

Abstract
Rationale:
Diabetes mellitus (DM) is a complex, multisystem disease, affecting large populations worldwide. Chronic CaMKII activation may occur in DM and be arrhythmogenic. Diabetic hyperglycemia was shown to activate CaMKII by (1) O-linked attachment of N-acetylglucosamine (O-GlcNAc) at S280 leading to arrhythmia and (2) a reactive-oxygen species (ROS) mediated oxidation of CaMKII, that can increase post-infarction mortality.
Objective:
To test whether high extracellular [glucose] (Hi-Glu) promotes ventricular myocyte ROS generation and the role played by CaMKII.
Methods and Results:
We tested how extracellular Hi-Glu influences ROS production in adult ventricular myocytes, using H2DCFDA and genetically targeted Grx-roGFP2 redox sensors. Hi-Glu (30 mmol/L) significantly increased the rate of ROS generation, an effect prevented in myocytes pretreated with CaMKII inhibitor KN-93 or from either global or cardiac specific CaMKIIδ knockout mice. CaMKII knockout or inhibition also prevented Hi-Glu induced sarcoplasmic reticulum (SR) Ca2+ release events (Ca2+ sparks). Thus, CaMKII activation is required for Hi-Glu induced ROS generation and SR Ca leak in cardiomyocytes. To test the involvement of O-GlcNAc-CaMKII pathway, we inhibited GlcNAcylation removal by Thiamet G (Thm-G), which mimicked the Hi-Glu-induced ROS production. Conversely, inhibition of GlcNAcylation (OSMI-1) prevented ROS induction in response to either Hi-Glu or Thm-G. Moreover, in a CRSPR-based knock-in mouse in which the functional GlcNAcylation site on CaMKIIδ was ablated (S280A) neither Hi-Glu nor Thm-G induced myocyte ROS generation. So CaMKIIδ-S280 is required for the Hi-Glu-induced (and GlcNAc-dependent) ROS production. To identify the ROS source(s), we used different inhibitors of NOX2 (Gp91ds-tat peptide), NOX4 (GKT137831), mitochondrial ROS (Mito-Tempo) and NOS pathway inhibitors (L-NAME, L-NIO and L-NPA). Only NOX2 inhibition or KO prevented Hi-Glu/Thm-G induced ROS generation.
Conclusions:
Diabetic hyperglycemia induces acute cardiac myocyte ROS production by NOX2 that requires O-GlcNAcylation of CaMKIIδ at S280. This novel ROS induction may exacerbate pathological consequences of diabetic hyperglycemia.
Subject Terms: Basic Science Research, Calcium Cycling/Excitation-Contraction Coupling, Cell Signaling/Signal Transduction
Keywords: Hyperglycemia, CaMKII, ROS, NOX2 complex, cell signaling, redox, cardiac myocyte
Graphical Abstract
Clinically, heart failure patients with diabetes mellitus show worse clinical outcomes than those without. Excessive ROS generation was considered a hallmark of diabetic cardiomyopathy. However, the origin of ROS generation in ventricular cardiomyocytes remained unclear. Here, we identified a novel molecular pathway by which acute hyperglycemia induces myocyte ROS generation, requiring both direct O-GlcNAcylation of Ser280 in CaMKIIδ and consequent activation of cytosolic NOX2. This finding provides foundational insight into diabetic cardiomyopathy progression.
INTRODUCTION
Globally, an estimated 422 million adults were living with diabetes in 2014, compared to 108 million in 1980. Diabetes caused 1.5 million deaths in 2012 according to WHO Global report on diabetes.1 Patients with DM also have higher propensity to develop heart failure.2–4 It is known that CaMKII, an important signaling kinase, is chronically activated in ventricular cardiomyocytes in both diseases and can directly induce pathological changes in Ca2+ handling and downstream effectors.5–9 Reactive oxygen species (ROS) signaling in DM and heart failure has also been proven to be a central node for diseases progression.10–13 Increasing evidence suggests that excessive ROS may lead to pathological conditions, such as cardiac hypertrophy, fibrosis, apoptosis and ventricular remodeling.14–17 However, the mechanisms that temporally and spatially link these two detrimental effectors remains unclear.
Our lab’s previous work has shown that CaMKII is highly GlcNAcylated in the hearts and brains of diabetic patients and rats.18 Furthermore, acute elevation of extracellular glucose concentration (Hi-Glu) caused O-GlcNAcylation of CaMKII that sensitized SR Ca2+ release channels (ryanodine receptors, RyRs) and increase the propensity for arrhythmias. While O-GlcNAcylation at a critical Ser280 on CaMKIIδ was shown to mediate this CaMKIIδ activation under hyperglycemia, CaMKIIδ autonomous activation can also be produced by other post-translational modifications (PTMs), including oxidation, nitrosylation and phosphorylation.19–23 Indeed, in preliminary experiments we found that acute Hi-Glu also promoted myocyte ROS production, raising the possibility that both Hi-Glu induced ROS and CaMKII activation could be related.
Anderson’s group previously used CaMKIIδ-MM281/282VV knock-in mice (MMVV-KI, that resist oxidation) which exhibited protection from sinoatrial node (SAN) dysfunction and atrial fibrillation in STZ-induced diabetes, suggesting oxidation of CaMKII in diabetes.8,24 Those two methionines are immediately adjacent to the Ser280 in CaMKIIδ that are O-GlcNAcylated in Hi-Glu and cause enhanced SR Ca2+ leak and arrhythmias.18 We recently made a O-GlcNAcylation resistant CaMKIIδ knock-in mouse expressing only S280A CaMKIIδ.25 These two knock-in mice can help to distinguish roles of CaMKII oxidation and O-GlcNAcylation in mediating acute effects of Hi-Glu on ROS production and SR Ca2+ leak in adult ventricular myocytes. CaMKII is also known to drive nuclear export of histone deacetylase 4 (HDAC4) in pathological nuclear signaling, but Backs’ group recently showed that O-GlcNAcylation of HDAC4 induced by Hi-Glu could partly counteract this transcriptional CaMKII effect and be protective.26 Thus, O-GlcNAcylation of CaMKII could be a potential turning point of disease progression.
We further tested the key molecular sources of ROS in this Hi-Glu-induced ROS production: NADPH oxidase (NOX) type 2 or 4 or ROS produced by mitochondria. We tested whether nitric oxide synthase (NOS) activity or direct oxidation of CaMKIIδ might be involved, because CaMKII can also be activated by direct oxidation and S-nitrosylation at C290 and MM281/282,19,23 respectively in the same regulatory domain in CaMKIIδ which are autophosphorylated and O-GlcNAcylated (Thr287 and Ser280).18 In addition, we used transgenic mice with either cytosolic- or mitochondrial-targeted redox sensors27 to test the temporal and spatial compartments of Hi-Glu induced ROS increase. Notably, both of these targeted sensors retain their rapid ROS response and wide dynamic range as described for the original sensors.28–30 This helped further distinguish between cytosolic and mitochondrial ROS production induced by acute hyperglycemia. Our results showed that acute Hi-Glu induces cardiac myocyte ROS production via O-GlcNAcylation of CaMKIIδ and consequent activation of NOX2 ROS production in the cytosol, but not in the mitochondria.
METHODS
The data and analytical methods that support the findings of this study are available from the corresponding author upon reasonable request.
Animals and cardiomyocytes isolation.
Several types of adult mice aged from 10 to 12 weeks were used, including: WT C57b/6J (Jackson Lab), WT C57b/6N (Jackson Lab), Ser280Ala KI,25,31 MMVV-KI,32 CaMKIIδ cardiac-specific and global KO,33,34 NOX2−/− mice (Jackson Lab) and Grx1-roGFP2 (targeted either to the cytosol or mitochondria).27 All the genetically modified mouse lines were bred on the C57b/6J background, except for the C57b/6N line. See Online Methods for more details.
Cardiac ventricular myocytes were isolated using previously described methods35 which were approved by the UC Davis Institutional Animal Care and Use Committee (IACUC). Freshly isolated myocytes were plated on laminin-coated glass cover slips for 30 min before dye loading. All experiments were performed at room temperature (RT) (22–23°C) with pH 7.4.
Cardiomyocyte ROS production measurement.
Freshly isolated ventricular cardiomyocytes were plated on laminin-coated glass cover slips in normal Tyrode’s buffer (NT). Intact cells were loaded with 1 μmol/L H2DCFDA (Thermo Fisher) for 10 min to measure reactive oxygen species. DCF loaded cells were imaged at 488nm using confocal 2D-scanning microscopy with (Nikon, ×40 objective) ~20 nW laser power to minimize laser induced artifacts. Cells were normally stimulated at 0.5 Hz with NT perfusion at 1 ml/min.
Ca2+ spark and transient measurements.
Intact ventricular myocytes were loaded with Fluo-4 AM dye (5 μmol/L) for 30 min, transients and sparks were recorded as previously described.6,18 Ca2+ transients were obtained by field stimulation at 0.5 Hz in normal Tyrode’s buffer. SR Ca2+ load was evaluated by Ca2+ transient amplitude induced upon rapid application of caffeine (10 mmol/L). Images were acquired with confocal microscopy (Nikon, ×40 objective) using line scan mode with excitation at 488 nm, emission at >505 nm. Image analysis used ImageJ software and Sparkmaster.36
Redox sensor measurements and EGSH calculation.
Isolated cardiac myocyte redox measurements were performed using a confocal microscope (Nikon). The roGFP2 sensor was excited at 405 and 488 nm, with emission detected at >530 nm. Images were acquired in every 20 secs. Sensor calibration was done according to a previous study with these sensors.29 Oxidation difference (OxD) and EGSH calculations were performed as described therein (see online Methods).
hiPSC-CMs redox and Ca2+ transient measurements.
Human induced pluripotent stem cell derived cardiac myocytes (iPSC-CMs) were prepared by Dr. Mercola’s lab by methods previously described.37,38 At day 25, cells were dissociated and plated onto Matrigel-coated coverslips in a 24-well plate.39 Then, hiPSC-CMs were subjected to adenoviral-mediated gene transfer of mito- and cyto-Grx1-roGFP2 and cultured for 24 hr. For Ca2+ transient measurement, cells were loaded with Fluo-4 AM dye (5 μmol/L) for 30 min and imaged with confocal microscopy (Biorad, 40X oil objective).
Solution and experimental protocols.
NT contained (mmol/L) 140 NaCl, 6 KCl, 10 HEPES, 1 MgCl2, 1.8 CaCl2 and 2, 5.5. or 30 glucose as below (RT, pH7.4). For Hi-Glu exposures, myocytes were usually paced at 0.5 Hz, and first equilibrated with NT containing 2 mmol/L glucose plus 28 mmol/L mannitol. Then an acute switch was made to NT in which the mannitol was replaced by glucose (bringing [glucose] to 30 mmol/L).
NT with 2 mmol/L glucose was chosen to minimize activation of O-GlcNAcylation at baseline, while still nourishing myocytes. To confirm that the central observations regarding HiGlu-induced ROS production occur in physiological NT ([glucose] =5.5 mM) we measured the increases in ROS production from either 2 or 5.5 mmol/L upon increase to HiGlu (30 mmol/L glucose; Online Figure 1C). The results were similar, except that when starting from 5.5 mmol/L glucose the mean ROS increases were slightly smaller. This is consistent with prior observations on the monotonic rise in CaMKII activation in going from 5 .5 to 33 mM (100–500 mg/dl).18
Protein samples of hearts were harvested after 20 min Langendorff perfusion with different treatments and frozen immediately.40
Statistics.
Pooled data are represented as the mean ± SEM. Statistical comparisons were made using unpaired Student t test, one-sample t test and ANOVA where applicable. Redox sensor calibration data was fitted with sigmoidal curve in GraphPad. P values <0.05 was considered significant.
RESULTS
Hi-Glu induces ROS production that requires O-GlcNAcylation and CaMKII activity in adult mouse ventricular myocytes.
Figure 1A shows a wild type (WT) isolated mouse ventricular myocyte loaded with H2DCFDA (DCF) to assess ROS generation during regular pacing at 0.5 Hz. DCF fluorescence (F) rises irreversibly at a linear rate at baseline during steady state in isolated myocytes, but that rate changes when ROS production is increased or decreased. We normalize the initial rate (dF/dt) after the intervention (30 mmol/L glucose in Figure 1B) to that during the first 3 min at the baseline steady state (Control). Thus, we readily detect relative changes in ROS production rate induced by acutely altered conditions.
Figure 1. Glucose induced ROS production without contractility alteration.
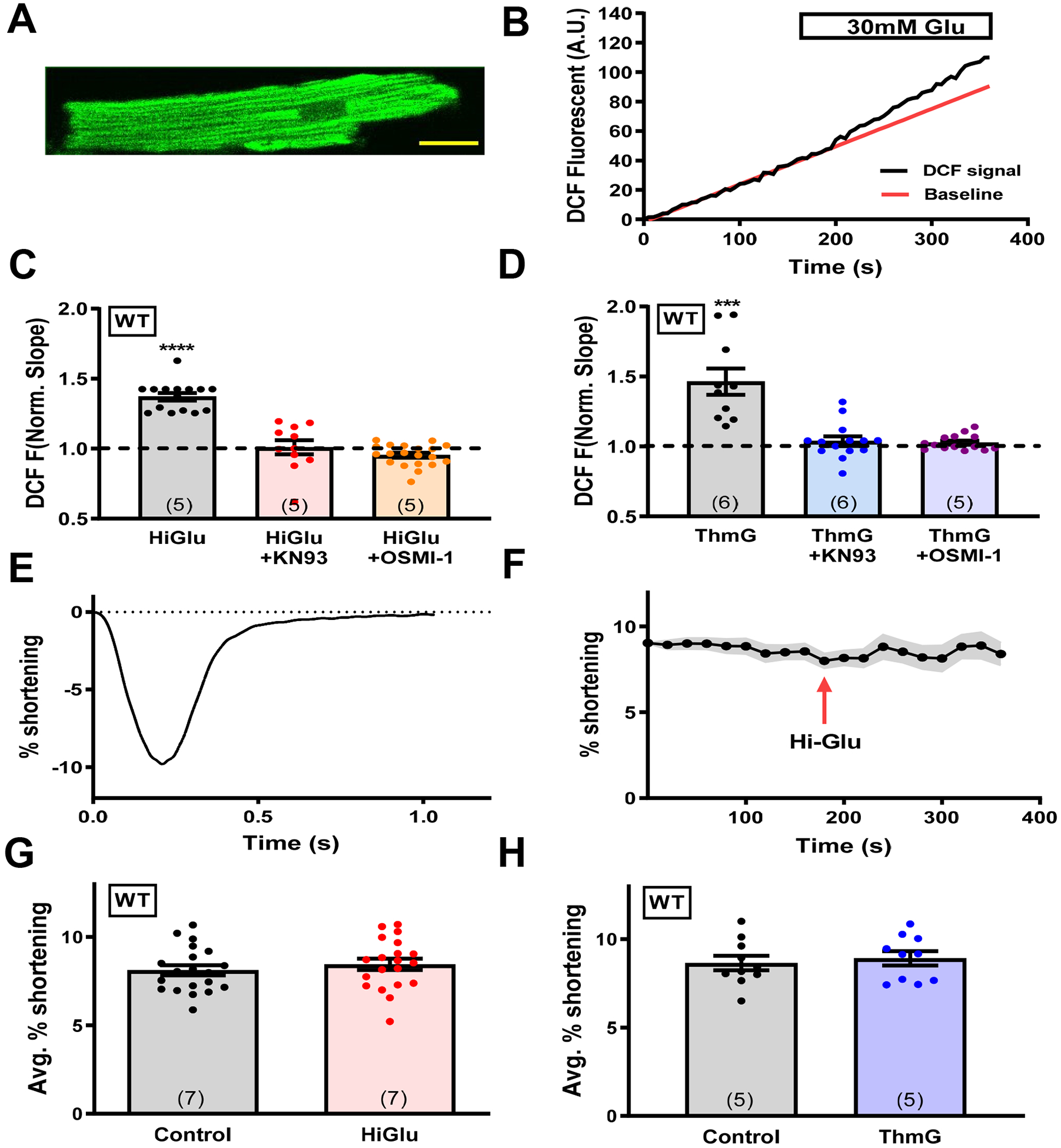
(A) Representative mouse cardiomyocyte loaded with DCF. Scale bar is 20μm. (B) DCF fluorescence change in response to Hi-Glu in myocytes. (C-D) Quantification of DCF slope in presence of Hi-Glu/ThmG with KN93 and OSMI-1, normalized to the slope prior to HiGlu or ThmG exposure. (E) Representative fractional shortening of unloaded cardiomyocytes.(F) Peak fractional shortening with Hi-Glu perfusion at 3 minutes. (G-H) Mean fractional shortening in control NT or with Hi-Glu or ThmG. (Animal number N is indicated in bars, while each individual myocyte (n) is shown as a data point, ***P<0.001, ****P<0.0001).
Increasing [glucose] from 2 to 30 mmol/L (Hi-Glu) acutely increased the rate of ROS generation by 37% vs. the Control at 2 mM glucose (p<0.05; Figure 1C). However, pre-incubation of myocytes with inhibitors of CaMKII (1 μmol/L KN-93) or O-GlcNAc transferase (50 μmol/L OSMI-1)41 for 30 min completely prevented the effect of Hi-Glu on ROS production (101% of control for KN-93, p = 0.85; 99% of control, for OSMI-1, p=0.31: Figure 1C). The pre-incubation itself did not significantly alter the Control ROS production rate, as shown for KN-93 and OSMI-1 pretreatment in Online Figure IA–B. Figure 1D shows that the Hi-Glu effect (in Figure 1C) could be quantitatively mimicked simply by blocking de-GlcNAcylation, which prevents O-GlcNAc cleavage from the target, by using Thiamet G (ThmG) in normal 2 mmol/L glucose. That is, ThmG raised the rate of ROS production by 42% (P<0.05). Furthermore, the ThmG-induced rise in ROS production was again completely prevented by either KN-93 or OSMI-1 pre-incubation (P<0.05; Figure 1D). These results suggest that both O-GlcNAcylation and CaMKII are required for the Hi-Glu-induced rise in ROS production in beating ventricular myocytes.
Hi-Glu promotes arrhythmogenic SR Ca2+ leak, without altering contraction.
To test whether Hi-Glu altered myocyte contraction, cell shortening was measured during these same protocols. Average myocyte shortening was not significantly altered by either Hi-Glu or ThmG (Figure 1E–H). In Figure 1G–H the Control values are from the same cells before the indicated HiGlu or ThmG treatment (as for normalization in Figure 1C–D), except that here the results are shown in absolute, rather than relative units. Thus, the levels of ROS achieved by the Hi-Glu exposure were not sufficient to alter myocyte contraction within this short time period. Similarly, Hi-Glu exposure did not significantly alter regular twitch Ca2+ transients amplitude or the time constant (tau) of twitch [Ca2+]i decline (Figure 2A,B), although SR Ca2+ content was significantly reduced, a scenario consistent with increased RyR sensitivity and potential diastolic SR Ca2+ leak.
Figure 2. Hi-Glu induced diastolic SR Ca2+ leak.
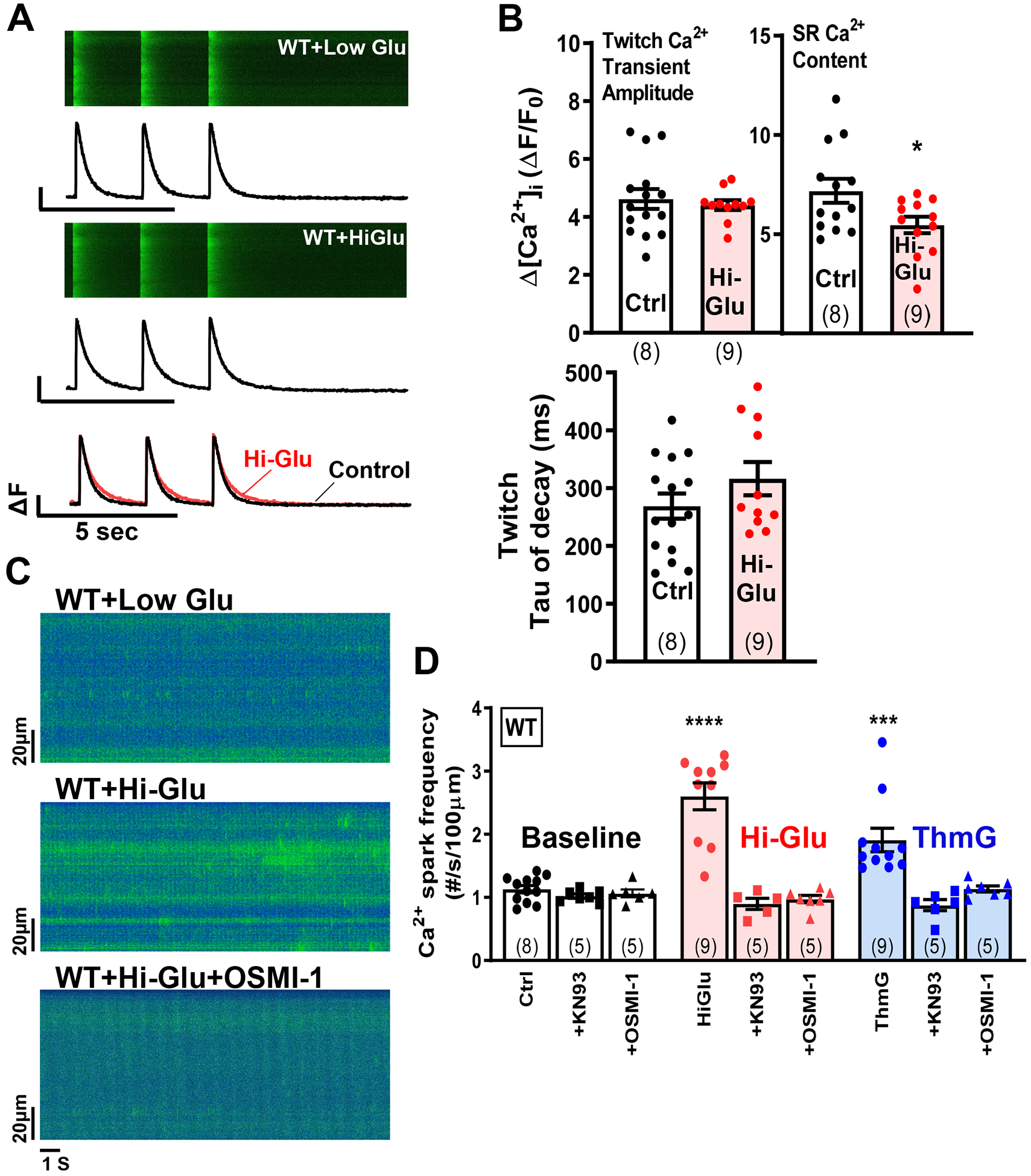
(A) Line scans show Ca2+ transients with line plots below. (B) Data show that Hi-Glu has no effect on Ca2+ transients with decreased SR load. (C-D) Hi-Glu/ThmG induced Ca2+ spark frequency is inhibited by KN93 and OSMI-1, respectively. (Animal number shown at bar, with points indicating individual myocytes; *P<0.05, ***P<0.001, ****P<0.0001)
Indeed, prior work has shown that Hi-Glu induces O-GlcNAcylation of CaMKII at Ser280 which activates CaMKII to phosphorylate RyR and enhance arrhythmogenic SR release events (Ca2+ sparks and waves at the myocyte level).18 Here we confirmed that, in mouse ventricular myocytes, Hi-Glu and ThmG both promote diastolic SR Ca2+ release events (Ca sparks), that was not a consequence of either increased [Ca2+]i or SR Ca2+ content (Figure 2C–D). Indeed, Hi-Glu or Thm-G increased Ca2+ spark frequency to 260 or 190% of control, respectively (P<0.05). As shown for ROS production in Figure 1, inhibition of either CaMKII or O-GlcNAcylation by KN-93 or OSMI-1 suppressed the Hi-Glu- or ThmG-induced enhancement of Ca2+ sparks to the baseline level (Figure 2D). Note that neither KN-93 nor OSMI-1 altered baseline Ca2+ spark frequency (Figure 2D, left), so this is consistent with a Hi-Glu-induced, O-GlcNAc- and CaMKII-dependent elevation in SR Ca2+ leak via RyR sensitization. Note also that such RyR sensitization can enhance diastolic leak and reduce SR Ca2+ content, while having little effect on Ca2+ transient properties.42 That is because the sensitized RyR may release a higher fraction of the lower SR Ca2+ content as the Ca2+ fluxes come to a new steady state (Figure 2B). ThmG also failed to alter SR Ca2+ content or steady state twitch Ca2+ transient amplitude (not shown).
Hi-Glu-induced ROS production requires myocyte Ca2+ transients and CaMKIIδ.
To test whether Ca2+ transients were required for enhanced ROS production, we tested quiescent myocytes pretreated with thapsigargin to prevent SR Ca2+ uptake and Nifedepine to prevent Ca2+ entry via L-type Ca2+ channels. Figure 3A shows that under these conditions neither Hi-Glu nor ThmG promoted ROS production, whereas normally paced myocytes done in parallel here still showed the ~34% increase in ROS production (Figure 3B). These results indicate that normal myocyte Ca2+ transients are required for this CaMKII- and GlcNAc-dependent ROS production, which is consistent with the requirement of initial CaMKII activation via Ca2+-CaM to enable autonomous CaMKII activation by O-GlcNAcylation.18
Figure 3. Acute hyperglycemia induced ROS production is Ca2+ and CaMKII dependent.
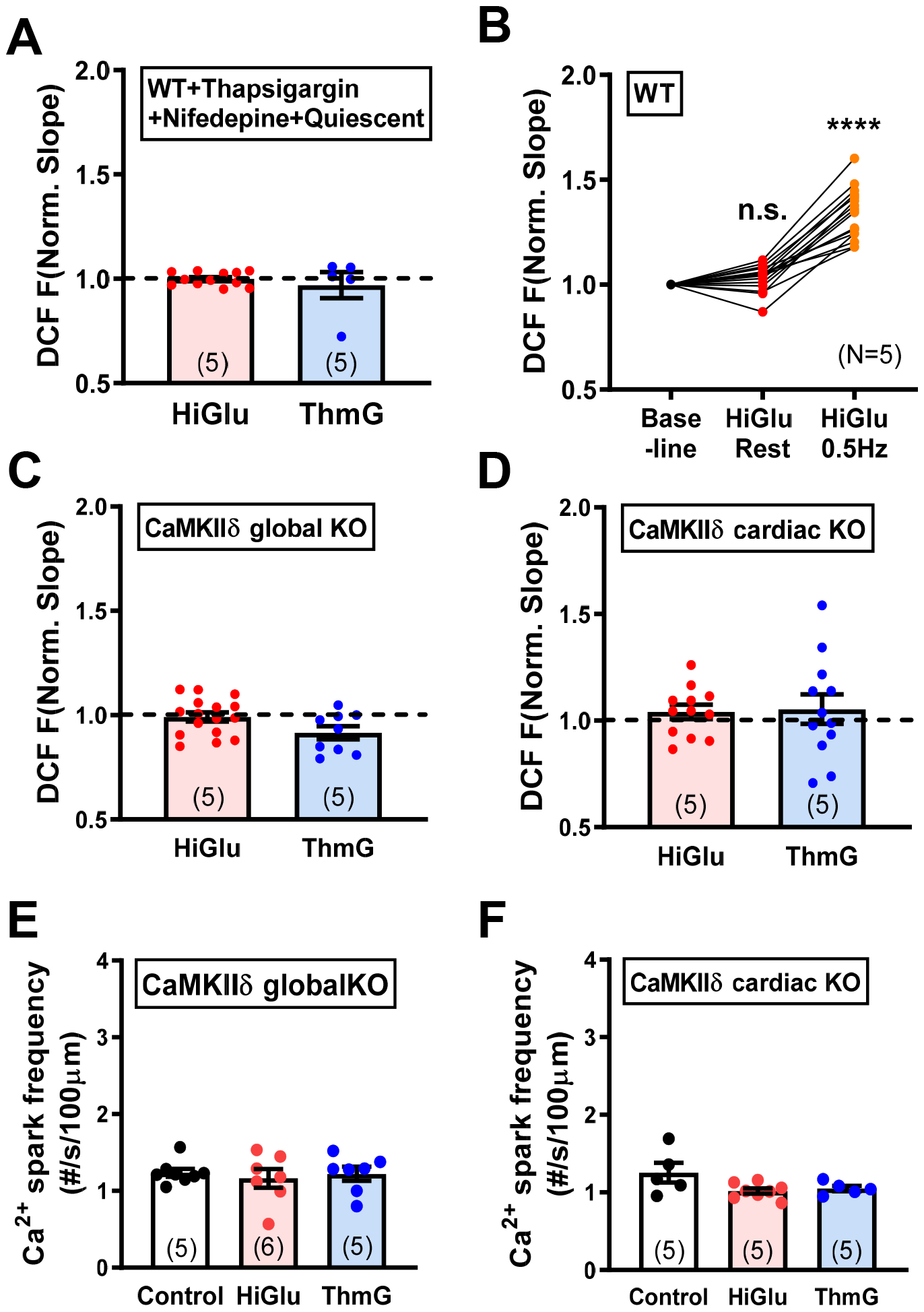
(A) Quiescent cells show no ROS accumulation in Hi-Glu/ThmG. (B) Pacing induce the ROS production with Hi-Glu. (C-D) Neither CaMKIIδ global KO nor cardiac specific KO induces ROS with Hi-Glu/ThmG. (E-F) Unaffected Ca2+ spark frequency in CaMKIIδ global- and cardiac- KO. (****P<0.0001).
The above CaMKII inhibition studies used KN-93, which inhibits CaMKII, but may also have off-target effects.43 Figure 3C–F shows that genetic deletion of CaMKIIδ in adult either globally (Figure 3C) or specifically in cardiac myocyte (Figure 3D) also completely prevented both the Hi-Glu- and ThmG-induced rise in myocyte ROS production, as well as their activation of Ca2+ sparks (Figure 3E–F) or effects on Ca2+ transients (Online Figure II). Thus, the endogenous CaMKIIδ isoform is specifically required to mediate this, and that the cardiac myocyte-specific CaMKIIδ-KO sufficed. So autonomous cardiomyocyte CaMKIIδ activation via GlcNAcylation is required for Hi-Glu induced ROS generation.
CaMKII S280 is required for Hi-Glu- and ThmG-induced ROS and Ca2+ sparks.
We recently generated a CRISPR-based knock-in mouse in which the functional O-GlcNAcylation site on CaMKIIδ was ablated (S280A).25 This allowed us to further test whether that specific CaMKIIδ site was required for the Hi-Glu-induced ROS production. Indeed, the CaMKIIδ-S280A knock-in entirely prevented both the Hi-Glu and Thm-G induced ROS production and Ca2+ sparks (Figure 4A–B) without significant perturbation on Ca2+ transient and SR load (Online Figure IIIA–D). Thus, this single O-GlcNAcylation site (S280) on CaMKIIδ is required for the Hi-Glu-induced ROS production and RyR sensitization. In contrast, robust ROS production was still induced by acute angiotensin II (Ang II) exposure, even in the S280A knock-in mice. Thus, Ang II (unlike Hi-Glu or ThmG) does not require O-GlcNAcylation of CaMKIIδ at Ser280 to increase myocyte ROS production. This also indicates that the myocyte ROS generation machinery is still present in the S280A knock-in mouse. Immunoprecipitation of CaMKII O-GlcNAcylation was further performed in the control and Hi-Glu perfused hearts (Online Figure IIIE). Figure 4C shows that the Hi-Glu induced O-GlcNAcylation of CaMKII was prevented in S280A mice.
Figure 4. CaMKIIδ Ser280 instead of Met281/282 is responsible for Hi-Glu induced ROS production.
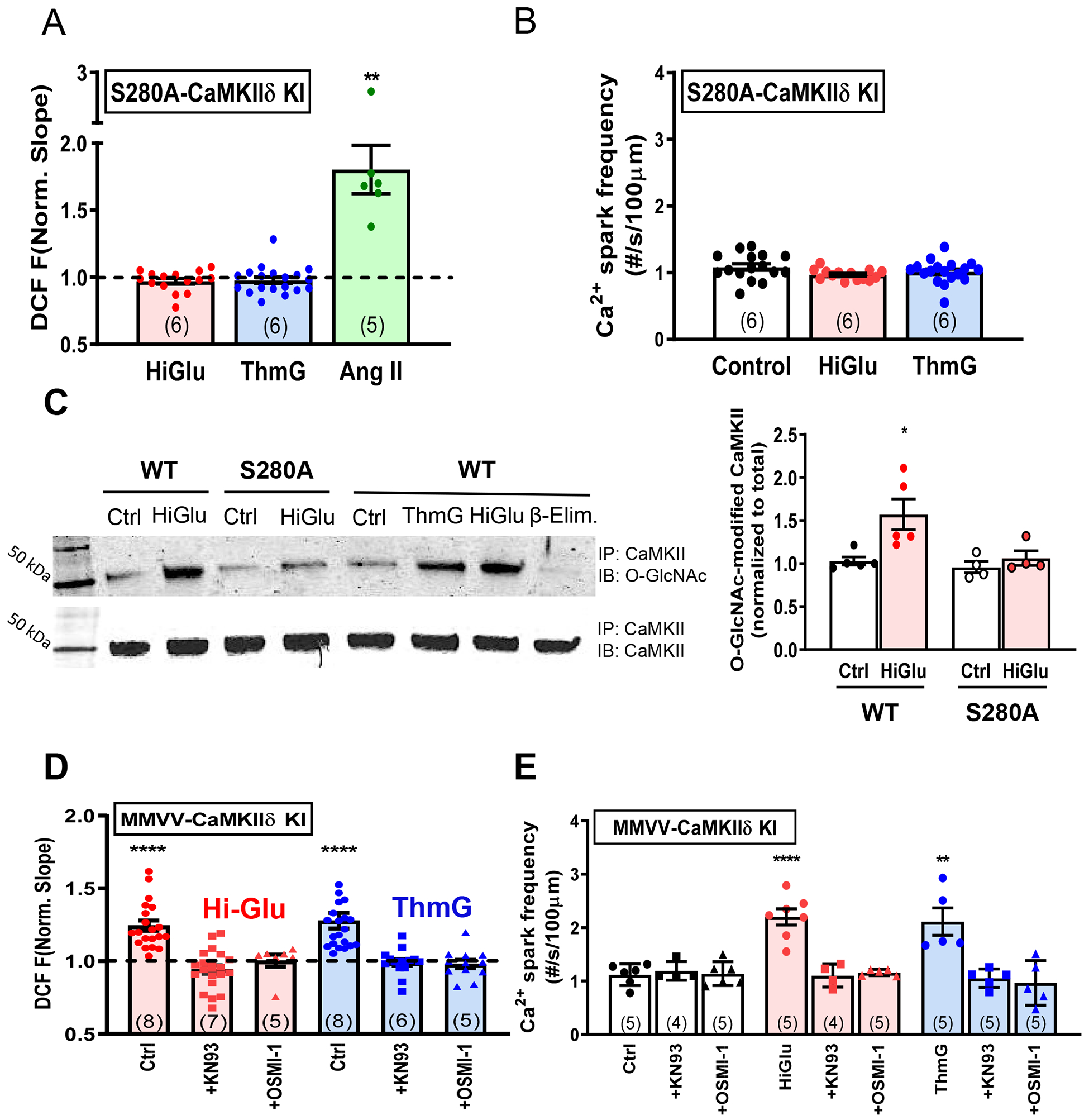
(A) S280A blocks the Hi-Glu/ThmG induced ROS, but AngII could still induce ROS production. (B) Ca2+ spark frequency remains unaffected in Hi-Glu/ThmG in S280A KI.(C) Immunoblot (IB) with O-GlcNAc-specific and CaMKIIδ antibodies show that Hi-Glu increases O-GlcNAcylation, but not in S280A CaMKII. IP, immunoprecipitation. (D)MMVV-CaMKIIδ fails to prevent Hi-Glu/ThmG induced ROS, which could be inhibited via KN93/ OSMI-1. (E) MMVV KI shows similar response to Hi-Glu/ThmG as WT in Ca2+ spark frequency. (*P<0.05, **P<0.01, **** P<0.0001).
We also used mice in which the oxidation-sensitive Met281/282 in CaMKIIδ were replaced by valines,8 because oxidation of these sites can also promote CaMKIIδ autonomy and might be critical to or a consequence of the Hi-Glu-induced ROS production. Figure 4D shows that Hi-Glu and ThmG both still promoted ROS production in MMVV-KI myocytes, and that was sensitive to inhibition of either CaMKII (KN-93) or O-GlcNAc transferase (OSMI-1). As in WT myocytes, Ca2+ spark frequency was also enhanced in MMVV-KI myocytes by Hi-Glu or ThmG, and this effect was again suppressed by KN-93 or OSMI-1 (Figure 4E). The stimulated Ca2+ transients and SR Ca2+ content in MMVV-KI myocytes were not significantly perturbed by Hi-Glu or ThmG (Online Figure IV). Thus, CaMKIIδ Met281/282 are not required for the Hi-Glu-induced ROS production or Ca2+ sparks in WT myocytes. However, the average increases of DCF dF/dt was slightly lower in MMVV than those observed in WT myocytes (24.4% vs. 37.2%). Thus, we cannot rule out the logical possibility that the CaMKIIδ O-GlcNAcylation induced ROS production also recruits CaMKIIδ oxidation to further strengthen the net CaMKII activation.
Cytosolic NOX2 is the ROS source recruited by Hi-Glu and Thm-G.
To determine the source of ROS, mitochondria and other cytosolic ROS generators were tested during acute hyperglycemia. Myocytes were pretreated with a cell-permeant NOX2 inhibitory peptide (GP91ds-tat) or a scrambled version (scrGP91ds-tat) for 30 min prior to exposure to Hi-Glu or Thm-G. NOX2 inhibitory peptide prevented the enhanced ROS production that was still observed with the scrambled peptide version (Figure 5A) and comparable to that seen in untreated WT myocytes. The membrane permeable ROS scavenger (TEMPOL) also prevented HiGlu and ThmG induced ROS production (Online Figure VE).
Figure 5. NOX2 complex inhibition could prevent Hi-Glu induced ROS generation.
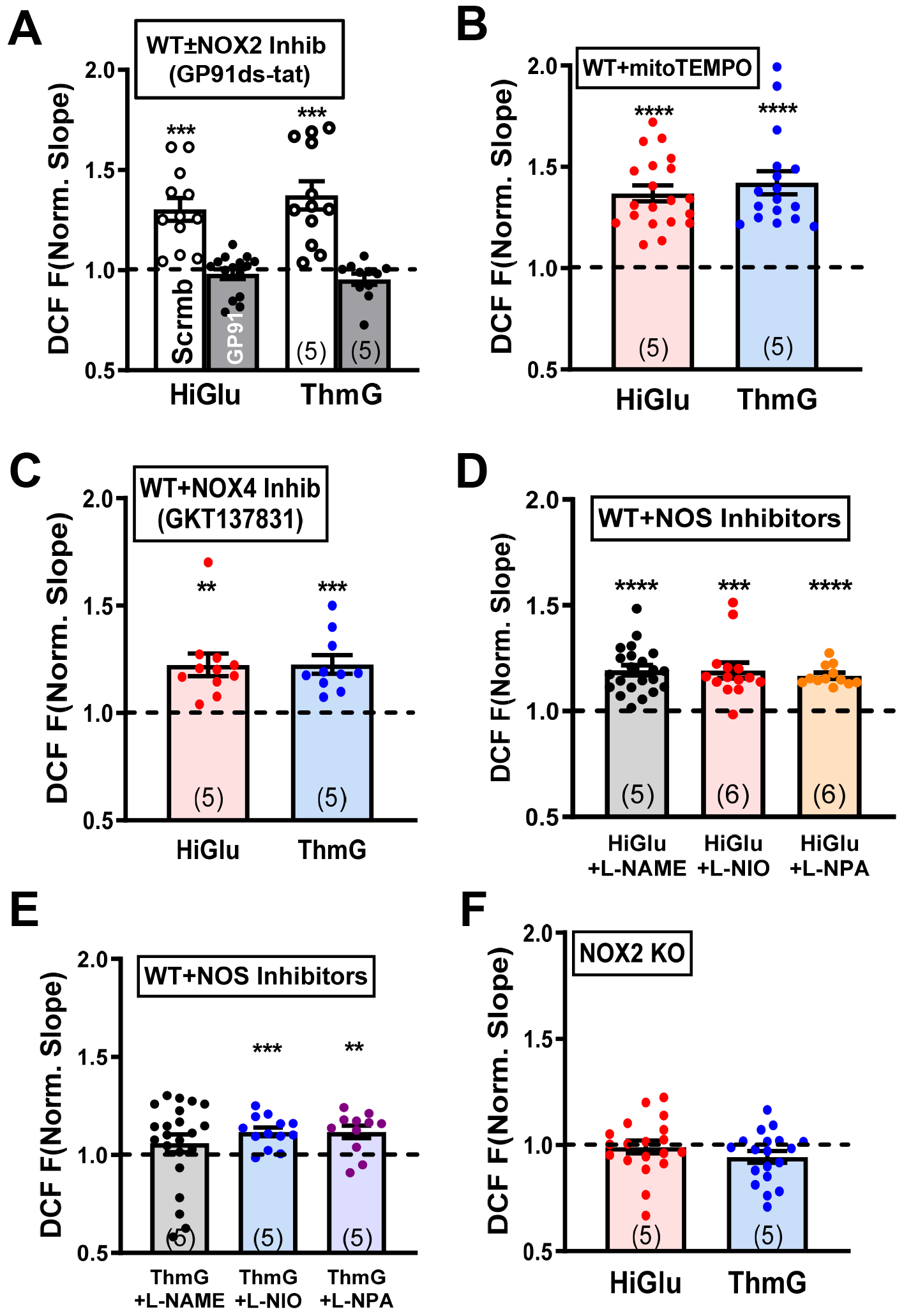
(A) NOX2 inhibitor blocks excessive ROS compared to scrambled peptide control. (B-E) Quantification of DCF slope change in WT with mitoTEMPO, GKT137831 and NOS inhibitors, respectively. (F) No obvious ROS generation under hyperglycemia in NOX2 KO. (**P<0.01, ***P<0.001,****P<0.0001).
Mitochondria are a major source of ROS production in cardiomyocytes. To target mitochondrial ROS, we used pre-incubation with the antioxidant MitoTEMPO. Figure 5B shows that MitoTEMPO did not prevent the increased ROS induced by acute Hi-Glu or ThmG. It was recently shown that C57BL/6J mice can be protected from pathological workload-related mitochondrial ROS production, because this strain lacks nicotinamide nucleotide transhydrogenase (NNT) and thus prevents backward flux (NADPH to NADH) and consequent ROS enhancement.44 So, we tested whether the presence of NNT in the C57BL/6N strain might reveal a mitochondrial ROS contribution to HiGlu or ThmG induced overall ROS production. Online Figure V (E) shows that the the HiGlu and ThmG responses were almost identical in C57BL/6N as in C57BL/6J (Figure 5A–B), including the full ROS inhibition by GP91ds-tat and lack of inhibition by MitoTEMPO. These results suggest that mitochondrial ROS production was not triggered by HiGlu. We also tested the involvement of NOX4 in this pathway, using the NOX4 inhibitor GKT137831 (Figure 5C), and also for NOS involvement using NOS, NOS1 and NOS2 selective inhibitors (L-NAME, L-NIO and L-NPA, respectively; Figure 5D–E). None of these inhibitors prevented the Hi-Glu- or ThmG-induced increase in ROS production, although L-NAME effects after ThmG were inconclusive. Thus, while NOS-related modulation of ROS production cannot be excluded, the effects are likely minor compared to NOX2. Neither GP91ds-tat or MitoTEMPO altered the effect of Hi-Glu on Ca sparks or Ca transient properties Online Figure V (E).
Since inhibitor studies suggested that NOX2 was the main ROS source, we further tested that in NOX2 KO mice (from Jackson Labs). Indeed, Figure 5F shows that neither Hi-Glu nor ThmG were able to increase ROS production in NOX2-KO mouse myocytes. Notably though, Ca2+ spark frequency was still strongly increased with Hi-Glu or Thm-G in NOX2-KO myocytes without alteration in SR Ca2+ content or Ca2+ transient amplitude (Figure 6A–C) very much like WT myocytes. As in WT myocytes, there was a hint of slowed twitch Ca2+ transient decline in NOX2-KO myocytes (Figures 6D vs. 2B), which could be a consequence of SR Ca2+ leak.45 Thus, the Hi-Glu-induced enhancement of RyR function that depends upon O-GlcNAcylation and activation of CaMKII remained intact in NOX2 KO mice. This suggests a pathway in which the CaMKII activation by O-GlcNAcylation is upstream of the NOX2 activation and that the ROS from NOX2 exerts little extra CaMKII activation.
Figure 6. Ca2+ sparks and SR Ca2+ handling in NOX2 KO.
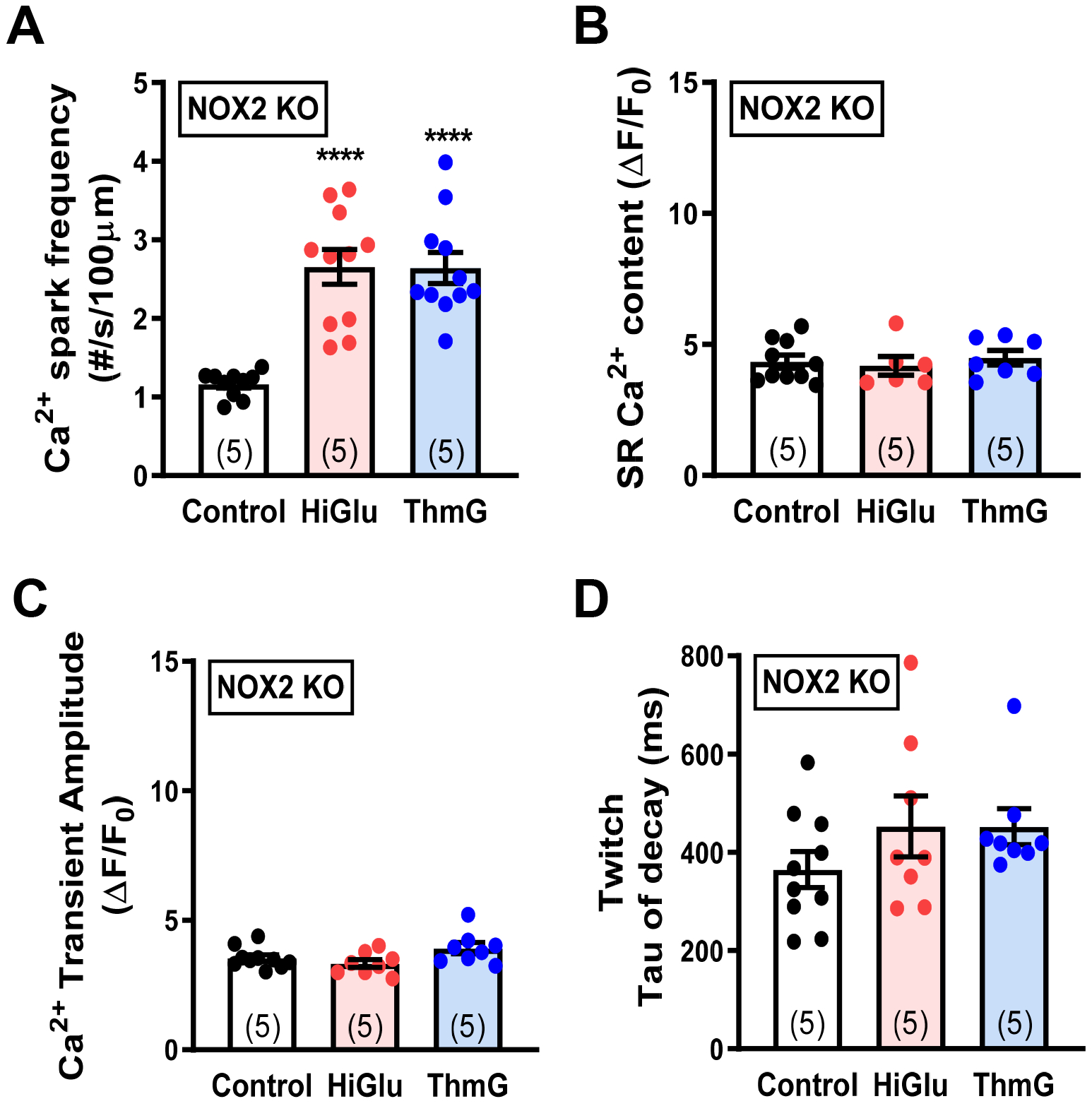
(A) Diastolic Ca2+ spark frequency in cardiomyocytes that were treated with control, Hi-Glu and ThmG. (B-D) SR Ca2+ content measurement, Ca2+ transient amplitude and twitch tau of decay are stable under treatments. (****P<0.0001).
Genetically encoded redox sensors confirm Hi-Glu-induced oxidative stress.
To confirm that the ROS measured by DCF (above) are observable using an independent sensor, we used transgenic mice with cardiac specific expression of a genetically encoded redox reporter Grx-roGFP2 that is targeted to either the mitochondrial matrix or cytosol (mito- and cyto-Grx1-roGFP2).27 First, we calibrated the Grx1-roGFP2 sensors in our imaging system. Figure 7–B demonstrates appropriate localization of the mitochondrial and cytosolic sensors and similar EC50 of cyto- and mito-roGFP2 (26.6 and 15.3 μM H2O2, respectively). In myocytes expressing the mitochondrial roGFP2 sensor, neither Hi-Glu nor ThmG caused a significant increase in mitochondrial oxidation levels (Figure 7C). In contrast, the cytosolic roGFP2 sensor sensed a substantial increase in oxidative stress in response to both Hi-Glu and ThmG (Figure 7D). The genetically encoded sensors also made it more feasible to measure ROS at physiological temperature. Online Fig VIA–B show results at 35°C which qualitatively recapitulate those in Figure 7, showing that HiGlu only caused a rise in ROS in the cytosol and not in mitochondria. At 35°C the rise in cytosolic ROS signal was much faster, and reached a lower amplitude, both of which may be attributable to faster enzymatic activity that produce and limit net ROS levels. Online Fig VIA–B also showed that pretreatment with either KN93 or OSMI-1 prevented the HiGlu-induced ROS increase. These observations confirm the above DCF results where the HiGlu-induced ROS production require both CaMKII and O-GlcNAcylation. Online Figure VI(C) also shows that the increase in ROS is graded as a function of extracellular [glucose], as previously shown for CaMKII activation by HiGlu.18
Figure 7. Redox sensor evaluation of ROS production in isolated myocytes.
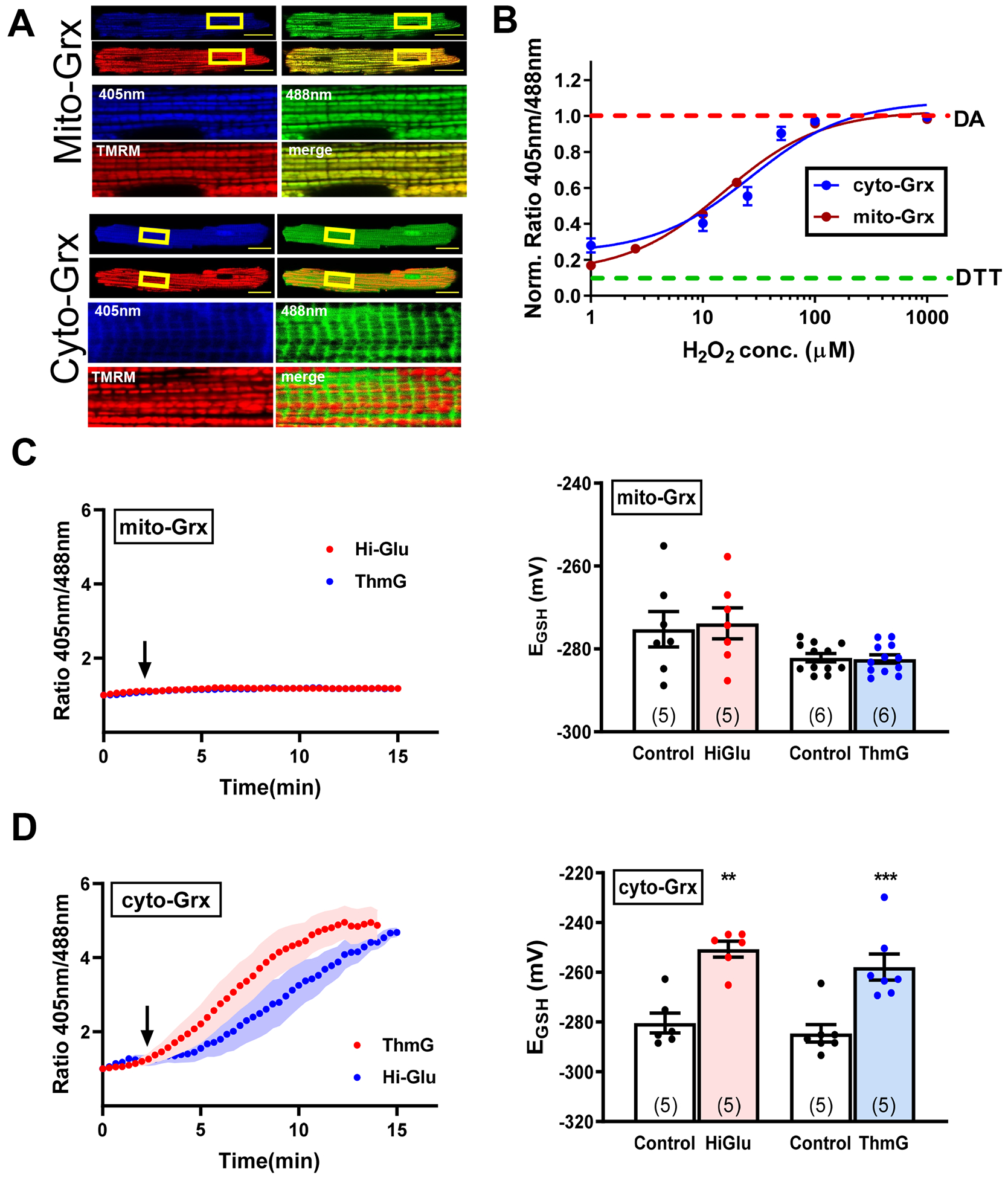
(A) Representative confocal images of myocytes expressing cyto- and mito-Grx redox sensors with 20 μm scale bar. (B) In situ redox sensors calibration. (C) Representative traces of mito-Grx ratio metric signal and converted EGSH in response to Hi-Glu/ThmG. (D) Representative traces of cyto-Grx ratio metric signal and converted EGSH in response to Hi-Glu/ThmG. (**P<0.01, ***P<0.001).
These Grx1-roGFP2 sensors also provide more dynamic and specific glutathione redox potential (EGSH) information in intact cardiomyocytes during Hi-Glu treatment. Initial mitochondrial EGSH was –280±0.6mV and –284±1.7mV at RT and 35°C, respectively (see Online Methods). Mito-Grx-roGFP2 also showed no change in EGSH upon Hi-Glu or Thm-G perfusion, while significant increases were seen with Cyto-Grx-roGFP2 at either RT (Figure 7D) or 35°C and were prevented by KN-93 or OSMI-1 (Online Figure VIA–B).
Human iPSC-CM exhibit Hi-Glu-induced oxidative stress and SR Ca2+ leak.
We also tested whether Hi-Glu would increase ROS production in human iPSC-CMs in which cyto-Grx1-roGFP2 was adenoviraly expressed (Figure 8A). With acute Hi-Glu exposure these iPSC-CMs showed an increased oxidative stress signal within 15 min (Figure 8B). Consistent with observations in adult mouse cardiomyocytes, Figure 9C shows that the Hi-Glu induced ROS increase in hiPSC-CMs was prevented by inhibiting either CaMKII or O-GlcNAcylation (by KN93 and OSMI-1, respectively).
Figure 8. ROS generation and SR Ca2+ handling in hiPSC-CMs.
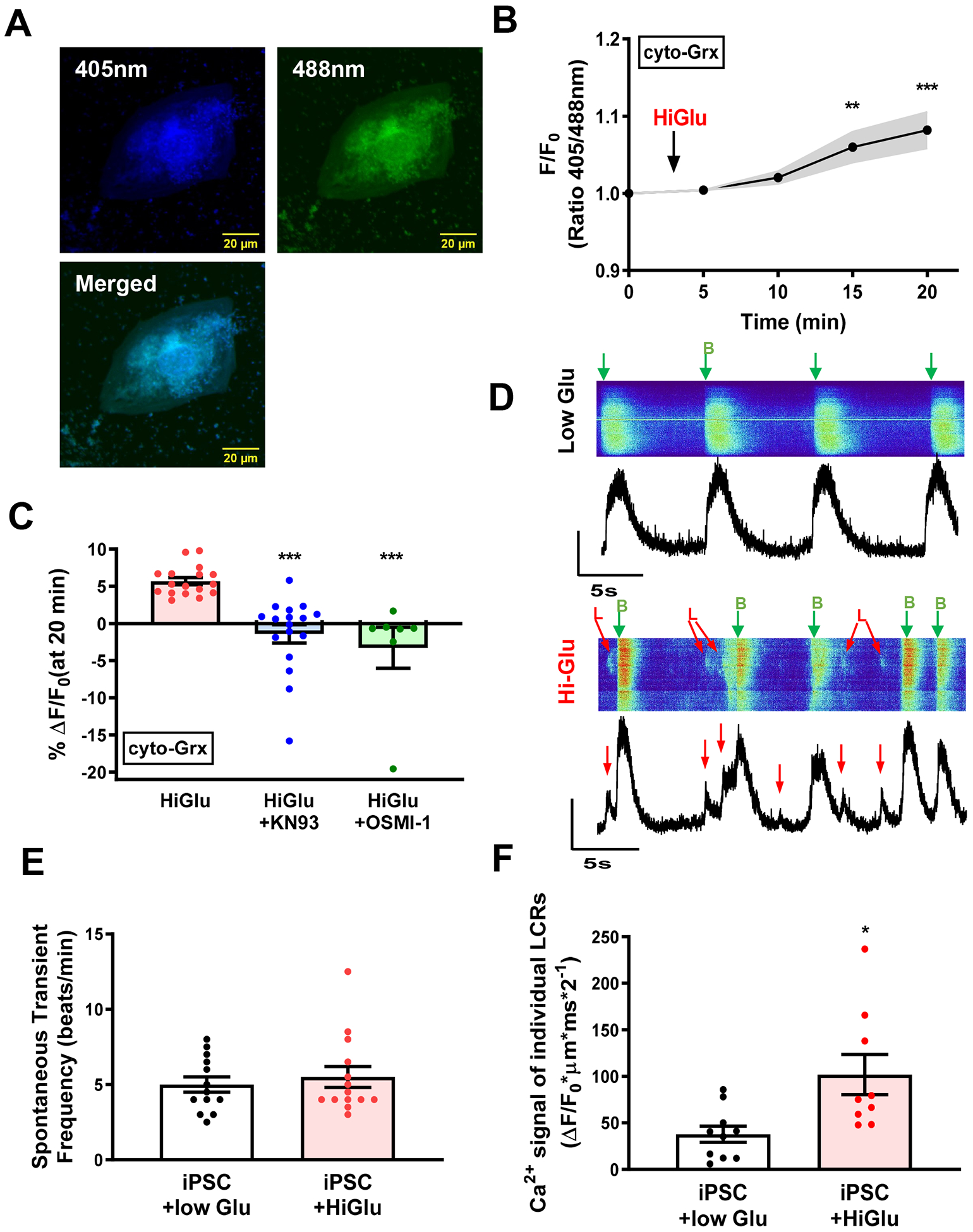
(A) Representative confocal images of hiPSC-CMs expressing adenovirus carried cyto-Grx redox sensor. (B) Average ratio metric change with Hi-Glu. (C) Hi-Glu induced ROS production could be inhibited by KN93 and OSMI-1, respectively. (D) Line-scan image of Ca2+ transients and LCRs. B, Transient; L, LCRs. (E) No change of spontaneous transient frequency with Hi-Glu. (F) Hi-Glu induced LCRs in hiPSC-CMs. (*P<0.05, **P<0.01, ***P<0.001)
Figure 9. Schematic representation of the proposed model.
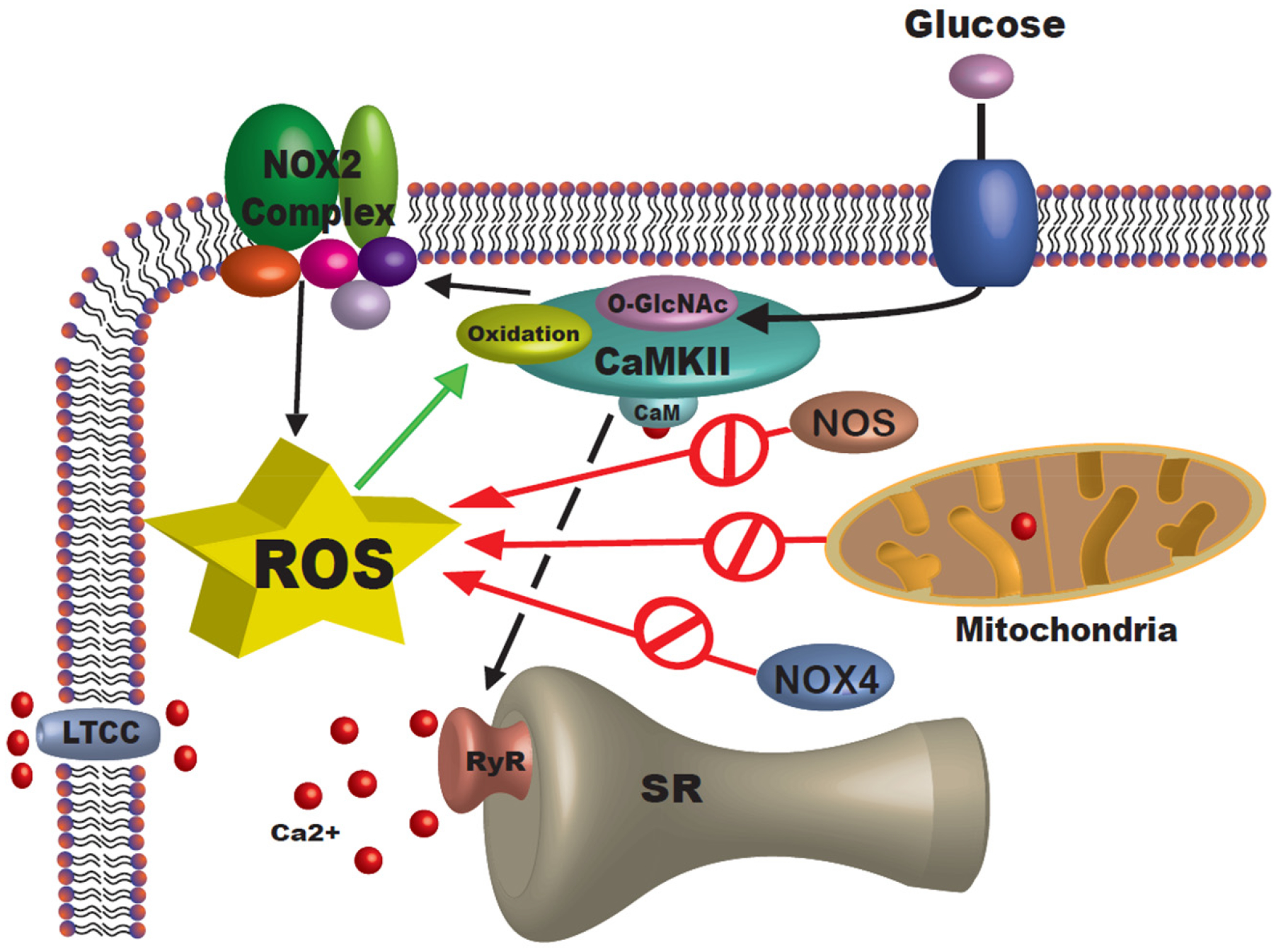
Cardiomyocytes submerged in acute Hi-Glu will trigger O-GlcNAc and activation of CaMKII. This will lead to NOX2 complex activation and increase cytosolic ROS accumulation.
We also assessed Ca2+ transients and local Ca2+ release events using Fluo 4-AM loaded iPSC-CMs (Figure 8D). The spontaneous overall beating rate (labeled with B and green arrows in Figure 8E) was not altered by Hi-Glu. These may represent the intrinsic pacemaking activity inherent in such iPSC-CMs. However, the frequency of local Ca2+ sparks and waves (labeled with L and red arrows in Figure 8D) was higher in Hi-Glu exposed iPSC-CMs (Figure 8F). The Hi-Glu induced CaMKII-dependent ROS production correlates with these local SR Ca2+ leak events (but not the apparent pacemaker rate). We conclude that the same Hi-Glu-CaMKII-ROS signaling pathway described in detail above for mouse myocytes is likely to be present in human iPSC-CM and adult myocytes.
DISCUSSION
CaMKII has been identified as a key regulator in cardiac physiology and pathology. Its upregulation and post-translational modification has been shown to be associated with cardiac pathology.22 In this study, we demonstrate that acute hyperglycemia drives O-GlcNAcylation of CaMKII (at S280) causing NOX2 activation and cytosolic ROS generation (Figure 9). Cardiomyocytes isolated from different mice were studied with traditional ROS indicator DCF. Cells showed nearly instantaneous ROS accumulation during acute Hi-Glu/ThmG challenge. This fast response is consistent with previous FRET-based measurements of rapid intracellular [glucose] changes upon Hi-Glu (τ ~11 sec).46 Meanwhile, myocyte contractility was unaltered by this short duration exposure to Hi-Glu or Thm-G, consistent with multicellular data in working preparatios.47 This also suggests that HiGlu is unlikely to alter myofilament properties on this time scale, but could during more prolonged Hi-Glu via other signaling cascades.47,48 Hi-Glu also had remarkably little effect on electrically evoked myocyte Ca2+ transients, but strongly promoted arrhythmogenic diastolic SR Ca2+ release, which also slightly reduces SR Ca2+ content. So, while systolic SR Ca2+ release may be normal, it may result from a larger fractional SR Ca2+ release, an intrinsic form of autoregulation that stabilizes E-C coupling,42 but this may be at the cost of higher arrhythmogenic propensity.18
DCF is a widely used ROS indicator, but requires careful use in the myocyte setting, and is neither reversible nor practically calibratable. Therefore, we also used genetically encoded redox sensors (Grx1-roGFP2) to confirm the Hi-Glu and Thm-G induced ROS production in both adult cardiac myocytes and human iPSC-CMs. This also confirmed that the Hi-Glu/ThmG induced ROS production occurs in the cytosol and not mitochondria in adult mouse myocytes and that CaMKII and O-GlcNAcylation are required.
In our study, both chemical inhibitors and transgenic knock-in mice provide consistent evidence for O-GlcNAc-CaMKII induced ROS generation. Ser280 on CaMKII, rather than its neighboring Methionine pair (281/282), is the main site responsible for the Hi-Glu induced ROS increase. Indeed, MMVV-KI mice, which suppress oxidative activation of CaMKII,19 failed to prevent the Hi-Glu-induced ROS rise in ventricular cells. WT littermate controls for these KI mice behaved like the other WT mice (Online Figure IIIF), helping to rule out off-target effects in both KI strains. While the O-GlcNAcylation at S280 in CaMKIIδ is compelling as the initiator of ROS production via NOX2, that ROS would also be expected to oxidize MM281/282 on CaMKIIδ to amplify both SR Ca2+ leak and further ROS production. We saw a hint of evidence for this synergy in that the Hi-Glu/Thm-G induced ROS was slightly lower in MMVV-KI than WT mice (Figure 4D vs. Figure 1C–D), but were frankly surprised that this feedback effect was not stronger.
Once activated and O-GlcNAcylated, CaMKII can not only phosphorylate numerous ion channels and transporters,9 it also appears to activate NOX2. NOX2 has multiple candidate subunits, each of which could be targets for direct phosphorylation49 and possibly by CaMKII. However, the molecular mechanism by which CaMKII may activate NOX2 is not known and potentially complex.
Interestingly, mitochondrial ROS did not change acutely in response to Hi-Glu (Figure 7, Online Figure VIB). That would be consistent with sarcolemmal NOX2 being the ROS source, and with the mitochondria maintaining their own redox-optimized ROS balance, that Aon et al. have described to maximize energy output while limiting ROS accumulation.50 That is, mitochondria may have substantial redox buffering capacity, such that the Hi-Glu induced ROS produced by NOX2 is not paralleled in the mitochondria. Taking both mitochondrial and cytosolic redox status into consideration, it is possible that Hi-Glu-induced cytosolic ROS could eventually initiate ROS induced ROS release (RIRR) and excessive ROS accumulation at longer times.
Anderson and colleagues have highlighted the importance of CaMKIIδ oxidation at MM281/282 in multiple cardiac pathologies in which the MMVV-KI CaMKIIδ has been protective.19,24,51 Our data here shows that acute hyperglycemia causes CaMKIIδ O-GlcNAcylation at S280 and consequent NOX2-dependent ROS production complements that work. It also demonstrates a strong connection between pathological CaMKII activation by O-GlcNAcylation and oxidation at three adjacent amino acids in this key regulatory domain of CaMKII. Further work will be required to more clearly elucidate how these post-translational modifications of CaMKII synergize in acute and/or chronic conditions. For example, the acute O-GlcNAcylation effect we report here may synergize with or even shift to a predominantly MM281/282 oxidized state in chronic disease states.
We also confirmed here our prior observations that Hi-Glu exposure enhances arrhythmogenic SR Ca2+ release via O-GlcNAcylation of CaMKII and consequent phosphorylation of RyRs.18 Moreover, here we show that this Hi-Glu (and Thm-G) induced SR Ca2+ leak is prevented by a single point mutation in CaMKIIδ (S280A) as well as global and cardiac-specific CaMKIIδ knockout, but not by the CaMKIIδ MMVV-KI, NOX2-KO or inhibition of NOX4 or mitochondrial ROS production. We showed that the direct O-GlcNAcyl transferase inhibitor OSMI1 prevents this effect and extends prior studies that relied on the inhibitor DON, which works a step upstream of the OSMI-1 target and can exhibit off-target effects on mitochondria.52 We also showed here that human iPSC-CMs also exhibit this same acute Hi-Glu induced ROS production and SR Ca2+ release events and are inhibited by KN-93 and OSMI-1. This suggests that this same arrhythmogenic pathway may also exist in adult human myocytes,53 but the immature phenotype of iPSC-CMs suggest that further study is required.54
Our working model of the Hi-Glu induced O-GlcNAc-CaMKII-NOX2-ROS pathway provides some of the molecular details that may help further test the importance of this pathway in the context of DM, where even acutely uncontrolled hyperglycemia could predispose patients to some increased oxidative stress as well as CaMKII-dependent mechanisms of arrhythmias and transcriptional regulation that can contribute to progresssion of heart failure.9,26,55 Indeed, the chronic CaMKII activation known to occur in heart failure may well synergize with this hyperglycemic effect in an increasing number of patients that have both heart failure and DM. While this study focuses on acute hyperglycemia, its specific role in these complex chronic disease co-morbidities will require future study.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
Diabetic hyperglycemia induces both excessive reactive oxygen species (ROS) and autonomous CaMKII activation which can be arrhythmogenic.
ROS generation and chronic CaMKII activation in cardiomyocytes play detrimental roles in the pathophysiology of cardiac remodeling and heart failure.
What New Information Does This Article Contribute?
We show that acute hyperglycemia promotes substantial ROS production in adult cardiac myocytes and arrhythmogenic sarcoplasmic reticulum Ca leak.
Hyperglycemia-induced myocyte ROS production requires direct O-linked attachment of N-acetylglucosamine (O-GlcNAc) at Serine 280 of CaMKIIδ and consequent activation of NOX2 (NADPH oxidase 2).
We identify a novel direct mechanistic pathway by which hyperglycemia activates cardiac myocyte ROS production.
ACKNOWLEDGMENTS
We thank Mark E. Anderson (Johns Hopkins University) for providing CaMKIIδ MMVV knock-in mice, Brian O’Rourke (Johns Hopkins University) for providing vectors for the Grx-ro-GFP reporter that we expressed in human iPSC-CMs, Dries Feyen (Stanford University) who helped with iPSC-CMs and Kenneth Ginsburg (University of California, Davis) for help with data analysis.
SOURCES OF FUNDING
Supported by NIH grants R01-HL030077 (DMB), R01-HL132831 (DMB and JDM) and R01-HL142282 (DMB and JB) and a Predoctoral Fellowship from the American Heart Association #17PRE33670858 (SL).
Nonstandard Abbreviations and Acronyms:
- Ca2+
Calcium
- CaMKII
Ca2+/calmodulin-dependent kinase II
- DCF
2’,7’-dichlorodihydrofluorescein diacetate
- Grx1
Glutaredoxin1
- Hi-Glu
High glucose (30 mM glucose)
- hiPSC-CM
Cardiomyocytes derived from human induced pluripotent stem cells
- KN93
2-[N-(2-hydroxyethyl)]-N-(4-methoxybenzenesulfonyl)]amino-N-(4-chlorocinnamyl)-N-methylbenzylamine)
- mitoTEMPO
(2-(2,2,6,6-Tetramethylpiperidin-1-oxyl-4-ylamino)-2-oxoethyl)triphenylphosphonium chloride
- MMVV
CaMKII with mutated Met281Val and Met282Val
- NOS
Nitric oxide synthase
- NOX
NADPH oxidase
- O-GlcNAc
β-linked N-acetylglucosamine
- OSMI-1
(αR)-α-[[(1,2-Dihydro-2-oxo-6-quinolinyl)sulfonyl]amino]-N-(2-furanylmethyl)-2-methoxy-N-(2-thienylmethyl)-benzeneacetamide
- PTMs
Post translational modifications
- roGFP
reduction oxidation sensitive green fluorescent protein
- ROS
Reactive oxygen species
- RyR
Ryanodine receptor
- S280A
CaMKII with mutated Ser280Ala
- SR
Sarcoplasmic reticulum
- TEMPOL
4-hydroxy-2,2,6,6-tetramethylpiperidin-1-oxyl
- Thm-G
2-(ethylamino)-5-(hydroxymethyl)-5,6,7,7a-tetrahydro-3aH-pyrano[3,2-d][1,3]thiazole-6,7-diol
- WT
Wild-type
Footnotes
DISCLOSURES
None.
REFERENCES
- 1.Abhary S, Hewitt AW, Burdon KP, Craig JE. A systematic meta-analysis of genetic association studies for diabetic retinopathy, Diabetes. 2009;58:2137–2147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.MacDonald MR, Petrie MC, Varyani F, Ostergren J, Michelson EL, Young JB, Solomon SD, Granger CB, Swedberg K, Yusuf S, Pfeffer MA, McMurray JJ. Impact of diabetes on outcomes in patients with low and preserved ejection fraction heart failure: An analysis of the candesartan in heart failure: Assessment of reduction in mortality and morbidity (charm) programme. Eur Heart J. 2008;29:1377–1385 [DOI] [PubMed] [Google Scholar]
- 3.Fadini GP, Avogaro A, Degli Esposti L, Russo P, Saragoni S, Buda S, Rosano G, Pecorelli S, Pani L. Risk of hospitalization for heart failure in patients with type 2 diabetes newly treated with dpp-4 inhibitors or other oral glucose-lowering medications: A retrospective registry study on 127,555 patients from the nationwide osmed health-db database. Eur Heart J. 2015;36:2454–2462 [DOI] [PubMed] [Google Scholar]
- 4.Rosano GM, Vitale C, Seferovic P. Heart failure in patients with diabetes mellitus. Cardiac failure review. 2017;3:52–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Anderson ME. Multiple downstream proarrhythmic targets for calmodulin kinase ii: Moving beyond an ion channel-centric focus. Cardiovasc Res. 2007;73:657–666 [DOI] [PubMed] [Google Scholar]
- 6.van Oort RJ, McCauley MD, Dixit SS, Pereira L, Yang Y, Respress JL, Wang Q, De Almeida AC, Skapura DG, Anderson ME, Bers DM, Wehrens XH. Ryanodine receptor phosphorylation by calcium/calmodulin-dependent protein kinase ii promotes life-threatening ventricular arrhythmias in mice with heart failure. Circulation. 2010;122:2669–2679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Curran J, Tang L, Roof SR, Velmurugan S, Millard A, Shonts S, Wang H, Santiago D, Ahmad U, Perryman M, Bers DM, Mohler PJ, Ziolo MT, Shannon TR. Nitric oxide-dependent activation of camkii increases diastolic sarcoplasmic reticulum calcium release in cardiac myocytes in response to adrenergic stimulation. PLOS ONE. 2014;9:e87495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Luo M, Guan X, Luczak ED, Lang D, Kutschke W, Gao Z, Yang J, Glynn P, Sossalla S, Swaminathan PD, Weiss RM, Yang B, Rokita AG, Maier LS, Efimov IR, Hund TJ, Anderson ME. Diabetes increases mortality after myocardial infarction by oxidizing camkii. J Clin Invest. 2013;123:1262–1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anderson ME, Brown JH, Bers DM. Camkii in myocardial hypertrophy and heart failure. J Mol Cell Cardiol. 2011;51:468–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Akki A, Zhang M, Murdoch C, Brewer A, Shah AM. Nadph oxidase signaling and cardiac myocyte function. J Mol Cell Cardiol. 2009;47:15–22 [DOI] [PubMed] [Google Scholar]
- 11.Heymes C, Bendall JK, Ratajczak P, Cave AC, Samuel JL, Hasenfuss G, Shah AM. Increased myocardial nadph oxidase activity in human heart failure. J Am Coll Cardiol. 2003;41:2164–2171 [DOI] [PubMed] [Google Scholar]
- 12.Yano M, Okuda S, Oda T, Tokuhisa T, Tateishi H, Mochizuki M, Noma T, Doi M, Kobayashi S, Yamamoto T, Ikeda Y, Ohkusa T, Ikemoto N, Matsuzaki M. Correction of defective interdomain interaction within ryanodine receptor by antioxidant is a new therapeutic strategy against heart failure. Circulation. 2005;112:3633–3643 [DOI] [PubMed] [Google Scholar]
- 13.Guo Z, Xia Z, Jiang J, McNeill JH. Downregulation of nadph oxidase, antioxidant enzymes, and inflammatory markers in the heart of streptozotocin-induced diabetic rats by n-acetyl-l-cysteine. Am J Physiol Heart Circ Physiol. 2007;292:H1728–1736 [DOI] [PubMed] [Google Scholar]
- 14.Takimoto E, Champion HC, Li M, Ren S, Rodriguez ER, Tavazzi B, Lazzarino G, Paolocci N, Gabrielson KL, Wang Y, Kass DA. Oxidant stress from nitric oxide synthase-3 uncoupling stimulates cardiac pathologic remodeling from chronic pressure load. J Clin Invest. 2005;115:1221–1231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cucoranu I, Clempus R, Dikalova A, Phelan PJ, Ariyan S, Dikalov S, Sorescu D. Nad(p)h oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ Res. 2005;97:900–907 [DOI] [PubMed] [Google Scholar]
- 16.von Harsdorf R, Li PF, Dietz R. Signaling pathways in reactive oxygen species-induced cardiomyocyte apoptosis. Circulation. 1999;99:2934–2941 [DOI] [PubMed] [Google Scholar]
- 17.Hori M, Nishida K. Oxidative stress and left ventricular remodelling after myocardial infarction. Cardiovasc Res. 2009;81:457–464 [DOI] [PubMed] [Google Scholar]
- 18.Erickson JR, Pereira L, Wang L, Han G, Ferguson A, Dao K, Copeland RJ, Despa F, Hart GW, Ripplinger CM, Bers DM. Diabetic hyperglycaemia activates camkii and arrhythmias by o-linked glycosylation. Nature. 2013;502:372–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Erickson JR, Joiner M-lA, Guan X, Kutschke W, Yang, Oddis CV, Bartlett RK, Lowe JS, O’Donnell SE, Aykin-Burns N, Zimmerman MC, Zimmerman K, Ham A-JL, Weiss RM, Spitz DR, Shea MA, Colbran RJ, Mohler PJ, Anderson ME. A dynamic pathway for calcium-independent activation of camkii by methionine oxidation. Cell. 2008;133:462–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gutierrez DA, Fernandez-Tenorio M, Ogrodnik J, Niggli E. No-dependent camkii activation during beta-adrenergic stimulation of cardiac muscle. Cardiovasc Res. 2013;100:392–401 [DOI] [PubMed] [Google Scholar]
- 21.Meyer T, Hanson PI, Stryer L, Schulman H. Calmodulin trapping by calcium-calmodulin-dependent protein kinase. Science. 1992;256:1199–1202 [DOI] [PubMed] [Google Scholar]
- 22.Erickson JR. Mechanisms of camkii activation in the heart. Frontiers in pharmacology. 2014;5:59–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Erickson JR, Nichols CB, Uchinoumi H, Stein ML, Bossuyt J, Bers DM. S-nitrosylation induces both autonomous activation and inhibition of calcium/calmodulin-dependent protein kinase ii delta. J Biol Chem. 2015;290:25646–25656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Anderson ME. Oxidant stress promotes disease by activating camkii. Journal of Molecular and Cellular Cardiology. 2015;89:160–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hegyi B, Borst JM, Lucena AJ, Bailey LRj, Bossuyt J, Bers DM. Diabetic hyperglycemia regulates potassium channels and arrhythmias in the heart via autonomous camkii activation by o-linked glycosylation. Biophys J. 2019;116:98A [Google Scholar]
- 26.Kronlage M, Dewenter M, Grosso J, Fleming T, Oehl U, Lehmann LH, Falcao-Pires I, Leite-Moreira AF, Volk N, Grone HJ, Muller OJ, Sickmann A, Katus HA, Backs J. O-glcnacylation of histone deacetylase 4 protects the diabetic heart from failure. Circulation. 2019;140:580–594 [DOI] [PubMed] [Google Scholar]
- 27.Swain L, Kesemeyer A, Meyer-Roxlau S, Vettel C, Zieseniss A, Guntsch A, Jatho A, Becker A, Nanadikar MS, Morgan B, Dennerlein S, Shah AM, El-Armouche A, Nikolaev VO, Katschinski DM. Redox imaging using cardiac myocyte-specific transgenic biosensor mice. Circ Res. 2016;119:1004–1016 [DOI] [PubMed] [Google Scholar]
- 28.Albrecht Simone C, Barata Ana G, Großhans J, Teleman Aurelio A, Dick Tobias P. In vivo mapping of hydrogen peroxide and oxidized glutathione reveals chemical and regional specificity of redox homeostasis. Cell Metabolism. 2011;14:819–829 [DOI] [PubMed] [Google Scholar]
- 29.Gutscher M, Pauleau AL, Marty L, Brach T, Wabnitz GH, Samstag Y, Meyer AJ, Dick TP. Real-time imaging of the intracellular glutathione redox potential. Nat Methods. 2008;5:553–559 [DOI] [PubMed] [Google Scholar]
- 30.Gutscher M, Sobotta MC, Wabnitz GH, Ballikaya S, Meyer AJ, Samstag Y, Dick TP. Proximity-based protein thiol oxidation by h2o2-scavenging peroxidases. J Biol Chem. 2009;284:31532–31540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ma X, Chen C, Veevers J, Zhou X, Ross RS, Feng W, Chen J. Crispr/cas9-mediated gene manipulation to create single-amino-acid-substituted and floxed mice with a cloning-free method. Sci Rep. 2017;7:42244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Purohit A, Rokita AG, Guan X, Chen B, Koval OM, Voigt N, Neef S, Sowa T, Gao Z, Luczak ED, Stefansdottir H, Behunin AC, Li N, El-Accaoui RN, Yang B, Swaminathan PD, Weiss RM, Wehrens XHT, Song L-S, Dobrev D, Maier LS, Anderson ME. Oxidized ca(2+)/calmodulin-dependent protein kinase ii triggers atrial fibrillation. Circulation. 2013;128:1748–1757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ling H, Zhang T, Pereira L, Means CK, Cheng H, Gu Y, Dalton ND, Peterson KL, Chen J, Bers D, Brown JH. Requirement for ca2+/calmodulin-dependent kinase ii in the transition from pressure overload-induced cardiac hypertrophy to heart failure in mice. J Clin Invest. 2009;119:1230–1240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ling H, Gray CBB, Zambon AC, Grimm M, Gu Y, Dalton N, Purcell NH, Peterson K, Brown JH. Ca2+/calmodulin-dependent protein kinase ii δ mediates myocardial ischemia/reperfusion injury through nuclear factor-κb. Circulation research. 2013;112:935–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pereira L, Cheng H, Lao DH, Na L, van Oort RJ, Brown JH, Wehrens XH, Chen J, Bers DM. Epac2 mediates cardiac beta1-adrenergic-dependent sarcoplasmic reticulum ca2+ leak and arrhythmia. Circulation. 2013;127:913–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Picht E, Zima AV, Blatter LA, Bers DM. Sparkmaster: Automated calcium spark analysis with imagej. American Journal of Physiology-Cell Physiology. 2007;293:C1073–C1081 [DOI] [PubMed] [Google Scholar]
- 37.Burridge PW, Matsa E, Shukla P, Lin ZC, Churko JM, Ebert AD, Lan F, Diecke S, Huber B, Mordwinkin NM, Plews JR, Abilez OJ, Cui B, Gold JD, Wu JC. Chemically defined generation of human cardiomyocytes. Nature Methods. 2014;11:855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cunningham TJ, Yu MS, McKeithan WL, Spiering S, Carrette F, Huang CT, Bushway PJ, Tierney M, Albini S, Giacca M, Mano M, Puri PL, Sacco A, Ruiz-Lozano P, Riou JF, Umbhauer M, Duester G, Mercola M, Colas AR. Id genes are essential for early heart formation. Genes Dev. 2017;31:1325–1338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McKeithan WL, Savchenko A, Yu MS, Cerignoli F, Bruyneel AAN, Price JH, Colas AR, Miller EW, Cashman JR, Mercola M. An automated platform for assessment of congenital and drug-induced arrhythmia with hipsc-derived cardiomyocytes. Frontiers in physiology. 2017;8:766–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang Z, Wang L, Tapa S, Pinkerton KE, Chen CY, Ripplinger CM. Exposure to secondhand smoke and arrhythmogenic cardiac alternans in a mouse model. Environ Health Perspect. 2018;126:127001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ortiz-Meoz RF, Jiang J, Lazarus MB, Orman M, Janetzko J, Fan C, Duveau DY, Tan ZW, Thomas CJ, Walker S. A small molecule that inhibits ogt activity in cells. ACS Chem Biol. 2015;10:1392–1397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Trafford AW, Diaz ME, Eisner DA. Coordinated control of cell ca(2+) loading and triggered release from the sarcoplasmic reticulum underlies the rapid inotropic response to increased l-type ca(2+) current. Circ Res. 2001;88:195–201 [DOI] [PubMed] [Google Scholar]
- 43.Rezazadeh S, Claydon TW, Fedida D. Kn-93 (2-[n-(2-hydroxyethyl)]-n-(4-methoxybenzenesulfonyl)]amino-n-(4-chlorocinnamyl)-n -methylbenzylamine), a calcium/calmodulin-dependent protein kinase ii inhibitor, is a direct extracellular blocker of voltage-gated potassium channels. J Pharmacol Exp Ther. 2006;317:292–299 [DOI] [PubMed] [Google Scholar]
- 44.Nickel AG, von Hardenberg A, Hohl M, Loffler JR, Kohlhaas M, Becker J, Reil JC, Kazakov A, Bonnekoh J, Stadelmaier M, Puhl SL, Wagner M, Bogeski I, Cortassa S, Kappl R, Pasieka B, Lafontaine M, Lancaster CR, Blacker TS, Hall AR, Duchen MR, Kastner L, Lipp P, Zeller T, Muller C, Knopp A, Laufs U, Bohm M, Hoth M, Maack C. Reversal of mitochondrial transhydrogenase causes oxidative stress in heart failure. Cell Metab. 2015;22:472–484 [DOI] [PubMed] [Google Scholar]
- 45.Guo T, Zhang T, Ginsburg KS, Mishra S, Brown JH, Bers DM. Camkiiδc slows [ca]i decline in cardiac myocytes by promoting ca sparks. Biophysical journal. 2012;102:2461–2470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.John SA, Ottolia M, Weiss JN, Ribalet B. Dynamic modulation of intracellular glucose imaged in single cells using a fret-based glucose nanosensor. Pflugers Archiv : European journal of physiology. 2008;456:307–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bhatt NM, Aon MA, Tocchetti CG, Shen X, Dey S, Ramirez-Correa G, O’Rourke B, Gao WD, Cortassa S. Restoring redox balance enhances contractility in heart trabeculae from type 2 diabetic rats exposed to high glucose. American journal of physiology. Heart and circulatory physiology 2015;308:H291–H302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Singh RM, Waqar T, Howarth FC, Adeghate E, Bidasee K, Singh J. Hyperglycemia-induced cardiac contractile dysfunction in the diabetic heart. Heart Failure Reviews. 2018;23:37–54 [DOI] [PubMed] [Google Scholar]
- 49.Belambri SA, Rolas L, Raad H, Hurtado-Nedelec M, Dang PM, El-Benna J. Nadph oxidase activation in neutrophils: Role of the phosphorylation of its subunits. Eur J Clin Invest. 2018;48 Suppl 2:e12951. [DOI] [PubMed] [Google Scholar]
- 50.Aon MA, Cortassa S, O’Rourke B. Redox-optimized ros balance: A unifying hypothesis. Biochim Biophys Acta. 2010;1797:865–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang Q, Quick AP, Cao S, Reynolds J, Chiang DY, Beavers D, Li N, Wang G, Rodney GG, Anderson ME, Wehrens XHT. Oxidized camkii (ca(2+)/calmodulin-dependent protein kinase ii) is essential for ventricular arrhythmia in a mouse model of duchenne muscular dystrophy. Circ Arrhythm Electrophysiol. 2018;11:e005682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu F, Lukinius A, Bergström M, Eriksson B, Watanabe Y, Långström B. A mechanism behind the antitumour effect of 6-diazo-5-oxo-l-norleucine (don): Disruption of mitochondria. European Journal of Cancer. 1999;35:1155–1161 [DOI] [PubMed] [Google Scholar]
- 53.Li Y, Sirenko S, Riordon DR, Yang D, Spurgeon H, Lakatta EG, Vinogradova TM. Camkii-dependent phosphorylation regulates basal cardiac pacemaker function via modulation of local ca2+ releases. Am J Physiol Heart Circ Physiol. 2016;311:H532–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bowman PRT, Smith GL, Gould GW. Glut4 expression and glucose transport in human induced pluripotent stem cell-derived cardiomyocytes. PLoS One. 2019;14:e0217885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dewenter M, von der Lieth A, Katus HA, Backs J. Calcium signaling and transcriptional regulation in cardiomyocytes. Circ Res. 2017;121:1000–1020 [DOI] [PubMed] [Google Scholar]
- 56.Ma X, Chen C, Veevers J, Zhou X, Ross RS, Feng W, Chen J. Crispr/cas9-mediated gene manipulation to create single-amino-acid-substituted and floxed mice with a cloning-free method. Sci Rep. 2017;7:42244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hegyi B, Borst JM, Lucena AJ, Bailey LRj, Bossuyt J, Bers DM. Diabetic hyperglycemia regulates potassium channels and arrhythmias in the heart via autonomous camkii activation by o-linked glycosylation. Biophys J. 2019;116:98A [Google Scholar]
- 58.Purohit A, Rokita AG, Guan X, Chen B, Koval OM, Voigt N, Neef S, Sowa T, Gao Z, Luczak ED, Stefansdottir H, Behunin AC, Li N, El-Accaoui RN, Yang B, Swaminathan PD, Weiss RM, Wehrens XHT, Song L-S, Dobrev D, Maier LS, Anderson ME. Oxidized ca(2+)/calmodulin-dependent protein kinase ii triggers atrial fibrillation. Circulation. 2013;128:1748–1757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ling H, Zhang T, Pereira L, Means CK, Cheng H, Gu Y, Dalton ND, Peterson KL, Chen J, Bers D, Brown JH. Requirement for ca2+/calmodulin-dependent kinase ii in the transition from pressure overload-induced cardiac hypertrophy to heart failure in mice. J Clin Invest. 2009;119:1230–1240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ling H, Gray CBB, Zambon AC, Grimm M, Gu Y, Dalton N, Purcell NH, Peterson K, Brown JH. Ca2+/calmodulin-dependent protein kinase ii δ mediates myocardial ischemia/reperfusion injury through nuclear factor-κb. Circulation research. 2013;112:935–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Swain L, Kesemeyer A, Meyer-Roxlau S, Vettel C, Zieseniss A, Guntsch A, Jatho A, Becker A, Nanadikar MS, Morgan B, Dennerlein S, Shah AM, El-Armouche A, Nikolaev VO, Katschinski DM. Redox imaging using cardiac myocyte-specific transgenic biosensor mice. Circ Res. 2016;119:1004–1016 [DOI] [PubMed] [Google Scholar]
- 62.Pereira L, Cheng H, Lao DH, Na L, van Oort RJ, Brown JH, Wehrens XH, Chen J, Bers DM. Epac2 mediates cardiac beta1-adrenergic-dependent sarcoplasmic reticulum ca2+ leak and arrhythmia. Circulation. 2013;127:913–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Suetomi T, Willeford A, Brand CS, Cho Y, Ross RS, Miyamoto S, Brown JH. Inflammation and NLRP3 Inflammasome Activation Initiated in Response to Pressure Overload by Ca2+/Calmodulin-Dependent Protein Kinase II δ Signaling in Cardiomyocytes Are Essential for Adverse Cardiac Remodeling. Circulation. 2018;138:2530–2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Erickson JR, Pereira L, Wang L, Han G, Ferguson A, Dao K, Copeland RJ, Despa F, Hart GW, Ripplinger CM, Bers DM. Diabetic hyperglycaemia activates camkii and arrhythmias by o-linked glycosylation. Nature. 2013;502:372–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.van Oort RJ, McCauley MD, Dixit SS, Pereira L, Yang Y, Respress JL, Wang Q, De Almeida AC, Skapura DG, Anderson ME, Bers DM, Wehrens XH. Ryanodine receptor phosphorylation by calcium/calmodulin-dependent protein kinase ii promotes life-threatening ventricular arrhythmias in mice with heart failure. Circulation. 2010;122:2669–2679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Picht E, Zima AV, Blatter LA, Bers DM. Sparkmaster: Automated calcium spark analysis with imagej. American Journal of Physiology-Cell Physiology. 2007;293:C1073–C1081 [DOI] [PubMed] [Google Scholar]
- 67.Gutscher M, Pauleau AL, Marty L, Brach T, Wabnitz GH, Samstag Y, Meyer AJ, Dick TP. Real-time imaging of the intracellular glutathione redox potential. Nat Methods. 2008;5:553–559 [DOI] [PubMed] [Google Scholar]
- 68.Burridge PW, Matsa E, Shukla P, Lin ZC, Churko JM, Ebert AD, Lan F, Diecke S, Huber B, Mordwinkin NM, Plews JR, Abilez OJ, Cui B, Gold JD, Wu JC. Chemically defined generation of human cardiomyocytes. Nature Methods. 2014;11:855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cunningham TJ, Yu MS, McKeithan WL, Spiering S, Carrette F, Huang CT, Bushway PJ, Tierney M, Albini S, Giacca M, Mano M, Puri PL, Sacco A, Ruiz-Lozano P, Riou JF, Umbhauer M, Duester G, Mercola M, Colas AR. Id genes are essential for early heart formation. Genes Dev. 2017;31:1325–1338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McKeithan WL, Savchenko A, Yu MS, Cerignoli F, Bruyneel AAN, Price JH, Colas AR, Miller EW, Cashman JR, Mercola M. An automated platform for assessment of congenital and drug-induced arrhythmia with hipsc-derived cardiomyocytes. Frontiers in physiology. 2017;8:766–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang Z, Wang L, Tapa S, Pinkerton KE, Chen CY, Ripplinger CM. Exposure to secondhand smoke and arrhythmogenic cardiac alternans in a mouse model. Environ Health Perspect. 2018;126:127001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Greis KD, Hayes BK, Comer FI, Kirk M, Barnes S, Lowary TL, Hart GW. Selective detection and site-analysis of o-glcnac-modified glycopeptides by β-elimination and tandem electrospray mass spectrometry. Analytical Biochemistry. 1996;234:38–49 [DOI] [PubMed] [Google Scholar]
- 73.Zachara NE, Vosseller K, Hart GW. Detection and analysis of proteins modified by o-linked n-acetylglucosamine. Current protocols in protein science. 2011;Chapter 12:Unit12.18–Unit12.18 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.