

Abstract
Literature methods to access gem-difluoroalkenes are largely limited to harsh, organometallic-based methods, and known photoredox mediated processes are not amenable to aryl radical addition to trifluoromethyl alkenes. A mild, metal-free, functional group-tolerant method for the preparation of benzylic gem-difluoroalkenes is described. The combination of organic dye photocatalyst and silanol reductant enables halogen atom abstration from (hetero)aryl halides to generate aryl radicals that undergo a defluorinative arylation of α-trifluoromethyl alkenes. A breadth of electronically disparate aryl radicals and α-trifluoromethyl alkenes are effective under the developed conditions, which demonstrates high functional group compatibility and allows access to structures previously untenable under traditional transition metal-mediated methods.
Introduction
The incorporation of fluorinated motifs into organic structures is an ever-growing field of organic chemistry. It is a well-established and often-utilized strategy to introduce fluorine into organic molecules to impart desirable pharmacological properties. These properties include increased metabolic stability, enhanced lipophilicity, and improved bioavailability.1 As such, methods for the synthesis of new and unusual fluorinated groups continue to increase in relevance.2 Most importantly, there is a great need for methods that afford the incorporation or modification of fluorinated groups under mild conditions to maximize potential feasibility in late-stage functionalization.
One functional group of interest for medicinal chemistry is the geminally-substituted difluoroalkene. These structures are postulated as isosteres for carbonyl groups, and are particularly attractive when metabolism at a carbonyl is a point of undesirable metabolic degradation.3 Although methods for the synthesis of gem-difluoroalkenes have been described,4 they rely on two-electron chemistry and can be largely classified as either the direct conversion of carbonyls into gem-difluoroalkenes or as the modification of an existing fluorinated group. In the context of the addition of aryl groups to trifluoromethyl alkenes, these methods are limited to two-electron additions of organometallic species such as phenyllithium and phenylcopper reagents.5 To date, milder, more functional group-tolerant arylation conditions have not been described, representing an important methodological gap.
Recent work by our group and others has included the development of photoredox-mediated approaches for the synthesis of gem-difluoroalkenes.6 Several classes of radical precursors have been leveraged through this approach, including alkyltrifluoroborates, alkylbis(catecholato)silicates, α–silylamines, and alkyl carboxylate salts (Scheme 1). These radical precursors are effective for the generation of alkyl radicals displaying diverse functional groups; however, these methods are not feasible for arylation, as aryl radicals have elevated oxidation potentials outside the range of many common photocatalysts.7 The present work demonstrates the first photoredox-catalyzed arylation of trifluoromethyl alkenes using (hetero)aryl halides as a radical source.
Scheme 1.
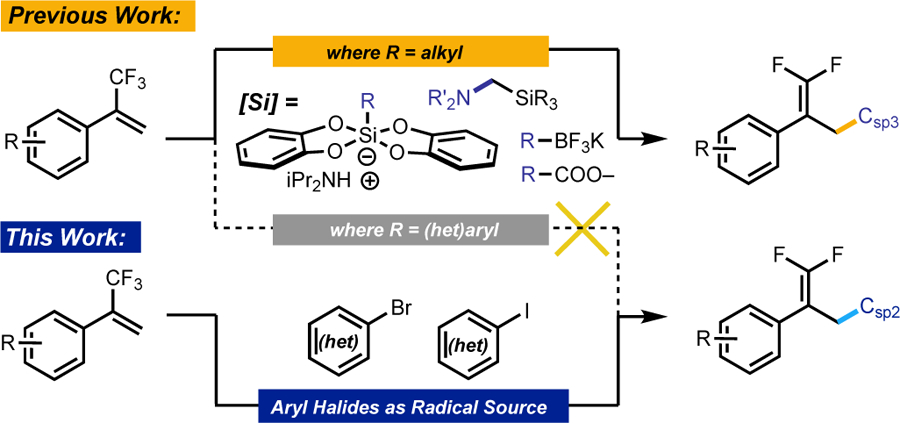
Arylation of trifluoromethyl alkenes through a new pathway
An established pathway for the formation of aryl radicals in photoredox catalysis follows a one-electron reduction of aryl halides (I), facilitated by oxidative quenching of an excited photocatalyst ([PC]*) (Scheme 2).8 As demonstrated by the groups of Jui, Lee, and others, these reductively generated radicals (II) can engage in Giese-type additions to alkenes.8,9 Then, in a redox-neutral radical polar crossover (RPC) manifold, the photocatalyst cycle is completed by oxidation of radical intermediate III to regenerate the photocatalyst and form cationic species IV.9,10 Intermediate IV could then react further by either a base-mediated elimination to re-form an alkene or by nucleophilic attack at the cationic carbon. However, a cationic intermediate would not be viable for the E1cb fluoride elimination needed to produce the desired gem-difluoroalkene product.
Scheme 2.
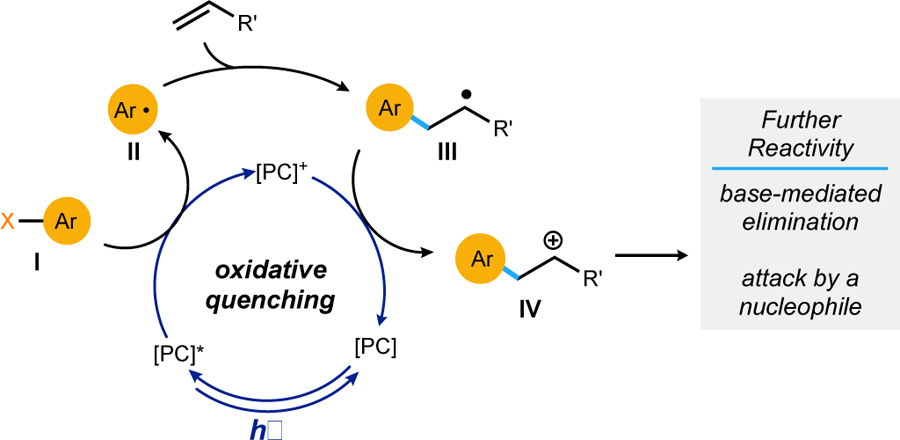
Net-neutral radical polar crossover via oxidative quenching photocatalysis
To access the anionic intermediate necessary for fluoride elimination (Scheme 3, XI) we envisioned generating aryl radicals through a net reductive pathway. A recent report by the MacMillan group demonstrated the feasibility of using tris(trimethylsilyl)silanol (V) as an activator of aryl halides (Ep = +1.54 V versus SCE).11 We proposed that use of V, which alters the sequence of single electron transfer (SET) events so that a reductive quenching photocatalytic cycle occurs, would generate aryl radicals in a reducing environment capable of accessing the anionic intermediate XI needed for fluoride elimination.
Scheme 3.

Mechanism for silanol-mediated radical polar crossover
We proposed that the catalytic cycle would be initiated by base-mediated deprotonation of silanol V to give VI. Then, oxidation by the photocatalyst followed by a radical Brook rearrangement generates silyl radical VIII. The silyl radical next abstracts a halogen atom from aryl halide I to afford aryl radical II, which adds to the trifluoromethyl alkene IX. The photocatalytic cycle is closed by a reduction of species X to the α–trifluoromethyl anion XI, and finally an E1cb elimination of fluoride generates the desired product XII.12
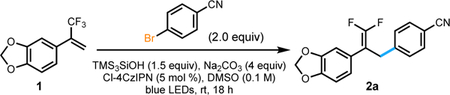
With the aid of high throughput experimentation, the reaction conditions were evaluated (Table 1) (see Supporting Information for additional details). Photocatalyst screening revealed the organic photocatalyst 2,4,5,6-tetrakis(3,6-dichloro-9H-carbazol-9-yl)isophthalonitrile (Cl-4CzIPN, Ep = +1.71 V versus SCE) gave the highest formation of desired product, even when compared to commonly used iridium photocatalysts.13 The evaluation of various solvents identified polar aprotic solvents as most suitable, with DMSO giving the highest conversion. A screening of inorganic bases saw Na2CO3 provide optimal product formation. Control studies confirmed the necessity of each reaction component. Without photocatalyst or silanol, no reaction, whether undesired or otherwise, was observed. Without base, which is important for silanol activation, only trace product was formed. Finally, when the reaction was allowed to run in ambient light, no reactivity was observed.
Table 1.
Reaction optimization and control studies
 | |||
|---|---|---|---|
| Entry | Deviation from Standard | Conversiona | |
| 1 | None | 92 |  |
| 2 | [Ir{dF(CF3)ppy}2(dCF3bpy)](PF6) | 59 | |
| 3 | acetone | 77 | |
| 4 | MeCN | 32 | |
| 5 | DMF | 32 | |
| 6 | NaOAc | 92b | |
| 7 | K3PO4 | 49 | |
| 8 | None | 98 | |
| 9 | NO CI-4CzIPN | 0 | |
| 10 | NO TMS3SiOH | 0 | |
| 11 | No base | < 5 | |
| 12 | No LEDs | 0 | |
Conversions determined by 19F NMR of crude reaction mixtures.
Conversion to product was observed with full consumption of starting materials with the formation of unidentified reaction byproducts.
Having identified optimal conditions, the scope of the reaction was evaluated (Table 2). The reaction was optimized with the electron deficient aryl nitrile (2a), and as expected good reactivity was observed with other electron-poor substrates such as an aryl ketone (2g) and ester (2l). The reaction was also amenable to the addition of several electron-rich aryl radicals (2d, 2h, 2i). Further experiments demonstrated moderate to good yields with meta- and para-substituted aryl methoxy halides (2h and 2i), albeit with marginally lower isolated yields. More impressively, the conditions were also amenable toward a variety of heteroaryl halides, including several substituted pyridines (2b, 2c, 2f, 2j, 2k) and even an unprotected indole (2d). Importantly, both aryl iodides and -bromides are substrates, allowing facile, direct access to numerous aryl radicals from commercially available reagents. Example 2k demonstrates the selectivity observed in the presence of both aryl bromides and -iodides, with radical generation in the ratio of 7.6:1 favoring reactivity at the iodide.
Table 2.
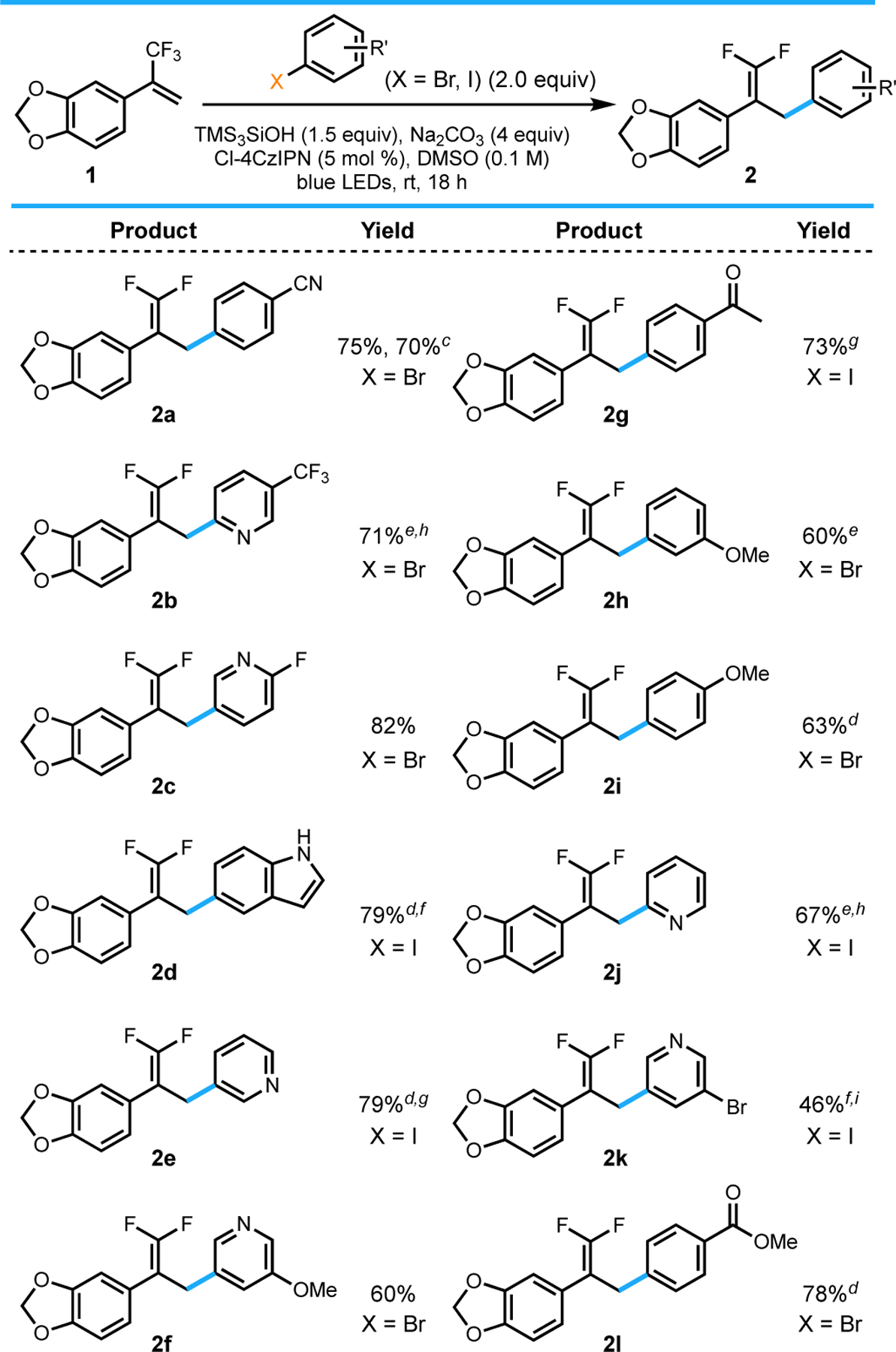 |
All values indicate the yield of the isolated product.
General reaction conditions: aryl halide (2.0 equiv, 1.0 mmol), alkene (1.0 equiv, 0.50 mmol), Cl-4CzIPN (5 mol %, 0.025 mmol), TMS3SiOH (1.5 equiv, 0.75 mmol), Na2CO3 (4 equiv, 2.0 mmol), DMSO (0.1 M), 18 h, irradiating with blue LEDs (30 W). See the Supporting Information for details.
Isolated yield on 2.5 mmol scale; reaction run for 5 d.
Isolated on 1.0 mmol scale.
Isolated yield on 0.3 mmol scale.
Conducted using 10 mol % Cl-4CzIPN.
Reaction run for 2 d.
Conducted using 1 equiv Ar–X. hIsolated with <5% impurity of alkene.
Isolated as a 7.6:1 mixture of Br:I arene.
We then sought to explore the scope of the method with respect to the trifluoromethyl alkene substrate. In addition to the dioxole substrate used in optimization, the standard conditions were applicable to other electron-rich trifluoromethylalkenes. These included dimethoxy- (2s), dimethylamino- (2t), thioether- (2o), and tert-butyl-substituted (2u) trifluoromethyl alkenes. Electron poor substrates were also reactive, affording the corresponding products in moderate to good yields. The dichloro-substituted alkene (2m) is one example of this, and also demonstrates that the method is chemoselective for the activation of aryl iodides and bromides in the presence of aryl chlorides.14 The reaction also tolerated an appended alkyne (2v) and carboxylic acid (2x), with no evidence of undesired side-reactivity. Additionally, this method is viable on substrates with unprotected alcohols (2p). Other notable examples of the substrate scope include the expansion of this method to heteroaryl trifluoromethyl alkenes (2q, 2r, 2w). The reaction was also successful when performed on a 2.5 mmol scale, although a longer reaction time was necessary (2a, Table 2).
During the course of the scope exploration, interesting reactivity was noted in the case of the exceptionally electron deficient example 2b (Scheme 4). When the standard conditions using two equivalents of aryl bromide were used, a significant portion of double addition product was observed (3a). This product results from the addition of a second aryl radical to the gem-difluoroalkene, where reduction and a second fluoride elimination furnishes the monofluoroalkene. Fluorinated triarylalkenes are themselves relevant to medicinal chemists as they are known anticancer agents in the family of tamoxifen derivatives.15 A second interesting observation was made while developing the reaction workup conditions. It was found that when electron poor compounds were eluted through a plug of KF/alumina, a rearrangement occurred to convert the gem-difluoroalkene to a difluoromethyl-substituted internal alkene (3b). Moderate amounts of this alkene rearrangement was observed even when exposure to the KF/alumina was limited to only 30 s. When the sample was stirred in KF/alumina for 3 h, complete conversion to the internal alkene was observed. These two products demonstrate the utility of the trifluoromethyl alkenes prepared here as starting materials for a diverse array of functionalization reactions.
Scheme 4.
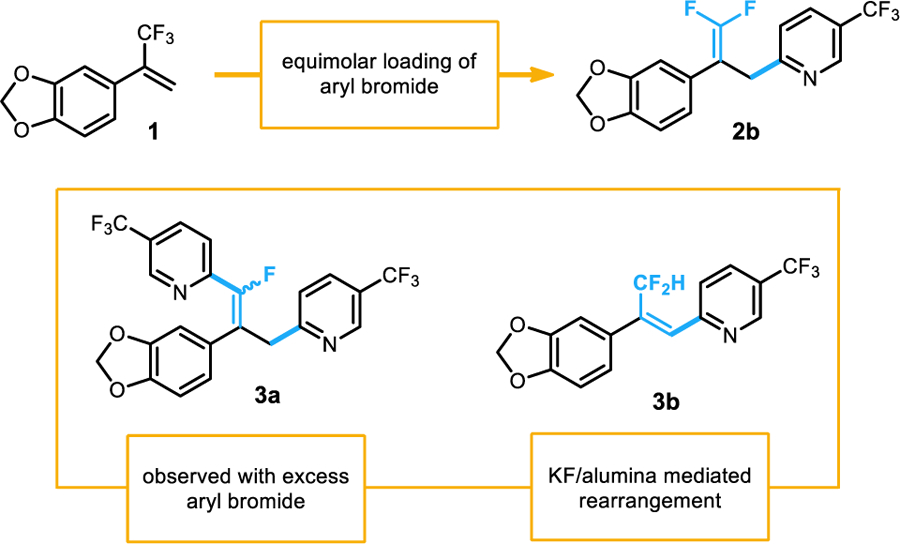
Unusual experimental observations and side reactivity
Conclusions
Methods to access structurally diverse fluorinated molecules have gained significant prominence in recent years. Photoredox catalysis has offered unique opportunities for achieving such transformations with mild reaction conditions and high functional group compatibility. The reported method offers an efficient, metal-free approach to achieve the arylation of trifluoromethyl alkenes to make a new class of substituted gem-difluoroalkenes that were previously inaccessible under such mild conditions, and thus represents a step forward in our ability to synthesize and modify useful fluorinated motifs.
Supplementary Material
Table 3.
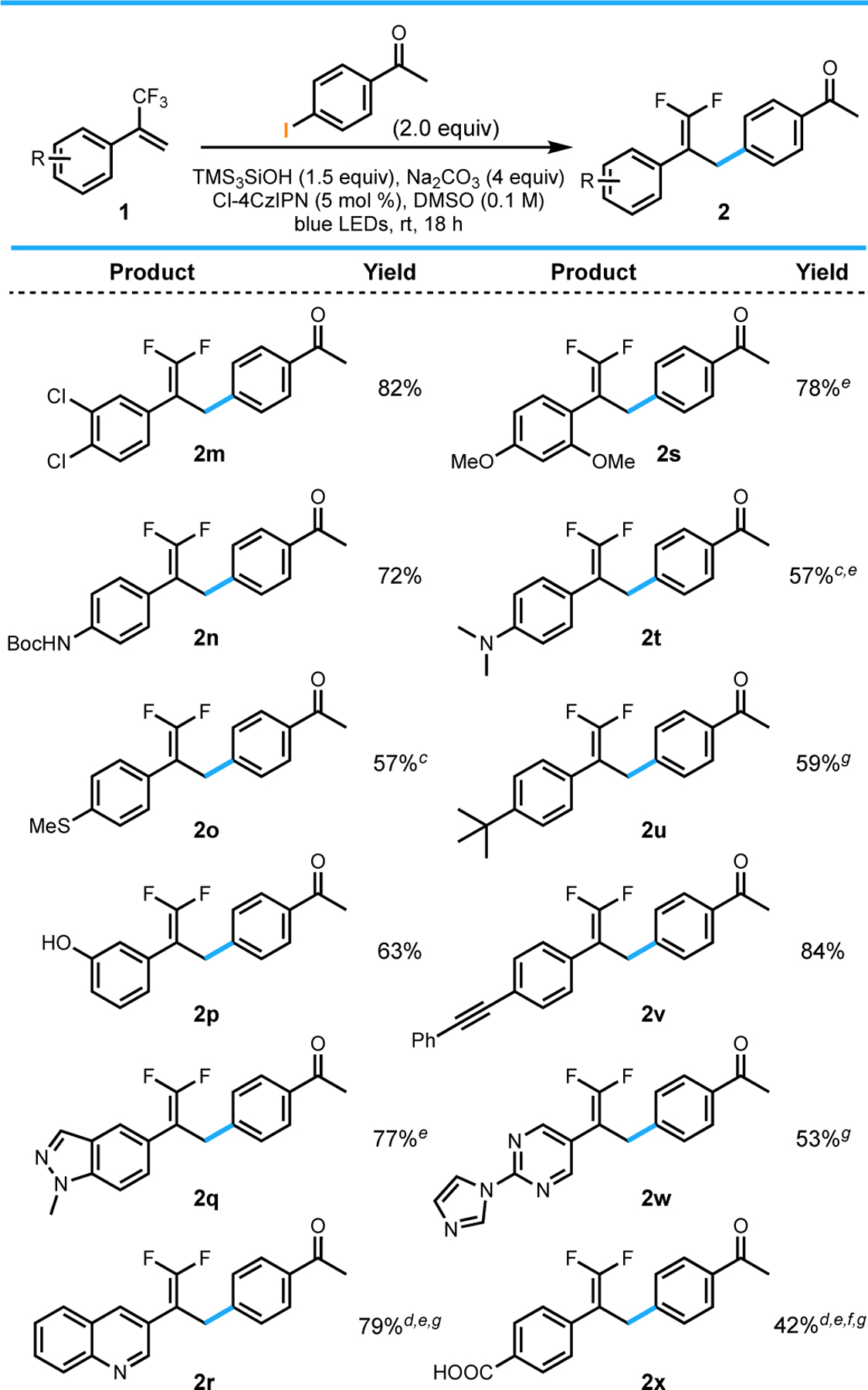 |
All values indicate the yield of the isolated product.
General reaction conditions: aryl halide (2.0 equiv, 1.0 mmol), alkene (1.0 equiv, 0.50 mmol), Cl-4CzIPN (5 mol %, 0.025 mmol), TMS3SiOH (1.5 equiv, 0.75 mmol), Na2CO3 (4 equiv, 2.0 mmol), DMSO (0.1 M), 18 h, irradiating with blue LEDs (30 W). See the Supporting Information for details.
Isolated on 1.0 mmol scale.
Conducted using 10 mol % Cl-4CzIPN.
Reaction run for 2 d.
Conducted using 3 equiv Ar–X and 2.0 equiv TMS3SiOH.
Isolated with <5% impurity of alkene.
Acknowledgements
We sincerely thank Dr. John Milligan of the University of Pennsylvania and Dr. Christopher Kelly of Virginia Commonwealth University for useful discussion. We thank Dr. Charles Hendrick for his assistance in the Penn Merck High Throughput Experimentation Center. We thank Dr. Charles W. Ross, III for assistance in obtaining HRMS data. We thank Dr. Jun Gu and Prof. Bill Dailey for helpful insight into 19F NMR data.
Footnotes
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/x0xx00000x
Conflicts of interest
There are no conflicts to declare.
Notes and references
- 1.(a) Meanwell NA, J. Med. Chem, 2011, 54, 2529; [DOI] [PubMed] [Google Scholar]; (b) Purser S, Moore PR, Swallow S, Gouverneur S,V, Chem. Soc. Rev, 2008, 37, 320; [DOI] [PubMed] [Google Scholar]; (c) Hagmann KW, J. Med. Chem, 2008, 51, 4359; [DOI] [PubMed] [Google Scholar]; (d) Müeller K, Faeh C, Diederich F, Science, 2007, 317, 1881; [DOI] [PubMed] [Google Scholar]; (e) Liang T, Neumann CN, Ritter T, Angew. Chem. Int. Ed, 2013, 52, 8214; [DOI] [PubMed] [Google Scholar]; (f) Ni CF, Hu JB, Chem. Soc. Rev, 2016, 45, 5441. [DOI] [PubMed] [Google Scholar]
- 2.(a) Magueur G, Crousse B, Ourévitch M, Bonnet-Delpon D, Bégué J-P, J. Fluorine Chem, 2006, 127, 637; [Google Scholar]; (b) Leriche C, He S, Chang C.-w., Liu H.-w., J. Am. Chem. Soc, 2003, 125, 6348. [DOI] [PubMed] [Google Scholar]
- 3.(a) Malátková P, Wsól V, Drug Metab. Rev, 2014, 46, 96; [DOI] [PubMed] [Google Scholar]; (b) Matsunaga T, Shintani S, Hara A, Drug Metab. Pharmacokinet, 2006, 21, 1; [DOI] [PubMed] [Google Scholar]; (c) Barski OA, Tipparaju SM, Bhatnagar A, Drug Metab. Rev 2008, 40, 553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Chelucci G, Chem. Rev, 2012, 112, 1344; [DOI] [PubMed] [Google Scholar]; (b) Zhang S, Cao S, Tetrahedron Lett. 2017, 58, 375; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Hu M, Ni C, Li L, Han Y, Hu J, J. Am. Chem. Soc, 2015, 137, 14496; [DOI] [PubMed] [Google Scholar]; (d) Zhao X, Chunmai L, Wang B, Cao S, Tetrahedron Lett. 2019, 60, 129; [Google Scholar]; (e) Lu X, Wang X-X, Gong T-J, Pi J-J, He S-J, Fu Y, Chem. Sci, 2019, 10, 809; [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Xinfei J, Yisen L, Hongyan S, Song C, Tetrahedron, 2018, 74, 4155; [Google Scholar]; (g) Lan Y, Yang F, Wang C, ACS Catal., 2018, 8, 9245; [Google Scholar]; (h) Fujita T, Takazawa M, Sugiyama K, Suzuki N, Ichikawa J, J. Org. Lett., 2017, 19, 588; [DOI] [PubMed] [Google Scholar]; (i) Liu Y, Zhou Y, Zhao Y, Qu J, Org. Lett, 2017, 19, 946; [DOI] [PubMed] [Google Scholar]; (j) Fuchibe K, Hatta H, Oh K, Oki R, Ichikawa J, Angew. Chem. Int. Ed, 2017, 56, 5890. [DOI] [PubMed] [Google Scholar]
- 5.(a) Begue J-P, Bonnet-Delpon D, Rock MH, Tetrahedron Lett, 1995, 36, 5003; [Google Scholar]; (b) Kitazume T, Ohnogi T, Synthesis, 1988, 8, 614; [Google Scholar]; (c) Miura T, Ito Y, Murakami M, Chem. Lett 2008, 37, 1006. [Google Scholar]
- 6.(a) Zhiyang L, Yun L, Wang C, ACS Catal, 2019, 9, 775; [Google Scholar]; (b) Wu L-H, Cheng J-K, Shen L, Shen Z-L, Loh T-P, Adv. Synth. Catal., 2018, 360, 3894; [Google Scholar]; (c) Xia P-J, Ye Z-P, Hu Y-Z, Song D, Xiang H-Y, Chen X-Q, Org. Lett, 2019, ASAP; DOI: 10.1021/acs.orglett.9b00651. [DOI] [PubMed] [Google Scholar]
- 7.(a) Prier CK, Rankic DA, MacMillan DWC, Chem. Rev, 2013, 113, 5322; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Romero NA, Nicewicz DA, Chem. Rev 2016, 116, 10075; [DOI] [PubMed] [Google Scholar]; (c) Shundrin LA, Bardin VV, Frohn H-J, Z. Anorg. Allg. Chem, 2004, 630, 1253. [Google Scholar]
- 8.(a) Ghost I, Marzo L, Das A, Shaikh R, König B, Acc. Chem. Res, 2016, 49, 1566; [DOI] [PubMed] [Google Scholar]; (b) Qiu G, Li Y, Wu J, Org. Chem. Front, 2016, 3, 1011; [Google Scholar]; (c) Studer A, Curran DP, Nat. Chem, 2014, 6, 765; [DOI] [PubMed] [Google Scholar]
- 9.(a) Aycock RA, Wang H, Jui NT, Chem. Sci, 2017, 8, 3121; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Seath CP, Vogt DB, Xu Z, Boyington AJ, Jui NT, J. Am. Chem. Soc, 2018, 140, 15525; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Boyington AJ, Seath CP, Zearfoss AM, Xu Z, Jui NA, J. Am. Chem. Soc 2019, 141, 4147; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Kim H, Lee C, Angew. Chem. Int. Ed, 2012, 51, 12303. [DOI] [PubMed] [Google Scholar]
- 10.(a) Romero NA, Nicewicz DA, Chem. Rev, 2016, 116, 10075; [DOI] [PubMed] [Google Scholar]; (b) Xie J, Jin H, Hashmi ASK, Chem. Soc. Rev, 2017, 46, 5193. [DOI] [PubMed] [Google Scholar]
- 11.(a) Le C, Chen TQ, Liang T, Zhang P, MacMillan DWC, Science, 2018, 360, 1010; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhang P, Le C, MacMillan DWC, J. Am. Chem. Soc, 2016, 138, 8084; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lucarini M, Marchesi E, Pedulli GF, J. Org. Chem, 1998, 63, 1687. [Google Scholar]
- 12.This mechanism was supported by computational studies in our recent report, where the energy barrier for anionic fluoride elimination was calculated as ~6 kcal mol−1, compared to ~63 kcal mol−1 for a radical mediated fluoride elimination. Phelan JP, Lang SB, Compton JS, Kelly CB, Molander GA, J. Am. Chem. Soc. 2018, 140, 8037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Le Vaillant F, Garreau M, Nicolai S, Gryn’ova G, Corminboeuf C, Waser J, Chem. Sci, 2018, 9, 5883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Enemaerke RJ, Christensen TB, Jensen J, Daasbjerg K, J. Chem. Soc., Perkin Trans 2, 2001, 1620 [Google Scholar]; (b) Andrieux CP, Blocman C, Dumas-Bouchiat JM, Saveant JM, J. Am. Chem. Soc, 1979, 101, 3431. [Google Scholar]
- 15.(a) Isanbor C, O’Hagan D, J. Fluorine Chem 2006, 127, 303; [Google Scholar]; (b) Govek SP, Nagasawa JY, Douglas KL, Lai AG, Kahraman M, Bonnefous C, Aparicio AM, Darimont BD, Grillot KL, Joseph JD, Kaufman JA, Lee K-J, Lu N, Moon M, Prudente RY, Sensintaffar J, Rix PJ, Hager JH, Smith ND, Bioorg. Med. Chem. Lett, 2015, 25, 5163. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.