

Abstract
Background
Chronic thromboembolic pulmonary hypertension (CTEPH) is a life-threatening condition arising from the thrombus and obstructive remodeling of the pulmonary arteries, which causes a significant morbidity and mortality. Although the modern treatment in CTEPH has been significant advanced both in surgical and medical treatment, none can claim to cure the disease, largely because of our limited understanding of the underlying pathogenesis of the disease and lack of a reliable CTEPH animal model to study for. Recently, inflammation has been accepted as a common pathway through which various risk factors trigger venous thrombo-embolism (VTE) formation, we describe a novel mouse model of CTEPH which reproduces a frequent trigger and resembles the time course, histological features, and clinical presentation of CTEPH in humans, to open a new horizons of inflammation in CTEPH.
Methods
By administering a pulmonary embolism (PE) protocol (comprising 3 sequential left jugular vein injections of autologous blood clots) to 8-week-old male Sprague Dawley (SD) rats using tranexamic acid (200 mg/kg.d) to inhibit fibrinolysis and injecting additional carrageenan (20 mg/kg, once a week) to create perivascular inflammation, we successfully generated a CTEPH animal model. By monitoring the mean pulmonary artery pressure (mPAP) and the histopathological change to evaluate the CTEPH model. By detecting the RT-PCR, western blot, TUNEL, and immunohistochemistry in the sub-groups to find the potential mechanism of inflammation may work in the pulmonary vascular remolding.
Results
In this study, rats with CTEPH exhibited pronounced pulmonary vascular remolding with higher vessel wall area/total area (WA/TA) ratio in comparison to the control rats (85.41%±7.37% vs. 76.41%±5.97%, P<0.05), the mPAP (25.51±1.13 vs. 15.92±1.13 mmHg, P<0.05). Significant differences in mean pulmonary artery pressure (mPAP) values were observed between rats injected solely with clots and those injected with both clots and carrageenan (25.51±1.13 vs. 29.82±1.26 mmHg, P<0.05, respectively). Furthermore, following the third embolization, thrombi and intimal hyperplasia occurred in the pulmonary artery. In addition, repeated embolization elevated mRNA and protein levels of tumor necrosis factor-α (TNF-α), NF-κB/p65, and B-cell lymphoma-2 (BCL-2), but decreased BAX expression in a time-dependent manner.
Conclusions
Take advantage of the inflammation to trigger VTE formation, we successfully generated a CTEPH animal model. Inflammation may play a crucial role in the pathogenesis and progression of CTEPH by inhibiting endothelial cell apoptosis. Understanding the role of inflammation in CTEPH may not only help to determine the optimal treatment options but also may aid in the development of future preventative strategies, since current anticoagulation treatment regimens are not designed to inhibit inflammation.
Keywords: Chronic thromboembolic pulmonary hypertension (CTEPH), thrombosis, carrageenan, inflammation, apoptosis
Introduction
Chronic thromboembolic pulmonary hypertension (CTEPH) is a distinct form of pulmonary hypertension (PH), which remains under-researched (1). It is characterized by incomplete thrombus resolution, which promotes a progressive pulmonary vascular obstruction and pulmonary vascular remodeling. The subsequent chronic increase in pulmonary vascular resistance (PVR), resulting in higher pulmonary artery pressure and a hypertrophic right ventricle (RV), ultimately causes RV failure and even death (2).
The advent of pulmonary endarterectomy (PEA), what was once thought to be the optimal treatment for the condition, has drawn more attention to this orphan disease, and there have been significant improvements in both mortality and morbidity as a result (3). However, a relevant proportion of patients, varying from 5% to 35%, present sustained small-vessel vasculopathy after PEA, and many affected individuals are poor candidates for PEA. For some, the blockage is inaccessible by surgery, whereas others may have severe comorbidities or high age, and a large number of patients opt not to have PEA (2,4).
In recent decades, our understanding of the pathophysiology of the disease has improved. This has resulted in the development of effective therapies targeting endothelial dysfunction (epoprostenol and derivatives, endothelin receptor antagonists, and phosphodiesterase type 5 inhibitors) and, owing to these treatments, a substantial progression in clinical function and hemodynamics has been made. However, to date, none of the current therapies have been curative, and prognosis for CTEPH patients remains poor (5). Thus, to promote the quality of life and, eventually, the survival of CTEPH patients, additional or alternative nonsurgical treatment is urgently needed.
Unfortunately, our limited cognitions of the pathophysiology of CTEPH act as a barrier in the development of new treatment strategies. For example, it is not yet understood how pulmonary embolism (PE) progresses to CTEPH, or how thrombi escape thrombolysis and the subsequent fibrotic transformation operates. Therefore, the creation of a simple and reproducible CTEPH animal model, which not only enables a study of the molecular signals involved in the pathology of this disease but also permits the evaluation of new therapeutic strategies to improve the fate of individuals diagnosed with CTEPH.
It is widely recognized that research in pathophysiology is lagging significantly behind other areas. In part, technical reasons are to blame, as is a lack of reliable CTEPH animal models. Unfortunately, generating a small CTEPH animal model has proven to be a challenging task due to efficient fibrinolysis, the remarkable adaptive capabilities of the pulmonary circulation, and the inability of the RV to overcome massive and sharp increases in afterload (6,7). Several types of embolus (e.g., ceramic beads and polystyrene microsphere) have been used to model CTEPH in dogs, pigs, and rodents (7,8), and even some complex surgical protocols like left pulmonary artery ligation were conducted (9). However, these all failed to mimic the key pathophysiological thrombogenesis processes observed in CTEPH patients (2,7,10,11).
Previously, we successfully established a rat model of CTEPH with repeated injections of autologous blood clots and the addition of tranexamic acid; however, this model had poor stability and lacked pronounced PH (12). Since there are numerous risk factors believed to be associated with the development of CTEPH, the acquired thrombophilias and chronic inflammatory states were included. To address this issue, we applied three sequential external jugular vein injections of autologous blood clot with tranexamic acid, and a clot persistence and organization by carrageenan-mediated, which enables interactions of the PE with its local cellular environment and opens new insights into its pathophysiology with inflammation.
Methods
Animals
All experimental and animal care protocols were approved by the Animal Ethics Committee of Fujian Medical University (SYXK2012-0001), the Institutional Animal Care and Use Committees (IACUCs), and The Guide for the Care and Use of Laboratory Animals (NIH, Bethesda, MD, USA). Eighty 8-week-old Sprague Dawley (SD) rats weighing 230±22 g were purchased from the Experimental Animal Center of the Fujian Medical University (Fujian, China) and acclimatized for 4–5 days prior to the experiments. All rats had free access to food and water throughout the duration of the study.
Study methods
The rats were weighed and anesthetized (10% chloral hydrate 0.3 mL/100 g) before the procedure. Pulmonary embolization was performed through 3 consecutive injections of autologous blood clots via the left external jugular vein at a dosage of 32±5 per animal on the 1st, 4th, and 11th days. Control animals underwent the same protocol but were treated with saline solution instead of the thrombotic material. Endogenous fibrinolysis was inhibited with an intraperitoneal injection of tranexamic acid (200 mg/kg). In addition, rats in PE group B and the carrageenan control group were intraperitoneally injected with 0.2% carrageenan (20 mg/kg, once a week) after the PE and, to prevent infection, penicillin (10,000 U/kg/day) was intramuscularly injected for 3 days following. After euthanasia, organs and plasma were collected under chloral hydrate anesthesia. The lung tissues and pulmonary arteries were prepared for histological analysis, RNA reverse transcriptase polymerase chain reaction array analysis, and Western blotting. We established a subgroup to help to determine the effects of apoptosis and inflammation.
Extended methods
Grouping
Eighty SD rats were randomly divided into the following four groups (n=20): normal saline (NS) control group, carrageenan control group, PE group A, and PE group B. The rats in the PE groups were injected with autologous blood clots, while the control animals underwent the same protocol but were treated with saline solution instead of autologous blood clots. Rats in the carrageenan control group and PE group B received additional injections with 0.2% carrageenan (20 mg/kg, once a week) after the embolism.
Thirty SD rats were randomly divided into five subgroups to explore the effect of carrageenan on CTEPH. The groups were characterized as follows: group I, control group; group II, one week after the first embolism; group III: one week after the second embolism; group IV, one week after the third embolism; group V, three weeks after the third embolism. The protocol in the sub-PE groups II–V) was the same as that in PE group B.
Establishment of CTEPH model
Autologous blood clot preparation
Blood was collected from the orbital vein after anesthesia one day before the embolism using a capillary glass tube and was placed into sterilized Petri dishes overnight at 4 °C. Clots were rushed out from the capillary glass tubes with NS and then trimmed to 1.1×3 mm clots on the embolism day (Figure 1).
Figure 1.
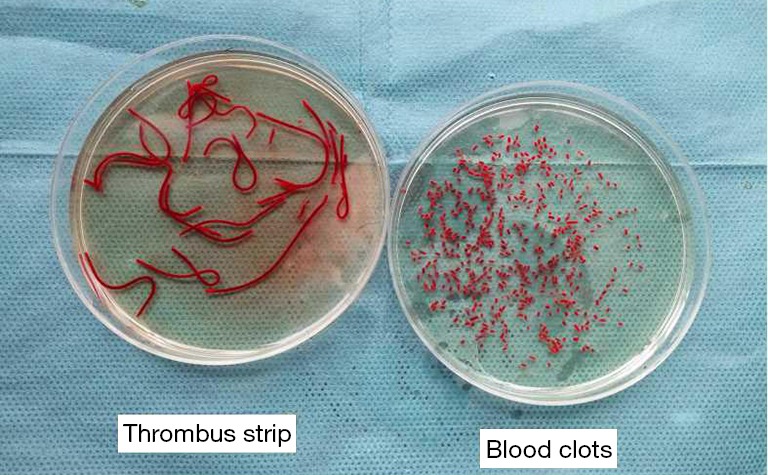
Preparation of autologous blood clots.
Modeling CTEPH
Rats were anesthetized with 10% chloral hydrate (0.3 mL/100 g) and, under sterile conditions, the left external jugular vein was exposed. Then, the blood clots that had been prepared (32±5 per animal) were injected into the left jugular vein at a rate of 0.5 mL/min (Figure 2) on the first day of the experiment. After the embolism, if the rats presented with cyanosis and accelerated breathing, then the injection was stopped. Equal amounts of NS (1.5 mL) were injected into the jugular vein of the control group rats. On the 4th and 11th days after the first embolization, the procedures were repeated under identical conditions, and endogenous fibrinolysis was inhibited with an intraperitoneal injection of tranexamic acid (200 mg/kg). In addition, rats in PE group B and the carrageenan control group were intraperitoneally injected with 0.2% carrageenan (20 mg/kg, once a week). After the PE, penicillin (10,000 U/kg/day) was intramuscularly injected for three days to prevent infection.
Figure 2.
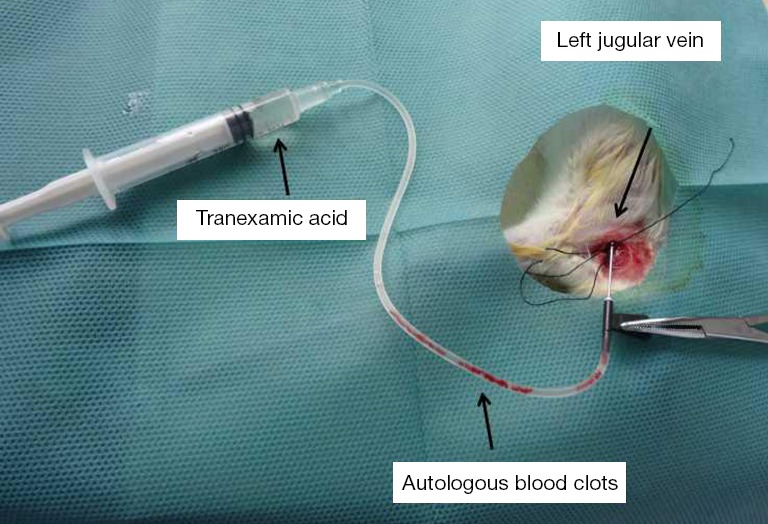
The procedure for embolization of autologous blood clots.
Three weeks after the last PE/sham embolism, the rats underwent hemodynamic analysis (mean pulmonary arterial pressure, Figure 3). Immediately after, rats were euthanized under chloral hydrate anesthesia, and their blood was collected and centrifuged to obtain serum. The serum was kept at −80 °C until measurements of tumor necrosis factor-α (TNF-α) (Abcam) were detected using ELISA. Lungs were excised in a block, the connective tissue was trimmed away, and the lung lobe was perfused with 10% formaldehyde with fixation continuing overnight in 10% formaldehyde solution.
Figure 3.

Procedures to measure the mPAP. (A) Isolation of the right external jugular vein and connecting the pressure transducer (A, white arrow); (B) recording the pulmonary arterial pressure (B, red arrow). mPAP, mean pulmonary arterial pressure.
Histological examination
After incubation in a 10% formaldehyde solution overnight, the lung lobes were embedded in paraffin. Transversal mid-sections were used to prepare 10 and 30 µm whole-lung paraffin sections. In line with standard protocols, the sections were stained with HE. Microphotographs of complete lung transversal sections stained with HE were obtained and used to count the persistent autologous blood clot/secondary in situ thrombosis, hypertrophied blood vessels, vessels with perivascular infiltration, and cellular/fibrotic lesions. The value of the pulmonary arterial vessel wall area/total area (WA/TA) ratio was used to evaluate the level of pulmonary arterial remodeling that had occurred.
Expression of factor VIII immunohistochemistry and TUNEL in pulmonary artery endothelial cells
After the pulmonary artery sections were deparaffinized and rehydrated, sections were incubated with rabbit anti-rat factor VIII polyclonal antibodies [at 1:50 Abcam (Shanghai) Trading Co. Ltd., Shanghai, China] according to standard protocol. The sections were treated with the biotin-streptavidin horseradish peroxidase (HRP) detection system (ZSGB-BIO, Shanghai, China), and immunoreactivity was visualized using 3,3'-diaminobenzidine (DAB). We used an apoptosis TUNEL detection kit to detect pulmonary artery endothelial apoptosis.
RT-PCR
RNA was extracted from the pulmonary artery tissue samples with the Trizol reagent (Invitrogen, Carlsbad, CA, USA), and the purity of the RNA was analyzed using a spectrophotometer (Macy Instruments Inc., Shanghai, China) and reversely transcribed into cDNA. Primer pairs were designed for TNF-α, NF-κB, and B-cell lymphoma-2 (BCL-2), BAX, and β-actin (internal control) using their DNA sequences (Sangon Biotech Co., Ltd., Shanghai, China). The primers used in this study were as follows: TNF-α sense primer: 5'-CTC AAG CCC TGG TAT GAG CC-3'; antisense primer: 5'-TGG ACC CAG AGC CAC AAT TC -3'; 295-bp PCR products. NF-κB sense primer: 5'-GTG CAG AAA GAA GAC ATT GAG GTG-3'; antisense primer: 5'-AGG CTA GGG TCA GCG TAT GG-3'; 290-bp PCR products. BCL-2 sense primer: 5'-CCC TGT TTT TGG CAG TGA GT-3'; antisense primer: 5'-TTC CCT TCT CCA TTT TGT GC-3'; BAX sense primer: 5'-GGA TCG AGC AGA GAG GAT GG-3'; antisense primer: 5'-CCA GTT GAA GTT GCC GTC TG-3'; β-actin sense primer: 5'-CGG GAA ATC GTG CGT GAC-3'; antisense primer: 5'-TGG AAG GTGGAC AGC GAG G-3'. PCR reaction was carried out in a 20-µL volume as follows: initial denaturation at 95 °C for 5 min was followed by 40 cycles of 95 °C denaturation for 10 s, 58 °C annealing for 30 s, and 72 °C extension for 30 s; PCR products were subjected to electrophoresis, and the bands were visualized using a gel imaging system.
Western blots
Using a cold saline solution, we briefly washed some parts of the lung lobe to prepare it for protein extraction. Western blotting was performed according to standard protocols using the following primary antibodies: anti-rat TNF-α, anti-NF-κB (p65), anti-BCL-2, and anti-BAX antibodies [dilutions of 1:1,000, 1:1,000, 1:2,000, and 1:2,000 respectively; Abcam (Shanghai) Trading Co., Ltd., Shanghai, China]. β-actin was employed as an internal standard, and immunoreactive proteins were visualized by HRP-coupled antibodies (Amersham) and ECL. The targeted proteins were analyzed using the Lab-work image analysis software (Gene Company Limited, Hong Kong, China).
Statistical analysis
Data are presented as the mean ± standard deviation (SD). Statistical analysis was carried out using SPSS 17.0 software. Differences between groups were analyzed by using the one-way analysis of variance and were considered statistically significant if P<0.05.
Results
PE time
Moser et al. (13) found that only 30% of the initial injected thrombus volume remained inside the dog pulmonary artery 3 hours after the acute PE. However, in our experiment, we found that the original thrombus was still present almost two hours after embolization (blue arrow in Figure 4A, and that, after a day, we could detect a new strip of fresh thrombus (red arrow in Figure 4B), while the original one could not be found. On the 3rd day, the new thrombus was dissolved or had disappeared (red arrow in Figure 4C); therefore, to delay the thrombolysis, we added tranexamic acid to the anti-fibrinolytic action to make the 4th day equivalent to the second embolization occurrence. Similarly, the new thrombus resorption was found on the 7th day (red arrow in Figure 4D) after the 2nd embolization (i.e., the 11th day), and so the 11th day was considered the 3rd embolization occurrence. Since repeated embolization can injure the jugular vein and cause difficulty in sustaining another embolization, we performed only 3 embolizations during our experiment.
Figure 4.
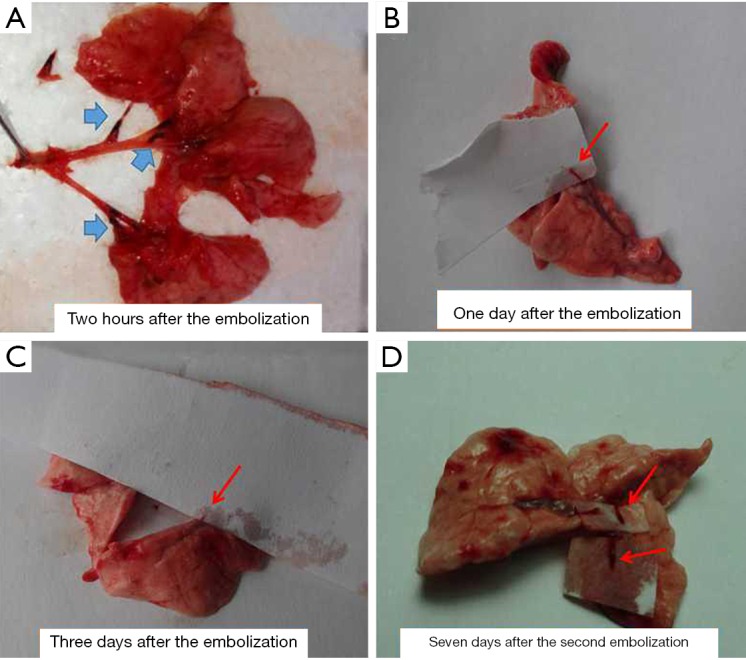
Different features following embolization. Blue arrow, autologous blood clot; red arrow, secondary thrombosis or a new thrombus.
Lung histomorphology
As shown in Figure 5, there were no obvious pathological changes found in the NS and carrageenan control groups, at either 1 week after the 2nd embolization or 3 weeks after the 3rd embolization. Interestingly, there were many changes observed in the PE group. As can be seen from Figure 5S3 (red arrows I, II), PE group A had small long-strip thrombi 1 week following the 2nd embolization, but the autologous clots had dissolved; thereafter, a reddish-brown infarct was observed 3 weeks after the 3rd embolization (red arrow III in Figure 5T3). In contrast to PE group A, the autologous clots were not completely resorbed in PE group B, and 1 week after the 2nd embolization, the newly formed thrombi were markedly larger than those in PE group A (red IV arrow in Figure 5S4). In addition, the pulmonary artery embolism was still observable 3 weeks after the 3rd embolization (red arrow V in Figure 5T4). These thrombi were formed mainly in the inferior lobe of the right lung, which resembled the localization of thrombi in patients with CTEPH.
Figure 5.

Pathological changes in the four groups one week after the second embolization (S) and three weeks following the third embolization (T). I,II: newly formed thrombi observed a week after the second embolization in PE group A; III: pulmonary infarction observed three weeks after the third embolization in PE group A; IV: newly formed thrombi observed a week after the second embolization in PE group B; V: pulmonary artery embolism occurred three weeks after the third embolization in PE group B. (1, normal saline control group; 2, carrageenan control group; 3, PE group A; 4, PE group B). PE, pulmonary embolism.
Microphotographs of lung transversal sections stained with HE show the same results (Figure 6), and we could detect the recanalization and invasive fibrous tissue 1 week after the 2nd embolization in PE group A (Figure 6S3), while PE group B exhibited infiltration of inflammatory cells and incomplete recanalization (Figure 6S4). The WA/TA ratio gradually increased following repeated embolization in the PE group 3 weeks after the 3rd embolization (P<0.05), as indicated by mPAP (Table 1). In addition, PE group B developed thicker vascular walls, more vascular remolding, and more intimal hyperplasia (Figure 6T4) than the other groups; and even onion skin-like hyperplasia was observed (Figure 6T4i-T4iii). However, no changes were found in the control group (P>0.05) in the WA/TA ratio or the mPAP.
Figure 6.

Pathological changes one week after the second embolization (S) and three weeks after the third embolization (T). S3: recanalization and invasive fibrous tissues were (red arrow) observed in PE group A one week after the second embolization. S4: inflammatory cell infiltration and incomplete recanalization in PE group B one week after the second embolization. T4–T4iii: thicker vascular walls, intimal hyperplasia (small red arrow in T4) and onion skin-like hyperplasia were found in PE group B three weeks after the third embolization. (1, normal saline control group; 2, carrageenan control group; 3, PE group A; 4, PE group B). PE, pulmonary embolism.
Table 1. mPAP and WA/TA ratio changes in the rat CTEPH model.
| Group | n | mPAP (mmHg) | WA/TA ratio (%) |
|---|---|---|---|
| NS control group | 20 | 13.73±1.54 | 32.94±2.75 |
| Carrageenan control group | 20 | 15.92±1.13▲ | 35.39±7.30▲ |
| PE group A | 20 | 25.51±1.37* | 76.41±5.97* |
| PE group B | 20 | 29.82±1.26*# | 85.41±7.37*# |
Data are expressed as (mean ± SD). The WA/TA ratio gradually increased following repeated embolization in the PE group 3 weeks after the 3rd embolization (P<0.05), as indicated by mPAP, but no changes occurred in the control group (P>0.05). *, P<0.05, compared to control group; #, P<0.05, compared to PE group A; ▲, P>0.05, compared to the NS control group. WA/TA, wall area/total area; CTEPH, chronic thromboembolic pulmonary hypertension.
Endothelial cell apoptosis and the expression of factor VIII were detected
In the sub-groups, endothelial cell apoptosis detected by TUNEL (red arrow shown in the brown result section in Figure 7A) gradually decreased (P<0.05) according to the number of embolizations and time after embolization, while the expression of endothelial marker VIII factor showed the opposite trend (the brown result section in Figure 7B, P<0.05). In addition, as the number of embolizations and embolization time increased, the vessel walls became thicker, and the lumen stenosis became smaller; thus, we speculated that membrane thickening is involved in vascular remodeling and is one of the causes of the narrowing of the lumen, while the decrease in endothelial cell apoptosis is of great importance. Table 2 depicts the endothelial cell apoptosis index and the OD rate of endothelial marker VIII factor.
Figure 7.

Endothelial cell apoptosis was detected by TUNEL and the expression of factor VIII was detected by immunohistochemistry. (A) Endothelial cell apoptosis was detected by TUNEL. TUNEL (red arrow shown in the brown result section) was gradually decreased (P<0.05) as indicated by the number of embolizations and time after embolization. (I, control group; II, one week after the first embolization; III, one week after the second embolization; IV, one week after the third embolization; V, three weeks after the third embolization); (B) the expression of factor VIII was detected by immunohistochemistry. Note: expression of factor VIII was gradually increased (P<0.05) as indicated by the number of embolizations and time after embolization (the brown result section). (I, control group; II, one week after the first embolization; III, one week after the second embolization; IV, one week after the third embolization; V, three weeks after the third embolization).
Table 2. Endothelial cell apoptosis index and the OD rate of endothelial marker VIII factor.
| Group | I | II | III | IV | V |
|---|---|---|---|---|---|
| TUNEL | 0.58±0.02 | 0.41±0.03* | 0.30±0.04*# | 0.21±0.03*#& | 0.12±0.03*#&▲ |
| VIII | 0.10±0.02 | 0.17±0.01* | 0.23±0.01*# | 0.28±0.01*#& | 0.35±0.02*#&▲ |
TUNEL gradually decreased as indicated by the number of embolizations and time after embolization, while the expression of endothelial marker VIII factor displayed an opposite trend. *, P<0.05 compared to group I; #, P<0.05 compared to group II; &, P<0.05 compared to group III; ▲, P<0.05 compared to group IV (I, control group; II, one week after the first embolization; III, one week after the 2nd embolization; IV, one week after the 3rd embolization; V, three weeks after the 3rd embolization). OD, optical density.
Pulmonary artery mRNA and protein expression pattern
Reverse transcriptase polymerase chain reaction array analysis identified an increased expression of the mRNA transcripts TNF-α, NF-κB/p65, and BCL-2. This was indicated by the number of embolizations and time after embolization. (Table 3, Figure 8), with statistically significant differences between the experimental groups (P<0.05). Meanwhile, the BAX mRNAs were significantly less abundant in the pulmonary arteries of PE when compared with the control group (P<0.05), and decreased in expression, as indicated by a number of embolizations and time after embolization. Similar results were obtained for protein level (Table 4, Figure 9).
Table 3. mRNA expression in different subgroups.
| Group | I | II | III | IV | V |
|---|---|---|---|---|---|
| TNF-a | 0.24±0.02 | 0.34±0.02* | 0.75±0.02*# | 0.84±0.31*#& | 1.13±0.09*#&▲ |
| NF-κB/p65 | 0.23±0.03 | 0.78±0.04* | 0.91±0.02*# | 1.07±0.04*#& | 1.14±0.04*#&▲ |
| BAX | 1.26±0.07 | 0.98±0.02* | 0.77±0.01*# | 0.65±0.02*#& | 0.36±0.04*##&▲ |
| BCL-2 | 0.23±0.02 | 0.36±0.02* | 0.60±0.04*# | 0.88±0.06*#& | 1.22±0.10*#&▲ |
TNF-α, NF-κB/p65, and BCL-2 levels were increased, as indicated by the number of embolizations and time after embolization, while BAX mRNAs expression showed the opposite trend, with statistically significant differences between the experimental groups. *, P<0.05 compared to group I; #, P<0.05 compared to group II; &, P<0.05 compared to group III; ▲, P<0.05 compared to group IV.
Figure 8.

mRNA levels in different sub- groups of rats. TNF-α, NF-κB/p65, and BCL-2 increased as indicated by the number of embolizations and time after embolization, but BAX mRNAs expression showed the opposite trend, with statistically significant differences between the experimental groups. TNF-α, tumor necrosis factor-α; BCL-2, B-cell lymphoma-2.
Table 4. Proteins expressed in different subgroups.
| Group | I | II | III | IV | V |
|---|---|---|---|---|---|
| TNF-a | 0.014±0.003 | 0.039±0.002* | 0.129±0.012*# | 0.384±0.021*#& | 0.671±0.036*#&▲ |
| NF-κB/p65 | 0.012±0.001 | 0.033±0.005* | 0.075±0.007*# | 0.182±0.145*#& | 0.191±0.013*#&▲ |
| BAX | 0.298±0.023 | 0.224±0.291* | 0.117±0.087*# | 0.055±0.051*#& | 0.022±0.006*#&▲ |
| BCL-2 | 0.126±0.101 | 0.186±0.015* | 0.325±0.022*# | 0.353±0.151*#& | 0.657±0.058*#&▲ |
TNF-α, NF-κB/p65, and BCL-2 levels were increased, as indicated by the number of embolizations and time after embolization, while BAX mRNAs expression showed the opposite trend, with statistically significant differences between the experimental groups. *, P<0.05 compared to group I; #, P<0.05 compared to group II; &, P<0.05 compared to group III; ▲, P<0.05 compared to group IV.
Figure 9.

Protein levels in different sub-groups. TNF-α, NF-κB/p65, and BCL-2 levels were increased as indicated by the number of embolizations and time after embolization, but BAX protein expression showed the opposite trend, with statistically significant differences between the experimental groups. *, P<0.05 compared to group I; #, P<0.05 compared to group II; &, P<0.05 compared to group III; ▲, P<0.05 compared to group IV. TNF-α, tumor necrosis factor-α; BCL-2, B-cell lymphoma-2.
Discussion
Considerable efforts have been made to facilitate the understanding of the pathogenesis of CTEPH, which is caused by residual thromboemboli with flaps and meshwork forming in pulmonary arteries. Unfortunately, the molecular pathogenesis of CTEPH has long been elusive (14), particularly the mechanisms by which the pulmonary emboli or thrombi fail to undergo lysis and instead organize into occluding fibrotic material. To the best of our knowledge, this is the first model that resembles the functional and histological key features of CTEPH that has been achieved completely through autologous blood clots.
Since the 1990s, several attempts have been made to induce animal models of CTEPH; ceramic beads, polystyrene microspheret, and other embolic materials have all been administered to rodents, piglets, sheep, and dogs and failed (7,8,15). Mercier et al. (16). established a reliable piglet model that replicated most of the clinical features of human CTEPH. The model demonstrated increased PVR, increased mean pulmonary artery pressure (mPAP), increased median thickness of distal pulmonary arteries in both obstructed and unobstructed territories, increased systemic blood supply through the bronchial arteries in the obstructed territories, RV dilatation, RV hypertrophy, and paradoxical septal motion. However, it failed to duplicate the impaired thrombus resolution seen in human CTEPH.
Our model is thus the first animal model that not only resembles CTEPH but is also much easier than previous attempts to establish and maintain. Furthermore, we can quantify the extent of PE postmortem simply by counting the microspheres deposited in the pulmonary vasculature. Moreover, the thrombotic material is simply autologous blood clots and the addition of tranexamic acid to delay thrombus resorption; the carrageenan is critical.
CTEPH is a unique subtype of PH which, depending on different clinical series, arises from an acute PE with an estimated clinical prevalence of 0.4–9.1%. However, studies have shown that only ~75% of patients presenting with CTEPH suffered prior acute thromboembolic events (17,18). Moreover, mimicking the progression of human CTEPH has been challenging as these components require large clot burden, chronic pulmonary-artery obstruction, PH, the development of systemic blood supply to ischemic lung regions, pulmonary vasculopathy in unobstructed territories, and RV remodeling (9). These features indicate why it has been so difficult to induce a reliable CTEPH animal model and why the previous attempts all failed. In our model, serial PEs help to demonstrate most of the key features of CTEPH including persistent and organized clots, the additional increase in PVR which translates from the combination of obstructive and non-obstructive microvascular disease, RV hypertrophy, RV dysfunction, perivascular inflammation, and the appearance of cellular-fibrotic lesions. We chose the left jugular vein as the administration route due to its accessibility and the anatomy of the circulatory system of the rat. In the rat, venous blood flow is directed from the caudal veins via the inferior vena cava directly to the right atrium, thus bypassing the portal circulation (19). Therefore, this administration route assures that the pulmonary arteries are the first to ramify, and the arterial tree filter retains the autologous blood clots, decreasing the risk of non-pulmonary bead deposition and reducing the damage of the blood flow. Our observations of the microspheres supported this concept.
Our previous study proved the important role apoptosis-resistant endothelial cells play in pulmonary vascular remolding, which results in intimal proliferation (20). In our present study, levels of mRNA and protein-encoding in the pro-apoptotic protein BAX gradually increased following repeated embolisms. In parallel, the levels of the anti-apoptotic protein BCL-2 decreased following PE induction. The decreased endothelial cell apoptosis detected by TUNEL and the increase in factor VIII in endothelial cells detected by immunohistochemistry support the findings of our previous. Carrageenan, an inflammatory polysaccharide made up of galactose and 3,6-anhydrogalactose, is widely used to induce inflammation (21). In this study, carrageenan was applied to establish an aseptic inflammation cellular environment. Although the functional role of inflammatory processes in the development of PH is not yet fully understood, the favorable effects when these processes are inhibited suggest their role is important in pulmonary vascular remodeling in animal and human PH (22). Our new CTEPH rat model is based on the original one but is much improved, mainly because of the massive and acute increases in inflammation induced by carrageenan. Namely, it may facilitate the understanding of why we are unable to attribute CTEPH pathogenesis to a single or recurrent PE. Pulmonary vasculopathy was found in the distal unobstructed territories and residual/recurrent PH after PEA. It has been reported that inhibiting the NF-κB activity can increase the cellular sensitivity to TNF-α (23), while NF-κB can withstand apoptosis and necrosis. Moreover, activation of NF-κB to induce apoptosis and TNF-α activation of NF-κB is crucial in host cell resistance to pathogens (24). In this study, we observed an increase of TNF-α alongside an increase in NF-κB. For this reason, inhibiting the activity of NF-κB may become a keystone in the treatment and development of new anti-inflammatory therapies for CTEPH.
Study limitations
Although we have successfully established a CTEPH model in rats in this study, isolating the pulmonary endothelial cells from pulmonary artery and performing in vitro assays was extremely challenging. In the future, isolated pulmonary artery endothelial cells from rats and patients of CTEPH will be conducted to explore the role of inflammation in the CTEPH in vitro, and then a new therapeutic strategy of CTEPH will be proceed.
Conclusions
In summary, we generated a small animal model that could replicate the key phenotype of CTEPH. This may not only facilitate experiments to reveal a better pathophysiological understanding of how PE transitions to CTEPH in humans, but may provide an animal model to evaluate novel treatment strategies. Inflammatory factor TNF-α activated via NF-κB may give rise to apoptosis-resistant endothelial cells resulting in the pulmonary vascular remolding. Activation of inflammatory pathways, which may be the result of systemic inflammation in combination with neurohumoral activation, presents a new understanding of CTEPH. Anti-inflammation may aid in the future preventative strategies of CTEPH.
Supplementary
The article’s supplementary files as
Acknowledgments
Funding: This research was supported by the National Natural Science Foundation of China (81770367), Projects of Financial Foundation On Health in Fujian Province (BPB-DCS2018).and Medical Innovation Project in Fujian Province (2019-CX-26).
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All experimental and animal care protocols were approved by the Animal Ethics Committee of the Fujian Medical University Institutional Animal Care (SYXK2012-0001) and the Use Committee and the Guide for the Care and Use of Laboratory Animals (NIH, Bethesda, MD, USA).
Footnotes
Conflicts of Interest: All authors have completed the ICMJE uniform disclosure form (available at http://dx.doi.org/10.21037/atm.2020.02.86). The authors have no conflicts of interest to declare.
References
- 1.Toshner M, Pepke-Zaba J. Chronic thromboembolic pulmonary hypertension: time for research in pathophysiology to catch up with developments in treatment. F1000Prime Rep 2014;6:38. 10.12703/P6-38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arias-Loza PA, Jung P, Abeßer M, et al. Development and Characterization of an Inducible Rat Model of Chronic Thromboembolic Pulmonary Hypertension. Hypertension 2016;67:1000-5. 10.1161/HYPERTENSIONAHA.116.07247 [DOI] [PubMed] [Google Scholar]
- 3.Kulkarni SK, Bhairappa S, Bishnoi A, et al. Chronic thromboembolic pulmonary hypertension: an enigma. BMJ Case Rep 2018. doi: . 10.1136/bcr-2018-225764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Freed DH, Thomson BM, Berman M, et al. Survival after pulmonary thromboendarterectomy: effect of residual pulmonary hypertension. J Thorac Cardiovasc Surg 2011;141:383-7 10.1016/j.jtcvs.2009.12.056 [DOI] [PubMed] [Google Scholar]
- 5.Montani D, Chaumais MC, Guignabert C, et al. Targeted therapies in pulmonary arterial hypertension. Pharmacol Ther 2014;141:172-91. 10.1016/j.pharmthera.2013.10.002 [DOI] [PubMed] [Google Scholar]
- 6.Diaz JA, Obi AT, Myers DD, Jr, et al. Critical review of mouse models of venous thrombosis. Arterioscler Thromb Vasc Biol 2012;32:556-62. 10.1161/ATVBAHA.111.244608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Runyon MS, Gellar MA, Sanapareddy N, et al. Development and comparison of a minimally-invasive model of autologous clot pulmonary embolism in Sprague-Dawley and Copenhagen rats. Thromb J 2010;8:3. 10.1186/1477-9560-8-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Courtney DM, Watts JA, Kline JA. End tidal CO(2) is reduced during hypotension and cardiac arrest in a rat model of massive pulmonary embolism. Resuscitation 2002;53:83-91. 10.1016/S0300-9572(01)00504-4 [DOI] [PubMed] [Google Scholar]
- 9.Mercier O, Fadel E. Chronic thromboembolic pulmonary hypertension: animal models. Eur Respir J 2013;41:1200-6. 10.1183/09031936.00101612 [DOI] [PubMed] [Google Scholar]
- 10.Grandi L, Grandi RA, Tomasi CD, et al. Acute and chronic consequences of polidocanol foam injection in the lung in experimental animals. Phlebology 2013;28:441-4. 10.1258/phleb.2012.012120 [DOI] [PubMed] [Google Scholar]
- 11.Mercier O, Tivane A, Dorfmüller P, et al. Piglet model of chronic pulmonary hypertension. Pulm Circ 2013;3:908-15. 10.1086/674757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deng C, Wu D, Yang M, et al. The role of tissue factor and autophagy in pulmonary vascular remodeling in a rat model for chronic thromboembolic pulmonary hypertension. Respir Res 2016;17:65. 10.1186/s12931-016-0383-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moser KM, Guisan M, Bartimmo EE, et al. In vivo and post mortem dissolution rates of pulmonary emboli and venous thrombi in the dog. Circulation 1973;48:170-8. 10.1161/01.CIR.48.1.170 [DOI] [PubMed] [Google Scholar]
- 14.Satoh T, Satoh K, Yaoita N, et al. Activated TAFI Promotes the Development of Chronic Thromboembolic Pulmonary Hypertension: A Possible Novel Therapeutic Target. Circ Res 2017;120:1246-62. 10.1161/CIRCRESAHA.117.310640 [DOI] [PubMed] [Google Scholar]
- 15.Sato H, Hall CM, Griffith GW, et al. Large animal model of chronic pulmonary hypertension. ASAIO J 2008;54:396-400. 10.1097/MAT.0b013e31817efa85 [DOI] [PubMed] [Google Scholar]
- 16.Mercier O, Tivane A, Raoux F, et al. A reliable piglet model of chronic thromboembolic pulmonary hypertension. Am J Respir Crit Care Med 2011;183:A2415. [DOI] [PubMed] [Google Scholar]
- 17.Aguero J, Hammoudi N, Bikou O, et al. Chronic Pulmonary Artery Embolization Models in Large Animals. Methods Mol Biol 2018;1816:353-66. [DOI] [PubMed]
- 18.Ende-Verhaar YM, Cannegieter SC, Vonk Noordegraaf A, et al. Incidence of chronic thromboembolic pulmonary hypertension after acute pulmonary embolism: a contemporary view of the published literature. Eur Respir J 2017. doi: . 10.1183/13993003.01792-2016 [DOI] [PubMed] [Google Scholar]
- 19.Popesko P, Rajtová V, Horák J. A Colour Atlas of Anatomy of the Small Laboratory Animals. Vol. 2: Rat, Mouse, and Hamster. London: Wolfe Publishing, 1990:46-89. [Google Scholar]
- 20.Deng C, Zhong Z, Wu D, et al. Role of FoxO1 and apoptosis in pulmonary vascular remolding in a rat model of chronic thromboembolic pulmonary hypertension. Sci Rep 2017;7:2270. 10.1038/s41598-017-02007-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cunha TM, Verri WA, Jr, Silva JS, et al. A cascade of cytokines mediates mechanical inflammatory hypernociception in mice. Proc Natl Acad Sci U S A 2005;102:1755-60. 10.1073/pnas.0409225102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stacher E, Graham BB, Hunt JM, et al. Modern age pathology of pulmonary arterial hypertension. Am J Respir Crit Care Med 2012;186:261-72. 10.1164/rccm.201201-0164OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Van Antwerp DJ, Martin SJ, Kafri T, et al. Suppression of TNF-alpha-induced apoptosis by NF-kappaB. Science 1996;274:787-9. 10.1126/science.274.5288.787 [DOI] [PubMed] [Google Scholar]
- 24.Smith ZR, Makowski CT, Awdish RL. Treatment of patients with chronic thromboembolic pulmonary hypertension: focus on riociguat. Ther Clin Risk Manag 2016;12:957-64. 10.2147/TCRM.S80131 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The article’s supplementary files as