

Abstract
Objective:
Chronic kidney disease (CKD) with tubular injury and fibrosis occurs in HIV infection treated with certain protease inhibitor (PI)-based antiretroviral therapies. The pathophysiology is unclear.
Design:
We hypothesized that fibrosis, mediated by platelet-derived transforming growth factor (TGF)-β1, underlies PI-associated CKD. We induced this in mice exposed to the PI ritonavir (RTV), and intervened with low-dose inhaled carbon monoxide (CO), activating erythroid 2-related factor (Nrf2)-associated anti-oxidant pathways.
Methods:
C57BL/6 mice, wild-type and deficient in platelet TGF-β1, were given RTV (10mg/kg) or vehicle daily for 8 weeks. Select groups were exposed to CO (250ppm) for 4 hours after RTV or vehicle injection. Renal pathology, fibrosis, and TGF-β1- and Nrf2-based signaling were examined by histology, immunofluorescence, and flow cytometry. Renal damage and dysfunction were assessed by KIM-1 and cystatin C ELISAs. Clinical correlations were sought among HIV-infected individuals.
Results:
RTV induced glomerular and tubular injury, elevating urinary KIM-1 (p=0.004). It enhanced TGF-β1-related signaling, accompanied by kidney fibrosis, macrophage polarization to an inflammatory phenotype, and renal dysfunction with cystatin C elevation (p=0.008). Mice with platelet TGF-β1 deletion were partially protected from these abnormalities. CO inhibited RTV-induced fibrosis and macrophage polarization in association with upregulation of Nrf2 and heme oxygenase-1 (HO-1). Clinically, HIV infection correlated with elevated cystatin C levels in untreated women (n=17) vs. age-matched controls (n=19; p = 0.014). RTV-treated HIV+ women had further increases in cystatin C (n=20; p=0.05), with parallel elevation of HO-1.
Conclusion:
Platelet TGF-β1 contributes to RTV-induced kidney fibrosis and dysfunction, which may be amenable to anti-oxidant interventions.
Keywords: chronic kidney disease, fibrosis, protease inhibitor, platelets, TGF-β
Introduction
The nature of HIV-related kidney disease has changed markedly following the introduction of combination antiretroviral therapy (cART) [1–3]. Pre-1995, HIV-associated nephropathy (HIVAN), characterized by heavy proteinuria and rapid progression to end-stage renal disease, occurred in up to 10% of HIV infected individuals, predominantly African-Americans with poorly controlled viremia [2,4,5]. It is a collapsing form of focal segmental glomerulosclerosis (FSGS) with tubulointerstitial inflammation and podocyte effacement, linked to interactions among HIV gene products, host proteins, and host genetic factors [4,6,7]. Over the past decade HIVAN has declined in incidence by up to 60%, in parallel with cART-mediated reduction of HIV viral loads to undetectable levels [7,8]. But other forms of kidney disease are now prevalent among the HIV-infected worldwide. Chronic kidney disease (CKD) occurs in 1.1% of cART-treated individuals per year of follow up, with a median annual decline in estimated glomerular filtration rate (eGFR) of 9.0 mL/min/1.73m2 [3]. Its pathology is distinct from HIVAN, in that non-collapsing forms of FSGS predominate, with less tubular damage but increased inflammation and interstitial fibrosis [7].
The nucleotide reverse transcriptase inhibitor tenofovir disoproxyl fumarate (TDF) and the protease inhibitor (PI) ritonavir (RTV), the latter used alone or in RTV boosted PI-based regimens, are most often associated with contemporary forms of HIV-linked kidney disease [7,9]. But there are critical distinctions between renal disorders in the context of TDF vs. PI-based cART. Acute tubular injury and tubulo-interstitial nephritis correlate with TDF use, but this pathology is reversible [9,10]. In the Aquitaine cohort of 4350 HIV-infected individuals followed for a median of 5.8 years, none developed CKD when exposure to TDF was the only risk factor [3]. This finding was replicated by an Australian cohort of 748 individuals followed for a median 4.7 years [11]. But pathologic fibrosis with inflammation characterizes PI-associated CKD [4,7], and urinary biomarkers suggest ongoing renal fibrosis in PI-treated individuals.
Measures of kidney injury (urine albumin/creatinine ratio, Kidney Injury Molecule (KIM)-1, IL-18) and fibrosis (pro-collagen type III N-terminal pro-peptide) were assessed in urines of 1234 men, 813 of whom were HIV infected, in a Multicenter AIDS Cohort Study (MACS) [5]. Those receiving PI-based cART had evidence of extensive fibrosis, independent of age, baseline eGFR, HIV load, or TDF exposure. Multivariate analysis distinguished RTV use from other risk factors for CKD, including alterations in HIV viral load, hypertension, diabetes, and hyperlipidemia [3].
In addition, PI-linked CKD may not be readily reversible by switching regimens. For example, in the NIH IMPAACT 219/219C study a lower resolution rate of renal laboratory abnormalities was found among HIV-infected children and adolescents treated with mainly PI-based cART, compared to other cART regimens [2]. Among HIV infected women receiving a PI-based regimen there was a marked increase in F2-isoprostanes and prostaglandin E2, urinary eicosanoids which reflect inflammation and oxidant stress linked to CKD [12]. These abnormalities failed to resolve after switching patients to HIV integrase inhibitor-based cART, which has not been associated with kidney disease [12].
In terms of preclinical models linking PI use with organ fibrosis, our group recently documented myocardial fibrosis and depressed cardiac function in mice exposed to pharmacologic doses of RTV [13]. We correlated these changes to RTV-induced production and activation of TGF-β1, a potent pro-fibrotic cytokine, and its dependence on platelet TGF-β1. Platelets contain 40–100 times the concentration of TGF-β1 as other cells, and it is rapidly released upon platelet activation [13]. We now hypothesized that RTV-exposed mice to pharmacologic doses of RTV would develop kidney dysfunction as a consequence of renal fibrosis. We also note that the transcription factor erythroid 2-related factor (Nrf2) has a central role in the coordinated induction of anti-oxidant products such as hemoxygenase-1 (HO-1) and endogenous carbon monoxide (CO) [14]. In terms of renal fibrosis, Nrf2 can suppress TGF-β1 signaling and fibrosis in kidney in the setting of diabetes [15] and unilateral ureteral obstruction in mice [16], through control of NFκB [17]. Low-dose exogenous CO can fully substitute for endogenous CO in mediating these salutary effects [17]. We thus further hypothesized that inhaled CO would suppress RTV-induced kidney injury in association with induction of Nrf2 and HO-1. Finally, we sought correlations between use of RTV and renal function and HO-1 expression in a clinical cohort. Our studies provide proof-of-concept for investigation of the importance of platelet activation with TGF-β1 production in HIV/cART-linked CKD, and intervention with Nrf2 activators.
Methods
Mouse cohorts and treatment
Animal studies were approved by the Institutional Animal Care and Use Committee of OMRF and WCMC. Two genotypes of mice of both sexes were used: C57Bl/6 wild-type (wt) mice and mice with specific deletion of TGF-β1 in megakaryocytes/platelets (PF4CreTgfb1flox/flox) [18]. The latter had lower platelet (by ~94%) and plasma (by ~50%) TGF-β1 levels than wt mice [19]. For both groups, one received intraperitoneal injections of vehicle (1% DMSO in PBS) or RTV (10mg/kg body weight in vehicle) daily, 5 days per week for 8 wks. The RTV dose used is typical of published preclinical studies and was designed to reflect levels achieved in RTV-boosted PI regimens, as U.S. FDA guidelines suggest that dosing should be ~6-fold higher in rats and mice than humans [20,21].
Select groups of mice were exposed to low-dose (250ppm) CO for 4 h daily, 5 days per week, administered as 1% CO mixed with room air and directed into a 3.7 ft3 plexiglass chamber at 12 L/min. A CO analyzer (Interscan, Chatsworth, CA) was used to maintain CO levels. Mice not undergoing CO exposure were maintained in ambient air.
Histologic analysis and fibrosis quantification
Mice were sacrificed and the kidneys removed. One kidney was fixed in neutral buffered formalin, processed, and embedded in paraffin blocks. Sections were cut at 4μM and stained with hematoxylin and eosin (H&E), Periodic Acid Schiff (PAS), Picrosirius red to evaluate fibrosis, and Jones methenamine silver to evaluate glomerular basement membrane. Immunofluorescence with antibodies against α-smooth muscle actin (α-SMA) and collagen α1 ((Col1α1) were used to identify extracellular matrix composition, as detailed elsewhere [13]. High magnification images were obtained using a Leica-Aperio CF5 whole slide scanner (Leica Biosystems) and imported into software for quantification of positive areas, based on analysis of edited images in which major blood vessels are deleted, as detailed in Supplemental Fig. 1. As the magnitude of fibrosis quantified on the basis of tissue sections can vary dependent on levels of background staining and tissue sectioning artefacts [22], in order to validate these findings we also established a fibrosis scoring system. This involved visualizing coded slides and independent assessment by four trained observers, including a nephropathologist who established a fibrosis score, graded 0 to 8, based on staining intensity, presence of tubular atrophy, and basement membrane thickening, consistent with published guidelines [22].
Macrophage phenotype determination
The second kidney was prepared for extraction of total CD45+ leukocytes and flow cytometry per established procedures [23]. Briefly, kidneys were minced, incubated with Liberase TM (0.25 mg/ml; Roche), deoxyribonuclease I (0.1mg/ml; Invitrogen), and Dispase (0.8 mg/ml; Roche) at 37°C for 30 min, and digestion stopped by adding a solution of 2% fetal bovine serum and 5mM EDTA in PBS. After lysis of erythrocytes with Lysis Buffer the remaining cells were incubated in Fc block (CD16/CD32; eBiosciences). Antibodies for flow cytometry analysis included: CD45 (clone I.3/2.3); CD11b (clone M1/70); CD11c (clone N418); F4/80 (clone BM8); MHC class II I-A/E (clone M5/114.15.2); CD206 (clone C068C2); and B7-H4 (clone HMH4–5G1). Stained cells were analyzed using a BD Fortessa flow cytometer (BD Biosciences). All antibodies were purchased from Biolegend, except for CD11b (BD Biosciences). Analysis was done using FlowJo software (Tree Star).
TGF-β1 determination
Murine blood samples were collected using our previously established method, with 0.1% sodium citrate as an anti-coagulant and 1uM PGE1 to prevent in vitro platelet activation [13]. Total plasma TGF-β1 was quantitated by ELISA using a TGF-β1 DuoSet kit (R&D Systems) after converting latent to active cytokine by acidification followed by neutralization, as previously described [13].
Assessment of TGF-β signaling, Nrf-2 and HO-1 induction, and renal function in mice
Paraffin-embedded sections were evaluated with an immunostain specific for phospho-Smad2 (EMD Millipore). Cell nuclei were stained with DAPI (Life Technologies). Images were taken by fluorescence microscopy and quantitation performed using the NIH ImageJ program. Nrf2 and HO-1 protein expression was examined by immunofluorescence of paraffin-embedded kidney sections per published methods [24]. Cystatin C was measured by ELISA (R&D Systems) in mouse plasma following the manufacturers’ instructions. Cystatin C changes, rather than determination of serum creatinine or urine creatinine: albumin ratio, were used as RTV, as well as cobicistat, a recent replacement for RTV in many PI-boosted regimens, is known to elevate serum creatinine via inhibition of creatinine efflux through the multidrug and toxin extrusion 1 (MATE1) transporter in the proximal tubule cell [25]. In a randomized trial to evaluate eGFR by creatinine vs. cystatin C in the HIV+/ART-naive vs. ART-treated, the latter emerged as a “more stringent” method [26]. KIM-1 was measured in mouse urine by ELISA (R&D Systems).
Assessment of renal function and HO-1 regulation in HIV-infected patients
Patient samples were obtained through study approval by the Institutional Review Board of WCMC. We utilized all available banked samples from an original cohort of 100 HIV+ and 100 HIV- postmenopausal women enrolled in an observational study of the metabolic effects of divergent cART regimens. Subjects were comparable in terms of age, ethnicity (all Hispanic or African-American), tobacco, hormone, ethanol, and illicit drug use, hypertension, and endocrine disease, as described in our prior publications [27,28]. Among the HIV-infected there were approximately equal numbers in the three groups examined here: HIV treatment naïve or off treatment; RTV-containing, PI-based regimens; and non PI, predominantly non-nucleoside reverse transcriptase inhibitor (NNRTI)-based cART. There were no significant differences in total time on cART, estimated total time of HIV infection, or immune status based on CD4+ T cell counts among those on cART [27,28]. HO-1 plasma levels (BioSource, Inc.) and serum cystatin C levels (BioSource, Inc.) were measured by ELISA following the manufacturers’ instructions. Cystatin C levels were used to calculate eGFR by the Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) equation, and kidney dysfunction classified as “mild” or “moderate” per guidelines established by the National Kidney Foundation [29].
Statistical analyses
Analysis of renal function by cystatin C measurement involved ANOVA and an alpha of 0.05. Analysis of macrophage subsets utilized a 2-tailed Student’s t test. Correlation between measures of renal function and TGF-β1 involved linear regression analyses.
Results
RTV induced renal pathology
Treatment of wt mice with RTV for eight wks led to multiple pathologic changes. H&E and PAS stained sections revealed focal segmental sclerotic thickening in approximately one-third of glomeruli, with some of them showing partial collapse (Fig. 1A,B). There was also significant widening of Bowman’s spaces, with a mean gap distance 8.2 ± 3.0 μM in PBS vs. 15.8 ± 8.0 μM in RTV (n=30 glomeruli/kidney; p <0.001; Fig. 1A) and tubular atrophy. Eosinophilic globules of mean diameter ~20–30 μm were prominent in Bowman’s spaces (Fig. 1B). These globules were negative on Picrosirius red and Jones stains. Examination of Picrosirius red stained sections under regular and polarized light (Fig. 1C,D) and direct examination of these slides by trained observers (Fig. 1E), documented statistically significant differences in fibrosis in RTV- vs. vehicle-exposed wt mice (p<0.001 and p = 0.01, respectively). The levels seen in RTV-exposed wt mice, a mean of 7.8%, are consistent with those in grade I interstitial disease of HIV-infected individuals on cART, 5–25% [30].
Figure 1.
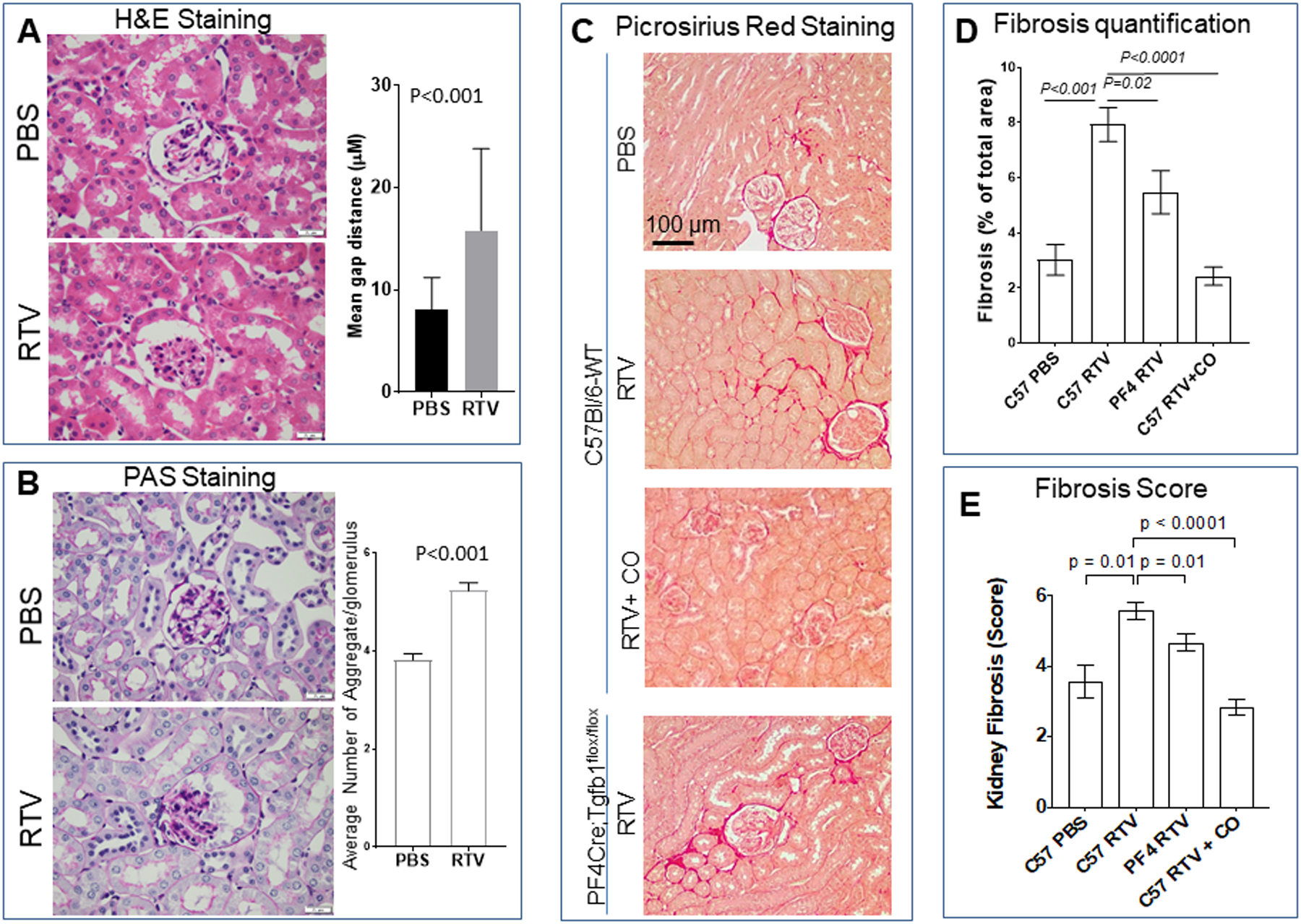
Pharmacologic levels of the HIV protease RTV induce renal pathology, including fibrosis. Higher levels of renal fibrosis are seen in RTV-exposed wt vs. platelet TGF- β1 deficiency PF4CreTgfb1flox/flox mice, and this can be suppressed by inhaled carbon monoxide. Wt and PF4CreTgfb1flox/flox mice were treated with RTV (10mg/kg) or vehicle (DMSO) for 8 wks. Kidneys were harvested and 4μM sections evaluated for fibrosis by staining with (A) H&E, (B) PAS, and (C) Picrosirius red, and quantification by Aperio whole slide scanning and Aperio image analysis, as described in the legend to Suppl. Fig. 1. In addition, independent evaluation of the extent of fibrosis was performed by four trained observers (E). Widening of Bowman’s spaces (A) with globular eosinophilic material (B), and prominent fibrosis (C-E) was characteristic of RTV exposure of wt mice. Fibrosis was attenuated in PF4CreTgfb1flox/flox mice. Exposure of wt mice to CO daily for 4 hrs (250ppm with each RTV injection) suppressed RTV-induced fibrosis (C-E).
RTV induced renal fibrosis was dependent on platelet-derived TGF-β1
The aforementioned histopathologic findings based on H&E and PAS staining were observed in both wt and PF4CreTgfb1flox/flox mice exposed to RTV. In contrast, RTV-exposed wt mice demonstrated much higher levels of fibrosis in the renal cortex, particularly surrounding the glomeruli (Fig. 1C,D,E). Consistent with this difference, immunofluorescence demonstrated marked increases in α-SMA (p <0.001) and collagen 1α (p = 0.033) in RTV-exposed wt mice, changes which were significantly suppressed in the PF4CreTgfb1flox/flox mice (p = 0.02 and p = 0.05, respectively) (Fig. 2A,B). This difference in extent of tissue fibrosis correlated with TGF-β-mediated Smad2 phosphorylation (Fig. 2C; p=0.001).
Figure 2.
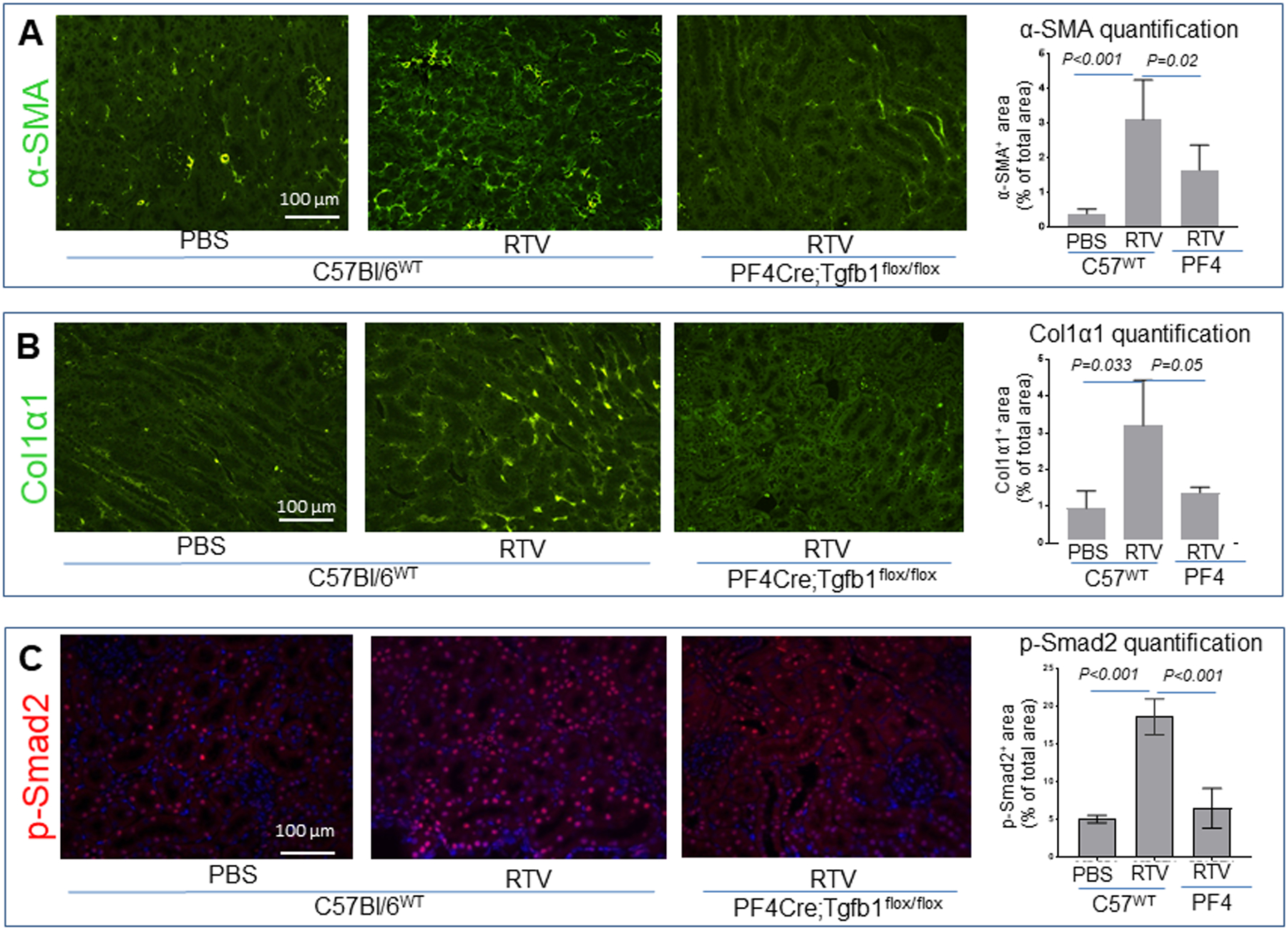
RTV-induced fibrosis involved over-expression of α-SMA and collagen α1 in kidneys, paralleled by TGF-β1-mediated pSmad2 signaling in wt but not platelet TGF-β1 deficient mice. Paraffin-embedded kidney sections were stained for α-SMA (A), collagen α1 (B), or phosphor-Smad2-specific antibodies. Photographs were taken using a Nikon Eclipse 80i fluorescence microscope with 20X magnification. Quantification of pSmad2 positive nuclei (red dots in blue nuclei counter-stained with DAPI) was determined using the NIH ImageJ program. Platelet TGF-β1 deficient PF4CreTgfb1flox/flox mice had lower levels of pSmad2 in kidney than wt mice after 8 wks of RTV exposure.
Correlation of RTV-induced renal fibrosis with kidney injury, kidney dysfunction, and circulating TGF-β1 levels
Plasma cystatin C levels (Fig. 3A), used as a marker of kidney dysfunction, and urinary KIM-1 levels (Fig. 3B), a measure of tubular damage, were significantly higher in RTV- vs. vehicle (control)-exposed wt mice (p = 0.008). In contrast, cystatin C levels were equivalent to control values in RTV-exposed PF4CreTgfb1flox/flox mice (Fig. 3A). As previously documented for our wt and transgenic mice [13], total TGF-β1 levels were increased nearly 5-fold in plasma of RTV-treated wt mice vs. vehicle controls. In contrast, there was no increase in TGF-β1 levels in PF4CreTgfb1flox/flox mice treated with RTV vs. vehicle. Linear regression analysis of cystatin C levels showed a positive correlation with total plasma TGF-β1 levels (Fig. 3C; R2 = 0.4210, p = 0.031).
Figure 3.
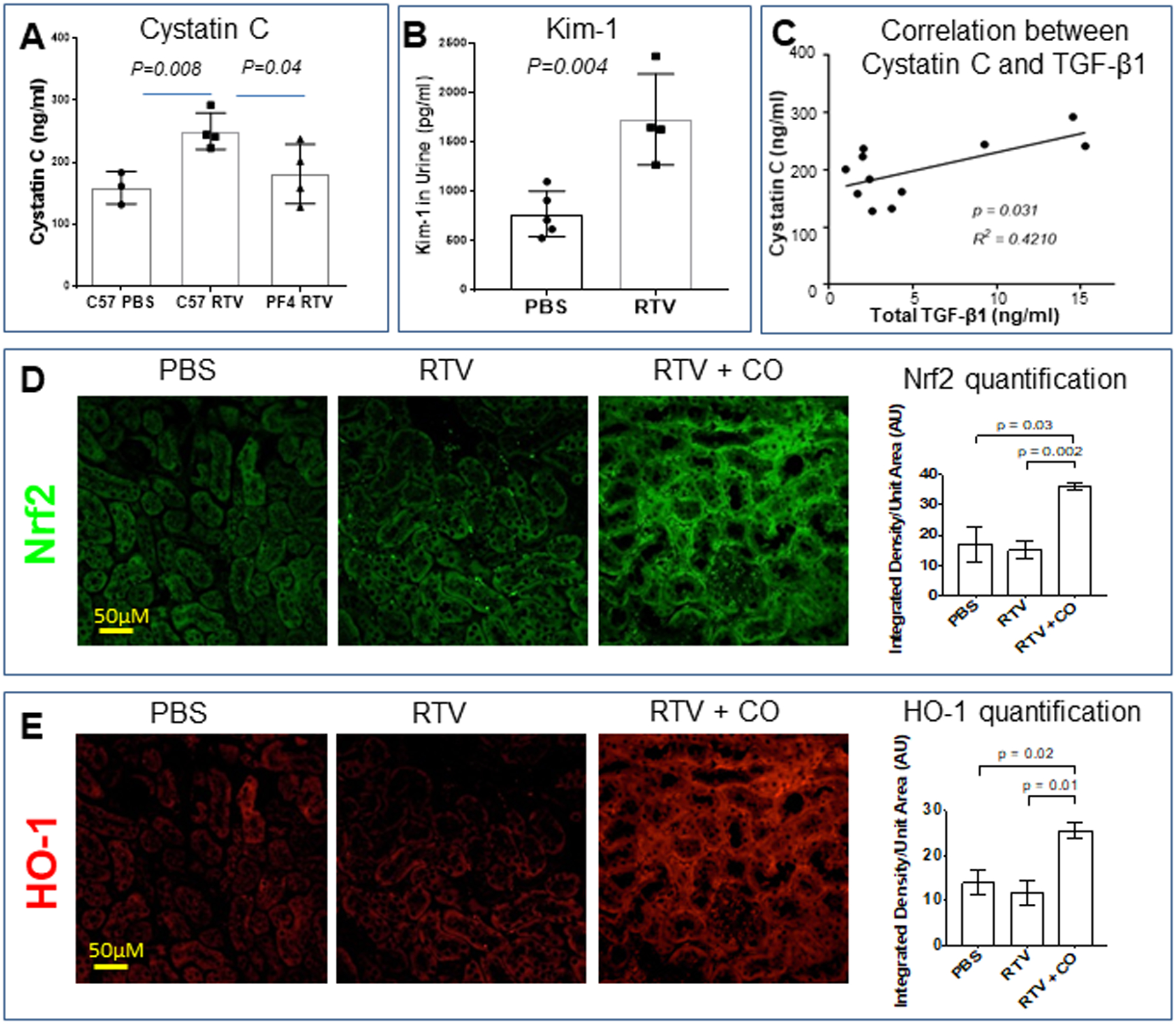
RTV induced renal dysfunction in association with tubular injury in wt mice, its dependence on platelet TGF-β1, and suppression by inhaled CO in association with Nrf2 activation. Cystatin C levels in plasma (A) and KIM-1 levels in urine (B) were assessed by ELISA following 8 wks of RTV exposure. They were significantly elevated in RTV-exposed wt mice. RTV-exposed PF4CreTgfb1flox/flox mice had cystatin C levels equivalent to control wt mice (A). Total plasma TGF-β1 levels correlated with degree of cystatin C elevation (C). Paraffin-embedded kidney sections from wt mice treated with RTV or RTV plus inhaled CO were then stained for Nrf2 (D) and HO-1 (E) and positive areas quantified by confocal scanning and ImageJ analysis. Both proteins, involved in anti-oxidant signaling, were markedly increased following CO exposure.
CO blocked RTV-mediated renal fibrosis in association with Nrf2 activation and macrophage polarization
We examined Nrf2 and HO-1 expression in kidneys of wt mice exposed to RTV vs. vehicle in the presence and absence of inhaled low-dose CO. RTV had no effect on expression of either molecule in the kidney (Fig. 3D,E). In contrast, CO treatment led to a significant increase in both Nrf2 (p = 0.03) and HO-1 (p = 0.02) in RTV-exposed wt mice (Fig. 3D,E).
We assessed macrophage phenotypes in kidneys of wt mice exposed to RTV vs. RTV plus CO. Divergent TGF-β1-dependent pathways can either augment collagen synthesis or promote its degradation [29]. Macrophage polarization may be involved in TGF-β1-dependent pathologic fibrosis through expansion of the pro-inflammatory M1 macrophage subset and suppression of M2c regulatory macrophages, the latter capable of promoting regulatory T cell (T-reg) development and collagen autophagy [31,32]. RTV had no effect on the level of total CD45+ mononuclear cells or total macrophages, quantitated as the F4/80+CD11b+ fraction of CD45+ cells in kidneys (Fig. 4A–C). However, the M1 macrophage subset was increased 3.5-fold following RTV exposure (p <0.0001), and this rise was reduced by CO (p = 0.001) (Fig. 4D). Co-exposure of RTV treated mice to CO increased the number of total M2 cells (p = 0.001) (Fig. 4E), with a marked increase in the M2c subset (12.5-fold increase over control levels; p = 0.006) (Fig. 4F). In parallel, CO blocked RTV-induced renal fibrosis, as defined by Picrosirius red staining (p <0.001; Fig. 1C,D) and fibrosis score (p <0.0001; Fig. 1E).
Figure 4.
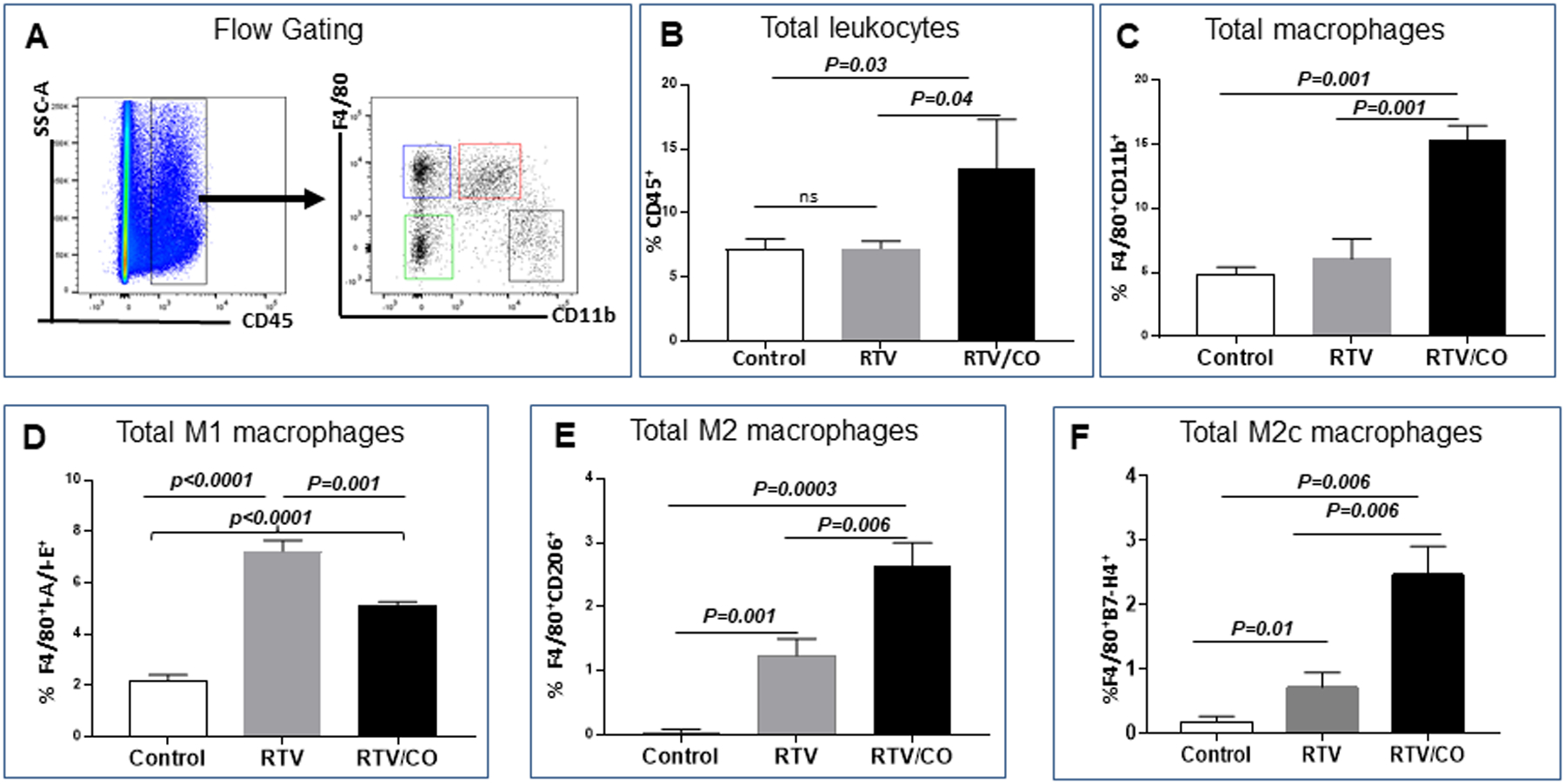
Low-dose CO modulates macrophage polarization in kidneys from RTV-exposed wt mice. Subsets of F4/80+ CD11b+ macrophages, expressed as a percentage of CD45+ leukocytes (A-C) were examined in RTV-exposed wt mice either untreated or following CO inhalation. RTV had no effect on the fraction of total leukocytes (B) or total macrophages (C), whereas CO inhalation significantly increased both populations (B,C). RTV markedly elevated the pro-inflammatory CD45+/F4/80+ I-A/E+ M1 subset, which was reduced significantly by CO (D). RTV also increased the number of CD45+/F4/80+ CD206+ M2 cells as a proportion of total macrophages, an effect amplified by CO (E). The CO-mediated alteration in M2 cells predominantly involved augmentation of regulatory, anti-inflammatory CD45+/F4/80+ B7-H4+ M2c cells (F).
Cystatin C levels and eGFR in HIV-infected individuals treated with RTV-based vs. non-PI-based cART
Cystatin C was significantly elevated in sera of HIV+/ART-naive (n = 17) vs. HIV- controls (n = 19; p=0.014), and further increased in those treated with RTV-containing cART (n = 20; p=0.05 compared to values with the HIV+/ART-naïve) (Fig. 5A). Cystatin C-based calculation of eGFR demonstrated that all HIV-infected subjects had significantly more renal dysfunction characterized as “mild” than HIV uninfected controls (n = 27), but only the RTV group (n= 20) had significantly more “moderate” renal disease than the HIV+/ART naïve (n = 27) or the non-PI-ART treated group (n = 30; p <0.02) (Fig. 5B).
Figure 5.
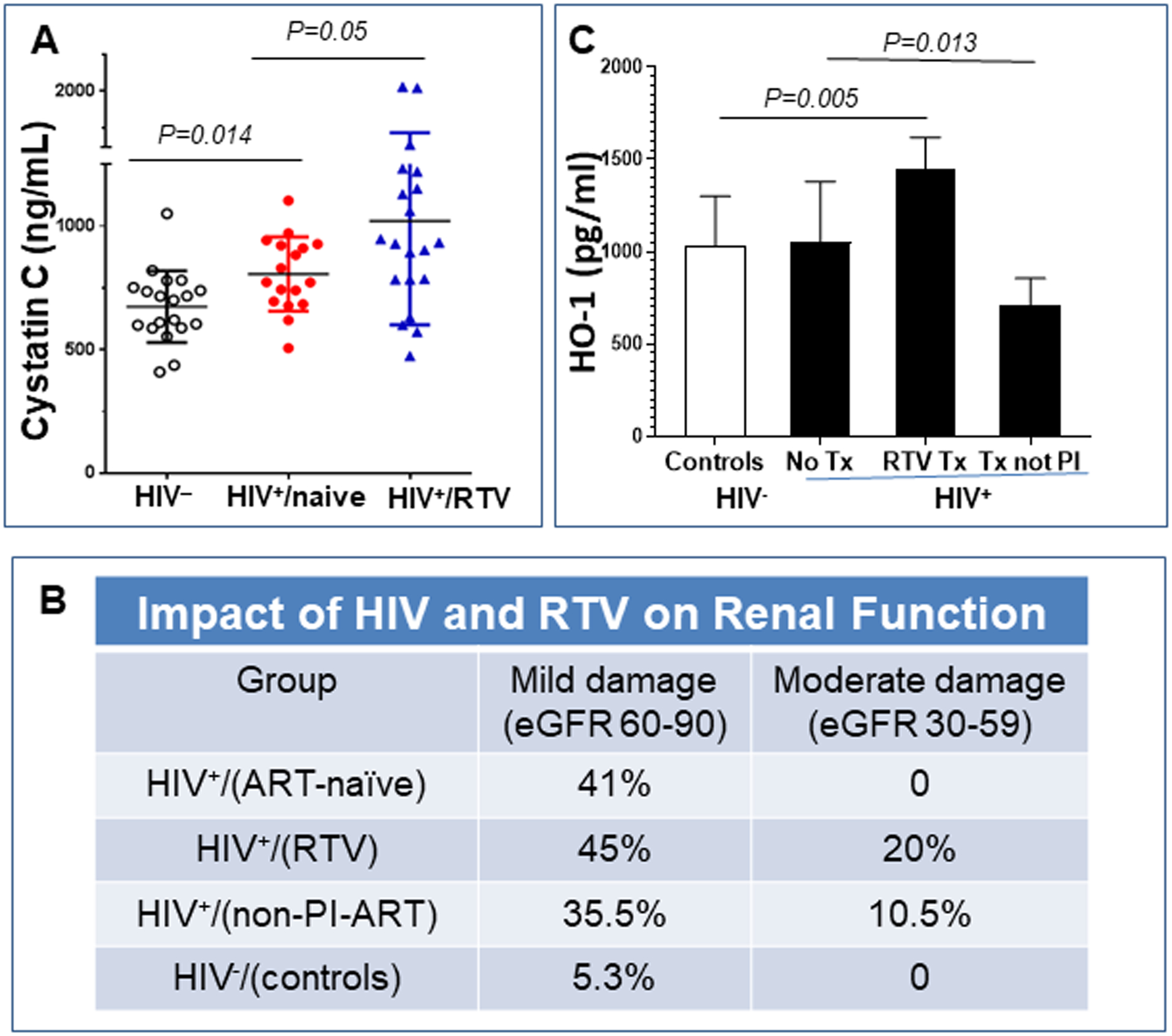
Impact of RTV on circulating cystatin C and HO-1 levels in cART-treated, HIV-infected postmenopausal women. Cystatin C was measured in sera and HO-1 in plasma from postmenopausal women, either HIV seronegative, HIV-positive on no antiretroviral (ART) therapy, or HIV-positive on a stable ART regimen for ≥two years. The later involved either a RTV-boosted PI or a non-PI based regimen (primarily non-nucleoside reverse transcriptase inhibitor- based). (A) HIV infection alone (n=17) was linked to an increase in cystatin C levels compared with age-matched controls (n=19), with a further increase in levels in those HIV+ women treated with RTV-based cART (n=20). (B) eGFR was calculated as described in Methods. The HIV+/RTV group (n= 20) had significantly more moderate renal disease than the HIV- matched controls (n = 27), the HIV+/no ART (n = 27), or the HIV+/non-PI-ART groups (n = 30) (p<0.02). (C) HO-1 levels were no different in women infected with HIV but on no therapy (n=5) vs. HIV negative control women (n=7). RTV-based cART led to a significant rise in HO-1 (n=7), not seen with other cART regimens.
Plasma HO-1 levels in HIV-infected individuals treated with RTV-based vs. non-PI-based cART
There was no difference in HO-1 levels between the HIV negative controls and untreated HIV+ women (Fig. 5C). In contrast, individuals on RTV-based regimens had significantly elevated levels of HO-1 (p = 0.005) while those on non-PI-based cART had significantly lower HO-1 levels than controls (p = 0.013; Fig. 5C).
Discussion
The incidence of CKD characterized by interstitial fibrosis is heightened in HIV+ individuals receiving RTV, either alone or as part of certain RTV-boosted PI cART regimens, vs. non-PI regimens. This has been documented by numerous observational studies conducted among diverse populations [3,10,33–35]. These data are paralleled by the cystatin C measurements and eGFR values in our cohort of HIV+ postmenopausal women on RTV-based vs. non-PI-based therapy. Our system to model such renal disease in mice is robust, as exposure of wt mice to pharmacologic doses of RTV alone induced renal dysfunction with fibrosis in association with predominance of pro-inflammatory M1 macrophages. Comparing these changes to clinical disease, HIVAN occurring in untreated HIV infection is a collapsing glomerulopathy with hyperplasia of glomerular epithelial cells and resulting pseudo-crescent formation, accompanied by tubular injury with microcyst formation [4,7]. Pathologic fibrosis is seen only in end-stage lesions [4,7]. By contrast, CKD in the setting of HIV/cART is a predominantly non-collapsing FSGS with significantly more fibrosis and inflammation [4], as reproduced in our RTV-exposed mice.
The degree of interstitial fibrosis seen in our RTV-exposed wt mice parallels levels described in grade I interstitial disease in the setting of HIV/cART [30]. These results parallel recent data from the MACS cohort showing that urinary markers of fibrosis are associated with PI use and kidney dysfunction, independent of other risk factors for CKD [3,5]. Our murine model also documents the capacity of RTV to induce renal pathology characteristic of HIV/cART in the absence of HIV infection. This addresses the question as to whether the renal histology characteristic of PI-based treatment in HIV infection represents attenuation of a developing HIVAN lesion by effective ART, as opposed to a direct insult by cART itself [7].
However, these data do not rule out the possibility that RTV-induced inflammation and inflammation induced by HIV infection alone--which persists, albeit at suppressed levels, in the setting of effective ART--could interact synergistically. The impact of RTV, or any other ART drug, tested in animals absent HIV may not accurately mimic all of the influences at play in the pathologic fibrosis of HIV/cART. We are now planning to model the effects of RTV and more contemporary ART regimens in HIV transgenic mice.
In terms of pathophysiology, our demonstration of the dependence of RTV-mediated renal fibrosis on platelet-derived TGF-β1 is consistent with the ability of RTV to induce release of TGF-β1 from platelets [36]. It is also consistent with the fact that TDF and the PI nelfinavir, antiretroviral drugs which have not been associated with CKD or renal fibrosis, do not activate platelets [37–39], although TGF-β production and release has not been specifically examined in that context. It had been hypothesized that several non-AIDS defining fibrotic disorders, including CKD in the cART-treated, may be induced or sustained by HIV-mediated upregulation of TGF-β1 in the setting of a persistent inflammatory milieu [40,41], but we document the independence of these changes from HIV infection itself. While TGF-β1 plasma levels are increased 2-fold in HIV+/ART-naïve asymptomatic individuals, with a further rise in advancing disease, these levels are not suppressed by cART [36, 42]. Accurate evaluation of in vivo changes in TGF-β1 has been confounded by the failure of many investigators to utilize controls for ex vivo platelet activation, and the fact that measures of functional TGF-β1, the activation product from its latent form [43], have not been assessed in the setting of divergent cART regimens.
Other inflammatory biomarkers have also proved of limited utility in defining fibrotic disease in the setting of fully suppressed HIV, as discussed in a recent review from our group [37]. For example, in the SPIRAL study of 273 HIV+ individuals switched from RTV-boosted PIs to integrase strand transfer inhibitor-based cART, there were marked declines in hsCRP, monocyte chemoattractant protein-1, osteoprotegerin, IL-6, and TNF-α, but no changes in related inflammatory biomarkers, including IL-10, sICAM-1, sVCAM-1, E-selectin, and P-selectin [44]. In addition, markers of inflammation and immune activation did not correlate with an MRI-based myocardial fibrosis index of HIV-infected individuals on cART [45].
Renal fibrosis is a pathologic process common to many forms of CKD [46]. In our study all of the changes associated with RTV exposure—renal dysfunction (cystatin C and KIM-1), renal inflammation (increase in M1 macrophages and pSmad2 staining), and renal fibrosis (Picrosirius red staining, fibrosis score, α-SMA expression, and Col1α1 expression)--were reversed by CO treatment. Given the limited number of animals studied we did not attempt to define the magnitude of specific inter-correlations among these measures. However, the minimal fibrosis, decreased presence of inflammatory macrophages, and preserved renal function seen in RTV-exposed mice deficient in platelet TGF-β1 suggests that RTV-associated fibrosis and inflammation was etiologic in the kidney dysfunction, and not simply a reaction to tissue damage from a nephrotoxic drug.
This is important, as RTV and certain other PIs concentrate in the urinary tract and may lead to crystalluria, with some nephrologists hypothesizing that PI irritant effects alone could cause renal dysfunction [47]. Indeed, while certain histopathologic changes were found in both wt and platelet TGF-β1 deficient mice exposed to RTV, only the former showed pathologic fibrosis and renal dysfunction. A recent update from the D:A:D (Data collection on Adverse events of Anti-HIV Drugs) study examining contemporary RTV-boosted atazanavir vs. darunavir regimens confirmed the association of RTV-atazanavir, but not RTV-darunavir, with CKD, despite the fact that both may precipitate crystals in urine [35]. This observation suggests the need to model interactions among classes of ART in terms of potentiation of RTV-induced nephrotoxicity. For example, initial but non-progressive increases in serum creatinine and corresponding decreases in eGFR have been observed with co-administration of TDF to individuals on RTV-boosted PI-regimens, consistent with effects on renal tubular transport of creatinine [48].
In terms of potential interventions, pan-neutralization of TGF-β fails to inhibit or reverse pathologic fibrosis clinically or in many mouse models, regardless of the organ involved [31]. This may relate to divergent TGF-β-dependent pathways which can either augment collagen synthesis or promote its degradation [31]. Indeed, knock-out of TGF-β1 leads to massive inflammatory disease in major organs, including the kidney [49]. We therefore sought to model an intervention based on the endogenous CO pathway.
CO is generated in mammalian cells through the catalysis of heme by HO, and the stress-inducible form HO-I has a key physiologic role in protecting against oxidative stress [50]. Exogenous CO administration at low-dose can fully substitute for the cytoprotective effects of endogenous CO in a variety of model systems. For example, inhaled low-dose CO blocked renal fibrosis in the unilateral ureteral obstruction mouse model in association with decreased expression of TGF-β receptors and suppression of TAK1/MKK3/p38 signaling [51]. CO may also facilitate collagen autophagy [52] as it is promoted by M2c macrophages [13]. Consistent with these activities, we found that inhaled low-dose CO blocked RTV-mediated renal fibrosis in association with promotion of the M2c macrophage regulatory subset. These latter changes also may be relevant to other forms of CKD, where the importance of macrophage polarization to fibrosis has been raised [53,54]. Exposure to even very low dose CO can cause myocardial damage, but the duration of such exposures (reviewed in [17]) was much longer than used here, and doses of inhaled CO equivalent to our study have been well-tolerated in two recent clinical trials [55,56]. In addition, intervention with exogenous CO may serve as a proof-of-concept for related pathways amenable to more pragmatic therapeutics. For example, Nrf2 is a key regulator of the expression of genes coding for the majority of endogenous anti-oxidant and anti-inflammatory proteins linked to fibrosis, including HO-1. As shown here, and by others, Nrf2 is activated by exogenous CO as well as by several orally bioavailable inducers [16,57]. We also found that RTV-based cART was associated with a significant elevation in HO-1 in our HIV-infected postmenopausal women, similar to a recent report from a European cohort [58]. This increase may be a physiologic attempt to reduce oxidative stress induced by RTV, as adenovirus-mediated enhancement of HO-1 blocked inflammation and endothelial cell injury induced by RTV and certain other PIs in vitro, while suppression of HO-1 augmented this pathology [59]. We had anticipated that a similar compensatory rise in HO-1 would be seen in our RTV-exposed mice. While CO did induce the expected and dramatic rise in HO-1 levels in the setting of RTV, this was not seen in mice exposed to RTV alone. This variance from the human cohort may represent one limitation of models lacking the full panoply of inflammatory changes occurring in the context of HIV/cART.
Conclusion
HIV-associated nephropathy has declined markedly following introduction of cART, but this has been paralleled by a rise in CKD characterized by renal fibrosis and inflammation in HIV+ individuals receiving PI-based therapy. Unlike TDF, which has been linked to kidney dysfunction but does not cause fibrosis or irreversible CKD in the absence concomitant use of RTV-boosted PIs [60], PI-induced renal injury may not be ameliorated by simple ART regimen switching [13,61]. We discovered that mice exposed to clinically relevant doses of the PI ritonavir developed renal tubular damage and renal dysfunction in association with macrophage polarization to an inflammatory subtype and pathologic renal fibrosis. It was dependent on release of platelet TGF-β1 and blocked by low-dose inhaled CO, a potent inducer of Nrf2-associated anti-oxidant pathways. These results document that certain PI-based cART may cause renal injury, inflammation, and dysfunction in the absence of HIV. If corroborated by additional studies, they offer proof-of-concept for involvement of platelet TGF-β1 in HIV/cART-associated kidney disease. This is consistent with the emerging importance of platelet TGF-β1 in other disorders [62]. Indeed, the parallel development of cardiac fibrosis in RTV-exposed mice, and its dependence on platelet TGF-β1, as previously documented by our group [13], suggests a similar mechanism of action for both renal and cardiac injury related to RTV, and a similar potential to respond to anti-platelet and/or anti-oxidant interventions. Given this insight, circulating biomarkers currently being assessed in clinical studies of HIV/cART-linked CVD with fibrosis should be examined in terms of renal and other organ (e.g., liver and lung) fibrosis as well, all of which may be part of morbidities linked to chronic cART use as the HIV population ages.
Supplementary Material
Supplemental Figure 1. Assessment and quantitation of renal fibrosis in mouse kidney by Picrosirius red staining. Whole kidney sections of 4μM thickness were stained with Picrosirius red and scanned using an Aperio Whole Slide Scanner (Leica Biosystems) at 20x magnification. Images were then analyzed in ImageScope using a modified “Positive Pixel Count Algorithm V9” with Hue Value of 0.38 and Hue Width of 0.72. Because large blood vessels naturally contain collagen fiber, those stained areas were deleted (black areas in the middle panel) using Photoshop or Paint programs before quantification. Fibrotic areas are depicted by the red color (red box insert), with a background threshold set such that fibrotic areas were a clearly visible (right panel, with a magnified insert in the green box). Picrosirius red labels collagen bundles red under natural light. Under polarized light these bundles appear as a mixture of green, red and yellow birefringence against a dark background and are more clearly differentiated from background. The percentage of fibrosis was calculated by dividing the fibrotic area (x) by the total area (y), then multiplying by 100.
Acknowledgements
We thank: Karim Kouzbari for performing the ELISAs; Tyler Robison for performing select experiments and their analysis; Luisa Williams for histology; Dr. Sandeep Subrahmanian and Pratibha Dube for assistance with animal harvesting and tissue processing; and Members of cardiovascular biology program at OMRF for assistance with microscopy. This work was supported by NIH grants R21 HL125044 (J.L. and J.A.), R01 HL123605 (J.A.), RO1 HL133801 and RO1 HL060234 (M.E.C.), COBRE grant GM114731, and funding from PHF and OCASCR (J.A.) and the Angelo Donghia Foundation (J.L.).
Sources of Funding:
This work was supported by NIH grants R21 HL125044 (J.L. and J.A.), R01 HL123605 (J.A.), RO1 HL133801 and RO1 HL060234 (M.E.C.), COBRE grant GM114731, and funding from PHF and OCASCR (J.A.) and the Angelo Donghia Foundation (J.L.).
Footnotes
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the PDF version of this article on the journal’s Web site.
Conflicts of Interest
The spouse of M.E.C is a co-founder, shareholder and serves on the Scientific Advisory Board of Proterris, Inc. The remaining authors have no conflicts.
References
- [1].Mocroft A, Kirk O, Reiss P, et al. Estimated glomerular filtration rate, chronic kidney disease and antiretroviral drug use in HIV-positive patients. AIDS 2010;24:1667–1678. [DOI] [PubMed] [Google Scholar]
- [2].Mitchell CD, Chernoff MC, Seage GR III, et al. Predictors of resolution and persistence of renal laboratory abnormalities in pediatric HIV infection. Pediatr Nephrol 2015;30:153–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Morlat P, Vivot A, Vandenhende M-A, et al. Role of traditional risk factors and antiretroviral drugs in the incidence of chronic kidney disease, ANRS CO3 Aquitane cohort, France, 2004–2012. PLoS One 2013;8:e66223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Rosenberg AZ, Naicker S, Winkler CA, et al. HIV-associated nephropathies: epidemiology, pathology, mechanisms and treatment. Nat Rev Nephrol 2015;11:150–160. [DOI] [PubMed] [Google Scholar]
- [5].Jotwani V, Scherzer R, Estrella MM, et al. Association of HIV infection with biomarkers of kidney injury and fibrosis in the Multicenter AIDS Cohort Study. Antivir Ther 2017;22:421–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Rednor SJ, Ross MJ. Molecular mechanisms of injury in HIV-associated nephropathy. Front Med 2018;5:177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Swanepoel CR, Atta MG, D’Agati VD, et al. Kidney disease in the setting of HIV infection: conclusions from a Kidney Disease Improving Global Outcomes (KDIGO) controversies conference. Kidney Int 2018;93:545–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Arendse CG, Wearne N, Okpechi IG, et al. The acute, the chronic and the news of HIV-related renal disease in Africa. Kidney Int 2010;78:239–245. [DOI] [PubMed] [Google Scholar]
- [9].Hamzah L, Booth JW, Jose S, et al. Renal tubular disease in the era of combination antiretroviral therapy. AIDS 2015;29:1831–1836. [DOI] [PubMed] [Google Scholar]
- [10].Ryom L, Mocroft A, Kirk O,et al. Predictors of estimated glomerular filtration rate progression, stabilization or improvement after chronic renal impairment in HIV-positive individuals. AIDS 2017;31:1261–1270. [DOI] [PubMed] [Google Scholar]
- [11].Woolnough EL, Hoy JF, Cheng AC, et al. Predictors of chronic kidney disease and utility of risk prediction scores in HIV positive individuals. AIDS 2018;32:1829–1835. [DOI] [PubMed] [Google Scholar]
- [12].Hulgan T, Boger MS, Liao DH, et al. Urinary eicosanolid metabolites in HIV-infected women with central obesity switching to raltegravir: an analysis from the women, integrase, and fat accumulation trial. Mediat Inflamm 2014;e803095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Laurence J, Elhadad S, Robison T, et al. HIV protease inhibitor-induced cardiac dysfunction and fibrosis is mediated by platelet-derived TGF-β1 and can be suppressed by exogenous carbon monoxide. PLoS One 2017;12:e0187185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ruiz S, Pergola PE, Zager RA, et al. Targeting the transcription factor Nrf2 to ameliorate oxidative stress and inflammation in chronic kidney disease. Kidney Intl 2013;83:1029–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Zoja C, Benigni A, Remuzzi G. The Nrf2 pathway in the progression of renal disease. Nephrol Dial Transplant 2014;29:i19–124. [DOI] [PubMed] [Google Scholar]
- [16].Oh CJ, Kim J-Y, Choi Y-K, et al. Dimethylfumarate attenuates renal fibrosis via NF-E2-related factor 2-mediated inhibition of transforming growth factor-β/Smad signaling. PLoS One 2012; 7:e45870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ahamed J, Laurence J. Role of platelet-derived transforming growth factor-β1 and reactive oxygen species in radiation-induced organ fibrosis. Antioxidants Redox Signaling 2017;27:977–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Meyer A, Wang W, Qu J, et al. Platelet TGF-β1, cardiac fibrosis, and systolic dysfunction in a mouse model of pressure overload. Blood 2012;119:1064–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ghafoory S, Varshney R, Robison T, et al. Platelet TGF-β1 deficiency decreases liver fibrosis in a mouse model of liver injury. Blood Adv 2018:2:470–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Mak IT, Chmielinska JJ, Spurney CE, et al. Combination ART-induced oxidative/nitrosative stress, neurogenic inflammation and cardiac dysfunction in HIV-1 transgenic (Tg) rats: protection by Mg. Int J Med Sci 2018;19:2409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].ElZohary L, Weglicki WB, Chmielinska JJ, et al. Mg-supplementation attenuated lipogenic and oxidative/nitrosative gene expression caused by combination antiretroviral therapy (cART) in HIV-1-transgenic rats. PLoS One 2019;14:e0210107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Farris AB, Adams CD, Brousaides N, et al. Morphometric and visual evaluation of fibrosis in renal biopsies. J Am Soc Nephrol 2011;22:176–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Epelman S, Lavine KJ, Beaudin AE, et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 2014;40:91–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Johnson ACM, Delrow JJ, Zager RA. The protoporphyrin activates the oxidant-dependent NRF2-cytoprotective pathway and mitigates acute kidney injury. Transl Res 2017;186:1–18. [DOI] [PubMed] [Google Scholar]
- [25].Lepist EI, Zhang X, Hao J, et al. Contribution of the organic anion transporter OAT2 to the renal active tubular secretion of creatinine and mechanism for serum creatinine elevations caused by cobicistat. Kidney Int 2014;86:350–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Albini L, Cesana BM, Mottia D, et al. A randomized, pilot trial to evaluate glomerular filtration rate by creatinine or cystatin C in naïve HIV-infected patients after tenofovir/emtricitabine in combination with atazanavir/ritonavir or efavirenz. J Acqui Immune Def Syndr 2012:59:18–24. [DOI] [PubMed] [Google Scholar]
- [27].Yin YT, Modarresi R, Shane E, et al. Effects of HIV infection and antiretroviral therapy with ritonavir on induction of osteoclast-like cells. Osteoporosis Intl 2011;22:1459–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Yin MT, McMahon D, Ferris DC, et al. Low bone mass and high bone turnover in postmenopausal HIV-infected women. J Clin Endocrinol Metab 2010;95:620–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Levey AS, Stevens LA, Schmid CH, et al. A new equation to estimate glomerular filtration rate. Ann Intern Med 2009;150:604–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Wearne N, Swanepoel CR, Boulle A, et al. The spectrum of histologies seen in HIV with outcomes, prognostic indicators and clinical correlations. Nephrol Dial Transplant 2012;27:4109–4118. [DOI] [PubMed] [Google Scholar]
- [31].Sureshbabu A, Muhsin SA, Choi ME. TGF-β signaling in the kidney: pro-fibrotic and protective effects. Am J Physiol Renal Physiol 2016;310:F596–F606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lu J, Cao Q, Zheng D,et al. Discrete functions of M2a and M2c macrophage subsets determine their relative efficacy in treating chronic kidney disease. Kidney Int 2013;84:745–755. [DOI] [PubMed] [Google Scholar]
- [33].Mocroft A, Lundgren JD, Fux CA, et al. Cumulative and current exposure to potentially nephrotoxic antiretrovirals and development of chronic kidney disease in HIV-positive individuals with a normal baseline estimated glomerular filtration rate: a prospective international cohort study. Lancet HIV 2016;3:e23–e32. [DOI] [PubMed] [Google Scholar]
- [34].Overton ET, Patel P, Mondy K, et al. Cystatin C and baseline renal function among HIV-infected persons in the SUN study. AIDS Res Hum Retrov 2012;28:148–155. [DOI] [PubMed] [Google Scholar]
- [35].Ryom L, Lundgren JD, Reiss P, et al. Use of contemporary protease inhibitors and risk of incident chronic kidney disease in HIV-positive persons; the D:A:D Study. J Infect Dis 2019;220:1629–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Ahamed J, Terry H, Choi ME, et al. Transforming growth factor-β1-mediated cardiac fibrosis: potential role in HIV and HIV/ART-linked cardiovascular disease. AIDS 2016;30:535–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Laurence J, Elhadad S, Ahamed J. HIV-associated cardiovascular disease: importance of platelet activation and cardiac fibrosis in the setting of specific antiretroviral therapies. Open Heart 2018,5:e000823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Falcinelli E, Francisci D, Belfiori B, et al. In vivo platelet activation and platelet hyperreactivity in abacavir-treated HIV-infected patients. Thromb Hemost 2013;110:349–357. [DOI] [PubMed] [Google Scholar]
- [39].Taylor KA, Smyth E, Rauzi F, et al. Pharmacological impact of antiretroviral therapy on platelet function to investigate human immunodeficiency virus-associated cardiovascular risk. Br J Pharmacol 2019;176:879–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Yamamoto T, Noble NA, Miller DE, et al. Increased levels of transforming growth factor-β in HIV-associated nephropathy. Kidney Intl 1999;55:579–592. [DOI] [PubMed] [Google Scholar]
- [41].Theron AJ, Anerson R, Rossouw TM, et al. The role of transforming growth factor beta-1 in the progression of HIV/AIDS and development of non-AIDS-defining fibrotic disorders. Front Immunol 2017;8:1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Liovat AS, Rey-Cuillé MA, Lécuroux C, et al. Acute plasma biomarkers of T cell activation set-point levels and of disease progression in HIV-1 infection. PLoS One 2012;7:e46143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Mancini D, Monteagudo J, Suarez-Farinas M, et al. New methodologies to accurately assess circulating active transforming growth factor-β1 levels: implications for evaluating heart failure and the impact of left ventricular assist devices. Transl Res 2018;192:15–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Martínez E, D’Albuquerque PM, Llibre JM, et al. Changes in cardiovascular biomarkers in HIV-infected patients switching from ritonavir-boosted protease inhibitors to raltegravir. AIDS 2012;26:2315–2326. [DOI] [PubMed] [Google Scholar]
- [45].Thiara DK, Liu CY, Raman F, et al. Abnormal myocardial function is related to myocardial steatosis and diffuse myocardial fibrosis in HIV-infected adults. J Infect Dis 2015;212:1544–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Klinkhammer BM, Goldschmeding R, Floege J, et al. Treatment of renal fibrosis—turning challenges into opportunities. Adv Chronic Kidney Dis 2017;24:117–129. [DOI] [PubMed] [Google Scholar]
- [47].Chu GJ, Henderson C, Evans L, et al. Chronic granulomatous interstitial nephritis and urothelial metaplasia associated with ritonavir-boosted atazanavir: a case study and literature review. Pathology 2018;50:565–568. [DOI] [PubMed] [Google Scholar]
- [48].Bagnis CI, Stellbrink H-J. Protease inhibitors and renal function in patients with HIV infection: a systematic review. Infect Dis Ther 2015;4:15–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Loboda A, Sobczak M, Jozkowicz A, et al. TGF-β1/Smads and miR-21 in renal fibrosis and inflammation. Mediat Inflamm 2016;8319283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Wang L, Lee J-YS, Kwak JH, et al. Protective effects of low-dose carbon monoxide against renal fibrosis induced by unilateral ureteral obstruction. Am J Physiol Renal Physiol 2008;F508–F517. [DOI] [PubMed] [Google Scholar]
- [51].Ding Y, Kim Sl, Lee SY, et al. Autophagy regulates TGF-β expression and suppresses kidney fibrosis induced by unilateral ureteral obstruction. J Am Soc Nephrol 2014;25:2835–2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Kim S II, Na H-J, Ding Y, et al. Autophagy promotes intracellular degradation of type I collagen induced by transforming growth factor (TGF)-β1. J Biol Chem 2012, 287:11677–11688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Cellular Liu Y. and molecular mechanisms of renal fibrosis. Nat Rev Nephrol 2011;7:684–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Chung S, Overstreet JM, Li Y, et al. TGF-β promotes fibrosis after severe acute kidney injury by enhancing renal macrophage infiltration. JCI Insight 2018;3(21):e123563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Rosas IO, Goldberg HJ, Collard HR, et al. A phase II clinical trial of low-dose inhaled carbon monoxide in idiopathic pulmonary fibrosis. Chest 2018;153:94–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Fredenburgh LE, Perrella MA, Barragan-Bradford D, et al. A phase I trial of low-dose inhaled carbon monoxide in sepsis-induced ARDS. JCI Insight 2018; 3(23):e124039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Loboda A, Damulewicz M, Pyza E, et al. Role of Nrf2/HO-1 system in development, oxidative stress responses and diseases: an evolutionarily conserved mechanism. Cell Mol Life Sci 2016;73:3221–3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Taylor N, Kremser I, Auer S, et al. Hemeoxygenase-1 as a novel driver in ritonavir-induced insulin resistance in HIV-1-infected patients. J Acquir Immune Defic Syndr 2017;75:e13–e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Liu X-M, Durante ZE, Peyton KJ, et al. Heme oxygenase-1-derived bilirubin counteracts HIV protease inhibitor-mediated endothelial cell dysfunction. Free Radic Biol Med 2016;94:218–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Suzuki S, Nishijima T, Kawasaki Y, et al. Effect of tenofovir disoproxil fumarate on incidence of chronic kidney disease and rate of estimated glomerular filtration rate decrement in HIV-1-infected treatment-naïve Asian patients: results from 12-year observational cohort. AIDS Patient Care STDs 2017;31:105–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Shafi T, Choi MJ, Racusen LC, et al. Ritonavir-induced acute kidney injury: kidney biopsy findings and review of the literature. Clin Nephrol 2011;75:60–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Varshney R, Murphy B, Woolington S, et al. Inactivation of platelet-derived TGF-β1 attenuates aortic stenosis progression in a robust murine model. Blood Adv. 2019;12:777–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Assessment and quantitation of renal fibrosis in mouse kidney by Picrosirius red staining. Whole kidney sections of 4μM thickness were stained with Picrosirius red and scanned using an Aperio Whole Slide Scanner (Leica Biosystems) at 20x magnification. Images were then analyzed in ImageScope using a modified “Positive Pixel Count Algorithm V9” with Hue Value of 0.38 and Hue Width of 0.72. Because large blood vessels naturally contain collagen fiber, those stained areas were deleted (black areas in the middle panel) using Photoshop or Paint programs before quantification. Fibrotic areas are depicted by the red color (red box insert), with a background threshold set such that fibrotic areas were a clearly visible (right panel, with a magnified insert in the green box). Picrosirius red labels collagen bundles red under natural light. Under polarized light these bundles appear as a mixture of green, red and yellow birefringence against a dark background and are more clearly differentiated from background. The percentage of fibrosis was calculated by dividing the fibrotic area (x) by the total area (y), then multiplying by 100.