D–2E). Moreover, in line with EP300 and CREBBP regulating PD-L1 expression, they also showed positive correlations with tumor infiltrating immune cells in PRAD (Supp. Fig. 2F and 2G). These results suggest that PD-L1 also plays a critical role in PCa progression, and targeting PD-L1 could be a promising treatment for PCa therapy if the resistance to PD-L1 blockade was overcome.
p300/CBP inhibition enhanced the efficacy of PD-L1 blockade in PCa
Given the crucial roles of p300/CBP in the regulation of CD274 transcription in PCa, we asked whether inhibition of p300/CBP could enhance the efficacy of PD-L1 blockade treatment in PCa by cutting off the secreted exosomal PD-L1. To test this, we used a syngeneic model of PCa, TRAMP-C2 model, which has shown resistance to anti-PD-L1 blockade by releasing exosomal PD-L1 [9]. As TRAMP-C2 cells need a long time to form tumors, we used TRAMP-C2 cells stably expressing Ras (TRAMP-C2-Ras) instead. Consistent with a previous study that Ras could stabilize PD-L1 mRNA [23], TRAMP-C2-Ras cells showed a higher level of cellular PD-L1 than that of TRAMP-C2 cells (Supp. Fig. 3A), which made it more appropriate for studying the response to anti-PD-L1 treatment. First, we confirmed that in TRAMP-C2-Ras cells, p300/CBP inhibitor A485 could also block the cellular (Fig. 7A), surface (Fig. 7B) and especially exosomal (Fig. 7C) PD-L1 expression, in both the intrinsic and IFN-γ/HDACs inhibitor-induced levels (Fig. 7A–C and Supp. Fig. 3B–D). Then we transplanted TRAMP-C2-Ras cells in the flank of C57BL/6J mice followed by the treatment with A485; anti-PD-L1 Ab; A485 with anti-PD-L1 Ab; LBH589 and LBH589 with A485 (Fig. 7D). As shown in Figure 7E–G, anti-PD-L1 Ab treatment had little effect on the tumor growth, but anti-PD-L1 Ab combined with A485 significantly limited tumor progression. Consistent with this, the combination group displayed lower levels of Ki-67 (Fig. 7H and 7I) and PCNA (Fig. 7J), two markers for cell proliferation. Moreover, without immune system, there was no combination effect of anti-PD-L1 Ab and A485 in vitro (Supp. Fig. 3E). In addition, A485 also enhanced the effects of HDACs inhibitor, LBH589, on the reduction of tumor growth (Fig. 7E–7G). In line with the results in vitro, A485 significantly reduced intrinsic or induced PD-L1 level in vivo (Fig. 7K), and increased the infiltrating CD8+ T cells especially when combined with anti-PD-L1 Ab (Fig. 7L). Finally, mice in all groups showed no significant weight loss during the treatment (Supp. Fig. 3F), suggesting no obvious toxicity with all the treatments.
Figure 7. p300/CBP inhibition enhanced the efficacy of anti-PD-L1 treatment in PCa.
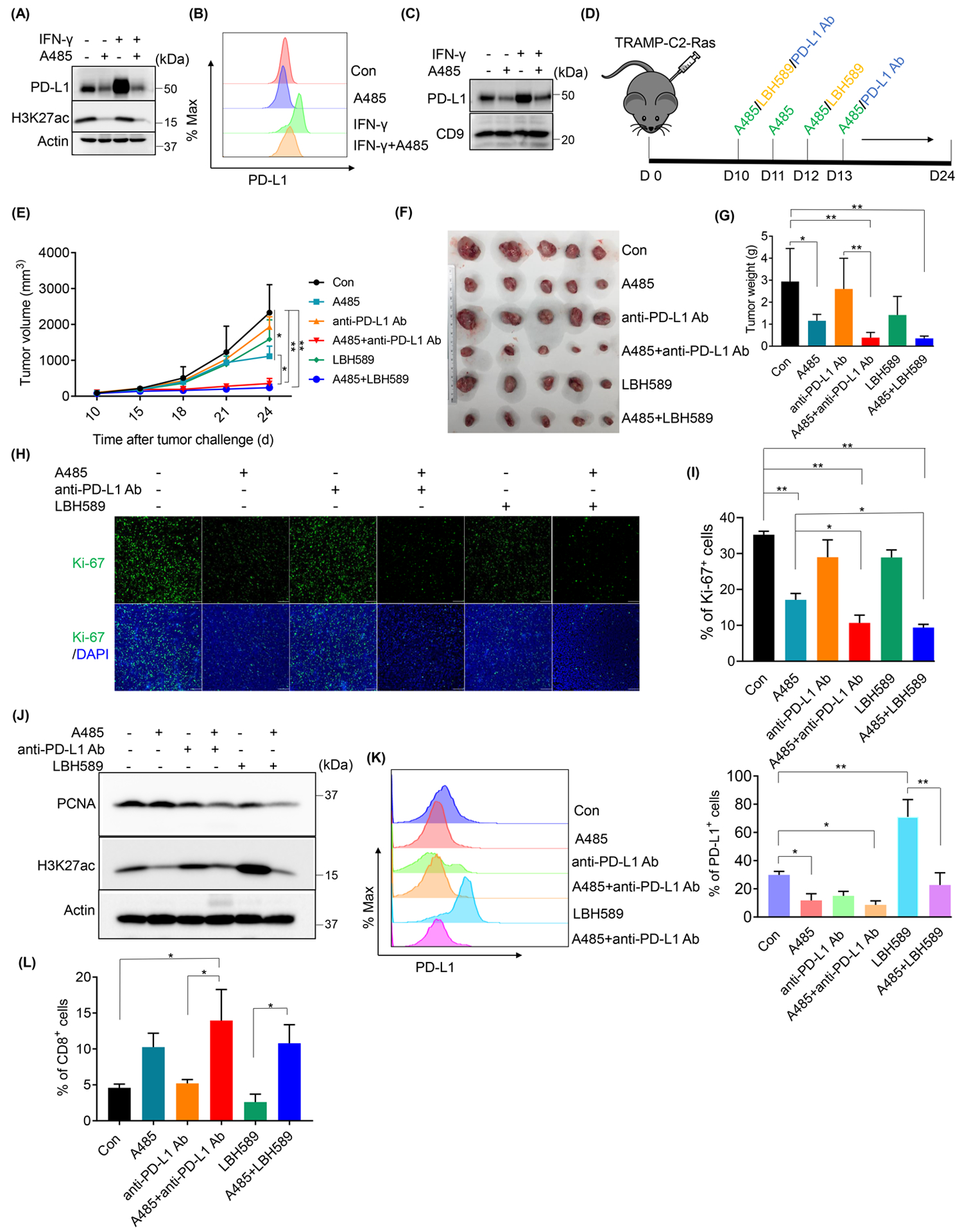
Expressions of cellular (A), surface (B) or exosomal PD-L1 were determined by IB (A and C) or FACS (B) after TRAMP-C2-Ras cells were treated with 10 μM A485 or 10 ng/ml IFN-γ for 24 h. (D-L) TRAMP-C2-Ras cells were transplanted in the flank of C57BL/6J mice and followed by the indicated treatment (D). Tumor growth was shown (E). After tumors were harvested and photographed at day 24 (F), tumors weight were determined (G). Ki-67 levels of tumors were determined by IF (H, scale bar= 100 μm) and quantified (I). The protein levels of PCNA and H3K27ac within the tumors were analyzed by IB (J). The levels of PD-L1 expression (K) and tumor infiltrating CD8+ T cells (L) were determined by FACS. All statistic data were shown as mean values ± SD (n = 5), *p < 0.05 **p<0.01.
DISCUSSION
p300, a coactivator of AR to regulate its transcriptional program and AR signaling axis, has been reported to be involved in recurrent PCa and therapy resistance during PCa treatment. More recently, A485 was identified as a new, potent, selective and drug-like catalytic inhibitor for p300/CBP; and was shown to selectively inhibit cell proliferation of several hematological malignancies and AR-positive PCa [13]. Here, we reported that combined treatment of A485 with anti-PD-L1 antibody significantly inhibited androgen-independent metastatic tumor growth in a syngeneic PCa model. Mechanistically, p300 could be recruited to CD274 promoter by the transcription factor IRF-1, consequently inducing acetylation of histone H3 at the promoter and elevating transcription of CD274. A485, a p300/CBP inhibitor, abrogated this process and enhanced the efficacy of PD-L1 blockade treatment by cutting off PD-L1 expression and reducing the secreted exosomal PD-L1 (Fig. 8). As p300/CBP is critical for progression of many types of cancers, other caners likely also benefit from this new combinatory treatment.
Figure 8. A working model of how p300/CBP inhibition enhanced the efficacy of PD-L1 blockade.
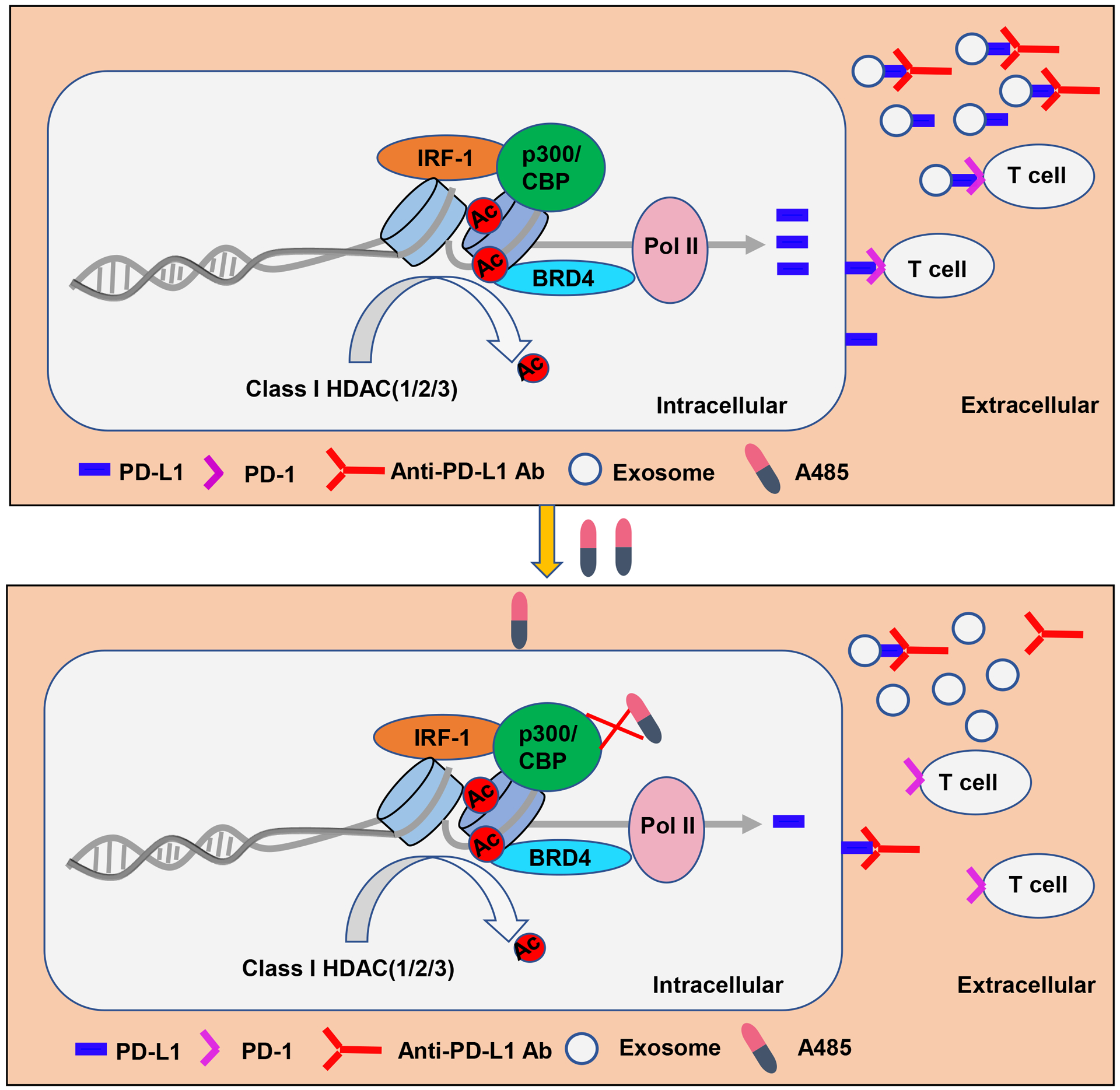
In PCa cells, most of the translated PD-L1 was secreted extracellularly by exosomes, which suppresses T cell function and antagonizes anti-PD-1 response. p300/CBP, as the histone acetyltransferase, directly binds to the promoter of CD274 and thus regulating its expression. A485, a p300/CBP inhibitor, abrogated this process and cut off the secretion of PD-L1 by blocking the transcription of CD274, which combined with the anti-PD-L1 antibody to reactivate T cell function for tumor attack.
It is reported that highly expressed p300 and its highly homologous histone acetyltransferase CBP were implicated in progression of PCa, and that deletion of p300 in mice limited PCa progression and extended mice survival [18]. The oncogenic roles of p300 in the progression of PCa was usually related to the regulation of AR, the key driver of PCa. p300 could directly acetylate AR or bind with AR to enhance AR transcriptional activity, consequently inducing the expression of oncogenes and promoting tumor growth [13, 18, 24]. In addition to enhancing AR transcriptional activity, p300 could also regulate AR protein level by preventing its degradation [18]. These findings highlight p300 to be a valid target for PCa treatment. Indeed, our and other studies have shown that targeting p300/CBP inhibited both androgen-sensitive and castration-resistant PCa cell growth (Supp. Fig. 3G) [12, 13]. Here, we report a novel mechanism underlying how p300 is involved in PCa progression by upregulating PD-L1 expression, thus creating an immune cell-free microenvironment for tumor progression. Our finding suggests that p300 is not only a modifier but also a co-driver for PCa progression, confirming that p300 could be a promising target for PCa treatment.
Early studies hypothesized that the failure of PD-1/PD-L1 pathway blockade in PCa treatment might be due, in part, to the lower expression of PD-L1 [5, 25]. However, to date, a number of reports have demonstrated that PD-L1 expression was also highly prevalent in both primary PCa and CRPC, and that PD-L1 level was associated with the Gleason Score and aggressive tumor behavior [26–28]. Maybe due to the use of different anti-PD-L1 antibody, even we tried two different antibody clones (E1L3N and 28–8), we found membranous PD-L1 expression in primary prostate cancer patients was weak. Whereas, our finding is consistent with recent studies reporting the mechanisms of resistance to PD-L1 blockade in PCa that most PD-L1 was secreted to extracellular instead on cell surface [9]. Of recent note, increasing studies showed PD-L1 level was not sufficient to predict clinical response to PD-1/PD-L1 blockade therapy, and patients with low or no PD-L1 expression (<1%) could also benefit from PD-1/PD-L1 blockade therapy [4, 29]. On the other hand, with the TCGA RNA sequence database, we demonstrated that PD-L1 and PD-1 were correlated with tumor purity and tumor infiltrating immune cells in PCa, comparable with what has been described in lung cancer and melanoma, the two PD-1/PD-L1 blockade well-responded cancer types. In addition, we also demonstrated that PD-L1, correlated with p300, was involved in the progression of PCa. These findings suggest that the PD-L1/PD-1 pathway could also be a promising target for PCa treatment. More recently, studies reported that tumor cells could release PD-L1 through exosomes or in variants manner, both directly inhibit lymphocyte function and interrupt the reactivation of T cells by PD-L1 blockade and could not be blocked by the current antibodies therapy [7–9, 30]. These studies suggest that blockade of the surface PD-L1 only might not be able to reactivate the inhibited immune functions in some cancer types, and that PD-L1 has to be cut off at the source. In this study, we identified p300/CBP, a general driver for multiple types of cancer, to be a promising target to address this issue, eventually enhancing the efficacy of PD-1/PD-L1 blockade therapy.
Recently, PD-L1-mediated evasion of tumor immunity was described as adaptive resistance [6]. INF-γ, the major driver of PD-L1 expression in cancer cells, was released by tumor infiltrating lymphocytes (TIL) to enhance TIL effector functions by stimulating antigen processing and increasing TIL differentiation, but was captured and used by tumor cells to paralyze T cells [6, 31]. Therefore, a way to block this process would return the function of IFN-γ in increasing TIL effector and avoid T cell exhaustion induced by PD-L1. Here we report that p300/CBP plays a critical role in IFN-γ induced PD-L1 expression and could be a good target to overcome the adaptive resistance induced by the PD-1/PD-L1 pathway.
Aberrant expression and activity of HDACs have been observed in various cancer types, suggesting that HDACs could be a good target for cancer therapy [32]. Indeed, HDAC inhibitors vorinostat (SAHA), belinostat and romidepsin have been approved for T cell lymphoma treatment and panobinostat (LBH589) for multiple myeloma treatment [33]. In parallel, multiple HDAC inhibitors are in clinical trials for treatment of solid malignancies, including PCa. Specifically, it was reported that HDAC inhibition could downregulate AR protein level, indicating that HDAC inhibitors may likely be effective for PCa treatment [34]. Nevertheless, none of these inhibitors were able to reach Phase III trial due to the poor response in clinic. In this study, we found that HDAC inhibition significantly induced PD-L1 expression by increasing the acetylation of CD274 promoter, consequently resulting in an immune evasive microenvironment for tumor progression. And, it might be due to the exosomal secretion of PD-L1, p300 inhibitor but not anti-PD-L1 Ab (Supp. Fig. 3H–J) significantly enhances the efficacy of HDAC inhibitor on limiting tumor progression by blocking HDAC inhibition-induced PD-L1. This may explain the failure of HDAC inhibitors in PCa clinical trial and offer a possible approach to enhance the efficacy of HDAC inhibitors in PCa treatment.
In summary, this study provides a strategy to overcome the resistance of PCa to PD-L1 blockade through the molecular targeting of p300/CBP to cut off the PD-L1 expression at the source. Considering the roles of p300/CBP in tumor progression and the promising effects of PD-L1 blockade in clinical cancer therapy, this combined treatment might also benefit patients with other types of cancer.
MATERIALS AND METHODS
Cell culture
Human PCa cell lines DU145, PC3, 22Rv1 and LNCaP were purchased from American Type Culture Collection, culturing respectively in Dulbecco’s modified Eagle’s medium (DMEM, DU145), Ham’s F-12K medium (PC-3) or RPMI-1640 medium (the remainder). C4–2 cells were obtained from MD Anderson Cancer Center via an MTA and cultured in RPMI-1640 medium. Tramp-C2-Ras, a murine origin PCa cell line and courtesy of Dr. Marxa L. Figueiredo (Purdue University, IN, USA), was maintained in DMEM/Nutrient F-12 Ham medium. HEK293T cells was a gift from Dr. Andrea Kasinski (Purdue University, IN, USA) and cultured in DMEM. All medium contained 10% of fetal bovine serum (Atlanta Biologicals, GA, USA), and cells were cultured in a humidified atmosphere at 37 °C, with 5% CO2.
Mouse tumor implantation and treatment
All animal experiments were approved by the Institutional Animal Care and Use Committee at University of Kentucky (KY, USA). C57BL/6J mice (male, 4–6 weeks) were obtained from Jackson Lab (MI, USA). After acclimating for one week, 2*105 TRAMP-C2-Ras cells were transplanted in the flank of each mouse. Tumor growth was monitored every three days by measuring the length and width of tumors. Tumor volume was evaluated using the formula: (length*width2)/2. Once the tumor volume reached 100 mm3, the mice were treated with the following strategies (as indicated in Fig. 7C and Fig. S4A): 200 μg anti-PD-L1 Ab per mouse (every three days) or the same amount of IgG as the control, 50 mg/kg A485 or the solvent as the control (every day), 15 mg/kg LBH589 or the solvent as the control (every other day). After two weeks treatment, the mice were sacrificed to harvest the tumors for experiments.
IB and IP analyses
Cell lysates were prepared as previously described [35]. Briefly, for cultured cells, cells were harvested and lysed with the lysis buffer (50 mM Tris, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.2% SDS, protease inhibitors, and phosphatase inhibitors). For tumors tissue, the lysates were prepared by homogenization using the above lysis buffer followed by sonication with a probe sonicator. After quantification with BCA assay, the equal amounts of protein were loaded on the SDS-PAGE gel and then transferred to PVDF membranes, followed by IB with the indicated antibodies. For IP analysis, 1 mg proteins collected with the lysis buffer (50 mM Tris, 150 mM NaCl, 0.5% sodium deoxycholate,1% NP-40, protease inhibitors) were incubated with the indicated antibodies overnight at 4°C followed by the protein A magnetic beads for 3 h at room temperature (RT) or antibody-conjugated beads overnight at 4 °C. The immunocomplexes were washed for three times with the lysis buffer and analyzed by IB.
Flow cytometry
For cultured cells, the cells were detached with trypsin and washed twice with ice-cold PBS. For tumor tissues, the tumors were homogenized with PBS and washed twice with ice-cold PBS. The prepared cells were then subjected to staining in Zombie Violet™ dye and PE anti-human or mouse PD-L1 antibodies for 30 min in ice-cold PBS. After staining, cells were washed and fixed with 2.5% formaldehyde in PBS. Expression of PD-L1 was analyzed with the BD LSRFortessa (BD Bioscience) or CytoFLEX (BECKMAN) cytometer. The results were analyzed by FlowJo software.
Statistical analysis
Statistical analyses were performed with Student t test for single comparison or One-way ANOVA for multiple comparisons. All data were shown as mean values ± SD (n > 3), *p < 0.05 **p<0.01.
All other methods are in Supplemental Materials.
Supplementary Material
Acknowledgments:
This work was supported by NIH grants R01 CA157429 (X. Liu), R01 CA192894 (X. Liu), R01 CA196835 (X. Liu), and R01 CA196634 (X. Liu). The work was also supported by Biospecimen Procurement and Translational Pathology, Biostatistics and Bioinformatics, Flow Cytometry and Immune Monitoring Shared Resources of the University of Kentucky Markey Cancer Center (P30CA177558). We thank Heather Russell-Simmons at Research Communications Office of Markey Cancer Center for proof-reading of the manuscript.
Footnotes
The authors declare that they have no conflicts of interest with the contents of this article.
References Cited
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin 2019; 69: 7–34. [DOI] [PubMed] [Google Scholar]
- 2.Seruga B, Ocana A, Tannock IF. Drug resistance in metastatic castration-resistant prostate cancer. Nat Rev Clin Oncol 2011; 8: 12–23. [DOI] [PubMed] [Google Scholar]
- 3.Claessens F, Helsen C, Prekovic S, Van den Broeck T, Spans L, Van Poppel H et al. Emerging mechanisms of enzalutamide resistance in prostate cancer. Nat Rev Urol 2014; 11: 712–716. [DOI] [PubMed] [Google Scholar]
- 4.Rittmeyer A, Barlesi F, Waterkamp D, Park K, Ciardiello F, von Pawel J et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. Lancet 2017; 389: 255–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012; 366: 2443–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen L, Han X. Anti-PD-1/PD-L1 therapy of human cancer: past, present, and future. J Clin Invest 2015; 125: 3384–3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen G, Huang AC, Zhang W, Zhang G, Wu M, Xu W et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 2018; 560: 382–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gong B, Kiyotani K, Sakata S, Nagano S, Kumehara S, Baba S et al. Secreted PD-L1 variants mediate resistance to PD-L1 blockade therapy in non-small cell lung cancer. J Exp Med 2019; 216: 982–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Poggio M, Hu T, Pai CC, Chu B, Belair CD, Chang A et al. Suppression of Exosomal PD-L1 Induces Systemic Anti-tumor Immunity and Memory. Cell 2019; 177: 414–427 e413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell 2012; 150: 12–27. [DOI] [PubMed] [Google Scholar]
- 11.Nebbioso A, Tambaro FP, Dell’Aversana C, Altucci L. Cancer epigenetics: Moving forward. PLoS Genet 2018; 14: e1007362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jin L, Garcia J, Chan E, de la Cruz C, Segal E, Merchant M et al. Therapeutic Targeting of the CBP/p300 Bromodomain Blocks the Growth of Castration-Resistant Prostate Cancer. Cancer Res 2017; 77: 5564–5575. [DOI] [PubMed] [Google Scholar]
- 13.Lasko LM, Jakob CG, Edalji RP, Qiu W, Montgomery D, Digiammarino EL et al. Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature 2017; 550: 128–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Asangani IA, Dommeti VL, Wang X, Malik R, Cieslik M, Yang R et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature 2014; 510: 278–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang YA, Yu J. EZH2, an epigenetic driver of prostate cancer. Protein Cell 2013; 4: 331–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Drazic A, Myklebust LM, Ree R, Arnesen T. The world of protein acetylation. Biochim Biophys Acta 2016; 1864: 1372–1401. [DOI] [PubMed] [Google Scholar]
- 17.Zhu H, Bengsch F, Svoronos N, Rutkowski MR, Bitler BG, Allegrezza MJ et al. BET Bromodomain Inhibition Promotes Anti-tumor Immunity by Suppressing PD-L1 Expression. Cell Rep 2016; 16: 2829–2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhong J, Ding L, Bohrer LR, Pan Y, Liu P, Zhang J et al. p300 acetyltransferase regulates androgen receptor degradation and PTEN-deficient prostate tumorigenesis. Cancer Res 2014; 74: 1870–1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haeussler M, Zweig AS, Tyner C, Speir ML, Rosenbloom KR, Raney BJ et al. The UCSC Genome Browser database: 2019 update. Nucleic Acids Res 2019; 47: D853–D858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smithy JW, Moore LM, Pelekanou V, Rehman J, Gaule P, Wong PF et al. Nuclear IRF-1 expression as a mechanism to assess “Capability” to express PD-L1 and response to PD-1 therapy in metastatic melanoma. J Immunother Cancer 2017; 5: 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA et al. Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep 2017; 19: 1189–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li T, Fan J, Wang B, Traugh N, Chen Q, Liu JS et al. TIMER: A Web Server for Comprehensive Analysis of Tumor-Infiltrating Immune Cells. Cancer Res 2017; 77: e108–e110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Coelho MA, de Carne Trecesson S, Rana S, Zecchin D, Moore C, Molina-Arcas M et al. Oncogenic RAS Signaling Promotes Tumor Immunoresistance by Stabilizing PD-L1 mRNA. Immunity 2017; 47: 1083–1099 e1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fu M, Wang C, Reutens AT, Wang J, Angeletti RH, Siconolfi-Baez L et al. p300 and p300/cAMP-response element-binding protein-associated factor acetylate the androgen receptor at sites governing hormone-dependent transactivation. J Biol Chem 2000; 275: 20853–20860. [DOI] [PubMed] [Google Scholar]
- 25.Martin AM, Nirschl TR, Nirschl CJ, Francica BJ, Kochel CM, van Bokhoven A et al. Paucity of PD-L1 expression in prostate cancer: innate and adaptive immune resistance. Prostate Cancer Prostatic Dis 2015; 18: 325–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gevensleben H, Dietrich D, Golletz C, Steiner S, Jung M, Thiesler T et al. The Immune Checkpoint Regulator PD-L1 Is Highly Expressed in Aggressive Primary Prostate Cancer. Clin Cancer Res 2016; 22: 1969–1977. [DOI] [PubMed] [Google Scholar]
- 27.Calagua C, Russo J, Sun Y, Schaefer R, Lis R, Zhang Z et al. Expression of PD-L1 in Hormone-naive and Treated Prostate Cancer Patients Receiving Neoadjuvant Abiraterone Acetate plus Prednisone and Leuprolide. Clin Cancer Res 2017; 23: 6812–6822. [DOI] [PubMed] [Google Scholar]
- 28.Massari F, Ciccarese C, Calio A, Munari E, Cima L, Porcaro AB et al. Magnitude of PD-1, PD-L1 and T Lymphocyte Expression on Tissue from Castration-Resistant Prostate Adenocarcinoma: An Exploratory Analysis. Target Oncol 2016; 11: 345–351. [DOI] [PubMed] [Google Scholar]
- 29.Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N Engl J Med 2015; 373: 1627–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mahoney KM, Shukla SA, Patsoukis N, Chaudhri A, Browne EP, Arazi A et al. A secreted PD-L1 splice variant that covalently dimerizes and mediates immunosuppression. Cancer Immunol Immunother 2019; 68: 421–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol 2004; 75: 163–189. [DOI] [PubMed] [Google Scholar]
- 32.Li Y, Seto E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb Perspect Med 2016; 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eckschlager T, Plch J, Stiborova M, Hrabeta J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int J Mol Sci 2017; 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaushik D, Vashistha V, Isharwal S, Sediqe SA, Lin MF. Histone deacetylase inhibitors in castration-resistant prostate cancer: molecular mechanism of action and recent clinical trials. Ther Adv Urol 2015; 7: 388–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu J, Wang B, Huang P, Wang H, Xu K, Wang X et al. Microcystin-LR promotes cell proliferation in the mice liver by activating Akt and p38/ERK/JNK cascades. Chemosphere 2016; 163: 14–21. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.