

Abstract
The CRISPR-Cas9 system is a powerful gene-editing tool with wide-ranging applications, but the safe and efficient intracellular delivery of CRISPR components remains a challenge. In this study, we utilized biodegradable poly(beta-amino ester) nanoparticles to codeliver plasmid DNA encoding Cas9 and short guide RNA (sgRNA), respectively, to enable gene knockout following a CRISPR-mediated cleavage at one genomic site (1-cut edit), as well as gene deletion following DNA cleavage at two sites flanking a region of interest (2-cut edits). We designed a reporter system that allows for easy evaluation of both types of edits: gene knockout can be assessed by a decrease in near-infrared fluorescent protein (iRFP) fluorescence, whereas deletion of an expression stop cassette turns on a red-enhanced nanolantern fluorescence/luminescence dual reporter. Nanoparticles enabled up to 70% gene knockout due to small indels, as well as 45% gain-of-function expression after a 600-bp deletion edit. The efficiency of 2-cut edits is more sensitive than 1-cut edits to Cas9 and the sgRNA expression level. We demonstrate promising biodegradable nanoparticle formulations for gene editing. Our findings also provide new insights into the screening and transfection requirements for different types of gene edits, which are applicable for designing nonviral delivery systems for the CRISPR-Cas9 platform.
Keywords: polymer, nanoparticle, non-viral, CRISPR-Cas9, gene editing, gene delivery, PBAE
Graphical Abstract
The development of safe and effective tools to deliver gene-editing complexes is crucial to realizing the full clinical potential of the CRISPR-Cas9 system. This study presents a systematic examination of delivery parameters needed to enable CRISPR gene disruption and gene deletion using biodegradable polymeric nanoparticles encapsulating CRISPR plasmids.
Introduction
The CRISPR-Cas9 gene-editing system consists of a short guide RNA (sgRNA) conferring target sequence specificity, which complexes with the Cas9 endonuclease to enable site-specific DNA cleavage.1, 2, 3 This could result in gene knockout following nonhomologous end joining (NHEJ) or in the presence of a repair template, gene knockin through homology-directed repair (HDR). The targeting of sgRNAs to two sites flanking a genomic region of interest can result in the complete removal of the gene segment following NHEJ, which could be important in the silencing of genetic elements with no open-reading frames, such as microRNAs or long noncoding RNAs.4,5 CRISPR-mediated gene editing is contingent upon nuclear colocalization of both the Cas9 protein and sgRNA, and efficient intracellular delivery of CRISPR components remains a challenge.
Viral vectors have been demonstrated to be effective for delivery but are more challenging to produce for both preclinical and clinical studies and restricted in cargo size. This is problematic, as the Cas9 gene is over 4 kb long, and delivery using adeno-associated viruses (AAVs; packaging capacity ∼4.7 kb) sometimes requires that different CRISPR components be packaged in separate viral particles, introducing complexity and potentially reducing efficacy.6,7 Synthetic vectors are largely agnostic to cargo size, and several recent reports have demonstrated strategies for nonviral intracellular delivery of the CRISPR-Cas9 gene-editing platform. These include nanoparticle delivery of Cas9 and sgRNA as a ribonucleoprotein (RNP) complex8, 9, 10, 11, 12 or in the form of Cas9 mRNA and sgRNA.13,14 Cas9 and sgRNA encoded in plasmid DNA are another delivery format for CRISPR gene editing. Plasmid DNA can be easily constructed using standard molecular cloning techniques to include different Cas9 structures,15,16 multiplex sgRNA,17 and transcriptional targeting elements for cell type-specific editing.18 Furthermore, large libraries of biomaterials previously used for plasmid DNA delivery can be screened for CRISPR gene editing in a high-throughput manner19 to yield optimal formulations for gene editing in different applications.
It is important to note that delivery of CRISPR gene-editing complexes in the form of plasmid DNA carries several potential safety concerns that must be taken into account when designing translationally relevant therapeutics. For example, there is a risk of plasmid DNA unintentionally integrating into the host genome, inducing insertional mutagenesis when highly active promoter elements are inserted into oncogenes or disrupt tumor-suppressor genes.20 Furthermore, plasmid DNA encoding Cas9 and sgRNA increases the persistence time of CRISPR RNPs inside the cell, which has been shown to increase off-target editing.21 In this respect, delivery of CRISPR gene-editing complexes in the form of mRNA or proteins has the benefit of low persistence time and reduced off-target editing. As effective nonviral delivery vehicles for mRNA or protein complexes are still somewhat lacking, however, CRISPR delivery in these formats can often suffer from low serum tolerance or poor in vivo efficacy.22
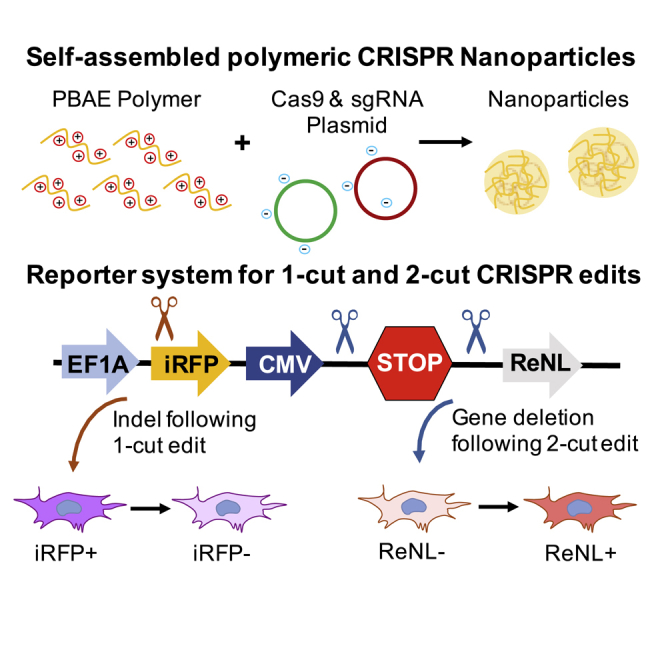
Although several studies have reported strategies for nonviral CRISPR plasmid delivery,18,23, 24, 25, 26 most involve gene-knockout applications using sgRNA designed to enable cleavage at a single site, and none, to our knowledge, have investigated the transfection requirements for gene deletion after cleavage at multiple sites. In this study, we designed a novel reporter system for easy detection of gene knockout following CRISPR-mediated cleavage at one genomic site (1-cut edit), as well as gene deletion following DNA cleavage at two sites flanking a region of interest (2-cut edit). We used poly (beta-amino esters) (PBAEs), a class of biodegradable cationic polymers that have been shown to be effective at plasmid DNA delivery27 for intracellular delivery of plasmid DNA encoding both the Cas9 endonuclease and sgRNA, respectively, and demonstrate that these polymeric nanoparticles enable efficient 1-cut, as well as 2-cut, edits. Moreover, we systematically varied transfection parameters to probe the relationship between the expression of CRISPR components and the subsequent efficacy of different types of CRISPR-mediated edits. Our results provide important insights on the threshold gene-expression levels required for 1- and 2-cut edits in easy-to-transfect, as well as hard-to-transfect, cell lines.
Results
Polymeric Nanoparticles for Gene Delivery
Polymer 446, which has been shown previously to be effective at plasmid DNA delivery to a variety of cells,28,29 was used to transfect HEK293T cells (Figure 1A). The newly developed branched polymer 7,8-4-J11 enabled higher transfection efficacy in B16-F10 murine melanoma cells30 (Figure S1) and was used to transfect these cells. Both polymers formed nanoparticles 100–200 nm in diameter with positive zeta potentials (12–25 mV) (Figure 1B). Transfection efficacy, as assessed with a GFP reporter plasmid, showed that >80% cells were transfected in both cell lines (Figure 1C). However, when geometric mean fluorescence was used to quantify expression, 293T cells achieved expression that was nearly 1 order of magnitude higher than B16 cells.
Figure 1.

PBAEs Form Nanoparticles with Plasmid DNA and Enable Transfection in HEK293T and B16-F10 Cells
(A) Polymer structures for 446 and 7,8-4-J11, which were used to transfect HEK293T and B16-F10 cells, respectively. (B) Nanoparticle hydrodynamic diameter and zeta potentials, as measured by dynamic light scattering. 446 nanoparticles were formulated at 60 w/w, whereas 7,8-4-J11 nanoparticles were formulated at 30 w/w. (C) Transfection efficacy, as measured by nanoparticles delivering GFP; 600 ng/well dose was used. Bars show mean + SEM; n = 4. (D) TEM image of 7,8-4-J11 nanoparticles. Scale bars, 100 nm.
Gene Knockout Following 1-Cut Edits
The efficiency of 1-cut edits was assessed in 293T cells constitutively expressing a destabilized form of GFP (GFPd2). GFPd2 is ubiquitinylated for rapid degradation and has a half-life (t1/2) of around 2 h (compared to a half-life of 26 h for wild-type GFP).31 This allows for rapid detection of gene knockout, which can be assessed by a decrease in GFP fluorescence following transfection with nanoparticles encapsulating two plasmids encoding the Cas9 endonuclease and a sgRNA targeting GFP, respectively. Nanoparticles codelivering both plasmids enabled coexpression, generating 70% gene knockout, as assessed by a decrease in GFP fluorescence, and 24% gene knockout, as assessed by Tracking of Indels by Decomposition (TIDE) analysis of genomic DNA; formulations delivering either component alone had negligible effects (Figure 2A). A kinetic study revealed that gene knockout reached maximal levels on day 3 and was maintained for over 3 weeks (Figure 2B). The Surveyor mutation detection assay was performed on cells treated with the combination nanoparticles or each component alone (Figure 2C) and confirmed that edits occurred only when both CRISPR components were delivered. Sanger sequencing revealed that most edits were single base-pair indels (Figure 2D; Figure S2), which likely caused frameshift mutations and subsequent gene silencing.
Figure 2.

446 Nanoparticles Enable Sustained and Robust Gene Knockout in HEK293T Cells
(A) GFP knockout experiments in HEK-GFPd2 cells showed gene knockout only when plasmids coding for both components were codelivered. Gene knockout was assessed by flow cytometry analysis of loss of GFP fluorescence and using TIDE analysis of Sanger sequencing data of genomic DNA of treated cells. n = 4; data presented as mean+ SEM. (B) Gene knockout was sustained over the 3-week experiment. n = 4. (C) Surveyor mutation detection assay confirms genomic DNA cleavage in the Cas9+/sgRNA+ treatment group. (D) Sanger sequencing of edited cells suggest that all edits were small indels. sgRNA targeting region highlighted in gray; protospacer adjacent motif (PAM) site highlighted in red.
Gain-of-Function Edits after 2-Cut Stop-Cassette Deletion
We designed a reporter system based on the Ai9 mouse32 in which an expression stop cassette consisting of two simian virus 40 (SV40) terminators in series was placed upstream of a red-enhanced nanolantern (ReNL) fluorescence-luminescence dual reporter33 (Figure 3A). This CRISPR-stop expression cassette was cloned into a piggyBac transposon plasmid to facilitate genomic integration at high efficiency after cotransfection with a piggyBac transposase plasmid.34 A near-infrared fluorescent protein (iRFP670)35 was also incorporated into the system as a selection marker for positively expressing cells during fluorescence-activated cell sorting (FACS). Thus, this system can be easily used to generate stably expressing reporter cell lines for rapid readout of knockout, as well as deletion mutations.
Figure 3.

Deletion of an Expression Stop Cassette Following 2-Cut Edits Results in Gain-of-Function ReNL Expression
(A) Schematic demonstrating that complete removal of the dual-SV40 stop cassette results in turning on ReNL. (B) 2-cut gene-deletion efficiency in cells treated with Cas9 and different sgRNAs, as assessed by flow cytometry, for turning on the ReNL fluorescence reporter (Fluorescent Reporter), gel electrophoresis-based genomic DNA analysis for total % editing (Total Edits), and % editing leading to ReNL expression (Effective Edits). n = 4; data presented as mean + SEM. (C) Gel electrophoresis of PCR products of the genomic region surrounding the stop cassette show differential banding patterns in treated cells. (D) Fluorescence microscopy image of untransfected control cells and cells transfected with Cas9 + sg1 or Cas9 + sg2 + sg3 plasmids, respectively. Scale bars, 200 μm.
The sgRNA sequence sg1, which was designed to remove both SV40 sequences (cut sites indicated by blue arrows in Figure 3A) via a 630-bp deletion, resulted in turning on ReNL expression in nearly 50% of cells when cotransfected with the Cas9 plasmid. In contrast, sgRNA sequences sg2 or sg3 were designed to remove only one SV40 sequence and yielded negligible levels of ReNL expression. A plasmid containing both sg2 and sg3 sequences governed by two U6 promoters (sg2 + sg3) also resulted in turning on expression through the deletion of both SV40 sequences (Figure 3B). Gain of ReNL fluorescence increased steadily after nanoparticle transfection, reaching a plateau at 3 days (Figure S3). Therefore, all experiments assessing the efficacy of 2-cut edits were performed at 3 days post-transfection. Genomic DNA of cells treated with each sgRNA was PCR amplified for the 800-bp region immediately surrounding the stop cassette. Gel electrophoresis of the PCR products revealed unique banding patterns for each sgRNA (Figure 3C). For cells treated with sg1, a faint band around 150 bp corresponded to the deletion of around 630 bp and the complete removal of both SV40 sequences. The banding pattern for cells treated with sg2 or sg3 showed a band around 500 bp, indicating the removal of only one SV40 sequence (270 bp deletion). As flow cytometry results showed negligible ReNL expression in these cells, this demonstrates that the remaining SV40 sequence was sufficient for blocking transcription of the downstream ReNL sequence and that removal of both SV40 sequences (>450 bp deletion) was necessary for gain-of-function ReNL expression. For cells treated with the combination sg2 + sg3 plasmid, a faint band around 500 bp was observed, indicating that only one SV40 sequence was deleted in a fraction of edits, and a second band around 300 bp indicated that both SV40 sequences were deleted in other cells. Thus, the level of editing that led to functional turning on of ReNL (termed Effective Edits in Figure 3B) was lower in these cells than the total editing level.
qRT-PCR of cells transfected with combination Cas9 and sg1 plasmids revealed that Cas9 mRNA levels stayed relatively constant throughout all time points evaluated (Figure 4A). Western blots over the same time course showed that Cas9 protein levels steadily accumulated after transfection; expression levels peaked at day 2 and became virtually undetectable after 11 days (Figure 4B; Figure S6). sgRNA levels plateaued after 48 h (Figure 4C), and the same trend was observed in ReNL mRNA levels after stop-cassette removal (Figure 4D).
Figure 4.

Expression Kinetics of CRISPR Components after Codelivery of Cas9 and sg1 Plasmids in 293T Cells
(A–D) Cas9 mRNA (A; red curve) and protein expression (A, blue curve; B) were measured over time in HEK293T cells. (C) sgRNA and (D) ReNL mRNA expression kinetics. n = 2 for qRT-PCR experiments; n = 1 for western blots.
Expression Thresholds for 1-Cut and 2-Cut Edits
In order to assess the expression levels necessary to achieve 1-cut knockout edits and 2-cut gain-of-function edits, respectively, we varied the dosage of plasmid DNA delivered in nanoparticles. In 293T cells engineered to express the CRISPR-stop gene construct, a GFP reporter was used to gauge transfection levels. Results showed that lowering the total DNA dose from 600 to 300 ng did not change the percentage of cells positively expressing GFP, but the geometric mean of fluorescence decreased by nearly 50% (Figure 5A). This effect can be observed in flow cytometry histograms as the 300-ng treatment yielded a larger population of cells with low GFP fluorescence compared to the 600-ng treatment (Figure 5D, left panel). The lowering of the total DNA dose significantly decreased levels of 2-cut deletion edits (Figure 5B) but did not significantly change the levels of 1-cut knockout edits (Figure 5C).
Figure 5.

DNA Dosage Titration Reveals Different Threshold Expression Requirements for 1-Cut and 2-Cut Edits
(A) DNA dosage decrease from 600 ng to 300 ng did not change the overall percentage of GFP-positive cells but significantly decreased the geometric mean of expression. (B and C) Dosage decrease significantly decreased the efficacy of 2-cut gene deletion edits (B) but not 1-cut iRFP knockout edits (C). Statistical significance determined by Holm-Sidak corrected multiple t tests; ∗∗p < 0.01, ∗∗∗p < 0.001. Data presented as mean + SEM; n = 4. (D) Flow cytometry plots of cells treated with different DNA doses.
We varied the total DNA dose delivered over a wider range in order to probe more thoroughly the effect of transfection efficacy on gene-editing levels (Figure 6A). The plotting of percent editing as a function of Cas9 mRNA expression levels revealed a logarithmic relationship for 1-cut edits (R2 = 0.9550) and a linear relationship for 2-cut edits (R2 = 0.9195). Transfection levels were further varied by manipulating cellular metabolic rates through incubation temperature variation (Figure 6B). Cells were transfected using the same nanoparticle formulation delivering the same DNA dose, after which, they were either incubated at standard 37°C or treated with a transient “cold shock” via incubation at 30°C. Transfection efficacy, as measured by Cas9 mRNA expression levels, increased significantly in cold-shocked cells; the same trend was observed for the level of 2-cut edits. Interestingly, cold shock treatment did not significantly change the level of 1-cut editing efficiency, which is consistent with the results from dose-titration experiments.
Figure 6.

1-Cut and 2-Cut Edits in Easy-to-Transfect HEK293T Cells and Hard-to-Transfect B16-F10 Cells
(A) 1-cut edit efficiency correlated logarithmically with level of transfection, as indicated by qRT-PCR measurement of Cas9 mRNA expression, whereas 2-cut edit efficiency correlated linearly in 293T cells. (B) In B16 cells, transient cold shock after transfection significantly increased transfection efficacy (measured by Cas9 mRNA expression levels), as well as 2-cut editing efficiency, but no significant change was seen in 1-cut editing efficiency, as assessed by Holm-Sidak corrected multiple t tests; ∗p < 0.05, ∗∗p < 0.01. (C) B16 cells achieved minimal levels of 2-cut edits; 1-cut edits were lower compared to 293T cells, but the difference is smaller. Data in (B) and (C) shown as mean + SEM; n = 4. (D and E) Differences in editing are observed in flow cytometry histograms.
1-cut knockout of iRFP expression and 2-cut gain-of-function edits were also performed on B16-F10 murine melanoma cells, which achieved lower levels of transfection compared to 293T cells. This was seen both when transfection was assessed using the geometric mean fluorescence of GFP expression when using a GFP reporter gene for nanoparticle screening (Figure 1), as well as the level of Cas9 mRNA expression (Figure 4; Figure S5). The lower transfection level in B16 cells was reflected, most notably, in the results for 2-cut editing, where the ReNL fluorescence observed in B16 cells was 1 order of magnitude lower than 293T cells (Figures 6C and 6D). Interestingly, the effect of lower transfection efficacy was less pronounced for 1-cut iRFP knockout experiments. Although lower knockout levels were observed in B16 compared to 293T cells, the difference was much smaller (12% for B16 and 33% for 293T). Representative flow cytometry histograms showed that in 293T cells, 1-cut editing produced a new iRFP-negative peak, whereas no such peak was observed in B16 cells, which showed an overall population shift of iRFP expression (Figure 6E). Since each cell very likely contains several copies of iRFP integrated into the genome,36 the peak in 293T cells indicates that 1-cut editing produced a small population of cells with complete iRFP knockout, whereas the edited population in B16 cells lost some but not all copies of iRFP, and the incomplete iRFP knockout in this population resulted in a general shift in fluorescence. Combined, this validates results seen earlier with dose titration, as well as temperature-modulation experiments, and confirms our hypothesis that 1-cut knockout edits require a lower expression threshold compared to 2-cut edits.
Standard transfection reagents were also used to assess 2-cut editing efficiency. For both cell lines, the commercially available cationic polymer transfection reagent jetPrime resulted in significantly lower editing levels than PBAE nanoparticles (Figure S7). The commercially available cationic lipid transfection reagent Lipofectamine 3000 enabled significantly higher editing than the earlier-generation linear PBAE polymer 446 in 293T cells but did not achieve significantly higher levels of editing than the newly developed next-generation branched PBAE polymer 7,8-4-J11 in harder-to-transfect B16 cells. Notably, Lipofectamine 3000 caused significantly higher levels of cytotoxicity than both PBAE nanoparticle formulations, further demonstrating the advantage of using a biodegradable gene-delivery system.
A Multiplex tRNA-Guide RNA (gRNA) Expression System
To facilitate a simpler method for multiplex CRISPR editing, we designed a tRNA-gRNA expression system,17 which utilizes the cell’s endogenous tRNA processing machinery to generate multiple sgRNAs (Figure 7A). With the use of a simple Golden Gate assembly strategy, we created a plasmid in which the targeting sequences and gRNA scaffolds of sg2 and sg3 are arrayed in tandem with pre-tRNA, with all components governed by a single U6 promoter. Mature sgRNA is released after processing of the primary RNA transcript by tRNA-processing RNases. When transfected into cells alongside the Cas9 plasmid, this tRNA-gRNA plasmid enabled similar levels of 2-cut editing as the plasmid in which a U6 promoter governed each sgRNA (Figure 7B). This demonstrates that the multiplex tRNA-gRNA expression system effectively expressed both sgRNAs required for 2-cut editing.
Figure 7.

A tRNA-gRNA Expression System for Multiplex Editing
(A) Schematic of a multiplex sgRNA expression system in which multiple tRNA-gRNA units are arrayed in tandem. The primary RNA transcript is processed by the endogenous tRNA machinery, releasing mature sgRNAs. (B) The tRNA-gRNA plasmid coding for sg2 and sg3 results in similar levels of 2-cut editing compared to a plasmid in which each sgRNA is governed by an individual U6 promoter (sg2 + sg3). Statistical analysis was assessed by one-way ANOVA with Tukey post hoc tests. Data presented as mean ± SEM; n = 4.
Discussion
In this work, we demonstrated that both linear and branched PBAE nanoparticles, codelivering two DNA plasmids encoding Cas9 and sgRNA, respectively, can achieve efficient gene editing in both 1-cut knockout, as well as 2-cut gene-deletion applications. We created a novel CRISPR-stop reporter system that can be used to assess both types of edits: an iRFP fluorescent reporter can be silenced by indels after 1-cut edits, whereas an expression stop cassette upstream of a ReNL reporter can be deleted using 2-cut edits for gain-of-function ReNL expression. This expression cassette was cloned into a piggyBac transposon system and can be used to generate stably expressing cell lines to investigate gene-editing efficacy in vitro, eliminating the need to culture primary cells from the Ai9 mouse,37 on which our reporter system is based. This system further has the potential to be used as an in vivo reporter for live-animal imaging studies of effective 2-cut gain-of-function ReNL expression using the red-shifted luminescent properties of ReNL. With the use of two cell lines stably expressing this construct—easy-to-transfect HEK293T and hard-to-transfect B16-F10—we further investigated the transfection requirements for each type of gene editing.
Several recent studies have demonstrated the feasibility of using polymeric nanoparticles, including a different PBAE formulation,38 to deliver CRISPR gene-editing components in the form of plasmid DNA.18,23, 24, 25 All of these systems have exclusively investigated the use of 1-cut editing to achieve gene knockout, and none has presented a systematic study of the expression levels required for 1-cut and 2-cut edits. The removal of a gene segment requires sgRNA to target two sites flanking the region of interest and is significantly more difficult than 1-cut knockout edits.5 To date, only 3 studies have reported the use of nonviral delivery vectors for 2-cut gene deletion by delivering Cas9 mRNA and sgRNA14 or RNP complexes,12,37 but plasmid delivery with polymeric nanoparticles to achieve this type of deletion has not been previously reported. The use of DNA plasmids to encode Cas9 overcomes the manufacturing challenges of producing large scales of Cas9 mRNA or Cas9 protein, but the intracellular delivery and expression of exogenous DNA can be more challenging than the delivery of its downstream products.
We evaluated two types of PBAE nanoparticles to encapsulate Cas9 and sgRNA plasmids for intracellular delivery of gene-editing complexes. One of these was the well-published linear PBAE polymer 446 that has shown efficacy in multiple cell types, and one was a newly developed branched PBAE polymer 7,8-4-J11; both were found to be useful for developing efficacious biodegradable nanoparticles for gene editing. The cationic polymer and anionic DNA self assembled into nanoparticles 100–200 nm in diameter with positive zeta potentials (12–25 mV) (Figure 1). Previous reports have shown that high levels of codelivery can be achieved by pre-mixing plasmids prior to nanoparticle assembly.39 With the use of this strategy, we showed successful codelivery of CRISPR plasmids that enabled robust 1-cut gene knockout (Figure 2). More importantly, we demonstrated a versatile gene-deletion platform in which a single sgRNA targeting site flanking the region of interest and a combination of sgRNA targeting sites throughout the region of interest both resulted in successful removal of the entire gene segment (Figure 3). Successful deletion of up to 630 bp could be easily visualized through the gain-of-function expression of a ReNL fluorescence/luminescence dual reporter.
Evaluation of the expression kinetics of Cas9 and sgRNA revealed that Cas9 mRNA was maintained at high levels throughout the time period tested (4.5–48 h), whereas sgRNA expression reached peak levels at 48 h and plateaued thereafter (Figure 4). The plateau observed in the expression of sgRNA (but not Cas9) in the short term is likely due to a difference in the expression kinetics of the U6 promoter driving sgRNA expression compared to the cytomegalovirus (CMV) promoter driving Cas9 expression. After a slower start, sgRNA rapidly accumulated inside the cell, eventually reaching a plateau, at which point, the expression of additional copies of sgRNA was likely balanced out by plasmid dilution through cellular division. This is a common pattern seen in transient gene expression induced by nanoparticle transfection,40 and we would expect expression levels of both Cas9 and sgRNA to drop at longer time points. Indeed, when we evaluated Cas9 protein accumulation at longer time periods, we found that Cas9 protein levels declined steadily after 48 h and were virtually undetectable at 11 days post-transfection (Figure S6). Compared to delivery of CRISPR components in mRNA or protein form, where Cas9 protein expression decreased to below levels of detection after 3 days,21 the long Cas9 persistence time following plasmid DNA delivery raises concerns of off-target editing. It is important to note that the risk of off-target editing following synthetic nanoparticle delivery of CRISPR plasmids is significantly lower than when viral vectors are used for gene editing. However, these risks can be further mitigated by using Cas9 variants41,42 that have been engineered to have enhanced proofreading and lower off-target editing rates compared to wild-type Cas9.
We further explored the transfection requirements for 1-cut and 2-cut edits by titrating the total DNA dose delivered. Interestingly, the decrease of total DNA dose from 600 ng to 300 ng significantly decreased the level of 2-cut editing but did not affect the level of 1-cut edits (Figure 5). The same trend was observed when transfection efficiency was varied by treating transfected cells with a minor cold shock (Figure 6). A brief cold shock slowed the rate of cellular division, which enhanced protein accumulation in expressing cells and decreased the rate of plasmid DNA dilution in the cell population. This increased transfection efficiency and the level of 2-cut edits, which is consistent with previous reports using cold shock treatment to enhance the editing efficiency of zinc-finger nuclease (ZFN)-mediated gene disruption43 or CRISPR-mediated, homology-directed repair.44 In contrast, cold shock treatment did not significantly change the efficiency of 1-cut edits. Recent studies on the enzyme kinetics of sgRNA-Cas9 RNPs have reported that whereas Cas9-sgRNA binding (k = 6.1 s−1), target DNA binding (t1/2 = 4–40 s), and DNA cleavage events (k = 25–90 s−1) happen very quickly,45 the release of DNA cleavage products is extremely slow (t1/2 = 43–91 h),46 causing Cas9 to be virtually a single turnover enzyme. Taken together with these results, our data suggest that 2-cut edits have a much higher expression threshold than 1-cut edits, because twice the number of DNA cleavage events are required for successful edits to occur.
The expression thresholds of 1-cut and 2-cut edits have important implications on gene editing in different cell types. To demonstrate this, we compared the gene-editing efficiency in easier-to-transfect HEK293T and harder-to-transfect B16-F10 cells. HEK293T cells were derived from the parent HEK293 human embryonic kidney cell line and further modified with the SV40 large T antigen.47 The SV40 large T antigen causes plasmid DNA containing the SV40 origin of replication to unwind inside HEK293T cells, allowing for plasmid DNA replication and high levels of transfection.48 HEK293T cells are widely known to be an easy-to-transfect cell line and are commonly used for the production of recombinant proteins49 and viral vectors50 after transient plasmid DNA transfection using commercially available transfection reagents. B16-F10 cells, commonly used for tumor inhibition studies, are a well-established murine melanoma cell line.51 B16-F10 cells have been reported to be much more difficult to transfect with nonviral vectors,52 which is, in part, due to significantly lower nanoparticle uptake levels (Figure S8).
Although the top nanoparticle formulation for each cell line achieved >80% transfection, as assessed by percentage of total cells transfected, the level of expression, as assessed by the normalized geometric mean of expression of a GFP reporter, was 1 order of magnitude higher for 293T cells (Figure 1). This discrepancy was reflected in the level of 2-cut edits, as B16 cells showed very minimal levels of ReNL expression after stop-cassette deletion (Figure 6). In contrast, the difference in editing efficiency between the two cell lines was much smaller for 1-cut iRFP knockout (<3-fold difference compared to nearly 44-fold difference for 2-cut edits). These results further validate our hypothesis that the efficiency of 2-cut edits correlates more strongly with the level of DNA expression. Taken together, we have demonstrated that low levels of DNA transfection severely limit 2-cut editing efficiency. One solution for improving 2-cut editing efficiency is to deliver CRISPR components in the RNP form, which we recently demonstrated enabled >40% 2-cut editing in B16 cells after nanoparticle delivery of CRISPR RNPs targeted to excise the transcription stop cassette.53 This demonstrates that bypassing limits in DNA transfection altogether may be a viable way to achieve efficient 2-cut editing in hard-to-transfect cell lines such as B16 cells.
Finally, we designed and implemented a tRNA-gRNA plasmid in which the expression of multiplex sgRNAs is governed under a single U6 promoter. The expression of two sgRNAs required for turning on ReNL fluorescence in these tRNA-gRNA tandem repeats enabled similar levels of editing compared to that of a plasmid in which each sgRNA is governed by its own U6 promoter (Figure 7). This expression system has the advantage of ease of synthesis, as upward of 6 sgRNAs can be arranged in tandem using a single Golden Gate assembly reaction.17 More importantly, the tRNA-gRNA system reduces the need for repeating U6 promoters, enabling the use of a much smaller plasmid construct, especially at high numbers of sgRNAs. Originally developed for use in rice plants,17 this system has also been adapted for use in yeast54 and zebrafish.55 To our knowledge, this is the first time it has been adapted for gene editing in mammalian cells.
In summary, we have demonstrated that PBAE nanoparticles codelivering plasmids encoding Cas9 and sgRNA can achieve 1-cut knockout, as well as 2-cut deletion edits. We designed a novel reporter system, whereby both modes of edits can be easily evaluated. 2-cut deletion events required much higher levels of transfection than 1-cut gene knockout edits, which we demonstrated by titrating the DNA dosage delivered, treating transfected cells with a transient cold shock, and comparing editing efficiencies in two cell lines with different transfection efficacy. The PBAE/DNA nanoparticles optimized here are promising for DNA-based nonviral gene editing. Further, the results presented herein have implications on the design and screening of next-generation nonviral delivery vehicles broadly for CRISPR-Cas9 gene editing.
Materials and Methods
Materials
Small molecules used as monomers for polymer synthesis were obtained as follows: bisphenol A glycerolate (1 glycerol/phenol) diacrylate (B7; 411167), trimethylolpropane triacrylate (B8; 246808), 2-(3-aminopropylamino)ethanol (E6; 09293), and N,N-diethyldiethylenetriamine (J11; 518832)56 were purchased from Sigma-Aldrich; 1,4-butanediol diacrylate (B4; 32780) and 4-amino-1-butanol (S4; A12680) were purchased from Alfa Aesar. The following plasmids were purchased from Addgene: hCas9 (41815),3 gRNA_GFP-T2 (41820),3 pCAG-GFPd2 (14760),31 PBCAG-EGFP (40973),57 piRFP670-N1 (45457),35 and tubulin-ReNL_pcDNA3 (89530).58 PB-CMV-MCS-EF1a-RFP PiggyBac plasmid (PB512B-1) and PiggyBac transposase expression plasmid (PB200A-1) were purchased from System Biosciences. sgRNA gBlock sequences were purchased from Integrated DNA Technologies (IDT), and the expression stop cassette was synthesized by Synbio Technologies (Monmouth Junction, NJ, USA). Restriction enzymes and T4 DNA ligase for molecular cloning were purchased from New England BioLabs.
Polymer Synthesis
Polymer 446 was synthesized by reacting monomers B4 and S4 at a molar ratio of 1.1:1 at 90°C with stirring overnight. The B4-S4 polymer was dissolved in anhydrous tetrahydrofuran (THF) at 167 mg/mL and added to monomer E6 (0.5 M in THF) at a 3:2-volume ratio and reacted at room temperature for 1 h. The end-capped polymer was washed in diethyl ether twice to remove unreacted monomers and oligomers. Solvents were removed in a vacuum desiccant chamber, and polymer was dissolved in DMSO at 100 mg/mL and then stored at −20°C with desiccant. Polymer 7,8-4-J11 was synthesized by reacting monomers B7, B8, and S4 at an overall vinyl:amine ratio of 2.2:1 and monomer concentration of 200 mg/mL in anhydrous DMSO at 90°C with stirring overnight; the acrylate monomer composition was 80% B7 and 20% B8 by mole fraction. Polymer end-capping and purification were done following the same procedure as polymer 446 but using monomer J11.
Nanoparticle Characterization
Nanoparticle hydrodynamic diameter was measured via dynamic light scattering (DLS) using a Malvern Zetasizer NanoZS (Malvern Instruments). Samples were prepared in 25 mM sodium acetate (NaAc) (pH 5.0) and then diluted 1:6 in 150 mM PBS to determine the hydrodynamic diameter in neutral, isotonic buffer. Zeta potential was measured by electrophoretic light scattering on the same instrument. Transmission electron microscopy (TEM) images were captured using a Philips CM120 (Philips Research) on 400 square mesh carbon-coated TEM grids. Samples were prepared at a polymer concentration of 1.8 mg/mL at 30 w/w in 25 mM NaAc, and 30 μL was allowed to coat TEM grids for 20 min. Grids were then rinsed with ultrapure water and allowed to dry fully before imaging.
Cell Culture and Cell Line Preparation
HEK293T and B16-F10 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Thermo Fisher Scientific), supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. Cells were induced to express fluorescent protein constructs constitutively using the PiggyBac transposon/transposase system. The GFPd2 gene was cloned into the PB512B-1 plasmid using restriction enzyme cloning to create a PiggyBac transposon plasmid containing the GFPd2 gene. A sequence containing iRFP and transcription stop sequences was cloned into the PBCAG-EGFP plasmid backbone, and the ReNL gene was inserted into this plasmid using restriction enzyme cloning to create a PiggyBac transposon plasmid containing the iRFP-STOP-ReNL sequence (plasmid available on Addgene). Each transposon plasmid was cotransfected with the PiggyBac transposase plasmid into HEK293T and/or B16-F10 cells using nanoparticles, as described below. Fluorescent protein signal from DNA not integrated into the cell genome was allowed to fade over 5 passages, after which, positive cells were isolated using FACS. Cells were further expanded for 3 more passages and sorted again to generate stably expressing cell lines.
sgRNA Design and Preparation
Single guide RNAs were designed using the http://chopchop.cbu.uib.no platform and ordered as gBlocks containing the U6 promoter, a unique 20-bp targeting sequence, and the duplex optimized sgRNA scaffold from IDT.5 The gBlocks were cloned into the pCAG-GFPd2 plasmid backbone using restriction enzyme cloning. sgRNA plasmids were transformed into DH5α-competent E. coli (NEB) and grown out overnight at 37°C in 5 mL Luria-Bertani (LB) broth liquid cultures, and plasmid DNA was harvested using the QIAprep Miniprep Kit (QIAGEN). Plasmid DNA was characterized using the NanoDrop spetrophotometer (Thermo Fisher Scientific) and sequence confirmed via Sanger sequencing before use in transfections. All sgRNA target sequences are listed in Table S2, and plasmids are available on Addgene.
The gRNA-tRNA plasmid containing multiplex sgRNA constructs under a single U6 promoter was synthesized according to the protocol by Xie et al.17 Briefly, the pGTR construct containing a sgRNA scaffold sequence, fused to a tRNA fragment, was synthesized as a gBlock from IDT and cloned into a plasmid via restriction enzyme cloning. This pGTR plasmid was used as the template DNA for PCR reactions that produced amplicons used in a hierarchical Golden Assembly process to generate a DNA fragment containing the tRNA-gRNA tandem arrays. This fragment was then cloned into a backbone plasmid containing a U6 promoter via restriction enzyme cloning. The sequences for the pGTR sequence and PCR primers used are listed in Table S3.
Transfection
Cells were plated at 15,000 cells per well (HEK293T) or 10,000 cells per well (B16-F10) in 100 μL complete medium in CytoOne 96-well plates (USA Scientific) and allowed to adhere overnight. Polymers and DNA were dissolved separately in 25 mM NaAc at the desired concentrations and then mixed together via pipetting. Nanoparticles were allowed to self-assemble for 10 min, and then 20 μL of the nanoparticle solution was added per well for a final volume of 120 μL and 600 ng DNA per well, unless otherwise noted; for transfection experiments using the CRISPR-Cas9 system, the Cas9 and sgRNA plasmids were used at a 1:1 weight ratio. For example, in order to formulate nanoparticles that would deliver 600 ng DNA per well in 96-well plates at a polymer-to-DNA weight ratio of 60 (60 w/w), plasmid DNA was first dissolved in 25 mM NaAc at 0.06 mg/mL and polymer dissolved at 3.6 mg/mL. These two solutions were then mixed at a 1:1-volume ratio and allowed to self-assemble into nanoparticles. The Nitrogen to Phosphate (N/P) ratio is 34.5 for the 446 60-w/w formulation and 14.5 for the 7,8-4-J11 30-w/w formulation (Calculation S1). Nanoparticles were incubated with cells for 2 h at 37°C, at which point, the media and nanoparticles were removed and replaced with fresh complete media. Commercially available transfection reagents jetPrime (Polyplus) and Lipofectamine 3000 (Thermo Fisher Scientific) were used as instructed by the manufacturer. For cold shock treatment, cells were transfected using standard transfection procedures and allowed to recover at 37°C after media change for 6 h before being moved to 30°C. Cells were maintained at 30°C for 3 days, after which time, they were moved back to 37°C.
Transfection and gene-editing efficacies were evaluated via flow cytometry using a BD Accuri C6 flow cytometer (BD Biosciences). For nanoparticle screening experiments using a GFP reporter gene, transfection was quantified via two methods: (1) the percentage of cells positively expressing GFP when gated against untreated cells was reported as % positive expression, and (2) the geometric mean fluorescence intensity in the FL1 channel (corresponding to GFP) for each treated well was normalized against that of untreated control wells, and the normalized geometric mean expression was reported. CRISPR knockout was quantified by normalizing the geometric mean fluorescence of treated wells to that of wells transfected with the Cas9 plasmid only. Gain of fluorescence was quantified as the percentage of cells positively expressing the fluorescent protein when gated against untreated control. Gene editing in gene-deletion experiments was also assessed by luminescence readings using the Promega Nano-Glo Luciferase assay system (Promega), measured with a Synergy2 plate reader (Biotek) with open optics and normalized to untreated control. Cell viability was assessed 24 h post-transfection using the MTS CellTiter 96 Aqueous One cell proliferation assay (Promega) (n = 4 ± SEM). Unless otherwise stated, flow cytometry to assess gene editing efficacy was performed on day 3 post-transfection.
Surveyor Assay
Genomic DNA from cells transfected with the combination Cas9-sgRNA plasmids and untransfected control was isolated using the GeneJET genomic DNA purification kit (Thermo Fisher Scientific). A 660-bp region flanking the predicted cut site was PCR amplified, and the PCR products were purified using the QIAquick PCR purification kit. 400 ng of PCR amplicons was hybridized, and the Surveyor assay was performed using the Surveyor mutation detection kit (IDT), following the manufacturer’s instructions. The uncut and cut DNA products were then run on a 2% agarose gel stained with ethidium bromide in Tris/borate/EDTA (TBE) buffer and imaged under UV light.
TIDE Analysis to Assess 1-Cut Editing Efficiency
Sanger sequencing was performed on purified PCR products from Surveyor assays. Sequencing data were uploaded to the online TIDE analysis tool (https://tide.deskgen.com) to assess 1-cut editing efficiency.
Gel Electrophoresis Assay to Assess 2-Cut Editing Efficiency
Genomic DNA from 293T-CRISPR-stop cells transfected with Cas9 and sgRNA plasmids or untransfected controls were isolated as described above. An 800-bp region flanking the predicted cut sites was PCR amplified, and PCR products were purified as described above. Standard gel electrophoresis was performed on PCR products using 2% agarose gel stained with ethidium bromide in TBE buffer at 80 V for 45 min and imaged under UV light to reveal unique banding patterns (Figure 3C). Band intensities were quantified using ImageJ image processing software, and % editing was calculated using the method reported by Schumann et al.59 Edits where >450 bp were deleted were quantified as Effective Edits, leading to gain-of-function ReNL expression, whereas all deletion edits were quantified as Total Edits for each sample.
Sanger Sequencing to Detect Gene Editing
PCR products for the Surveyor assay were cloned into plasmid vectors using the NEB PCR Cloning kit and transformed into DH5α-competent E. coli (NEB). 30 colonies were grown out in 5 mL liquid cultures overnight, and the plasmid DNA was isolated and characterized by Sanger sequencing.
qRT-PCR
Cells transfected with the combination Cas9-sgRNA plasmids in a 12-well plate were collected, and total RNA, including small RNAs (<100 nt), were extracted using the miRNeasy Mini Kit (QIAGEN). RNA was reverse transcribed using the iScript cDNA synthesis kit (Bio-Rad), and qRT-PCR was run on a StepOnePlus Real-Time PCR system (Thermo Fisher Scientific) using SYBR Green PCR Master Mix (Thermo Fisher Scientific). The qPCR program is as follows: 95°C for 10 min; 95°C 15 s, 55°C 30 s, and 60°C 30 s for 40 cycles. Primers used for qRT-PCR are listed in Table S1. Results are shown as fold expression over β-actin.
Western Blotting
Transfected cells in 12-well plates were lysed in a solution of 1× radioimmunoprecipitation assay (RIPA) buffer and 1× Protease/Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific). The lysate was cleared by centrifugation, protein concentration was determined using Pierce Micro BCA assay (Thermo Fisher Scientific), and samples were denatured in Laemmli sample buffer (Bio-Rad) in the presence of DTT. 50 μg proteins was loaded into 4%–15% Mini-PROTEAN TGX Precast Protein Gels (Bio-Rad). Proteins were then transferred to a polyvinylidene fluoride (PVDF) membrane using a Pierce Power Blotter (Thermo Fisher Scientific). Membranes were blocked in 5% nonfat milk for 1 h at room temperature (RT) and probed with primary antibodies against Cas9 (Cell Signaling Technologies; 14697; 1:500) or β-actin (Abcam; ab8226; 1:10,000) at 4°C overnight. Secondary antibodies were applied at RT for 1 h (mouse immunoglobulin G kappa binding protein conjugated to horseradish peroxidase [m-IgGκ BP-HRP]; Santa-Cruz; Sc-516102; 1:1,000). The membrane was developed with Amersham ECL (enhanced chemiluminescence) Western Blotting Detection Reagent (GE Healthcare) and imaged using an ImageQuant LAS 4000 CCD (charge-coupled device) imager (GE Healthcare). Semiquantitative analysis of Cas9 protein expression was done by calculating band intensities using ImageJ image analysis software and normalizing the intensity of Cas9 bands to that of β-actin.
Statistics
Prism 8 (GraphPad, La Jolla, CA, USA) was used for all statistical analyses and curve plotting. The statistical test used and number of experimental replicates are detailed in the captions for each figure. Statistical significance was denoted as follows: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Author Contributions
Conceptualization, Y.R., D.R.W., and J.J.G.; Methodology, Y.R., D.R.W., and J.J.G.; Investigation, Y.R., M.V., S.M., H.M.Y., and D.R.W.; Resources and Funding Acquisition, J.J.G.; Writing and Editing, Y.R., M.V., S.M., H.M.Y., D.R.W., and J.J.G.; Supervision and Administration, J.J.G.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
J.J.G. thanks the Bloomberg∼Kimmel Institute for cancer immunotherapy and the Research to Prevent Blindness James and Carole Free Catalyst Award for support. The authors thank the following organizations for financial support: NSF Graduate Research Fellowship (DGE-0707427 to D.R.W. and DGE-1232825); NIH (grants R01CA228133 and R01EB022148 to J.J.G.); Microscopy Core (grant S10 OD016374); and Wilmer Core (grant P30 EY001765).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtn.2020.04.005.
Supplemental Information
References
- 1.Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jinek M., East A., Cheng A., Lin S., Ma E., Doudna J. RNA-programmed genome editing in human cells. Elife. 2013;2:e00471. doi: 10.7554/eLife.00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mali P., Yang L., Esvelt K.M., Aach J., Guell M., DiCarlo J.E., Norville J.E., Church G.M. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ho T.-T., Zhou N., Huang J., Koirala P., Xu M., Fung R., Wu F., Mo Y.-Y. Targeting non-coding RNAs with the CRISPR/Cas9 system in human cell lines. Nucleic Acids Res. 2015;43:e17. doi: 10.1093/nar/gku1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dang Y., Jia G., Choi J., Ma H., Anaya E., Ye C., Shankar P., Wu H. Optimizing sgRNA structure to improve CRISPR-Cas9 knockout efficiency. Genome Biol. 2015;16:280. doi: 10.1186/s13059-015-0846-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang Y., Wang L., Bell P., McMenamin D., He Z., White J., Yu H., Xu C., Morizono H., Musunuru K. A dual AAV system enables the Cas9-mediated correction of a metabolic liver disease in newborn mice. Nat. Biotechnol. 2016;34:334–338. doi: 10.1038/nbt.3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yu W., Mookherjee S., Chaitankar V., Hiriyanna S., Kim J.-W., Brooks M., Ataeijannati Y., Sun X., Dong L., Li T. Nrl knockdown by AAV-delivered CRISPR/Cas9 prevents retinal degeneration in mice. Nat. Commun. 2017;8:14716. doi: 10.1038/ncomms14716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun W., Ji W., Hall J.M., Hu Q., Wang C., Beisel C.L., Gu Z. Self-assembled DNA nanoclews for the efficient delivery of CRISPR-Cas9 for genome editing. Angew. Chem. Int. Ed. Engl. 2015;54:12029–12033. doi: 10.1002/anie.201506030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zuris J.A., Thompson D.B., Shu Y., Guilinger J.P., Bessen J.L., Hu J.H., Maeder M.L., Joung J.K., Chen Z.-Y., Liu D.R. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat. Biotechnol. 2015;33:73–80. doi: 10.1038/nbt.3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang M., Zuris J.A., Meng F., Rees H., Sun S., Deng P., Han Y., Gao X., Pouli D., Wu Q. Efficient delivery of genome-editing proteins using bioreducible lipid nanoparticles. Proc. Natl. Acad. Sci. USA. 2016;113:2868–2873. doi: 10.1073/pnas.1520244113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee K., Conboy M., Park H.M., Jiang F., Kim H.J., Dewitt M.A., Mackley V.A., Chang K., Rao A., Skinner C. Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair. Nat. Biomed. Eng. 2017;1:889–901. doi: 10.1038/s41551-017-0137-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mout R., Ray M., Yesilbag Tonga G., Lee Y.-W., Tay T., Sasaki K., Rotello V.M. Direct Cytosolic Delivery of CRISPR/Cas9-Ribonucleoprotein for Efficient Gene Editing. ACS Nano. 2017;11:2452–2458. doi: 10.1021/acsnano.6b07600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jiang C., Mei M., Li B., Zhu X., Zu W., Tian Y., Wang Q., Guo Y., Dong Y., Tan X. A non-viral CRISPR/Cas9 delivery system for therapeutically targeting HBV DNA and pcsk9 in vivo. Cell Res. 2017;27:440–443. doi: 10.1038/cr.2017.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miller J.B., Zhang S., Kos P., Xiong H., Zhou K., Perelman S.S., Zhu H., Siegwart D.J. Non-Viral CRISPR/Cas Gene Editing In Vitro and In Vivo Enabled by Synthetic Nanoparticle Co-Delivery of Cas9 mRNA and sgRNA. Angew. Chem. Int. Ed. Engl. 2017;56:1059–1063. doi: 10.1002/anie.201610209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shen B., Zhang W., Zhang J., Zhou J., Wang J., Chen L., Wang L., Hodgkins A., Iyer V., Huang X., Skarnes W.C. Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects. Nat. Methods. 2014;11:399–402. doi: 10.1038/nmeth.2857. [DOI] [PubMed] [Google Scholar]
- 16.Konermann S., Brigham M.D., Trevino A.E., Joung J., Abudayyeh O.O., Barcena C., Hsu P.D., Habib N., Gootenberg J.S., Nishimasu H. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature. 2015;517:583–588. doi: 10.1038/nature14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xie K., Minkenberg B., Yang Y. Boosting CRISPR/Cas9 multiplex editing capability with the endogenous tRNA-processing system. Proc. Natl. Acad. Sci. USA. 2015;112:3570–3575. doi: 10.1073/pnas.1420294112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Luo Y.-L., Xu C.-F., Li H.-J., Cao Z.-T., Liu J., Wang J.-L., Du X.-J., Yang X.-Z., Gu Z., Wang J. Macrophage-Specific in Vivo Gene Editing Using Cationic Lipid-Assisted Polymeric Nanoparticles. ACS Nano. 2018;12:994–1005. doi: 10.1021/acsnano.7b07874. [DOI] [PubMed] [Google Scholar]
- 19.Steyer B., Carlson-Stevermer J., Angenent-Mari N., Khalil A., Harkness T., Saha K. High content analysis platform for optimization of lipid mediated CRISPR-Cas9 delivery strategies in human cells. Acta Biomater. 2016;34:143–158. doi: 10.1016/j.actbio.2015.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yin H., Kanasty R.L., Eltoukhy A.A., Vegas A.J., Dorkin J.R., Anderson D.G. Non-viral vectors for gene-based therapy. Nat. Rev. Genet. 2014;15:541–555. doi: 10.1038/nrg3763. [DOI] [PubMed] [Google Scholar]
- 21.Liang X., Potter J., Kumar S., Zou Y., Quintanilla R., Sridharan M., Carte J., Chen W., Roark N., Ranganathan S. Rapid and highly efficient mammalian cell engineering via Cas9 protein transfection. J. Biotechnol. 2015;208:44–53. doi: 10.1016/j.jbiotec.2015.04.024. [DOI] [PubMed] [Google Scholar]
- 22.Rui Y., Wilson D.R., Green J.J. Non-Viral Delivery To Enable Genome Editing. Trends Biotechnol. 2019;37:281–293. doi: 10.1016/j.tibtech.2018.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liang C., Li F., Wang L., Zhang Z.-K., Wang C., He B., Li J., Chen Z., Shaikh A.B., Liu J. Tumor cell-targeted delivery of CRISPR/Cas9 by aptamer-functionalized lipopolymer for therapeutic genome editing of VEGFA in osteosarcoma. Biomaterials. 2017;147:68–85. doi: 10.1016/j.biomaterials.2017.09.015. [DOI] [PubMed] [Google Scholar]
- 24.Kim S.M., Yang Y., Oh S.J., Hong Y., Seo M., Jang M. Cancer-derived exosomes as a delivery platform of CRISPR/Cas9 confer cancer cell tropism-dependent targeting. J. Control. Release. 2017;266:8–16. doi: 10.1016/j.jconrel.2017.09.013. [DOI] [PubMed] [Google Scholar]
- 25.Timin A.S., Muslimov A.R., Lepik K.V., Epifanovskaya O.S., Shakirova A.I., Mock U., Riecken K., Okilova M.V., Sergeev V.S., Afanasyev B.V. Efficient gene editing via non-viral delivery of CRISPR-Cas9 system using polymeric and hybrid microcarriers. Nanomedicine (Lond.) 2018;14:97–108. doi: 10.1016/j.nano.2017.09.001. [DOI] [PubMed] [Google Scholar]
- 26.Rui Y., Wilson D.R., Sanders K., Green J.J. Reducible Branched Ester-Amine Quadpolymers (rBEAQs) Codelivering Plasmid DNA and RNA Oligonucleotides Enable CRISPR/Cas9 Genome Editing. ACS Appl. Mater. Interfaces. 2019;11:10472–10480. doi: 10.1021/acsami.8b20206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sunshine J.C., Peng D.Y., Green J.J. Uptake and transfection with polymeric nanoparticles are dependent on polymer end-group structure, but largely independent of nanoparticle physical and chemical properties. Mol. Pharm. 2012;9:3375–3383. doi: 10.1021/mp3004176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sunshine J.C., Akanda M.I., Li D., Kozielski K.L., Green J.J. Effects of base polymer hydrophobicity and end-group modification on polymeric gene delivery. Biomacromolecules. 2011;12:3592–3600. doi: 10.1021/bm200807s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wilson D.R., Mosenia A., Suprenant M.P., Upadhya R., Routkevitch D., Meyer R.A., Quinones-Hinojosa A., Green J.J. Continuous microfluidic assembly of biodegradable poly(beta-amino ester)/DNA nanoparticles for enhanced gene delivery. J. Biomed. Mater. Res. A. 2017;105:1813–1825. doi: 10.1002/jbm.a.36033. [DOI] [PubMed] [Google Scholar]
- 30.Wilson D.R., Rui Y., Siddiq K., Routkevitch D., Green J.J. Differentially Branched Ester Amine Quadpolymers with Amphiphilic and pH-Sensitive Properties for Efficient Plasmid DNA Delivery. Mol. Pharm. 2019;16:655–668. doi: 10.1021/acs.molpharmaceut.8b00963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matsuda T., Cepko C.L. Controlled expression of transgenes introduced by in vivo electroporation. Proc. Natl. Acad. Sci. USA. 2007;104:1027–1032. doi: 10.1073/pnas.0610155104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Madisen L., Zwingman T.A., Sunkin S.M., Oh S.W., Zariwala H.A., Gu H., Ng L.L., Palmiter R.D., Hawrylycz M.J., Jones A.R. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 2010;13:133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Suzuki K., Tsunekawa Y., Hernandez-Benitez R., Wu J., Zhu J., Kim E.J., Hatanaka F., Yamamoto M., Araoka T., Li Z. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature. 2016;540:144–149. doi: 10.1038/nature20565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yusa K., Zhou L., Li M.A., Bradley A., Craig N.L. A hyperactive piggyBac transposase for mammalian applications. Proc. Natl. Acad. Sci. USA. 2011;108:1531–1536. doi: 10.1073/pnas.1008322108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shcherbakova D.M., Verkhusha V.V. Near-infrared fluorescent proteins for multicolor in vivo imaging. Nat. Methods. 2013;10:751–754. doi: 10.1038/nmeth.2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kettlun C., Galvan D.L., George A.L., Jr., Kaja A., Wilson M.H. Manipulating piggyBac transposon chromosomal integration site selection in human cells. Mol. Ther. 2011;19:1636–1644. doi: 10.1038/mt.2011.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Staahl B.T., Benekareddy M., Coulon-Bainier C., Banfal A.A., Floor S.N., Sabo J.K., Urnes C., Munares G.A., Ghosh A., Doudna J.A. Efficient genome editing in the mouse brain by local delivery of engineered Cas9 ribonucleoprotein complexes. Nat. Biotechnol. 2017;35:431–434. doi: 10.1038/nbt.3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhu D., Shen H., Tan S., Hu Z., Wang L., Yu L., Tian X., Ding W., Ren C., Gao C. Nanoparticles based on poly (β-amino ester) and HPV16 targeting CRISPR/shRNA as potential drugs for HPV16 related cervical malignancy. Mol. Ther. 2018;26:2443–2455. doi: 10.1016/j.ymthe.2018.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bhise N.S., Shmueli R.B., Gonzalez J., Green J.J. A novel assay for quantifying the number of plasmids encapsulated by polymer nanoparticles. Small. 2012;8:367–373. doi: 10.1002/smll.201101718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tzeng S.Y., Guerrero-Cázares H., Martinez E.E., Sunshine J.C., Quiñones-Hinojosa A., Green J.J. Non-viral gene delivery nanoparticles based on poly(β-amino esters) for treatment of glioblastoma. Biomaterials. 2011;32:5402–5410. doi: 10.1016/j.biomaterials.2011.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen J.S., Dagdas Y.S., Kleinstiver B.P., Welch M.M., Sousa A.A., Harrington L.B., Sternberg S.H., Joung J.K., Yildiz A., Doudna J.A. Enhanced proofreading governs CRISPR-Cas9 targeting accuracy. Nature. 2017;550:407–410. doi: 10.1038/nature24268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kleinstiver B.P., Pattanayak V., Prew M.S., Tsai S.Q., Nguyen N.T., Zheng Z., Joung J.K. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature. 2016;529:490–495. doi: 10.1038/nature16526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Doyon Y., Choi V.M., Xia D.F., Vo T.D., Gregory P.D., Holmes M.C. Transient cold shock enhances zinc-finger nuclease-mediated gene disruption. Nat. Methods. 2010;7:459–460. doi: 10.1038/nmeth.1456. [DOI] [PubMed] [Google Scholar]
- 44.Guo Q., Mintier G., Ma-Edmonds M., Storton D., Wang X., Xiao X., Kienzle B., Zhao D., Feder J.N. ‘Cold shock’ increases the frequency of homology directed repair gene editing in induced pluripotent stem cells. Sci. Rep. 2018;8:2080. doi: 10.1038/s41598-018-20358-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mekler V., Minakhin L., Semenova E., Kuznedelov K., Severinov K. Kinetics of the CRISPR-Cas9 effector complex assembly and the role of 3′-terminal segment of guide RNA. Nucleic Acids Res. 2016;44:2837–2845. doi: 10.1093/nar/gkw138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Raper A.T., Stephenson A.A., Suo Z. Functional Insights Revealed by the Kinetic Mechanism of CRISPR/Cas9. J. Am. Chem. Soc. 2018;140:2971–2984. doi: 10.1021/jacs.7b13047. [DOI] [PubMed] [Google Scholar]
- 47.DuBridge R.B., Tang P., Hsia H.C., Leong P.M., Miller J.H., Calos M.P. Analysis of mutation in human cells by using an Epstein-Barr virus shuttle system. Mol. Cell. Biol. 1987;7:379–387. doi: 10.1128/mcb.7.1.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dean F.B., Bullock P., Murakami Y., Wobbe C.R., Weissbach L., Hurwitz J. Simian virus 40 (SV40) DNA replication: SV40 large T antigen unwinds DNA containing the SV40 origin of replication. Proc. Natl. Acad. Sci. USA. 1987;84:16–20. doi: 10.1073/pnas.84.1.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mancia F., Patel S.D., Rajala M.W., Scherer P.E., Nemes A., Schieren I., Hendrickson W.A., Shapiro L. Optimization of protein production in mammalian cells with a coexpressed fluorescent marker. Structure. 2004;12:1355–1360. doi: 10.1016/j.str.2004.06.012. [DOI] [PubMed] [Google Scholar]
- 50.Pear W.S., Nolan G.P., Scott M.L., Baltimore D. Production of high-titer helper-free retroviruses by transient transfection. Proc. Natl. Acad. Sci. USA. 1993;90:8392–8396. doi: 10.1073/pnas.90.18.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.van Deventer H.W., Serody J.S., McKinnon K.P., Clements C., Brickey W.J., Ting J.P.Y. Transfection of macrophage inflammatory protein 1 α into B16 F10 melanoma cells inhibits growth of pulmonary metastases but not subcutaneous tumors. J. Immunol. 2002;169:1634–1639. doi: 10.4049/jimmunol.169.3.1634. [DOI] [PubMed] [Google Scholar]
- 52.Reynier P., Briane D., Coudert R., Fadda G., Bouchemal N., Bissieres P., Taillandier E., Cao A. Modifications in the head group and in the spacer of cholesterol-based cationic lipids promote transfection in melanoma B16-F10 cells and tumours. J. Drug Target. 2004;12:25–38. doi: 10.1080/10611860410001683040. [DOI] [PubMed] [Google Scholar]
- 53.Rui Y., Wilson D.R., Choi J., Varanasi M., Sanders K., Karlsson J., Lim M., Green J.J. Carboxylated branched poly(β-amino ester) nanoparticles enable robust cytosolic protein delivery and CRISPR-Cas9 gene editing. Sci. Adv. 2019;5:eaay3255. doi: 10.1126/sciadv.aay3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang Y., Wang J., Wang Z., Zhang Y., Shi S., Nielsen J., Liu Z. A gRNA-tRNA array for CRISPR-Cas9 based rapid multiplexed genome editing in Saccharomyces cerevisiae. Nat. Commun. 2019;10:1053. doi: 10.1038/s41467-019-09005-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shiraki T., Kawakami K. A tRNA-based multiplex sgRNA expression system in zebrafish and its application to generation of transgenic albino fish. Sci. Rep. 2018;8:13366. doi: 10.1038/s41598-018-31476-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mishra B., Wilson D.R., Sripathi S.R., Suprenant M.P., Rui Y., Wahlin K.J., Berlinicke C., Green J.J., Zack D.J. Combinatorial library of biodegradable polyesters enables delivery of plasmid DNA to polarized human RPE monolayers for retinal gene therapy. bioRxiv. 2018 doi: 10.1101/264390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen F., LoTurco J. A method for stable transgenesis of radial glia lineage in rat neocortex by piggyBac mediated transposition. J. Neurosci. Methods. 2012;207:172–180. doi: 10.1016/j.jneumeth.2012.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Suzuki K., Kimura T., Shinoda H., Bai G., Daniels M.J., Arai Y., Nakano M., Nagai T. Five colour variants of bright luminescent protein for real-time multicolour bioimaging. Nat. Commun. 2016;7:13718. doi: 10.1038/ncomms13718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schumann K., Lin S., Boyer E., Simeonov D.R., Subramaniam M., Gate R.E., Haliburton G.E., Ye C.J., Bluestone J.A., Doudna J.A., Marson A. Generation of knock-in primary human T cells using Cas9 ribonucleoproteins. Proc. Natl. Acad. Sci. USA. 2015;112:10437–10442. doi: 10.1073/pnas.1512503112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.