

Abstract
Pain is among the most common symptoms in cancer and approximately 90% of patients experience end-stage cancer pain. The management of cancer pain is challenging due to the significant side effects associated with opioids, and novel therapeutic approaches are needed. MMG22 is a bivalent ligand containing MOR agonist and mGluR5 antagonist pharmacophores joined by a 22-atom spacer. MMG22 exhibited extraordinary analgesia following intrathecal administration in a mouse model of bone cancer pain. Here, we assessed the effectiveness of systemic administration of MMG22 in reducing cancer pain and evaluated whether MMG22 displays side effects associated with opioids. Fibrosarcoma cells were injected into and around the calcaneus bone in C3H mice. Mechanical hyperalgesia was defined as an increase in the paw withdrawal frequencies (PWFs) evoked by application of a von Frey monofilament (3.9 mN bending force) applied to the plantar surface of the hind paw
Subcutaneous (s.c.), intramuscular (i.m.), and oral (p.o.) administration of MMG22 produced robust dose-dependent antihyperalgesia, whose ED50 was orders of magnitude lower than morphine. Moreover, the ED50 for MMG22 decreased with disease progression. Importantly, s.c. administration of MMG22 did not produce acute (24 h) or long-term (9 days) tolerance, was not rewarding (conditioned place preference test), and did not produce naloxone-induced precipitated withdrawal or alter motor function. A possible mechanism of action of MMG22 is discussed in terms of inhibition of spinal NMDAR via antagonism of its co-receptor, mGluR5, and concomitant activation of neuronal MOR. We suggest that MMG22 may be a powerful alternative to traditional opioids for managing cancer pain.
1. Introduction
Pain is among the most common symptoms in cancer patients and is estimated to affect 90% of patients with end-stage cancer (Marcus, 2011). Of the millions of patients diagnosed with cancer, approximately 58% suffer from intolerable pain, which increases to 85% of the population as the disease becomes terminal (Marcus, 2011).
Pain is usually associated with emotional distress and decreased function, and negatively affects the patient’s quality of life (Abram, 1989; Marcus, 2011). Although opioids are the primary class of analgesics used to treat severe cancer pain, these drugs have adverse side effects that include nausea, sedation, constipation, tolerance, dependence, respiratory depression and overdose-related death that limit their use (Mantyh, 2006). Therefore, there is an urgent need to develop new and effective treatments for cancer pain that lack the serious side effects associated with opioids.
Earlier studies showed that co-administration of a mu opioid receptor (MOR) agonist and metabotropic glutamate receptor 5 (mGluR5) antagonist reduced morphine analgesic tolerance and dependence, and augmented its antinociceptive properties (Kozela et al., 2003; Sotgiu et al., 2003). The interaction between MOR and mGluR5, their expression in astrocytes and neurons, and evidence that MOR/mGluR5 heteromers exist in cultured cells (Brasseur, 1997; Schröder et al., 2009), led to the development of MMG22. MMG22 is a bivalent ligand consisting of an oxymorphone-derived MOR agonist and the mGluR5 antagonist, M-MPEP, tethered by a 22-atom spacer (Akgün et al., 2013). Significantly, intrathecal (i.t.) administration of MMG22 exhibited thousands of fold greater potency based on ED50 in murine models of LPS-induced inflammatory pain relative to naïve mice. The necessity of inflammation as a condition for efficacy also was observed in a murine model of cancer pain in which osteolytic fibrosarcoma cells were implanted into and around the calcaneus bone (Smeester et al., 2014).
Intrathecal administration of MMG22 afforded antinociception that was three orders of magnitude more effective than morphine, a gold standard for reducing tumor-evoked hyperalgesia (Wacnik et al., 2001). That MMG22 exhibited 38,000-times greater potency than a mixture of the individual monovalent ligands containing MOR agonist and mGluR5 antagonist pharmacophores supports the notion that MMG22 interacts with a MOR-mGluR5 heteromer (Akgün et al., 2013). The exceptional potency of MMG22 may be a result of optimal bridging of the two protomers of the putative MOR-mGluR5 heteromer.
In the present study, we show that systemic administration of MMG22 is highly effective at reducing cancer pain. The extraordinary potency of MMG22 and lack of side effects typically associated with opioids, suggests that MMG22 is an attractive alternative to morphine in managing cancer pain.
2. Methods
2.1. Subjects
Adult male C3H/HeNCr MTV mice (Charles River; 25–30 g) were used. Mice were housed four per cage, allowed free access to food and water, and maintained on a 12-h light/dark schedule. All protocols and procedures were approved by the University of Minnesota Institutional Animal Care and Use Committee and were conducted according to the guidelines established by the International Association for the Study of Pain (Zimmermann, 1983).
2.2. Cancer cell implantation
NCTC clone 2472 fibrosarcoma cells (American Type Culture Collection, Manassas, VA, USA) were maintained as described previously (Khasabova et al., 2007). This clone was derived from a connective tissue tumor in a C3H mouse, thus the fibrosarcoma cells are syngeneic with C3H/He mice (Wacnik et al., 2001). Mice were anesthetized with isoflurane (2%) and fibrosarcoma cells (2 × 105 cells in 10 μL PBS, pH 7.3) were injected into and around the calcaneus bone of the animal’s left hind paw using a 29-g needle. This approach produces a tumor with bone osteolysis (Wacnik et al., 2001).
2.3. Drug preparation and administration
All bivalent ligands were synthesized as described previously (Akgün et al., 2013). Compounds and Morphine sulfate (Mallinckrodt Inc., Hazelwood, MO) were all dissolved in 1.0% DMSO. Homologs of MMG22 with spacer lengths of 10 and 21 atoms were compared. Fig. 1 shows the structure of MMG22.
Fig. 1.
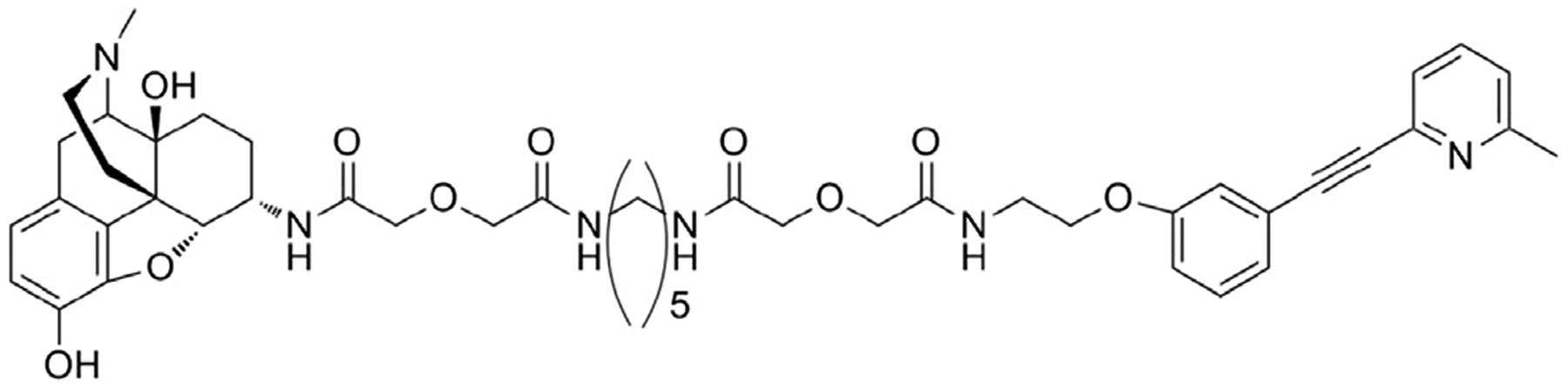
Chemical structure of MMG22.
2.4. Behavioral studies of mechanical hyperalgesia
Mice were placed on an elevated wire mesh platform, covered individually with glass containers, and allowed to habituate to their surroundings for 30 min before testing. A calibrated von Frey monofilament (3.9 mN) was used to measure mechanical sensitivity of the hind paw and was applied to the plantar surface of each hind paw ten times. Mechanical hyperalgesia is defined as a significant increase in the paw withdrawal frequency (PWF), which is calculated as the (number of withdrawal responses/total stimuli) X 100% for each paw (Smeester et al., 2014).
Mice were tested for three consecutive days before cancer cell implantation to confirm the absence of hyperalgesia and to acclimate to the testing procedure. PWFs were determined on post-implantation day (PID) 3–17 during which time mechanical hyperalgesia is maximal. After baseline testing, mice were divided randomly into groups of 6–8 mice per group. On the test day, animals received MMG22 administered s.c., i.m., or p.o. at escalating doses until the percent maximum possible effect (%MPE) was achieved. The %MPE was calculated from paw withdrawal frequencies using the following formula.
These values were adjusted to 100 and 0, respectively in order to address only the antihyperalgesic drug effects (Harris and Pierson, 1964; Hamamoto et al., 2008). A separate group of tumor-bearing mice received morphine (5 mg/kg, s.c.) for comparison. PWFs were determined before and every 30 min for 4 h following drug administration. The experimenter conducting the behavioral experiments was blinded to the treatment for all experiments, and at least two drug groups were tested in each session. Data were analyzed using multivariate analysis of variance (MANOVA) to compare differences in the PWF between groups and post-hoc comparisons were made using Bonferroni t-tests. ED50 values with 95% confidence intervals (C.I.) were obtained using nonlinear regression in GraphPad Prism v4.
2.5. Conditioned place preference
The conditioned place preference (CPP) test (Lax et al., 2014) was used to determine whether MMG22 was rewarding in naïve mice. The CPP apparatus consists of two chambers with walls containing either horizontal or vertical black lines (visual stimuli). Naïve mice (n = 8/group) were given access to the entire compartment for 30 min/day for 3 days before conditioning in order to acclimate to the testing environment. Baseline measures were taken one day before conditioning, where animals received six counter-balanced conditioning trials (3 drug and 3 vehicle). Conditioning consisted of mice receiving vehicle and MMG22 once per day (morning and afternoon) respectively for three consecutive days where they were restricted to one side (vertical vs. horizontal black lines on the walls) for 30 min and paired with drug or vehicle. On the test day, mice were given access to both compartments and the time spent in each compartment was determined for 30 min. The mean amount of time spent in each compartment (drug-paired vs. vehicle-paired) was compared between drug and vehicle using one-way ANOVA.
2.6. Naloxone-induced jumping response
Naloxone-induced jumping (Alder et al., 2002; Elkadi & Sharif, 1994) was used to determine whether MMG22 produces physical dependence. Testing of tumor-bearing mice began on PID17, and the mean PWF was determined. Mice were given three s.c. injections of morphine (n = 5) or MMG22 (n = 6) per day 4 h apart. Morphine doses were escalated on day 1 to day 4 (5, 10 and 20 mg/kg). On the fifth day, mice received a single injection of 20 mg/kg. Similarly, MMG22 was given in escalating doses of 0.1, 0.2, and 0.4 mg/kg for the first four days, and mice received 0.4 mg/kg on the fifth day.
At 3 h after the final injection of morphine or MMG22, all mice received a single bolus of naloxone (50 mg/kg, s.c.). Mice were placed in a Plexiglas observation cylinder and the number of jumps counted for 10 min by two independent investigators. Differences in the mean number of jumps were analyzed by student’s t-test.
2.7. Effects of MMG22 on motor coordination
The rotarod test was used to determine whether MMG22 alters motor function. Naïve C3H mice (n = 8/group) were acclimated for 3 days before drug administration. The treadmill was gradually accelerated from 3.75 to 5 rpm, with a maximum cutoff time of 300 s. On the test day, mice received one s.c. injection of 1 mg/kg MMG22, or the 5 mg/kg clonidine which served as a positive control (Stone et al., 2014; Yoon et al., 2015). Testing was done 60 min after drug administration and the time when the mouse fell off the treadmill was recorded. Mean times spent on the treadmill were compared between groups using two-way ANOVA with post-hoc Bonferroni analysis (p < 0.05) considered significant.
3. Results
3.1. MMG22 dose-dependently reduces tumor-evoked hyperalgesia
Systemic administration of MMG22 potently reduced tumor-evoked mechanical hyperalgesia as defined by MPE% derived from the formula above (Fig. 2). Subcutaneous, intramuscular, and oral administration each reduced hyperalgesia dose dependently. Depending on the dose, the antihyperalgesia peaked at 30–60 min after administration and persisted for at least 4 h. Hyperalgesia returned to baseline levels by 24 h. Interestingly, s.c. administration not only produced potent anti-hyperalgesia, but also became more potent with continued tumor growth as evidenced by a profound leftward shift in the dose response curve with increasing PID. This is best illustrated by the ED50 values at various times after tumor cell implantation (Table 1). For example, at PID3, the ED50 (confidence interval) for MMG22 was 1.16 (0.15–8.90) mg/kg, and this decreased to 0.00096 (0.0003–0.003) mg/kg at PID17. At this time, the ED50 for morphine was 2.37 mg/kg (CI = 1.93–2.9). The ED50 values for MMG22 given i.m. and p.o. decreased over time, but not as dramatically as for s.c. administration (Table 1). The antihyperalgesic effect of morphine given at a dose of 2.5 mg/kg s.c. was tested at different PIDs (Fig. 3). Although it reduced hyperalgesia, it did not show potentiation over time (1-way ANOVA with repeated measures, F(2, 11) = 0.206, P = 0.82). Compared to this dose of morphine, which consistently reduced hyperalgesia by approximately 50% at all PIDs (estimated ED50), the efficacy of the ED50 dose for MMG22 was approximately 250 times more potent at PID 17 (see Fig. 2).
Fig. 2.
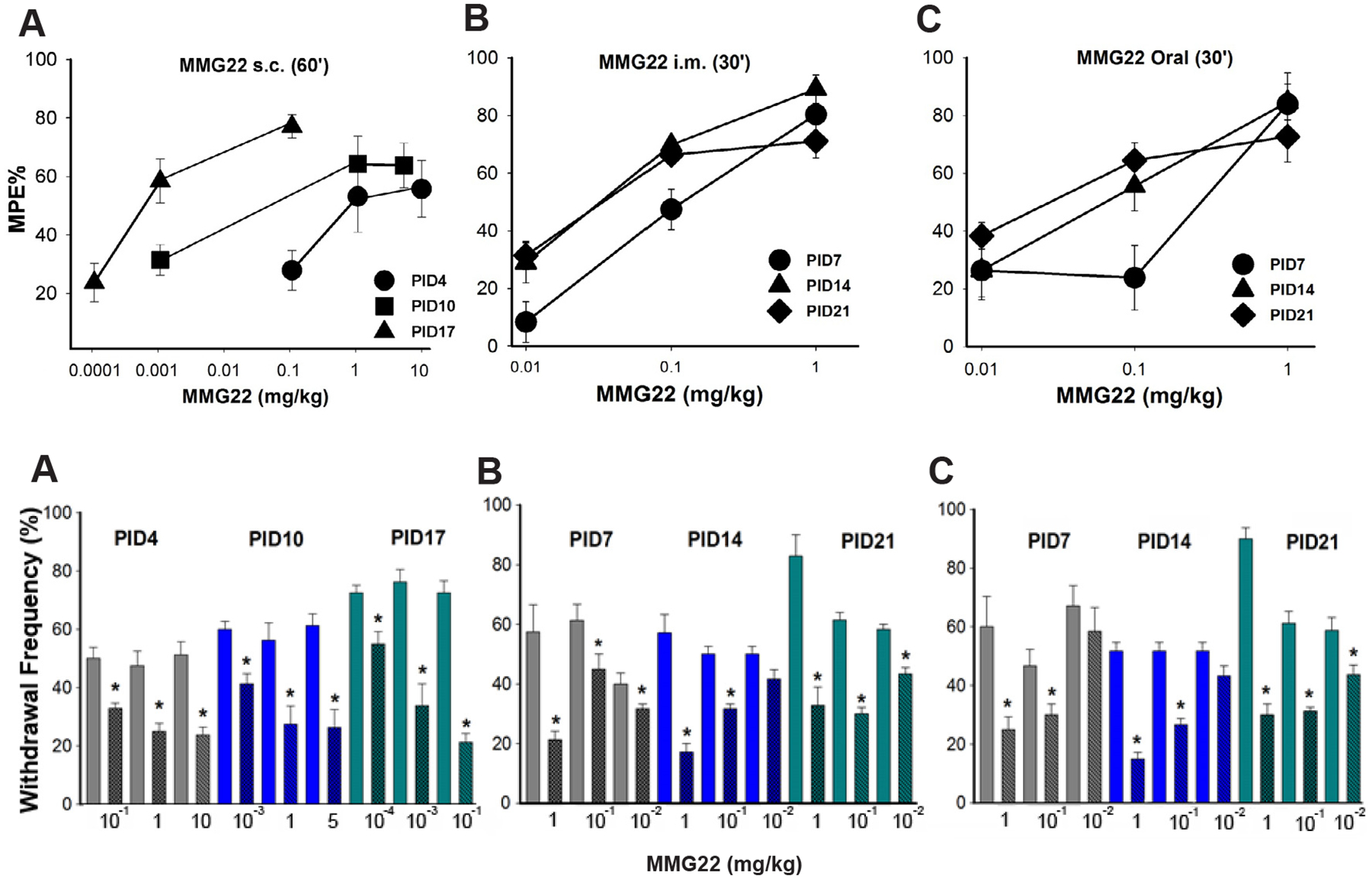
Dose-response curves illustrating the reduction in tumor-evoked mechanical hyperalgesia following systemic administration of MMG22 (s.c., i.m., and oral) on different post-implantation days (PID). Data in upper panels show mean (± SEM) % maximum possible effect. A) Antinociception s.c. administration of MMG22 increased from PID4–17. The increase in potency occurred at all subsequent time points tested. B) Reduction in mechanical hyperalgesia on different PIDs following i.m. administration of MMG22. C) Reduction in mechanical hyperalgesia on different PIDs following oral administration of MMG22. The X-axis scale in A differs from those in B and C because of the greater number of doses used. Lower panels show mean paw withdrawal frequencies that correspond to the MPE% in the panels above.
Table 1.
ED50 for MMG22 (mg/kg ± CI) when given by different routes of administration on different post-implantation days.
| Route | ED50 (mg/kg ± CI) | |||||
|---|---|---|---|---|---|---|
| PID3 | PID7 | PID10 | PID14 | PID17 | PID21 | |
| s.c. | 1.16 (0.15–8.9) | – | 0.15 (0.01–1.47) | 0.00096 (0.0003–0.003) | – | |
| oral | – | 0.025 (0.007–0.1) | – | 0.03 (0.021–0.056) | – | 0.04 (0.01–0.13) |
| i.m | – | 0.16 (0.05–0.45) | – | 0.06 (0.03–0.15) | – | 0.03 (0.01–0.1) |
Fig. 3.
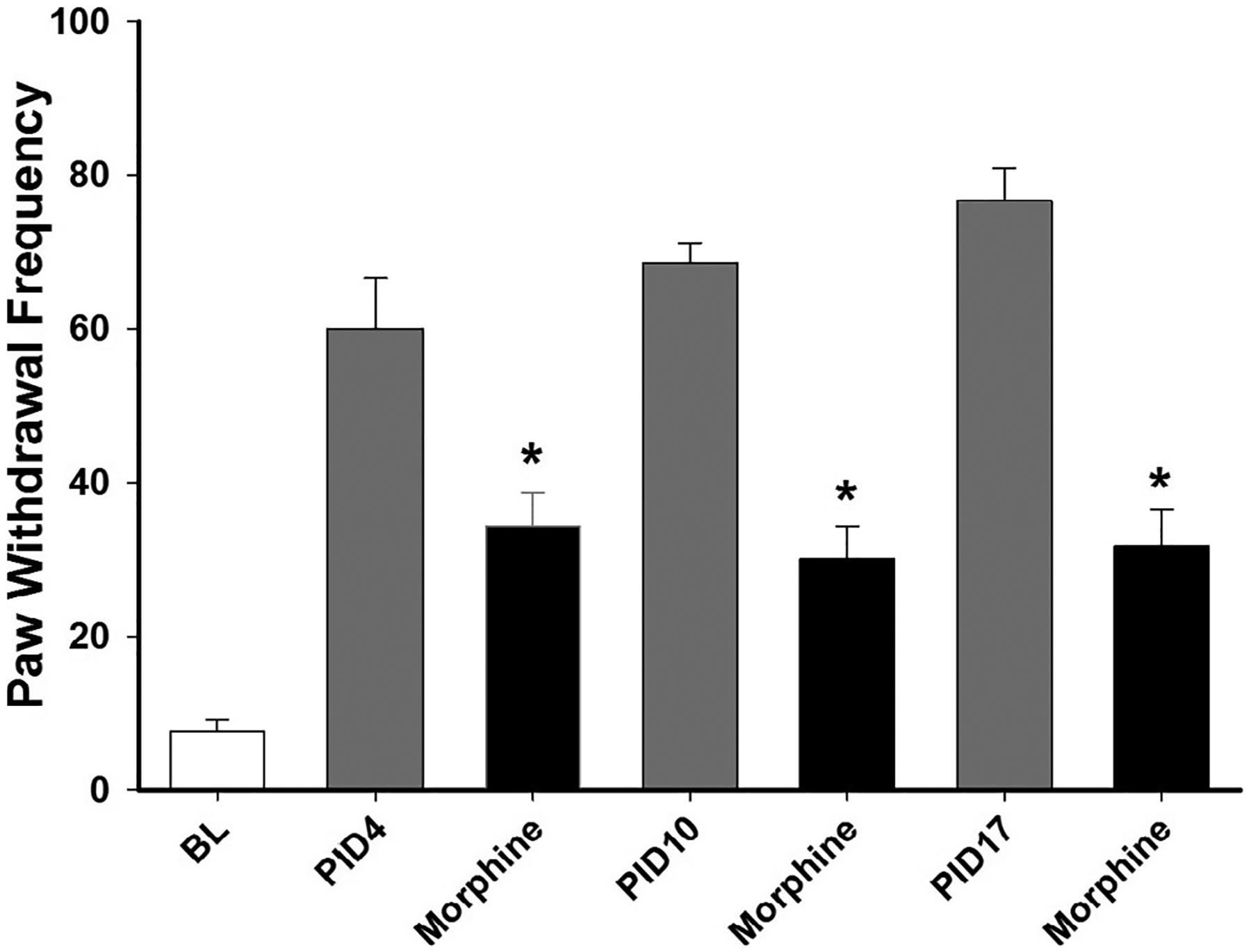
Antihyperalgesia produced by 2.5 mg/kg morphine (s.c.) at different PIDs. PWF was determined before (BL) cancer cell implantation and at 60 min after morphine. The white bar shows the mean paw withdrawal frequency before implantation of cancer cells. The grey and black bars show bars show mean paw withdrawal frequencies before and after morphine, respectively, on PIDs 4, 10 and 17. * indicates a significant difference from pre-drug values (Student-Newman Keuls test, p = 0.001.
To determine if shorter (MMG10, MMG21) spacer lengths had effects similar to that of MMG22, tumor-bearing mice (PID4) were injected with each and were tested for acute tolerance the following day. The short spacer MMG10 resulted in acute tolerance for all routes of administration, s.c. (1-way ANOVA with repeated measures, F(1,10) = 6.98, p < 0.05), o.p (1-way ANOVA with repeated measures, F (1, 10) = 6.52, p < 0.05) and i.m. (1-way ANOVA with repeated measures, F(1, 8) = 8.09, p < 0.05).
3.2. MMG22 did not produce acute or chronic tolerance
To determine whether MMG22 produced acute tolerance, the antihyperalgesic effects of MMG22 were determined for two consecutive days beginning on PID4. As shown in (Fig. 4), MMG22 (10 mg/kg, s.c., n = 8) reduced mechanical hyperalgesia similarly on each day. The decrease in paw withdrawal frequency at 120 min after injection did not differ between the two days (1-way ANOVA with repeated measures, F(4, 7) = 61.2, p < 0.001). Mice were then given a second injection of MMG22 (10 mg/kg, s.c.) 24hrs (PID5) after the first injection and produced a similar antihyperalgesic effect MMG22 peaked at 60–120 min after injection for PID5. Hyperalgesia returned to baseline values at 4 h after injection on both days, indicating that the time course of antihyperalgesia produced by MMG22 was not altered following the second administration. These data show that MMG22 produces long-lasting anti-hyperalgesia without acute tolerance.
Fig. 4.
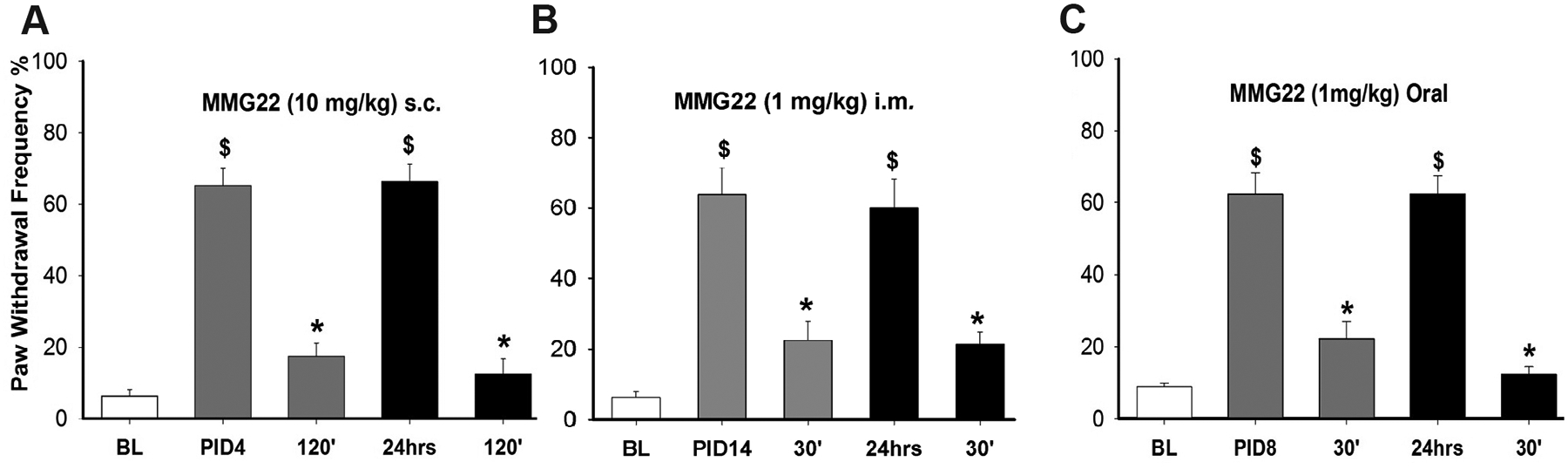
MMG22 did not produce acute tolerance to its analgesic effects when administered s.c. (A), i.m. (B). or orally (C). Anti-hyperalgesia peaked at 60–120 min after MMG22 and was just as effective when administered 24 h after the first administration. $ indicates a significant difference from baseline. * indicates a significant difference from pre-drug values. All comparisons were made using Bonferroni t-tests, p < 0.001).
Similarly, acute tolerance for 1 mg/kg of MMG22 (ED90) was measured when the drug was administered i.m. and p.o. No acute tolerance to the analgesic effect was detected for i.m. administration (1-way ANOVA with repeated measures, F(1, 7) = 0.046, p = 0.836) or p.o. administration (1-way ANOVA with repeated measures, F(1, 8) = 4.00, p = 0.081).
Tolerance usually develops with repeated administration of morphine resulting in the need for higher doses. We therefore examined whether tolerance occurred after recurrent administration of MMG22 and compared it with morphine. We compared the antihyperalgesia produced by repeated administration of the ED80 dose for MMG22(0.1 mg/kg) with that produced by repeated administration of the ED80 dose for morphine (5 mg/kg) in separate groups of C3H mice. Beginning on PID10, mice were given twice daily s.c. injections of either MMG22 or morphine for nine consecutive days. Withdrawal response frequencies were determined before and at 60 min after injection of MMG22 or morphine on day 1, 3, 6, and 9 of treatment. Tolerance was not observed after MMG22 which produced maximal antihyperalgesia over the 9-day time course (1-way ANOVA with repeated measures, F (3, 21) = 0.605, P = 0.62) (Fig. 5). However, morphine demonstrated tolerance as early as day 6 (1-way ANOVA with repeated measures, F(3,21) = 37.658, p < 0.001), and did not produce any antihyperalgesia by day 9 of treatment (Bonferroni t-test, t = 9.453, p= < 0.001).
Fig. 5.
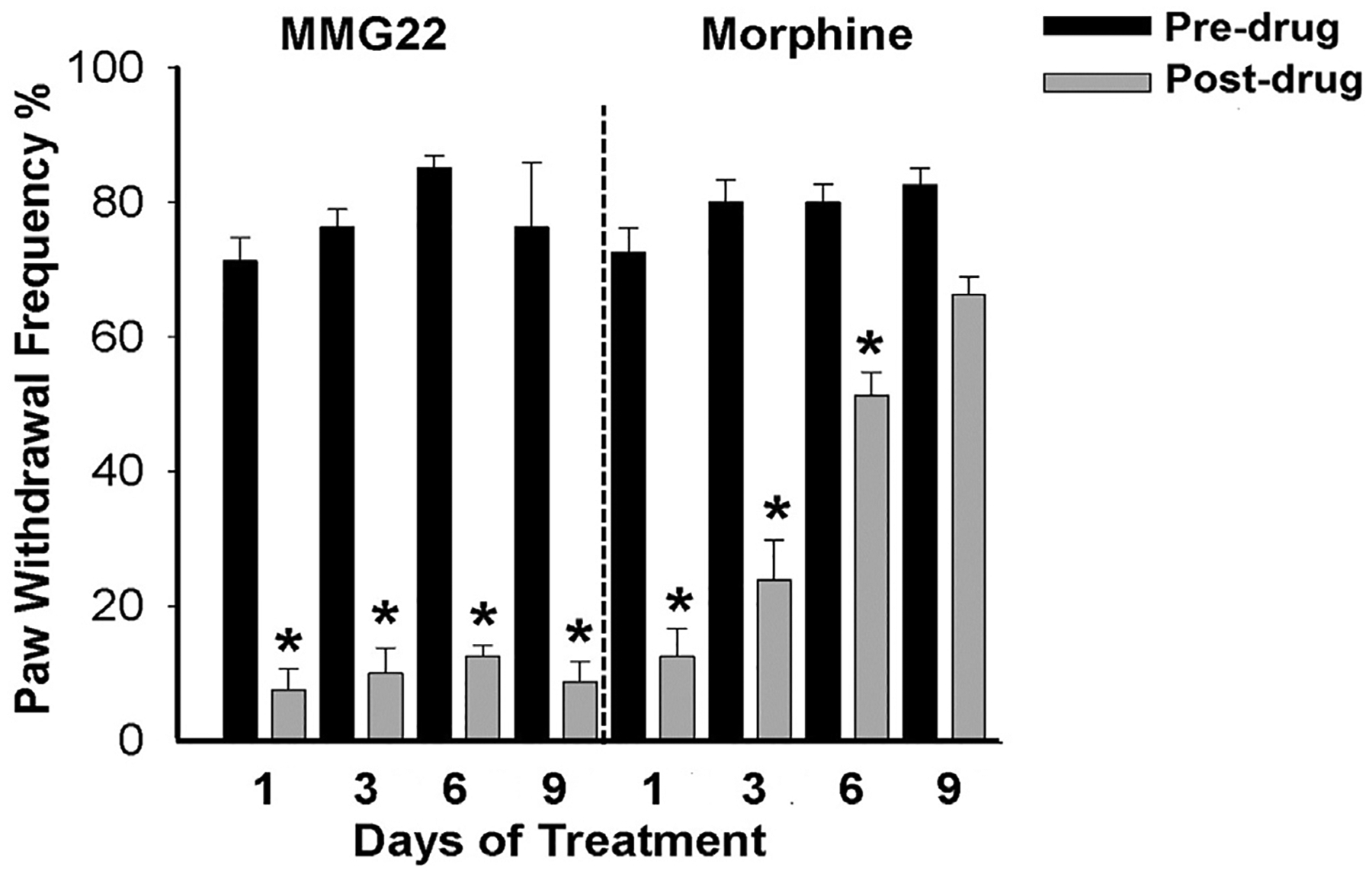
MMG22 does not produce tolerance to its analgesic effect following prolonged administration. Tumor-bearing mice were treated twice daily with s.c. injections of MMG22 (0.1 mg/kg; n = 8) (left panel) or morphine (5 mg/kg; n = 8) (right panel) for nine consecutive days, and paw withdrawal frequency was determined before and at 60 min after the injection on days 1, 3, 6, and 9. No analgesic tolerance occurred in mice given MMG22. Black bars are paw withdrawal frequencies before injection and grey bars are withdrawal frequencies after injection. * indicates a significant difference from pre-injection values (Student-Newman Keuls test, p < 0.001.
3.3. MMG22 did not produce conditioned place preference
Conditioned place preference test was used to determine whether MMG22 is rewarding in naïve mice. Vehicle or MMG22 (10 mg/kg, s.c.) did not increase the amount of time mice spent on the drug-paired side of the chamber (1-Way ANOVA, F(3, 35) = 1.58, p = 0.21). The mean amount of time (min) spent in the vehicle-paired chamber was14.9 ± 1.4 before conditioning and 12.9 ± 1.2 min. Similarly, the amount of time spent in the MMG22-paired chamber was 15.0 ± 1.4 before conditioning and 17.0 ± 1.2 min.
3.4. MMG22 did not alter motor function
The rotarod test was used to assess whether MMG22 produced sedation and/or motor deficits. Rotarod testing revealed differences between the groups (MMG22 vs. clonidine) in the amount of time mice remained on the treadmill. Whereas mice remained on the treadmill for less time following clonidine (5 mg/kg) as compared to baseline values, MMG22 (1 mg/kg) did not alter the amount of time spent on the treadmill (2-way ANOVA with repeated measures F(1, 31) = 9.74, p = 0.004). These data indicate that MMG22 did not cause sedation or impair motor function (Fig. 6).
Fig. 6.
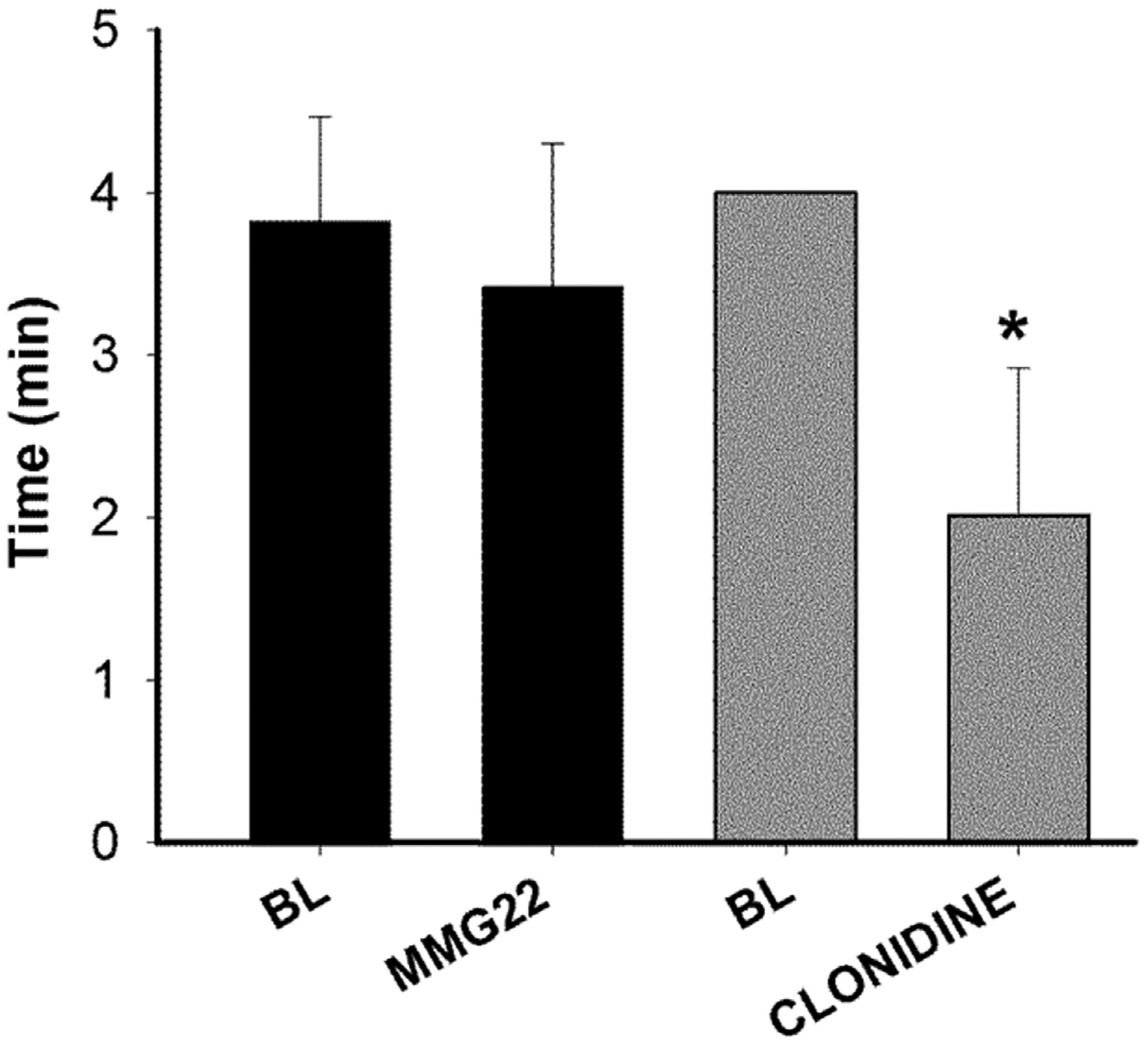
MMG22 did not produce sedation or motor impairment. Naïve mice were given either MMG22 (1 mg/kg, s.c.) or Clonidine (5 mg/kg, s.c.) and then place on a treadmill before and at 1 h after injection. Data show the amount of time mice spent on the treadmill. MMG22 did not alter the amount of time spent on the treadmill whereas this was reduced following clonidine. * Indicates the difference between time spent on the treadmill before and after clonidine (Bonferroni t-test, p < 0.001).
3.5. Naloxone did not precipitate withdrawal following MMG22
We determined whether naloxone produced precipitated withdrawal by determining naloxone-induced jumping in mice treated with morphine or MMG22 for four consecutive days. Naloxone was given 3 h after the final dose of MMG22 or morphine and produced jumping responses in mice treated with morphine (Mean ± SEM = 32.2 ± 10.1 jumps), but not in those that received MMG22 (Mean = 0.0; t = 45.0, p = 0.004).
4. Discussion
The design of the bivalent ligand MMG22 was based on studies showing that opioid receptors can form heteromers with multiple types and classes of GPCRs (Costantino et al., 2012), and on evidence indicating that MOR and mGluR5 interact functionally (Schröder et al., 2009). Receptor dimerization can alter receptor function, ligand pharmacology, signal transduction, and cellular trafficking (Hiller et al., 2013). Importantly, the formation of heteromers may be modulated in pathological states (Gomes et al., 2013). Thus, MMG22 consisting of pharmacophores derived from oxymorphone (MOR agonist) and MMPEP (mGluR5 antagonist) and linked through a 22-atom spacer was developed in an effort to target putative MOR-mGluR5 heteromers (Akgün et al., 2013). The presence of MOR-mGluR5 heteromers has been suggested in cultured cells (Schröder et al., 2009), where phosphorylation, internalization, and desensitization of MOR is reduced following mGluR5 antagonism (Schröder et al., 2009). Importantly, pain and opioid administration have both been shown to be associated with increased mGluR5 expression in the spinal cord dorsal horn (Abbadie et al., 2001; Coggeshall and Carlton, 1997; Jia et al., 1999; Pitcher et al., 2007). In this regard, mGluR5 upregulation is thought to contribute to the development of analgesic tolerance of opioids (Huang et al., 2005; Osikowicz et al., 2008; Valerio et al., 1997; Zhou et al., 2013a).
However, the increased expression of mGluR5 in the inflammatory state alone does not explain the ultra-high efficacy of MMG22 in reducing hyperalgesia, given that a mixture of the monovalents of oxymorphone and MPEP did not enhance antinociception (Akgün et al., 2013). Another possibility is induction of heteromer formation by a bivalent ligand, (Portoghese et al., 2017; Zheng et al., 2009). Regardless of how heteromers are formed, a spacer of specific length (22-atom) that links the pharmamacophores plays a crucial role in the enhancement of antinociceptive potency of MMG22. Homologues with shorter or longer spacers than 22 atoms were reported to have 3 orders of magnitude lower potency with respect to antinociception in LPS inflamed mice as compared to the 22-atom spacer (Akgün et al., 2013), suggesting interaction with a heteromer.
In prior studies (Smeester et al., 2014) the efficacy of MMG22 administered i.t. in mice with fibrosarcoma increased with respect to tumor growth and was 572-times greater on PID21 relative to PID3 (ED50: 5.7 to 0.01 fmol/mouse). Moreover, MMG22 was 23,000 times more potent than morphine on PID10 and 3.6 million times more potent on PID21 (Smeester et al., 2014). Furthermore, i.t. administration of MMG22 did not cause tolerance or respiratory depression (Akgün et al., 2013). The present study extends these findings and reveals that systemic administration (subcutaneous, intramuscular, and oral) is highly efficacious in producing progressive, potent antihyperalgesia without tolerance in tumor-bearing mice.
Although the precise mechanisms by which MMG22 reduces hyperalgesia is not clear, recent studies (Akgün et al., 2019) suggest that spinal astrocytes are one of the likely targets of i.t. MMG22, given that the specific astrocyte toxin, L-α aminoadipipic acid (LAA), selectively reduced antinociception of MMG22 in inflamed mice. Both MOR and mGluR5 are found on the peripheral and central terminals of primary afferent nociceptors (Abbadie et al., 2001; Bhave et al., 2001; Jia et al., 1999; Pitcher et al., 2007), post-synaptically on neurons in the superficial dorsal horn, and on astrocytes (Abbadie et al., 2001; Coggeshall and Carlton, 1997; Huang et al., 2005; Jia et al., 1999; Pitcher et al., 2007; Valerio et al., 1997; Vidnyánszky et al., 1994). It has been reported that antagonism of cancer-mediated pain associated with upregulated mGluR5 is decreased by the administration of a selective mGluR5 antagonist (Ren et al., 2012). Moreover, such treatment (Osikowicz et al., 2008; Picker et al., 2011; Sevostianova and Danysz, 2006) decreases place preference and morphine self-administration (Brown et al., 2012).
In considering the reported linkage of mGluR5 to the NR2 subunit of the N-methyl-D-aspartate receptor (NMDAR) (Perroy et al., 2008), it was determined whether MMG22 selectively inhibits this ionotropic receptor via antagonism of the mGluR5 co-receptor. Significantly, pre-treatment of mice with the specific NMDAR ion channel blocker, MK801, reduced the antinociception of MMG22 by 2700-fold, suggesting that the antinociception produced by MMG22 is due, at least in part, to NMDAR inhibition via MMG22 antagonism of mGluR5 (Akgün et al., 2019). This is consistent with the necessity for inflammation-induced activation of the NMDAR in order for MMG22 to be effective, as MMG22 was not effective in naïve mice. Opioid receptor involvement in MMG22 antinociception was established by irreversible blockage of antinociception using the selective MOR irreversible antagonist, β-FNA (Akgün et al., 2019).
The antinociception following systemic administration of MMG22 was not associated with sedation or motor impairment when compared to clonidine as a positive control. Unlike morphine, MMG22 did not produce tolerance or naloxone-precipitated withdrawal, nor did it exhibit rewarding properties as suggested by the lack of conditioned place preference. Interestingly, MMG22 given to mice without bone cancer at a dose that potently reduced hyperalgesia, did not produce analgesic place preference. In addition, high doses of MMG22 given systemically did not cause respiratory depression (unpublished observations). Consistent with earlier studies using i.t. administration of MMG22 (Smeester et al., 2014), we found that longer and shorter spacer lengths (data not shown) were less potent than MMG22 and caused acute tolerance to its antihyperalgesic effect, further demonstrating the importance of the optimal 22-atom spacer length.
In summary, the present study shows that systemic administration of MMG22 potently reduces cancer pain without adverse effects. Moreover subcutaneous, intramuscular, and oral routes of administration are substantially more potent and effective compared to that of morphine. The use of bivalent ligands offer a novel and effective approach to treat pain and these data suggest that MMG22 may be advantageous for long-term clinical usage. Given the effectiveness and ED50 dose ranges for MPE suggest that despite its relatively high molecular weight, systemic bioavailability of MMG22 does not appear to be a problem.
HIGHLIGHTS.
Systemic administration OF MMG22 targets a MOR-mGluR5 heteromer and potently reduces bone cancer pain in mice.
MMG22 does not possess adverse effects associated with traditional opioids.
MMG22 activates MOR and antagonizes mGluR5.
Acknowledgements
This work was supported by NIH grants HL135895 (DAS), DA030316 (PSP), and 2T32 DA007234-31 (SJE and RS). We thank Mr. Malcolm Johns for assistance in culturing the cancer cells and Dr. Iryna Khasabova for statistical advice.
Footnotes
Conflicts of interest
The authors have no conflicts of interest to disclose.
References
- Abbadie C, Pasternak GW, Aicher SA, 2001. Presynaptic localization of the carboxy-terminus epitopes of the μ opioid receptor splice variants MOR-1C and MOR-1D in the superficial laminae of the rat spinal cord. Neuroscience 106, 833–842. [DOI] [PubMed] [Google Scholar]
- Abram SE, 1989. Perceived dangers from intraspinal steroid injections. Arch. Neurol 46, 719–721. [DOI] [PubMed] [Google Scholar]
- Akgün E, Javed MI, Lunzer MM, Smeester BA, Beitz AJ, Portoghese PS, 2013. Ligands that interact with putative MOR-mGluR5 heteromer in mice with inflammatory pain produce potent antinociception. Proc. Natl. Acad. Sci. Unit. States Am 110, 11595–11599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akgün E, Lunzer MM, Portoghese PS, 2019. Combined glia inhibition and opioid receptor agonism afford highly potent analgesics without tolerance. ACS Chem. Neurosci 10, 2004–2011. [DOI] [PubMed] [Google Scholar]
- Bhave G, Karim F, Carlton SM, Gereau Iv RW, 2001. Peripheral group I metabotropic glutamate receptors modulate nociception in mice. Nat. Neurosci 4, 417–423. [DOI] [PubMed] [Google Scholar]
- Brasseur L, 1997. [Review of current pharmacologic treatment of pain]. Drugs 53(Suppl. 2), 10–17. [DOI] [PubMed] [Google Scholar]
- Brown RM, Mustafa S, Ayoub MA, Dodd PR, Pfleger KDG, Lawrence AJ, 2012. mGlu5 receptor functional interactions and addiction. Front. Pharmacol 7 (3), 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coggeshall R, Carlton S, 1997. Receptor localization in the mammalian dorsal horn and primary afferent neurons. Brain Res. Rev 24, 28–66. [DOI] [PubMed] [Google Scholar]
- Costantino CM, Gomes I, Stockton SD, Lim MP, Devi LA, 2012. Opioid receptor heteromers in analgesia. Expert Rev. Mol. Med 10, 14 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes I, Fujita W, Chandrakala MV, Devi LA, 2013. Disease-specific heteromerization of G-protein-coupled receptors that target drugs of abuse In: Progress in Molecular Biology and Translational Science. Elsevier, pp. 207–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamamoto DT, Khasabov SG, Cain DM, Simone DA, 2008. Tumor -evoked sensitization of C nociceptors: a role for endothelin. J. Neurophysiol 100, 2300–2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris LS, Pierson AK, 1964. Some narcotic antagonists in the benzomorphan series. J.Pharmacol. Exp. Ther 143, 141–148. [PubMed] [Google Scholar]
- Hiller C, Kühhorn J, Gmeiner P, 2013. Class A G-protein-coupled receptor (GPCR) dimers and bivalent ligands. J. Med. Chem 56, 6542–6559. [DOI] [PubMed] [Google Scholar]
- Huang H-Y, Cheng J-K, Shih Y-H, Chen P-H, Wang C-L, Tsaur M-L, 2005. Expression of A-type K+channel α subunits Kv4.2 and Kv4.3 in rat spinal lamina II excitatory interneurons and colocalization with pain-modulating molecules. Eur. J. Neurosci 22, 1149–1157. [DOI] [PubMed] [Google Scholar]
- Jia H, Rustioni A, Valtschanoff JG, 1999. Metabotropic glutamate receptors in superficial laminae of the rat dorsal horn. J. Comp. Neurol 410, 627–642. [PubMed] [Google Scholar]
- Khasabova IA, Stucky CL, Harding-Rose C, Eikmeier L, Beitz AJ, Coicou LG, Hanson AE, Simone DA, Seybold VS, 2007. Chemical interactions between fibrosarcoma cancer cells and sensory neurons contribute to cancer pain. J. Neurosci 27, 10289–10298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozela E, Pilc A, Popik P, 2003. Inhibitory effects of MPEP, an mGluR5 antagonist, and memantine, an N-methyl-D-aspartate receptor antagonist, on morphine antinociceptive tolerance in mice. Psychopharmacology 165, 245–251. [DOI] [PubMed] [Google Scholar]
- Lax NC, George DC, Ignatz C, Kolber BJ, 2014. The mGluR5 antagonist fenobam induces analgesic conditioned place preference in mice with spared nerve injury. PLoS One 9 e103524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantyh PW, 2006. Cancer pain and its impact on diagnosis, survival and quality of life. Nat. Rev. Neurosci 7, 797–809. [DOI] [PubMed] [Google Scholar]
- Marcus DA, 2011. Epidemiology of cancer pain. Curr. Pain Headache Rep 15, 231–234. [DOI] [PubMed] [Google Scholar]
- Osikowicz M, Mika J, Makuch W, Przewlocka B, 2008. Glutamate receptor ligands attenuate allodynia and hyperalgesia and potentiate morphine effects in a mouse model of neuropathic pain. Pain 139, 117–126. [DOI] [PubMed] [Google Scholar]
- Perroy J, Raynaud F, Homburger V, Rousset M-C, Telley L, Bockaert J, Fagni L, 2008. Direct interaction enables cross-talk between ionotropic and group I metabotropic glutamate receptors. J. Biol. Chem 283, 6799–6805. [DOI] [PubMed] [Google Scholar]
- Picker MJ, Daugherty D, Henry FE, Miller LL, Dykstra LA, 2011. Metabotropic glutamate antagonists alone and in combination with morphine. Behav. Pharmacol 22, 785–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitcher MH, Ribeiro-Da-Silva A, Coderre TJ, 2007. Effects of inflammation on the ultrastructural localization of spinal cord dorsal horn group I metabotropic glutamate receptors. J. Comp. Neurol 505, 412–423. [DOI] [PubMed] [Google Scholar]
- Portoghese PS, Akgun E, Lunzer MM, 2017. Heteromer induction: an approach to unique pharmacology? ACS Chem. Neurosci 8, 426–428. [DOI] [PubMed] [Google Scholar]
- Ren B. x., Gu X. p., Zheng Y. g., Liu C. l., Wang D, Sun Y. e., Ma Z. l., 2012. Intrathecal injection of metabotropic glutamate receptor subtype 3 and 5 agonist/antagonist attenuates bone cancer pain by inhibition of spinal astrocyte activation in a mouse model. Anesthesiology 116, 122–132. [DOI] [PubMed] [Google Scholar]
- Schröder H, Wu DF, Seifert A, Rankovic M, Schulz S, Höllt V, Koch T, 2009. Allosteric modulation of metabotropic glutamate receptor 5 affects phosphorylation, internalization, and desensitization of the μ-opioid receptor. Neuropharmacology 56, 768–778. [DOI] [PubMed] [Google Scholar]
- Sevostianova N, Danysz W, 2006. Analgesic effects of mGlu1 and mGlu5 receptor antagonists in the rat formalin test. Neuropharmacology 51, 623–630. [DOI] [PubMed] [Google Scholar]
- Smeester BA, Lunzer MM, Akgün E, Beitz AJ, Portoghese PS, 2014. Targeting putative mu opioid/metabotropic glutamate receptor-5 heteromers produces potent antinociception in a chronic murine bone cancer model. Eur. J. Pharmacol 743, 48–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotgiu ML, Bellomi P, Biella GEM, 2003. The mGluR5 selective antagonist 6- methyl-2-(phenylethynyl)-pyridine reduces the spinal neuron pain-related activity in mononeuropathic rats. Neurosci. Lett 342, 85–88. [DOI] [PubMed] [Google Scholar]
- Stone LS, German JP, Kitto KF, Fairbanks CA, Wilcox GL, 2014. Morphine and clonidine combination therapy improves therapeutic window in mice: synergy in antinociceptive but not in sedative or cardiovascular effects. 9. PLoS One 9 (10) e109903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valerio A, Rizzonelli P, Paterlini M, Moretto G, Knöpfel T, Kuhn R, Memo M, Spano P, 1997. mGluR5 metabotropic glutamate receptor distribution in rat and human spinal cord: a developmental study. Neurosci. Res 28, 49–57. [DOI] [PubMed] [Google Scholar]
- Vidnyánszky Z, Hamori J, Négyessy L, Rüegg D, Knopfel T, Kuhn R, Görcs TJ, 1994. Cellular, and subcellular localization of the mGluR5a metabotropic glutamate receptor in rat spinal cord. Neuroreport 6, 209–213. [DOI] [PubMed] [Google Scholar]
- Wacnik PW, Eikmeier LJ, Ruggles TR, Ramnaraine ML, Walcheck BK, Beitz AJ, Wilcox GL, 2001. Functional interactions between tumor and peripheral nerve: Morphology, algogen identification, and behavioral characterization of a new murine model of cancer pain. J. Neurosci 21, 9355–9366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon SY, Kang SY, Kim HW, Kim HC, Roh DH, 2015. Clonidine reduces nociceptive responses in mouse orofacial formalin model: potentiation by sigma-1 receptor antagonist BD1047 without impaired motor coordination. Biol. Pharm. Bull 38, 1320–1327. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Akgün E, Harikumar KG, Hopson J, Powers MD, Lunzer MM, Miller LJ, Portoghese PS, 2009. Induced association of μ opioid (MOP) and type 2 cholecystokinin (CCK2) receptors by novel bivalent ligands. J. Med. Chem 52, 247–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Wang J, Zhang X, Zeng L, Wang L, Jiang W, 2013. Effect of metabotropic glutamate 5 receptor antagonists on morphine efficacy and tolerance in rats with neuropathic pain. Eur. J. Pharmacol 718, 17–23. [DOI] [PubMed] [Google Scholar]
- Zimmermann M, 1983. Ethical guidelines for investigations of experimental pain in conscious animals. Pain 16, 109–110. [DOI] [PubMed] [Google Scholar]