

Abstract
Muscle atrophy is the loss of skeletal muscle mass and strength in response to diverse catabolic stimuli. At present, no effective treatments except exercise have been shown to reduce muscle atrophy clinically. Here, we report that CRISPR/Cas9-mediated genome editing through local injection into gastrocnemius muscles or tibialis anterior muscle efficiently targets the biogenesis processing sites in pre-miR-29b. In vivo, this CRISPR-based treatment prevented the muscle atrophy induced by angiotensin II (AngII), immobilization, and denervation via activation of the AKT-FOXO3A-mTOR signaling pathway and protected against AngII-induced myocyte apoptosis in mice, leading to significantly increased exercise capacity. Our work establishes CRISPR/Cas9-based gene targeting on miRNA as a potential durable therapy for the treatment of muscle atrophy and expands the strategies available interrogating miRNA function in vivo.
Keywords: muscle atrophy, miR-29b, CRISPR/Cas9, AKT-FOXO3A-mTOR signaling pathway
Graphical Abstract
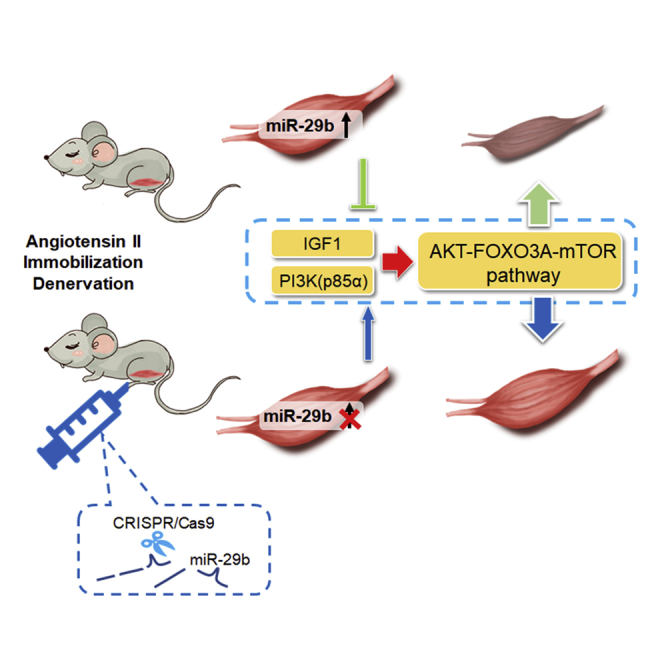
Xiao and colleagues report that CRISPR/Cas9-mediated miR-29b editing prevented the muscle atrophy induced by angiotensin II, immobilization, and denervation via activation of the AKT-FOXO3A-mTOR signaling pathway in mice, leading to significantly increased exercise capacity. Their work establishes CRISPR-based miRNA targeting as a potential therapy for the treatment of muscle atrophy.
Introduction
Muscle atrophy is the loss of muscle mass and strength caused by a variety of stimuli such as inactivity, denervation, glucocorticoid treatment, fasting, and various systemic diseases including heart failure and cancer.1, 2, 3, 4, 5 The common underlying pathways in muscle atrophy result in acceleration of protein degradation and a decrease in protein synthesis, giving rise to increased disability, loss of mobility, and premature fatality.6, 7, 8, 9 Although progress has been made in understanding the pathogenesis of atrophy, except exercise, no other effective treatments are currently available. However, compliance with a regular exercise program is difficult or not feasible in patients who suffer from severe systemic disease or are on long-term bed rest. Therefore, providing therapeutic strategies to combat muscle atrophy is urgent and necessary.
MicroRNAs (miRNAs, miRs) are a large group of short, small non-coding RNAs that control gene expression at a posttranscriptional level.10 Dysregulation of miRNA expression is associated with numerous human diseases.3,11 Some muscle-specific miRNAs, such as miR-1 and miR-133, have been previously reported to play an essential role in the regulation of muscle energy and protein metabolism.12, 13, 14 We previously reported that miR-29b plays an important role in the pathogenesis of muscle atrophy in diverse models.11,15 Overexpression of miR-29b promotes muscle atrophy, while reducing miR-29b attenuates muscle atrophy.15 Consequently, disrupting the expression of miR-29b would be a therapeutic approach for muscle atrophy in several diverse models.
The CRISPR/Cas9 system allows precise genome modification and has been widely explored as a genome-editing tool for genetic modification and human therapeutic applications.16, 17, 18, 19, 20, 21, 22 In the specific application for miRNA loss-of-function studies, CRISPR/Cas9 is an approach that directly introduces mutations in pre-miRNA sequences to disrupt the processing of miRNA biogenesis.23, 24, 25 Considering the importance of miRNAs in regulating gene expression, the utilization of CRISPR-based manipulation tools both for investigation of miRNA function as well as for therapeutic purposes would be a significant advance.
In this study, we packaged Streptococcus pyogenes CRISPR-associated nuclease Cas9 (SpCas9) and guide RNA construct into lentiviral vector and adeno-associated virus 8 (AAV8) with Staphylococcus aureus CRISPR-associated nuclease Cas9 (SaCas9) driven by a skeletal muscle cell-specific double muscle creatine kinase (dMCK) promoter vector and locally injection them into mice skeletal muscle, respectively.17 For the first time we demonstrated that CRISPR-based genome editing in miR-29b could specifically target miR-29b rather than miR-29a and miR-29c, leading to the prevention of the muscle atrophy induced by angiotensin II (AngII), immobilization or denervation in mice models via activation of AKT-FOXO3A-mTOR signaling pathway and protection against AngII-induced apoptosis of myocytes in mice models. The treatment effects are evidenced by muscle morphology, wheat germ agglutinin (WGA) staining and hematoxylin and eosin (H&E) staining for muscle fibers as well as exercise capacity. In this study, we show that targeted CRISPR disruption could be a valuable tool for miRNA loss-of-function studies in animal models. The present data further support the notion and feasibility that miRNA-based in vivo genome editing may provide a platform for the therapeutic intervention of muscle atrophy.
Results
CRISPR/Cas9-Mediated In Vivo Mutagenesis of miR-29b through Intramuscular Administration
Previously, we have reported that miR-29b contributes to multiple types of muscle atrophy.15 To translate this into a therapeutic strategy, we first developed a lentiviral CRISPR/Cas9-based system for the treatment of muscle atrophy in mouse model by disrupting the expression miR-29b. We hypothesized that CRISPR-mediated mutagenesis of primary miR-29b from genomic DNA would restore or partially restore muscle atrophy. As shown in Figure 1A, the mature miR-29b sequence was highlighted as magenta in the pre-miR-29b hairpin structure. A panel of guide RNAs (gRNAs) were designed by the CRISPR design server (http://zlab.bio/guide-design-resources) for the SpCas9 protospacer adjacent motif (PAM) (5′-NGG-3′) with close proximity to the Drosha and Dicer processing site and prioritized according to predicted specificity by minimizing potential off-target sites in the mouse genome (http://zlab.bio/guide-design-resources) (Table S1). We next assessed candidate gRNAs targeting the pre-miR-29b stem in C2C12 myotubes. Mature miR-29b expression level was detected by quantitative real-time reverse transcription PCR (quantitative real-time RT-PCR) and was significantly reduced with gRNA-miR-29b-D treatment compared to control (Figure 1B). We also performed T7EI assay and found that gRNA-miR-29b-D displayed a significant mutagenesis rate at the on-target site as judged (Figure 1C). We then accessed the potential off-target mutagenesis at 10 sites that are mostly matched to the gRNA-miR-29b-D targeting sequences by genome-wide prediction using the CRISPR design server (http://zlab.bio/guide-design-resources) and found no significant off-target mutagenesis (Figure S1).
Figure 1.

CRISPR/Cas9-Mediated Mutagenesis of miR-29b through Intramuscular CRISPR Administration
(A) Diagram of miR-29b pre-miRNA hairpin structure. (B) Real-time PCR analysis of the expression of miR-29b in C2C12 myotubes after treatment with lentiCRISPRv2, gRNA-miR-29b-A, gRNA-miR-29b-B, gRNA-miR-29b-C, and gRNA-miR-29b-D, respectively (n = 5, 6, 6, 6, 5). (C) T7EI assays performed at the targeting loci with genomic DNA from C2C12 myotubes transfected with Cas9 and gRNAs. Arrows indicate the expected bands after gene editing. (D) Real-time PCR analysis of the expression of mature miR-29a, miR-29b, and miR-29c in C2C12 myotubes after CRISPR-mediated (gRNA-miR-29b-D) gene editing (n = 3). (E) Real-time PCR analysis of the expression of mature miR-29a, miR-29b, and miR-29c in SpCRISPR-gRNA-miR-29b-D-treated mice compared with control (n = 5). CRISPRv2(lentiCRISPRv2) is a control Cas9 vector with no gRNA cassette, and gRNA-miR-29b-A, gRNA-miR-29b-B, gRNA-miR-29b-C, and gRNA-miR-29b-D are CRISPRv2 with the gRNA-miR-29b-A, gRNA-miR-29b-B, gRNA-miR-29b-C, and gRNA-miR-29b-D inserted into them, respectively. An unpaired, two-tailed Student’s t test was used for comparisons between two groups (B, D, and E). ∗p < 0.05, ∗∗p < 0.01 versus respective control. Data are represented as mean ± SEM. See also Figures S1 and S2.
We next tested gRNA-miR-29b-D for its off-target effects on miR-29a and miR-29c in C2C12 myotubes; quantitative real-time PCR analysis also showed that the gRNA-miR-29b-D we used in this study is specific in silencing miR-29b without affecting miR-29a and miR-29c (Figure 1D). To explore the therapeutic potential of gRNA-miR-29b-D, we further evaluated the protective effects of gRNA-miR-29b-D in atrophy in cultured C2C12 myotubes (Figure S2). In dexamethasone-induced atrophy, upon gRNA-miR-29b-D treatment, as detected by cell morphology, the diameters of C2C12 myotubes were markedly thicker than those treated by lentiCRISPRv2 vector (Figure S2A). Consistently, the protein levels of muscle specific RING-finger 1 (MuRF-1) and Atrogin-1 were significantly reduced (Figure S2B). Besides, the AKT-FOXO3A-mTOR signaling pathway was activated as evidenced by the restored phosphorylation of AKT(S473), FOXO3A(S253), mTOR, P70S6K, and EIF-4EBP1 (Figure S2C). These data suggest that gRNA-miR-29b-D treatment can alleviate atrophy in C2C12 myotubes.
To investigate whether those observed effects worked well in vivo, we therefore selected the most potent gRNA-miR-29b-D for further in vivo targeting analysis. We use the denotation gRNA for gRNA-D unless otherwise specified. We next adapted CRISPR for delivery by means of lentivirus, using the lentiCRISPRv2 plasmid.26 Seven days after injection, RNAs were extracted from muscle tissues to examine the expression level of mature miR-29b. The expression levels of mature miR-29b declined in the muscle with the designated CRISPR/Cas9 constructs when compared to the control lentiCRISPRv2 vectors, while they did not affect miR-29a and miR-29c expression levels (Figure 1E), supporting that the lentiviral CRISPR/Cas9 system is effective in abrogating miR-29b expression in vivo.
CRISPR/Cas9-Mediated miR-29b Editing Prevents AngII-Induced Muscle Atrophy In Vivo
Muscle atrophy is a significant comorbidity that commonly accompanies end-stage congestive heart failure. The elevated level of AngII that is a hallmark of congestive heart failure is thought to be an important mediator of muscle atrophy.27,28 The AngII-induced skeletal muscle atrophy model is considered to be an important model to mimic muscle atrophy in heart failure.27,29,30 To further investigate whether CRISPR-mediated gene editing of miR-29b could ameliorate muscle atrophy in vivo, we administered the lentiviral CRISPR/Cas9 vector and gRNA into the gastrocnemius (GA) muscles of 10-week-old male mice in the AngII-infusion-mediated skeletal muscle atrophy models that were induced by AngII-infusion. The mice were then sacrificed after 7 days, and the beneficial effects were evaluated (Figure 2A). In CRISPR-miR-29b-gRNA-treated mice, compared to control, the expression level of miR-29b was significantly decreased in AngII-treated mice (Figure 2B). AngII-induced muscle atrophy as determined by gastrocnemius muscle morphology and the reduced gastrocnemius muscle weight/body weight (GW/BW) ratio was attenuated in the CRISPR-miR-29b-gRNA-treated mice (Figure 2C). We next checked the protein levels of atrophy-linked ubiquitin ligases, including MuRF-1 and Atrogin-1. As shown in Figure 2D, CRISPR-miR-29b-gRNA-treated mice abolished the AngII-induced muscle atrophy, as indicated by reduced protein levels of MuRF-1 and Atrogin-1. Also, WGA staining and H&E staining showed that the reduction in the cross-sectional area of myofibers in AngII-treated mice was attenuated by CRISPR-miR-29b-gRNA treatment compared with lentiCRISPRv2-treated mice (Figures 2E and 2F). Finally, to evaluate beneficial effects in CRISPR-miR-29b-gRNA-treated mice, we measured muscle performance (grams of force) by subjecting mice to grip-strength testing. Compared to lentiCRISPRv2-treated mice, CRISPR-miR-29b-gRNA-treated mice demonstrated a significant improvement in grip strength (Figure 2G). Taken together, compared to CRISPRv2-treated mice, CRISPR-miR-29b-gRNA treatment not only alleviated AngII-induced muscle atrophy, but also restored mouse exercise capacity as assessed by grip strength.
Figure 2.

CRISPR/Cas9-Mediated miR-29b Editing Prevents AngII-Induced Muscle Atrophy and Maintains Mouse Exercise Capacity In Vivo
(A) The schedule of virus injection and AngII-induced atrophy mouse model establishment. (B) RT-PCR analysis of the expression of mature miR-29b in AngII-treated mice with or without SpCRISPR-gRNA-miR-29b treatment (n = 4, 5, 6, 6). (C) Gastrocnemius muscle morphology (scale bar, 1 cm) and gastrocnemius muscle weight/body weight (GW/BW) ratio are shown in AngII-treated mice with or without SpCRISPR-gRNA-miR-29b treatment (n = 6, 6, 6, 7). (D) Western blot and quantitative analysis of protein levels of MuRF-1 and Atrogin-1 in AngII-treated mice with or without SpCRISPR-gRNA-miR-29b treatment (n = 3). GAPDH protein and total proteins are stained for control. (E) Representative WGA staining photomicrographs and quantification of cross-sectional area of myofibers in AngII-treated mice with or without SpCRISPR-gRNA-miR-29b treatment (n = 5; scale bar, 20 μm). (F) Representative H&E staining photomicrographs and quantification of cross-sectional area of myofibers of gastrocnemius muscle in AngII-treated mice with or without SpCRISPR-gRNA-miR-29b treatment (n = 4; scale bar, 50 μm). (G) Grip strength of limbs measured in mice from indicated groups (n = 4, 5, 4, 4). Con, control; AngII, angiotensin II. One-way ANOVA test was performed to compare multiple groups followed by Bonferroni or Dunnett T3’s post hoc test based on homogeneity of variance test. ∗p < 0.05, ∗∗p < 0.01 versus respective control. Data are represented as mean ± SEM. See also Figures S3 and S4.
We next investigated AngII-induced apoptosis in the CRISPR-miR-29b-gRNA-treated mice. Western blot analysis for the ratios of Bax/Bcl-2 and cleaved-caspase 3/caspase 3 showed that CRISPR-miR-29b-gRNA treatment led to reduced ratios of Bax/Bcl-2 and cleaved-caspase 3/caspase 3, indicating a protective effect against apoptosis in mice (Figure S3A). This protective effect was also evidenced by TUNEL staining (Figure S3B). Then, we stained frozen sections of gastrocnemius muscles with fiber-type-specific antibodies, as shown in Figure S4A, in AngII-induced atrophy, gastrocnemius muscles presented fiber type composition changes of type IIa and type IIb fibers, while CRISPR-miR-29b-gRNA treatment prevented these alterations. Neither the regenerative fibers nor the inflammation factors (transforming growth factor α [TNF-α], interleukin-6 [IL-6], and IL-1b) were affected during this process (Figures S4B and S4C).
Previously, insulin growth factor 1 (IGF1) and phosphatidylinositol 3-kinase (PI3K)(p85α) have been identified as the target genes of miR-29b.15 The AngII-mediated decrease in the IGF1 and PI3K(p85α) protein levels in the lentiCRISPRv2-treated mice was prevented in the CRISPR-miR-29b-gRNA-treated mice (Figure 3A). The AKT-FOXO3A-mTOR signaling pathway is activated to stimulate protein synthesis and inhibits protein degradation. Therefore, we asked whether the prevented muscle atrophy treated by CRISPR administration was also a result of activating the AKT/FOXO3A/mTOR pathway, which are the downstream effectors of IGF1. Western blot analysis demonstrated that the phosphorylation of AKT(S473), FOXO3A(S253), mTOR, P70S6K, and EIF-4EBP1 were decreased in AngII-treated mice, while after CRISPR-miR-29b-gRNA treatment, the reduction of phosphorylation levels of AKT(S473), FOXO3A(S253), mTOR, P70S6K, and EIF-4EBP1 were all prevented (Figure 3B). These data suggest that the AKT-FOXO3A-mTOR signaling pathway is indeed activated by miR-29b-targeted editing, which is consistent with the prevented atrophy effects in intramuscular CRISPR-miR-29b-gRNA-treated mice.
Figure 3.

CRISPR/Cas9-Mediated miR-29b Editing Prevents AngII-Induced Atrophy Effects through Activation of AKT-FOXO3A-mTOR Signaling Pathway
(A) Western blot and quantitative analysis of extracts from gastrocnemius muscle to detect IGF1 and PI3K(p85α) protein levels in AngII-treated mice with or without SpCRISPR-gRNA-miR-29b treatment (n = 3). GAPDH protein and total proteins are stained for control. (B) Western blot and quantitative analysis of relative phosphorylation levels of AKT(S473), AKT(T308), FOXO3A(S253), FOXO3A(T32), mTOR, P70S6K, and EIF-4EBP1 (n = 3). Con, control; AngII, angiotensin II. One-way ANOVA test was performed to compare multiple groups followed by Bonferroni or Dunnett T3’s post hoc test based on homogeneity of variance test. ∗p < 0.05, ∗∗p < 0.01 versus respective control. Data are represented as mean ± SEM.
CRISPR/Cas9-Mediated miR-29b Editing Prevents Immobilization-Induced Muscle Atrophy
Clinically, muscle atrophy often occurs when patients are confined to bed rest. To explore whether the CRISPR-based treatment could work in muscle atrophy induced by long-term bed rest, we investigated the effect of CRISPR treatment on muscle atrophy induced by immobilization of limbs (Figure 4A). Immobilization-induced muscle atrophy was verified by increased miR-29b, tibialis anterior muscle morphology consistent with atrophy, and decreased tibialis anterior weight/body weight (TAW/BW) ratio, together with elevated protein levels of MuRF-1 and Atrogin-1 (Figures 4B–4D). On the contrary, lentiviral CRISPR-miR-29b-gRNA administration revealed that declined miR-29b accompanied with the recovery of tibialis anterior muscle morphology, TAW/BW ratio, and MuRF-1 and Atrogin-1 protein expression levels (Figures 4B–4D). Moreover, compared to CRISPRv2-treated mice, CRISPR-miR-29b-gRNA-treated mice showed clearly prevented atrophy of myofibers, as indicated by WGA staining and H&E staining after performing immobilization-induced muscle atrophy (Figures 4E and 4F). Western blot of whole muscle lysates showed that CRISPR-miR-29b-gRNA treatment upregulated the baseline expression of IGF1 and PI3K(p85α), which led to the attenuation of atrophy effect and substantial activation of AKT-FOXO3A-mTOR signaling pathway in the immobilization-treated mice compared with lentiCRISPRv2-treated mice (Figure S5). Besides, CRISPR-miR-29b-gRNA treatment in immobilization-induced muscle atrophy did not affect the fiber type composition, muscle regeneration, and inflammation as presented in Figure S6.
Figure 4.

CRISPR/Cas9-Mediated miR-29b Editing Prevents Immobilization-Induced Muscle Atrophy
(A) The schedule of virus injection and immobilization (Imo)-induced atrophy mouse model establishment. (B) Real-time PCR analysis of the expression of mature miR-29b in immobilization-treated mice with or without SpCRISPR-miR-29b treatment (n = 5, 4, 4, 4). (C) Tibialis anterior muscle morphology (scale bar, 1 cm) and tibialis anterior weight/body weight (TAW/BW) ratio are shown in immobilization-treated mice with or without SpCRISPR-gRNA-miR-29b treatment (n = 6, 10, 6, 9). (D) Western blot and quantitative analysis of protein levels of MuRF-1 and Atrogin-1 in immobilization-treated mice with or without SpCRISPR-gRNA-miR-29b treatment (n = 3). GAPDH protein and total proteins are stained for control. (E) Representative WGA staining photomicrographs and quantification of cross-sectional area of myofibers in immobilization-treated mice with or without SpCRISPR-gRNA-miR-29b treatment (n = 4, 5, 5, 5; scale bar, 20 μm). (F) Representative H&E staining photomicrographs and quantification of cross-sectional area of myofibers of tibialis anterior in immobilization-treated mice with or without SpCRISPR-gRNA-miR-29b treatment (n = 3, 4, 3, 4; scale bar, 50 μm). Sham, sham operation; Imo, immobilization. One-way ANOVA test was performed to compare multiple groups followed by Bonferroni or Dunnett T3’s post hoc test based on homogeneity of variance test. ∗p < 0.05, ∗∗p < 0.01 versus respective control. Data are represented as mean ± SEM. See also Figures S5 and S6.
CRISPR/Cas9-Mediated miR-29b Editing Prevents Denervation-Induced Muscle Atrophy
Since miR-29b contributes to multiple types of muscle atrophy, we next explored the effect of CRISPR treatment on muscle atrophy induced by denervation in mouse models (Figure 5A). The results showed that, similar to the AngII and immobilization models, lentiviral CRISPR-miR-29b administration was effective in preventing muscle atrophy caused by denervation. These prevention effects were evidenced by reduced miR-29b expression levels, restored muscle morphological characteristics, elevated GW/BW ratio, decreased expression of atrogene protein levels, and ameliorated atrophy of myofibers (Figures 5B–5F). Furthermore, western blot demonstrated that the therapeutic effect of CRISPR-miR-29b on muscle atrophy in denervation mouse was also through upregulation of IGF1 and P85α, followed by activation of the AKT-FOXO3A-mTOR signaling pathway (Figure S7). As demonstrated in the immobilization-induced muscle atrophy model, neither denervation induced muscle atrophy nor did CRISPR-miR-29b-gRNA treatment modify the fiber type composition, muscle regeneration, and inflammation as presented in Figure S8. These data indicate that inhibition of miR-29b by CRISPR/Cas9 could prevent denervation-induced muscle atrophy.
Figure 5.

CRISPR/Cas9-Mediated miR-29b Editing Prevents Denervation-Induced Muscle Atrophy
(A) The schedule of virus injection and denervation (Den)-induced atrophy mouse model establishment. (B) RT-PCR analysis of the expression of mature miR-29b in denervation-treated mice with or without SpCRISPR-gRNA-miR-29b treatment (n = 12, 10, 14, 10). (C) Gastrocnemius muscle morphology (scale bar, 1 cm) and GW/BW ratio is shown in denervation-treated mice with or without SpCRISPR-gRNA-miR-29b treatment (n = 6). (D) Western blot and quantitative analysis of protein levels of MuRF-1 and Atrogin-1 in denervation-treated mice with or without SpCRISPR-gRNA-miR-29b treatment (n = 3). GAPDH protein and total proteins are stained for control. (E) Representative WGA staining photomicrographs and quantification of cross-sectional area of myofibers in denervation-treated mice with or without SpCRISPR-gRNA-miR-29b treatment (n = 4; scale bar, 100 μm). (F) Representative H&E staining photomicrographs and quantification of cross-sectional area of myofibers of gastrocnemius muscle in denervation-treated mice with or without SpCRISPR-gRNA-miR-29b treatment (n = 4; scale bar, 50 μm). Sham, sham operation. One-way ANOVA test was performed to compare multiple groups followed by Bonferroni or Dunnett T3’s post hoc test based on homogeneity of variance test. ∗p < 0.05, ∗∗p < 0.01 versus respective control. Data are represented as mean ± SEM. See also Figures S7 and S8.
AAV8-CRISPR/SaCas9-Mediated miR-29b Editing Prevents AngII-Induced Muscle Atrophy In Vivo
To confirm the CRISPR-based genome-editing efficiency and decipher whether the editing specifically occurred in the skeletal myofibers, we sought to apply an independent method to further explore the function of CRISPR-miR-29b-gRNA in muscle atrophy. We used an AAV8-CRISPR backbone with SaCas9 driven by a skeletal-muscle-cell-specific dMCK promoter to examine the therapeutic role of CRISPR-miR-29b-gRNA-D in AngII-induced muscle atrophy mouse model (Figure 6A).17 We injected the AAV8-SaCRISPR-miR-29b-gRNA and AAV8-GFP-2a virus locally to gastrocnemius muscle, and after AAV8 injection 21 days, AngII infusion occurred for another 7 days to generate the AngII-induced muscle atrophy in mice (Figure 6B). Consistent with the above results demonstrated by lentiviral CRISPR-miR-29b treatment, AAV8-SaCRISPR-miR-29b administration exhibited protection effects in the AngII-induced muscle-atrophy mice, as evidenced by lower expression levels of mature miR-29b, recovered muscle morphological characteristics, increased GW/BW ratio, and reduced atrogene protein levels (Figures 6C–6E). Also, WGA staining and H&E staining suggested the prevented atrophy of myofibers (Figures 6F and 6G). Consistently, after AAV8-SaCRISPR-miR-29b-gRNA treatment, muscle performance in AngII-induced muscle atrophy exhibited a marked improvement in grip strength (Figure 6H). In addition, both the anti-apoptosis effects and activation of the AKT-FOXO3A-mTOR signaling pathway were also detected by TUNEL staining and western blot (Figures S9A and S9B). Similar to lentiviral CRISPR-miR-29b treatment in AngII-induced atrophy (Figure S4A), the shift in muscle fiber types was prevented after AAV8-SaCRISPR-miR-29b administration (Figure S10A). The regenerative fibers and the inflammation factors (TNFα, IL-6, and IL-1b) were not affected as well (Figures S10B and S10C).
Figure 6.

AAV8-CRISPR/SaCas9-Mediated miR-29b Editing Prevents AngII-Induced Muscle Atrophy In Vivo
(A) The backbone of AAV8-CRISPR/SaCas9-miR-29b with SaCas9 driven by a skeletal-muscle-cell-specific dMCK promoter. (B) The schedule of AAV8 virus injection and AngII-induced atrophy mouse model establishment. (C) RT-PCR analysis of the expression of mature miR-29b in AngII-treated mice with or without AAV8-SaCRISPR-miR-29b administration (n = 6, 5, 8, 5). (D) Gastrocnemius muscle morphology (scale bar, 1 cm) and GW/BW ratio is shown in AngII-treated mice with or without AAV8-SaCRISPR-miR-29b injection (n = 7, 5, 10, 6). (E) Western blot and quantitative analysis of MuRF-1 and Atrogin-1 protein levels in AngII-treated mice with or without AAV8-SaCRISPR-miR-29b treatment (n = 3). GAPDH protein and total proteins are stained for control. (F) Representative WGA staining photomicrographs and quantification of cross-sectional area of myofibers in AngII-treated mice with or without AAV8-SaCRISPR-miR-29b treatment (n = 4; scale bar, 50 μm). (G) Representative H&E staining photomicrographs and quantification of cross-sectional area of myofibers of gastrocnemius muscle in AngII-treated mice with or without AAV8-SaCRISPR-miR-29b treatment (n = 4; scale bar, 100 μm). (H) Grip strength of limbs measured in mice from indicated groups (8, 7, 10, 8). AAV8-GFP-2a-Luci is a control vector with dMCK promoter, and AAV8-SaCRISPR-miR-29b is AAV8/dMCK promoter with the SaCas9 and gRNA-miR-29b-D inserted into it. One-way ANOVA test was performed to compare multiple groups followed by Bonferroni or Dunnett T3’s post hoc test based on homogeneity of variance test. ∗p < 0.05, ∗∗p < 0.01 versus respective control. Data are represented as mean ± SEM. See also Figures S9 and S10.
Discussion
Previous studies by other investigators have demonstrated that the CRISPR technology could be used to affect pre-miRNA processing in cultured cells.23, 24, 25 As to in vivo, to our knowledge, only one study has been reported using this strategy to generate miR-21 knockdown phenotype HT-29 cells in vitro and then subcutaneously inject into xenograft tumors of nude mice.25 However, none have taken this strategy applied to in situ disrupting miRNA expression in live animals. In our work, we take advantage of the lentivirus-delivered CRISPR/SpCas9 system or AAV8-dMCK promoter-delivered CRISPR/SaCas9 together with recently adapted CRISPR/Cas9 targeting the pre-miRNA sequences within biogenesis processing sites.31,32 Our data demonstrate that CRISPR mediated gene editing efficiently and specifically disrupts miR-29b expression in mice and prevents both the muscle atrophy and exercise capacity. Further, to examine whether this CRISPR-based miR-29b editing could be a potential therapy to patients, we also conducted a therapy experiment based on immobilization-induced muscle atrophy, which is a common and serious problem in medicine and surgery. As shown in Figure S11, for AAV8-SaCRISPR treatment beginning at 7 days after immobilization, mice with AAV8-CRISPR-miR-29b-gRNA administration displayed the alleviation of muscle weight loss, as demonstrated by TAW/BW ratio after 28 days. These results provide the first evidence that CRISPR-mediated miRNA gene editing can be directly achieved in the muscle tissues of live mice. This targeting miRNA strategy not only provides a promising method for therapeutic strategy for muscle atrophy but may also be a platform for general application to other miRNA-associated diseases.
Loss of function is the most common strategy used to study the biological roles of miRNAs in animal models.15,33 However, the widely used antisense inhibitors and sponges in miRNA functional studies have limitations, such as low binding affinity, incomplete masking of miRNAs, short-term studies, and off-target effects on miRNAs in the seed region. Locked nucleic acid (LNA) is a class of nucleic acid analogs that presented high binding affinity toward complementary nucleic acids based on Watson-Crick base pairing.34 LNA could use a shorter sequence to target the miRNA’s seed region to affect a whole family of related miRNAs.35 To increase the specificity, antisense the full length of miRNA often used.36 Considering the short sequence of mature miRNAs, antisense inhibitors that target mature miRNAs are only loss of function while barely leading to degradation of mature miRNAs; thus, it is difficult to precisely evaluate the inhibition efficiency, and therefore the potential side-effects cannot be accurately evaluated. Homologous-recombination-based miRNA knockout methodology is the most reliable and specific technique to generate definitive loss-of-function, but has been limited due to its time-consuming and expensive nature. Evaluating and increasing the specificity is the important concern of the gene-editing tools application. Tremendous progress has been made in addressing the challenges of conventional gene therapy by developing technologies for precise modification of genome since the concept of gene therapy has been raised.37, 38, 39, 40 CRISPR systems have demonstrated very prospective therapeutic potential with low off-target incidents and high efficiency.41, 42, 43, 44 Here, using CRISPR-based genome editing, we achieve efficient knockdown of miR-29b by targeting the endogenous loci in mice. Moreover, we also show that gRNA-miR-29b can specifically edit miR-29b rather than miR-29a and miR-29c. Our strategy overcomes the previous limitations and demonstrates that CRISPR-mediated miRNA gene editing is a feasible and convenient technique for investigating the function studies of miRNAs in vivo.
The emergence of CRISPR/Cas9 has created new possibilities for gene therapy by making precise genome modifications possible in cultured cells and animal studies.17,20, 21, 22,45, 46, 47, 48, 49 Currently, gene editing based on CRISPR-Cas9 has been widely investigated to correct mutations of the dystrophin gene in Duchenne muscular dystrophy (DMD) patients and demonstrated a promising option for treatment of DMD in the future.16,18, 19, 20,50, 51, 52, 53, 54, 55, 56, 57, 58, 59 Affection of grip strength by intramuscular injection of virus or recombinant protein into gastrocnemius muscle has been previously reported.17,60,61 However, considering the complexity of the cause of muscle atrophy, no gene-editing treatment strategies have been reported to target various types of muscle atrophy. Previously, miR-29b, which has been reported to contribute to multiple types of muscle atrophy, could be an attractive therapeutic target.15 With a view to establishing a therapeutic modality toward multiple types of muscle atrophy, we reasoned that disrupting the function of miR-29b specifically would be sufficient to prevent muscle atrophy. In this study, we show the therapeutic benefit of CRISPR/Cas9 genome editing on miR-29b in mouse model of muscle atrophy induced by AngII, immobilization, and denervation. Our data demonstrate that CRISPR-miR-29b gene-editing efficiently prevents multiple types of muscle atrophy.
Safety and efficiency of gene editing are the main concerns that limit its promise for the treatment of human disease. In our work, we did not observe off-target mutations at 10 potential off-target sites in the mouse genome (Figure S1). However, off-target effects may occur at sites beyond those. Another important consideration for the therapeutic applications of the CRISPR-Cas9 system is its potential immunogenicity.62,63 In our systems, either SpCas9 or SaCas9 was detected high expression throughout the experimental period, interferon gamma (IFNγ) displayed no change after 2 weeks for lentivirus-delivered CRISPR/SpCas9 or 4 weeks for AAV8-delivered CRISPR/SaCas9 (Figure S12). Previously have been reported that long-term therapeutic of AAV-CRISPR-SaCas9 system, after stimulated with SaCas9, IFNγ markedly increased compared to control.48 This inconsistency might be due to different experimental cycles. To clarify this in-depth, further systematic evaluation of the host immune response to the CRISPR/Cas9 system will be needed. Of note, in our work, mice were injected intramuscularly. To make it more clinically relevant to be able to target all muscles, it would be interesting to show the protective effects on muscle atrophy after systemic, intravenous injections. Also, much consideration should be taken before this approach is translated into clinical application, such as the optimization of the delivery system, an unbiased whole-genome analysis, and the immune response, as well as ethical issues.
Elevated AngII concentrations often occur in congestive heart patients. Our current work takes advantage of the AngII-induced muscle atrophy as one model to investigate the effect of miR-29b and the benefits of CRISPR-based miR-29b editing on muscle atrophy. In this study, we also detected the prevented phenotypes of the leg we interfered in AngII-induced muscle atrophy. Therefore, at present, our work is a beneficial attempt to translate a potential therapeutic target to combat muscle atrophy into a clinically relevant approach for genome editing in muscle in a mouse model. Further evaluation about the systemic effects and comprehensive assessment of the ability to correct muscle atrophy features by intramuscular injection of a gene-editing vector after establishment of muscle atrophy models is necessary before this approach is translated into clinical application. Nonetheless, our data raise the possibility that, in the future, human beings can benefit from CRISPR-based miRNAs targeting through muscular administration alone or in combination with other therapies.
Materials and Methods
Animal Experiments
Ten-week-old male C57BL/6 mice were purchased from Cavens Laboratory Animal (Changzhou, China) and maintained in an specific pathogen-free (SPF) laboratory animal facility of Shanghai University (Shanghai, China). All procedures with animals were in accordance with the guidelines on the use and care of laboratory animals for biomedical research published by the National Institutes of Health (no. 85-23, revised 1996), and the experimental protocol was reviewed and approved by the ethical committees of the School of Life Science, Shanghai University.
For lentiCRISPR experiments, a single intramuscular injection to gastrocnemius muscles (for AngII-induced and denervation-induced muscle atrophy) or tibialis anterior muscle (for immobilization-induced muscle atrophy) of lentiviral particles was performed at the dose of 108 transduction unit (TU) per mouse. Then muscle atrophy models were introduced after a week. For AngII-induced muscle atrophy, AngII (Sigma, Darmstadt, Germany; 1.5 μg/kg/min) was delivered chronically by an implanted osmotic minipump (ALZET model 2001, CA, USA). Pair feeding of AngII and vehicle-treated mice was conducted to correct for AngII-induced anorexia. For immobilization-induced muscle atrophy, the right ankle joint of C57BL/6 mice was fixed at 90° of flexion by insertion of a screw (0.4 × 8 mm) through the calcaneus and talus into the shaft of the tibia. For denervation-induced muscle atrophy, the mid-thigh region of right sciatic nerve of C57BL/6 mice was cut off. The sham was generated by same process except without cutting off the sciatic nerve. Mice were sacrificed 7 days after AngII administration, immobilization, and denervation. Gastrocnemius and tibialis anterior were harvested, and muscle weight and body weight were measured. Muscle specimens were immediately snap frozen in liquid nitrogen and stored at −80°C for further analyses and embedded for histological analyses.
Production of Cas9 mRNA and Single Guide RNA (sgRNA)
CRISPR design (http://zlab.bio/guide-design-resources) was used to choose gRNA targets of miR-29b. Four gRNA oligonucleotide pairs for each targeting site were annealed and inserted into lentiCRISPRv2 according to the lentiviral CRISPR toolbox.26,50 lentiCRISPRv2 was a gift from Feng Zhang (Addgene plasmid # 52961); a brief schematic diagram of lentiCRISPRv2 is displayed in Figure S1A, and sequences of sgRNA used in this study are listed in Table S1.
AAV8 Virus Packaging and Local Injection into Muscles of AAV8
We generated AAV8-SaCRISPR-miR-29b-gRNA, in which a skeletal-muscle-cell-specific dMCK promoter is used to drive the expression of SaCas9 in skeletal muscle.17 AAV8-GFP-2a was used as control. The AAV8 was packaged in 293T cells as previously described.64 The AAV8 virus was purified using iodixanol gradient density centrifugation. Absolute quantification of viral titers of the AAV8 virus after titer determination can be directly used in animal experiments or frozen at −80°C. A single intramuscular injection to gastrocnemius muscles at the dose of 1011 vector genome (vg)/mice, after 3 weeks, AngII (Sigma, Darmstadt, Germany; 1.5 μg/kg/min) was delivered chronically by an implanted osmotic minipump (ALZET model 2001, CA, USA) to establish the AngII-induced muscle atrophy model.
Cell Culture
C2C12 cells (mouse skeletal myoblasts) and 293T cells (human embryonic kidney) were obtained from the ATCC and cultured in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum at 37°C supplemented with 5% CO2.
T7 Endonuclease I (T7EI) Assay
Targeted miR-29b-1 and miR-29b-2 genome DNA fragments were amplified using 100 ng genomic DNA as template (KOD plus high-fidelity DNA polymerase, Toyobo, Osaka, Japan). The primers used are as follows: miR-29b-1, F 5′-GCTGCACCGTGAATGTGTAA-3′, R 5′-AGGTCTTCATCCGAGCATGG-3′; miR-29b-2, F 5′-TGTACATATGTTGAATGGATTTGGT-3′, R 5′-TGCTGCAACCAGGACTGAAT-3′. The purified PCR products were denatured and reannealed in NEB buffer 2 (NEBuffer 2, NEB, MA, USA) in 20 μL volume under the following conditions: 95°C, 5 min; 95°C–75°C, −0.1°C/cycle, 200 times; 75°C–15°C, −0.1°C/cycle, 600 times; hold at 4°C. Then, 1 U of T7EN1 enzymes (T7 endonuclease I, NEB, MA, USA) were added to hybridized PCR products, and reaction mix was incubated at 37°C for 1 h. The products were separated by 2% agarose, stained with GelRED, and images were captured by ChemiDoc XRS (Bio-Rad, PA, USA).
RNA Isolation and Quantitative Real-Time Polymerase Chain Reactions
Total RNA was extracted from cells and muscle tissues by using RNAiso plus (Takara, Shiga, Japan). Total RNA was reverse transcribed using a RevertAid first-strand cDNA synthesis kit (Thermos Fisher, MA, USA), according to the manufacturer’s instructions. PCR reactions were performed using SYBR premix Ex Taq (Takara, Shiga, Japan) and the Roche real-time PCR detection system (Roche, Basel, Switzerland). The relative expression level of mRNA or miRNA was calculated using the 2−ΔΔCt method. miRNAs were quantified by 5S, and mRNAs were quantified by 18S. For miRNA assay, the bulge-loop miRNA qPCR primer set (RiboBio, Guangzhou, China) was used. The primers sequences used in this study are listed in Table S2.
Grip-Strength Test
A digital grip strength (Yiyan Technology, Jinan, China) was used to measure the grip strength of mice as following protocol. The mice adjusted to the meter for 10 min before the grip-strength test began. The legs of mice with virus injected were allowed to grab the metal pull bar and pulled backward by the tail. The force at the time of release was recorded as the peak tension. Each mouse was tested five times with a 30 s break between tests, and average values were used to represent the grip strength of one mouse. Investigators were blinded to the animal groups.
Western Blot
Protein samples were extracted from mouse muscle tissues using western and immunoprecipitation lysis buffer (KeyGEN, Nanjing, China) with a protease inhibitor cocktail (KeyGEN, Nanjing, China). Equal amounts of protein samples was employed by bicinchoninic acid (BCA) protein assay kit (Takara, Shiga, Japan) and separated by SDS-PAGE electrophoresis. Then the protein was transferred to polyvinylidene difluoride (PVDF) membrane (Millipore, MA, USA). After that, the membrane was blocked in 5% BSA at room temperature. Then primary antibodies were incubated at 4°C overnight. A horseradish-peroxidase-conjugated secondary antibody was used to amplify the signal. All proteins were visualized by ECL chemiluminescent kit (Thermo Fisher, MA, USA) and chemical luminescence of membranes was detected by Bio-Rad luminescent imaging system. The primary antibodies used in this study were as follows: IGF1 (1:1,000; Bioworld Technology, Nanjing, China), PI3K(p85α) (1:500; Cell Signaling Technology, MA, USA), FOXO3A (1:1,000; Abclonal Technology, Wuhan, China), P-AKT(T308) (1:1,000; Cell Signaling Technology, MA, USA), P-AKT(S473) (1:1,000; Cell Signaling Technology, MA, USA), AKT (1:1,000; Proteintech, Wuhan, China), P-FOXO3A(S253) (1:1,000; Cell Signaling Technology, MA, USA), P-FOXO1(T24)/FOXO3A(T32) (1:1,000; Cell Signaling Technology, MA, USA), P-mTOR (1:1,000; Cell Signaling Technology, MA, USA), mTOR (1:1,000; Cell Signaling Technology, MA, USA), P-P70S6K (1:1,000; Cell Signaling Technology, MA, USA), P70S6K (1:1,000; Cell Signaling Technology, MA, USA), P-4EBP1 (1:1,000; Abclonal Technology, Wuhan, China), 4EBP1 (1:1,000; Abclonal Technology, Wuhan, China), bax (Abclonal Technology, Wuhan, China), Bcl2 (Abclonal Technology, Wuhan, China), caspase-3 (Cell Signaling Technology, MA, USA), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (1:10,000; Bioworld Technology, Nanjing, China). In the western blots that use one reference protein for quantification, total proteins are also stained by Light GREEN SF Yellowwish (AMRESCO, USA).
Stainings
WGA, which binds to glycosaminoglycans, is used to stain sarcolemma to quantify the size of myofibers.65,66 The gastrocnemius and tibialis anterior muscles were dissected, embedded in optimal cutting temperature (OCT) compound, snap frozen in cold liquid nitrogen, and cut at 10 μm per section. Then, WGA staining was used to determine the cross-sectional area of muscle fibers. The sections were dried at room temperature for 30 min and then incubated with WGA and DAPI at room temperature for 30 min. Images of Figures 2E and 4E were captured by confocal microscope (Zeiss, Oberkochen, Germany); images of Figures 5E and 6F were captured by fluorescence microscope (Leica, Wetzlar, Germany). The area of muscle fibers was measured by ImageJ. Terminal dexynucleotidyl transferase (TdT)-mediated 2'-deoxyuridine 5'-triphosphate (dUTP) nick end labeling (DeadEnd fluorometric TUNEL system, Promega, WI, USA) staining was used to determine the apoptosis of muscle cells. The sections were dried, fixed with 4% paraformaldehyde at room temperature for 30 min, permeabilized with 0.1% Triton X-100 in PBS for 20 min, and blocked with 5% BSA for 20 min. Then, sections were stained with a TUNEL fluorescein isothiocyanate (FITC) apoptosis detection kit (DeadEnd fluorometric TUNEL system, Promega, WI, USA) according to the manufacturer’s instructions and next were incubated with WGA conjugates (Invitrogen, USA) at room temperature (RT) for 2 h. Then nuclei were stained with DAPI. Images were captured by fluorescent microscope (Zeiss, Oberkochen, Germany). For H&E staining, gastrocnemius muscle and tibial anterior muscle were fixed in 4% paraformaldehyde (PFA) and then embedded in paraffin. 10-μm-thick sections of muscle tissues were subjected to H&E staining following a standard procedure (KeyGEN, China). Images were captured by fluorescent microscope (Nikon, Japan), the area of myofibers was measured by ImageJ, and a minimum of 500 fibers per mouse was analyzed.
Statistical Analysis
Results were presented as mean ± SEM. An unpaired, two-tailed Student’s t test was used for comparisons between two groups. One-way ANOVA test was performed to compare multiple groups followed by Bonferroni or Dunnett T3’s post hoc test based on homogeneity of variance test. All analyses were performed using SPSS statistics 20. All statistical graphs performed by GraphPad Prism 8.0. Differences were considered significant with p < 0.05.
Author Contributions
J.X. designed the study, instructed all experiments, and drafted the manuscript. J.L., L.W., X.H., H.T., R.C., and T.Y. performed the experiments and analyzed the data. S.D. provided technical assistance and revised the manuscript.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
This work was supported by the grants from the National Natural Science Foundation of China (81722008 and 81911540486 to J.X., 81800358 to L.W., and 81900359 to J.L.); the Innovation Program of Shanghai Municipal Education Commission (2017-01-07-00-09-E00042 to J.X.); the National Key Research and Development Project (2018YFE0113500 to J.X.); the “Dawn” Program of Shanghai Education Commission (19SG34 to J.X.); a grant from the Science and Technology Commission of Shanghai Municipality (17010500100, 18410722200 to J.X.); and the NIH (grants UG3 TR002463 and HL122547 to S.D.).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2020.03.005.
Supplemental Information
References
- 1.Wallace G.Q., McNally E.M. Mechanisms of muscle degeneration, regeneration, and repair in the muscular dystrophies. Annu. Rev. Physiol. 2009;71:37–57. doi: 10.1146/annurev.physiol.010908.163216. [DOI] [PubMed] [Google Scholar]
- 2.Cohen S., Nathan J.A., Goldberg A.L. Muscle wasting in disease: molecular mechanisms and promising therapies. Nat. Rev. Drug Discov. 2015;14:58–74. doi: 10.1038/nrd4467. [DOI] [PubMed] [Google Scholar]
- 3.von Haehling S., Ebner N., Dos Santos M.R., Springer J., Anker S.D. Muscle wasting and cachexia in heart failure: mechanisms and therapies. Nat. Rev. Cardiol. 2017;14:323–341. doi: 10.1038/nrcardio.2017.51. [DOI] [PubMed] [Google Scholar]
- 4.Andres-Mateos E., Brinkmeier H., Burks T.N., Mejias R., Files D.C., Steinberger M., Soleimani A., Marx R., Simmers J.L., Lin B. Activation of serum/glucocorticoid-induced kinase 1 (SGK1) is important to maintain skeletal muscle homeostasis and prevent atrophy. EMBO Mol. Med. 2013;5:80–91. doi: 10.1002/emmm.201201443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xiao J., editor. Muscle Atrophy. Springer; 2018. [Google Scholar]
- 6.Jagoe R.T., Lecker S.H., Gomes M., Goldberg A.L. Patterns of gene expression in atrophying skeletal muscles: response to food deprivation. FASEB J. 2002;16:1697–1712. doi: 10.1096/fj.02-0312com. [DOI] [PubMed] [Google Scholar]
- 7.Lecker S.H., Jagoe R.T., Gilbert A., Gomes M., Baracos V., Bailey J., Price S.R., Mitch W.E., Goldberg A.L. Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J. 2004;18:39–51. doi: 10.1096/fj.03-0610com. [DOI] [PubMed] [Google Scholar]
- 8.Gomes M.D., Lecker S.H., Jagoe R.T., Navon A., Goldberg A.L. Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc. Natl. Acad. Sci. USA. 2001;98:14440–14445. doi: 10.1073/pnas.251541198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sacheck J.M., Hyatt J.P., Raffaello A., Jagoe R.T., Roy R.R., Edgerton V.R., Lecker S.H., Goldberg A.L. Rapid disuse and denervation atrophy involve transcriptional changes similar to those of muscle wasting during systemic diseases. FASEB J. 2007;21:140–155. doi: 10.1096/fj.06-6604com. [DOI] [PubMed] [Google Scholar]
- 10.Ivey K.N., Srivastava D. microRNAs as Developmental Regulators. Cold Spring Harb. Perspect. Biol. 2015;7:a008144. doi: 10.1101/cshperspect.a008144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bei Y., Xiao J. MicroRNAs in muscle wasting and cachexia induced by heart failure. Nat. Rev. Cardiol. 2017;14:566. doi: 10.1038/nrcardio.2017.122. [DOI] [PubMed] [Google Scholar]
- 12.Horak M., Novak J., Bienertova-Vasku J. Muscle-specific microRNAs in skeletal muscle development. Dev. Biol. 2016;410:1–13. doi: 10.1016/j.ydbio.2015.12.013. [DOI] [PubMed] [Google Scholar]
- 13.Kukreti H., Amuthavalli K., Harikumar A., Sathiyamoorthy S., Feng P.Z., Anantharaj R., Tan S.L., Lokireddy S., Bonala S., Sriram S. Muscle-specific microRNA1 (miR1) targets heat shock protein 70 (HSP70) during dexamethasone-mediated atrophy. J. Biol. Chem. 2013;288:6663–6678. doi: 10.1074/jbc.M112.390369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rau C.S., Jeng J.C., Jeng S.F., Lu T.H., Chen Y.C., Liliang P.C., Wu C.J., Lin C.J., Hsieh C.H. Entrapment neuropathy results in different microRNA expression patterns from denervation injury in rats. BMC Musculoskelet. Disord. 2010;11:181. doi: 10.1186/1471-2474-11-181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li J., Chan M.C., Yu Y., Bei Y., Chen P., Zhou Q., Cheng L., Chen L., Ziegler O., Rowe G.C. miR-29b contributes to multiple types of muscle atrophy. Nat. Commun. 2017;8:15201. doi: 10.1038/ncomms15201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nelson C.E., Hakim C.H., Ousterout D.G., Thakore P.I., Moreb E.A., Castellanos Rivera R.M., Madhavan S., Pan X., Ran F.A., Yan W.X. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science. 2016;351:403–407. doi: 10.1126/science.aad5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wei Y., Chen Y., Qiu Y., Zhao H., Liu G., Zhang Y., Meng Q., Wu G., Chen Y., Cai X. Prevention of Muscle Wasting by CRISPR/Cas9-mediated Disruption of Myostatin In Vivo. Mol. Ther. 2016;24:1889–1891. doi: 10.1038/mt.2016.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu L., Park K.H., Zhao L., Xu J., El Refaey M., Gao Y., Zhu H., Ma J., Han R. CRISPR-mediated Genome Editing Restores Dystrophin Expression and Function in mdx Mice. Mol. Ther. 2016;24:564–569. doi: 10.1038/mt.2015.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tabebordbar M., Zhu K., Cheng J.K.W., Chew W.L., Widrick J.J., Yan W.X., Maesner C., Wu E.Y., Xiao R., Ran F.A. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science. 2016;351:407–411. doi: 10.1126/science.aad5177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Long C., Amoasii L., Mireault A.A., McAnally J.R., Li H., Sanchez-Ortiz E., Bhattacharyya S., Shelton J.M., Bassel-Duby R., Olson E.N. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science. 2016;351:400–403. doi: 10.1126/science.aad5725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Platt R.J., Chen S., Zhou Y., Yim M.J., Swiech L., Kempton H.R., Dahlman J.E., Parnas O., Eisenhaure T.M., Jovanovic M. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell. 2014;159:440–455. doi: 10.1016/j.cell.2014.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ran F.A., Cong L., Yan W.X., Scott D.A., Gootenberg J.S., Kriz A.J., Zetsche B., Shalem O., Wu X., Makarova K.S. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520:186–191. doi: 10.1038/nature14299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lataniotis L., Albrecht A., Kok F.O., Monfries C.A.L., Benedetti L., Lawson N.D., Hughes S.M., Steinhofel K., Mayr M., Zampetaki A. CRISPR/Cas9 editing reveals novel mechanisms of clustered microRNA regulation and function. Sci. Rep. 2017;7:8585. doi: 10.1038/s41598-017-09268-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huo W., Zhao G., Yin J., Ouyang X., Wang Y., Yang C., Wang B., Dong P., Wang Z., Watari H. Lentiviral CRISPR/Cas9 vector mediated miR-21 gene editing inhibits the epithelial to mesenchymal transition in ovarian cancer cells. J. Cancer. 2017;8:57–64. doi: 10.7150/jca.16723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chang H., Yi B., Ma R., Zhang X., Zhao H., Xi Y. CRISPR/cas9, a novel genomic tool to knock down microRNA in vitro and in vivo. Sci. Rep. 2016;6:22312. doi: 10.1038/srep22312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sanjana N.E., Shalem O., Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods. 2014;11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Song Y.H., Li Y., Du J., Mitch W.E., Rosenthal N., Delafontaine P. Muscle-specific expression of IGF-1 blocks angiotensin II-induced skeletal muscle wasting. J. Clin. Invest. 2005;115:451–458. doi: 10.1172/JCI22324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Du Bois P., Pablo Tortola C., Lodka D., Kny M., Schmidt F., Song K., Schmidt S., Bassel-Duby R., Olson E.N., Fielitz J. Angiotensin II Induces Skeletal Muscle Atrophy by Activating TFEB-Mediated MuRF1 Expression. Circ. Res. 2015;117:424–436. doi: 10.1161/CIRCRESAHA.114.305393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shen C., Zhou J., Wang X., Yu X.Y., Liang C., Liu B., Pan X., Zhao Q., Song J.L., Wang J. Angiotensin-II-induced Muscle Wasting is Mediated by 25-Hydroxycholesterol via GSK3β Signaling Pathway. EBioMedicine. 2017;16:238–250. doi: 10.1016/j.ebiom.2017.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kinugawa S., Takada S., Matsushima S., Okita K., Tsutsui H. Skeletal Muscle Abnormalities in Heart Failure. Int. Heart J. 2015;56:475–484. doi: 10.1536/ihj.15-108. [DOI] [PubMed] [Google Scholar]
- 31.Nguyen T.A., Jo M.H., Choi Y.G., Park J., Kwon S.C., Hohng S., Kim V.N., Woo J.S. Functional Anatomy of the Human Microprocessor. Cell. 2015;161:1374–1387. doi: 10.1016/j.cell.2015.05.010. [DOI] [PubMed] [Google Scholar]
- 32.Auyeung V.C., Ulitsky I., McGeary S.E., Bartel D.P. Beyond secondary structure: primary-sequence determinants license pri-miRNA hairpins for processing. Cell. 2013;152:844–858. doi: 10.1016/j.cell.2013.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu X., Xiao J., Zhu H., Wei X., Platt C., Damilano F., Xiao C., Bezzerides V., Boström P., Che L. miR-222 is necessary for exercise-induced cardiac growth and protects against pathological cardiac remodeling. Cell Metab. 2015;21:584–595. doi: 10.1016/j.cmet.2015.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kauppinen S., Vester B., Wengel J. Locked nucleic acid (LNA): High affinity targeting of RNA for diagnostics and therapeutics. Drug Discov. Today. Technol. 2005;2:287–290. doi: 10.1016/j.ddtec.2005.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Obad S., dos Santos C.O., Petri A., Heidenblad M., Broom O., Ruse C., Fu C., Lindow M., Stenvang J., Straarup E.M. Silencing of microRNA families by seed-targeting tiny LNAs. Nat. Genet. 2011;43:371–378. doi: 10.1038/ng.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Beermann J., Piccoli M.T., Viereck J., Thum T. Non-coding RNAs in Development and Disease: Background, Mechanisms, and Therapeutic Approaches. Physiol. Rev. 2016;96:1297–1325. doi: 10.1152/physrev.00041.2015. [DOI] [PubMed] [Google Scholar]
- 37.Friedmann T., Roblin R. Gene therapy for human genetic disease? Science. 1972;175:949–955. doi: 10.1126/science.175.4025.949. [DOI] [PubMed] [Google Scholar]
- 38.Cox D.B., Platt R.J., Zhang F. Therapeutic genome editing: prospects and challenges. Nat. Med. 2015;21:121–131. doi: 10.1038/nm.3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nelson C.E., Robinson-Hamm J.N., Gersbach C.A. Genome engineering: a new approach to gene therapy for neuromuscular disorders. Nat. Rev. Neurol. 2017;13:647–661. doi: 10.1038/nrneurol.2017.126. [DOI] [PubMed] [Google Scholar]
- 40.Pickar-Oliver A., Gersbach C.A. The next generation of CRISPR-Cas technologies and applications. Nat. Rev. Mol. Cell Biol. 2019;20:490–507. doi: 10.1038/s41580-019-0131-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Knott G.J., Doudna J.A. CRISPR-Cas guides the future of genetic engineering. Science. 2018;361:866–869. doi: 10.1126/science.aat5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang H.X., Li M., Lee C.M., Chakraborty S., Kim H.W., Bao G., Leong K.W. CRISPR/Cas9-Based Genome Editing for Disease Modeling and Therapy: Challenges and Opportunities for Nonviral Delivery. Chem. Rev. 2017;117:9874–9906. doi: 10.1021/acs.chemrev.6b00799. [DOI] [PubMed] [Google Scholar]
- 43.Veres A., Gosis B.S., Ding Q., Collins R., Ragavendran A., Brand H., Erdin S., Cowan C.A., Talkowski M.E., Musunuru K. Low incidence of off-target mutations in individual CRISPR-Cas9 and TALEN targeted human stem cell clones detected by whole-genome sequencing. Cell Stem Cell. 2014;15:27–30. doi: 10.1016/j.stem.2014.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang L., Grishin D., Wang G., Aach J., Zhang C.Z., Chari R., Homsy J., Cai X., Zhao Y., Fan J.B. Targeted and genome-wide sequencing reveal single nucleotide variations impacting specificity of Cas9 in human stem cells. Nat. Commun. 2014;5:5507. doi: 10.1038/ncomms6507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jinek M., East A., Cheng A., Lin S., Ma E., Doudna J. RNA-programmed genome editing in human cells. eLife. 2013;2:e00471. doi: 10.7554/eLife.00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cho S.W., Kim S., Kim J.M., Kim J.S. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat. Biotechnol. 2013;31:230–232. doi: 10.1038/nbt.2507. [DOI] [PubMed] [Google Scholar]
- 47.Beyret E., Liao H.K., Yamamoto M., Hernandez-Benitez R., Fu Y., Erikson G., Reddy P., Izpisua Belmonte J.C. Single-dose CRISPR-Cas9 therapy extends lifespan of mice with Hutchinson-Gilford progeria syndrome. Nat. Med. 2019;25:419–422. doi: 10.1038/s41591-019-0343-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nelson C.E., Wu Y., Gemberling M.P., Oliver M.L., Waller M.A., Bohning J.D., Robinson-Hamm J.N., Bulaklak K., Castellanos Rivera R.M., Collier J.H. Long-term evaluation of AAV-CRISPR genome editing for Duchenne muscular dystrophy. Nat. Med. 2019;25:427–432. doi: 10.1038/s41591-019-0344-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Santiago-Fernández O., Osorio F.G., Quesada V., Rodríguez F., Basso S., Maeso D., Rolas L., Barkaway A., Nourshargh S., Folgueras A.R. Development of a CRISPR/Cas9-based therapy for Hutchinson-Gilford progeria syndrome. Nat. Med. 2019;25:423–426. doi: 10.1038/s41591-018-0338-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shalem O., Sanjana N.E., Hartenian E., Shi X., Scott D.A., Mikkelson T., Heckl D., Ebert B.L., Root D.E., Doench J.G., Zhang F. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liao H.K., Hatanaka F., Araoka T., Reddy P., Wu M.Z., Sui Y., Yamauchi T., Sakurai M., O’Keefe D.D., Nunez-Delicado E. In Vivo Target Gene Activation via CRISPR/Cas9-Mediated Trans-epigenetic Modulation. Cell. 2017;171:1495–1507.e15. doi: 10.1016/j.cell.2017.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mendell J.R., Rodino-Klapac L.R. Duchenne muscular dystrophy: CRISPR/Cas9 treatment. Cell Res. 2016;26:513–514. doi: 10.1038/cr.2016.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.El Refaey M., Xu L., Gao Y., Canan B.D., Adesanya T.M.A., Warner S.C., Akagi K., Symer D.E., Mohler P.J., Ma J. In Vivo Genome Editing Restores Dystrophin Expression and Cardiac Function in Dystrophic Mice. Circ. Res. 2017;121:923–929. doi: 10.1161/CIRCRESAHA.117.310996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhu P., Wu F., Mosenson J., Zhang H., He T.C., Wu W.S. CRISPR/Cas9-Mediated Genome Editing Corrects Dystrophin Mutation in Skeletal Muscle Stem Cells in a Mouse Model of Muscle Dystrophy. Mol. Ther. Nucleic Acids. 2017;7:31–41. doi: 10.1016/j.omtn.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bengtsson N.E., Hall J.K., Odom G.L., Phelps M.P., Andrus C.R., Hawkins R.D., Hauschka S.D., Chamberlain J.R., Chamberlain J.S. Muscle-specific CRISPR/Cas9 dystrophin gene editing ameliorates pathophysiology in a mouse model for Duchenne muscular dystrophy. Nat. Commun. 2017;8:14454. doi: 10.1038/ncomms14454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ousterout D.G., Kabadi A.M., Thakore P.I., Majoros W.H., Reddy T.E., Gersbach C.A. Multiplex CRISPR/Cas9-based genome editing for correction of dystrophin mutations that cause Duchenne muscular dystrophy. Nat. Commun. 2015;6:6244. doi: 10.1038/ncomms7244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Min Y.L., Li H., Rodriguez-Caycedo C., Mireault A.A., Huang J., Shelton J.M., McAnally J.R., Amoasii L., Mammen P.P.A., Bassel-Duby R., Olson E.N. CRISPR-Cas9 corrects Duchenne muscular dystrophy exon 44 deletion mutations in mice and human cells. Sci. Adv. 2019;5 doi: 10.1126/sciadv.aav4324. eaav4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Duchêne B.L., Cherif K., Iyombe-Engembe J.P., Guyon A., Rousseau J., Ouellet D.L., Barbeau X., Lague P., Tremblay J.P. CRISPR-Induced Deletion with SaCas9 Restores Dystrophin Expression in Dystrophic Models In Vitro and In Vivo. Mol. Ther. 2018;26:2604–2616. doi: 10.1016/j.ymthe.2018.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Koo T., Lu-Nguyen N.B., Malerba A., Kim E., Kim D., Cappellari O., Cho H.Y., Dickson G., Popplewell L., Kim J.S. Functional Rescue of Dystrophin Deficiency in Mice Caused by Frameshift Mutations Using Campylobacter jejuni Cas9. Mol. Ther. 2018;26:1529–1538. doi: 10.1016/j.ymthe.2018.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Benoit B., Meugnier E., Castelli M., Chanon S., Vieille-Marchiset A., Durand C., Bendridi N., Pesenti S., Monternier P.A., Durieux A.C. Fibroblast growth factor 19 regulates skeletal muscle mass and ameliorates muscle wasting in mice. Nat. Med. 2017;23:990–996. doi: 10.1038/nm.4363. [DOI] [PubMed] [Google Scholar]
- 61.Vinel C., Lukjanenko L., Batut A., Deleruyelle S., Pradère J.P., Le Gonidec S., Dortignac A., Geoffre N., Pereira O., Karaz S. The exerkine apelin reverses age-associated sarcopenia. Nat. Med. 2018;24:1360–1371. doi: 10.1038/s41591-018-0131-6. [DOI] [PubMed] [Google Scholar]
- 62.Charlesworth C.T., Deshpande P.S., Dever D.P., Camarena J., Lemgart V.T., Cromer M.K., Vakulskas C.A., Collingwood M.A., Zhang L., Bode N.M. Identification of preexisting adaptive immunity to Cas9 proteins in humans. Nat. Med. 2019;25:249–254. doi: 10.1038/s41591-018-0326-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chew W.L., Tabebordbar M., Cheng J.K., Mali P., Wu E.Y., Ng A.H., Zhu K., Wagers A.J., Church G.M. A multifunctional AAV-CRISPR-Cas9 and its host response. Nat. Methods. 2016;13:868–874. doi: 10.1038/nmeth.3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Grieger J.C., Choi V.W., Samulski R.J. Production and characterization of adeno-associated viral vectors. Nat. Protoc. 2006;1:1412–1428. doi: 10.1038/nprot.2006.207. [DOI] [PubMed] [Google Scholar]
- 65.Wright C.S. Structural comparison of the two distinct sugar binding sites in wheat germ agglutinin isolectin II. J. Mol. Biol. 1984;178:91–104. doi: 10.1016/0022-2836(84)90232-8. [DOI] [PubMed] [Google Scholar]
- 66.Guo Y., Wang H., Tang Y., Wang Y., Zhang M., Yang Z., Nyirimigabo E., Wei B., Lu Z., Ji G. GCN2 deficiency protects mice from denervation-induced skeletal muscle atrophy via inhibiting FoxO3a nuclear translocation. Protein Cell. 2018;9:966–970. doi: 10.1007/s13238-018-0504-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.