

Abstract
Tumor-targeting oncolytic viruses such as vaccinia virus (VV) are attractive cancer therapeutic agents that act through multiple mechanisms to provoke both tumor lysis and anti-tumor immune responses. However, delivery of these agents remains restricted to intra-tumoral administration, which prevents effective targeting of inaccessible and disseminated tumor cells. In the present study we have identified transient pharmacological inhibition of the leukocyte-enriched phosphoinositide 3-kinase δ (PI3Kδ) as a novel mechanism to potentiate intravenous delivery of oncolytic VV to tumors. Pre-treatment of immunocompetent mice with the PI3Kδ-selective inhibitor IC87114 or the clinically approved idelalisib (CAL-101), prior to intravenous delivery of a tumor-tropic VV, dramatically improved viral delivery to tumors. This occurred via an inhibition of viral attachment to, but not internalization by, systemic macrophages through perturbation of signaling pathways involving RhoA/ROCK, AKT, and Rac. Pre-treatment using PI3Kδ-selective inhibitors prior to intravenous delivery of VV resulted in enhanced anti-tumor efficacy and significantly prolonged survival compared to delivery without PI3Kδ inhibition. These results indicate that effective intravenous delivery of oncolytic VV may be clinically achievable and could be useful in improving anti-tumor efficacy of oncolytic virotherapy.
Keywords: vaccinia virus, phosphoinositide 3-kinase δ, intravenous delivery, macrophage, colorectal cancer, breast cancer, oncolytic virus, immunotherapy
Graphical Abstract
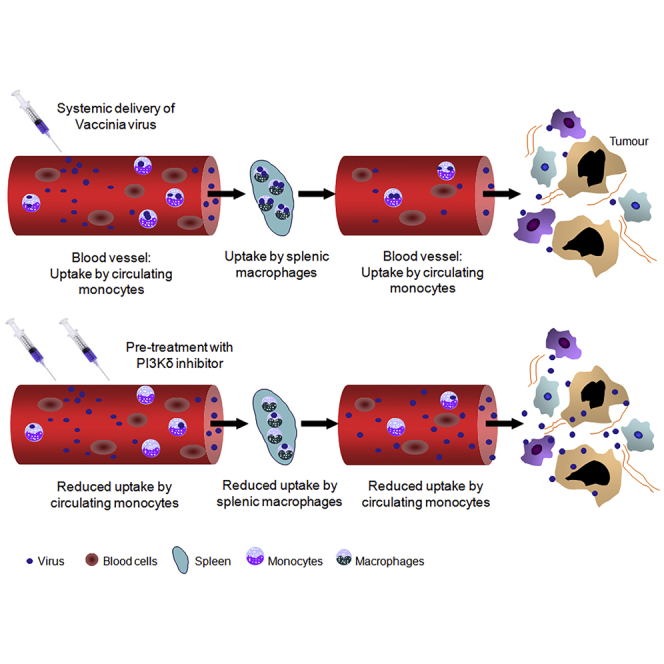
Transient pharmacological inhibition of the PI3 kinase δ isoform using CAL-101 allows for improved vaccinia virus accumulation in tumor tissues after intravenous delivery by preventing macrophage uptake of the virus. This platform allows for effective anti-tumor activity after systemic administration of oncolytic vaccinia virus.
Introduction
Oncolytic virus (OV) therapy has emerged as an attractive immunotherapeutic approach for cancer, but although intra-tumoral (i.t.) delivery of a variety of OVs has demonstrated a measure of success, culminating in the recent approval of T-VEC (talimogene laherparepvec) for treatment of melanoma,1 treatment often fails as inaccessible or metastatic tumor deposits are left untargeted. As such, methods for successful and therapeutically meaningful systemic delivery of OV are required. While a number of OVs have been investigated for their suitability for systemic delivery,2 vaccinia virus (VV) has emerged as one of the most promising agents, with clinical studies demonstrating tumor infection and safety upon high-dose systemic administration.3 However, trials demonstrating measurable anti-tumor activity after systemic delivery remain elusive.
Intravenous (i.v.) rather than i.t. delivery of OVs would be expected to confer a therapeutic advantage by simultaneous targeting of the primary tumor and metastases. In order for this strategy to be effective, the virus must persist for some time in the circulation and maintain the ability to infect tumor cells. The main obstacles to successful systemic delivery of OVs are immunological barriers, such as complement, antibodies, antiviral T cells, and anti-viral cytokines,2,3 as well as non-specific uptake by other tissues (including lung, liver, and spleen by tissue-resident macrophages) and poor viral transport out of the circulation to the tumor.4 VV is able to overcome some immunological barriers,5 as it expresses proteins designed to blunt the effects of anti-viral immune responses. In addition, VV produces multiple virion forms, including intracellular mature virions (IMVs) and extracellular enveloped virions (EEVs). Of these, EEVs are the most important for dissemination within the host. These virions are produced as a result of IMV wrapping with a second membrane that confers resistance to neutralizing antibodies and complement.6,7 However, EEVs represent less than 1% of progeny virions, and the good manufacturing practice (GMP)-standard VV used in the clinic is the IMV form, which is highly immunogenic and rapidly cleared from the host. Thus, an effective strategy to overcome the rapid clearance of the prevalent IMV is required.
Here, using pre-clinical models in mice, we show that macrophages are key players hampering the efficacy of i.v.-delivered oncolytic VV, clearing virus from the circulation before it can reach the tumor. However, clearance can be overcome by pharmacological targeting of a class IA family member of phosphatidylinositol 3-kinase (PI3K) prior to i.v. delivery. The PI3K signaling pathway controls numerous intracellular pathways, including cytoskeletal rearrangements necessary for macrophage phagocytosis.8 Three isoforms of class IA PI3Ks exist, of which PI3Kα and PI3Kβ are ubiquitously expressed, whereas PI3Kδ expression is highly enriched in leukocytes, with roles in immune signaling.9 In preclinical mouse models, we show that after i.v. delivery, macrophage uptake of VV prevents therapeutically effective viral doses from reaching the tumor. However, transient and specific pharmacological inhibition of PI3Kδ significantly reduces virus attachment to macrophages, resulting in increased anti-tumor immune responses and therapeutic efficacy associated with oncolytic VV treatment. These findings present a potential approach to improve systemic delivery of oncolytic VV, enhancing its potential as an anti-tumor immunotherapeutic.
Results
Macrophages Are the Key Cell Type Hindering i.v. Delivery of VV in an Immunocompetent Mouse Model
Using a subcutaneous CT26 colorectal cancer model in immunocompetent mice, we found that i.v. delivery of 1 × 108 plaque-forming units (PFU) of VV (VVL15, a Lister strain thymidine kinase (TK)-deleted VV expressing fLUC) did not lead to efficient infection of the tumor cells (Figure 1A). To determine the fate of i.v. delivered VV, we examined the spleen of infected mice and found that the red pulp displayed considerable VV protein staining at early time points post-infection (Figure 1B). Further analysis demonstrated strong co-localization of VV coat protein with splenic macrophages (Figure 1C), prompting us to speculate that these cells might be involved in the early clearance of VV from circulation. This hypothesis was substantiated by experiments using in vivo administration of clodronate liposome, which irreversibly depletes macrophage populations by inducing apoptosis.10 Indeed, tumors in mice pre-treated with clodronate liposomes had a significantly higher virus load compared to control mice (phosphate-buffered saline [PBS] treatment or control liposomes) after i.v. delivery of VV (Figure 1D), indicating that macrophages play a key role in the clearance of systemically delivered VV in immunocompetent mice.
Figure 1.

Macrophages Prevent Successful i.v. Delivery of VVL15 to Tumors
(A) Quantification of whole-body IVIS imaging, in which luminescence was observed and quantified from tumors of VVL15-infected immunocompetent BALB/c mice bearing CT26 flank tumors (n = 3/group) following single i.v. administration of 1 × 108 PFU of VVL15, containing a luciferase reporter protein, or PBS. (B) Immunohistochemical staining for VV coat protein in the spleen, 3 and 8 h after i.v. administration of 1 × 108 PFU of VVL15 (dark brown areas). Stained spleens from PBS-infected animals are shown for reference. Original magnification, ×200 (n = 3/group). (C) Representative confocal image of a VV-infected spleen of an immunocompetent mouse showing the co-localization of VV and macrophages. Macrophages and VV were detected by staining for CD68 and VV coat protein, respectively, 48 h after i.v. delivery of VVL15. Three mice per group were analyzed. Scale bars, 10 μm. (D) Quantification of tumor-specific VVL15 using whole-body IVIS imaging of BALB/c mice with subcutaneous tumors after intra-peritoneal (i.p.) injection of PBS, PBS/liposomes, or clodronate liposomes at 24, 72, and 144 h prior to i.v. injection of 1 × 108 PFU of VVL15 (n = 5/group). Significance of groups in comparison to clodronate liposomes + VVL15 is shown. Data are presented as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 (one-way ANOVA with Newman-Keuls multiple comparison test).
PI3Kδ Activity Regulates Monocyte Uptake of VV
Given that a successful strategy for the viral treatment of cancer is likely to consist of both viral oncolysis and immune modulation, irreversible depletion of macrophage populations could diminish beneficial activities of these cells in the tumor microenvironment (TME). We therefore explored more selective and transient macrophage-targeting agents to enhance systemic delivery of VV. Several lines of evidence support the importance of the PI3K signaling enzymes in macrophage phagocytosis.8,11,12 Among the class I PI3K isoforms (PI3Kα, PI3Kβ, PI3Kδ, PI3Kγ), PI3Kδ and PI3Kγ are highly enriched in leukocytes, including in macrophages.13 In light of the approval of several PI3Kδ-targeted inhibitors for the treatment of B cell malignancies,14 including idelalisib/CAL-101 from Gilead, we focused our study on PI3Kδ inhibition as a potentially clinically feasible approach to reduce viral attachment to macrophages.
Systemic delivery of VV enhanced PI3Kδ mRNA expression in splenocytes of mice (Figure 2A), implying a possible virus-induced upregulation of PI3Kδ mRNA. In vitro, direct infection of murine macrophages, matured from monocytes isolated from immunocompetent mice, with or without treatment using a highly selective inhibitor of PI3Kδ, IC87114,15 did not affect protein expression of PI3Kα, PI3Kγ, or PI3Kδ (Figure 2B; Figure S1A), despite the increased mRNA detected. However, phosphorylation of AKT, a downstream target of PI3Kδ, was prevented after infection, demonstrating an inhibitory effect on the pathway associated with PI3Kδ signaling (Figure 2B; Figure S1B).
Figure 2.

Pharmacological Inhibition of PI3Kδ Prevents VVL15 Attachment to Macrophages
(A) Quantitative RT-PCR detection of PI3Kδ mRNA expression at 0 and 3 h in splenocytes of CT26 tumor-bearing immunocompetent BALB/c mice injected with 1 × 108 PFU of VVL15 or PBS (n = 3/group). Data are presented as mean ± SEM. ∗p < 0.05 (Student’s two-tailed paired t test). (B) Immunoblot showing expression of PI3Kα, PI3Kδ, PI3Kγ, and p-AKT and total-AKT proteins in macrophages derived from immunocompetent mice (n = 3) pre-treated with IC87114 (1 μM) or vehicle buffer for 2 h before infection with VVL15 at an MOI of 5 or mock infected for 1 h. PCNA was used to verify consistent loading. (C) Representative confocal microscopic images showing a reduction in in vitro attachment of VVL15 to macrophages matured from monocytes of immunocompetent mice pre-treated with IC87114 (1 μM) (left panels) compared to pre-treatment with vehicle buffer (right panels) for 2 h. The viral attachment assay was undertaken using RFP-labeled VVL15 (red) and the cells were stained with DAPI (blue) for nuclei and tubulin for cytoplasm (green) (top right). A merge is shown in the bottom right of each panel. Scale bars, 10 μm. The experiment was conducted in triplicate for each condition. (D) Semi-quantitative analysis of confocal microscopy (C); six high-power fields (HPFs) were counted for each condition, and the percentage co-localization of VV and monocytes was recorded. Data are presented as mean ± SEM. ∗∗∗∗p < 0.0001 (Student’s unpaired t test). (E) Quantitative RT-PCR detection of VVL15 in macrophages derived from immunocompetent mice (n = 3/group) pre-treated with 1 μM IC87114 or 5 μM idelalisib or vehicle buffer for 2 h (n = 4/group). Data are presented as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01 (one-way ANOVA with Newman-Keuls multiple comparison test).
Pre-treatment of macrophages with a PI3Kδ inhibitor reduced in vitro VV particle attachment to these cells (Figures 2C–2E), but it did not affect their phagocytic activity, with all attached particles being internalized in the presence of PI3Kδ inhibitor (Figures S1C and S1D). These data demonstrate that PI3Kδ signaling is required for attachment of virus to the cell membrane, but not the subsequent internalization of attached particles. These results were mirrored using the related, clinically approved PI3Kδ-selective inhibitor idelalisib (CAL-101)16 that prevented virus attachment, but not internalization, by macrophages (Figure 2E; Figure S1E) and inhibited AKT phosphorylation (Figures S1F and S1G). Interestingly, PI3K inhibition had no effect on the attachment or internalization of another OV, adenovirus (AdV), to macrophage populations (Figure S2). Systemic delivery of AdV is severely hampered by non-specific uptake of the virus in the liver, and mechanisms to avoid such clearance are being actively sought in order to improve the therapeutic potential of oncolytic AdV.17
Downstream Effectors of PI3Kδ Involved in VV Attachment to Macrophages
We next explored possible mechanisms by which PI3Kδ regulates virus binding to macrophages. Expression of a constitutively active RhoA mutant in HeLa cells has previously been shown to reduce viral entry.18,19 Given that PI3Kδ activity inhibits cellular RhoA activity13 and the VV F11L protein inhibits RhoA signaling,20, 21, 22 we investigated the involvement of PI3Kδ-RhoA signaling in virus attachment to macrophages. In line with previous results,13 treatment with IC87114 led to an increase in the levels of activated RhoA in macrophages compared to vehicle treatment (Figure 3A). Inhibition of the downstream target of RhoA, the Rho-associated protein kinase (ROCK), using the ROCK inhibitors Y-27632 or GSK429286, neutralized the IC87114-induced decrease in viral attachment (Figures 3B and 3C), demonstrating that activation of RhoA-ROCK signaling is involved in reduced viral attachment when PI3Kδ is inhibited. Contained within the Rho GTPase superfamily are the RhoA, Cdc42, and Rac subfamilies, with Cdc42 and Rac signaling often counteracting the RhoA axis and vice versa.23 Rac signaling is also responsive to PI3K.24 VV infection has been shown to activate Rac1, and prevention of Rac1 activation leads to reduced viral infection.18,19,25 In line with these reports, the Rac1 inhibitor NSC23766 significantly reduced VV attachment to macrophages (Figure 3B).
Figure 3.

Inhibition of PI3Kδ Affects a Signaling Axis Involved in Cytoskeletal Rearrangements
(A) qPCR quantification of activated GTP-bound RhoA normalized against the total RhoA at different time intervals in macrophages pooled from two immunocompetent BALB/c mice pre-treated with vehicle or IC87114 (1 μM) (n = 4/group). Data are presented as mean ± SEM. ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001 (Student’s two-tailed paired t test). (B) Quantitative RT-PCR detection of VVL15 attachment to macrophages pooled from two immunocompetent BALB/c mice pre-treated with vehicle or IC87114 (1 μM) with or without Y-27632 (ROCK inhibitor) (10 μM) or the Rac1 inhibitor NSC23766 (50 μM) (n = 4/group). Data are presented as mean ± SEM. ∗p < 0.05; ns, not significant (one-way ANOVA with Newman-Keuls multiple comparison test). (C) Quantitative RT-PCR detection of VVL15 attachment to macrophages pooled from two immunocompetent BALB/c mice pre-treated with vehicle or IC87114 (1 μM), or IC87114 in combination with the ROCK kinase inhibitor GSK429286 (100 μM) (n = 4/group). Data are presented as mean ± SEM. ∗p < 0.05 (one-way ANOVA with Newman-Keuls multiple comparison test). (D) Confocal microscopic images of macrophages treated for 2 h at 37°C with vehicle buffer or IC87114, in which the cells were stained with DAPI (blue), F-actin (red), and α-tubulin (green). Condensation of α-tubulin is indicated by the arrows. Scale bars, 10 μm.
RhoA belongs to the Rho GTPase superfamily, key regulators of actin and microtubule dynamics and organization.23 Given that treatment with IC87114 resulted in reduction of F-actin and condensation of α-tubulin (Figure 3D), it is likely that activation of the RhoA-ROCK signaling cascade by PI3Kδ inhibition prevents virus attachment to macrophages via a perturbation of the cytoskeletal dynamics required for effective attachment.8,26
In Vivo Inhibition of PI3Kδ Increases Tumor Localization of i.v.-Delivered VV
We next explored the impact of oral administration of IC8711427 on the i.v. delivery of VVL15 to subcutaneous tumors in immunocompetent mice. Several doses of IC87714 have been used in the literature.28,29 Preliminary dose escalation studies and a pharmacokinetic study performed in rats demonstrate that the time to maximum plasma concentration after oral administration of IC87114 was 4 h.27 We performed a dose escalation study using colorectal CT26 subcutaneous tumors implanted into BALB/c mice that demonstrated 75 mg/kg as the optimal in vivo dose for maximal viral delivery to tumors (Figure S3A). Three hours after oral administration of IC87114, CT26 tumor-bearing mice were given a single i.v. injection of 1 × 108 PFU of VVL15. Tumor luminescence demonstrated a significant advantage to viral delivery when IC87114 was administered (Figure 4A). We have previously reported that systemically delivered VVL15 has few off-target effects, and using this model, we confirmed that i.v. delivered VVL15 was only detected in the spleen and lungs of mice. We were only able to recover minimal virus from other organs (brain, ovaries, liver) and in no case did pre-treatment with IC87115 affect viral biodistribution (Figures S3B–S3F). Of note, idelalisib was able to enhance in vivo delivery of VV to tumors to the same extent as IC87114 at all doses tested using the same model (Figure S3G). Using the same model, mice were treated three times (days 1, 3, and 5) with a therapeutically effective dose of 1 × 108 PFU of VVL15 post-IC87114 treatment, and tumors were examined 3 or 7 days post-treatment. Hematoxylin and eosin (H&E) staining demonstrated that VV infection resulted in necrosis and eosinophilic degeneration of tumor cells, indicative of virus infection, with the combination of IC87114 with VVL15 inducing these features to a greater extent than treatment with VVL15 alone (Figure 4B). VV protein expression within the tumor was significantly enhanced by IC87114 at both 3 days (Figures 4C and 4E) and 7 days (Figures 4D and 4E) post-infection. Analysis of the spleen of mice treated with one dose of virus 3 h post-infection demonstrated an enhanced accumulation of VV protein in mice treated with vehicle buffer compared to IC87114 (Figures 4F–4H), consistent with the notion that IC87114 leads to a reduced early clearance of VVL15 by splenic macrophages and indicative of a dampening of the innate immune response to viral infection by inhibition of PI3Kδ.
Figure 4.

Inhibition of PI3Kδ Results in a Reduced Interaction of VV with Splenic Macrophages and Increased Infection of Tumor Cells In Vivo
(A) Quantification of IVIS imaging to detect intra-tumoral VVL15 in BALB/c mice bearing CT26 flank tumors. Mice were pre-treated with 75 mg/kg IC87714 or vehicle buffer 3 h before i.v. injection of 1 × 108 PFU of VVL15 (n = 11/group). Shown are the combined results of three experiments. Data are presented as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01 (two-way ANOVA with Bonferonni multiple test correction). (B) CT26 tumors were established in the flanks of immunocompetent BALB/c mice and treated three times (days 1, 3, and 5) with IC87114 via oral gavage at 75 mg/kg. Three hours later, mice were treated i.v. with PBS or 1 × 108 PFU of VVL15. Animals were sacrificed 3 days post-infection and snap-frozen tumors were stained using H&E to examine the pathology of treated tumors (n = 3/group). Eosinophilic degranulation, indicative of viral infection, is shown by the arrows. (C and D) Animal models were performed as for (B), and tumors were examined for VV protein expression via immunohistochemical staining for VV coat protein at 3 (C) and 7 (D) days post-treatment. Representative images are shown for each group. (E) Quantification of VV expression was performed by counting VV-positive cells in 10 high-power fields (HPFs) for each tumor from (C). Data are presented as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01 (one-way ANOVA with Bonferonni multiple test correction). (F–H) Animal models were performed as for (B), but only one dose of virus was delivered. 3 h post-delivery, animals were sacrificed and spleens analyzed for the presence of VV coat protein using immunohistochemistry. Representative images of mice treated with vehicle buffer (F) or IC87114 (G) prior to virus delivery are shown at ×40 and ×200 (inset) original magnification. (H) Cells positively staining for VV coat protein in 10 randomly selected HPFs from each tumor (n = 3/group) were quantified. The number of cells immunoreactive for VV coat protein was manually counted using random fields at ×200 original magnification. Data are presented as mean ± SEM. ∗∗∗p < 0.001 (Student’s two-tailed unpaired t test).
In Vivo Inhibition of PI3Kδ Enhances Anti-tumor Efficacy of i.v.-Delivered VV
We next examined whether the combination of IC87114 and VVL15 could result in enhanced anti-tumor efficacy after i.v. delivery in immunocompetent murine cancer models. Using both subcutaneous CT26 colorectal cancer (Figures 5A and 5B) and orthotopic 4T1 breast cancer (Figures 5C and 5D) models, we found that IC87114 enhanced the in vivo anti-tumor efficacy of i.v. delivered VVL15, reducing the tumor burden and prolonging survival. IC87114 did not have direct effects on replication and cytotoxicity of VVL15 in these murine tumor cell lines in vitro (Figures S4A and SB), which is not surprising, given that the solid tumor cells tested in this study do not express PI3Kδ (Figures S4C). In addition, no clinical or pathological evidence of treatment toxicity was observed.
Figure 5.

Pharmacological Inhibition of PI3Kδ Enhances the Anti-tumor Effects of i.v. Administered VVL15
(A and B) Tumor growth (A) and survival (B) of BALB/C mice bearing CT26 tumors treated with 75 mg/kg IC87114 or vehicle buffer 3 h prior to i.v. injection of 1 × 108 PFU of VVL15 (n = 10/group). Mice were treated three times, at the days indicated by arrows on the x axis. Day 0 represents the day of tumor cell inoculation into the animals. Data are presented as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 (two-way ANOVA with Bonferonni multiple test correction). Survival analysis data are represented in a Kaplan-Meier plot and the significance (comparing VVL15 alone to IC87114 + VVL15) was assessed using the log-rank (Mantel-Cox) test. (C and D) Tumor growth (C) and survival (D) of BALB/c mice bearing orthotopic 4T1 tumors treated with 75 mg/kg IC87114 or PBS 3 h prior to i.v. injection of 1 × 108 PFU of VVL15 (n = 9/group). Mice were treated three times, at the days indicated by arrows on the x axis. Day 0 represents the day of tumor cell inoculation into the animals. Data are presented as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 (two-way ANOVA with Bonferonni multiple test correction). Survival analysis data are represented in a Kaplan-Meier plot and the significance (comparing VVL15 alone to IC87114 + VVL15) was assessed using the log-rank (Mantel-Cox) test.
In Vivo Inhibition of PI3Kδ Enhances T Cell Infiltration into Tumors and Engages Anti-tumor Immune Responses
Systemic inactivation of PI3Kδ has been shown to give rise to increased adaptive anti-tumor immunity, by preferentially inhibiting immunosuppressive regulatory T cells (Tregs), allowing the development of a host CD8+ cytotoxic cell response.30, 31, 32 PI3Kδ is involved in a range of T cell biological responses and cytokine production in both mice and humans.30,32, 33, 34. Using the subcutaneous CT26 model, we found that virus infection was able to significantly enhance CD4+ T cell infiltration into the tumor (Figure 6A), although we were unable to efficiently detect FoxP3+ CD4+ T cells in this tumor model and as such cannot clarify the contribution of pro-tumor and anti-tumor CD4+ T cells to this increase. CD8+ T cell infiltration into tumors was only enhanced when virus was delivered in combination with IC87114 (Figure 6B), demonstrating that transient inhibition of PI3Kδ can potentiate virus-induced immune responses within the tumor. Interestingly, single agent treatment with either IC87114 or i.v. administered VVL15, while not able to significantly raise i.t. CD8+ T cell levels, was able to enhance interferon (IFN)-γ production by ex vivo explanted splenocytes isolated 7 days after treatment, indicative of an enhanced adaptive host anti-tumor immune response (Figure 6C). However, splenocytes isolated from mice treated with the combination of IC87114 and VVL15 produced significantly more IFN-γ upon ex vivo stimulation with mitomycin-C-treated CT26 cells compared to single agent treatment. These data are consistent with a scenario whereby inhibition of PI3Kδ results in increased viral load within the tumor, potentiating the development of anti-tumor immunity.
Figure 6.

Pre-treatment with IC87114 Results in Increased T Cell Infiltration and Anti-tumor Immunity In Vivo
CT26 tumors were established in the flanks of immunocompetent BALB/c mice and treated three times 14 days after tumor inoculation (days 1, 3, and 5) with IC87114 (or vehicle buffer) via oral gavage at 75 mg/kg. Three hours later, mice were treated i.v. with PBS or 1 × 108 PFU of VVL15. Animals were sacrificed 3 days post-infection and snap-frozen tumors were stained using antibodies reactive to CD4 (A) or CD8 (B) T cells. Representative images for each condition are shown. 6–12 HPFs were examined for each condition and the CD4+ or CD8+ immune infiltrate was quantified (n = 3/group). Data are presented as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01 (one-way ANOVA with Tukey multiple test correction). (C) Tumors were established and treated as in (A). 7 days post-treatment, spleens were harvested and IFN-γ production by splenocytes was assessed ex vivo (n = 5/group) after stimulation for 7 days with mitomycin-treated CT26 cells. Data are presented as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 (one-way ANOVA with Newman-Keuls multiple comparison test).
Discussion
In the era of cancer immunotherapy, OVs have emerged as powerful potential anti-tumor agents, due to their ability to target and lyse tumor cells, also providing in situ vaccination that leads to host anti-tumor immune responses. Thus, the efficacy of OV therapy depends on both the oncolytic action of the virus and the stimulation immune responses to locally released tumor-associated antigens (TAAs).35 While the clinical impact of viral vectors used has been modest to date, an increased understanding of the complex interplay between tumors and the immune system is allowing the development of therapeutically more powerful agents, for example by incorporating exogenous agents in the virus that can be expressed in the TME to potentiate an anti-tumor response.36,37 However, successful delivery of OV agents remains an important barrier to realize their full potential for cancer treatment.
VV is one of the most promising viral agents for OV. In this study, we present evidence that macrophages are key players in the early clearance of VV from the circulation of immunocompetent mice and that transient, pharmacological inhibition of PI3Kδ can enhance systemic delivery of oncolytic VV to tumors. Pre-treatment of mice with the IC87114 inhibitor, highly selective for the PI3Kδ isoform,15 increases RhoA levels within macrophages, correlating with (1) reduced viral attachment in vitro, (2) reduced VV coat protein association with macrophages in the red pulp of the spleen, (3) effective infection of tumors, (4) enhanced tumor necrosis, (5) enhanced anti-tumor adaptive immune responses, and (6) prolonged survival of tumor-bearing mice. The potentiation of VV treatment by PI3Kδ inhibition is most likely the result of a combined action on several immune cell types, including macrophages and an enhanced T cell-mediated anti-tumor response.30,32,38,39
Most previous studies analyzing VV-induced PI3K signaling have been undertaken in HeLa cells, with only one group investigating VV entry in dendritic cells.40 Our work expands previous studies that have shown that a pan-class I PI3K inhibitor can reduce VV attachment and infection in HeLa cells in vitro18,19,25,41 and for the first time shows that PI3K signaling is involved in viral attachment in macrophages. Izmailyan et al.41 have suggested that VV associates with glycosaminoglycans and laminin on the cell membrane and induces the recruitment of integrins such as β1, with subsequent recruitment of CD98 (a heterodimeric membrane transport protein) and activation of PI3K signaling, resulting in endocytic internalization of VV. Others have implicated EGFR as a key protein involved in the induction of intracellular signaling leading to viral attachment and internalization.19 Although we have not elucidated potential transmembrane proteins involved in the attachment of the VV, we have shown that PI3Kδ inhibition reduces viral attachment to, but not internalization by, macrophages, through several complementary pathways involving RhoA/ROCK, AKT, and Rac signaling (Figure 7). PI3Kδ has been shown to downregulate RhoA and ROCK via activation of PYK/Src and AKT that lead to cytoplasmic accumulation of p27, inhibiting the conversion of guanosine diphosphate (GDP)-bound RhoA to RhoA-GTP.9,13 Our results confirm previous studies that have demonstrated expression of constitutively active RhoA prevents VV infection of tumor cells25 and that the PI3K/AKT signaling axis is required for viral infection of cells,41 likely by preventing the process of actin remodeling required for pseudopod extensions that allow binding of large viruses such as VV (around 360 nm in size) to the cell surface.8,19,25 PI3Kδ is also involved in activation of Rac1, which has previously been shown to be required for VV infection of cells.19,25 Rac1 signaling opposes RhoA signaling to the actin network and is required for actin ruffling and filopodia formation involved in macropinocytosis of VV particles.19,23,25 It is noteworthy that we found that PI3Kδ inhibition only affected the attachment of the virus to the cell surface and not the subsequent internalization steps. Previous studies have indicated a role for PI3K in micropinocytosis and phagocytosis by macrophages;11 however, our results suggest a functional dissociation between attachment and internalization. Cox et al.8 have reported that PI3K inhibition in macrophages prevented pseudopod extension, but not phagosomal closure, supporting a role for PI3Kδ in the early stages of phagocytosis. Our results support this conclusion and suggest that PI3Kδ is not required to complete the process of virus internalization, but for appropriate reorganization of the cell membrane required for viral attachment. Noteworthy also is the observation that AdV attachment to and internalization by macrophages is unaffected in the presence of PI3Kδ inhibition, although the difference in biology between AdV and VV suggests this result as unsurprising. For VV, a large particle (360 × 270 × 250 nm), attachment to macrophages seems to be dependent on the PI3Kδ-dependent increase in cell surface area and is receptor-independent. Conversely, AdV particles are much smaller (90–100 nm), attaching to cells in a receptor-dependent manner and not reliant on membrane rearrangements required by VV.
Figure 7.

Signaling Axis Affected by PI3Kδ Inhibition
The AKT, RhoA, and Rac signaling pathways are shown, and the effect on individual proteins after pharmacological inhibition of PI3Kδ is indicated by red arrows.
Although IC87114 is not being developed for clinical use, its derivative idelalisib (previously known as GS-1101 or CAL-101) has now been approved for therapy of B cell malignancies such as chronic lymphocytic leukemia (CLL). Whereas idelalisib is relatively well tolerated in heavily pre-treated cancer patients, with colitis as a main side effect, front-line therapy poses more risks, and determination of tolerable dose regimens is being evaluated in ongoing clinical trials using PI3Kδ inhibitors. In our study, PI3Kδ inhibitors were only used 3 h before each i.v. injection of VV, which resulted in no apparent adverse side effects. Recent data have shown that short-term PI3Kδ inhibitor treatment had effective anti-tumor activity in preclinical studies of solid tumor growth in mice.42 Coupled with the notion that PI3Kδ inhibitors are clinically approved, and the accumulating evidence that inhibition of PI3Kδ has broad anti-cancer activities, including a dampening of Treg function, reduced expansion of myeloid-derived suppressor cells (MDSCs), and improved generation of long-lived memory T cells,39 our data provide a strong rationale for the clinical exploration of PI3Kδ inhibition as a means to enhance the delivery of oncolytic VV.
In summary, this study provides a proof of concept of a new therapeutic regimen for clinical translation of i.v. injected VV for treatment of cancer.
Materials and Methods
Cell Lines
All cell lines were cultured in Dulbecco’s modified Eagle’s medium (DMEM) with 5%–10% fetal calf serum (FCS) except macrophages, which were cultured in RPMI 1640 with 10% FCS, 28.6 μM β-mercaptoethanol, and 1% streptomycin/penicillin and maintained at 37°C with 5% CO2. CT26 cells were obtained from the Cancer Research UK cell bank. The CV1 and 4T1 cell lines were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA).
Reagents
PI-103 (PI3Kα inhibitor), TGX221 (PI3Kβ inhibitor), and pan-PI3K inhibitor wortmannin were from obtained Merck. The Rac1 inhibitor NSC23766, GSK429286, and Y-27632 were from Tocris Bioscience. Sigma-Aldrich supplied the PI3Kγ isoform inhibitor AS604850. The PI3Kδ inhibitor IC87114 was purchased from SYNkinase and Selleck Chemicals, and CAL-101 (idelalisib) was obtained from Selleck Chemicals. Drug concentrations used for in vitro experiments were as follows: PI-103 at 80 nM,43 TGX-221 at 0.1 μM,44 NSC23766 at 50 μM,45 Y-27632 at 10 μM,46 and AS604850 at 1 μM.47 The PI3Kδ inhibitor IC87114 was used at 1 μM,29 and CAL-101 (PI3Kδ-specific) was used at 5 μM for all in vitro assays unless otherwise stated. The antibodies used for western blot and immunofluorescence studies were anti-PI3Kδ and anti-PI3Kα from Santa Cruz Biotechnology, anti-PI3Kγ, anti-AKT, anti-phosphorylated (p-)AKT, and anti-proliferating cell nuclear antigen (PCNA) from Cell Signaling Technology, anti-CD68 and anti-phalloidin from Invitrogen, and anti-α−tubulin and anti-vinculin from Sigma-Aldrich. The VV antibody used for the immunohistochemical experiments was a rabbit anti-VV polyclonal antibody supplied by AbD Serotec. F4/80 rat anti-mouse from AbD Serotec was used at a 1:1,000 dilution to detect macrophages. Rat anti-mouse CD8 and rat anti-mouse CD4 were both supplied by BioLegend. Pronase was supplied by Roche Applied Science and dissolved in PBS at 1 mg/mL. MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt) and PMS (phenazine methosulfate) were provided by Promega. T cell culture medium (TCM) was RPMI 1640, 10% FCS, 1% streptomycin, 1% penicillin, and 1% sodium pyruvate. Macrophage colony stimulating factor (M-CSF) was supplied by R&D Systems. d-Luciferin was supplied by Caliper Life Sciences. Clodronate liposomes were supplied by Dr. Nico van Rooijen (http://clodronateliposomes.org/ashwindigital.asp?docid=26). Vehicle buffer, used as either a control or to dissolve IC87114, was made by the addition of 0.25% Tween 20 and 0.5% carboxymethylcellulose to PBS. CAL-101 was re-suspended at 30 mg/mL with 30% polyethylene glycol (PEG) 400, 0.5% Tween 80, and 5% propylene glycol and administered via oral gavage at 10 mg/kg.
Viruses
VVL15 was a kind gift from Prof. Istvan Fodor (Loma Linda University Campus, Loma Linda, CA, USA). It was constructed by the insertion of the firefly luciferase and the lacZ reporter genes into the TK region of VV Lister under the control of the early-late vaccinia p7.5 promoter and IMV particles purified using CV1 cells as described previously.48,49 Adv type 5 has been described previously.37
DNA/RNA Extraction
DNA extraction was performed using the QIAGEN DNeasy blood and tissue kit. RNA was converted to cDNA using the SuperScript II RT system (Thermo Fisher Scientific).
Quantitative Polymerase Chain Reaction (qPCR)
Quantification of viral genome copy number was achieved using the TaqMan PCR system provided by Applied Biosystems as previously described.50 For VV quantification, the primers and probe were designed for the VV late transcription factor 1 (VLTF-1) gene as follows: forward, 5′-AACCATAGAAGCCAACGAATCC-3′ (code SY100203462-026), reverse, 5′-TGAGACATACAAGGGTGGTGAAGT-3′ (code SY100203462-027), and probe, 5′-ATTTTAGAACAGAAATACCC-3′. The primers were supplied by Sigma-Aldrich. The standard was VVL15 DNA, and 40 ng of DNA was used per sample as the template. Viral genome copy number was normalized by total DNA loaded. The PI3Kδ qPCR assay was carried out with a pre-designed kit from Applied Biosystems using PI3Kδ mouse cDNA as a standard.
Culture of Bone Marrow-Derived Macrophages
M-CSF was added to the suspension of bone marrow-derived monocytes and mixed several times. The cells were then plated at 10 mL/plate on 10-cm Petri dishes. The medium and M-CSF were replaced on alternative days, and the macrophages were considered fully matured by day 7 and were plated for experimentation on either day 7 or 8. Confirmation of the macrophage population was routinely undertaken by fluorescence-activated cell sorting (FACS) using CD11b and F4/80 antibodies.
Ex Vivo Infection of Isolated Blood Cells from BALB/c Mice
The blood from three mice was pooled and then diluted 1:1 with RPMI 1640 and buffy coat isolated. Monocytes were infected with VVL15 at a multiplicity of infection (MOI) of 0.01. The estimate of the total number of blood cells/mL in a mouse was obtained from https://phenome.jax.org/ with an assumption of a total circulating volume of 1.5 mL. The following formula was used to determine the ex vivo experimental MOI: MOI = a/bc, where a represents the standard in vivo dose of VV of 1 × 108 PFU/mouse, b indicates the total number of blood cells/mL, and c indicates total circulating volume (mL).
After infection, the monocytes were washed, DNA was extracted from all samples, and qPCR was performed to quantify the amount of viral DNA present.
Viral Attachment and Internalization Assays
Cultured monocytes were seeded in 24-well plates at 1 × 105 cells per well in 500 μL of macrophage medium. The next day, the cells were treated with inhibitors at required concentration for 2 h at 37°C. The cells were cooled and infected with VVL15 at an MOI of 5 (or Ad5 at an MOI of 5 × 103 particles/cell) and shaken slowly for 1 h at 4°C. The cells were washed twice with chilled PBS. For the attachment assay, the cells were detached and stored at −20°C ready for subsequent DNA extraction and qPCR. For the RhoA experiment the cells were mock infected or infected for 0, 3, 6, 12, and 30 min rather than 1 h and the samples were stored at −20°C ready for quantification of total RhoA and activated GTP-bound RhoA levels using commercially available ELISA and G-LISA kits, respectively, from Cytoskeleton.
For internalization assays, the virus-containing medium was removed and the cells were warmed at 37°C. At the desired time points, the cells washed with ice-cold PBS and treated with 500 μL of Pronase (1 mg/mL) per well on ice for 30 min. The cells were then collected, washed, and frozen for subsequent DNA extraction and qPCR. When these experiments were done using transgenic knock-in macrophages, the addition of any inhibitor was omitted. All experiments were performed in triplicate and repeated at least twice.
In Vitro Cell Survival Assay
For cell survival assays, 1 × 103 to 5 × 103 cells were seeded in each well of 96-well plates in 90 μL of medium with or without IC87114 and on the next day infected with VV at a serial dilution. Cell survival was determined by MTS assay (Promega) 6 days after infection, and the 50% effective concentration (EC50) values were obtained as previously described.51 All experiments were performed in triplicate and repeated twice.
Viral Replication Assay (TCID50)
The TCID50 (tissue culture inhibitory dose 50%) assay is a limiting dilution assay, which quantifies any infectious particles in the test sample. The calculation of TCID50 values was conducted as previously described.51 Titrations were performed in duplicate.
Confocal Microscopy
The macrophages were seeded into chamber slides (Lab-Tek II chamber slides) in complete medium and the next day the cells were pre-treated with IC87114 at 1 μM or vehicle (PBS) for 2 h and incubated at 4°C for 30 min before being infected with red fluorescent protein (RFP)-expressing VVL15B5R mutant VV at an MOI of 5 or mock infected. The cells were incubated at 4°C with shaking for 1 h. The cells were washed twice with cold PBS and fixed in methanol for 8 min at −20°C. For F-actin staining, the cells were fixed in 4% formaldehyde. Subsequently, the cells were incubated with a 1:500 dilution of mouse anti-α-tubulin (Sigma-Aldrich) for 1 h at room temperature. After three washes in PBS, the primary antibody was detected with Alexa Fluor 488-conjugated donkey anti-rabbit (Molecular Probes/Invitrogen) at a dilution of 1:1,000. Nuclear DNA was stained with DAPI (Molecular Probes/Invitrogen). Phalloidin-Alexa Fluor 594 (Invitrogen) was used to visualize F-actin. The slides were mounted using Permafluor aqueous mounting medium (Immunotech). Immunofluorescence staining on frozen tissue was carried out after the slides were placed at 37°C for 8 min and fixed in ice-cold methanol for 5 min at room temperature. The slides were permeabilized with 0.2% Triton X-100 in PBS and incubated with anti-CD68 and anti-VV primary antibodies, and the above procedures were followed. The CD68 primary antibody was detected with Alexa Fluor 488-conjugated chicken anti-rat, and the VV primary antibody was detected with Alexa Fluor 546-conjugated goat anti-rabbit (Molecular Probes/Invitrogen) at a dilution of 1:1,000. A series of optical sections were taken with a Zeiss LSM 510 confocal microscope and projected to single images using LSM 510 software (Zeiss). Identical acquisition methods were used for all samples to allow direct comparison of the resulting images. Photomontages were created using Adobe Photoshop 5.1 software. All conditions were assayed in triplicate.
Western Blotting
The monocytes were plated in six-well plates at 2 × 105 cells per well. On the next day, the cells were pre-treated with IC87114 at 1 μM or vehicle (PBS) for 2 h at 37°C. The cells were cooled and infected with VVL15 at an MOI of 5 or mock infected and incubated at 4°C for 1 h and the lysates were harvested in PhosphoSafe extraction reagent (Merck Millipore). For immunoblotting, 40–80 μg of protein was resolved in 8% SDS polyacrylamide gel and transferred to nitrocellulose membrane at 100 V for 60 min. The blocking was performed using 5% milk before incubation overnight with the primary antibodies at 4°C. The secondary antibodies were anti-rabbit, anti-goat, or anti-mouse horseradish peroxidase (HRP) conjugated from Santa Cruz Biotechnology Inc. Detection of the bound antibody was carried out using enhanced chemiluminescence (ECL) western blotting detection reagents (Amersham Biosciences, GE Healthcare). Anti-PCNA antibody was used as a loading control (Santa Cruz Biotechnology).
Animal Studies
All animal experiments were performed in accordance with UK Home Office regulations and approved by the Institutional Committee. BALB/c mice were supplied by the Queen Mary University of London Biological Services Unit (Charterhouse Square, London, UK). The animal studies were undertaken by the CRUK Biological Resources Unit at Clare Hall and by M.F., G.M., and L.S.C.D. at the QMUL Biological Services Unit at Charterhouse Square. Animals were killed when the width × length of tumors was greater than 1.4 cm2 or any signs of ulceration or distress appeared. Tumor volumes were calculated according to the following formula: Tumor volume = πw2l/6, where w is width and l is the length.
In efficacy studies, line graphs are used to present tumor volumes against time until the first mouse in the group died (or was culled). Similarly, Kaplan-Meier plots represent the survival of each mouse in each group and continue until the termination of the experiment. Functional analysis of immune cells and tumor pathology was performed at days 3 and 7 post-injection to allow viral infection of tumor cells to occur and for the induction of early innate and late adaptive immune responses.
2 × 106 CT26 cells were implanted subcutaneously into the left flank of 8-week-old female BALB/c mice. 5 × 105 4T1 cells were implanted orthotopically in the lower left thoracic mammary gland in 8-week-old female BALB/c mice. Animals were treated when tumors were palpable on days 0, 3, and 5, which were chosen to augment natural peaks of viral infection historically noted in tumors on days 3 and 5 after one i.v. injection of virus, with 1 × 108 PFU/injection VVL15 (or PBS) in 100 μL via tail vein injection. Virus was administered 3 h after IC87114, CAL-101, or vehicle buffer treatment, which was given via oral gavage at a concentration of 75 mg/kg (IC87114) or 10 mg/kg (CAL-101) in 200 μL of vehicle buffer. Group sizes are indicated in the figure legends.
Immunohistochemistry
Appropriately harvested samples from in vivo studies were immediately frozen using 2-methylbutane. Samples were then cut at 5-μm frozen sections. H&E as well as immunohistochemistry staining were performed by Pathology Services, Barts Cancer Institute as described previously.48 Each sample was assessed in duplicate.
Imaging
Whole-body IVIS (in vivo imaging system) imaging was performed under inhalation anesthetic 10 min following intra-peritoneal injection of d-luciferin (15 mg/mL). The luciferase signal was only observed in the subcutaneous tumor in all animals examined.
Ex Vivo Splenocyte Restimulation Assays
Harvested spleens were flushed through 70-μm BD Falcon cell strainers with complete T cell media (RPMI 1640 medium [Sigma-Aldrich], 10% FCS, 1% streptomycin/penicillin, 1% sodium pyruvate, and 1% non-essential amino acids (Gibco) and 0.1% β-mercaptoethanol). Red blood cells (RBCs) were lysed using RBC lysis buffer (Sigma-Aldrich) and re-suspended in complete T cell medium.
Splenocytes were re-suspended in TCM to a final concentration of 5 × 106 cells/mL. CT26 cells were growth arrested using mitomycin C (Roche) at a final concentration of 100 mg/mL in a humidified incubator at 37°C with 5% CO2 for 2 h. Cells were washed twice with PBS and re-suspended in TCM at a final concentration of 5 × 105 cells/mL. 100-μL aliquots of splenocyte suspensions were co-cultured with 100 μL of mitomycin C-treated CT26 cells in a 96-well plate. Plates were incubated at 37°C with 5% CO2 for 3 days, centrifuged at 1,200 rpm for 5 min, and the supernatant was collected. The concentration of IFN-γ was determined using a murine-specific IFN-γ ELISA kit from BioLegend.
Statistical Analysis
All statistical analysis was undertaken in Prism 5 except the calculation of TCID50 values using the Reed-Muench method, which was performed in Microsoft Excel. EC50 values were determined from dose-response curves created using non-linear regression analysis. Experiments with more than two groups were analyzed by either one-way ANOVA with Newman-Keuls or Tukey comparison post-tests or two-way ANOVA with Bonferroni post-tests depending on the number of variables. Experiments with two groups were analyzed by a paired or unpaired Student’s t test. Survival data were represented in a Kaplan-Meier plot, and log rank analysis was used to determine if differences between groups were significant. Significance is depicted on graphs (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001).
Author Contributions
Y.W. conceived and supervised this study. M.F. and Y.W. were responsible for all aspects of study design, M.F. conducted most of the experiments and the analyses of results, L.S.C.D. did extra in vivo experiments to consolidate the conclusion of this manuscript, and R.G., G.M., J.R.T., E.B., M.A.W., E.A., C.H., J.A., L.S.C.D. contributed to study design and conducted some experiments. B.V. and L.S.C.D. contributed to study design, analysis of results, and writing the manuscript. R.G., L.S.C.D., and N.R.L. contributed in analysis of results and reviewing of the manuscript. M.F., M.A.W., L.S.C.D, B.V., and Y.W. interpreted all results and wrote the manuscript, L.S.C.D. and Y.W. finalized the manuscript.
Conflicts of Interest
M.F., N.R.L., and Y.W. are inventors of a filed patent in the relevant field. B.V. is a consultant to Karus Therapeutics (Oxford, UK), iOnctura (Geneva, Switzerland), and Venthera (Palo Alto, CA, USA) and has received speaker fees from Gilead (Foster City, CA, USA). The remaining authors declare no competing interests.
Acknowledgments
M.F. received funding from the Joint Royal College of Surgeons of England and British Association of Surgical Oncology-Association of Cancer Surgery Research Fellowship and Cancer Research UK Clinical Research Fellowship (C16420/A16373). The authors are grateful to George Elia for histopathology staining. Work in the N.R.L. laboratory was supported by Cancer Research UK (A12008 and A25137). Work in the B.V. laboratory was supported by Cancer Research UK (C23338/A25722) and the UK NIHR University College London Hospitals Biomedical Research Centre. G.M. and L.S.C. are funded by the MRC (MR/M015696/1 and MR/N027655/1).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2020.02.017.
Supplemental Information
References
- 1.Andtbacka R.H., Kaufman H.L., Collichio F., Amatruda T., Senzer N., Chesney J., Delman K.A., Spitler L.E., Puzanov I., Agarwala S.S. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J. Clin. Oncol. 2015;33:2780–2788. doi: 10.1200/JCO.2014.58.3377. [DOI] [PubMed] [Google Scholar]
- 2.Ferguson M.S., Lemoine N.R., Wang Y. Systemic delivery of oncolytic viruses: hopes and hurdles. Adv. Virol. 2012;2012:805629. doi: 10.1155/2012/805629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Breitbach C.J., Burke J., Jonker D., Stephenson J., Haas A.R., Chow L.Q., Nieva J., Hwang T.H., Moon A., Patt R. Intravenous delivery of a multi-mechanistic cancer-targeted oncolytic poxvirus in humans. Nature. 2011;477:99–102. doi: 10.1038/nature10358. [DOI] [PubMed] [Google Scholar]
- 4.Wong H.H., Lemoine N.R., Wang Y. Oncolytic viruses for cancer therapy: overcoming the obstacles. Viruses. 2010;2:78–106. doi: 10.3390/v2010078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Al Yaghchi C., Zhang Z., Alusi G., Lemoine N.R., Wang Y. Vaccinia virus, a promising new therapeutic agent for pancreatic cancer. Immunotherapy. 2015;7:1249–1258. doi: 10.2217/imt.15.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ichihashi Y. Extracellular enveloped vaccinia virus escapes neutralization. Virology. 1996;217:478–485. doi: 10.1006/viro.1996.0142. [DOI] [PubMed] [Google Scholar]
- 7.Vanderplasschen A., Mathew E., Hollinshead M., Sim R.B., Smith G.L. Extracellular enveloped vaccinia virus is resistant to complement because of incorporation of host complement control proteins into its envelope. Proc. Natl. Acad. Sci. USA. 1998;95:7544–7549. doi: 10.1073/pnas.95.13.7544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cox D., Tseng C.C., Bjekic G., Greenberg S. A requirement for phosphatidylinositol 3-kinase in pseudopod extension. J. Biol. Chem. 1999;274:1240–1247. doi: 10.1074/jbc.274.3.1240. [DOI] [PubMed] [Google Scholar]
- 9.Tzenaki N., Papakonstanti E.A. p110δ PI3 kinase pathway: emerging roles in cancer. Front. Oncol. 2013;3:40. doi: 10.3389/fonc.2013.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van Rooijen N., Hendrikx E. Liposomes for specific depletion of macrophages from organs and tissues. Methods Mol. Biol. 2010;605:189–203. doi: 10.1007/978-1-60327-360-2_13. [DOI] [PubMed] [Google Scholar]
- 11.Araki N., Johnson M.T., Swanson J.A. A role for phosphoinositide 3-kinase in the completion of macropinocytosis and phagocytosis by macrophages. J. Cell Biol. 1996;135:1249–1260. doi: 10.1083/jcb.135.5.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cox D., Dale B.M., Kashiwada M., Helgason C.D., Greenberg S. A regulatory role for Src homology 2 domain-containing inositol 5′-phosphatase (SHIP) in phagocytosis mediated by Fcγ receptors and complement receptor 3 (αMβ2; CD11b/CD18) J. Exp. Med. 2001;193:61–71. doi: 10.1084/jem.193.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Papakonstanti E.A., Ridley A.J., Vanhaesebroeck B. The p110δ isoform of PI 3-kinase negatively controls RhoA and PTEN. EMBO J. 2007;26:3050–3061. doi: 10.1038/sj.emboj.7601763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vanhaesebroeck B., Khwaja A. PI3Kδ inhibition hits a sensitive spot in B cell malignancies. Cancer Cell. 2014;25:269–271. doi: 10.1016/j.ccr.2014.02.012. [DOI] [PubMed] [Google Scholar]
- 15.Berndt A., Miller S., Williams O., Le D.D., Houseman B.T., Pacold J.I., Gorrec F., Hon W.C., Liu Y., Rommel C. The p110δ structure: mechanisms for selectivity and potency of new PI(3)K inhibitors. Nat. Chem. Biol. 2010;6:117–124. doi: 10.1038/nchembio.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang Q., Modi P., Newcomb T., Quéva C., Gandhi V. Idelalisib: first-in-class PI3K delta inhibitor for the treatment of chronic lymphocytic leukemia, small lymphocytic leukemia, and follicular lymphoma. Clin. Cancer Res. 2015;21:1537–1542. doi: 10.1158/1078-0432.CCR-14-2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khare R., May S.M., Vetrini F., Weaver E.A., Palmer D., Rosewell A., Grove N., Ng P., Barry M.A. Generation of a Kupffer cell-evading adenovirus for systemic and liver-directed gene transfer. Mol. Ther. 2011;19:1254–1262. doi: 10.1038/mt.2011.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Locker J.K., Kuehn A., Schleich S., Rutter G., Hohenberg H., Wepf R., Griffiths G. Entry of the two infectious forms of vaccinia virus at the plasma membane is signaling-dependent for the IMV but not the EEV. Mol. Biol. Cell. 2000;11:2497–2511. doi: 10.1091/mbc.11.7.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mercer J., Knébel S., Schmidt F.I., Crouse J., Burkard C., Helenius A. Vaccinia virus strains use distinct forms of macropinocytosis for host-cell entry. Proc. Natl. Acad. Sci. USA. 2010;107:9346–9351. doi: 10.1073/pnas.1004618107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Valderrama F., Cordeiro J.V., Schleich S., Frischknecht F., Way M. Vaccinia virus-induced cell motility requires F11L-mediated inhibition of RhoA signaling. Science. 2006;311:377–381. doi: 10.1126/science.1122411. [DOI] [PubMed] [Google Scholar]
- 21.Cordeiro J.V., Guerra S., Arakawa Y., Dodding M.P., Esteban M., Way M. F11-mediated inhibition of RhoA signalling enhances the spread of vaccinia virus in vitro and in vivo in an intranasal mouse model of infection. PLoS ONE. 2009;4:e8506. doi: 10.1371/journal.pone.0008506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Handa Y., Durkin C.H., Dodding M.P., Way M. Vaccinia virus F11 promotes viral spread by acting as a PDZ-containing scaffolding protein to bind myosin-9A and inhibit RhoA signaling. Cell Host Microbe. 2013;14:51–62. doi: 10.1016/j.chom.2013.06.006. [DOI] [PubMed] [Google Scholar]
- 23.Van den Broeke C., Jacob T., Favoreel H.W. Rho’ing in and out of cells: viral interactions with Rho GTPase signaling. Small GTPases. 2014;5:e28318. doi: 10.4161/sgtp.28318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Welch H.C., Coadwell W.J., Stephens L.R., Hawkins P.T. Phosphoinositide 3-kinase-dependent activation of Rac. FEBS Lett. 2003;546:93–97. doi: 10.1016/s0014-5793(03)00454-x. [DOI] [PubMed] [Google Scholar]
- 25.Mercer J., Helenius A. Vaccinia virus uses macropinocytosis and apoptotic mimicry to enter host cells. Science. 2008;320:531–535. doi: 10.1126/science.1155164. [DOI] [PubMed] [Google Scholar]
- 26.Liu B.P., Chrzanowska-Wodnicka M., Burridge K. Microtubule depolymerization induces stress fibers, focal adhesions, and DNA synthesis via the GTP-binding protein Rho. Cell Adhes. Commun. 1998;5:249–255. doi: 10.3109/15419069809040295. [DOI] [PubMed] [Google Scholar]
- 27.Thappali S.R., Varanasi K.V., Veeraraghavan S., Vakkalanka S.K., Mukkanti K. Simultaneous quantitation of IC87114, roflumilast and its active metabolite roflumilast N-oxide in plasma by LC-MS/MS: application for a pharmacokinetic study. J. Mass Spectrom. 2012;47:1612–1619. doi: 10.1002/jms.3103. [DOI] [PubMed] [Google Scholar]
- 28.Durand C.A., Hartvigsen K., Fogelstrand L., Kim S., Iritani S., Vanhaesebroeck B., Witztum J.L., Puri K.D., Gold M.R. Phosphoinositide 3-kinase p110δ regulates natural antibody production, marginal zone and B-1 B cell function, and autoantibody responses. J. Immunol. 2009;183:5673–5684. doi: 10.4049/jimmunol.0900432. [DOI] [PubMed] [Google Scholar]
- 29.Sadhu C., Dick K., Tino W.T., Staunton D.E. Selective role of PI3Kδ in neutrophil inflammatory responses. Biochem. Biophys. Res. Commun. 2003;308:764–769. doi: 10.1016/s0006-291x(03)01480-3. [DOI] [PubMed] [Google Scholar]
- 30.Ali K., Soond D.R., Pineiro R., Hagemann T., Pearce W., Lim E.L., Bouabe H., Scudamore C.L., Hancox T., Maecker H. Inactivation of PI(3)K p110δ breaks regulatory T-cell-mediated immune tolerance to cancer. Nature. 2014;510:407–411. doi: 10.1038/nature13444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ahmad S., Abu-Eid R., Shrimali R., Webb M., Verma V., Doroodchi A., Berrong Z., Samara R., Rodriguez P.C., Mkrtichyan M., Khleif S.N. Differential PI3Kδ Signaling in CD4+ T-cell Subsets Enables Selective Targeting of T Regulatory Cells to Enhance Cancer Immunotherapy. Cancer Res. 2017;77:1892–1904. doi: 10.1158/0008-5472.CAN-16-1839. [DOI] [PubMed] [Google Scholar]
- 32.Patton D.T., Garden O.A., Pearce W.P., Clough L.E., Monk C.R., Leung E., Rowan W.C., Sancho S., Walker L.S., Vanhaesebroeck B., Okkenhaug K. Cutting edge: the phosphoinositide 3-kinase p110δ is critical for the function of CD4+CD25+Foxp3+ regulatory T cells. J. Immunol. 2006;177:6598–6602. doi: 10.4049/jimmunol.177.10.6598. [DOI] [PubMed] [Google Scholar]
- 33.Okkenhaug K., Patton D.T., Bilancio A., Garçon F., Rowan W.C., Vanhaesebroeck B. The p110δ isoform of phosphoinositide 3-kinase controls clonal expansion and differentiation of Th cells. J. Immunol. 2006;177:5122–5128. doi: 10.4049/jimmunol.177.8.5122. [DOI] [PubMed] [Google Scholar]
- 34.Soond D.R., Bjørgo E., Moltu K., Dale V.Q., Patton D.T., Torgersen K.M., Galleway F., Twomey B., Clark J., Gaston J.S. PI3K p110δ regulates T-cell cytokine production during primary and secondary immune responses in mice and humans. Blood. 2010;115:2203–2213. doi: 10.1182/blood-2009-07-232330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thorne S.H. Immunotherapeutic potential of oncolytic vaccinia virus. Immunol. Res. 2011;50:286–293. doi: 10.1007/s12026-011-8211-4. [DOI] [PubMed] [Google Scholar]
- 36.Bartee M.Y., Dunlap K.M., Bartee E. Tumor-localized secretion of soluble PD1 enhances oncolytic virotherapy. Cancer Res. 2017;77:2952–2963. doi: 10.1158/0008-5472.CAN-16-1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang P., Li X., Wang J., Gao D., Li Y., Li H., Chu Y., Zhang Z., Liu H., Jiang G. Re-designing Interleukin-12 to enhance its safety and potential as an anti-tumor immunotherapeutic agent. Nat. Commun. 2017;8:1395. doi: 10.1038/s41467-017-01385-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Abu-Eid R., Samara R.N., Ozbun L., Abdalla M.Y., Berzofsky J.A., Friedman K.M., Mkrtichyan M., Khleif S.N. Selective inhibition of regulatory T cells by targeting the PI3K-Akt pathway. Cancer Immunol. Res. 2014;2:1080–1089. doi: 10.1158/2326-6066.CIR-14-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Okkenhaug K., Graupera M., Vanhaesebroeck B. Targeting PI3K in cancer: impact on tumor cells, their protective stroma, angiogenesis, and immunotherapy. Cancer Discov. 2016;6:1090–1105. doi: 10.1158/2159-8290.CD-16-0716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sandgren K.J., Wilkinson J., Miranda-Saksena M., McInerney G.M., Byth-Wilson K., Robinson P.J., Cunningham A.L. A differential role for macropinocytosis in mediating entry of the two forms of vaccinia virus into dendritic cells. PLoS Pathog. 2010;6:e1000866. doi: 10.1371/journal.ppat.1000866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Izmailyan R., Hsao J.C., Chung C.S., Chen C.H., Hsu P.W., Liao C.L., Chang W. Integrin β1 mediates vaccinia virus entry through activation of PI3K/Akt signaling. J. Virol. 2012;86:6677–6687. doi: 10.1128/JVI.06860-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carnevalli L.S., Sinclair C., Taylor M.A., Gutierrez P.M., Langdon S., Coenen-Stass A.M.L., Mooney L., Hughes A., Jarvis L., Staniszewska A. PI3Kα/δ inhibition promotes anti-tumor immunity through direct enhancement of effector CD8+ T-cell activity. J. Immunother. Cancer. 2018;6:158. doi: 10.1186/s40425-018-0457-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Raynaud F.I., Eccles S., Clarke P.A., Hayes A., Nutley B., Alix S., Henley A., Di-Stefano F., Ahmad Z., Guillard S. Pharmacologic characterization of a potent inhibitor of class I phosphatidylinositide 3-kinases. Cancer Res. 2007;67:5840–5850. doi: 10.1158/0008-5472.CAN-06-4615. [DOI] [PubMed] [Google Scholar]
- 44.Lu X.Y., Ciraolo E., Stefenia R., Chen G.Q., Zhang Y., Hirsch E. Sustained release of PI3K inhibitor from PHA nanoparticles and in vitro growth inhibition of cancer cell lines. Appl. Microbiol. Biotechnol. 2011;89:1423–1433. doi: 10.1007/s00253-011-3101-1. [DOI] [PubMed] [Google Scholar]
- 45.Gao Y., Dickerson J.B., Guo F., Zheng J., Zheng Y. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc. Natl. Acad. Sci. USA. 2004;101:7618–7623. doi: 10.1073/pnas.0307512101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Uehata M., Ishizaki T., Satoh H., Ono T., Kawahara T., Morishita T., Tamakawa H., Yamagami K., Inui J., Maekawa M., Narumiya S. Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature. 1997;389:990–994. doi: 10.1038/40187. [DOI] [PubMed] [Google Scholar]
- 47.Camps M., Rückle T., Ji H., Ardissone V., Rintelen F., Shaw J., Ferrandi C., Chabert C., Gillieron C., Françon B. Blockade of PI3Kγ suppresses joint inflammation and damage in mouse models of rheumatoid arthritis. Nat. Med. 2005;11:936–943. doi: 10.1038/nm1284. [DOI] [PubMed] [Google Scholar]
- 48.Chard L.S., Maniati E., Wang P., Zhang Z., Gao D., Wang J., Cao F., Ahmed J., El Khouri M., Hughes J. A vaccinia virus armed with interleukin-10 is a promising therapeutic agent for treatment of murine pancreatic cancer. Clin. Cancer Res. 2015;21:405–416. doi: 10.1158/1078-0432.CCR-14-0464. [DOI] [PubMed] [Google Scholar]
- 49.Dénes B., Yu J., Fodor N., Takátsy Z., Fodor I., Langridge W.H. Suppression of hyperglycemia in NOD mice after inoculation with recombinant vaccinia viruses. Mol. Biotechnol. 2006;34:317–327. doi: 10.1385/MB:34:3:317. [DOI] [PubMed] [Google Scholar]
- 50.Wang Y., Gangeswaran R., Zhao X., Wang P., Tysome J., Bhakta V., Yuan M., Chikkanna-Gowda C.P., Jiang G., Gao D. CEACAM6 attenuates adenovirus infection by antagonizing viral trafficking in cancer cells. J. Clin. Invest. 2009;119:1604–1615. doi: 10.1172/JCI37905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang Y., Hallden G., Hill R., Anand A., Liu T.C., Francis J., Brooks G., Lemoine N., Kirn D. E3 gene manipulations affect oncolytic adenovirus activity in immunocompetent tumor models. Nat. Biotechnol. 2003;21:1328–1335. doi: 10.1038/nbt887. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.